
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
HFIP promoted carbo-lactonisation as a new route to functionalised lactones†
Daniya
Aynetdinova‡
 a,
Megan
Jaschinski‡
a,
Timothy C.
Jenkins
ab and
Timothy J.
Donohoe
*a
a,
Megan
Jaschinski‡
a,
Timothy C.
Jenkins
ab and
Timothy J.
Donohoe
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: timothy.donohoe@chem.ox.ac.uk
bChemical Crystallography, Chemistry Research Laboratory, Oxford, UK
First published on 15th August 2024
Abstract
We have been able to induce a series of lactonisation reactions of unsaturated acids using a carbocationic intermediate formed in situ. The transformation generates five and six membered lactone rings AND forms a new exocyclic C–C bond at the same time, thus removing the need for further derivatisation of typical lactonisation products (such as from iodo lactonisation). The reaction is operationally straightforward, does not employ any metals and utilises readily accessible alcohol and unsaturated acid substrates.
γ-Lactones are a privileged scaffold in natural products and biologically active compounds.1 Their wide range of biological properties, such as antimicrobial,2a anti-inflammatory2b and cytotoxic activities,2c have established 5- and 6-membered lactones as a common structural motif in drug design. Lactones possessing a quaternary centre adjacent to the oxygen atom, are widespread in natural products and have been extensively studied in medicinal chemistry.3
The use of heteroatom containing electrophiles to induce the lactonisation of an unsaturated carboxylic acid is commonplace in organic synthesis.4 Typically, an electrophile such as iodine promotes cyclisation by initial reaction with an alkene followed by cyclisation via nucleophilic attack of the carboxylic acid (Scheme 1A).4 One disadvantage of this approach is that it is often necessary to further transform the newly formed C-heteroatom bond in the product, especially if further C–C bond forming sequences are desired. Lactonisation reactions that are triggered by the formation of a new C–C bond are rare,5 and often rely on transition-metal catalysis.6 In this regard, we wondered if carbocations7 might play a role in initiating lactonisation reactions. The reaction of an alkene with a carbocation initiator and a carboxylic acid should allow the formation of ring systems (here lactones) and form a new exocyclic C–C bond in the same reaction (Scheme 1B). This mode of lactonisation can be facilitated by the reaction of allylic or benzylic alcohols with Lewis or Brønsted acid in HFIP solvent.8 With this approach, a set of highly functionalised lactones could be prepared rapidly from readily available unsaturated acids and activated alcohols. The role of HFIP is likely essential in facilitating ionisation of an alcohol without the need to activate the hydroxyl group separately.9 Furthermore, the new method reported here enables an unconventional transformation to take place wherein combining a carboxylic acid partner and an alcohol starting material in the presence of a Brønsted acid (with no requirement for a Lewis acid additive) leads to the formation of a functionalised lactone, instead of the more traditional Fischer esterification product.10
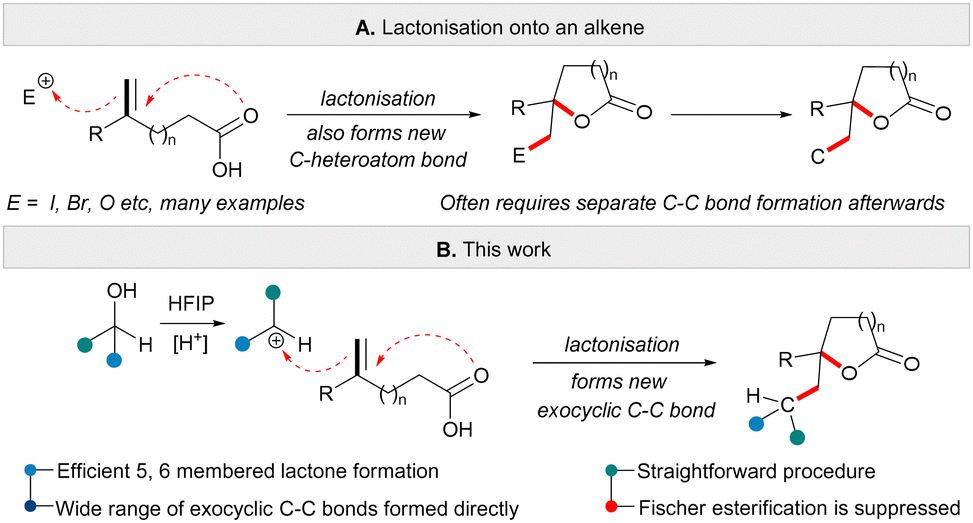 | ||
| Scheme 1 Direct carbo-lactonisation of unsaturated acids. | ||
First, an extensive reaction optimisation was carried out to identify the optimum system for the formation of 2a (see Scheme 2 and ESI† for full details). With the optimised metal-free conditions available (TFA in HFIP at RT gave 2a in 94% yield), the scope of the method was then studied with regards to both the alcohol and the alkene component (Scheme 2). A range of activated benzylic and allylic alcohols were shown to be viable in this reaction, forming the corresponding 5-membered lactones 2a–k in good to excellent yields. Free hydroxyl (2b), amine (2c), halogen (2e) and acetal (2d) groups on the benzylic alcohols were well tolerated under the reaction conditions. Activated allylic alcohols could also be engaged, furnishing products 2f, 2g and 2i bearing a versatile exocyclic alkene functionality in high yields. The presence of an aryl substituent on the allylic alcohol was essential, as unsatisfactory results were obtained with alkyl substituted allylic alcohols (eg prenyl alcohol). Asymmetric secondary alcohols furnished products 2i–j in excellent yields but as a mixture of diastereomers. In contrast, excellent diastereoselectivity was observed for carboxylic acid substrates possessing a Ph substituent adjacent to the alkene; γ-lactones 2g and 2h were both isolated in good yields and as single diastereomers. The reliability of this transformation was demonstrated by a scale-up experiment: carboxylic acid 1-Ph and benzhydrol afforded excellent yields of lactone 2k when performed on both a 0.12 mmol and 1.2 mmol scale. Notably, the products of this transformation could be further derivatised to install versatile functional groups. For example, RuCl3 mediated oxidative cleavage of the electron-rich PMP group in lactone 2a furnished carboxylic acid 4 in 27% yield.11 Alternatively, lactones formed from reaction with allylic alcohols were elaborated at the exocyclic position using ozonolysis;12 lactone 2f was subjected to ozonolysis to afford aldehyde 5 in 67% yield.
 | ||
Scheme 2 Alcohol and aryl group reaction scope. (a) Relative stereochemistry of 2h is assigned based on the X-ray crystal structure of 2g;14 (b) dr was calculated based on NMR analysis of the crude reaction mixture; (c) relative stereochemistry at the exocyclic position could not be determined. Reaction conditions: (A) RuCl3 (5 mol%), NaIO4 (20 equiv.), CCl4/CH3CN/pH 7 buffer; 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3, 0 °C to rt, overnight; (B) O2/O3, CH2Cl2, −78 °C, 5 min then DMS (excess), −78 °C to rt, 3 h. :2:3, 0 °C to rt, overnight; (B) O2/O3, CH2Cl2, −78 °C, 5 min then DMS (excess), −78 °C to rt, 3 h. | ||
The developed carbo-lactonisation methodology could be extended to 6-membered lactones. Products 2l–n were isolated in moderate to good yields, Scheme 2. Here, cyclisation yields to access δ-lactones were found to be somewhat lower compared to γ-lactones, which is consistent with previous results for other HFIP-promoted heterocyclisations.8
Next, we evaluated the scope of this method with regard to the terminal alkene substituent. We were pleased to observe that inductively electron-donating (2r) and mesomerically electron-donating (2o–q) substituents in the arene ortho- and meta-positions were well tolerated, furnishing γ-lactones in up to 66% yield. Alkenes conjugated with aryl groups bearing inductively electron-withdrawing groups could also be engaged, as demonstrated with 2s and 2t which were isolated in 77% and 67% yield respectively. We were also pleased to find that a trifluoromethyl functional group,13a was well tolerated. Furthermore, a carboxylic acid bearing a benzothiophene13b could be engaged to furnish γ-lactone 2u in 63% using benzhydrol as the electrophile. Notably, the reaction is not limited to aryl substituted terminal alkenes. Aliphatic carboxylic acids could also be efficiently engaged furnishing lactones 2v–x in good to excellent yields using benzylic and allylic alcohols.
The limitation of this methodology is represented by carboxylic acid substrates bearing aryl groups with strong mesomerically electron-withdrawing groups in the para and meta-position (eg. CN), as well as substrates possessing electron-rich five-membered rings (eg thiophene, see ESI† for details).
Given the successful formation of alkyl substituted lactones 2v–x, and to further explore the breadth of this transformation, we applied the carbo-lactonisation methodology to the synthesis of bridged, fused and spirolactones (Scheme 3). To our delight, aliphatic carboxylic acid substrates possessing exocyclic alkenes furnished products 9a–d in moderate to good yields (Scheme 3A). The reaction between carboxylic acid substrate 6 and 1,1-diphenylprop-2-en-1-ol afforded bridged lactone 9a in 46% yield (structure confirmed by X-ray). Carbo-lactonisation of five-membered carboxylic acid 7 proceeded with excellent diastereoselectivity to furnish the cis-fused [5,5] bicyclic lactone 9b in 65% yield. The levels of diastereoselectivity dropped with six-membered substrate 8; products 9c and 9d were isolated in 71% and 64% yield respectively as a 70:30 mixture of diastereomers. NOESY analysis indicated trans-stereochemistry at the ring fusion for the major diastereomer (see ESI† for details). Next, carbo- lactonisation of carboxylic acids bearing aliphatic endocyclic alkenes were explored (Scheme 3B). Substrate 10 and 1,1-diphenylprop-2-en-1-ol furnished [5,5] spirolactone 12a in 38% yield with moderate diastereoselectivity. [6,5] Spirolactone 12b could also be accessed from cyclisation of acid 11 in 75% yield and in a similar diastereomeric ratio. The use of an allylic alcohol for cyclisation of substrates 12a and 12b allowed for the installation of an exocyclic alkene that could be used for further transformations. Thus, we performed ozonolysis of an exocyclic alkene to afford aldehyde 13 in 63% yield isolated as a 74:26 mixture of diastereomers. This material was used to determine the relative stereochemistry of products 12a and 12b based on NOESY analysis of the mixture of diastereomers 13. The observed trans-configuration for the major isomer is consistent with a predominate trans-addition of a carbocation across the double bond of the nucleophile.8
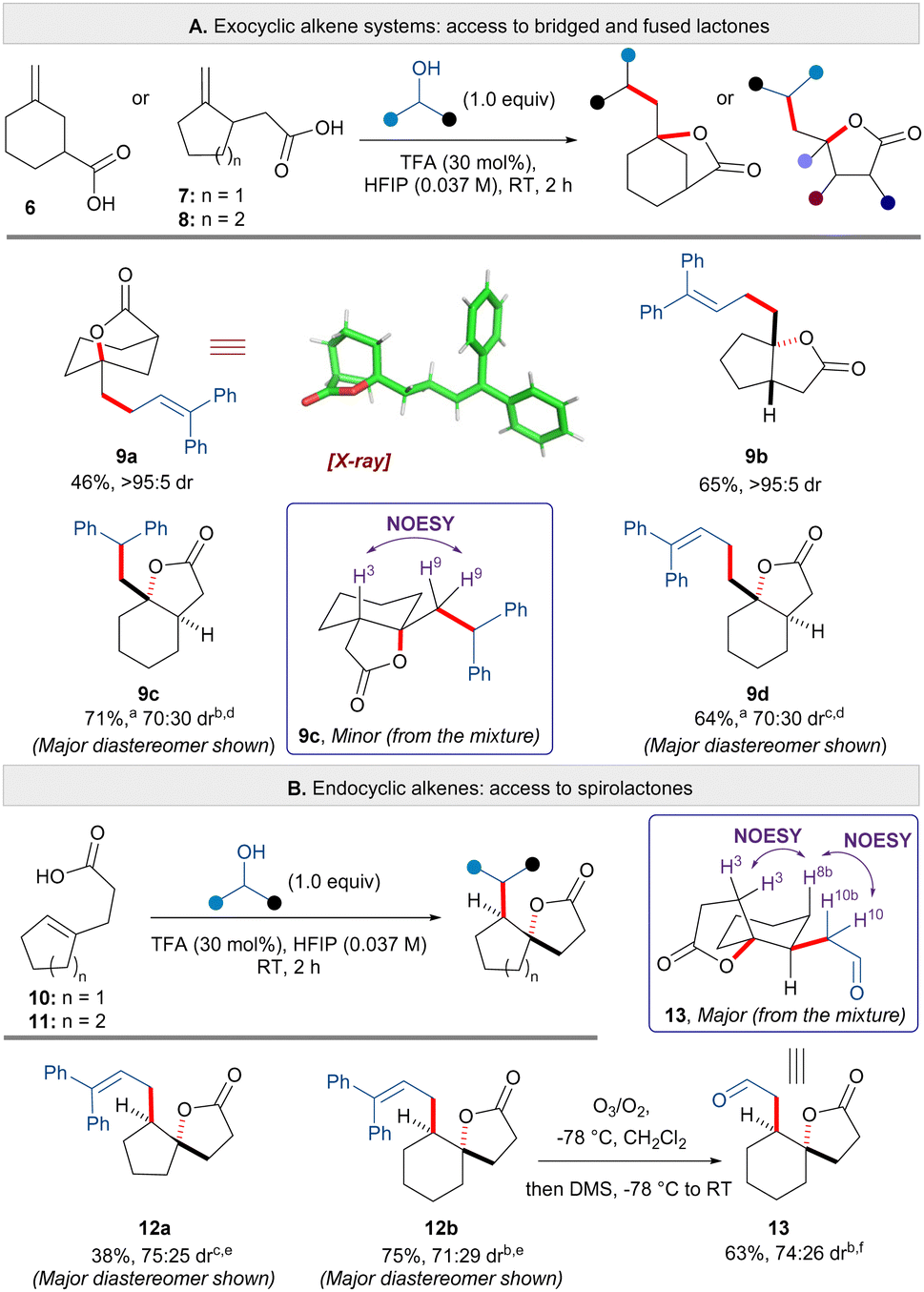 | ||
| Scheme 3 Synthesis of bridged, fused and spirolactones. (a) Reaction performed at 0 °C; (b) dr calculated after column chromatography; (c) dr calculated based on the crude reaction mixture; (d) relative stereochemistry of the diastereomers is assigned based on NOESY analysis (see ESI†); (e) relative stereochemistry is assigned based on aldehyde 13; (f) relative stereochemistry of the major diastereomer is assigned based on NOESY analysis of the mixture. | ||
Finally, we explored the mechanism of the carbo-lactonisation reaction. Firstly, to show that the reaction indeed does proceed via carbocation formation, the synthesis of lactone 2f was carried out using transposed isomers of allylic alcohol 14a and 14b (Scheme 4A). In both cases, the same regioisomer of lactone 2f was observed and isolated, which is consistent with the formation of a delocalised carbocation. Next, to shed light on whether the C–C or C–O bond is formed in a concerted or stepwise manner, and also to probe the possible intermediacy of esters,15 the reaction was attempted using unsaturated ester 15 and 4-methoxybenzyl alcohol (Scheme 4B). Compound 15, bearing a terminal alkene but lacking a carboxylic acid functional group, yielded a mixture of unsaturated esters 16a, 16b and 16c in 51% yield. Here it appears that a benzylic cation has undergone reaction with the alkene but that cyclisation has not occurred from an oxygen nucleophile.
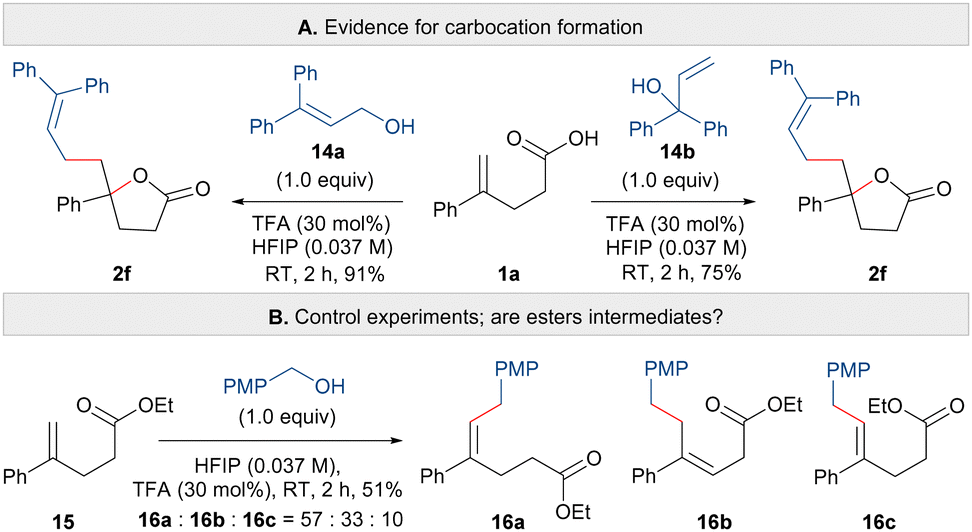 | ||
| Scheme 4 Control experiments. | ||
These results support the hypothesis that the carbo-lactonisation proceeds in a stepwise fashion in which the carbocation generated from the alcohol reacts with the terminal alkene, forming a new C–C bond and another stabilised cation; this is followed by intramolecular attack of the carboxylic acid group. This mechanism could also explain the moderate diastereoselectivities observed in the synthesis of spirolactones (12a and 12b) where a concerted (trans) addition of a heteroatom and a cation across the substrate alkene would be expected to form a single diastereoisomer.
In conclusion, this work details the development of a carbo-lactonisation methodology to form a diverse array of substituted γ- and δ-lactones. Activated benzylic and allylic alcohols were engaged in the cyclisation to form new C–C and C–O bonds and to set-up tetrasubstituted stereogenic centres. The reaction has a broad substrate scope with yields of up to 98%.
D. A. is grateful for Clarendon Scholarship and 1851 Royal Commission for the Exhibition for funding. D. A. is grateful to the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) for a studentship, generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- (a) J. M. Schomakera and B. Borhan, Org. Biomol. Chem., 2004, 2, 621 RSC; (b) S. P. Gunasekera, R. A. Isbrucker, R. E. Longley, A. E. Wright, S. A. Pomponi and J. K. Reed, J. Nat. Prod., 2004, 67, 110 CrossRef CAS PubMed; (c) D. K. Mohapatra, C. Pramanik, M. S. Chorghade and M. K. Gurjar, Eur. J. Org. Chem., 2007, 5059 CrossRef CAS; (d) S. K. Sartori, M. A. N. Diaz and G. Diaz-Munoz, Tetrahedron, 2021, 84, 132001 CrossRef CAS.
- (a) M. Mazur and D. Masłowiec, Antibiotics, 2022, 11, 1327 CrossRef CAS PubMed; (b) M. S. Matos, J. D. Anastácio and C. Nunes dos Santos, Pharmaceutics, 2021, 13, 991 CrossRef CAS PubMed; (c) K.-H. Lee and B.-R. Huang, Eur. J. Med. Chem., 2002, 37, 333 CrossRef CAS PubMed.
- (a) G. Chamorro, F. Vega, E. Madrigal, E. Mercado and M. Salazar, Toxicol. Lett., 2003, 142, 37 CrossRef CAS PubMed; (b) D. A. Mulholland, K. McFarland and M. Randrianarivelojosia, Biochem. Syst. Ecol., 2006, 34, 365 CrossRef CAS.
- For examples see: (a) M. D. Dowle and D. I. Davies, Chem. Soc. Rev., 1979, 8, 171 RSC; (b) S. Ranganathan, K. M. Muraleedharan, N. K. Vaisha and N. Jayaraman, Tetrahedron, 2004, 60, 5273 CrossRef CAS; (c) H.-Y. Luo, Y.-Y. Xie, X.-F. Song, J.-W. Dong, D. Zhu and Z.-M. Chen, Chem. Commun., 2019, 55, 9367 RSC; (d) J. Yan, Z. Zhou, Q. He, G. Chen, H. Wei and W. Xie, Org. Chem. Front., 2022, 9, 499 RSC; (e) M. J. S. Smith, W. Tu, C. M. Robertson and J. F. Bower, Angew. Chem., Int. Ed., 2023, 62, e202312797 CrossRef CAS PubMed.
- (a) D. Just, C. R. Gonçalves, U. Vezonik, D. Kaiser and N. Maulide, Chem. Sci., 2023, 14, 10806 RSC; (b) D. M. Fischer, M. Freis, W. M. Amberg, H. Lindner and E. M. Carreira, Chem. Sci., 2023, 14, 7256 RSC.
- (a) Y. Yasu, Y. Arai, R. Tomita, T. Koike and M. Akita, Org. Lett., 2014, 16, 780 CrossRef CAS PubMed; (b) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 8069 CrossRef CAS PubMed; (c) W. Sha, W. Zhang, S. Ni, H. Mei, J. Han and Y. Pan, J. Org. Chem., 2017, 82, 9824 CrossRef CAS PubMed; (d) E. Hoque and J.-Q. Yu, Angew. Chem., Int. Ed., 2023, 62, e202312331 CrossRef PubMed.
- (a) M. Niggemann and M. J. Meel, Angew. Chem., Int. Ed., 2010, 49, 3684 CrossRef CAS PubMed; (b) K. V. Wagh and B. M. Bhanage, RSC Adv., 2014, 4, 22763 RSC; (c) M. Saito, N. Tsuji, Y. Kobayashi and Y. Takemoto, Org. Lett., 2015, 17, 3000 CrossRef CAS PubMed; (d) J. M. Pérez, C. Maquilón, D. J. Ramón and A. Baeza, Asian J. Org. Chem., 2017, 6, 1440 CrossRef; (e) V. D. Vukovic, E. Richmond, E. Wolf and J. Moran, Angew. Chem., Int. Ed., 2017, 56, 3085 CrossRef CAS PubMed; (f) Y. Chen, Y. Wang, R. Zhong and J. Li, J. Org. Chem., 2020, 85, 10638 CrossRef CAS PubMed.
- (a) Y. Zhu, I. Colomer, A. L. Thompson and T. J. Donohoe, J. Am. Chem. Soc., 2019, 141, 6489 CrossRef CAS PubMed; (b) L. Cox, Y. Zhu, P. J. Smith, K. E. Christensen, M. S. Portela and T. J. Donohoe, Angew. Chem., 2022, 134, e202206800 CrossRef; (c) D. Aynetdinova, R. Jacques, K. E. Christensen and T. J. Donohoe, Chem. – Eur. J., 2023, 29, e202203732 CrossRef CAS PubMed.
- (a) I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef CAS; (b) H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood and J. Aubé, Chem. Rev., 2022, 122, 12544 CrossRef CAS PubMed.
- (a) A. G. M. Barrett and D. C. Braddock, Chem. Commun., 1997, 351 RSC; (b) K. Ishihara, M. Nakayama, S. Ohara and H. Yamamoto, Tetrahedron, 2002, 58, 8179 CrossRef CAS; (c) C.-T. Chen and Y. S. Munot, J. Org. Chem., 2005, 70, 8625 CrossRef CAS PubMed.
- (a) P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. B. Sharpless, J. Org. Chem., 1981, 46, 3936 CrossRef CAS; (b) L. S. M. Miranda and M. L. A. A. Vasconcellos, Synth, 2004, 1767 CAS; (c) Y. Zhu, I. Colomer and T. J. Donohoe, Chem. Commun., 2019, 55, 10316 RSC.
- (a) R. Criegee, Angew. Chem., Int. Ed. Engl., 1975, 14, 745 CrossRef; (b) C. Geletneky and S. Berger, Eur. J. Org. Chem., 1998, 1625 CrossRef CAS; (c) S. G. Van Ornum, R. M. Champeau and R. Pariza, Chem. Rev., 2006, 106, 2990 CrossRef CAS PubMed.
- (a) A. Abula, Z. Xu, Z. Zhu, C. Peng, Z. Chen, W. Zhu and H. A. Aisa, J. Chem. Inf. Model., 2020, 60, 6242 CrossRef CAS PubMed; (b) R. S. Keri, K. Chand, S. Budagumpi, S. B. Somappa, S. A. Patil and B. M. Nagaraja, Eur. J. Med. Chem., 2017, 138, 1002 CrossRef CAS PubMed.
- Single crystal X-ray diffraction data were collected using a (Rigaku) Oxford Diffraction SuperNova A diffractometer and reduced using CrysAlisPro. Structures were solved using Super-Flip [L. Palatinus, G. Chapuis J. Appl. Cryst. 2007, 40, 786-790] and refined using CRYSTALS [P. Parois, R. Cooper, A. L. Thompson, Chem. Cent. J. 2015, 9, 30; R. Cooper, A. L. Thompson, D. J. Watkin, J. Appl. Cryst. 2010, 43, 1100–1107] as per the ESI† (CIF). Crystallographic data for compounds 2a, 2g, 2t, 2k, 4 and 9a have been deposited with the Cambridge Crystallographic Data Centre with deposition numbers: 2349117 (2a); 2349118 (2g); 2349119 (2k); 2349120 (2t); 2349121 (4); 2349122 (9a).
- (a) J. Wennerberg, L. Eklund, M. Polla and T. Frejd, Chem. Commun., 1997, 445 RSC; (b) J. Wennerberg, C. Olofsson and T. Frejd, J. Org. Chem., 1998, 63, 3595 CrossRef CAS; (c) J. Wennerberg, F. Ek, A. Hansson and T. Frejd, J. Org. Chem., 1999, 64, 54 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of all experimental procedures, 1H/13C NMR spectroscopic characterisation and X-ray data. CCDC 2349117 (2a), 2349118 (2g), 2349119 (2k), 2349120 (2t), 2349121 (4), 2349122 (9a). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02472a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |