
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew trends in nanoparticle exsolution
Alfonso J.
Carrillo
 *,
Andrés
López-García
,
Blanca
Delgado-Galicia
and
Jose M.
Serra
*
*,
Andrés
López-García
,
Blanca
Delgado-Galicia
and
Jose M.
Serra
*
Instituto de Tecnología Química, Universitat Politècnica de València, Consejo Superior de Investigaciones Científicas, 46022 Valencia, Spain. E-mail: alcardel@itq.upv.es; jmserra@itq.upv.es
First published on 11th June 2024
Abstract
Many relevant high-temperature chemical processes require the use of oxide-supported metallic nanocatalysts. The harsh conditions under which these processes operate can trigger catalyst degradation via nanoparticle sintering, carbon depositions or poisoning, among others. This primarily affects metallic nanoparticles created via deposition methods with low metal–support interaction. In this respect, nanoparticle exsolution has emerged as a promising method for fabricating oxide-supported nanocatalysts with high interaction between the metal and the oxide support. This is due to the mechanism involved in nanoparticle exsolution, which is based on the migration of metal cations in the oxide support to its surface, where they nucleate and grow as metallic nanoparticles partially embedded in the oxide. This anchorage confers high robustness against sintering or coking-related problems. For these reasons, exsolution has attracted great interest in the last few years. Multiple works have been devoted to proving the high catalytic stability of exsolved metallic nanoparticles in several applications for high-temperature energy storage and conversion. Additionally, considerable attention has been directed towards understanding the underlying mechanism of metallic nanoparticle exsolution. However, this growing field has not been limited to these types of studies and recent discoveries at the forefront of materials design have opened new research avenues. In this work, we define six new trends in nanoparticle exsolution, taking a tour through the most important advances that have been recently reported.
 Alfonso J. Carrillo | Alfonso J. Carrillo holds a PhD in chemical engineering from Universidad Rey Juan Carlos (Spain) – research conducted at the IMDEA Energy Institute. Then, he moved to the Electrochemical Materials Laboratory, first at ETH Zurich (Switzerland), and later at MIT (USA), where he was a 2018 Eni-MIT Energy Fellow. He has been awarded with the Energy and Environmental Research Grant by Fundación Iberdrola, Juan de la Cierva Formación by the Spanish Ministry of Science, and Junior Leader Fellowship by Fundación LaCaixa. He has been working at ITQ (Spain) since January 2019, focusing on the functionalization of redox oxides for energy storage and the production of renewable fuels. |
 Andrés López-García | Andrés López-García obtained his BSc degree in chemistry and MSc degree in applied chemistry from Universidad Autónoma de Madrid. During this period, he was a research intern at ICMM and ICP institutes (both in Madrid, Spain) thanks to the scholarships granted by the Spanish Society of Catalysis (SECAT). In 2020, he joined ITQ (Valencia, Spain) to conduct his PhD studies, focusing on alloyed nanocatalyst exsolution for energy applications. He has recently received the Young Researcher Award 2023 by E-MRS (Strasbourg, France) and the Best Oral Presentation Award at the Autumn School on Inorganic Electrochemical Reactors (Valencia, Spain). |
 Blanca Delgado-Galicia | Blanca Delgado-Galicia received her bachelor's degree in chemistry from Universidad Autónoma de Madrid. She continued her education at Universitat Politècnica de València where she pursued a Master's degree in Sustainable Chemistry. She obtained a scholarship from the Spanish Society of Catalysis (SECAT) to develop her Master Thesis, focused on the study of nanoparticle exsolution on ceramic materials. This project was carried out at the Energy Conversion and Storage Group at the Instituto de Tecnología Química, where she is currently employed as a research assistant. Recently, she has received the USC-Genesal Energy award for the best Master Thesis on energy transition. |
 Jose M. Serra | Prof. José M. Serra completed his PhD at the Institute of Chemical Technology (ITQ) under the guidance of Prof. A. Corma, in collaboration with the Institute Français du Petrole, followed by a 2-year postdoctoral stage at the Forschungszentrum Jülich, Germany. Since 2006 he has been the Head of the Energy Conversion and Storage group and, currently he is the Director of ITQ. For his contribution to the field, he has received the ExxonMobil Chemical European Science and Engineering Award, Spanish Catalysis Society PhD Award, Christian Friedrich Schönbein Medal, European Ceramic Society Young Scientist Award, Medal of the Royal Spanish Engineering Academy and Air Liquide Scientific Prize. |
Introduction
Oxide-supported metallic nanoparticles are at the core of a plethora of technologies, including traditional thermocatalytic processes for producing fuels and chemicals,1 fuel cells and electrolyzers,2–4 sensors5 or even memory storage devices.6 Specifically, in catalytic and electro-catalytic processes occurring at high temperatures (500–1000 °C), metallic nanoparticles can easily suffer from sintering-related phenomena if they are not firmly attached to the oxide support.7 Eventually, metallic nanoparticle growth and agglomeration will increase the nanoparticle size and lower their dispersion over the oxide support, which will dramatically lower the catalyst activity. Thus, preventing nanoparticle sintering-related phenomena has been a major field of study in the catalysis and materials science communities to obtain robust and durable catalysts with enhanced lifetimes.8 Based on this premise, in the last decade, nanoparticle exsolution has emerged as a promising alternative to conventional impregnation methods, allowing the fabrication of highly resistant metallic nanoparticles.9 This is mainly due to the mechanism through which exsolved nanoparticles grow.Exsolution involves the migration of metallic cations contained in the crystal lattice of the bulk oxide, which, under reductive conditions, nucleate as nanoparticles on the oxide surface. The fact that the nanoparticles grow from the bulk rather than from a deposited precursor, as in impregnation, endows them with high resistance to sintering since the particles remain “anchored” to the oxide.10 Additionally, the benefits of nanoparticle anchoring in preventing carbon deposition have been demonstrated.10 These two beneficial properties of exsolved nanoparticles, among others, have prompted great interest in the scientific community, leading to exponential growth in the number of publications over the last decade1,11 (Fig. 1a). However, it should be highlighted that pioneering works demonstrating nanoparticle exsolution were pointed out two decades ago, exemplified by the seminal article of Nishihata et al.12 In this first decade, many contributions can be found, especially on precious metal (Pd, Pt, and Ru) nanocatalyst exsolution from perovskite oxides.13–30 Since then, many works have been devoted to mechanistic studies in which thermodynamics and kinetics of metallic nanoparticle exsolution have been assessed. These works lay the foundation for more efficient materials design, where a profound understanding of the physicochemical phenomena controlling exsolution is critical. In parallel, and according to the inherent emerging nature of this field, research efforts have also been put into expanding the limits of nanoparticle exsolution to new avenues, enlarging the materials design space of this novel nanocatalyst fabrication route.
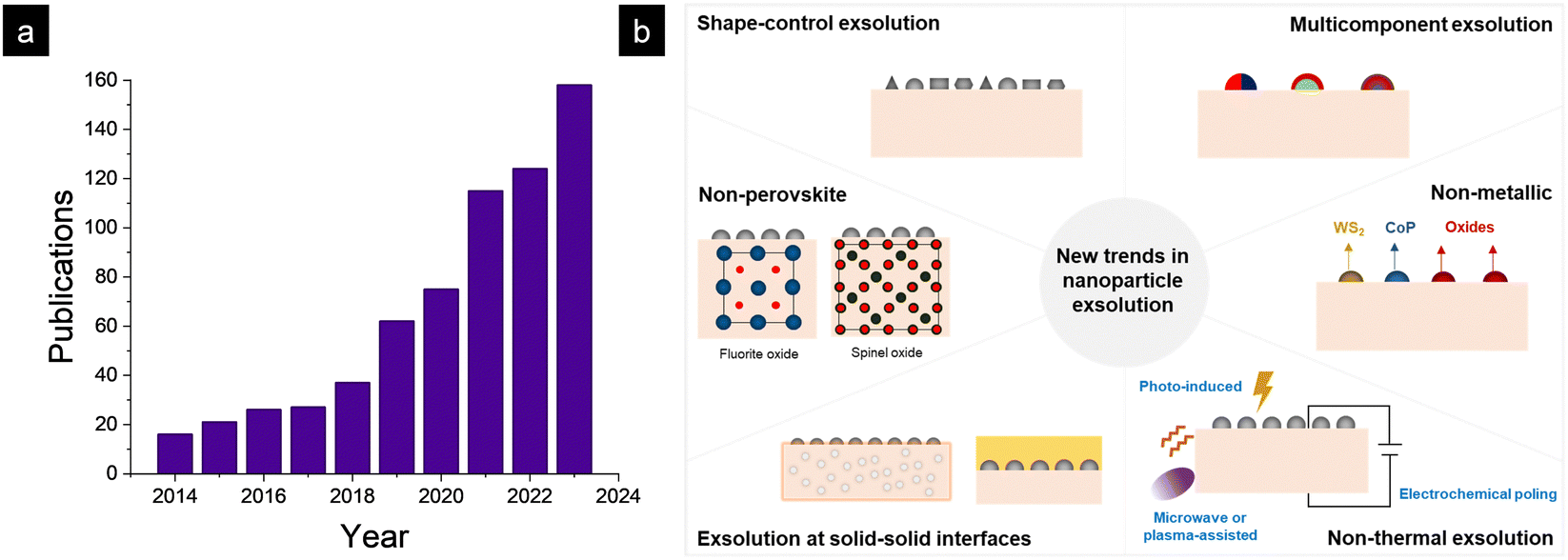 | ||
| Fig. 1 (a) Evolution of publications on nanoparticle exsolution in the last decade. (b) The six new trends in nanoparticle exsolution that are covered in this article. | ||
Background: how to control nanoparticle exsolution?
Besides its high robustness, one of the significant advantages of exsolution is its excellent capability for facile control over nanoparticle characteristics (morphological and compositional), which is an additional advantage of this fabrication method. This has attracted great interest in the community, which is focused on understanding the underlying mechanisms of this promising nano-fabrication method and finding structure–property relationships that could lead to more efficient catalysts or devices. Nanoparticle exsolution can be controlled by adjusting the external (conditions under which exsolution is performed) or internal factors (related to the oxide properties that serve as the host for nanoparticle exsolution). Regarding the former, parameters such as temperature, time and gas composition6,31–34 have resulted in changes in nanoparticle size and dispersion. Longer exposure times and lower temperatures generally lead to larger nanoparticles and lower dispersions. These trends are directly related to the kinetics of nanoparticle nucleation and growth, which, under the aforementioned conditions, favour the latter. Longer exposure times have also been recently proven to alter the shape of exsolved nanoparticles.11 The design of shape-controlled exsolved nanoparticles and their implications in the catalytic activity of heterogeneous reactions will be discussed in Section 1. It is important to emphasize here that morphological changes achieved via variations in the exsolution fabrication route should ensure an improvement in the catalytic (or device) performance while assuring long-term stability.Additionally, changes in the conditions under which exsolution is performed can greatly impact the composition of the nanoparticles. A recent work illustrated that the exsolution temperature influences the composition of exsolved ternary alloy metallic nanoparticles.31 The possibility of exsolving multiple B-site cations, leading to the formation of alloys or multi-elemental nanoparticles, is, undoubtedly, one of the main focuses of recent research in nanoparticle exsolution. Exsolved alloy nanocatalysts can unlock unprecedented catalytic properties, leading to higher activity while guaranteeing robustness over prolonged operation. The latest discoveries in the conceptual design of multi-elemental (alloy) nanocatalysts will be covered in Section 2.
Time and temperature can be used to alter the composition of the exsolved nanoparticles, as well as the gas atmosphere. Traditionally, the exsolution field has focused on fabricating metallic nanoparticles. However, in many chemical reactions or physicochemical processes, other chemistries such as oxides, sulphides or phosphides are the active species. The exsolution field has not put aside the catalytic potential of these compounds, and lately, the exsolution of oxide35 nanoparticles or sulphides36 has been reported. Section 3 will analyze the recent advances in the design of non-metallic exsolved nanoparticles.
Temperature, time, and gas partial pressure are the external parameters commonly changed to control exsolution. A recent review by Ruh et al. has covered these aspects.11 However, in recent years, metallic nanoparticle exsolution has been induced via non-thermal treatments such as electrochemical poling,37 laser induction,38 or applying plasma39 or microwaves.40 These methods have led to promising results in achieving higher exsolution extents or milder exsolution conditions in H2-free atmospheres, opening new avenues away from conventionally heating in reductive atmospheres. These latest trends will be discussed in Section 4.
Modifications in the material intrinsic (or internal) factors have also been widely studied in recent years to improve the exsolution capacity of oxide materials. One aspect that has greatly impacted the amount and shape of exsolved nanoparticles is oxide strain. Strain engineering has been mainly studied in thin films due to the relative ease with which strain can be induced in a thin film oxide by a lattice mismatch with the support in which it is deposited.41 Namely, for La0.2Sr0.7Ni0.1Ti0.9O3−δ, compressive strain was found to increase the exsolved nanoparticle population, leading to the exsolution of smaller nanoparticles compared to tensile strain.41 Interestingly, the relationship between strain and nanoparticle exsolution can be reverse-engineered as it has been recently demonstrated in bulk oxides.42–44 Here, exsolution is explicitly aimed at forming endogenous nanoparticles that precipitate inside the oxide bulk together with conventional surface exsolution. These metallic precipitates induce tensile strain in the perovskite oxide, which significantly enhances the oxygen exchange capacity of these materials, a fundamental property in the chemical looping production of syngas.43 The design of oxides functionalized with endogenous nanoparticles will be covered in Section 5.
The influence of the plane direction on nanoparticle exsolution has been explored using thin film exsolution. Kim et al. discovered that nanoparticle population and shape strongly correlate with the plane orientation in perovskite titanates.45 In particular, the (111) plane led to higher populations, whereas the (001) plane led to larger, more elongated particles.45 Here, the authors demonstrated the relevance of interfacial energies in the shape and dispersion of exsolved nanoparticles.
A-site deficiency has been conventionally employed as a compositional modification to enhance the number of exsolved nanoparticles.46 In a recent work, O’Leary and co-authors shed more light on the influence of A-site deficiency and composition on the energetics of Ni nanoparticle exsolution from perovskite titanates.47 However, in some cases, A-site deficiency is not favourable since it can lead to compositional instabilities, eventually resulting in the segregation of transition metal oxides such as NiO in non-stoichiometric double perovskites.31 This affects the extent of exsolution since not all the exsolvable B-site cations are in the perovskite oxide crystal lattice, causing a decrease in the nanoparticle surface dispersion compared to stoichiometric materials. Our group recently found that by partially replacing Sr in the A-site with trivalent lanthanides, it was possible to reduce the amount of NiO segregations in Sr1.9Fe1.3Ni0.2Mo0.5O6−δ double perovskites.48 We also found that there was a correlation between the lattice parameters induced by this lanthanide-doping strategy and the extent of exsolution, in which incorporation of larger cations, e.g., La3+ resulted in a decrease of the exsolved nanoparticle population.48 These results emphasize how alterations in material properties can directly affect the exsolution behavior of the pervovskite family.
Finally, in the last section, we will assess the recent progress on metallic nanoparticle exsolution from non-perovskite oxide materials (Section 6). This is quite a relevant topic since it can expand the realm of exsolution to other oxides conventionally used as supports in catalysis, such as fluorites, e.g., ceria or zirconia. In a recent work, we demonstrated that Ru exsolution from CeO2 significantly boosted the chemical looping performance of this type of materials for syngas production via methane partial oxidation.49 Exsolution from other oxides such as yttria-stabilized zirconia, pyrochlores and spinels will also be covered in this section, emphasizing the main differences that arise when comparing with perovskite exsolution.
This Feature Article presents a brief but thorough analysis of six trends in nanoparticle (Fig. 1b) exsolution based on the latest discoveries in this emerging field, emphasizing the discoveries led by ITQ. This work aims to connect these recent findings with future directions that will help designing durable materials with high catalytic activity by applying the exsolution method, regardless of the application or end-use of these catalysts. Specific applications and detailed thermodynamic or kinetic aspects will not be covered here, although their importance is key for more efficient exsolution design.
New trends in nanoparticle exsolution
1. Shape-control exsolved nanoparticles
Most exsolution studies focus on the size or population of nanoparticles as these are straightforward factors to control. These are usually modified by changing the temperature (or time) of reduction. For instance, increasing the temperature leads to an increase in the size of the nanoparticles while reducing their dispersion. Besides these morphological properties, modifying other characteristics of the exsolved nanoparticles would also be interesting, like the acquired shape during exsolution. This can, indeed, be modified by adjusting the process conditions and may significantly impact the catalytic performance of these nanoparticles. For example, for polyhedral-shaped nanoparticles, the exposed surface is higher than that of spherical ones for the same volume.50Conventionally, exsolution of metallic species tends to generate spherical-shaped nanoparticles. However, several works report alternative shapes, even demonstrating strategies to control the geometry of the exsolved metallic nanoparticles. To date, and the best of our knowledge, only two reports show how changing exsolution parameters can tune the exsolved nanoparticle's shape. The first of these two studies, by Neagu et al.,51Fig. 2a–d, demonstrates how the exsolution gas atmosphere plays an important role in shaping the emerging nanoparticles. Usually, as previously mentioned, spherical-shaped nanoparticles are expected when exsolving metallic species, like Ni from La0.8Ce0.1Ni0.4Ti0.6O3−δ, as shown in Fig. 2b after 650 °C, 10 hours under H2 flow reduction treatment. However, cubic-shaped nanoparticles were formed when this same material was subjected to 5% CO at 900 °C for 10 h (Fig. 2c). Remarkably, applying humidified (2.5% H2O) 5% H2 flow at 1000 °C for 10 h generated ellipsoidal-shaped nanoparticles interfaced with La2TiO5 oxide (Fig. 2d). In this same work, even vacuum conditions were explored for exsolution. From La0.43Ca0.37Ni0.03Ti0.94O3−δ perovskite, strongly faceted nanoparticles emerged upon heating up to 900 °C under vacuum (Fig. 2a). These results show the high impact that the exsolution treatment gas atmosphere and/or pressure can have on the shape of the exsolved nanoparticles, in this case, Ni.
 | ||
| Fig. 2 (a) ETEM micrographs of La0.43Ca0.37Ni0.03Ti0.94O3−δ faceted exsolved nanoparticle generated in vacuum at 900 °C. (b)–(d) ETEM micrographs of La0.8Ce0.1Ni0.4Ti0.6O3−δ exsolution: (b) spherical-shaped nanoparticles (H2, 650 °C); (c) cubic-shaped nanoparticles (5% CO, 900 °C, 10 h); (d) ellipsoidal-shaped nanoparticles (2.5% H2O/5% H2 1000 °C, 10 h). Reprinted (adapted) with permission from D. Neagu, V. Kyriakou, I. Roiban, M. Aouine, C. Tang, A. Caravaca, K. Kousi, I. Schreur-Piet, I. S. Metcalfe, P. Vernoux, M. C. M. van de Sanden and M. N. Tsampas, ACS Nano, 2019, 13, 12996–13005. Copyright 2019 American Chemical Society. High-voltage transmission electron microscopy micrographs showing the evolution with time and temperature of Ni nanoparticles from La0.7Ca0.2Ni0.25Ti0.75O3−δ under a H2 gas flow: (e) 800 °C, 24 h; (f) 900 °C, 3 h; and (g) 900 °C 24 h. Reproduced with permission from ref. 50. | ||
Kim et al. also conducted their work on Ni-doped lanthanum titanate, reporting shape alterations with temperature variation.50 La0.7Ca0.2Ni0.25Ti0.75O3−δ under a dry H2 atmosphere at temperatures ranging from 600–800 °C, either for short durations (3 h) or longer ones (24 h), resulted in the exsolution of spherical-shaped nanoparticles (Fig. 2e). But, when the temperature was raised up to 1000 °C, faceted nanoparticles were obtained, Fig. 2f and g. They proved the importance of exploring different nanoparticle shapes since the faceted Ni nanoparticles exhibited a longer and higher conversion of CH4 in dry reforming of methane reaction compared to spherical-shaped and infiltrated Ni catalysts.50
As previously mentioned, there are some other studies where non-spherical shaped nanoparticles are exsolved without reporting control over their shape. For instance, Fe-based nanorod formation was observed when La0.6Sr0.4FeO3−δ was exposed to H2 at 600 °C.52 For the same composition, flattened particles emerged when subjected to a 3% H2 (650 °C, 5 h) treatment,53 proving how the atmosphere plays, indeed, a relevant role modify specific morphological properties of the exsolved nanoparticles.
Finally, since exsolution can potentially be reverted under oxidizing conditions, partial or complete redissolution of the exsolved nanoparticles is expected under certain adequate conditions. For instance, one of the requirements for exsolved NP re-dissolution is that the perovskite is A-site soichiometric.12,14,16,54 However, Neagu et al. published some interesting results involving morphological changes affecting Ni and Co-Ni exsolved nanoparticles when subjected to a CO + O2 flow for carbon monoxide combustion. Under these conditions, more faceted shapes were first obtained, which led to cubic (Co,Ni)3O4 spinels instead of redissolution.55
2. Multi-elemental metallic nanoparticle exsolution
One fascinating aspect of metallic exsolution relies on the possibility of simultaneously exsolving several species and, thereby, creating multi-elemental nanoparticles in a relatively facile manner. This is mainly due to the easy compositional tunability of perovskite oxides, facilitating the incorporation of a wide variety of cations in both the A and B sites of the ABO3 perovskite structure. Specifically, the target has been to combine several cations with adequate thermodynamic properties for exsolution from the B-site. Here, it should be kept in mind that some metals, namely, Ag, usually occupy the A-site and have been shown to generate nanoparticles upon exsolution, although to date,56,57 metallic multi-elemental exsolution has mainly focused on metals located at the B-site of the perovskite. In this context, as metioned earlier, it is crucial to properly select metals for partial B-site substitution, as not every element is thermodynamically favored to exsolve. This matter has already been studied by Carrillo et al., showing the thermodynamic tendency for reduction (and, thereby, for exsolution) via calculations of Gibbs free energy (ΔG) for several metallic species.58 These studies reveal that metallic cations such as Ni, Fe, Co or several noble metals (Ru, Rh, Pt…) are thermodynamically favored for exsolution (ΔG < 0). In contrast, Mn, Ti or Mg are not expected to exsolve as metals. Therefore, accurate materials design via partial substitution of B-site atoms with exsolvable species can lead to a multicomponent exsolution process. Mainly, this phenomenon has been studied for bimetallic alloy exsolution, but in the past few years multi-elemental exsolution has experimented a remarkable development, aiming not only to achieve more complex alloys, but also to reveal the possibility of generating non-alloyed multicomponent nanoparticles.The exsolution of multicomponent nanoparticles has usually been related to binary alloys, and nowadays a major part of the published studies deals with this matter. In fact review articles about bimetallic alloy exsolution have already been published. For instance, the study by Tang et al. not only explores different exsolvable alloys (mainly concerning Fe, Co and Ni), but also investigates the mechanisms behind this process.59 Previous DFT studies performed by Kwon et al. focusing on Ni–Co alloy exsolution suggested that the most thermodynamically favored pathway for alloyed nanoparticle exsolution relies on the independent co-segregation to the surface of each metal together with oxygen vacancies 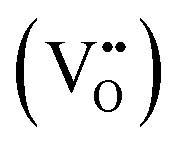 . Later, aggregation of Ni and Co species results in the formation of alloyed nanoparticles. This surface alloy formation (Fig. 3a) would be preferential over the bulk alloy formation, followed by segregation to the surface, according to the Gibbs energies calculated in this work.60
. Later, aggregation of Ni and Co species results in the formation of alloyed nanoparticles. This surface alloy formation (Fig. 3a) would be preferential over the bulk alloy formation, followed by segregation to the surface, according to the Gibbs energies calculated in this work.60
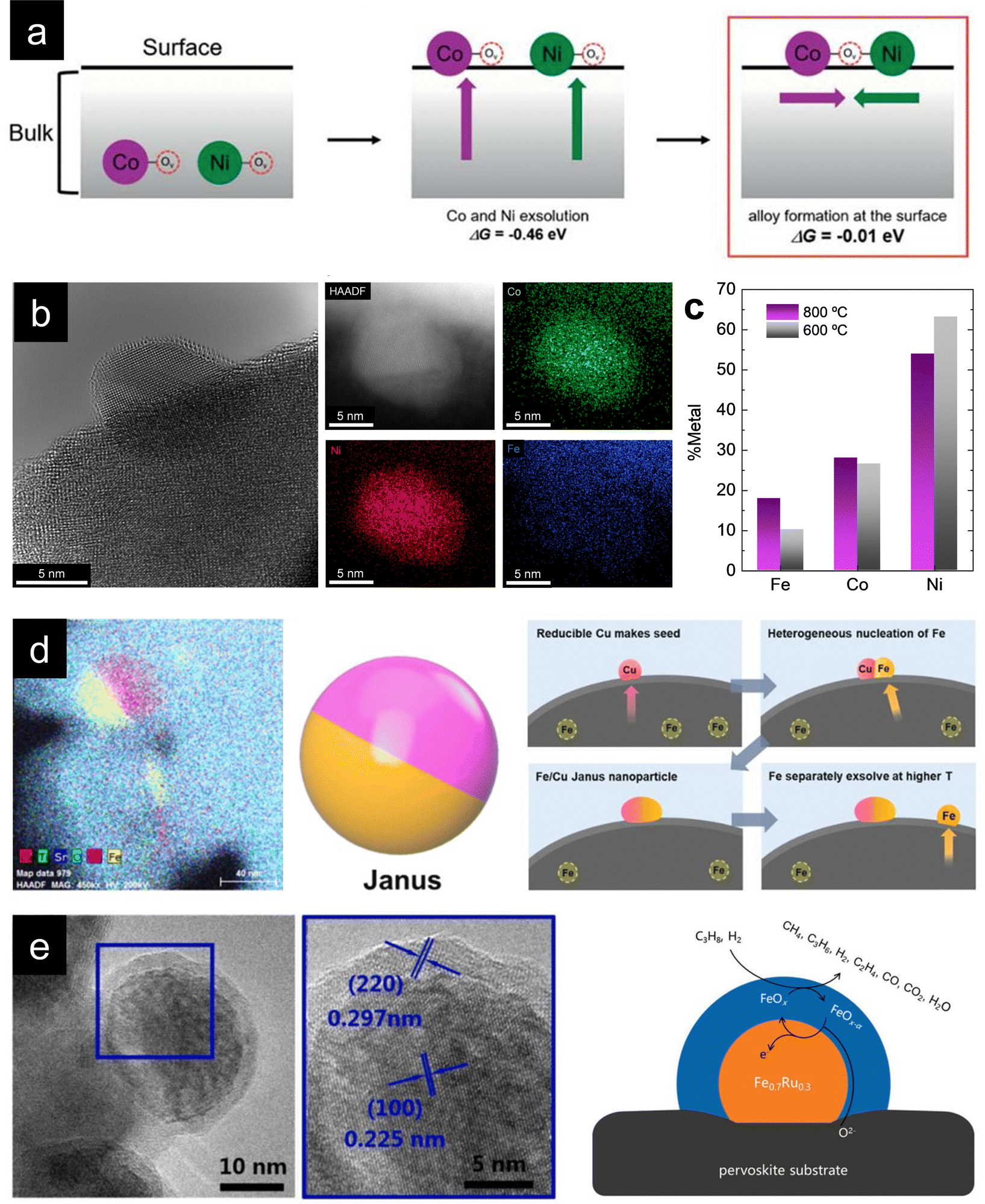 | ||
Fig. 3 (a) Thermodynamically favored mechanism for alloy exsolution. Co-segregation of each metal together with oxygen vacancies 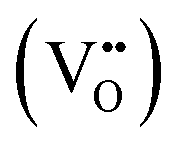 followed by superficial aggregation of metallic species (surface alloy formation) is preferential over bulk alloy formation. Reproduced with permission from ref. 60 with permission from the Royal Society of Chemistry. (b) High-resolution TEM and HAADF-STEM images, including EDXS mapping analyses, of ternary-alloyed exsolved FeCoNi nanoparticles from Sr2FeCo0.2Ni0.2Mn0.1Mo0.5O6−δ at 600 °C, 4 h, and a 5% H2/Ar atmosphere. (c) Compositional comparison of FeCoNi ternary alloys exsolved for 4 h, in a 5% H2/Ar atmosphere, at 600 and 800 °C. Exsolution temperature proved to be crucial for tuning the metallic rates in the generated alloyed nanoparticles. (d) Multi-elemental non-alloyed Fe–Cu exsolution of Janus type nanoparticles and co-segregation mechanism from La0.43Sr0.37Fe0.09Cu0.03Ti0.88O3−δ exsolution. Adapted with permission from ref. 61. (e) Multi-elemental exsolution of core–shell nanoparticles, consisting of a metallic Fe0.7Ru0.3 core and a FeOx shell. This structure has a key role in the reaction mechanism when employing these functionalized (Pr0.5Sr0.5)0.9Fe0.8Ru0.1Nb0.1O3−δ perovskite materials as anodes in an H2 or C3H8 SOFC. Adapted with permission from ref. 62. followed by superficial aggregation of metallic species (surface alloy formation) is preferential over bulk alloy formation. Reproduced with permission from ref. 60 with permission from the Royal Society of Chemistry. (b) High-resolution TEM and HAADF-STEM images, including EDXS mapping analyses, of ternary-alloyed exsolved FeCoNi nanoparticles from Sr2FeCo0.2Ni0.2Mn0.1Mo0.5O6−δ at 600 °C, 4 h, and a 5% H2/Ar atmosphere. (c) Compositional comparison of FeCoNi ternary alloys exsolved for 4 h, in a 5% H2/Ar atmosphere, at 600 and 800 °C. Exsolution temperature proved to be crucial for tuning the metallic rates in the generated alloyed nanoparticles. (d) Multi-elemental non-alloyed Fe–Cu exsolution of Janus type nanoparticles and co-segregation mechanism from La0.43Sr0.37Fe0.09Cu0.03Ti0.88O3−δ exsolution. Adapted with permission from ref. 61. (e) Multi-elemental exsolution of core–shell nanoparticles, consisting of a metallic Fe0.7Ru0.3 core and a FeOx shell. This structure has a key role in the reaction mechanism when employing these functionalized (Pr0.5Sr0.5)0.9Fe0.8Ru0.1Nb0.1O3−δ perovskite materials as anodes in an H2 or C3H8 SOFC. Adapted with permission from ref. 62. | ||
In this context, a wide variety of bimetallic alloys have been exsolved from a diverse range of materials, including not only perovskites (ABO3) and double perovskites (A2B2O6), but also other structures, like fluorite-based structures, (i.e., CeO2). The following sections will further discuss this matter in depth (see Section 6). Since bimetallic exsolution has already been widely explored by other authors, and this work focuses on the most recent trends in nanoparticle exsolution, this subsection will not delve into deep detail. However, it is essential to highlight the wide variety of applications in which bimetallic exsolution proved to be useful. For instance, Fe–Ni alloyed exsolved nanoparticles proved to perform better than non-alloyed Ni nanoparticles in CO oxidation reactions.63 Fe–Ni alloys have also been exsolved from perovskites to significantly improve the catalytic activity of (La0.75Sr0.25)(Cr0.5Fe0.5−xNix)O3 perovskites for methane reforming reaction.64 In fact, Fe–Ni alloyed nanoparticles (which generally form FeNi3 compositions) have been commonly explored as promising candidates for dry reforming of methane.65,66 In particular, our group found that increasing the Ni/Fe ratio on the B-site of Sr2Fe1−xNixMoO6−δ double perovskites enabled a greater extent of nanoparticle exsolution, which eventually increased CO2 and CH4 conversion.65 This result corroborates that Ni exhibits more favorable thermodynamics and kinetics for metal nanoparticle exsolution. Fe–Ni alloy exsolution also has a great interest in electrocatalysis applications, especially in solid oxide cells (SOCs).67–71
Other bimetallic alloys have proved to be versatile for applications, like FeCo exsolved nanoparticles. Just as in the FeNi case, materials functionalized with FeCo bimetallic exsolution exhibited enhanced catalytic activity in several applications, like in PrBaFeCoO5+δ double perovskite for dry reforming of methane.72 FeCo exsolution has also been employed successfully in solid oxide fuel cells (SOFCs),73 proton-conducting fuel cells (PCFC)74,75 or in double perovskite cathodes for CO2 electrolysis in solid oxide electrolytic cells (SOECs).76 This last work reported remarkable results regarding the possibility of redissolving FeCo alloyed exsolved nanoparticles from Co-doped Sr2Fe1.5Mo0.5O6−δ, extending the possibility of reversible exsolution to alloyed nanoparticles.
Most recently, alternative alloys involving noble metals have also been explored, mainly with Ru. Based on the same Sr2Fe1.5Mo0.5O6−δ double perovskite structure, Lv et al. obtained FeRu alloyed nanoparticles to functionalize CO2 electrolysis fuel electrodes.77 Nevertheless, the most remarkable result in this work was the demonstration of the promotion of exsolution through repeated redox manipulations. This work proved an increase in the extent of exsolution after several reduction–oxidation cycles using in-situ STEM. In addition, FeRu alloy exsolution caused a notable improvement in the SOEC performance. Guo et al. also obtained alloyed nanoparticles from double perovskites, namely, NiRu alloys from A-site deficient La2−xNiRuO6−δ for their application in symmetrical cells. Interestingly, this work showed the lowest thermal exsolution temperature achieved to date (350–450 °C).78
Besides this work, limited publications concerning the exsolution of ternary alloys have been published. Maybe the first direct exsolution of a ternary alloy was achieved by Santaya et al., who were able to exsolve ternary FeCoNi nanoparticles at 700 °C and 10% wet H2 in an Ar atmosphere from Sr0.93(Ti0.3Fe0.56Ni0.07Co0.07)O3−δ perovskites.80 These functionalized materials were tested as electrodes in symmetrical SOFCs, exhibiting a significant improvement in their performance after exsolution, indicating an enhancement in electrocatalytic activity. Most recently, another work has reported exsolution of FeCoNi from Sr0.9Co0.5Fe0.35Ni0.15O3−δ to improve the electrocatalytic performance for oxygen evolution reaction (OER), with satisfactory results.81
In this context, in our previous work, we wanted to achieve direct ternary alloyed nanoparticles from a double perovskite system based on Sr2Fe1.5Mo0.5O6−δ.31 The physicochemical properties of this family of materials make them exciting candidates for their use as electrodes for both SOEC82 and SOFC83 technologies or even symmetrical cells.84 As discussed in the binary alloy subsection, B-site partial substitution of Fe with other elements has already led to the direct exsolution of alloyed nanoparticles, for instance, FeCo, FeNi or FeRu. Moreover, B-site substitution of Fe with non-exsolvable elements, like Mn, demonstrated the possibility of improving specific properties of these materials, as Jiang et al. showed that in their work with Sr2Fe1.4Mn0.1Mo0.5O6−δ double perovskites.82 For all these reasons, we developed Sr2FeCo0.2Ni0.2Mn0.1Mo0.5O6−δ fuel electrodes for CO2 electrolysis in SOEC devices.31
With this material, FeCoNi exsolution was achieved (Fig. 3b) for the first time in this kind of double perovskites at different treatment temperatures (600, 700 and 800 °C) to investigate morphological changes of the nanoparticles with variations in external parameters. Lower exsolution temperatures led to higher population rates and smaller nanoparticles, namely, 1800, 1200 and 550 μm−2 (dispersion) and 8.9, 11.6 and 21.3 nm (mean sizes) at 600, 700 and 800 °C, respectively. These results showed that the morphological properties of the exsolved nanoparticles can be tuned by changing external treatment parameters, in this case, temperature. However, during this research, we wanted to achieve more profound control over the exsolution of ternary alloys, more specifically, over the composition of the exsolved alloyed nanoparticles. For this purpose, compositional analyses of the obtained nanoparticles via EDXS were carried out, comparing the generated nanoparticles at 600 and 800 °C. The question was if, besides morphological properties, the composition of the alloys could also be controlled and tuned by changing the exsolution treatment parameters, focusing on the applied temperature. Santaya et al. showed that the nanoparticles contained similar amounts of the three metals, whereas in our case, Ni is the main component of the alloy for both 600 and 800 °C exsolutions, followed by Co and Fe (Fig. 3c). Nevertheless, interesting changes can be observed with temperature variation during exsolution since Fe content experienced a twofold increase at 800 °C compared to the 600 °C treatment. Co amounts remain relatively unaltered, while Ni amount decreases after 600 °C reduction. These results proved, for the first time to our knowledge, that in addition to morphology, the composition of exsolved alloyed nanoparticles can be easily modified by adjusting external parameters of the reduction treatment, which can lead to a more rational and efficient design of catalysts via in-situ exsolution.
Other alternative multi-elemental nanoparticles exsolved are core–shell structures. Several works deal with this matter, such as Qin et al. publication, in which exsolved (Pr0.5Sr0.5)0.9Fe0.8Ru0.1Nb0.1O3−δ perovskite was employed as an electrocatalyst for SOFC anodes.62 After 900 °C and 2 h under wet H2 treatment (about 50 nm of mean size), the exsolved nanoparticles exhibited a clear core–shell structure, as seen in Fig. 3e. This core consisted of a FeRu alloy (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 molar ratio) and a thinner shell of Fe oxides (FeOx). This core–shell structure seems to be crucial for explaining the reaction process, and the authors proposed a reaction mechanism involving both the core and shell of the exsolved nanoparticles, exhibiting promising electrocatalytic results when using wet H2 or wet C3H8 as fuel. In addition, and as expected due to exsolution intrinsic advantages, the functionalized material showed a notable resistance against coke deposition. Other works have also demonstrated this core–shell (metal–metal oxide) exsolution. Namely, NiFe/FeOx nanoparticles from Pr0.4Sr1.6(NiFe)1.5Mo0.5O6−δ double perovskites for their use as CO2 electrolysis cathodes in SOECs89 or even Co/CoOx in an interesting work involving porous nanotubular webs of double perovskite materials (PrBa0.5Sr0.5Co0.2Fe1.8O5+δ).90
:30 molar ratio) and a thinner shell of Fe oxides (FeOx). This core–shell structure seems to be crucial for explaining the reaction process, and the authors proposed a reaction mechanism involving both the core and shell of the exsolved nanoparticles, exhibiting promising electrocatalytic results when using wet H2 or wet C3H8 as fuel. In addition, and as expected due to exsolution intrinsic advantages, the functionalized material showed a notable resistance against coke deposition. Other works have also demonstrated this core–shell (metal–metal oxide) exsolution. Namely, NiFe/FeOx nanoparticles from Pr0.4Sr1.6(NiFe)1.5Mo0.5O6−δ double perovskites for their use as CO2 electrolysis cathodes in SOECs89 or even Co/CoOx in an interesting work involving porous nanotubular webs of double perovskite materials (PrBa0.5Sr0.5Co0.2Fe1.8O5+δ).90
In summary, although multi-elemental exsolution is in its initial stages of development, it brings a promising opportunity for a more rational and efficient design of nanocatalysts via in-situ exsolution. The intrinsic advantages of exsolution, together with the ability to control the morphological and/or compositional properties of the exsolved nanoparticles, either alloys or non-alloyed multi-elemental structures, pave the path for expanding the potential applications of this kind of catalysts, leading to more efficient, highly active and resistant functionalized materials.
3. Exsolution of non-metallic nanoparticles
 | ||
| Fig. 4 (a) and (b) SEM micrographs of Ba0.5La0.4Lu0.1CoO3 depicting the oxide nanoparticle decoration over this double perovskite. (c) HAADF and EDXS mappings of an exsolved nanoparticle composed of Co, La, Ba and O from Ba0.85La0.15Co0.75Lu0.25O3−δ. Reproduced from ref. 35 with permission from the Royal Society of Chemistry. | ||
The thermodynamics of oxide exsolution were initially assessed by Tsvetkov et al.93 for Pr1−xBa1−xCo2O6−δ, a much more well-known cobalt-based double perovskite.94–96 In their work, Tsvetkov et al. achieved oxide exsolution after treating the PrBaCo2O6−δ sample at 1000 °C in an atmosphere with pO2 = 10−3 atm, whereas in their case, oxygen annealing did not lead to any exsolution. Based on thermodynamic calculations and oxygen content thermogravimetric data under different conditions, the authors identified these 30–40 nm nanoparticles as CoO, although depending on the temperature and pO2 used, Co3O4 could also be formed.93 However, recent works illustrate that this type of preliminary calculations might not be enough to predict the nature of the oxide-exsolved nanoparticles. In this sense, results found in the literature show that different compositions can be obtained from almost identical perovskite materials. For instance, Zhou et al. obtained BaCoO3−δ exsolved nanoparticles (identified through Raman spectroscopy) from PrBa0.8Ca0.2Co2O5+δ used as positrodes in PCECs.97 In a work by the same group, Hyuk Kim et al. also obtained exsolved BaCoO3−δ, which greatly promoted the electrocatalytic activity of these positrodes, through a water-induced surface modification.91 The treatment involved exposure to 3 vol% H2O/air for 3 h at 600 °C, resulting in particles smaller than 100 nm, which remained stable after operation at a constant cell voltage (0.65 V) for 230 h.91 Despite the promise, it remains unclear why Tsvetkov's predictions pointed to Co3O4/CoO exsolution from Pr1−xBa1−xCo2O6−δ, whereas Liu's group obtained BaCoO3 exsolutions from PrBa0.8Ca0.2Co2O5+δ. The same group also obtained BaCoO3 exsolution from Pr1−xBaxCoO3−δ thin-film coatings used for enhancing the electrocatalytic activity of (La0.6Sr0.4)0.95Co0.2Fe0.8O3−δ.98 Thus, Ca-substitution at the A-site could be ruled out as a possible reason for forming BaCoO3 instead of binary cobalt oxides. More recently, water-mediated exsolution has also been employed to functionalize alkali-metal doped BCFZY AxBa1−xCo0.4Fe0.4Zr0.2−yYyO3−δ (A = Li, Na, and K) with BaOx nanoparticles.99 Here, the choice of dopant highly affected the morphology of the exsolved nanoparticles. Li-doped and K-doped resulted in nanoparticles of 166 and 84 nm, respectively, whereas Na-doped size was too small for proper image analysis. This water-mediated exsolution (under 3% H2O wet air, 600 °C) resulted in faceted nanoparticles. The authors ascribed the formation of BaOx exsolved nanoparticles to the creation of oxygen and barium vacancies resulting from the acceptor doping strategy on the A-site. Based on DFT calculations, the authors demonstrated that Li and K doping decreased the energetics of barium and oxygen vacancy formation, thus increasing the driving force for BaOx exsolution. In particular, the K-doped sample showed a remarkable decrease in polarization resistance (44%) after 5 h of treatment in wet conditions, revealing the positive impact of BaOx exsolution on the electrochemical performance of PCFC.99
Based on these results, it is clear that more systematic experimental studies and computational calculations on oxide exsolution from perovskites are needed to discern the effects of the different processing conditions (pO2, moisture, temperature) and material-related aspects (strain, A-site deficiency) on oxide exsolution. It will be critical to obtain processing guidelines to aid a targeted exsolution of a specific compound.
Oxide exsolution has also been achieved from non-perovskite materials. Namely, PrOx was exsolved from (Ce0.9Gd0.1)1−xPrxO2−δ, significantly reducing the polarization resistance of these electrodes used in solid-oxide electrochemical cells.100 PrOx-exsolved nanoparticles were obtained after thermal treatments of 800 °C for 50 h in air. The particles are transformed into elongated needle-like nanoparticles by increasing the treatment time to 200 h.100
These works have served as a proof of concept for the exsolution of new chemistries (oxides, sulphides, and phosphides), which could greatly expand the benefits of these methods to other catalytic processes. However, conducting more systematic analyses, where the treatments are evaluated under different conditions, will be crucial for a better understanding of this process, unveiling the mechanisms behind the direct exsolution of these non-metallic compounds.
4. Non-thermally driven nanoparticle exsolution
Metallic nanoparticle exsolution has been conventionally triggered by thermal reduction treatments in a (diluted) H2 atmosphere. Different times, temperatures and H2 partial pressures have been utilized, which, depending on the perovskite system, have resulted in varying particle sizes, nanoparticle dispersion or even particle morphology (see Section 1). Probably the most studied perovskite system in exsolution has been La1−yAyTi1−xNixO3−δ in which A = Sr, Ca or Ce, often with A-site deficiency to favor the formation of exsolved nanoparticles. Anyhow, this perovskite class requires high temperatures and long treatments to reach nanoparticle exsolution. For instance, La0.43Ca0.37Ni0.06Ti0.94O3−γ electrodes required reduction with hydrogen at 900 °C for 20 h to reach a population of around 100 particles per μm2.37 These dispersion values are low compared to other perovskite systems in which at similar temperature conditions and shorter exposure times show a 6-fold improvement.31 In this sense, several studies have attempted to reach higher nanoparticle populations by using unconventional approaches in which exsolution is triggered by other phenomena different from thermal reduction. These are electrochemical-driven, plasma and microwave-assisted exsolution (Fig. 5a) and laser-induced exsolution. Most of these treatments allow for exsolution in the absence of an H2 atmosphere and at milder temperatures, reaching remarkable nanoparticle dispersions.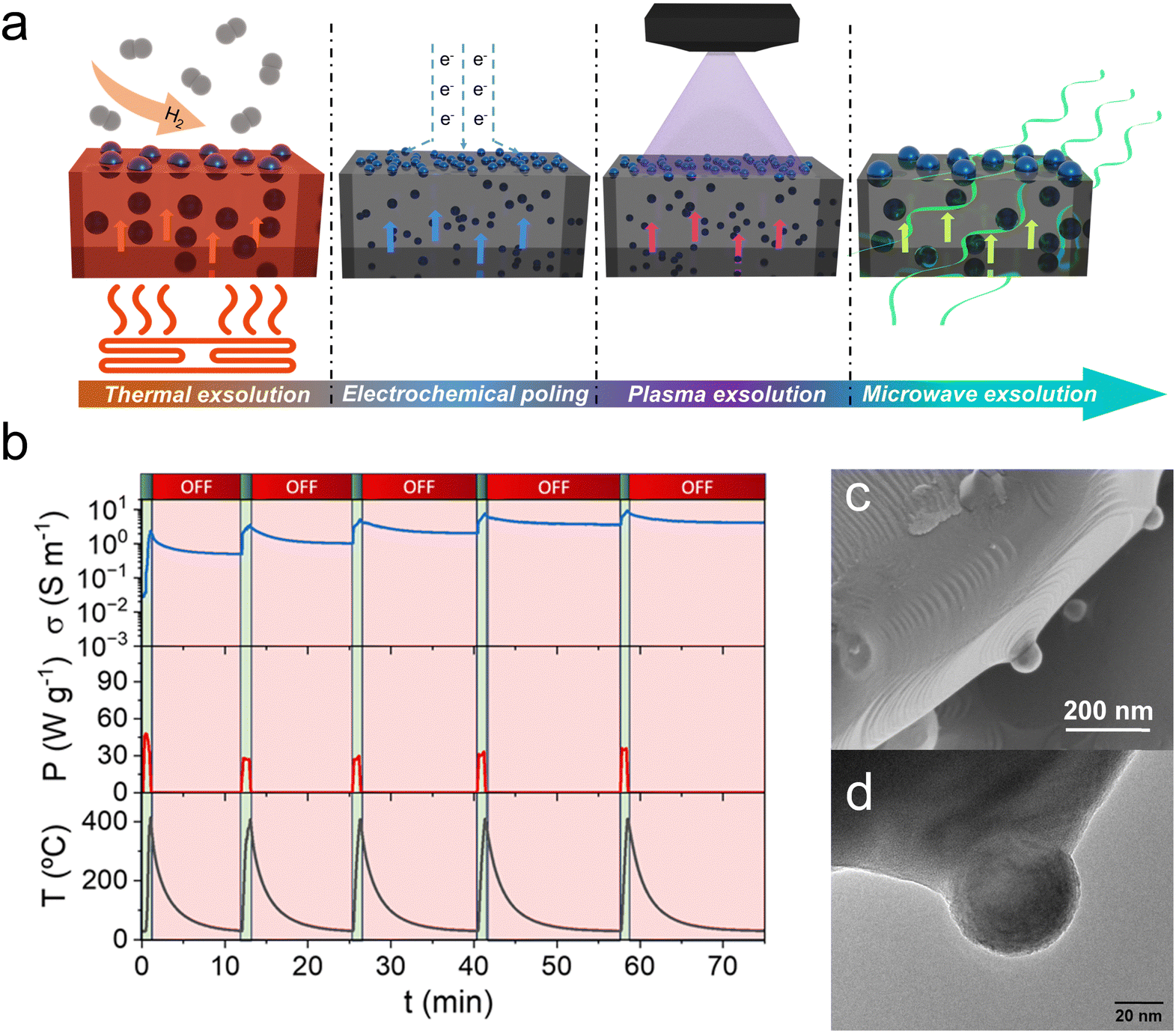 | ||
| Fig. 5 (a) Schematic of the different approaches used to drive the exsolution of metallic nanoparticles. (b) Plot of five microwave pulses utilized to drive the exsolution of Ni nanoparticles from La0.43Ca0.37Ni0.06Ti0.94O3−δ. The microwave-cavity reactor in which these experiments were performed allowed measuring the materials electrical conductivity during the microwave-driven exsolution process. This process was performed at low temperatures (400 °C) and just in the presence of inert N2 flow, avoiding the use of H2. (c) SEM and (d) TEM micrographs depicting spherical Ni nanoparticles exsolved by applying microwave radiation. Adapted from ref. 40. | ||
Lu and co-authors have studied the effects of the applied potential on the exsolved nanoparticle dispersion in more detail using La0.43Ca0.37Ti0.94Ni0.06O3−δ electrodes.106 The uniqueness of this work is that the authors designed an electrochemical device in such a way that the applied voltage was spatially graded. At 0 V (900 °C, 5 min exsolution), which correlates with pO2 < 10−20 atm (5% H2 atmosphere), the exsolved nanoparticle population was equal to 90 particles per μm2. However, in the region where a higher cathodic potential of −1.37 V was applied (equivalent to a pO2 of ca. 10−45 atm), the nanoparticle population increased to 205 particles per μm2.106 Interestingly, nanoparticle size did not suffer alterations when varying the voltage, indicating that on increasing the cathodic voltage, nucleation of the exsolved nanoparticles is promoted but not the growth of the exsolved nanoparticles. This fast nucleation driven by electrochemical switching was also confirmed in La0.52Ca0.28Ni0.06Ti0.94O3 fibers compared to conventional exsolution, which helped increase the activity of these electrodes.107 As commented above, in the work of Lu et al., the particle size of exsolved Ni in La0.43Ca0.37Ti0.94Ni0.06O3−δ electrodes was not affected by the applied potential.106 However, this effect might be material-specific and/or heavily influenced by the gas atmosphere conditions. In a recent work by Kim et al., electrochemical-driven exsolution was performed in La0.7Sr0.2Co0.2Fe0.8O3 electrodes for SOECs.108 Here, the authors report Co exsolution under cathodic bias. Quite interestingly, CoFe alloys were not formed. Moreover, the authors observed that increasing the applied voltage induces the growth of the exsolved nanoparticles.108 At low cell voltages, −0.5 V for 10 min, exsolved nanoparticles of around 30 nm appeared on the surface of La0.7Sr0.2Co0.2Fe0.8O3.108 The authors suggest that at such low voltages and before the point at which the electrochemical switching happens (−0.98 V) these Co nanoparticles are partially oxidized by H2O and/or CO2 used for electrolysis. After this point, completely reduced Co nanoparticles are obtained, however, with a remarkable increase in nanoparticle size up to 200 nm observed at −1.5 V.108
In summary, electrochemical-driven exsolution is a promising tool for (but limited to) the functionalization of electrodes in SOFCs, electrolyzers or other electrochemical devices. It has been proven under different atmospheres and applied cathodic voltages, and even using anodic shocks.109 The reported scientific evidence clearly indicates that electrochemical switching accelerates exsolved nanoparticle exsolution, obtaining remarkable dispersion in a few minutes. However, more fundamental works are required to decipher the underlying mechanisms of electrochemical poling, unveiling the role of process conditions and material properties on the exsolution extent.
A second work that deals with plasma-driven Ni exsolution is by Khalid et al. using an identical composition La0.43Ca0.37Ti0.94Ni0.06O2.955.110 Here, the exsolution was performed in Ar plasma (0.3 mbar vacuum pressure), applying a power of 40 W for times ranging from 10–20 min, and importantly, at room temperature. Even with just a 10 min treatment, it was possible to exsolve nanoparticles of ca. 20 nm and 60 particles per μm2 dispersion, which increased to 510 particles per μm2 by increasing the exposure time to 20 min.110 This remarkable result achieved in such a short exsolution time and at such low temperatures illustrated the potential of plasma-assisted exsolution. In both works, the plasma-treated samples presented stable catalytic activity for CO2 hydrogenation39 and CO oxidation reactions.110
More recently, a similar approach was applied to a non-perovskite oxide (in section 6 this topic is covered in more detail). In this case, photo-thermal exsolution was utilized to exsolve precious metals, viz. Pt, Rh, and Ir, from WO3.111 By applying an intense pulsed light-derived momentary photothermal treatment (reaching temperatures higher than 1000 °C), the exsolution process was achieved in an unprecedented short period of time of less than 20 ms.111 This method resulted in exsolved nanoparticles of around 5 nm, which presented remarkable sensing performance in the detection of H2S.111
5. Exsolution in solid–solid interfaces, endogenous nanoparticles and embedded metallic clusters
Nanoparticle exsolution has gained attention mostly due to its robustness, which provides high stability in high-temperature thermo- or electrochemical catalytic processes. Thus, it has been mainly employed to enhance the catalytic activity in gas-solid interfaces. However, recent works have shown that exsolved nanoparticles can also be employed to functionalize materials when completely embedded in solid–solid interfaces. There are two main paths to fabricate exsolved nanoparticles uniquely surrounded by one or more solid phases. First, by intrinsically generating the nanoparticles during the exsolution process that do not reach the surface generating sub-surface metallic nanoclusters or endogenous nanoparticles in the oxide bulk. The second approach involves the decoration of a surface via a conventional exsolution process followed by deposition of an oxide (or any other solid compound) over the functionalized surface, see Fig. 6a. While the former case was already reported a decade ago,114,115 for the second approach, there is a unique report developed by Spring et al. from Prof. Rupp's group.6 This specific work aimed at functionalizing the bottom electrode/switching oxide interface of a resistive switching device (Fig. 6b) to favor filament confinement, eventually improving their performance. The strategy here was to perform Ni exsolution, Fig. 6c, from La0.2Sr0.7Ti0.9Ni0.1O3−δ at different times and temperatures, resulting in thin films (fabricated through pulsed laser deposition (PLD) on a LaAlO3 substrate) with exsolved nanoparticles of various sizes and dispersions. The next step was depositing, also through PLD, a layer of amorphous SrTiO3 and, on top, a Pt electrode via sputtering. This nano-fabrication route was effective in improving cycle-to-cycle variability, which is one of the issues that this type of devices encounter.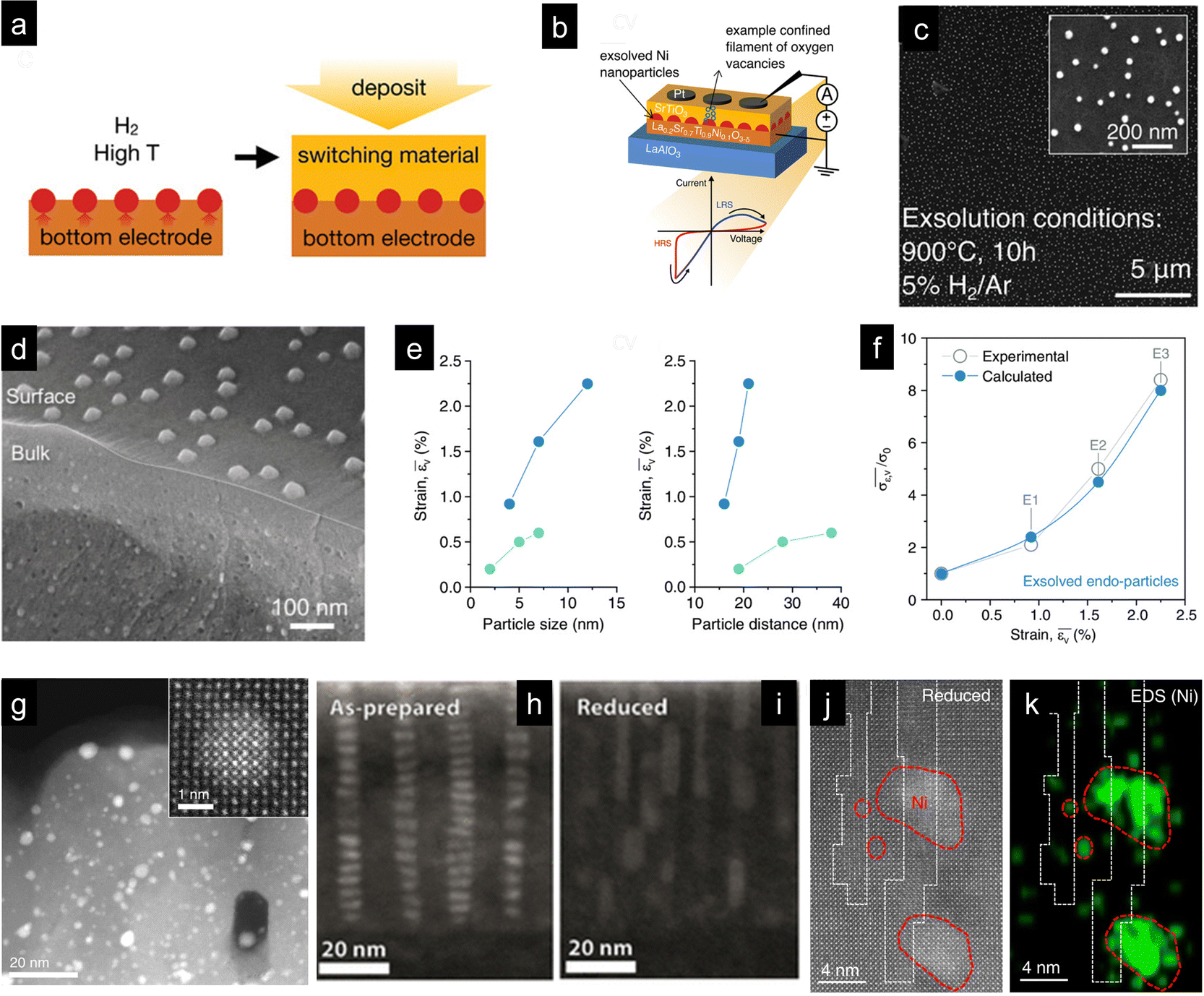 | ||
| Fig. 6 (a) Schematic depicting the fabrication strategy to functionalize the perovskite-based bottom electrode in resistive switches (b) with exsolved metallic nanoparticles. In the first step, the perovskite thin film, La0.2Sr0.7Ti0.9Ni0.1O3−δ, fabricated through pulsed laser deposition (PLD) on a LaAlO3 substrate was reduced to generate exsolved Ni nanoparticles. Afterwards, a second PLD thin film was deposited on top of the exsolved perovskite, rendering a solid–solid interface in which exsolved Ni nanoparticles were embedded. (c) SEM micrograph of Ni nanoparticle exsolution from the bottom electrode thin-film La0.2Sr0.7Ti0.9Ni0.1O3−δ thin film after reduction for 10 h at 900 °C. Reprinted with permission from ref. 6. (d) SEM micrograph depicting bulk and surface exsolution of Ni nanoparticles in La0.8Ce0.1Ni0.4Ti0.6O3 perovskite powders used as oxygen carriers in chemical looping syngas production. (e) Influence of particle size and interparticle distance of endogenous nanoparticles on strain. Dark blue dots represent endogenous nanoparticles created via exsolution, whereas light blue dots represent nanoparticles created by assembling and sintering. (f) Influence of strain generated with the exsolved endo-particles on the conductivity. Reproduced from ref. 44. (g) STEM-HAADF micrograph of a thin film with Pt exsolved clusters formed in a Pt-doped CaTiO3 film after a reduction treatment. The inset shows a metallic Pt cluster totally embedded in the perovskite thin film. Reproduced with permission from ref. 115. STEM-HAADF micrographs of SrTi0.9Nb0.05Ni0.05O3−δ thin films before (h) and after reduction (i). Reprinted (adapted) with permission from M. L. Weber, M. Wilhelm, L. Jin, U. Breuer, R. Dittmann, R. Waser, O. Guillon, C. Lenser and F. Gunkel, ACS Nano, 2021, 15, 4546–4560. Copyright 2021 American Chemical Society. (j) STEM-HAADF and (k) EDXS mapping of Ni clusters formed in a La0.2Sr0.7Ni0.1Ti0.9O3−δ thin film in anti-phase boundaries. Reproduced with permission from ref. 116. | ||
Besides this work, most of the reports on metallic nanoparticle exsolution in solid–solid interphases are related to nanoparticles formed during the exsolution process or sometimes by carefully adjusting the process conditions. Perhaps the most relevant work covering this latter aspect is by Kousi et al., who were able to exsolve Ni nanoparticles both at the perovskite surface and at the bulk of La0.8Ce0.1Ni0.4Ti0.6O3 perovskite powders (Fig. 6c). Interestingly, as depicted in Fig. 6c, the particles in the bulk, named “endogenous”, are smaller than the particles at the surface, namely, 13 and 45 nm, respectively.43 To obtain a material with such morphology, authors strategized a set of intrinsic and extrinsic parameters that effectively resulted in the targeted objective: a perovskite with relatively low cationic diffusion (to favor bulk versus surface exsolution), a high content of exsolvable metal transition cations on the B-site, A-site deficiency, and high exsolution temperatures (1000 °C) and exposure times (10 h) to help the nucleation and growth in the bulk, since this latter process has higher energy requirements than surface exsolution.43 However, the beauty of this work relies not only on the smart materials design using the aforementioned combination of material-related properties and treatment conditions but also on the effect caused by the endogenous nanoparticles. By forming the endogenous Ni NPs, the authors induced tensile strain in the perovskite oxide, leading to an increase in the oxygen exchange capacity of these oxides. This enhancement in lattice oxygen release improved the methane partial oxidation activity in the chemical looping syngas production reactions in which these materials were tested. This enabled working at unprecedented low temperatures in methane-assisted chemical looping (450 °C).42 These works unlocked a new functionality of nanoparticle exsolution, in which the nanoparticles are utilized to induce tensile strain in the oxide, where more conventionally, exsolution is aimed at catalyzing surface reactions.42 This could be considered a major breakthrough in the field since there are not many available technologies to induce tensile strain in bulk oxides. Only a few works by Ishihara's group, in which gold nanoparticles were embedded in perovskite electrodes in SOFCs, have been reported to date.117,118 In a recent work, Dalton and Neagu have evaluated the strain effects of both the exsolution-driven pathway and the route proposed by Ishihara's group.44 Their modelling results allowed identifying the amount of strain produced when varying the nanoparticle size and the nanoparticle distance for both approaches and the impact of strain on the ion conductivity.44 Strain increased with both increasing nanoparticle size and the interparticle distance (Fig. 6e), and the strain determined for the exsolution approach helped in enhancing the conductivity exponentially (Fig. 6f).
These works have revealed the great potential of using exsolution of endogenous nanoparticles as a strain-engineering strategy in metal oxides, which could be easily scaled up. Most works on strain engineering in oxides have been devoted to thin films, in which strain is usually induced by a lattice mismatch between the deposited oxide and the oxide substrate used. Indeed, this approach has been utilized to demonstrate the effects of strain on nanoparticle exsolution in thin films. For instance, Han et al. proved that compressive strain helped in increasing the nanoparticle dispersion compared with tensile strain,41 revealing the benefits of precise strain engineering in the development of exsolved nanocatalysts.
Thin films have served as a magnificent playground for fundamental studies that helped in unveiling the factors governing metallic nanoparticle exsolution.119 Moreover, the first reports on “bulk” exsolution of metallic nanoclusters were first observed in Pt-doped CaTiO3 thin films by Katz et al.,115Fig. 6g. This work pioneered in revealing that Pt clusters not only formed as exsolved nanoparticles on the perovskite surface but could also form metallic clusters fully embedded in the oxide sub-surface.115 Interestingly, after treatment in oxidation conditions, not all of these Pt nanoclusters re-dissolve back in the perovskite bulk, indicating a low extent of reversibility of these “bulk” exsolution processes.
In some perovskite systems, nanoclusters have been identified even under pristine conditions after thin-film deposition. This is observed in the case of NiOx clusters that were observed in SrTi0.9Nb0.05Ni0.05O3−δ thin films.120 This phase separation, Fig. 6h and i, occurred even before the reduction treatment, in which these B-site dopants formed columns of nanoclusters spontaneously.120 According to the authors, these columns of oxidized nickel served as preferential spots for the exsolution of metallic Ni.120 These results revealed an alternative mechanism for metallic nanoparticle exsolution in stoichiometric thin films, in which these capsule-like nanoclusters served as nuclei for metallic nanoparticle exsolution.120 More recently, Guo et al. proved that by varying the deposition conditions (vacuum pressure) it was possible to also obtain different nanostructures embedded in the LaFe0.7Co0.1Ni0.1Ru0.05Pd0.05O3−δ thin-films. Namely, with pressures of 0.15 mTorr in the PLD process, self-assembled nanorods were obtained, whereas at 0.015 mTorr, core–shell NPs were observed. The outter shell was formed by a Ni–Co oxide, whereas the core was composed of Pd.121
Following thin-film studies on titanate-based perovskites, works have also focused on A-site deficient perovskites, such as (La0.2Sr0.7Ni0.1Ti0.9O3−δ).116 In this case, the amount of exsolved nanoparticles was one order of magnitude higher (795 vs. 51 NPs per μm2) when comparing the non-stoichiometric and stoichiometric perovskites.116 The authors ascribed this higher nucleation extent to the formation of anti-phase boundaries in the perovskite, which facilitated the diffusion of Ni to the surface.116Fig. 6j and k depict the HAADF-STEM and EDXS mappings for Ni, in which the antiphase boundaries in the perovskite are marked by a dashed line.116 Authors observed the formation of Ni clusters in these regions that served as paths for the migration of Ni toward the surface of the perovskite in which they nucleate as exsolved nanoparticles, exhibiting high dispersion.116 These studies on thin films revealed the concomitant formation of metallic clusters or endogenous nanoparticles as well as exsolved nanocatalysts on the surface for both stoichiometric and non-stoichiometric perovskite thin films. Even though, for each case, a different mechanism seems to determine the exsolution kinetics, it is clear that crystal defects are crucial in accelerating the exsolution of metallic nanoparticles. Thus, defect engineering emerges as a major tool for improving the extent of metallic exsolution, which could be smartly used for generating endogenous exsolved nanoparticles. Despite the promise, scaling up the observations obtained in thin-film fundamental studies might not be straightforward for bulk oxide materials. However, the promising results regarding exsolved endogenous nanoparticle formation in bulk perovskite oxygen carriers and its superior oxygen exchange capacity, demonstrated for chemical looping syngas production, illustrate the potential of this novel approach.
6. Exsolution from non-perovskite oxides
Exsolution from non-perovskite oxide materials has fewer publications dedicated to it,1 yet it is a promising field of research. For instance, it is especially relevant in oxides commonly used as supports for metal nanoparticles in thermocatalytic processes, such as CO2 hydrogenation or hydrocarbon reforming. The latter are endothermic reactions that occur at high temperatures, usually resulting in nanoparticle agglomeration. Therefore, improving materials functionalization is crucial for developing more efficient and resistant catalysts, where the exsolution method can emerge as a promising alternative.In addition, these materials are widely employed in high-temperature electrocatalytic proceses.122 Conventionally, doping is a usual procedure to improve the electrochemical properties of these materials. For instance, doping ceria with trivalent cations such as gadolinium increases its ionic conductivity.124,125 These materials are employed in a wide range of high-temperature processes in which surface reactions are crucial. Thus, functionalization with robust exsolved nanoparticles would be highly convenient. However, the low solubility of transition metal cations in ceria and other fluorite structures can limit the compositional tunability of these materials.
One of the first reports of exsolution in fluorite-type oxides was probably on Ru exsolution from pyrochlores.126 This exsolution process was performed on Sm2RuxCe2−xO7 (with x = 0, 0.1, 0.2, and 0.4), where 10% H2 was employed to exsolve metallic Ru nanoparticles, showing a mean size of about 1–2 nm (Fig. 7a). This exsolved material was tested for dry methane reforming reactions, showing higher catalytic activity and stability when compared to impregnated catalysts, highlighting the potential benefits of exsolution in high-temperature thermocatalytic processes for syngas production.
 | ||
| Fig. 7 (a) TEM micrographs of Ru exsolution (10% H2, 700 °C, 1 h) from Sm2Ru0.2Ce1.8O7. Reprinted (adapted) with permission from M. A. Naeem, P. M. Abdala, A. Armutlulu, S. M. Kim, A. Fedorov and C. R. Müller, ACS Catal., 2020, 10, 1923–1937. Copyright 2020 American Chemical Society. (b) and (c) TEM micrographs of Ru exsolution (5% H2, 900 °C, 2 h) from Ru-doped CeO2. Reproduced from ref. 49 with permission from the Royal Society of Chemistry (d) SEM micrograph and XRD diffraction pattern of Ni nanoparticles exsolved from CeO2 monoliths. Reproduced with permission from ref. 127. (e) STEM and EDXS map analysis of Cu exsolution from CuFe2O4 spinels. Reproduced with permission from ref. 128. | ||
Our group also reported Ru exsolution from ruthenium-doped ceria (Ru0.01Ce0.99O2−δ), which is the first publication on exsolution in CeO2.49 Ru nanoparticles (5 nm) were exsolved with a 5% H2 flow in Ar at 900 °C for 2 hours (Fig. 7b and c). This new catalyst was tested in chemical looping reforming of CH4 coupled with CO2 splitting, exhibiting higher CO2 conversion at the same temperature as the undoped material and stable syngas production during 20 cycles. Remarkably, the well-anchored Ru nanoparticles remained morphologically unaltered (nanoparticle size of ca. 5 nm) after prolonged high-temperature treatment, evidencing that exsolution could be a promising superficial functionalization method for CeO2. Regarding Ru as the exsolvable cation, Hu et al., tested a composite material consisting of mixed Sr2Fe1.5Mo0.5O6−δ with Gd0.1Ce0.9O2−δ to increase ionic conductivity, both doped with Ru.129 They demonstrated that, under an H2 or CH4 atmosphere at 800 °C, Ru metallic nanoparticles exsolved over the surface of both materials, generating a carbon high-resistant anode with better performance compared to unaltered perovskites for methane conversion. This was the first work in which dual exsolution has been proven. On the other hand, changing the doping metal to Rh can generate efficient catalysts for hydrogen production through hydrocarbon steam reforming. Kim et al. used a rhodium-doped (8 wt%) ceria to generate exsolved nanoparticles (∼3 nm) under a 10% H2 atmosphere.130
As described in previous sections, Ni is the most common cation for exsolution in perovskites. Nevertheless, doping ceria or zirconia with transition metals such as Ni, Fe, or Co can be challenging due to the notable difference in ionic radius with host cations (Ce4+ or Zr4+vs. Ni2+).131 This size variation may limit the solubility and usually triggers the formation of secondary phases.132 However, two works report Ni introduction in ceria oxides, using different conditions and showing some drawbacks. Qi et al.133 reached 1000 °C to achieve metallic Ni exsolution under 5% H2 in Ar flows, while Xiao et al.127 tests involved the use of pure hydrogen. Although both reports represent a significant advance in Ni exsolution from fluorite-type oxides, the first approach133 suffers from Ni agglomeration when a 0.2% (wt) doped catalyst is used for CO2 direct electrolysis. The second approach127 reports the partial oxidation of Ni when exposed to CH4, reducing the amount of active catalyst available. One exciting aspect of the work of Xiao et al. is that they fabricated porous single-crystalline NixCe1−xO2 monoliths with high porosity (ca. 68%), from which they exsolved 4 nm Ni nanoparticles (Fig. 7d).127 This is relevant since obtaining highly porous single-phase oxides remains a challenge, especially when several cations are employed for the exsolution of multicomponent metallic nanoparticles. Finding a fabrication route that can yield exsolved materials with high specific surface areas will be a breakthrough in the field since it will expand the use of exsolution to other low-temperature processes in which the control over porosity is crucial.
Furthermore, NiO exsolution on CeO2-based structures was achieved thermally without reducing agents. In this case, Gd0.2−xNixCe0.8O2−δ heated to 1250 °C exsolves Ni-rich nanoparticles, which, due to the high-temperature annealing under oxidizing conditions (air), resulted in NiO exsolution. Further reduction in hydrogen at 650 °C is needed (as in other common functionalization methods) to generate the final Ni nanoparticles (∼15 nm). This could be a promising approach to lower the operation temperature in fuel cell performance (up to 500 °C).134
Finally, as discussed in Section 2, binary alloy exsolution has recently been achieved by Lee et al.135,136 More specifically, PtRu alloyed nanoparticles were obtained from Ce0.9Gd0.1O2−δ (CGO) for its use as a catalyst in the commercial diesel reforming process. Interestingly, after demonstrating the effective introduction of both Pt and Ru into the CGO lattice, exsolution of ∼10 nm nanoparticles occur during the reaction, resulting in an in-situ exsolution. The generated PtRu alloy exhibited high conversions (even at low reaction temperatures) and excellent stability during long operation times (up to 200 h).
Regarding other fluorite structures, such as the previously stated ZrO2, exsolution of Ni metal has also been reported. It is common to find the ZrO2 structure doped with Y3+ (YSZ) as it imparts phase stability to the ceramic material at high temperatures.131 Joo et al. demonstrated that YSZ can be doped up to 3% with Ni and, as in perovskites, an H2 atmosphere would generate Ni metallic nanoparticles. These nanoparticles, between 20–50 nm size, were tested as catalysts for dry reforming of methane, exhibiting twofold higher activity than conventional Ni-YSZ cermets at 800 °C.131
Outlook and future perspectives
Nanoparticle exsolution has gained considerable attention in the last decade as a catalyst fabrication route, assuring high structural robustness and long-term catalytic activity. It was initially explored mainly for high-temperature electrochemical conversion and storage devices, but rapidly, its applications expanded to other relevant thermocatalytic reactions, such as hydrocarbon reforming, CO2 hydrogenation, chemical looping syngas production or ammonia synthesis, just to name a few. Furthermore, it has been employed to functionalize other electrochemical devices, such as gas sensors or resistive switches, illustrating the broad applicability of this method. Regardless of the field of application, in this work, we have summarized the most promising research avenues in which efforts have been put in the last few years. We have identified six new trends, but other relevant aspects of nanoparticle exsolution are also currently under investigation. Our focus has been on identifying those technological modifications that we consider will have a high impact in the following years. Some of them are related to improving the exsolution extent by lowering the energy input required to generate nanoparticles or by making the process much faster under milder conditions. These non-thermal technologies show great promise for achieving high exsolution extents in very short exposure times. Despite the promise, more systematic and thorough experiments are required to better understand these new physicochemical processes and their interaction with the exsolution mechanism. Regarding materials design, it is crucial to gain further understanding of the evolution of nanoparticles over time and the influence that intrinsic properties have on the stability of exsolution. As recently pointed out by Weber and co-authors, Ni exsolution from p-type and n-type perovskites have different coalescence dynamics,138 which might in turn pose limitations on the long-term durability of the nanoparticles. Therefore, it is crucial to obtain materials design guidelines that ensure that NPs are properly anchored, avoiding isolated growth on specific spots, e.g., grain boundaries that in turn will limit the catalytic activity of the exsolved nanoparticles. In this respect, high throughput synthesis could be a useful tool to accelerate materials discoveries and obtain the most efficient synthesis protocols, which might be material and application specific.A second important aspect considered in this work is the expansion of nanoparticle exsolution to other chemistries. We have focused both on the support, non-perovskite oxides, and the nature of the exsolved nanoparticles, i.e., non-metallic nanoparticles. This direction will have a high impact since many catalytic processes do not rely on metallic nanoparticles or perovskite supports. Furthermore, exsolution from non-oxide supports will be essential to advance in this direction. Their development, combined with computational tools139 that accelerate materials discoveries and spectroscopic operando techniques to unveil the mechanism of nanoparticle exsolution from other non-oxide compounds, will be essential in this matter.
Finally, it is crucial to continue advancing metallic exsolution to unlock further functionalities. Fabrication of shape-controlled or multicomponent nanoparticles is a major aspect requiring more fundamental investigation to better understand and control these processes. The same applies to bulk exsolution, which has emerged as a remarkable tool for strain-engineering oxides on a large scale. This fact proves that exsolution can be successfully employed beyond surface modifications. In this respect, new synthetic routes that will allow for the fabrication of complex oxides with high surface area while guaranteeing phase purity will be essential for further expanding the exsolution method to other processes. This will be especially relevant in mild temperature processes where fine-tuning the textural properties is paramount for achieving relevant catalytic activities. However, especially in the case of perovskite oxides, high temperatures (>1000 °C) are required to avoid phase segregation, which usually results in low specific surface areas (<5 m2 g−1), limiting its applicability in many chemical reactions occurring at low temperatures.
These six research directions are expected to impact the fields of materials science, electrochemistry and catalysis in a wide range of applications. In several cases, many of them will be developed in combination, unlocking unprecedented material functionalities. These new research trends, at the forefront of materials design, combined with more conventional developments will help in expanding the knowledge and applicability of nanoparticle exsolution in the following years.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The project that gave rise to these results received the support of a fellowship from the “la Caixa” Foundation (ID 100010434). The fellowship code is LCF/BQ/PI20/11760015. Financial support by the Spanish Ministry of Science and Innovation (PID2022-139663OB-100 and CEX2021-001230-S grant funded by MCIN/AEI/10.13039/501100011033) and by Generalitat Valenciana (CIPROM/2022/10) is gratefully acknowledged. Blanca Delgado-Galicia acknowledges the funding provided by the SECAT Grants Program for the Completion of Master's Thesis in Catalysis.References
- K. Kousi, C. Tang, I. S. Metcalfe and D. Neagu, Small, 2021, 2006479 CrossRef CAS PubMed.
- J. H. Kim, J. K. Kim, J. Liu, A. Curcio, J. Jang, I. Kim, F. Ciucci and W. Jung, ACS Nano, 2021, 15, 81–110 CrossRef CAS PubMed.
- E. Vøllestad, R. Strandbakke, M. Tarach, D. Catalán-Martínez, M.-L. L. Fontaine, D. Beeaff, D. R. Clark, J. M. Serra and T. Norby, Nat. Mater., 2019, 18, 23–26 CrossRef PubMed.
- C. Solís, V. B. Vert, M. Balaguer, S. Escolástico, S. Roitsch and J. M. Serra, ChemSusChem, 2012, 5, 2155–2158 CrossRef PubMed.
- X. Li, L. Dai, Z. He, W. Meng, Y. Li and L. Wang, Sens. Actuators, B, 2019, 298, 126827 CrossRef CAS.
- J. Spring, E. Sediva, Z. D. Hood, J. C. Gonzalez-Rosillo, W. O’Leary, K. J. Kim, A. J. Carrillo and J. L. M. Rupp, Small, 2020, 16, 2003224 CrossRef CAS PubMed.
- T. W. Hansen, A. T. DeLaRiva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- G. Prieto, J. Zečević, H. Friedrich, K. P. De Jong and P. E. De Jongh, Nat. Mater., 2013, 12, 34–39 CrossRef CAS PubMed.
- D. Neagu, J. T. S. Irvine, J. Wang, B. Yildiz, A. K. Opitz, J. Fleig, Y. Wang, J. Liu, L. Shen, F. Ciucci, B. A. Rosen, Y. Xiao, K. Xie, G. Yang, Z. Shao, Y. Zhang, J. Reinke, T. A. Schmauss, S. A. Barnett, R. Maring, V. Kyriakou, U. Mushtaq, M. N. Tsampas, Y. Kim, R. O’Hayre, A. J. Carrillo, T. Ruh, L. Lindenthal, F. Schrenk, C. Rameshan, E. I. Papaioannou, K. Kousi, I. S. Metcalfe, X. Xu and G. Liu, J. Phys. Energy, 2023, 5, 031501 CrossRef.
- D. Neagu, T. S. Oh, D. N. Miller, H. Menard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. Irvine, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- T. Ruh, D. Berkovec, F. Schrenk and C. Rameshan, Chem. Commun., 2023, 59, 3948–3956 RSC.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Nature, 2002, 418, 164–167 CrossRef CAS PubMed.
- Y. Nishihata, J. Mizuki, H. Tanaka, M. Uenishi and M. Kimura, J. Phys. Chem. Solids, 2005, 66, 274–282 CrossRef CAS.
- H. Tanaka, M. Uenishi, M. Taniguchi, I. Tan, K. Narita, M. Kimura, K. Kaneko, Y. Nishihata and J. Mizuki, Catal. Today, 2006, 117, 321–328 CrossRef CAS.
- H. Tanaka, I. Tan, M. Uenishi, M. Taniguchi, M. Kimura, Y. Nishihata and J. Mizuki, J. Alloys Compd., 2006, 408–412, 1071–1077 CrossRef CAS.
- H. Tanaka, M. Taniguchi, M. Uenishi, N. Kajita, I. Tan, Y. Nishihata, J. Mizuki, K. Narita, M. Kimura and K. Kaneko, Angew. Chem., 2006, 118, 6144–6148 CrossRef.
- U. G. Singh, J. Li, J. W. Bennett, A. M. Rappe, R. Seshadri and S. L. Scott, J. Catal., 2007, 249, 349–358 CrossRef CAS.
- J. Li, U. G. Singh, J. W. Bennett, K. Page, J. C. Weaver, J. Zhang, T. Proffen, A. M. Rappe, S. Scott and R. Seshadri, Chem. Mater., 2007, 19, 1418–1426 CrossRef CAS.
- B. D. Madsen, W. Kobsiriphat, Y. Wang, L. D. Marks and S. A. Barnett, J. Power Sources, 2007, 166, 64–67 CrossRef CAS.
- J. Li, U. G. Singh, T. D. Schladt, J. K. Stalick, S. L. Scott and R. Seshadri, Chem. Mater., 2008, 20, 6567–6576 CrossRef CAS.
- T. Z. Sholklapper, C. P. Jacobson, S. J. Visco and L. C. Dejonghe, Fuel Cells, 2008, 8, 303–312 CrossRef CAS.
- S. Miyoshi and M. Martin, Phys. Chem. Chem. Phys., 2009, 11, 3063 RSC.
- W. Kobsiriphat, B. D. Madsen, Y. Wang, L. D. Marks and S. A. Barnett, Solid State Ionics, 2009, 180, 257–264 CrossRef CAS.
- X. Ouyang and S. L. Scott, J. Catal., 2010, 273, 83–91 CrossRef CAS.
- W. Kobsiriphat, B. D. Madsen, Y. Wang, M. Shah, L. D. Marks and S. A. Barnett, J. Electrochem. Soc., 2010, 157, B279 CrossRef CAS.
- T. Jardiel, M. T. Caldes, F. Moser, J. Hamon, G. Gauthier and O. Joubert, Solid State Ionics, 2010, 181, 894–901 CrossRef CAS.
- X. Lou, Z. Liu, S. Wang, Y. Xiu, C. P. Wong and M. Liu, J. Power Sources, 2010, 195, 419–424 CrossRef CAS.
- D. M. Bierschenk, E. Potter-Nelson, C. Hoel, Y. Liao, L. Marks, K. R. Poeppelmeier and S. A. Barnett, J. Power Sources, 2011, 196, 3089–3094 CrossRef CAS.
- I. Hamada, A. Uozumi, Y. Morikawa, A. Yanase and H. Katayama-Yoshida, J. Am. Chem. Soc., 2011, 133, 18506–18509 CrossRef CAS PubMed.
- W. Zhou, Z. Shao, F. Liang, Z. G. Chen, Z. Zhu, W. Jin and N. Xu, J. Mater. Chem., 2011, 21, 15343–15351 RSC.
- A. López-García, L. Almar, S. Escolástico, A. B. Hungría, A. J. Carrillo and J. M. Serra, ACS Appl. Energy Mater., 2022, 5, 13269–13283 CrossRef.
- Y. Gao, D. Chen, M. Saccoccio, Z. Lu and F. Ciucci, Nano Energy, 2016, 27, 499–508 CrossRef CAS.
- S. Otto, K. Kousi, D. Neagu, L. Bekris, J. Janek and I. S. Metcalfe, ACS Appl. Energy Mater., 2019, 2, 7288–7298 CrossRef CAS.
- W. O’Leary, L. Giordano and J. L. M. Rupp, J. Mater. Chem. A, 2023, 21429–21442 RSC.
- D. Balcerzak, I. Szpunar, R. Strandbakke, S. W. Saeed, C. Bazioti, A. Mielewczyk-Gryń, P. Winiarz, A. J. Carrillo, M. Balaguer, J. M. Serra, M. Gazda and S. Wachowski, CrystEngComm, 2023, 25, 4306–4316 RSC.
- S. Chattopadhyay, A. B. Naden, W. S. J. Skinner, G. Kerherve, D. J. Payne and J. T. S. Irvine, Adv. Energy Mater., 2023, 13, 1–9 CrossRef.
- J. Myung, D. Neagu, D. N. Miller and J. T. S. Irvine, Nature, 2016, 537, 528–531 CrossRef CAS PubMed.
- Y. Lu, Q. Shen, Q. Yu, F. Zhang, G. Li and W. Zhang, J. Phys. Chem. C, 2016, 120, 28712–28716 CrossRef CAS.
- V. Kyriakou, R. K. Sharma, D. Neagu, F. Peeters, O. De Luca, P. Rudolf, A. Pandiyan, W. Yu, S. W. Cha, S. Welzel, M. C. M. Sanden and M. N. Tsampas, Small Methods, 2021, 2100868 CrossRef CAS PubMed.
- A. López-García, A. Domínguez-Saldaña, A. J. Carrillo, L. Navarrete, M. I. Valls, B. García-Baños, P. J. Plaza-Gonzalez, J. M. Catala-Civera and J. M. Serra, ACS Nano, 2023, 17, 23955–23964 CrossRef PubMed.
- H. Han, J. Park, S. Y. Nam, K. J. Kim, G. M. Choi, S. S. P. Parkin, H. M. Jang and J. T. S. Irvine, Nat. Commun., 2019, 10, 1471 CrossRef PubMed.
- K. Kousi, D. Neagu, L. Bekris, E. Calì, G. Kerherve, E. I. Papaioannou, D. J. Payne and I. S. Metcalfe, J. Mater. Chem. A, 2020, 8, 12406–12417 RSC.
- K. Kousi, D. Neagu, L. Bekris, E. I. Papaioannou and I. S. Metcalfe, Angew. Chem., Int. Ed., 2020, 59, 2510–2519 CrossRef CAS PubMed.
- S. J. Dalton and D. Neagu, Adv. Energy Sustainability Res., 2022, 3, 2200054 CrossRef CAS.
- K. J. Kim, H. Han, T. Defferriere, D. Yoon, S. Na, S. J. Kim, A. M. Dayaghi, J. Son, T.-S. Oh, H. M. Jang and G. M. Choi, J. Am. Chem. Soc., 2019, 141, 7509–7517 CrossRef CAS PubMed.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Menard and J. T. Irvine, Nat. Chem., 2013, 5, 916–923 CrossRef CAS PubMed.
- W. O’Leary, L. Giordano, J. Park, S. S. Nonnenmann, Y. Shao-Horn and J. L. M. Rupp, J. Am. Chem. Soc., 2023, 145, 13768–13779 CrossRef PubMed.
- B. Delgado-Galicia, A. López-García, A. J. Carrillo and J. M. Serra, Solid State Ionics, 2024, 410, 116532 CrossRef CAS.
- A. J. Carrillo, L. Navarrete, M. Laqdiem, M. Balaguer and J. M. Serra, Mater. Adv., 2021, 2, 2924–2934 RSC.
- Y. H. Kim, Y. Kang, S. Jo, H. Jeong, D. Neagu and J. Ha Myung, Chem. Eng. J., 2022, 441, 136025 CrossRef CAS.
- D. Neagu, V. Kyriakou, I. Roiban, M. Aouine, C. Tang, A. Caravaca, K. Kousi, I. Schreur-Piet, I. S. Metcalfe, P. Vernoux, M. C. M. van de Sanden and M. N. Tsampas, ACS Nano, 2019, 13, 12996–13005 CrossRef CAS PubMed.
- R. Thalinger, M. Gocyla, M. Heggen, B. Klötzer and S. Penner, J. Phys. Chem. C, 2015, 119, 22050–22056 CrossRef CAS PubMed.
- J. Wang, J. Yang, A. K. Opitz, W. Bowman, R. Bliem, G. Dimitrakopoulos, A. Nenning, I. Waluyo, A. Hunt, J. J. Gallet and B. Yildiz, Chem. Mater., 2021, 33, 5021–5034 CrossRef CAS.
- V. B. Vert, F. V. Melo, L. Navarrete and J. M. Serra, Appl. Catal., B, 2012, 115–116, 346–356 CrossRef CAS.
- D. Neagu, E. I. Papaioannou, W. K. W. W. Ramli, D. N. Miller, B. J. Murdoch, H. Ménard, A. Umar, A. J. Barlow, P. J. Cumpson, J. T. S. S. Irvine and I. S. Metcalfe, Nat. Commun., 2017, 8, 1855 CrossRef PubMed.
- R. Sažinas, K. B. Andersen and K. K. Hansen, J. Solid State Electrochem., 2020, 24, 609–621 CrossRef.
- Y. Zhu, W. Zhou, R. Ran, Y. Chen, Z. Shao and M. Liu, Nano Lett., 2016, 16, 512–518 CrossRef CAS PubMed.
- A. J. Carrillo, K. J. Kim, Z. D. Hood, A. H. Bork and J. L. M. Rupp, ACS Appl. Energy Mater., 2020, 3, 4569–4579 CrossRef CAS.
- C. Tang, K. Kousi, D. Neagu and I. S. Metcalfe, Chem. – Eur. J., 2021, 27, 6666–6675 CrossRef CAS PubMed.
- O. Kwon, K. Kim, S. Joo, H. Y. Jeong, J. Shin, J. W. Han, S. Sengodan and G. Kim, J. Mater. Chem. A, 2018, 6, 15947–15953 RSC.
- S. Jo, Y. Han Kim, H. Jeong, C. Ho Park, B.-R. R. Won, H. Jeon, K. Taek Lee and J. Ha Myung, Appl. Energy, 2022, 323, 119615 CrossRef CAS.
- M. Qin, Y. Xiao, H. Yang, T. Tan, Z. Wang, X. Fan and C. Yang, Appl. Catal., B, 2021, 299, 120613 CrossRef CAS.
- E. I. Papaioannou, D. Neagu, W. K. W. Ramli, J. T. S. Irvine and I. S. Metcalfe, Top. Catal., 2019, 62, 1149–1156 CrossRef CAS.
- D. Papargyriou, D. N. Miller and J. T. Sirr Irvine, J. Mater. Chem. A, 2019, 7, 15812–15822 RSC.
- A. J. Carrillo and J. M. Serra, Catalysts, 2021, 11, 741 CrossRef CAS.
- S. Joo, A. Seong, O. Kwon, K. Kim, J. H. Lee, R. J. Gorte, J. M. Vohs, J. W. Han and G. Kim, Sci. Adv., 2020, 6, 1–9 Search PubMed.
- H. M. Ansari, A. S. Bass, N. Ahmad and V. I. Birss, J. Mater. Chem. A, 2022, 10, 2280–2294 RSC.
- X. Meng, Y. Wang, Y. Zhao, T. Zhang, N. Yu, X. Chen, M. Miao and T. Liu, Electrochim. Acta, 2020, 348, 136351 CrossRef CAS.
- H. Lv, L. Lin, X. Zhang, D. Gao, Y. Song, Y. Zhou, Q. Liu, G. Wang and X. Bao, J. Mater. Chem. A, 2019, 7, 11967–11975 RSC.
- Y. Wang, T. Liu, M. Li, C. Xia, B. Zhou and F. Chen, J. Mater. Chem. A, 2016, 4, 14163–14169 RSC.
- J. Xu, M. Wu, Z. Song, Y. Chen, L. Zhang, L. Wang, H. Cai, X. Su, X. Han, S. Wang and W. Long, J. Eur. Ceram. Soc., 2021, 41, 4537–4551 CrossRef CAS.
- P. B. Managutti, S. Tymen, X. Liu, O. Hernandez, C. Prestipino, A. Le Gal La Salle, S. Paul, L. Jalowiecki-Duhamel, V. Dorcet, A. Billard, P. Briois and M. Bahout, ACS Appl. Mater. Interfaces, 2021, 13, 35719–35728 CrossRef CAS PubMed.
- Z. Du, Y. Gong, H. Zhao, Y. Zhang, S. Yi and L. Gu, ACS Appl. Mater. Interfaces, 2021, 13, 3287–3294 CrossRef CAS PubMed.
- C. Solís, M. Balaguer, F. Bozza, N. Bonanos and J. M. Serra, Appl. Catal., B, 2014, 147, 203–207 CrossRef.
- S. Liu, K. T. Chuang and J. L. Luo, ACS Catal., 2016, 6, 760–768 CrossRef CAS.
- H. Lv, L. Lin, X. Zhang, Y. Song, H. Matsumoto, C. Zeng, N. Ta, W. Liu, D. Gao, G. Wang and X. Bao, Adv. Mater., 2020, 32, 1906193 CrossRef CAS PubMed.
- H. Lv, L. Lin, X. Zhang, R. Li, Y. Song, H. Matsumoto, N. Ta, C. Zeng, Q. Fu, G. Wang and X. Bao, Nat. Commun., 2021, 12, 5665 CrossRef CAS PubMed.
- J. Guo, R. Cai, E. Cali, G. E. Wilson, G. Kerherve, S. J. Haigh and S. J. Skinner, Small, 2022, 18, 1–10 Search PubMed.
- S. Joo, K. Kim, O. Kwon, J. Oh, H. J. Kim, L. Zhang, J. Zhou, J. Wang, H. Y. Jeong, J. W. Han and G. Kim, Angew. Chem., Int. Ed., 2021, 19104 Search PubMed.
- M. Santaya, H. E. Troiani, A. Caneiro and L. V. Mogni, ACS Appl. Energy Mater., 2020, 3, 9528–9533 CrossRef CAS.
- T. Chen, S. Sun, Y. He, H. Leng, C. Sun and C. Wu, J. Alloys Compd., 2023, 960, 170974 CrossRef CAS.
- Y. Jiang, Y. Yang, C. Xia and H. J. M. Bouwmeester, J. Mater. Chem. A, 2019, 7, 22939–22949 RSC.
- Q. Liu, D. E. Bugaris, G. Xiao, M. Chmara, S. Ma, H. C. Zur Loye, M. D. Amiridis and F. Chen, J. Power Sources, 2011, 196, 9148–9153 CrossRef CAS.
- Q. Liu, X. Dong, G. Xiao, F. Zhao and F. Chen, Adv. Mater., 2010, 22, 5478–5482 CrossRef CAS PubMed.
- S. Hou, X. Ma, Y. Shu, J. Bao, Q. Zhang, M. Chen, P. Zhang and S. Dai, Nat. Commun., 2021, 12, 5917 CrossRef CAS PubMed.
- M. Najimu, S. Jo and K. L. Gilliard-AbdulAziz, Acc. Chem. Res., 2023, 56, 3132–3141 CrossRef CAS PubMed.
- A. Nenning and J. Fleig, Surf. Sci., 2019, 680, 43–51 CrossRef CAS.
- L. Chen, J. Xu, X. Wang and K. Xie, Int. J. Hydrogen Energy, 2020, 45, 11901–11907 CrossRef CAS.
- T. Tan, Z. Wang, M. Qin, W. Zhong, J. Hu, C. Yang and M. Liu, Adv. Funct. Mater., 2022, 32, 2202878 CrossRef CAS.
- F. Fang, N. Feng, P. Zhao, C. Chen, X. Li, J. Meng, G. Liu, L. Chen, H. Wan and G. Guan, Chem. Eng. J., 2019, 372, 752–764 CrossRef CAS.
- J. H. Kim, S. Yoo, R. Murphy, Y. Chen, Y. Ding, K. Pei, B. Zhao, G. Kim, Y. Choi and M. Liu, Energy Environ. Sci., 2021, 14, 1506–1516 RSC.
- C. Duan, R. Kee, H. Zhu, N. Sullivan, L. Zhu, L. Bian, D. Jennings and R. O’Hayre, Nat. Energy, 2019, 4, 230–240 CrossRef CAS.
- D. S. Tsvetkov, I. L. Ivanov, D. A. Malyshkin and A. Y. Zuev, J. Mater. Chem. A, 2016, 4, 1962–1969 RSC.
- R. Strandbakke, V. A. Cherepanov, A. Y. Zuev, D. S. Tsvetkov, C. Argirusis, G. Sourkouni, S. Prünte and T. Norby, Solid State Ionics, 2015, 278, 120–132 CrossRef CAS.
- I. Szpunar, R. Strandbakke, M. H. Sørby, S. L. Wachowski, M. Balaguer, M. Tarach, J. M. Serra, A. Witkowska, E. Dzik, T. Norby, M. Gazda and A. Mielewczyk-Gryń, Materials, 2020, 13, 1–18 CrossRef PubMed.
- S. L. Wachowski, I. Szpunar, M. H. Sørby, A. Mielewczyk–Gryń, M. Balaguer, C. Ghica, M. C. Istrate, M. Gazda, A. E. Gunnæs, J. M. Serra, T. Norby and R. Strandbakke, Acta Mater., 2020, 199, 297–310 CrossRef CAS.
- Y. Zhou, E. Liu, Y. Chen, Y. Liu, L. Zhang, W. Zhang, Z. Luo, N. Kane, B. Zhao, L. Soule, Y. Niu, Y. Ding, H. Ding, D. Ding and M. Liu, ACS Energy Lett., 2021, 6, 1511–1520 CrossRef CAS.
- Y. Niu, Y. Zhou, W. Zhang, Y. Zhang, C. Evans, Z. Luo, N. Kane, Y. Ding, Y. Chen, X. Guo, W. Lv and M. Liu, Adv. Energy Mater., 2022, 12, 1–9 Search PubMed.
- K. Park, M. Saqib, H. Lee, D. Shin, M. Jo, K. M. Park, M. Hamayun, S. H. Kim, S. Kim, K. Lee, R. O’Hayre, M. Choi, S.-J. Song and J.-Y. Park, Energy Environ. Sci., 2024, 17, 1175–1188 RSC.
- P. K. Dubey, J. Hong, M. R. Anisur, K. Lee, S. Belko and P. Singh, ACS Appl. Energy Mater., 2023, 6, 657–666 CrossRef CAS.
- Y.-F. Sun, Y.-L. Yang, J. Chen, M. Li, Y.-Q. Zhang, J.-H. Li, B. Hua and J.-L. Luo, Chem. Commun., 2018, 54, 1505–1508 RSC.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878–3888 RSC.
- Y. Q. Zhang, H. B. Tao, Z. Chen, M. Li, Y. F. Sun, B. Hua and J. L. Luo, J. Mater. Chem. A, 2019, 7, 26607–26617 RSC.
- A. K. Opitz, A. Nenning, V. Vonk, S. Volkov, F. Bertram, H. Summerer, S. Schwarz, A. Steiger-Thirsfeld, J. Bernardi, A. Stierle and J. Fleig, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- H. Summerer, A. Nenning, C. Rameshan and A. K. Opitz, EES Catal., 2023, 1, 274–289 RSC.
- Y. Lu, Y. Huang, Z. Xu, K. Yang, W. Bao and Q. Lu, ACS Nano, 2023, 17, 14005–14013 CrossRef CAS PubMed.
- M. Xu, R. Cao, S. Wu, J. G. Lee, D. Chen and J. T. S. Irvine, J. Mater. Chem. A, 2023, 11, 13007–13015 RSC.
- J. Kim, S. Gunduz, A. C. Co and U. S. Ozkan, Appl. Catal., B, 2024, 344, 123603 CrossRef CAS.
- W. Fan, B. Wang, R. Gao, G. Dimitrakopoulos, J. Wang, X. Xiao, L. Ma, K. Wu, B. Yildiz and J. Li, J. Am. Chem. Soc., 2022, 144, 7657–7666 CrossRef CAS PubMed.
- H. Khalid, A. ul Haq, B. Alessi, J. Wu, C. D. Savaniu, K. Kousi, I. S. Metcalfe, S. C. Parker, J. T. S. Irvine, P. Maguire, E. I. Papaioannou and D. Mariotti, Adv. Energy Mater., 2022, 12, 2201131 CrossRef CAS.
- E. Shin, D. H. Kim, J. H. Cha, S. Yun, H. Shin, J. Ahn, J. S. Jang, J. W. Baek, C. Park, J. Ko, S. Park, S. Y. Choi and I. D. Kim, ACS Nano, 2022, 16, 18133–18142 CrossRef CAS PubMed.
- X. Hu, J. Qi, S. Qiao, W. Yu, J. Shang, L. Liu, Z. Zhao, L. Tang and W. Zhang, Nanotechnology, 2023, 34, 105709 CrossRef CAS PubMed.
- J. Wang, K. B. Woller, A. Kumar, Z. Zhang, H. Zhou, I. Waluyo, A. Hunt, J. M. LeBeau and B. Yildiz, Energy Environ. Sci., 2023, 16, 5464–5478 RSC.
- M. B. Katz, G. W. Graham, Y. Duan, H. Liu, C. Adamo, D. G. Schlom and X. Pan, J. Am. Chem. Soc., 2011, 133, 18090–18093 CrossRef CAS PubMed.
- M. B. Katz, S. Zhang, Y. Duan, H. Wang, M. Fang, K. Zhang, B. Li, G. W. Graham and X. Pan, J. Catal., 2012, 293, 145–148 CrossRef CAS.
- H. Han, Y. Xing, B. Park, D. I. Bazhanov, Y. Jin, J. T. S. Irvine, J. Lee and S. H. Oh, Nat. Commun., 2022, 13, 6682 CrossRef CAS PubMed.
- S. J. Kim, T. Akbay, J. Matsuda, A. Takagaki and T. Ishihara, ACS Appl. Energy Mater., 2019, 2, 1210–1220 CrossRef CAS.
- J. Hyodo, K. Tominaga, S. Ida and T. Ishihara, J. Mater. Chem. A, 2016, 4, 3844–3849 RSC.
- M. L. Weber, B. Šmíd, U. Breuer, M.-A. Rose, N. H. Menzler, R. Dittmann, R. Waser, O. Guillon, F. Gunkel and C. Lenser, Nat. Mater., 2024, 1–26 CAS.
- M. L. Weber, M. Wilhelm, L. Jin, U. Breuer, R. Dittmann, R. Waser, O. Guillon, C. Lenser and F. Gunkel, ACS Nano, 2021, 15, 4546–4560 CrossRef CAS PubMed.
- H. Guo, C. Mead, M. Balingit, S. Shah, X. Wang, M. Xu, I. Tran, T. Aoki, J. D. Samaniego, K. L. Gilliard-AbdulAziz, L. J. Lauhon and W. J. Bowman, Matter, 2024, 7, 1002–1017 CrossRef CAS.
- M. Melchionna and P. Fornasiero, RSC Appl. Interfaces, 2024, 1, 70–79 RSC.
- C. Bozo, N. Guilhaume and J. M. Herrmann, J. Catal., 2001, 203, 393–406 CrossRef CAS.
- M. Balaguer, C. Solís and J. M. Serra, J. Phys. Chem. C, 2012, 116, 7975–7982 CrossRef CAS.
- M. Laqdiem, A. J. Carrillo, G. Dimitrakopoulos, M. Balaguer, J. Garcia-Fayos, A. F. Ghoniem and J. M. Serra, Solid State Ionics, 2023, 394, 116192 CrossRef CAS.
- M. A. Naeem, P. M. Abdala, A. Armutlulu, S. M. Kim, A. Fedorov and C. R. Müller, ACS Catal., 2020, 10, 1923–1937 CrossRef CAS.
- Y. Xiao and K. Xie, Angew. Chem., Int. Ed., 2022, 61, 1–8 Search PubMed.
- K. Wu, J. Matsuda, A. Staykov and T. Ishihara, Adv. Energy Mater., 2023, 13, 1–16 Search PubMed.
- F. Hu, K. Chen, Y. Ling, Y. Huang, S. Zhao, S. Wang, L. Gui, B. He and L. Zhao, Adv. Sci., 2023, 2306845, 1–11 Search PubMed.
- M. Kim, G. Kwon, W.-G. Jung, Y. Choi, B.-J. Kim and H. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 48508–48515 CrossRef CAS PubMed.
- S. Joo, C. Lim, O. Kwon, L. Zhang, J. Zhou, J.-Q. Wang, H. Y. Jeong, Y. Sin, S. Choi and G. Kim, Mater. Rep.: Energy, 2021, 100021 CAS.
- W. Yang, X. Wang, S. Song and H. Zhang, Chem, 2019, 5, 1743–1774 CAS.
- W. Qi, K. Xie, M. Liu, G. Wu, Y. Wang, Y. Zhang and Y. Wu, RSC Adv., 2014, 4, 40494–40504 RSC.
- J. Tan, D. Lee, J. Ahn, B. Kim, J. Kim and J. Moon, J. Mater. Chem. A, 2018, 6, 18133–18142 RSC.
- J. Lee, M. Bae and J. Bae, Int. J. Hydrogen Energy, 2022, 47, 29327–29336 CrossRef CAS.
- J. Lee, C. Yeon, J. Oh, G. Han, J. Do Yoo, H. J. Yun, C. W. Lee, K. T. Lee and J. Bae, Appl. Catal., B, 2022, 316, 121645 CrossRef CAS.
- L. Z. Gan, L. T. Ye, C. Ruan, S. G. Chen and K. Xie, Adv. Sci., 2015, 3, 1–7 Search PubMed.
- M. L. Weber, D. Jennings, S. Fearn, A. Cavallaro, M. Prochazka, A. Gutsche, L. Heymann, J. Guo, L. Yasin, S. J. Cooper, J. Mayer, W. Rheinheimer, R. Dittmann, R. Waser, O. Guillon, C. Lenser, S. J. Skinner, A. Aguadero, S. Nemšák and F. Gunkel, ChemRxiv, 2023, preprint DOI:10.26434/chemrxiv-2023-8k11w.
- S. Kang, J. K. Kim, H. Kim, Y. Son, J. Chang, J. Kim, D. Kim, J. Lee and H. J. Kwon, Nano Lett., 2024, 24, 4224–4232 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |