
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChronicles of plutonium peroxides: spectroscopic characterization of a new peroxo compound of Pu(IV)†
Julien
Margate
a,
Simon
Bayle
a,
Thomas
Dumas
 b,
Elodie
Dalodière
a,
Christelle
Tamain
b,
Denis
Menut
c,
Paul
Estevenon
b,
Philippe
Moisy
b,
Sergey I.
Nikitenko
a and
Matthieu
Virot
*a
b,
Elodie
Dalodière
a,
Christelle
Tamain
b,
Denis
Menut
c,
Paul
Estevenon
b,
Philippe
Moisy
b,
Sergey I.
Nikitenko
a and
Matthieu
Virot
*a
aICSM, Univ Montpellier, CEA, CNRS, ENSCM, Marcoule, France. E-mail: matthieu.virot@cea.fr
bCEA, DES, ISEC, DMRC, Univ. Montpellier, Marcoule, France
cSynchrotron SOLEIL, L’Orme des Merisiers, Saint-Aubin, France
First published on 2nd May 2024
Abstract
Although hydrogen peroxide (H2O2) has been highly used in nuclear chemistry for more than 75 years, the preparation and literature description of tetravalent actinide peroxides remain surprisingly scarce. A new insight is given in this topic through the synthesis and thorough structural characterization of a new peroxo compound of Pu(IV).
Peroxo-based hexavalent actinide (An) compounds have been extensively studied with the synthesis of uranyl (and neptunyl to a less extent) cage clusters and mineral compounds.1–8 By contrast, the literature description of tetravalent An peroxides is surprisingly lacking. To date, their structural characterisation is only limited to one plutonium9,10 and two thorium11,12 compounds. Nevertheless, interactions between H2O2 and An(IV), specifically Pu, have been extensively applied since the Manhattan Project for adjusting the oxidation states, purifying the solutions, and precipitating peroxide precursors dedicated to the preparation of actinide oxides and metallic plutonium.13
Stoichiometric addition of H2O2 to acidic solutions of Pu(IV) results in the formation of the so-called “brown peroxo complex”, which transitions to the “red peroxo complex” when the amount of H2O2 increases.13–18 The main absorption bands for these species are located at ca. 494 and 658 nm for the former, and ca. 506 and 540 nm for the latter (Fig. 1).13 Both complexes have been postulated to be dimers ([Pu2(O2)(OH)]5+ and [Pu2(O2)2]4+ as the respectively proposed structures) that experimentally decompose in acidic solutions to yield Pu(III).14,15,17 Further addition of H2O2 to Pu(IV) solutions leads to the formation of precipitates that have been used as precursors for the preparation of PuO2 or for waste management processes.18–20 To the best of our knowledge, structural characterisations of these compounds have never been reported in the literature. More generally, the structures and relationships occurring between Pu(IV) peroxides in solution and in the solid precipitates remain unclear to date. The only resolved structure available in the literature deals with a dimeric peroxo-carbonato compound of 10-fold coordinated Pu(IV) exhibiting two bridging μ2:η2-O2 ligands and six bidentate carbonato ligands.9,10
 | ||
| Fig. 1 Vis-NIR abs. spectra acquired in solution for the (A) brown (494, 541 and 658 nm), (B) red (506 and 540 nm) and (C) green (455, 654–660 and 821 nm) peroxo species of 5 mM Pu(IV) in nitric media. Inset: Photo of the respective solutions. | ||
In this work, the stepwise addition at room temperature of aqueous solutions of H2O2to Pu(IV) solutions previously stabilized in 2 M HNO3 resulted in the successive formation of both the brown and red complexes ([Pu] = 5 mM, Fig. 1). The corresponding UV-Vis spectra were in good agreement with those previously reported in the literature (ESI†). Surprisingly, the reverse approach, which involved diluting an aliquot of an acidic Pu(IV) solution in a large volume of an aqueous solution of concentrated H2O2, generated a green, limpid and very stable solution devoid of any precipitate ([H2O2]/[Pu] > 10, pH = 1–2). The corresponding electronic spectrum strongly differed not only from the spectra of the reported Pu(IV) peroxo complexes, but also from the well-known colloidal or hexanuclear cluster structures of Pu(IV) that could have been expected in such conditions as a result of hydrolysis and condensation reactions (Fig. S1, ESI†).16,21–24 The principal features for this spectrum consist of two sharp bands attributed to f–f transitions of Pu(IV) and located at 455 and 821 nm, and a larger one standing at 654–660 nm (Fig. 1). The absence of absorption bands around 602 and 830 nm, respectively related to Pu(III) and Pu(VI), implies the absence of Pu(IV) disproportionation in the studied conditions.22,25,26 Most probably, the band located at 455 nm is a hypsochromic shift of the Pu(IV) band generally located at 476 nm in dilute aqueous solutions of HNO3 (Fig. S1, ESI†).
For a better understanding about the structure of this new green Pu(IV) compound, the species was precipitated at higher concentration ([H2O2]/[Pu] > 10, Pu > 10 mM, [HNO3] > 50 mM) before drying under Ar flow. Diffuse Reflectance Spectra (DRS) acquired for the powdered solid were consistent with the optical spectrum observed in the solution (Fig. 2). The good correlation between the electronic transitions observed in both the liquid and solid systems evidenced the similarity in the Pu(IV) coordination spheres for both species. This observation has led us to question the exact nature of the species in solution. Complementary filtration and ultrafiltration experiments carried out on a 1 mM Pu solution suggested the absence of a water-soluble complex, but rather the presence of a solid species with a size larger than 100 kDa (approx. 3–4 nm, Fig. S2, ESI†) that could aggregate into bigger structures (<100 nm) when increasing the concentration.
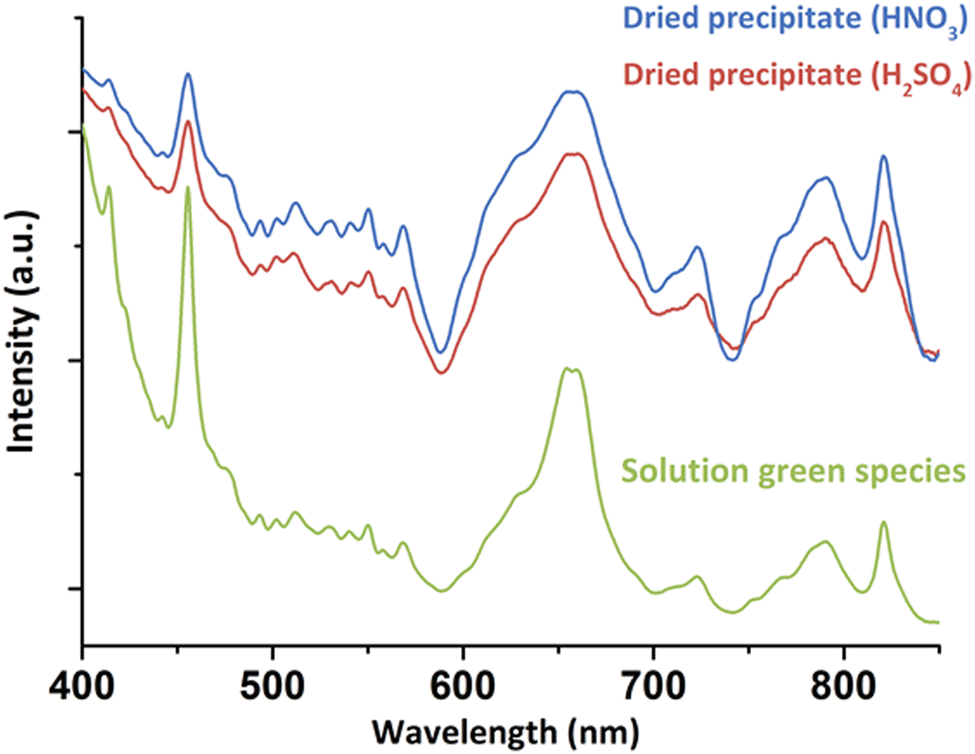 | ||
| Fig. 2 DRS acquired on the solids precipitated from nitric or sulfuric Pu(IV) solutions in comparison to the green species observed in solution (nitric system). | ||
Raman spectroscopy carried out on a drop of a suspended solid in solution evidenced the characteristic signature of an excess of free H2O2 at 877 cm−1 and free nitrates at 1049 cm−1 arising from the Pu mother solution (Fig. 3 and Fig. S3, ESI†).11,27–31 Three additional bands located at 423, 550 and 835 cm−1 were investigated using mixtures of 17O-marked water and H2O2 solutions. The use of the H217O/H217O2 mixture involved the red shift of all of the vibration bands, except for the nitrate one (red spectrum, Fig. 3). By contrast, a H217O/H216O2 mixture only induced the shift of a unique band initially located at 550 cm−1 (green spectrum, Fig. 3). This approach allowed assignment of the vibrational band located at 835 cm−1 (811 cm−1 with 17O) to the ν(O–O) symmetric stretching vibration mode of the peroxide ligand that shifts in comparison to pure H2O2 because of Pu atom(s) coordination (ESI†).
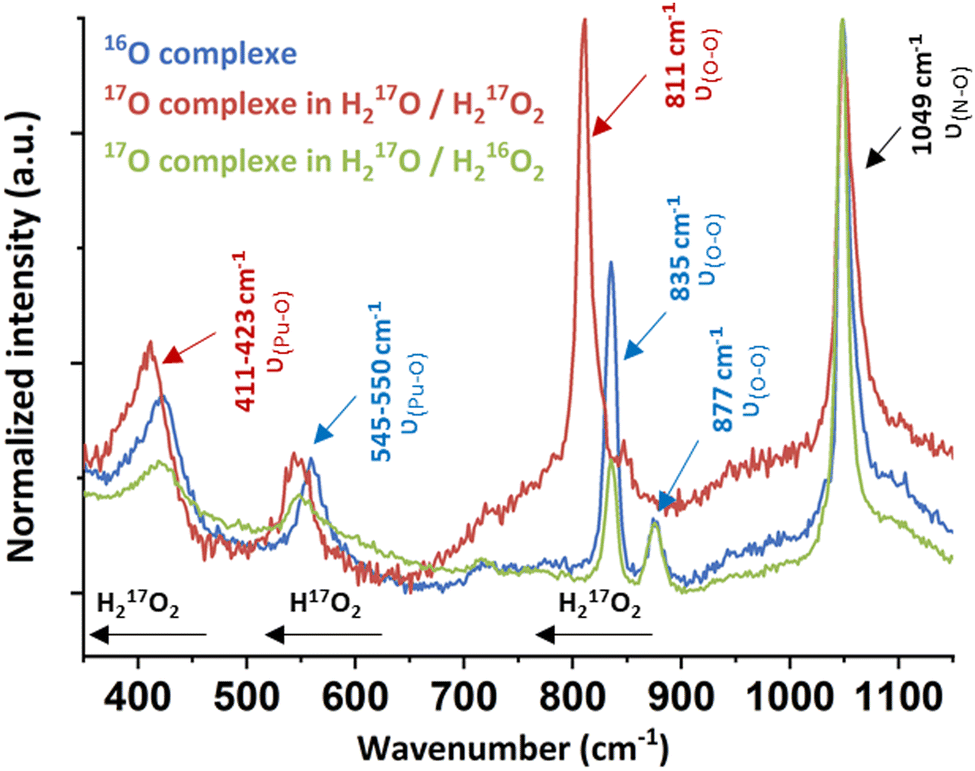 | ||
| Fig. 3 Raman spectra of the green peroxo compound of Pu(IV) (nitric system) prepared in H216O/H216O2 (blue spectrum), H217O/H217O2 (red spectrum) and H217O/H216O2 (green spectrum). | ||
The Raman band located at 423 cm−1 (411 cm−1 with 17O) was attributed to the ν(Pu–O) vibrational mode of the peroxide group. A similar signature has been observed for other An or Ce peroxo species exhibiting bridging μ2-η2:η2 or μ3-η2:η2:η2 coordination modes.10,11,27 The band located at 550 cm−1 could not be clearly identified using this approach, but it is not related to the peroxide group(s) due to the significant shift of the band when using a H217O/H216O2 mixture. Most probably, this band relates to oxo or hydroxo group(s) coordinating Pu atoms. Synthesizing the green Pu compound in a D2O/H2O mixture did not help in the functional group(s) identification. Nevertheless, we note that the T2g band for An dioxides is located around this area (465 cm−1 for ThO2, 445 cm−1 for UO2 or 478 cm−1 for PuO2).32 The T2g band represents the antisymmetric stretching of the Pu–μ4O bonds in the cubic structure and is highly affected by the bond strength and local environment, suggesting a possible assignment of this band to oxo group(s).33
Interestingly, the synthesis of the green peroxo compound at pH = 2.3 with a Pu(IV) solution previously stabilized in sulphuric medium did not lead to a stable solution, but quickly precipitated. DRS and Raman spectroscopies showed similar band positions with the nitrate system for both solid and concentrated liquid states (Fig. 2 and Fig. S3, ESI†). One exception is the vibrational band located at 994 cm−1 in Raman spectroscopy and attributed to ν1(SO4) of free sulphate ions.34 These observations suggested similar structures for both nitric and sulfuric systems, but also a possible absence of participation of the counter-ion in the coordination sphere of Pu in the solution or in the precipitate. Fourier Transform Infra-Red (FT-IR) spectroscopy on the solids obtained in both nitric and sulfuric media showed the characteristic signatures of nitrates,29,35 sulphates34,36 and interstitial and/or coordinated water molecules (Fig. S4, ESI†).37,38 The presence of the peroxo group(s) was confirmed with the weak bands located at 832–833 cm−1 for both media.11,39 The observation of peroxo bands for both vibrational spectroscopies (assigned to the ν(O–O) symmetric stretching vibration mode) agrees with the bridging coordination modes already observed for other Zr(IV), Ce(IV), U(VI), Th(IV) and Pu(IV) peroxo compounds.7,9–11,40,41
Solids obtained in the presence of sulphate and nitrate ions provided similar X-ray diffraction patterns, indicating similar crystalline structures (Fig. 4(a)), finally tending to stipulate (considering Raman and UV-vis spectroscopies) that the counter ions do not enter the Pu coordination spheres. Thermogravimetric analyses carried out on both solids obtained from the nitric and sulphuric systems (Fig. S5, ESI†) evidenced three domains attributed to the release of H2O molecules (<125 °C), followed by the decomposition of O22− ligands (125–225 °C) and the decomposition of nitrates (>225 °C) or sulfates (>550 °C).11,13,38,42 Mass loss calculations confirmed a double amount of NO3− in comparison to SO42−, assuming that the Pu core of the green peroxo compound exhibits a positive charge of +2 (ESI†).
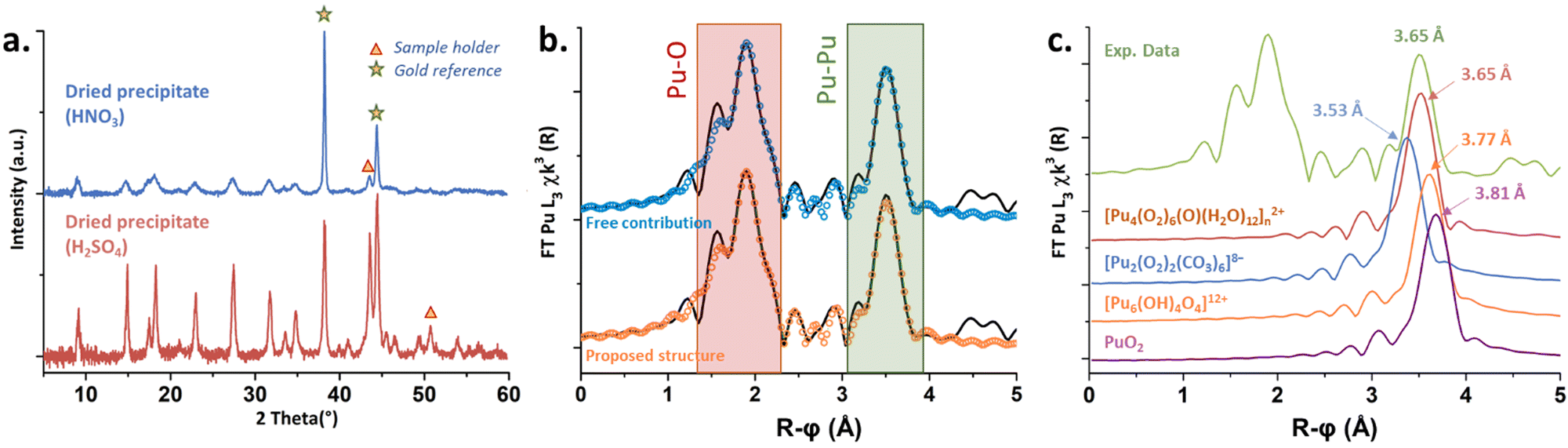 | ||
| Fig. 4 (a) XRD diffractograms for the green peroxo precipitated from Pu(IV) stabilized in HNO3 or H2SO4 media. (b) FT of the experimental k3-weighted EXAFS spectrum (black) measured on the green compound in solution (5 mM). Blue and orange circles stand for the fits. (c) Plot of the normalized Pu–Pu contributions observed on the FT EXAFS spectra for different Pu-based polynuclear structures in comparison to the experimental data (green) and the hypothetical structure (red). | ||
X-ray absorption near edge structure (XANES) obtained at the Pu L3-edge in solution for the green peroxo compound evidenced the predominance of the +IV oxidation state (Fig. S6, ESI†). The experimental k3-weighted extended X-ray absorption fine structure (EXAFS) spectrum obtained in the 3–16 Å−1 interval is provided in Fig. S7a (ESI†). The corresponding Fourier transform (FT) magnitude in black in Fig. 4(b) (uncorrected for phase shift) showed two large peaks standing at R − ϕ = 1.9 and 3.5 Å attributed to the Pu–O and Pu–Pu coordination spheres. The latter was adjusted at 3.65 Å (CN = 3.6(7)), indicating the formation of a polynuclear structure. Such a Pu–Pu distance does not correspond to any structure available in the literature (Fig. 4(c)), thus excluding peroxide dimers9 (3.53 Å), oxo-hydroxo hexamers24 (3.77 Å) or PuO2-like structures43 (3.81 Å). The absence of a secondary Pu–Pu distance combined with the data acquired using different techniques (charge +2, several Pu–O bonds, CNPu–Pu, bridging mode peroxides, no counter-ion in the structure) led us to propose a hypothetical structure inspired by Th, U and Pa tetrahedral arrangements.44,45 Consequently, three Pu–O sub-shells were adjusted using a short distance (ca. 2.2 Å) corresponding to a μ4-O group, an intermediate distance for the peroxide groups (ca. 2.3 Å agreeing with Runde et al.9) and a longer one (ca. 2.45 Å) for H2O molecules. The successful fit for this spectrum was achieved using free contributions (blue fit, Fig. 4(b)) with structural parameters at 2.14 Å (CN = 1.3(5)), 2.32 Å (CN = 6.5(6)) and 2.45 Å (CN = 3.5(7)) for the Pu–O sphere (ESI†). Hence, the resulting hypothetical unit pattern agrees with the formula [Pu4(O2)6(O)(H2O)12]n2+ provided in Fig. S8 (ESI†).
The latter is a unitary tetrameric pattern involving four Pu atoms located at the corner of a tetrahedron. The unit incorporates a μ4-O atom and is coordinated with six μ2-O2 ligands standing on each edge. The μ4-oxo position is reminiscent of the one observed in bulk PuO2. H2O molecules then stand around the Pu atoms to complete the coordination sphere, in agreement with previous Th and Pu peroxo structures (CN = 10).9,46 By using the atom coordinates (CrystalMaker Software Ltd), scattering paths were generated and structural parameters were calculated (ESI†). The as-obtained parameters and related FT (Fig. 4(b), orange fit and Fig. S7b, ESI†) were in very good agreement with the experimental EXAFS spectrum giving weight to the hypothetical structure. The most intriguing question deals with the Pu–O distance observed for the μ4-oxo group (2.13 Å), which is much smaller than that observed in bulk PuO2 (2.33 Å).43 The tetrahedral units reported for both Th(IV) and U(IV) compounds (ESI†) showed strongly split distances for the An–μ4O group incorporating at the same time shorter and longer distances when compared to the respective bulk oxides. In addition, such structures allow the possible observation of much shorter An–An distances in comparison to the one observed in the respective bulk oxides (3.74 Å and 3.52 Å for Th and U, against 3.97 Å and 3.87 Å for the respective oxides). These observations could agree and explain the nature of the shifted Raman band located at 550 cm−1 on Fig. 3, thus strengthening our proposition. Furthermore, it is worth noting that the mass losses calculations performed with the hypothetical and (+2) positively charged structure (Table S4, ESI†) well agreed with the experimental thermograms for both nitric and sulfuric systems with 2% or 0.4% error, respectively (can be improved with 10 H2O molecules instead of 12).
This newly-discovered green Pu(IV) compound fills the poor library available about tetravalent An peroxides. The distinct spectroscopic and structural characteristics of the associated polynuclear structure of Pu(IV) shed a new light on our overall knowledge on An peroxides and their potential applications in nuclear chemistry and materials preparation. Further investigations are imperative to unravel the precise structures and properties of the Pu(IV) peroxide species, including the brown and red ones. While these results offer insights into the first coordination sphere of Pu for the green peroxo compound, the long-range structure in the solution (cluster, polymer, colloid…) or in the precipitated solid remains undefined and deserves further investigation.
Conflicts of interest
There are no conflicts to declare.References
- P. C. Burns, K. A. Kubatko, G. Sigmon, B. J. Fryer, J. E. Gagnon, M. R. Antonio and L. Soderholm, Angew. Chem., Int. Ed., 2005, 44, 2135–2139 CrossRef CAS PubMed.
- B. Vlaisavljevich, L. Gagliardi and P. C. Burns, J. Am. Chem. Soc., 2010, 132, 14503–14508 CrossRef CAS PubMed.
- K. A. H. Kubatko, K. B. Helean, A. Navrotsky and P. C. Burns, Science, 2003, 302, 1191–1193 CrossRef.
- F. Clarens, J. De Pablo, I. Diez-Perez, I. Casas, J. Gimenez and M. Rovira, Environ. Sci. Technol., 2004, 38, 6656–6661 CrossRef CAS PubMed.
- C. R. Armstrong, M. Nyman, T. Shvareva, G. E. Sigmon, P. C. Burns and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1874–1877 CrossRef CAS PubMed.
- M. Nyman, M. A. Rodriguez and C. F. Campana, Inorg. Chem., 2010, 49, 7748–7755 CrossRef CAS PubMed.
- J. Qiu and P. C. Burns, Chem. Rev., 2013, 113, 1097–1120 CrossRef CAS.
- S. Hickam, D. Ray, J. E. S. Szymanowski, R. Y. Li, M. Dembowski, P. Smith, L. Gagliardi and P. C. Burns, Inorg. Chem., 2019, 58, 12264–12271 CrossRef CAS.
- W. Runde, L. F. Brodnax, G. S. Goff, S. M. Peper, F. L. Taw and B. L. Scott, Chem. Commun., 2007, 1728–1729 RSC.
- L. E. Sweet, J. F. Corbey, F. Gendron, J. Autschbach, B. K. McNamara, K. L. Ziegelgruber, L. M. Arrigo, S. M. Peper and J. M. Schwantes, Inorg. Chem., 2017, 56, 791–801 CrossRef CAS PubMed.
- L. Bonato, M. Virot, T. Dumas, A. Mesbah, P. Lecante, D. Prieur, X. Le Goff, C. Hennig, N. Dacheux, P. Moisy and S. I. Nikitenko, Chem. – Eur. J., 2019, 25, 9580–9585 CrossRef CAS PubMed.
- S. S. Galley, C. E. Van Alstine, L. Maron and T. E. Albrecht-Schmitt, Inorg. Chem., 2017, 56, 12692–12694 CrossRef CAS PubMed.
- J. A. Leary, A. N. Morgan and W. J. Maraman, Ind. Eng. Chem., 1959, 51, 27–31 CrossRef CAS.
- A. S. Mazumdar, P. R. Nataraja and S. Vaidyana, J. Inorg. Nucl. Chem., 1970, 32, 3363–3367 CrossRef.
- C. Maillard and J. M. Adnet, Radiochim. Acta, 2001, 89, 485–490 CrossRef CAS.
- R. E. Connick and W. H. Mcvey, J. Am. Chem. Soc., 1949, 71, 1534–1542 CrossRef CAS.
- A. Ekstrom and A. McLaren, J. Inorg. Nucl. Chem., 1972, 34, 1009–1016 CrossRef CAS.
- G. Daniel, Literature review of PuO2 calcination time and temperature data for specific surface area, United States, 2012.
- S. F. Marsh and T. D. Gallegos, Chemical treatment of plutonium with hydrogen peroxide before nitrate anion exchange processing. [Reduction to (IV)], Report LA-10907, 1987.
- P. G. Hagan and F. J. Miner, Plutonium peroxide precipitation: review and current research, American Chemical Society, United States, 1980 Search PubMed.
- T. Dumas, M. Virot, D. Menut, C. Tamain, C. Micheau, S. Dourdain and O. Diat, J. Synchrotron Radiat., 2022, 29, 30–36 CrossRef CAS PubMed.
- M. Cot-Auriol, M. Virot, T. Dumas, O. Diat, D. Menut, P. Moisy and S. I. Nikitenko, Chem. Commun., 2022, 58, 13147–13150 RSC.
- G. Chupin, C. Tamain, T. Dumas, P. L. Solari, P. Moisy and D. Guillaumont, Inorg. Chem., 2022, 61, 4806–4817 CrossRef CAS PubMed.
- C. Tamain, T. Dumas, D. Guillaumont, C. Hennig and P. Guilbaud, Eur. J. Inorg. Chem., 2016, 3536–3540 CrossRef CAS.
- D. Clark, S. Hecker, G. Jarvinen and M. Neu, in The Chemistry of the Actinide and Transactinide Elements, ed. L. Morss, N. Edelstein and J. Fuger, Springer, Netherlands, 2011, ch. 7, pp. 813–1264 DOI:10.1007/978-94-007-0211-0_7.
- E. Dalodière, M. Virot, T. Dumas, D. Guillaumont, M. C. Illy, C. Berthon, L. Guerin, A. Rossberg, L. Venault, P. Moisy and S. I. Nikitenko, Inorg. Chem. Front., 2018, 5, 100–111 RSC.
- B. T. McGrail, G. E. Sigmon, L. J. Jouffret, C. R. Andrews and P. C. Burns, Inorg. Chem., 2014, 53, 1562–1569 CrossRef CAS.
- A. A. Mikhaylov, A. G. Medvedev, A. V. Churakov, D. A. Grishanov, P. V. Prikhodchenko and O. Lev, Chem. – Eur. J., 2016, 22, 2980–2986 CrossRef CAS PubMed.
- U. Casellato, P. A. Vigato and M. Vidali, Coord. Chem. Rev., 1981, 36, 183–265 CrossRef CAS.
- S. Gajaraj, C. Fan, M. Lin and Z. Hu, Environ. Monit. Assess., 2013, 185, 5673–5681 CrossRef CAS PubMed.
- Y. Suffren, F.-G. Rollet and C. Reber, Comments Inorg. Chem., 2011, 32, 246–276 CrossRef CAS.
- G. M. Begun, R. G. Haire, W. R. Wilmarth and J. R. Peterson, J. Less-Common Met., 1990, 162, 129–133 CrossRef CAS.
- E. Epifano, M. Naji, D. Manara, A. C. Scheinost, C. Hennig, J. Lechelle, R. J. M. Konings, C. Guéneau, D. Prieur, T. Vitova, K. Dardenne, J. Rothe and P. M. Martin, Commun. Chem., 2019, 2, 59 CrossRef.
- K. Ben Mabrouk, T. H. Kauffmann, H. Aroui and M. D. Fontana, J. Raman Spectrosc., 2013, 44, 1603–1608 CrossRef CAS.
- M. Y. Mihaylov, V. R. Zdravkova, E. Z. Ivanova, H. A. Aleksandrov, P. S. Petkov, G. N. Vayssilov and K. I. Hadjiivanov, J. Catal., 2021, 394, 245–258 CrossRef CAS.
- D. E. Chasan and G. Norwitz, Infrared determination of inorganic sulfates and carbonates by the pellet technique, Report Test Report T69-10-1, 1969.
- C. R. Bhattacharjee, M. K. Chaudhuri, S. K. Chettri and J. J. Laiwan, J. Fluorine Chem., 1994, 66, 229–231 CrossRef CAS.
- M. Falk and T. A. Ford, Can. J. Chem., 1966, 44, 1699–1707 CrossRef CAS.
- J. A. Leary, U. S. A. E. Commission and L. A. S. Laboratory, Studies on the Preparation, Properties, and Composition of Plutonium Peroxide, Los Alamos Scientific Laboratory of the University of California, 1954.
- G. V. Jere and G. D. Gupta, J. Inorg. Nucl. Chem., 1970, 32, 537–542 CrossRef CAS.
- G. V. Jere and M. T. Santhamma, Inorg. Chim. Acta, 1977, 24, 57–61 CrossRef CAS.
- N. Hibert, B. Arab-Chapelet, M. Rivenet, L. Venault, C. Tamain and O. Tougait, Dalton Trans., 2022, 51, 12928–12942 RSC.
- M. Virot, T. Dumas, M. Cot-Auriol, P. Moisy and S. I. Nikitenko, Nanoscale Adv., 2022, 4, 4938–4971 RSC.
- K. E. Knope and L. Soderholm, Chem. Rev., 2013, 113, 944–994 CrossRef CAS PubMed.
- R. E. Wilson, S. De Sio and V. Vallet, Nat. Commun., 2018, 9, 622 CrossRef PubMed.
- L. Bonato, M. Virot, X. Le Goff, P. Moisy and S. I. Nikitenko, Ultrason. Sonochem., 2020, 69, 105235 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, methods, additional details and spectra. See DOI: https://doi.org/10.1039/d4cc01186d |
| This journal is © The Royal Society of Chemistry 2024 |