
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A dinuclear nickel peroxycarbonate complex: CO2 addition promotes H2O2 release†
Hayley L.
Lillo
 and
Joshua A.
Buss
*
and
Joshua A.
Buss
*
Willard Henry Dow Laboratory, Department of Chemistry, University of Michigan 930 N. University Avenue, Ann Arbor, MI 48109, USA. E-mail: jbuss@umich.edu
First published on 8th July 2024
Abstract
Nickel coordination compounds featuring Ni–O bonds are key structural motifs in both bioinorganic and synthetic chemistries. They serve as precursors for organic substrate oxidation and are commonly invoked intermediates in water oxidation and oxygen reduction schemes. Herein, we disclose a series of well-defined dinuclear nickel complexes that, upon treatment with CO2 and H2O2, afford the first nickel-bound peroxycarbonate. This unprecedented nickel–oxygen intermediate is stabilized by hydrogen bonding templated across the bimetallic core. Contrasting copper and iron analogues, the nickel peroxycarbonate reversibly dissociates H2O2, a process that is shown to be accelerated by exogenous CO2.
The ubiquity of Cu, Fe, and Mn enzymes has led to extensive studies interrogating metal-oxygen intermediates of mono- and bimetallic complexes of these ions.1 In contrast, Ni is generally employed by Nature for C1 conversion chemistry,2 but has recently received attention with respect to oxidation reactivity given its role in the active sites of superoxide dismutase (NiSOD; Fig. 1A)3 and dioxygenase enzymes.4 Ni(II) complexes are typically inert to dioxygen, however modifications to the supporting ligand(s) have been demonstrated to override this tendency.5 Even so, the vast majority of nickel–oxygen complexes are accessed via addition of strong oxidants, including H2O2, peroxyacids, and alkylperoxides.6 Continued preparation and study of new classes of nickel–oxygen intermediates is critical to elucidating the chemistry of nickel metalloenzymes, informing fundamental mechanisms in synthetic oxidation catalysis, and providing a basis for comparison to copper and iron homologs.6c
 | ||
| Fig. 1 Proposed mechanism for bioinorganic superoxide reduction to H2O2 (A) and representative examples of the diverse nickel–oxygen intermediates accessed synthetically via both O2 activation and H2O2/base addition (B). | ||
Given the high electronegativity of Ni, its low oxophilicity, and O(π)/Ni(d) repulsions, nickel–oxygen species can demonstrate potent oxidative reactivity.7 Mononuclear nickel oxidants—generated from the treatment of Ni(II) precursors with peroxyacids—are capable of oxidizing unactivated aliphatic C–H bonds, yet the nature of the active oxidant remains unknown.8 Tyrosinase-like NiIII-μ(O)2-NiIII diamond cores are competent for intramolecular aliphatic9 and aromatic10 C–H oxygenation, ultimately sourcing the oxidizing equivalents from hydrogen peroxide (H2O2). They are likewise proposed to be the active oxidants in catalytic aromatic hydroxylation protocols.1a,11 Recently, this has been scrutinized;12 more exotic nickel–oxygen species have been reported under similar reaction conditions and cannot be strictly ruled out as reaction-relevant intermediates (Fig. 1B).9,13
Given that many mechanistic proposals rely on self-assembly of monometallic precursors to dinuclear nickel–oxygen species, our lab has targeted well-defined bimetallic model systems supported by a bicompartmental ligand scaffold. Herein, we describe the synthesis of a series of multinuclear nickel complexes that serve as precursors for base-free H2O2 activation chemistry. These Ni(II) compounds were found to readily react with carbon dioxide (CO2), affording a dinickel complex bridged by κ2-bicarbonate linkages. The CO2 in these ligands plays a key role in subsequent H2O2 reactivity, yielding the first structurally characterized bimetallic peroxycarbonate complex, [LNi2(O2CO2)(H2O)2]+. Contrasting previously reported peroxycarbonates,14[LNi2(O2CO2)(H2O)2]+ releases H2O2 when warmed to 0 °C. In addition to expanding the library of characterized nickel–oxygen intermediates (Fig. 1B), this work lends credence to proposed peroxyacid adducts that are relevant to nickel-mediated alkane and alkene oxidations.8a,c,d
Treating lithium phenolate pro-ligand, LiL, with two equiv. of Ni(OTf)2, generates a dinickel complex15 featuring both inner- (19F δ = 0.81 ppm) and outer-sphere (19F δ = –74.42 ppm) triflate anions. The nickel(II) centres afford sharp paramagentically shifted resonances by 1H NMR spectroscopy, a fingerprint correlated to the bis(triflate) cation [LNi2(OTf)2]+ (Scheme 1), via complementary single crystal X-ray diffraction (SCXRD) analysis. μ-OH ligands are purported intermediates and/or precursors in H2O2-mediated oxidation chemistry;16 these groups were targeted via treatment of [LNi2(OTf)2]+ with NaOH. Under anaerobic conditions, addition of base results in generation of a tetranuclear complex with μ-OH ligands linking two distinct LNi2 motifs, the formulation of which was corroborated as [L2Ni4(OH)4]2+ by SCXRD. The four metal ions adopt a roughly tetrahedral arrangement (τ4 = 0.97),17 with bridging hydroxide and aryloxide ligands capping each of the six edges. The Ni–Ni distances across the aryloxide bridges are slightly elongated (3.90 Å) relative to those spanned by the hydroxide ligands (3.75 Å), suggesting that the intermolecular μ-hydroxo is more accessible with the semi-rigid dinucleating scaffold. When the same reaction is performed open to air, a distinct tetranickel complex results, with two bridging carbonate and two aquo ligands, [L2Ni4(CO3)2(H2O)2]2+, via the uptake of CO2 from ambient air (Scheme 1). All four Ni(II) centres remain six-coordinate with C2 symmetry and μ-κ2O,O:κO carbonate binding. The solid-state structure of this species highlights expansion of the inter-nickel distances between the two bimetallic units (Ni⋯Niave. = 4.96 Å), and distinct Ni–Ni contacts within the two bicompartmental chelates, with the κ2-carbanato moieties enforcing a more compact Ni2 spacing (viz. 3.45 vs. 3.81 Å). The solid state asymmetry observed for [L2Ni4(CO3)2(H2O)2]2+ is affirmed by the solution spectroscopy, and electrospray ionization mass spectrometry (ESI-MS) shows parent ion peaks consistent with both tetranuclear units, [L2Ni4(OH)4]2+ and [L2Ni4(CO3)2(H2O)2]2+.
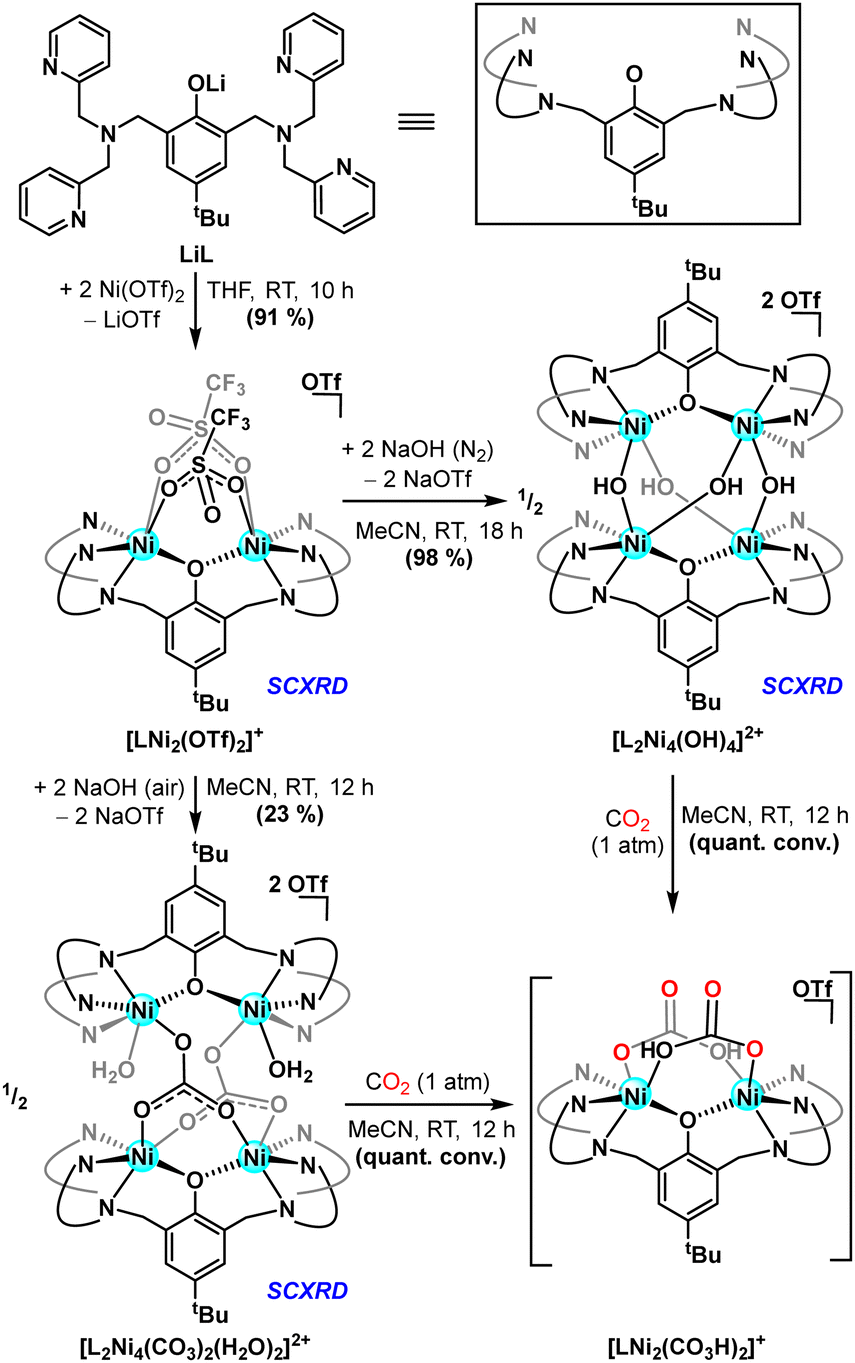 | ||
| Scheme 1 Synthesis of well-defined multinuclear nickel hydroxide complexes and subsequent reactivity with carbon dioxide. | ||
Whereas solutions of [L2Ni4(OH)4]2+ prove air-stable, they readily react with concentrated CO2 (1 atm) to generate a new species, as indicated by a slight blue shift in the UV-visible spectrum (Fig. 2, left) and a distinct paramagnetically shifted signature in the 1H NMR spectrum.18 The same spectral features are reproduced when CO2 (1 atm) is added to [L2Ni4(CO3)2(H2O)2]2+. Vibrational spectroscopy shows a sharp carbonyl stretch at 1687 cm−1 that is sensitive to 13CO2 isotopic labelling (Fig. 2, right), most consistent with a terminal (bi)carbonate assignment.14h Whereas this reaction product has eluded characterization in the solid state, ESI-MS of reactions stemming from the treatment of either precursor with 12/13CO2 corroborates generation of a bis(bicarbonate) cation, [LNi2(CO3H)2]+, the dinuclear nature of which is further supported by DOSY NMR experiments (Scheme 1).
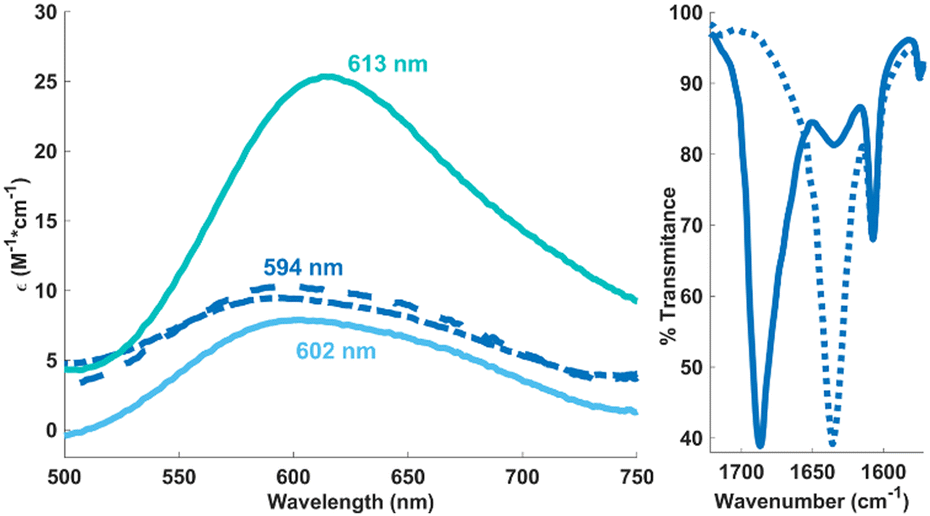 | ||
Fig. 2 Electronic absorption spectra evidencing conversion of [L2Ni4(OH)4]2+ ( ) and [L2Ni4(CO3)2(H2O)2]2+ ( ) and [L2Ni4(CO3)2(H2O)2]2+ ( ) to [LNi2(CO3H)2]+ ( ) to [LNi2(CO3H)2]+ ( & &  , respectively) in MeCN solution (left). Solution IR spectra (MeCN) corroborating isotopic sensitivity of the bicarbonate (12C , respectively) in MeCN solution (left). Solution IR spectra (MeCN) corroborating isotopic sensitivity of the bicarbonate (12C  ;13C ;13C  ) carbonyl stretch (right). ) carbonyl stretch (right). | ||
As an entry point to oxidative chemistry, we pursued H2O2 addition to these multinuclear Brønsted-basic nickel species. Peroxycarbonate ligands are shown to form from both CO2 addition to reactive metal peroxo species14b,d,f–k and H2O2 insertion into metal carbonatos.14a Treating [L2Ni4(CO3)2(H2O)2]2+ or [LNi2(CO3H)2]+ with excess aqueous H2O2 (50 wt%) at –30 °C resulted in an immediate colour change to pale purple. Low temperature 1H NMR spectroscopy supported the formation of a mixture of paramagnetic species; cleaner reactivity to a single major product was observed when the oxidant addition was carried out in the presence of exogenous CO2. Crystalline purple plates were formed when Et2O/MeCN mixtures of this new product were maintained at low temperature (−17 °C). SCXRD analysis revealed generation of a dinuclear peroxycarbonate complex, [LNi2(O2CO2)(H2O)2]+ (Fig. 3); the first nickel peroxycarbonate and the first structurally characterized bimetallic example.
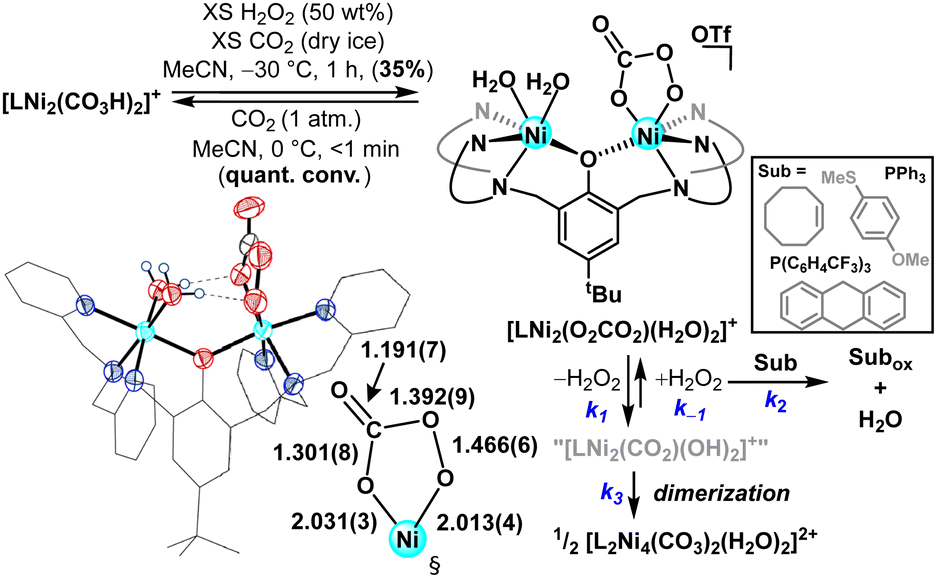 | ||
| Fig. 3 Synthesis, solid-state structure,‡ and reactivity of a nickel peroxycarbonate complex. Bond metrics are reported in angstroms. Substrate oxidations were conducted at 0 °C for 4 hrs in either MeCN or MeCN/DCM. | ||
Contrasting what was proposed for dicopper complexes on a similar phenolate-bridged ancillary ligand,14h the present dinickel peroxycarbonate shows preferential binding to a single Ni centre (Fig. 3). The metrical parameters highlight that the O2CO2-bound Ni ion in [LNi2(O2CO2)(H2O)2]+ adopts a distorted octahedral coordination environment, with peroxycarbonate bond lengths similar to Suzuki's related monometallic Fe analogue.14e The O–O bond is slightly elongated in the present case (cf. 1.466(6) vs. 1.455(5) Å) and more closely matches that of the recently reported peroxybicarbonate anion (1.469(2) Å).19 The second nickel centre in [LNi2(O2CO2)(H2O)2]+ is likewise pseudo-octahedral with two water molecules completing the coordination sphere. A striking feature of the solid-state structure is the orientation and proximity of the aquo ligands to the formally anionic oxygen atoms of the peroxycarbonate, which are well within the range of H-bonding contacts (OH2⋯Oave. = 2.682(5) Å). H-bonding is a proven strategy for stabilizing reactive metal-oxygen species;20 we hypothesize that nickel templated intramolecular H-bonding imbues added stability to the peroxycarbonate moiety in the present system.
The reported reactivity of base-metal peroxycarbonate complexes varies, with documented examples of both O–O bond scission14a,c and thermal disproportionation to liberate O2.14h Moreover, peroxycarbonate complexes have been shown to be competent oxidants in their own right, demonstrating O-atom14a,b,e–h and H-atom transfer14a,f reactivity. In light of this precedent, we were keen to explore the thermal (in)stability of [LNi2(O2CO2)(H2O)2]+, which demonstrated relatively slow conversion to [L2Ni4(CO3)2(H2O)2]2+ in MeCN solution at 0 °C (t1/2ca. 40 min.). This is markedly different from Karlin's dicopper analogue, which reacts rapidly via thermal disproportionation at temperatures as low as –50 °C,14h a distinction attributed to the intramolecular H-bonds (vide supra). A balanced reaction for formation of [L2Ni4(CO3)2(H2O)2]2+ necessitates the loss of H2O2—efforts to spectroscopically identify/quantify H2O2 were unsuccessful, but reactivity probes with phosphine reagents corroborated nearly quantitative peroxide release (92%; Fig. S21, ESI†).§ In this way, [LNi2(O2CO2)(H2O)2]+ demonstrates novel reactivity for this ligand motif, acting as a reservoir for H2O2.
We next sought to explore the potential of [LNi2(O2CO2)(H2O)2]+ to serve as a bona fide oxidant via low-temperature reactions with various substrates. Kinetic assays at 0 °C, under pseudo first-order conditions, displayed non-integer rate laws (Fig. 4 and
and  ) for two electronically differentiated phosphines. These results are more consistent with reversible H2O2 binding than the direct reaction of PR3 with [LNi2(O2CO2)(H2O)2]+. A kinetic regime in which H2O2 binding to form [LNi2(O2CO2)(H2O)2]+ (Fig. 3, k–1) is rate competitive with phosphine oxidation (k2) rationalizes the observed reaction profiles.¶ To further bolster this hypothesis, the rate of carbonate formation from [LNi2(O2CO2)(H2O)2]+ in the presence (
) for two electronically differentiated phosphines. These results are more consistent with reversible H2O2 binding than the direct reaction of PR3 with [LNi2(O2CO2)(H2O)2]+. A kinetic regime in which H2O2 binding to form [LNi2(O2CO2)(H2O)2]+ (Fig. 3, k–1) is rate competitive with phosphine oxidation (k2) rationalizes the observed reaction profiles.¶ To further bolster this hypothesis, the rate of carbonate formation from [LNi2(O2CO2)(H2O)2]+ in the presence ( ) and absence (
) and absence ( ) of H2O2 was likewise investigated. The zero-order dependence on Ni observed for formation of [L2Ni4(CO3)2(H2O)2]2+ supports rate determining H2O2 release (rather than dimerization), and addition of exogenous H2O2 suppresses conversion (Fig. 4
) of H2O2 was likewise investigated. The zero-order dependence on Ni observed for formation of [L2Ni4(CO3)2(H2O)2]2+ supports rate determining H2O2 release (rather than dimerization), and addition of exogenous H2O2 suppresses conversion (Fig. 4 and
and  , respectively). A comparison of three additional substrates (Fig. 3, inset) demonstrated no significant difference in reactivity relative to hydrogen peroxide controls, further supporting [LNi2(O2CO2)(H2O)2]+ acts as a storehouse for H2O2.
, respectively). A comparison of three additional substrates (Fig. 3, inset) demonstrated no significant difference in reactivity relative to hydrogen peroxide controls, further supporting [LNi2(O2CO2)(H2O)2]+ acts as a storehouse for H2O2.
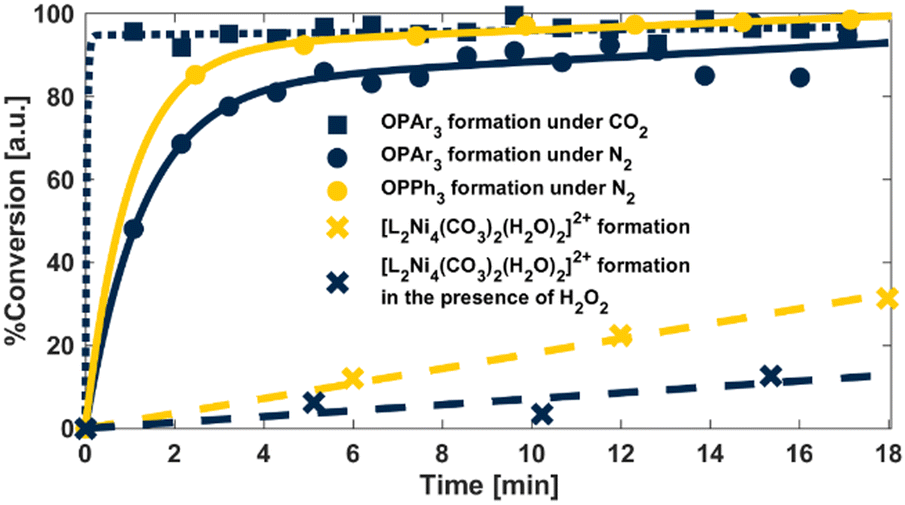 | ||
| Fig. 4 Reaction kinetics tracking [LNi2(O2CO2)(H2O)2]+ reactivity under conditions outlined in the accompanying legend. Ar = tris(4-trifluoromethylphenyl). | ||
Reaction atmosphere—N2vs. CO2vs. air—has been shown to play a role in peroxycarbonate reactivity.14a Under CO2, the decomposition of [LNi2(O2CO2)(H2O)2]+ is immediate, even at low temperature (thawing MeCN), quantitatively furnishing bicarbonate complex [LNi2(CO3H)2]+. Again, this chemistry proceeds via the release of H2O2 with concomitant CO2 uptake. This reversible sequestration of H2O2 at a base-metal-bound peroxycarbonate is unprecedented, differentiating the present Ni reactivity from that of both its Fe14a,c and Cu14g congeners. Subsequent chemical steps—dimerization to form [L2Ni4(CO3)2(H2O)2]2+, substrate oxidation, or CO2 addition (generating [LNi2(CO3H)2]+)—serve to drive the equilibrium toward exhaustive H2O2 release.
In summary, well-defined dinuclear nickel complexes have been accessed by employing a phenolate-bridged bicompartmental ligand scaffold. Hydroxide installation results in highly Lewis basic moieties that favour intermolecular bridging ([L2Ni4(OH)4]2+) or CO2 uptake ([L2Ni4(CO3)2(H2O)2]2+). Ensuing hydrogen peroxide chemistry affords the first reported example of a nickel peroxycarbonate complex, the stability of which is attributed to metal-templated intramolecular H-bonding interactions. Unlike previously reported metal peroxycarbonates,14[LNi2(O2CO2)(H2O)2]+ reacts via reversible H2O2 release, as established by kinetics assays. H2O2 dissociation is shown to be driven by CO2 addition, accelerating oxidant release. Compounding ambiguity regarding the exact nature of reactive nickel–oxygen intermediates accessed from H2O2 and base (cf.Fig. 1B), this work demonstrates that CO2 may likewise play a non-innocent role in altering the nickel speciation. Further studies investigating the fundamental chemistry of multinuclear nickel precursors with myriad catalysis-relevant oxidants are underway in our laboratory.
This work was supported by the University of Michigan and the NSF (XRD Instrumentation–CHE-0840456). We thank Dr. Eugenio Alvarado for NMR spectroscopy expertise, Drs. Jeff Kampf and Fengrui Qu for assistance with SCXRD and Claire R. Patterson for XRD data of [LNi2(OTf)2]+. Roy Wentz aided in the design and fabrication of custom glassware and the Szymczak lab shared instrumentation that made this work possible.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data have been deposited in the CCDC (2348000–2348003)—https://www.ccdc.cam.ac.uk/structures.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) L. Vicens, et al. , ACS Catal., 2020, 10, 8611 CrossRef CAS; (b) L. Que and W. B. Tolman, Nature, 2008, 455, 333 CrossRef CAS PubMed; (c) E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047 CrossRef CAS; (d) L. M. Mirica, et al. , Chem. Rev., 2004, 104, 1013 CrossRef CAS PubMed.
- S. W. Ragsdale, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 165 CrossRef CAS.
- (a) D. P. Barondeau, et al. , Biochem., 2004, 43, 8038 CrossRef CAS; (b) J. Wuerges, et al. , Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8569 CrossRef CAS.
- (a) J.-H. Jeoung, et al. , Angew. Chem., Int. Ed., 2016, 55, 3281 CrossRef CAS; (b) T. C. Pochapsky, et al. , Nat. Struct. Bio., 2002, 9, 966 CrossRef CAS.
- (a) D. M. Beagan, et al. , J. Am. Chem. Soc., 2024, 146, 12375 CrossRef CAS PubMed; (b) A. J. McNeece, et al. , J. Am. Chem. Soc., 2020, 142, 10824 CrossRef CAS; (c) N. Zhao, et al. , J. Am. Chem. Soc., 2020, 142, 21634 CrossRef CAS PubMed; (d) P.-C. Duan, et al. , J. Am. Chem. Soc., 2018, 140, 4929 CrossRef CAS PubMed.
- (a) H. Noh and J. Cho, Coord. Chem. Rev., 2019, 382, 126 CrossRef CAS; (b) T. Corona and A. Company, Chem. Eur. J., 2016, 22, 13422 CrossRef CAS; (c) M. R. Halvagar, et al., 3.17-Small Molecule Models: Cu, Ni, Co, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, 2nd edn, Elsevier, Amsterdam, 2013, p. 455 Search PubMed.
- J. R. Winkler and H. B. Gray, in Molecular Electronic Structures of Transition Metal Complexes I, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer, Berlin, Heidelberg, 2012, 17 Search PubMed.
- (a) J. Nakazawa, et al. , J. Am. Chem. Soc., 2013, 135, 6010 CrossRef CAS PubMed; (b) S. Hikichi, et al. , Dalton Trans., 2013, 42, 3346 RSC; (c) T. Nagataki, et al. , Dalton Trans., 2007, 1120 RSC; (d) T. Nagataki, et al. , Chem. Commun., 2006, 4016 RSC.
- (a) J. Cho, et al. , Inorg. Chem., 2006, 45, 2873 CrossRef CAS; (b) S. Itoh, et al. , J. Am. Chem. Soc., 2001, 123, 11168 CrossRef CAS PubMed; (c) K. Shiren, et al. , J. Am. Chem. Soc., 2000, 122, 254 CrossRef CAS.
- (a) T. Tano, et al. , Bull. Chem. Soc. Jpn., 2010, 83, 530 CrossRef CAS; (b) K. Honda, et al. , Angew. Chem., Int. Ed., 2009, 48, 3304 CrossRef CAS; (c) A. Kunishita, et al. , Inorg. Chem., 2009, 48, 4997 CrossRef CAS.
- (a) S. Muthuramalingam, et al. , Catal. Sci. Tech., 2019, 9, 5991 RSC; (b) Y. Morimoto, et al. , J. Am. Chem. Soc., 2015, 137, 5867 CrossRef CAS.
- K. Farshadfar and K. Laasonen, Inorg. Chem., 2024, 63, 5509 CrossRef CAS.
- (a) M. T. Kieber-Emmons, et al. , J. Am. Chem. Soc., 2006, 128, 14230 CrossRef CAS PubMed; (b) E. J. Brown, et al. , Angew. Chem., Int. Ed., 2005, 44, 1392 CrossRef CAS; (c) S. Hikichi, et al. , J. Am. Chem. Soc., 1998, 120, 10567 CrossRef CAS.
- (a) T. Tsugawa, et al. , Chem. Lett., 2015, 44, 330 CrossRef CAS; (b) G. Meier and T. Braun, Angew. Chem., Int. Ed., 2012, 51, 12564 CrossRef CAS; (c) H. Furutachi, et al. , J. Am. Chem. Soc., 2005, 127, 4550 CrossRef CAS PubMed; (d) M. Yamashita, et al. , J. Am. Chem. Soc., 2005, 127, 7294 CrossRef CAS PubMed; (e) K. Hashimoto, et al. , Angew. Chem., Int. Ed., 2002, 41, 1202 CrossRef CAS; (f) M. Aresta, et al. , Inorg. Chem., 1996, 35, 4254 CrossRef CAS PubMed; (g) I. Sanyal, et al. , J. Am. Chem. Soc., 1993, 115, 11259 CrossRef CAS; (h) P. P. Paul, et al. , J. Am. Chem. Soc., 1991, 113, 5322 CrossRef CAS; (i) M. Schappacher and R. Weiss, Inorg. Chem., 1987, 26, 1189 CrossRef CAS; (j) L. Dahlenburg and C. Prengel, J. Organomet. Chem., 1986, 308, 63 CrossRef CAS; (k) P. J. Hayward, et al. , J. Chem. Soc. D, 1969, 987b RSC.
- (a) H. T. Zhang, et al. , Angew. Chem., Int. Ed., 2023, 62, e202218859 CrossRef CAS PubMed; (b) S. S. Massoud, et al. , Dalton Trans., 2015, 44, 2110 RSC; (c) Y. Gultneh, et al. , Polyhedron, 1998, 17, 3351 CrossRef.
- A. Rajeev, et al. , ACS Catal., 2022, 12, 9953 CrossRef CAS.
- L. Yang, et al. , Dalton Trans., 2007, 955 RSC.
- D. Huang, et al. , Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1222 CrossRef CAS PubMed.
- Z. Yan, et al. , J. Am. Chem. Soc., 2023, 145, 22213 CrossRef CAS PubMed.
- (a) E. W. Dahl, et al. , Chem. Commun., 2018, 54, 892 RSC; (b) E. W. Dahl, et al. , J. Am. Chem. Soc., 2018, 140, 10075 CrossRef CAS; (c) C. L. Ford, et al. , Science, 2016, 354, 741 CrossRef CAS; (d) A. S. Borovik, Acc. Chem. Res., 2005, 38, 54 CrossRef CAS PubMed; (e) C. E. MacBeth, et al. , Science, 2000, 289, 938 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2348000–2348003. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02241f |
| ‡ Thermal anisotropic displacement ellipsoids are shown at the 50% probability level; non-aquo hydrogen atoms and the triflate counterion are omitted for clarity. The hydrocarbyl ligand backbone is depicted as a wireframe for simplicity. |
| § Control reactions of [L2Ni4(CO3)2(H2O)2]2+ and phosphine under O2 did not result in any detectable oxidation to phosphine oxide on the timescale of these experiments. |
| ¶ The difference in phosphine oxidation rate is insufficient to rule out direct reaction of the phosphine with [LNi2(O2CO2)(H2O)2]+; however, in totality, the data are inconsistent with this mechanism. |
| This journal is © The Royal Society of Chemistry 2024 |