
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and stability of the [45Ti]Ti–DOTA complex: en route towards aza-macrocyclic 45Ti-based radiopharmaceuticals†‡
Tamal
Roy§
 a,
Eduard
Pogorilyy§
a,
Chubina P.
Kumarananthan
b,
Unni A.
Kvitastein
b,
Marco
Foscato
a,
Karl W.
Törnroos
a,
Tom C. H.
Adamsen
*ab and
Erwan
Le Roux
*a
a,
Eduard
Pogorilyy§
a,
Chubina P.
Kumarananthan
b,
Unni A.
Kvitastein
b,
Marco
Foscato
a,
Karl W.
Törnroos
a,
Tom C. H.
Adamsen
*ab and
Erwan
Le Roux
*a
aDepartment of Chemistry, University of Bergen, Allégaten 41, Bergen, Norway. E-mail: Erwan.LeRoux@uib.no; Tom.Adamsen@uib.no
bDepartment of Radiology, Haukeland University Hospital, Centre for Nuclear Medicine and PET, Jonas Lies vei 65, Bergen, Norway
First published on 11th June 2024
Abstract
We report the use of DOTA as a chelator for titanium. The resulting complex is fully characterised and in vitro stability studies reveal its high kinetic inertness against transmetallation and transchelation. The radiolabeling of DOTA with 45Ti, via a guaiacol-based liquid–liquid extraction method, leads to a high radiochemical conversion up to 98%.
Titanium radionuclides were first synthesized over 80 years ago2 and are currently being revived due to their promising characteristics in nuclear medicine, especially for positron emission tomography (PET) imaging.3–7 From a cyclotron, two types of Ti isotopes can be produced: a long-lived titanium-44 (44Ti: t1/2 = 60 years)8 which is not-well suited for direct PET applications, and titanium-45 (45Ti: t1/2 = 3.08 h, β+: 84.8%, Eβ+avg = 439 keV)3 with negligible secondary γ-emission (0.154%) and a low β+ maximum endpoint energy of 1040.4 keV, which are favourable features for ensuring good image quality at low dosing for PET imaging.6,7,9,10 With such a half-life, its distribution can also be facilitated by shipping it away from the cyclotron production site, in contrast to the commonly used radiopharmaceuticals with short-lived radionuclides such as 11C (t1/2 = 20.4 min), 18F (t1/2 = 109.7 min) and 68Ga (t1/2 = 67.6 min), for instance.5,10 A further favourable aspect of 45Ti is its low production cost as it can be obtained by irradiating naturally monoisotopic scandium via a 45Sc(p,n)45Ti transmutation reaction.11
However, owing to titanium's high oxophilicity, low hydrolytic stability, and high propensity in forming titanyl species in aqueous environments, the radiochemistry of 45Ti currently remains underexploited.7 The first labelled compounds of 45Ti reported were based on [45Ti]Ti–Cl4 and [45Ti]Ti–ascorbate. In 1982, Ishiwata reported a series of titanyl compounds made of [45Ti]Ti–Cl2(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and [45Ti]Ti–phytate(O) along with other chelators based on diethylenetriamine pentaacetic acid, citric acid and human serum albumin.4,12 Recent reports also include mesoporous silica nanoparticles and proteins such as transferrin radiolabelled with 45Ti, and employed as radiopharmaceutical vectors with a limited efficiency in tumour uptake.13 Due to nonideal chelator–metal interactions, most of the in-vivo studies showed similar tissue biodistribution profiles of 45Ti indicating that these Ti-complexes have low kinetic stability towards transchelation, and thus leading to poor radiotracer performances.14 A handful of 45Ti based radiometal involving acyclic chelators (open-chain) such as [45Ti]Ti–salan, [45Ti]Ti–DFO/LDFC and [[45Ti]Ti–THP]+ linked to various targeting vectors (bioconjugates such as PSMA and/or DUPA),15–19 and without such as [[45Ti]Ti–THP]+ and [[45Ti]Ti–TREN–CAM]2− were recently investigated.20 While the “chelate effect” of these acyclic chelators has successfully led to a fast complexation, in most cases maximizing their thermodynamic and kinetic stabilities along with high radiolabelling, demetallation and degradation are still commonly observed to some extent under physiological conditions due to the intrinsic low kinetic inertness for this category of chelators.7,17,18,21 To date, no attempts have been made to use macrocyclic chelators for titanium, specifically with aza-macrocycles such as the DOTA chelator, which is widely utilised in the radiopharmaceutical field,22 and well-known for leading to more kinetically inert metal-complexes.7,10,23 The main reason might be their alleged ineffectiveness in forming stable complexes with titanium, as quoted ad verbum “the cyclen family of popular chelators, such as 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and its derivatives, has not demonstrated any utility for chelating Ti4+”.17 Contrary to the idea that DOTA and its derivatives are not suitable chelators for titanium, it remains very puzzling considering their recent successful application for zirconium-89.7,9,24–26 Here, we report the synthesis of Ti–DOTA, its full characterization and stability, including against other competitive metals and the EDTA chelator. Additionally, we have successfully produced its corresponding radio-analogue, i.e., [45Ti]Ti–DOTA.
O) and [45Ti]Ti–phytate(O) along with other chelators based on diethylenetriamine pentaacetic acid, citric acid and human serum albumin.4,12 Recent reports also include mesoporous silica nanoparticles and proteins such as transferrin radiolabelled with 45Ti, and employed as radiopharmaceutical vectors with a limited efficiency in tumour uptake.13 Due to nonideal chelator–metal interactions, most of the in-vivo studies showed similar tissue biodistribution profiles of 45Ti indicating that these Ti-complexes have low kinetic stability towards transchelation, and thus leading to poor radiotracer performances.14 A handful of 45Ti based radiometal involving acyclic chelators (open-chain) such as [45Ti]Ti–salan, [45Ti]Ti–DFO/LDFC and [[45Ti]Ti–THP]+ linked to various targeting vectors (bioconjugates such as PSMA and/or DUPA),15–19 and without such as [[45Ti]Ti–THP]+ and [[45Ti]Ti–TREN–CAM]2− were recently investigated.20 While the “chelate effect” of these acyclic chelators has successfully led to a fast complexation, in most cases maximizing their thermodynamic and kinetic stabilities along with high radiolabelling, demetallation and degradation are still commonly observed to some extent under physiological conditions due to the intrinsic low kinetic inertness for this category of chelators.7,17,18,21 To date, no attempts have been made to use macrocyclic chelators for titanium, specifically with aza-macrocycles such as the DOTA chelator, which is widely utilised in the radiopharmaceutical field,22 and well-known for leading to more kinetically inert metal-complexes.7,10,23 The main reason might be their alleged ineffectiveness in forming stable complexes with titanium, as quoted ad verbum “the cyclen family of popular chelators, such as 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and its derivatives, has not demonstrated any utility for chelating Ti4+”.17 Contrary to the idea that DOTA and its derivatives are not suitable chelators for titanium, it remains very puzzling considering their recent successful application for zirconium-89.7,9,24–26 Here, we report the synthesis of Ti–DOTA, its full characterization and stability, including against other competitive metals and the EDTA chelator. Additionally, we have successfully produced its corresponding radio-analogue, i.e., [45Ti]Ti–DOTA.
Non-radioactive Ti–DOTA can be obtained by stirring a mixture of DOTA in non-protic organic solvents or in methanol, followed by the addition of Ti(OiPr)4 at 70 °C for 16 h (Scheme 1 and Table S1, ESI‡). In all cases, a white solid is obtained in relatively high yield (67–89%). An alternative starting precursor as TiCl4 can also be used for forming Ti–DOTA in the presence of NEt3 but leads to an inferior yield (27%) (Table S1, ESI‡).
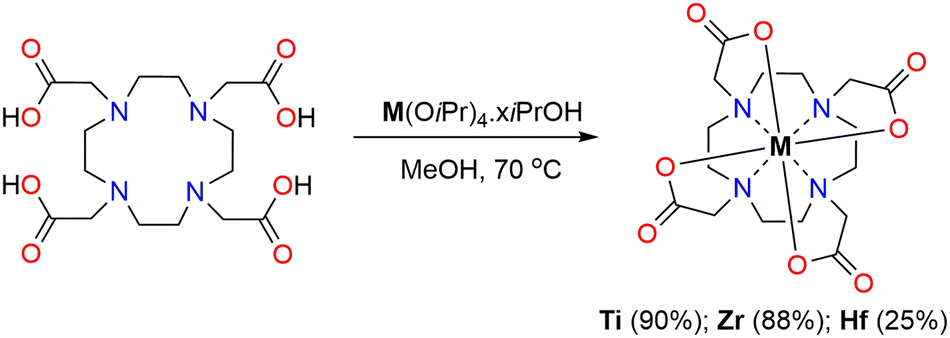 | ||
| Scheme 1 Synthesis of M–DOTA complexes. | ||
Solid-state analysis of Ti–DOTA through diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy indicates the disappearance of the broad ν(OH) stretching bands between 3200–2500 cm−1, and ν(CO) stretching band at 1744 cm−1 corresponding to the carboxylic acid groups, and the appearance of two νas/s(COO−) stretching vibrations at 1679 and 1329 cm−1, respectively. The difference of Δν = 350 cm−1 between the νas and νs stretching bands is characteristic and consistent with the monodentate chelating mode (Fig. S1, ESI‡).27 HR-MS analysis of this complex gives a (M + H)+ signal at 449.1149 m/z with the highest abundance, which supports the formation of the Ti–DOTA complex (Fig. S2, ESI‡).
Given that the Ti–DOTA complex exhibits poor solubility in classical polar and non-polar solvents except water, which sparingly solubilizes this complex, the solution-state NMR spectroscopy was performed in D2O. Owing to its C4-symmetry, a well-resolved 1H NMR spectrum was obtained, especially upon increasing the signal-to-noise ratio (NS = 128) without using a water suppression sequence (Fig. S3 (ESI‡) and Fig. 1). The 1H NMR data show a spectral pattern comparable to Zr–DOTA (Fig. S9, ESI‡),24,28 implying not only successful formation of the Ti–DOTA complex but also towards its structural similarity to Zr–DOTA. The assignments of the protons are further confirmed using 2D 1H–1H correlation (COSY, NOESY) and 1H–13C correlation (HSQC, HMBC) spectra, and the results agree with the presence of only one type of major conformer in solution (Fig. 1 and Fig. S4–S6, ESI‡), i.e., belonging either to a square antiprism (SAP) or twisted square antiprism (TSAP), including their enantiomers Δ(λλλλ)/Λ(δδδδ) and Λ(λλλλ)/Δ(δδδδ), respectively, as reported for other M–DOTA (M = Ln, Zr, Tl, Bi).25,28–31 A complete major conformer determination, as previously established for Ln–DOTA complexes29 by mean of variable temperature NMR experiments, was not possible due to the limited solubility of this Ti-complex. The 13C NMR spectrum of Ti–DOTA shows characteristic chemical resonances at δ 180.1 (–OCO), 69.7 (–CH2–COO) and 59.3/58.2 (–CH2CH2–) ppm (Fig. S7, ESI‡) with narrow line widths corroborating the presence of only one conformer in solution, unless the exchange rate between the two conformers SAP ↔ TSAP is very fast at the NMR timescale.
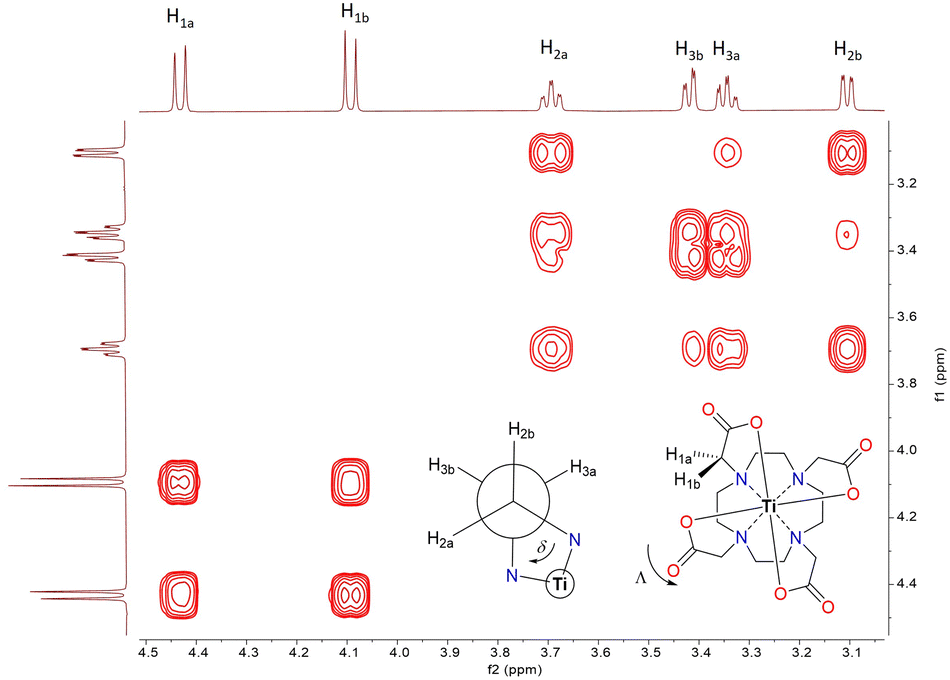 | ||
| Fig. 1 2D 1H–1H COSY spectrum of Ti–DOTA in D2O. | ||
Ti–DOTA crystallizes in aqueous solution as long colourless needles, but the obtained crystals were ill-suitable for single X-ray diffraction due to consistent merohedral rotational twinning. Nevertheless, we recorded Powder X-ray Diffraction (PXRD) for the synthesized Ti–DOTA complex, and the pattern was compared with the patterns of rutile, anatase and brookite to eliminate any doubt of the presence of crystalline TiO2 in addition to Ti–DOTA in the solid-state (Fig. S8, ESI‡). To gain further insight into the structural aspect of Ti–DOTA, we synthesized Hf–DOTA and the previously reported Zr–DOTA24 under identical reaction conditions (Scheme 1). HR-MS analyses indicate the formation of both Zr- and Hf–DOTA with the base peak at 491.0712 and 581.1136 m/z, respectively (Fig. S9 and S16, ESI‡). Both Zr- and Hf–DOTA displayed similar patterns in 1H, 13C and 2D NMR and DRIFT spectra like for Ti–DOTA (Fig. S1, S10–S15 and S17–S22, ESI‡), which essentially indicates that all three complexes have similar structural aspects in solution and in the solid-state. Similarly to Ti–DOTA, crystals of limited quality, yet again merohedrally twinned, of Hf–DOTA were obtained from an aqueous solution at room temperature, but in this case allowing deduction of the connectivity around the Hf-centre (Fig. S23, ESI‡). Hf–DOTA crystallizes as two overlapping 8-coordinate enantiomers, namely Δ(λλλλ) and Λ(δδδδ), in which both exhibit an SAP geometry identical to Zr–DOTA.24
To gather further insights into the structural aspects and relative stabilities of the M–DOTA complexes (M = Ti, Zr, Hf), we opted for DFT studies of these complexes (PBE-GD3MBJ-SMD(water)/def2-TZVP/D, see ESI‡). The crystallographic structure reported for Zr–DOTA shows a co-crystallized enantiomeric pair of κ8 conformations Λ(δδδδ) and Δ(λλλλ), i.e., only as SAP conformers.24 Notably, DOTA can also coordinate metals in κ8 chelation mode with the diastereomeric conformations leading to complexes Δ(δδδδ) and Λ(λλλλ) as experimentally shown by the crystal structure of the [Bi–DOTA]− complex where the geometry around Bi adopts a TSAP.30 Both geometries were modelled with DFT for Ti(IV), Zr(IV), and Hf(IV), but Λ(δδδδ) is found to be more stable than Δ(δδδδ) by 4–5 kcal mol−1 for all three metals (Fig. S24, S25 and Table S2, ESI‡), which is consistent with the geometry observed in the crystal structures of Zr–DOTA.24 Notably, while the Λ(δδδδ) M–DOTA geometries of the three metals are all similar (Fig. 2), the Ti complexes appear slightly more compact than the Zr and Hf analogues. In fact, the M–O distances in the DFT-modelled structures are 2.03–2.04 Å in Ti–DOTA and 2.17–2.20 Å in Zr–DOTA. Instead, the Ti–N distances are 2.39 Å and the Zr–N distances are 2.47–2.49 Å. Overall, these distances show that the κ8-metal-coordinating environment of Ti–DOTA is more compact than those of Zr–DOTA and Hf–DOTA (M–N4/M–O4 distances: Ti: 1.278/0.977 Å; Zr: 1.382/0.915 Å and Hf: 1.351/0.937 Å, Table S3, ESI‡). The index parameters (τ8), indicating the degree of idealised SAP (τ8 = 0) and TSAP (τ8 = 1) geometries (Table S3, ESI‡), for the non-capped 8-coordinate Zr–DOTA complexes determined from the X-ray structures and obtained from DFT are also in line with an SAP geometry with τ8 of 0.17 and 0.14/0.15, respectively. Furthermore, Ti–DOTA with its calculated τ8 of 0.07 is the lowest of all known M–DOTA complexes, indicating that the Ti centre adopts a nearly ideal SAP geometry, and thus is somehow shift-locked within the M–DOTA structure compared to Zr and Hf atoms or other larger metal atoms.
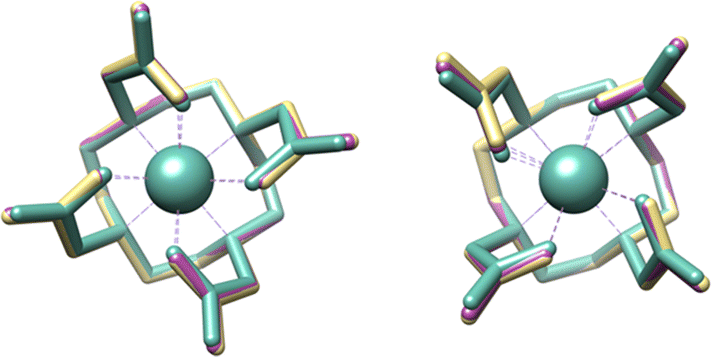 | ||
| Fig. 2 DFT-optimized geometries of Ti–DOTA (green), Zr–DOTA (gold), and Hf–DOTA (purple) in the SAP/Λ(δδδδ) (left) and TSAP/Δ(δδδδ) (right) configuration. | ||
Next, we evaluate the in vitro stability of the Ti–DOTA complex due to the perceived idea of aza-macrocycles being labile chelates for titanium ions. Accordingly, the kinetic stability of Ti–DOTA was studied at physiological pH (7.4) and temperature (37 °C) in the presence of competitive metal ions (Mg2+, Fe3+, Co2+, Cu2+ and Zn2+) as well as with an excess of EDTA chelator. Under these conditions, Ti–DOTA was found to be highly kinetically inert against transmetallation and DOTA dissociation (Table S4, ESI‡). The thermodynamic stability of the M–DOTA complexes was also assessed by DFT studies using two different methods. We first considered the free energy of complexation of the metal-aquo cation [M(H2O)19]4+ (M = Ti, Zr, Hf) by the anionic free DOTA4−, similarly to previous work in the field.32 The resulting thermodynamic preference for the complex is much higher for Ti than for Zr and Hf (Table S5, ESI‡). Notably, the Hf complex is predicted to be more stable than the Zr analogue, but overall, this trend correlates well with the effective ionic radii for 8-coordinate compounds: Ti(IV) = 0.74 Å, Zr(IV) = 0.84 Å, Hf(IV) = 0.83 Å.33 This trend is also confirmed by the estimation of the free energy for the exchange of EDTA4− with DOTA4− (Table S5, ESI‡). The Ti–DOTA complex is confirmed to be the most stable in the series, and significantly separated from Hf, i.e., the second most stable, and Zr.
The salt-based liquid target production of 45Ti was performed using natSc(NO3)3 dissolved in ultrapure HNO3 and irradiated with a proton beam current of 10–25 μA for 1.5–3 h (Table S6, ESI‡). The irradiations resulted in a 45Ti-activity at the end of bombardment (EOB) ranging from 0.31 to 1.25 GBq with an average radionuclidic purity of 98.0% (Table S6, ESI‡).
Initial attempts were made to separate [45Ti]Ti-HNO3 from the scandium matrix using a cation-exchange hydroxamate-modified resin (ZR-resin) as previously reported (Scheme S1, ESI‡).11 An extraction efficiency (EE) of only ca. 5% was achieved during the solid-phase extraction with the ZR-resin using 1 M oxalic acid as an eluent. Most importantly, no subsequent ligand exchange with the presumed [45Ti]Ti–oxalate species was identified in the presence of DOTA by radio-HPLC/HPLC with Ti–DOTA as a reference. Trying to remediate this drawback, a ligand exchange was attempted through a QMA anion exchange resin as successfully reported for the conversion of [89Zr]Zr–(Ox)2 to [89Zr]Zr–DOTA in high yield via a more reactive intermediate [89Zr]Zr–Cl4.24,34 In our case, the ligand exchange with QMA-resin did not proceed as for its Zr analogue (Scheme S1, ESI‡), presumably due to the high stability of [45Ti]Ti–oxalate species in aqueous media.35 Other eluants such as 0.1/1 M citric acid, and 1 M ascorbic acid were also tested but resulted in poor extraction efficiency <6%, and subsequent formation of [45Ti]Ti–DOTA was not observed by reacting the eluates with DOTA. These results indicate the slow kinetic formation of Ti–DOTA, contrasting the rapid complex formation with acyclic chelators such as DFO/LDFC, TREN–CAM2− and THP, where the 45Ti radiolabelling is straightforwardly obtained via the intermediate [45Ti]Ti–citrate, for instance.18–20 To mimic the reactivity of Ti–oxalate species with DOTA, we attempted to form a non-radioactive Ti–DOTA complex by refluxing DOTA with bis(ammonium lactato)dihydroxide titanium for 24 h in water. Only a 2% yield was achieved, indicating a remarkable stability of such derived Ti–oxalate species (Table S1, ESI‡).35 Alternatively, a liquid–liquid extraction (LLE) method was carried out for purifying 45Ti from its natSc(NO3)3 liquid matrix, hence avoiding strongly binding chelators and for replacing them by a more labile chelator such as guaiacol.36 Following the procedure reported by Zhuravlev,16,17 a mixture of guaiacol and anisole (9/1 v/v) was used for the separation of Sc from [45Ti]Ti–HNO3 (Scheme S2, ESI‡). With an optimized ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3 (aqueous/organic phase) (v/v), an extraction efficiency of 81% (d.c.) was obtained comparable to the range of EEs (75–85%) obtained by LLE protocols in continuous flow using a membrane separator module.17 The radiolabelling of DOTA was achieved by mixing the organic phase solution containing 93.6 MBq (2.53 mCi) of [45Ti]Ti–guaiacolate with a solution of DOTA (50 mM) in DMSO in the presence of an excess of pyridine. The reaction mixture was incubated at 60 °C for 15 min and a radiochemical conversion of 98% was determined (Fig. S26 and S27, ESI‡). Subsequently, the complex was purified using preparative radio-HPLC to obtain 15% RCY (d.c.) of the pure [45Ti]Ti–DOTA complex (Fig. S28, ESI‡).
:1.3 (aqueous/organic phase) (v/v), an extraction efficiency of 81% (d.c.) was obtained comparable to the range of EEs (75–85%) obtained by LLE protocols in continuous flow using a membrane separator module.17 The radiolabelling of DOTA was achieved by mixing the organic phase solution containing 93.6 MBq (2.53 mCi) of [45Ti]Ti–guaiacolate with a solution of DOTA (50 mM) in DMSO in the presence of an excess of pyridine. The reaction mixture was incubated at 60 °C for 15 min and a radiochemical conversion of 98% was determined (Fig. S26 and S27, ESI‡). Subsequently, the complex was purified using preparative radio-HPLC to obtain 15% RCY (d.c.) of the pure [45Ti]Ti–DOTA complex (Fig. S28, ESI‡).
In summary, we now report the first example of a DOTA complex of titanium, contradicting the established opinion that aza-macrocycles such as DOTA are not suitable chelators for Ti4+ ions. To the contrary, our DFT studies show a remarkable stability of the Ti–DOTA complex, even superior to their Zr and Hf analogues and supported experimentally by in vitro stability studies. In addition, our preliminary results on the radiolabelling of DOTA demonstrate that [45Ti]Ti–DOTA can be efficiently obtained via a LLE method. Albeit the radiolabelling with 45Ti isotope was hitherto limited to acyclic chelators, this example using an aza-macrocyclic chelator opens new opportunities for 45Ti-based radiopharmaceutical development applied to PET imaging.
This work was supported by the Trond Mohn Foundation, project no. TMS2019TMT07 and the University of Bergen. The Research Council of Norway is thanked for CPU (NN2506K) and storage resources (NS2506K). We would like to acknowledge the Haukeland University Hospital cyclotron team for enabling the production of 45Ti, and C. Bruhn for QMA-resin experiments. Prof. Dr N. Å. Frøystein and Dr D. Kratzert are thanked for their fruitful discussions on NMR spectroscopy and on crystallographic issues, respectively.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Álvarez-Moreno, C. de Graaf, N. López, F. Maseras, J. M. Poblet and C. Bo, J. Chem. Inf. Model., 2015, 55, 95–103 CrossRef PubMed.
- J. S. V. Allen, M. L. Pool, J. D. Kurbatov and L. L. Quill, Phys. Rev., 1941, 60, 425–429 CrossRef CAS; C. Ishii and K. Takahashi, J. Phys. Soc. Jpn., 1960, 15, 736–737 CrossRef; D. Gföller and A. Flammersfeld, Z. Phys., 1965, 187, 490–494 CrossRef; F. T. Porter, M. S. Freedman, F. Wagner and K. A. Orlandini, Phys. Rev., 1966, 146, 774–780 CrossRef; W. M. Zuk, W. F. Davidson, M. R. Najam and M. A. Awal, Z. Phys., 1971, 242, 93–101 CrossRef.
- J. C. Merrill, R. M. Lambrecht and A. P. Wolf, Int. J. Appl. Radiat. Isot., 1978, 29, 115–116 CrossRef CAS.
- K. Ishiwata, T. Ido, M. Monma, M. Murakami, M. Kameyama, H. Fukuda and T. Matsuzawa, J. Labelled Compd. Radiopharm., 1982, 19, 539–1541 Search PubMed.
- A. L. Vāvere, R. Laforest and M. J. Welch, Nucl. Med. Biol., 2005, 32, 117–122 CrossRef PubMed.
- P. Costa, L. F. Metello, F. Alves and M. Duarte Naia, Instruments, 2018, 2, 8 CrossRef CAS.
- E. Boros and A. B. Packard, Chem. Rev., 2019, 119, 870–901 CrossRef CAS PubMed.
- T. Hashimoto, K. Nakai, Y. Wakasaya, I. Tanihata, Z. Fulop, H. Kumagai, A. Ozawa, K. Yoshida and R. Goswarmi, Nucl. Phys. A, 2001, 686, 591–599 CrossRef.
- M. Brandt, J. Cardinale, M. L. Aulsebrook, G. Gasser and T. L. Mindt, J. Nucl. Med., 2018, 59, 1500 CrossRef CAS PubMed.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- I. F. Chaple, K. Thiele, G. Thaggard, S. Fernandez, E. Boros and S. E. Lapi, Appl. Radiat. Isot., 2020, 166, 109398 CrossRef CAS PubMed.
- M. Kawamura, K. Inoue, S. Kimura, T. Ido, K. Ishiwata, K. Kawashima, K. Matsuda and M. Kameyama, J. Labelled Compd. Radiopharm., 1986, 23, 1360–1362 Search PubMed; K. Ishiwata, T. Ido, M. Monma, M. Murakami, H. Fukuda, M. Kameyama, K. Yamada, S. Endo, S. Yoshioka, T. Sato and T. Matsuzawa, Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot., 1991, 42, 707–712 CrossRef CAS PubMed.
- A. L. Vavere and M. J. Welch, J. Nucl. Med., 2005, 46, 683–690 Search PubMed; F. Chen, H. F. Valdovinos, R. Hernandez, S. Goel, T. E. Barnhart and W. Cai, Acta Pharmacol. Sin., 2017, 38, 907–913 CrossRef CAS PubMed.
- S. Saini and S. E. Lapi, Pharmaceuticals, 2024, 17, 479 CrossRef CAS PubMed.
- G. W. Severin, C. H. Nielsen, A. I. Jensen, J. Fonslet, A. Kjær and F. Zhuravlev, J. Med. Chem., 2015, 58, 7591–7595 CrossRef CAS PubMed; K. Giesen, I. Spahn and B. Neumaier, J. Radioanal. Nucl. Chem., 2020, 326, 1281–1287 CrossRef.
- K. S. Pedersen, J. Imbrogno, J. Fonslet, M. Lusardi, K. F. Jensen and F. Zhuravlev, React. Chem. Eng., 2018, 3, 898–904 RSC.
- S. K. Pedersen, C. Baun, M. K. Nielsen, H. Thisgaard, I. A. Jensen and F. Zhuravlev, Molecules, 2020, 25, 1104 CrossRef PubMed.
- I. F. Chaple, H. A. Houson, A. Koller, A. Pandey, E. Boros and S. E. Lapi, Nucl. Med. Biol., 2022, 108–109, 16–23 CrossRef CAS PubMed.
- S. Saini, G. E. D. Mullen, P. J. Blower and S. E. Lapi, Mol. Pharmaceutics, 2024, 21, 822–830 CrossRef CAS PubMed.
- A. J. Koller, S. Saini, I. F. Chaple, M. A. Joaqui-Joaqui, B. M. Paterson, M. T. Ma, P. J. Blower, V. C. Pierre, J. R. Robinson, S. E. Lapi and E. Boros, Angew. Chem., Int. Ed., 2022, 61, e202201211 CrossRef CAS PubMed; A. J. Koller, L. Wang, M. Deluca, O. Glaser, M. J. Robis, J. C. Mixdorf, M. N. Chernysheva, I. A. Guzei, E. Aluicio-Sarduy, T. E. Barnhart, J. W. Engle and E. Boros, Inorg. Chem., 2023, 62, 20655–20665 CrossRef PubMed.
- R. Lengacher, A. Marlin, D. Śmiłowicz and E. Boros, Chem. Soc. Rev., 2022, 51, 7715–7731 RSC.
- N. Herrero Álvarez, D. Bauer, J. Hernández-Gil and J. S. Lewis, ChemMedChem, 2021, 16, 2909–2941 CrossRef PubMed.
- J. J. R. Frausto da Silva, J. Chem. Educ., 1983, 60, 390 CrossRef CAS; R. D. Hancock, Pure Appl. Chem., 1986, 58, 1445–1452 CrossRef; R. D. Hancock, J. Chem. Educ., 1992, 69, 615–621 CrossRef; N. Viola-Villegas and R. P. Doyle, Coord. Chem. Rev., 2009, 253, 1906–1925 CrossRef; Z. Baranyai, G. Tircsó and F. Rösch, Eur. J. Inorg. Chem., 2020, 36–56 CrossRef.
- D. N. Pandya, N. Bhatt, H. Yuan, C. S. Day, B. M. Ehrmann, M. Wright, U. Bierbach and T. J. Wadas, Chem. Sci., 2017, 8, 2309–2314 RSC.
- D. N. Pandya, K. E. Henry, C. S. Day, S. A. Graves, V. L. Nagle, T. R. Dilling, A. Sinha, B. M. Ehrmann, N. B. Bhatt, Y. Menda, J. S. Lewis and T. J. Wadas, Inorg. Chem., 2020, 59, 17473–17487 CrossRef CAS PubMed.
- J. K. Yoon, B. N. Park, E. K. Ryu, Y. S. An and S. J. Lee, Int. J. Mol. Sci., 2020, 21, 4309 CrossRef CAS PubMed.
- C. C. R. Sutton, G. da Silva and G. V. Franks, Chem. – Eur. J., 2015, 21, 6801–6805 CrossRef CAS PubMed.
- D. Parker, K. Pulukkody, F. C. Smith, A. Batsanov and J. A. K. Howard, J. Chem. Soc., Dalton Trans., 1994, 689–693 RSC.
- S. Aime, M. Botta and G. Ermondi, Inorg. Chem., 1992, 31, 4291–4299 CrossRef CAS.
- É. Csajbók, Z. Baranyai, I. Bányai, E. Brücher, R. Király, A. Müller-Fahrnow, J. Platzek, B. Radüchel and M. Schäfer, Inorg. Chem., 2003, 42, 2342–2349 CrossRef PubMed.
- C. Chen, C. Sommer, H. Thisgaard, V. McKee and C. J. McKenzie, RSC Adv., 2022, 12, 5772–5781 RSC.
- J. P. Holland, Inorg. Chem., 2020, 59, 2070–2082 CrossRef CAS PubMed.
- R. Shannon, Acta Cryst., 1976, A32, 751–767 CrossRef CAS.
- J. P. Holland, Y. Sheh and J. S. Lewis, Nucl. Med. Biol., 2009, 36, 729–739 CrossRef CAS PubMed.
- G. M. H. Van de Velde, J. Inorg. Nucl. Chem., 1977, 39, 1357–1362 CrossRef CAS; P. Chaudhuri and H. Diebler, J. Chem. Soc., Dalton Trans., 1977, 596–601 RSC.
- P. Sobota, K. Przybylak, J. Utko, L. B. Jerzykiewicz, A. J. L. Pombeiro, M. F. C. Guedes da Silva and K. Szczegot, Chem. – Eur. J., 2001, 7, 951–958 CrossRef CAS PubMed.
Footnotes |
| † A data set collecting the results from the DFT modelling is available from the ioChem-BD repository and it can be accessed viahttps://doi.org/10.19061/iochem-bd-6-344, see ref. 1. |
| ‡ Electronic supplementary information (ESI) available: Experimental section, DRIFT, 1–2D NMR and HR-MS spectra of M–DOTA complexes. Stability, DFT and 45Ti-radiolabeling studies. See DOI: https://doi.org/10.1039/d4cc01800a |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |