
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystallographic and spectroscopic studies on persistent triarylpropargyl cations†
Takuya
Shimajiri‡
 *ab,
Taiga
Tsue
a,
Shumpei
Koakutsu
a,
Yusuke
Ishigaki
a and
Takanori
Suzuki
*a
*ab,
Taiga
Tsue
a,
Shumpei
Koakutsu
a,
Yusuke
Ishigaki
a and
Takanori
Suzuki
*a
aDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Japan. E-mail: tak@sci.hokudai.ac.jp
bCreative Research Institution, Hokkaido University, Sapporo, Hokkaido 001-0021, Japan
First published on 30th May 2024
Abstract
By acid treatment of precursor alcohols, mesitylethynyl-substituted diarylmethyl cations were isolated as stable solids, X-ray structural analyses of which revealed a planar geometry. Furthermore, the ion pairs including these triarylpropargyl cations form charge-segregated assemblies in the crystal, and effective intermolecular interaction induces a red-shift of absorption in the crystal.
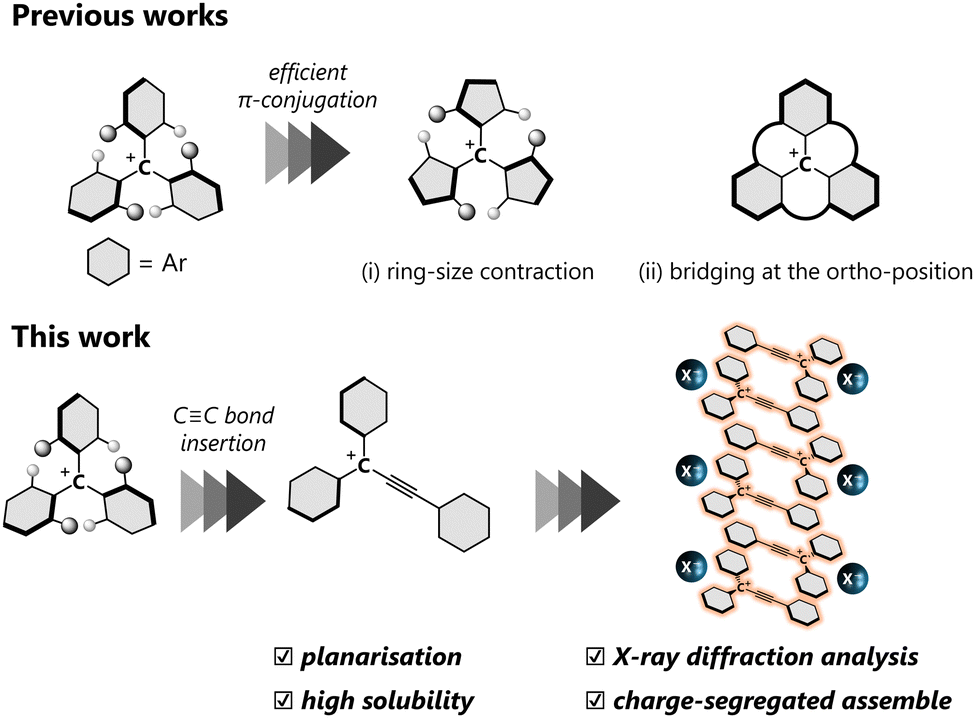
A carbocation is one of the most important chemical species in organic chemistry. In contrast to the intrinsic high reactivity of non-conjugated carbocations, a triarylmethyl cation with a skewed three-fold symmetry is a relatively stable classical carbocation since a positive charge can be delocalised over the entire molecule, which allows molecules to be obtained as stable entities. Thanks to the intense absorption in the visible region and electron-accepting ability, triarylmethyl cations have been used as organic dyes and oxidants. The triarylmethyl cation with a higher planar geometry, characterised by smaller dihedral θ values, facilitates effective π-conjugation, thereby endowing attractive properties such as near-IR absorption,1 emission2,3 and catalytic behaviour.4 Thus far, various approaches have been explored to achieve planar triarylmethyl cations, by (i) ring-size contraction5 and (ii) bridging at the ortho-position;6,7 the acquisition of perfect planarity is most effectively achieved in the latter approach. In general, the high planarity of the molecules also leads to more effective intermolecular interactions such as electrostatic and dispersion effects, resulting in unique properties in the solid state, such as red-shifted photoabsorption8 and charge transport.9
Incorporation of a –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– bond between the cationic centre and one of the aryl groups on the triarylmethyl cation offers another approach to achieve a planar geometry by reducing steric repulsion around the cationic centre, with the formation of a triarylpropargyl cation (Fig. 1). Reports on the triarylpropargyl cations are elusive due to the transient nature, stemming from the presence of an electron-withdrawing –CC– moiety attached to the cationic centre. While several attempts have been made to elucidate its electronic state,10–14 there are currently no examples of triarylpropargyl cations that are stable enough for investigation by X-ray crystallography.
C– bond between the cationic centre and one of the aryl groups on the triarylmethyl cation offers another approach to achieve a planar geometry by reducing steric repulsion around the cationic centre, with the formation of a triarylpropargyl cation (Fig. 1). Reports on the triarylpropargyl cations are elusive due to the transient nature, stemming from the presence of an electron-withdrawing –CC– moiety attached to the cationic centre. While several attempts have been made to elucidate its electronic state,10–14 there are currently no examples of triarylpropargyl cations that are stable enough for investigation by X-ray crystallography.
 | ||
| Fig. 1 Planarisation approaches in triarylpropargyl cations. | ||
Here, π-conjugated planar molecules are potential candidates for applications such as semi-conductors15 and ferroelectric materials.16 The arrangement of π-systems in their assemblies is crucial for controlling their electronic properties in the solid state. In ionic π-molecules, there are two classes of assembling modes, “charge-by-charge assemblies”, where anions and cations are stacked alternately, and “charge segregated assemblies”, where each charged species is stacked separately.17 Among them, the construction of charge-segregated assemblies, which would possess semi-conductive properties,18 is still challenging since the proximity of ionic molecules with the same charge leads to large electrostatic repulsion. Thus, guidelines for establishing charge-segregated assemblies are still elusive, and reliable building blocks have been sought.
In this work, we present a comprehensive investigation of triarylpropargyl cations with electronic stabilisation and steric protection, which were readily prepared by treatment of the corresponding triarylpropargyl alcohols with acid. Their structural parameters and electronic properties were examined by X-ray diffraction and IR and UV/Vis spectroscopy. In the crystal, the ion pair of the triarylpropargyl cation 2+ and common anions forms consistently charge-segregated assemblies for a series of small counter anions, and a subtle change in the packing arrangement causes a drastic change in solid-state absorption due to the different effectiveness of π–π interaction in a crystal.
Triarylpropargyl cations have hitherto been successfully prepared only by the introduction of strong electron-donating substituents. Molecular design that does not rely solely on strong electron-donating groups is required, which would not only allow for accurate elucidation of the electronic properties but also pave the way for further developments in the chemistry of propargyl cations. The major degradation processes of propargyl cations involve nucleophilic attack on the C3 atom corresponding to the cationic centre of the allenyl cation form, and subsequent tautomerisation into thermodynamically stable vinyl ketone derivatives.19,20 Thus, by sterically protecting the C3 position, we conceived that stable triarylpropargyl cations could be made without strong electron-donating groups, and newly designed triarylpropargyl cations 1+, 2+: mesitylethynyl-substituted bis(4-methoxyphenyl)methyl cation and xanthenyl cation.
Structural optimisation of 1+,2+ by DFT calculations at the B3LYP-D3/6-31G** level of theory predicted small θ values compared to the triarylmethyl cations 1+,2+ with similar substituents (θ1 = 31°, θ2 = 38° for R1a+; θ1 = 28°, θ2 = 56° for R1b+; θ1 = 2°, θ2 = 57° for R2a+; θ1 = 1°, θ2 = 78° for R2b+) (Fig. 2). Especially, the optimised structure of 2+ shows a completely planar shape. This planarisation enhances the efficiency of π-conjugation, as reflected by the coefficients of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) being located on the entire molecule (Fig. S25, ESI†). A natural population analysis revealed concentration of the positive charge at the C3 atom (0.133 for 1+; 0.150 for 2+) rather than the C1 atom (0.089 for 1+; 0.070 for 2+). Protection of the C3 position would warrant stability of the cation.
 | ||
| Fig. 2 X-ray structures of (A) 1+TfO− and (B) 2+TfO− at 150 K with thermal ellipsoids at 50% probability and (C) their structural parameters. | ||
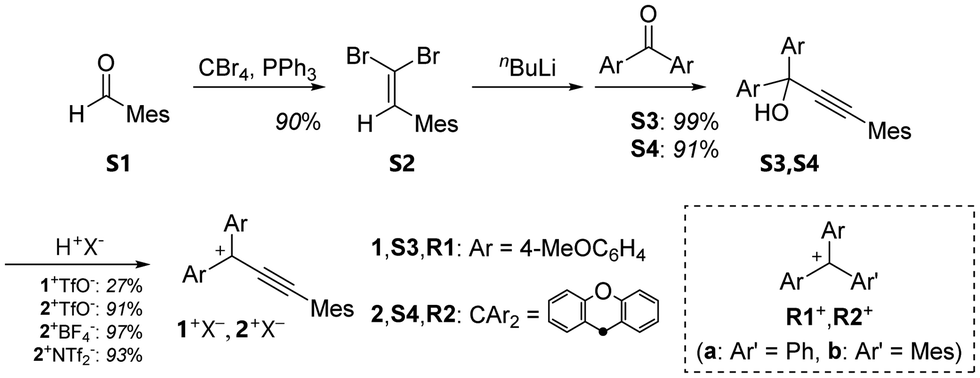
As shown in Scheme 1, the propargyl alcohols S3,S4 were prepared by the nucleophilic reaction of lithium acetylide, which was generated via a Corey–Fuchs reaction from benzaldehyde S1 and the corresponding benzophenones. The resulting propargyl alcohols S3,S4 were treated with CF3SO3H (TfOH) to give the desired triarylpropargyl cation salts 1+TfO−,2+TfO− as red solids. Although the addition of water causes degradation of the molecules, the salts of propargyl cations are thermally stable and can be handled in an ambient atmosphere. In particular, 1+TfO−,2+TfO− can be stored for more than two months in the solid state at −20 °C. Its non-acidic solution in an organic solvent remained intact for at least 2 days at room temperature without showing any degradation. The solids are readily soluble in common organic solvents such as CH2Cl2 and MeCN.
 | ||
| Scheme 1 Preparation of triarylpropargyl cations. | ||
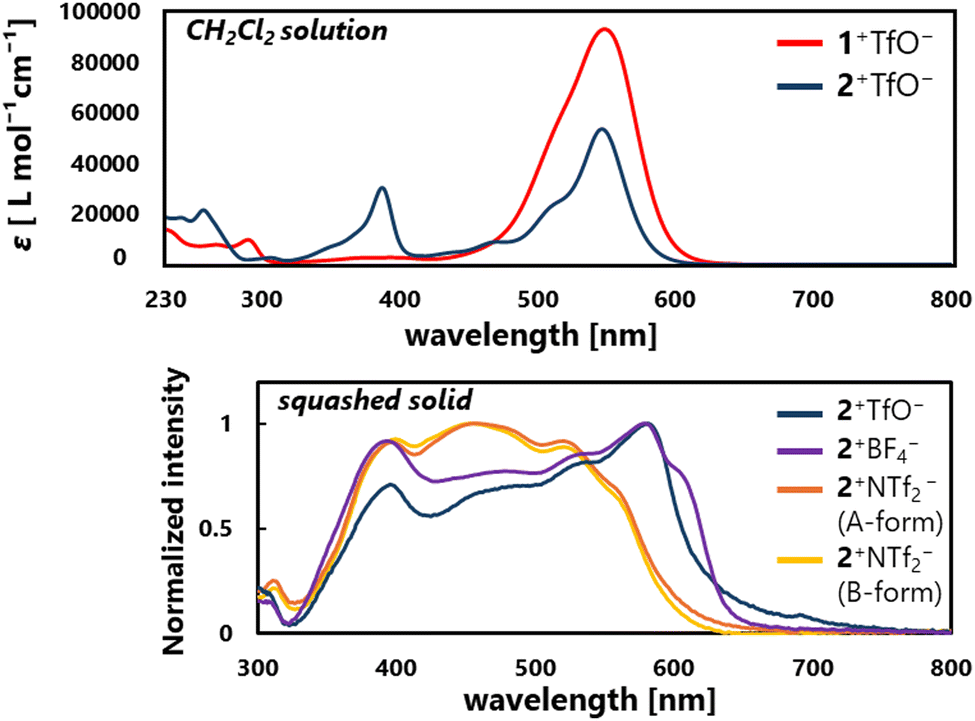
In the UV/Vis spectra of 1+TfO−,2+TfO− in CH2Cl2, the strong first absorption band which extends to 650 nm (1+TfO−: λmax = 543 nm, ε = 8.48 × 104; 2+TfO−: λmax = 547 nm, ε = 5.60 × 104) is observed, exhibiting a 50–100-nm red-shift compared to R1+,R2+ salts (R1a+BF4−: λmax = 497 nm, ε = 6.30 × 104; R2a+ClO4−: λmax = 453 nm, ε = 7.08 × 103; R2b+ClO4−: λmax= 452 nm, ε = 3.80 × 103).21,22 The intense first absorption is ensured by the high probability of the HOMO–LUMO transition since both are distributed due to the higher planarity.
In the IR spectra, the sharp peaks at 2115 cm−1 for 1+TfO− and 2131 cm−1 for 2+TfO− were assigned to the symmetric CC stretching vibration, which is in good agreement with the calculated values 2150 cm−1 and 2161 cm−1, respectively, simulated at the B3LYP-D3/6-31G** level with a scaling factor23 of 0.9648. These values fall within the standard range (2260–2100 cm−1) for the symmetric CC stretching vibration, but the relatively small values indicate the slight contribution of a canonical butatriene structure.
Finally, we determined the molecular structure in detail via X-ray analysis. Recrystallisation of triarylpropargyl cations 1+TfO−,2+TfO− afforded red single crystals suitable for X-ray diffraction measurements. In the case of 1+TfO−, two crystallographically independent molecules 1+ (mol.1 and mol.2) exist, each of which adopts a similar structure. The X-ray structures of triarylpropargyl cations 1+TfO−,2+TfO− exhibit a unique isosceles triangle-like structure as shown in Fig. 2A and B. The dihedral angles θ1 and θ2 for 1+TfO−,2+TfO− determined by X-ray analyses are small as predicted by calculations (Fig. 2C). Especially, 2+TfO− adopts a completely planar geometry. While the counter anion is positioned near the cation due to electrostatic stabilisation, formation of a strong ion pair with the C1 and C3 atoms was not observed. This molecular design based on the insertion of a –CC– bond into the triarylmethyl cation is useful for the construction of stable acceptor molecules with higher planarity.
Fig. 2C shows each bond length of 1+,2+ in 1+TfO−,2+TfO−. Compared to that of the reported triarylpropargyl alcohols, there is a slight change in bond lengths, with elongation of C2C3 bond and contraction of C3–C4 bond, thus crystallographically demonstrating the reduced bond alternation in triarylpropargyl cations. This is consistent with a low-wavenumber shift of the CC stretching vibration. These results revealed that, in alkynyl carbocations, the delocalised nature of the electronic structure is a more significant characteristic than the degree of bond-length alternation14 as determined experimentally via spectroscopic and crystallographic measurements.
Based on the above background, we shifted our focus to crystal structures of triarylpropargyl cations. In the crystal of 1+TfO− (including two crystallographically independent molecules, orthorhombic, Pna21, Z = 8), mol.1 and mol.2 dovetailed each other to form a ribbon-like structure (Fig. 3). The ribbons partially overlap with the shortest C⋯C contact of 3.44 Å and the counter anions are positioned between them, resulting in the intermediary packing of charge-by-charge and charge-segregated assemblies. On the other hand, in the crystal packing of 2+TfO− (monoclinic, Cc, Z = 4), 2+ forms brick layer stacking with the shortest C⋯C contact of 3.39 Å, as the completely planar shape of 2+ does not hinder the proximity of the cations. Counter anions are situated on both sides of the cationic layer, leading to charge-segregated assemblies.
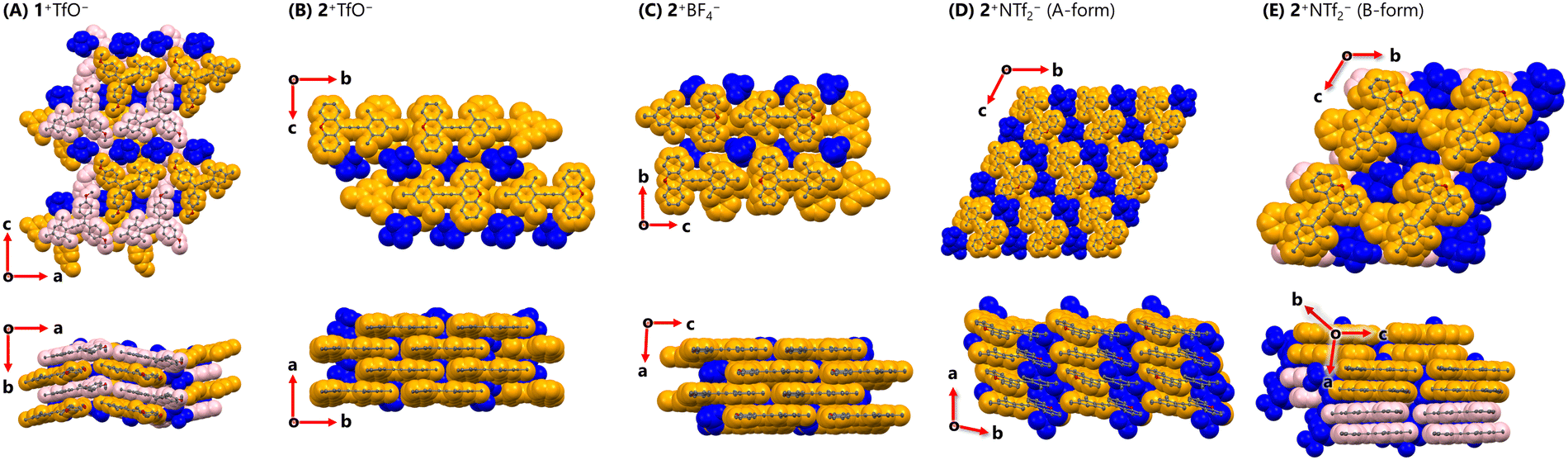 | ||
| Fig. 3 Crystal structures of (A) 1+TfO− (B) 2+TfO− (C) 2+BF4− (D) 2+NTf2− (A-form) and (E) 2+NTf2− (B-form) at 150 K. (orange: mol.1, pink: mol.2) | ||
Given that 2+TfO− forms charge-segregated assemblies in its single-crystal state, we explored salts of 2+ with different counter anions. Upon treatment of triarylpropargyl alcohol S4 with an alternative acid such as HBF4 or HNTf2, 2+BF4− (97% yield) and 2+NTf2− (93% yield) were obtained as red crystals.
In the crystal of 2+BF4− (monoclinic, P21/n, Z = 4), brick layer stacking with the shortest C⋯C contact of 3.40 Å is observed as in 2+TfO−. In single crystals of 2+NTf2−, two different polymorphs were observed. One contains only one crystallographically independent molecule (A-form, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2), while the other contains two crystallographically independent molecules (mol.1 and mol. 2) of 2+ (B-form, triclinic, P, Z = 4). In the A-form, 2+ exists in a slipped stacking manner with the shortest C⋯C contact of 3.47 Å. In the B-form, 2+ adopts a cofacial stacking mode with the shortest C⋯C contact of 3.48 Å, where neighbouring mol.1 and mol.2 face each other in a head-to-tail manner (shortest C⋯C contact: 3.41 Å), and the same molecules are oriented in a head-to-head manner (shortest C⋯C contact, mol.1⋯mol.1
, Z = 2), while the other contains two crystallographically independent molecules (mol.1 and mol. 2) of 2+ (B-form, triclinic, P, Z = 4). In the A-form, 2+ exists in a slipped stacking manner with the shortest C⋯C contact of 3.47 Å. In the B-form, 2+ adopts a cofacial stacking mode with the shortest C⋯C contact of 3.48 Å, where neighbouring mol.1 and mol.2 face each other in a head-to-tail manner (shortest C⋯C contact: 3.41 Å), and the same molecules are oriented in a head-to-head manner (shortest C⋯C contact, mol.1⋯mol.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.42 Å, mol.2⋯mol.2:3.48 Å). While the stacking modes of the A-form and B-form were different, they were both considered to give charge-segregated assemblies. Therefore, triarylpropargyl cations could consistently provide charge-segregated assemblies based on the ion pair despite the absence of strong intermolecular interactions such as metal⋯metal interactions.24 In this case, charge-segregated assemblies consisting of ion pairs including the triarylpropargyl cation, the molecular geometry, π–π interaction and dispersion force based on planar π-molecules with C2v symmetry would be key factors in controlling the mode of assembly.
:3.42 Å, mol.2⋯mol.2:3.48 Å). While the stacking modes of the A-form and B-form were different, they were both considered to give charge-segregated assemblies. Therefore, triarylpropargyl cations could consistently provide charge-segregated assemblies based on the ion pair despite the absence of strong intermolecular interactions such as metal⋯metal interactions.24 In this case, charge-segregated assemblies consisting of ion pairs including the triarylpropargyl cation, the molecular geometry, π–π interaction and dispersion force based on planar π-molecules with C2v symmetry would be key factors in controlling the mode of assembly.
According to the distance of the shortest C⋯C contact, strong intermolecular interaction between the cations would be present in a crystal of 2+TfO− and 2+BF4−, and thus the solid-state properties such as absorption should be affected. In fact, the red-shift of the absorption edge was recorded in a solid-state UV/Vis spectrum with a squashed single crystal of 2+TfO− and 2+BF4−, while that of 2+NTf2− appeared in a similar region to that for the solution of 2+TfO− (Fig. 4). Therefore, the solid-state properties of ion pairs containing triarylpropargyl cations such as assembly modes and photophysical properties can be easily manipulated by changing the counter anion, ensuring high tunability and modularity.
 | ||
| Fig. 4 UV/Vis absorption spectra of triarylpropargyl cations. | ||
In summary, we newly designed and synthesised triarylpropargyl cations 1+TfO−,2+TfO− with electronic stabilisation and steric protection. The obtained triarylpropargyl cations 1+TfO−,2+TfO− are stable enough to investigate their properties by using a wide range of techniques including spectroscopic and crystallographic measurements. Notably, this represents the first example of structural determination of the triarylpropargyl cation via X-ray analysis. Thanks to the planar geometry and π-extension, the red-shifted and strong absorption band extending to 650 nm was remarkable compared to those of the corresponding triarylmethyl cations. Among them, the ion pairs of triarylpropargyl cation 2+ and common anions form charge-segregated assemblies in a crystal. Red-shifted solid-state absorption can be induced by enhanced π–π stacking under the proper packing. The novel charge-segregated crystal motif presented herein can potentially facilitate the fabrication of new charge-transport materials. Further exploration of triarylpropargyl cations may lead to the development of a new platform for the dyes and electric materials.
We thank Dr E. Fukushi and Mr Y. Takata (Hokkaido University) for mass spectra and Ms A. Yamazaki (Hokkaido University) for UV/Vis measurements in the solid-state. Part of the theoretical calculations was carried out at the Research Center for Computational Science, Okazaki, Japan (Project 23-10 IMS-C218). T. Sh and Y. I. acknowledge Toyota Riken Scholar. This work was supported by Grant-in-Aid from MEXT and JSPS (No. JP23K13726 to T. Sh., JP20H02719 to T. Su., and JP21H01912 and JP23H04011 to Y. I.), JST PRESTO (JPMJPR23Q1 to Y. I.) and Research Program of “Five-star Alliance” in “NJRC Mater. & Dev”. MEXT.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Koide, Y. Urano, K. Hanaoka, W. Piao, M. Kusakabe, N. Saito, T. Terai, T. Okabe and T. Nagano, J. Am. Chem. Soc., 2012, 134, 5029–5031 CrossRef CAS PubMed.
- H. Noguchi, T. Hirose, S. Yokoyama and K. Matsuda, CrystEngComm, 2016, 18, 7377–7383 RSC.
- B. W. Laursen, K. L. Martinez, M. Rosenberg, T. Vosch, Z. Liao, A. Madsen and K. R. Rostgaard, Chem. Sci., 2018, 9, 3122–3130 RSC.
- A. C. Shaikh, J. M. Veleta, J. Moutet and T. L. Gianetti, Chem. Sci., 2021, 12, 4841–4849 RSC.
- A. Noack, A. Schröder and H. Hartmann, Angew. Chem., Int. Ed., 2001, 40, 3008–3011 CrossRef CAS PubMed.
- J. C. Martin and R. G. Smith, J. Am. Chem. Soc., 1964, 86, 2252–2256 CrossRef CAS.
- B. W. Laursen and F. C. Krebs, Angew. Chem., 2000, 39, 3432–3434 CrossRef CAS.
- G. Qian and Z. Y. Wang, Chem. – An Asian J., 2010, 5, 1006–1029 CrossRef CAS.
- S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Nat. Mater., 2020, 19, 491–502 CrossRef CAS.
- S. Akiyama, K. Yoshida, M. Hayashida, K. Nakashima, S. Nakatsuji and M. Iyoda, Chem. Lett., 1981, 311–314 CrossRef CAS.
- S. Akiyama, S. Nakatsuji, K. Nakashima and M. Watanabe, J. Chem. Soc., Chem. Commun., 1987, 710–711 RSC.
- K. Komatsu, T. Takai, S. Aonuma and K. Takeuchi, Tetrahedron Lett., 1988, 29, 5157–5160 CrossRef CAS.
- K. J. Thorley, J. M. Hales, H. Kim, S. Ohira, J. Brédas, J. W. Perry and H. L. Anderson, Chem. – Eur. J., 2013, 19, 10370–10377 CrossRef CAS PubMed.
- S. Ohira, J. M. Hales, K. J. Thorley, H. L. Anderson, J. W. Perry and J.-L. Brédas, J. Am. Chem. Soc., 2009, 131, 6099–6101 CrossRef CAS PubMed.
- D. Jérome, Chem. Rev., 2004, 104, 5565–5592 CrossRef.
- F. Kagawa, S. Horiuchi, H. Matsui, R. Kumai, Y. Onose, T. Hasegawa and Y. Tokura, Phys. Rev. Lett., 2010, 227602, 227602 CrossRef PubMed.
- Y. Haketa, K. Yamasumi and H. Maeda, Chem. Soc. Rev., 2023, 52, 7170–7196 RSC.
- K. Yamasumi, K. Ueda, Y. Haketa, Y. Hattori, M. Suda, S. Seki, H. Sakai, T. Hasobe, R. Ikemura, Y. Imai, Y. Ishibashi, T. Asahi, K. Nakamura and H. Maeda, Angew. Chem., Int. Ed., 2023, 62, e202216013 CrossRef CAS.
- H. G. Richey, J. C. Philips and L. E. Rennick, J. Am. Chem. Soc., 1965, 87, 1381–1382 CrossRef CAS.
- S. Nakatsuji, K. Nakashima, M. Iyoda and S. Akiyama, Bull. Chem. Soc. Jpn., 1988, 61, 2253–2255 CrossRef CAS.
- T. Erabi, M. Asahara, M. Miyamoto, K. Goto and M. Wada, Bull. Chem. Soc. Jpn., 2002, 75, 1325–1332 CrossRef CAS.
- M. Horn and H. Mayr, Chem. – Eur. J., 2010, 16, 7478–7487 CrossRef CAS PubMed.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 11683–11700 CrossRef CAS.
- W. Lu, K. T. Chan, S.-X. Wu, Y. Chen and C.-M. Che, Chem. Sci., 2012, 3, 752–755 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2346149–2346153. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01786b |
| ‡ Present address: Department of Applied Chemistry, Graduate School of Engineering, Kyushu University, 819-0395, Japan. E-mail: shimajiri.takuya.062@mkyushu-u.ac.jp |
| This journal is © The Royal Society of Chemistry 2024 |