
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peroxodicarbonate – a renaissance of an electrochemically generated green oxidizer
Theresa
Rücker†
 ab,
Niclas
Schupp†
b,
Fiona
Sprang†
b,
Tomas
Horsten
b,
Bernd
Wittgens
a and
Siegfried R.
Waldvogel
*bc
ab,
Niclas
Schupp†
b,
Fiona
Sprang†
b,
Tomas
Horsten
b,
Bernd
Wittgens
a and
Siegfried R.
Waldvogel
*bc
aProcess Technology, SINTEF Industry, Trondheim, Norway
bMax Planck Institute for Chemical Energy Conversion, Mülheim an der Ruhr, Germany. E-mail: siegfried.waldvogel@cec.mpg.de
cInstitute of Biological and Chemical Systems – Functional Molecular Systems (IBCS-FMS), Karlsruher Institut für Technologie (KIT), Karlsruhe, Germany
First published on 17th June 2024
Abstract
The direct anodic conversion of alkali carbonates in aqueous media provides access to peroxodicarbonate, which is a safe to use and green oxidizer. Although first reports date back around 150 years, its low concentrations and limited thermal stability have consigned this reagent to oblivion. Boron-doped diamond anodes, novel electrolyser concepts for heat dissipation, and the mixed cation trick allow record breaking peroxodicarbonate concentrations >900 mM. The electrochemical generation of peroxodicarbonate was already demonstrated on a pilot scale. The inherent safety is ensured by the limited stability of the peroxodicarbonate solution, which decomposes under ambient conditions to oxygen and facilitates subsequent downstream processing. This peroxide has, in particular at higher concentrations, an unusual reactivity and seems to be an ideal reagent when peroxo-equivalents in combination with alkaline base are required. The conversions with peroxodicarbonate include the Dakin reaction, epoxidation, oxidation of amines (aliphatic and aromatic) and sulfur compounds, deborolative hydroxylation reactions, and many more. Since the base equivalents also represent the makeup chemical for pulping plants, peroxodicarbonate is an ideal reagent for the selective degradation of lignin to vanillin. Moreover, peroxodicarbonate can be used as a halogen-free bleaching agent. The emerging electrogeneration and use of this green platform oxidizer are surveyed for the first time.
 Theresa Rücker | Theresa Rücker, born in Dresden, holds a Dipl. Ing. degree in process engineering and natural product technology from TU Dresden (Germany). She has been working in SINTEF Industry (Norway) since 2020. Theresa recently started as an external PhD candidate in the group of Prof. Siegfried Waldvogel at the Max-Planck-Institute for Chemical Energy Conversion on the topic of scale-up of electro-organic synthesis. |
 Niclas Schupp | Niclas Schupp was born in Wiesbaden in 1991. In 2011, he finished his apprenticeship as a laboratory technician at Johannes Gutenberg-University Mainz, and in 2020, he obtained his master's degree in chemistry at the University Mainz. Afterward, he started a PhD at the Department of Chemistry, Johannes Gutenberg-University Mainz, under the supervision of Prof. Dr S. R. Waldvogel. In 2024, Niclas moved to Max Planck Institute for chemical energy conversion, Mülheim an der Ruhr, to finish the PhD under Prof. Waldvogel's supervision. |
 Fiona Sprang | Fiona Sprang received her Master of Science degree in chemistry from Johannes Gutenberg University (Germany). During her studies, she conducted a research internship at the University of Toronto (Canada) and worked at the Max Planck Institute for Chemistry (Germany). Subsequently, she started to pursue PhD studies under the supervision of Professor S. R. Waldvogel at the Johannes Gutenberg University (Germany). Her research interests include the development of sustainable access to biologically relevant quinone scaffolds and related motifs. |
 Tomas Horsten | Tomas Horsten, born in Turnhout, obtained his MSc degree and PhD in Chemistry at KU Leuven Belgium in November 2021, under the supervision of Prof. Wim Dehaen. In 2022, he performed postdoctoral research at the Department of Green Chemistry and Technology, Ghent University Belgium, under the supervision of Prof. Christian Stevens. In 2023, Tomas joined the group of Prof. Siegfried Waldvogel at Johannes Gutenberg University Mainz, Germany. Tomas has conducted postdoctoral studies at the Max-Planck-Institute for Chemical Energy Conversion in Mülheim an der Ruhr under Prof. Waldvogel's supervision from February 2024 until now. |
 Bernd Wittgens | Bernd Wittgens, born in Cologne, Germany, obtained his PhD in Chemical Engineering at NTNU in 1999, Norway, under the supervision of Prof. S. Skogestad. From 1998 to 2005, he worked for ENERGOS ASA on the design, building and commissioning of waste incineration plants. In 2005, he joined the SINTEF Industry and worked initially with microreactors for process intensification and recently in the area of green chemistry and biorefineries, primarily on process design, piloting, technical and economic evaluations. |
 Siegfried R. Waldvogel | Siegfried R. Waldvogel studied chemistry in Konstanz and received his PhD in 1996 from the University of Bochum/Max-Planck-Institute for Coal Research (Prof. M. T. Reetz). After postdoctoral research in La Jolla, CA (Prof. J. Rebek, Jr.), he started his own research at the Universities of Münster, Bonn and Mainz. In 2023, he became director of the Max-Planck Institute for Chemical Energy Conversion. Prof. Waldvogel is heading large research clusters such as ETOS – Electrifying Technical Organic Synthesis. His research interests are novel electro-organic transformations including bio-based feedstock. In 2018, he cofounded ESy-Labs GmbH, which provides custom electrosynthesis and contract R&D. |
Introduction
To tackle the challenges of the prevailing climate crisis, novel technologies as well as a sustainable mindset must be adopted.1–5 A central step is the de-fossilisation of chemical processes by alternatives like molecular “green” hydrogen, accessible by electrochemical water-splitting.6,7 Substitution of the energy consuming, techno-economically insignificant and sluggish oxygen evolution reaction is desirable and would grant access to value-added products and intermediates.8,9 Electrochemistry provides a favourable and sustainable alternative to conventional synthetic approaches, as electrons are directly used, instead of stoichiometric reagents.8,9 This opens the field of ex-cell oxidizers wherein usually peroxides are applied. An on-demand electrochemical generation thereof decreases the typical dangers associated with the storage and handling of oxidizers on industrial scales.10–17 Typical electrochemically generated high performance oxidizers are periodate,18–22 peroxodisulfate,23–25 di(methanesulfonyl)-peroxide,26,27 and peroxodicarbonate 1. The latter is a good and safe surrogate for hydrogen peroxide combined with a base. This oxidizer has attracted attention in the past decade and is surveyed in this review.History
Peroxodicarbonate formally consists of two carbonate moieties that are linked by a peroxide bond. This is constitutionally different from commercially available percarbonate, which only presents an adduct of hydrogen peroxide to carbonate. In general, peroxodicarbonate can only be accessed via direct electrochemical oxidation of aqueous carbonates (Scheme 1).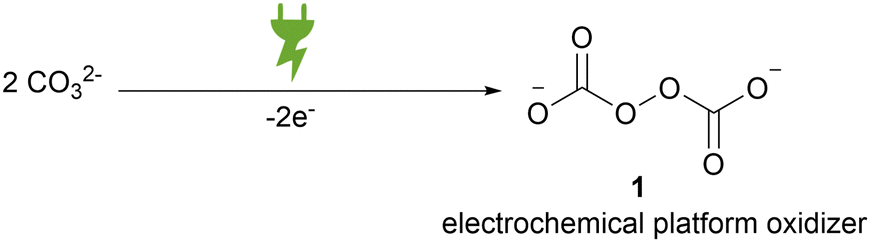 | ||
| Scheme 1 Electrochemical oxidation of two carbonates to peroxodicarbonate. | ||
The limited solubility of carbonates in aqueous media and the labile nature of peroxodicarbonate made it for a long time a curiosity research in chemistry.28 In 1869, the first report on the synthesis of peroxodicarbonate was given by Hansen and Constam.29 An aqueous potassium carbonate solution was electrolysed at −40 °C in a divided batch-type cell equipped with two platinum electrodes. A couple of decades later, a method to differentiate hydrogen peroxide adducts from factual peroxodicarbonate by its redox behaviour was proposed.30,31 The thermal sensitivity of peroxodicarbonate inhibits the complete removal of water and more specific analysis of the peroxide moiety.32–34 In 2002, Jansen et al. synthesised potassium peroxodicarbonate by electrolysis of a saturated potassium carbonate solution at −20 °C on platinum electrodes under galvanostatic conditions in a divided batch-type cell. The authors managed to isolate a microcrystalline, light blue powder and subjected it to high resolution powder X-ray diffraction at −123 °C.32 This analysis confirmed the previously attributed structure.31 The anion features a central C–O–O–C moiety with a dihedral angle of 93°.
In between the initial reports and structural elucidation, peroxodicarbonate faded into oblivion. In parallel, boron-doped diamond (BDD) emerged as a novel electrode material for the generation of oxidizers.35–37 BDD features an outstanding mechanical stability and a wide electrochemical window due to its high overpotentials for oxygen and hydrogen evolution.38,39 Moreover, BDD can be made from renewable carbon sources and is in contrast to platinum that id considered as a sustainable electrode material.37 In 2003, peroxodicarbonate was brought back into focus by Nishiki and co-workers (Fig. 1).34 The electrolysis of 1 M Na2CO3 was conducted at 0 °C in a divided batch-type cell applying a BDD anode. Using this anode material over platinum improved the Faraday efficiency from 16% to 82%, respectively. Noteworthily, the peroxodicarbonate concentration increases at higher current density, at the expense of the Faraday efficiency. Moreover, the synthesis of peroxodicarbonate above 0 °C is feasible but is accompanied by a decrease in Faraday efficiency. A maximum peroxodicarbonate concentration of 0.045 M was achieved at 0.5 A cm−2. This seminal work attracted several groups performing batch-type electrolysis to peroxodicarbonate with Faraday efficiencies of up to 98%. However, the peroxodicarbonate concentrations remained rather low (<0.1 M).40–43
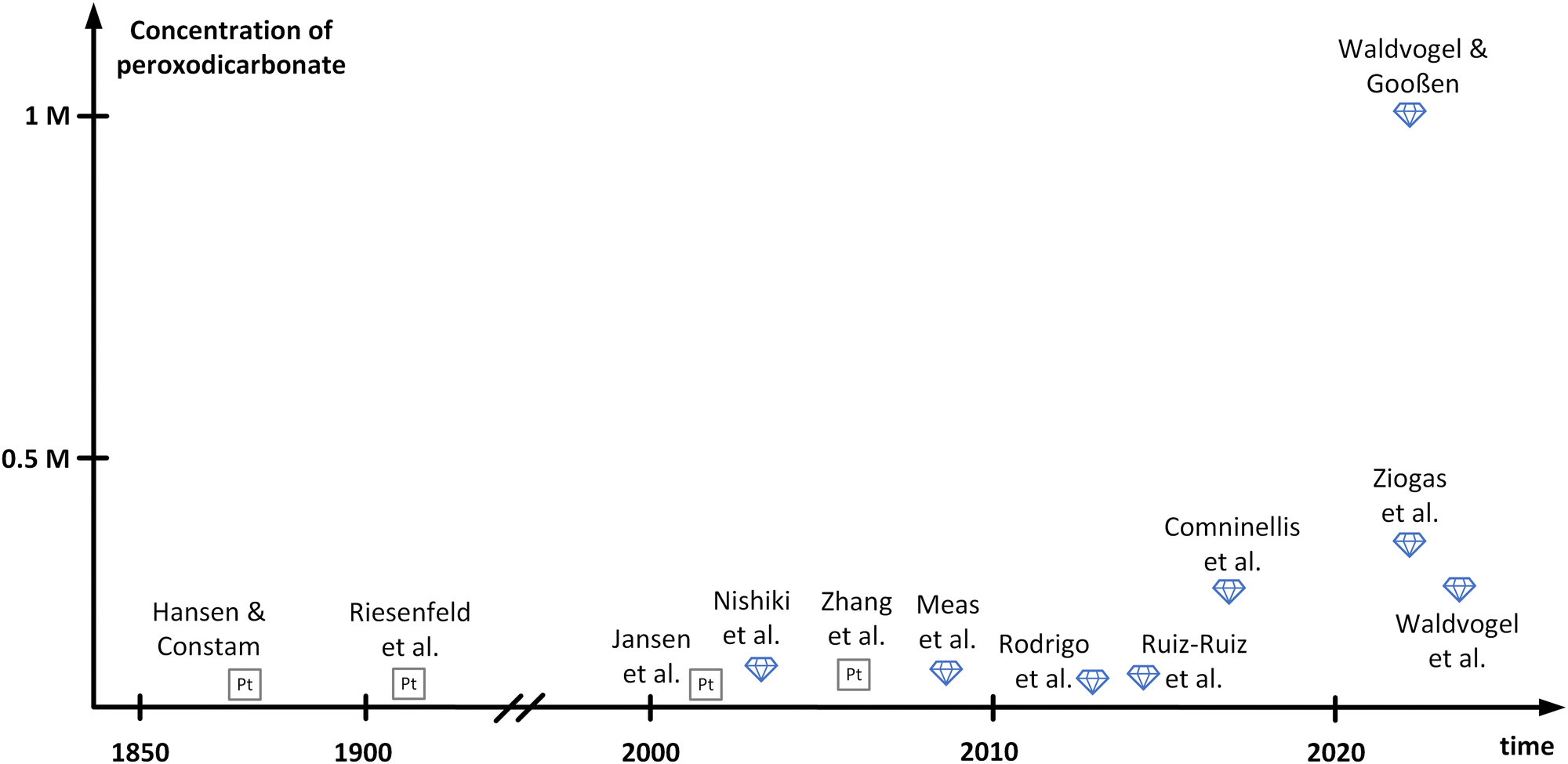 | ||
| Fig. 1 Summary of development of different peroxodicarbonate concentrations over time. The names mentioned in this figure refer to the corresponding authors of referenced publications. | ||
Remarkable progress was accomplished in 2018 by Comninellis and co-workers,33 who comprehensively investigated in flow electrolysers the influence of the carbonate concentration, temperature, current density, pH, and flow rate. An undivided cyclic flow electrolysis cell was operated, equipped with a stainless-steel cathode and a BDD anode. Since a sodium carbonate concentration of 1.5 M turned out to be beneficial,28 they had to operate at the edge of maximum solubility at 18 °C. Moreover, high current densities of 0.72 A cm−2, a pH of 11.4 and high flow rates were beneficial. Under these conditions, the peroxodicarbonate concentration of 0.28 M became accessible for the first time at a satisfying Faraday efficiency of 76%. With the same Faraday efficiency, Ziogas et al. were able to increase the concentration to 0.38 M, operating two in-house developed electrolyser cells in parallel mode.44 Furthermore, the stability of the obtained peroxodicarbonate solution was investigated. While the tremendous potential of peroxodicarbonate as a sustainable oxidant was recognized early on, reports on applications up until 2022 have been sparse, since the challenging synthesis and the low overall concentrations of peroxodicarbonate posed large obstacles.33 This changed with the publication of the landmark paper by Waldvogel and Gooßen.45 One important aspect which was solved is the enhancement of the carbonate concentration in aqueous media. When using a mixture of sodium and potassium carbonate, a peroxodicarbonate concentration of up to 0.337 M was obtained in a similar setup to that of Comninellis and co-workers.33 By adding bicarbonate to the solution the concentration of peroxodicarbonate could be further enhanced to 0.406 M. The authors assumed that bicarbonate balances the hydroxide ions released over the course of the electrolysis. Consequently, a composition of 1.125 M K2CO3, 0.9 M Na2CO3 and 0.225 M KHCO3 provided optimal results. To achieve even higher concentrations, not only was the composition of the electrolyte crucial, but also the heat dissipation during and after the electrolysis event. It was found that efficient cooling by addition of an in-line heat exchanger resulted in a concentration of 0.588 M.45 Based on this finding, a new electrolysis cell design with an elaborate heat-transfer system was developed, enabling the production of peroxodicarbonate with a concentration of up to 0.919 M. Replacing water in the electrolyte with D2O finally afforded concentrations over 1 M. This can be attributed to a solvent isotope effect, which has previously been reported in the electrolysis of chloride salts and results from weaker hydrogen bonds between anions and D2O compared to H2O.46 Recently, Hamm et al. investigated the parallel paired electrolysis of the green oxidizing agents, peracetic acid and peroxodicarbonate. The latter was produced on BDD electrodes at a concentration of 0.043 M, while peracetic acid was generated from oxygen reduction through a gas diffusion electrode at a concentration of 0.023 M, with a good Faraday efficiency of up to 56%.47 A graphical summary of the history of peroxodicarbonate including the respective concentrations achieved by the different research groups is displayed in Fig. 1. It must be noted that work published as patents has not been included herein.
Characteristics of peroxodicarbonate
Albeit a powerful oxidizer, peroxodicarbonate is not thermally stable. However, this instability has a significant safety aspect as it decomposes rapidly at room temperature (r.t.) and thus becomes non-hazardous. The remaining residual alkaline solution can be used as e.g., a makeup chemical in the paper industry.48 The total content of oxidizing agents can be determined by adding H2SO4 and converting all peroxodicarbonate species into hydrogen peroxide. After that an iodometric or cerimetric titration can be conducted. This is an established method but does not allow differentiation between the original types of oxidizing agent.33 Unambiguous proof for the formation of peroxodicarbonate can be found in the 13C NMR spectrum since its signal has been identified at δ = 162 ppm.45 When peroxodicarbonate is produced at cooling temperatures below 0 °C, carbonates will precipitate and form a sluggish mass with high viscosity.45 Since no critical elements are involved in the generation of peroxodicarbonate, it is particularly suitable for scale-up and use on a larger scale.49 The high ionic strength might be a disadvantage that cannot be concealed. However, its salty nature also provides options to facilitate downstream processing. The salty character of peroxodicarbonate solutions makes it difficult, for example, to convert lipophilic substrates, which is a challenge for synthetic chemists. Regarding the limited thermal stability of peroxodicarbonate, there is differing information in the literature (Table 1). For concentrations of 0.390 M and 0.282 M half-lives of 60 min and 69 min were subsequently reported, respectively.33,44 For a significantly lower initial concentration of peroxodicarbonate of 0.045 M, 835 min were specified. It is noteworthy that with decreasing concentrations the half-life extends in a clear trend. Using a low-concentration solution, Ruiz et al. carried out further investigations into the stability of peroxodicarbonate using sodium metasilicate as the stabiliser. The half-life increased significantly from 835 min to 6.5 days.50 Interestingly, it was found that no degradation was observed when peroxodicarbonate was shock frozen and stored below −18 °C.44Design of electrolysis cells
The design of electrochemical systems offers a multitude of adjustments.11 In the history of peroxodicarbonate generation, both divided cells32,34,50 and undivided setups33,51 have been used. Increasing attention is also being paid to the use of flow cells as a continuous process for later industrial applications.33,44,45,48 Here, it is favourable to target a stacked electrode setup in which the potential and current density are evenly distributed between the electrode plates without each of them having to be contacted individually.52 Besides these individually developed flow electrolysers, there are also commercially available setups like ElectraSyn Flow sold by IKA-Werke GmbH & Co. KG or Synthesis StarterKit sold by CONDIAS GmbH.Based on the highly modular flow cell concept of Gütz et al., Waldvogel and co-workers added an external cooling circuit to the half-cell on the cathode side, which is made entirely of stainless steel (Fig. 2).48,53 The BDD anode is embedded in a TeflonTM half-cell, which allows a quick exchange of the electrode material if needed. The active anode area in this setup is 12 cm2. Larger cells in this design exhibit even back-side cooling of the anode.54–56 However, the limited thermal conductivity of TeflonTM limits its applications and requires more dissipative systems.
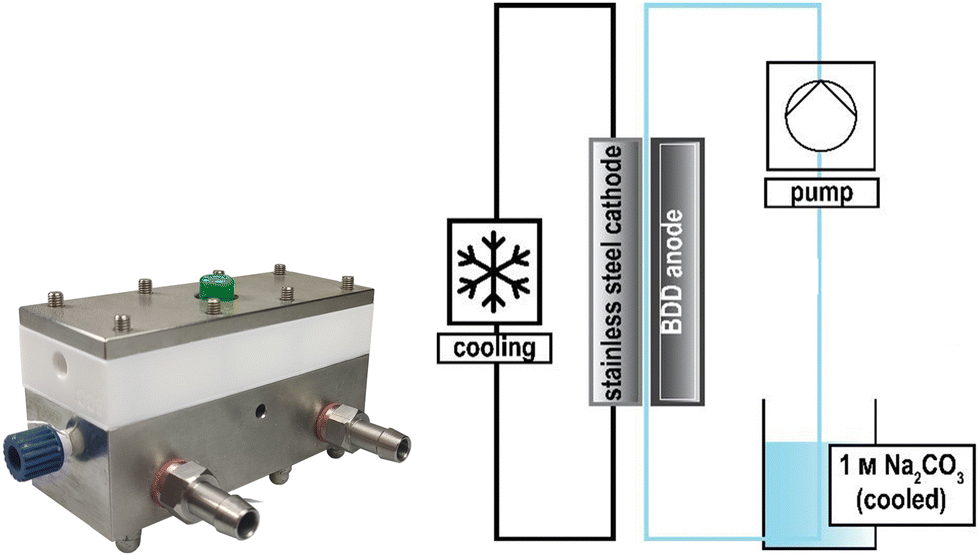 | ||
| Fig. 2 Flow electrolyser developed by Zirbes et al.48 Copyright (2023) Wiley. Used with permission from Zirbes et al. Peroxodicarbonate as a green oxidizer for the selective degradation of kraft lignin into vanillin, Angew. Chem. Int. Ed., Wiley. | ||
A different concept was designed by Ziogas et al.44 The authors developed an electrochemical micro-reactor with integrated heat exchangers (Fig. 3). The reactor consists of two milled cathode plates made of stainless steel, arranged in a sandwich configuration with a double-sided BDD anode in the middle. The geometric anode surface area per cell is 24.8 cm2 per electrode side, resulting in a total anodic area of 49.6 cm2. The heat dissipation in this system was realized by means of a thermostat at 7 °C, which was used for cooling both cathodes, serving as heat exchangers, and for pre-cooling the electrolyte solution.44
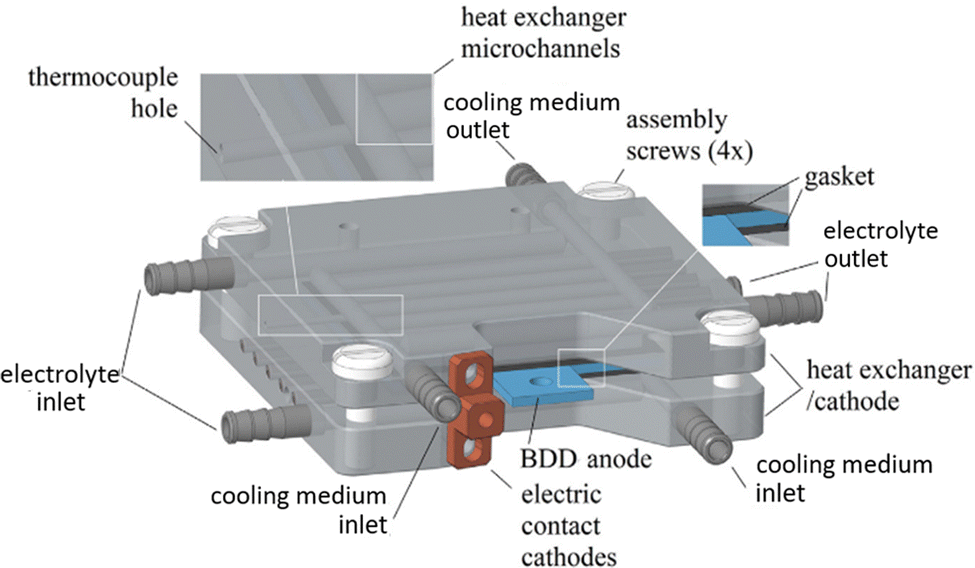 | ||
| Fig. 3 CAD image of the ECMR assembly.44 Copyright (2023) Elsevier. Used with permission from Ziogas et al. Peroxodicarbonate: Electrosynthesis and first directions to green industrial applications, Curr. Res. Green Sustainable Chem., Elsevier. | ||
An electrolyser design in which the cooling of both the cathode and anode has been implemented was described by Seitz et al. (Fig. 4). The new cell layout, based on a copper casing as a heat sink, significantly improved the efficiency of peroxodicarbonate formation and enabled the production of high concentrations. On the anodic side, a BDD anode on the Si support was inserted into the copper casing, while on the cathodic side, a stainless-steel plate was glued onto the copper casing. The anode surface in this setup is 3 cm2. Additional internal cooling channels were milled in the copper heat sink including a so-called microchannel structure to enhance the cooling efficiency further.45
 | ||
| Fig. 4 Custom-developed electrolyser based on copper-based cooling cell.45 Copyright (2023) Wiley. Used with permission from Seitz et al. Concentrated aqueous peroxodicarbonate: efficient electrosynthesis and use as an oxidizer in epoxidations, S- and N-oxidations, Angew. Chem. Int. Ed., Wiley. | ||
Applications
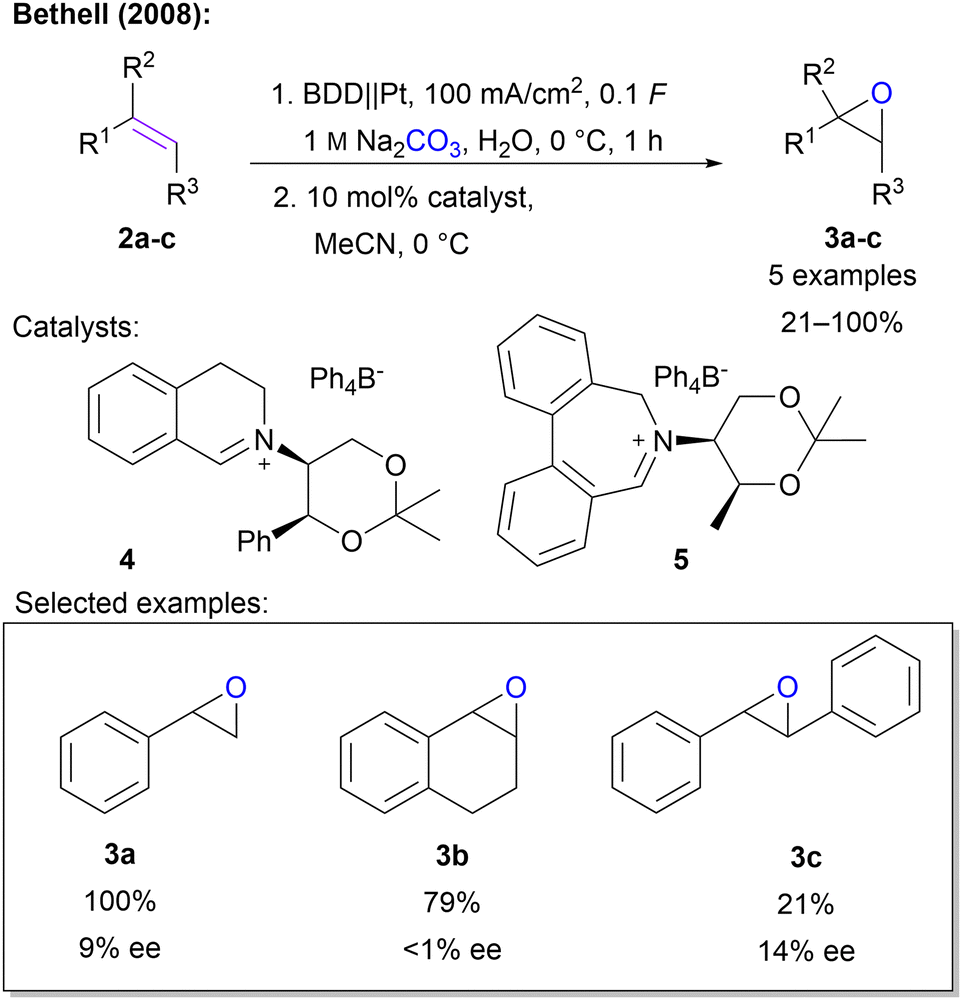 | ||
| Scheme 2 Epoxidation of alkenes with peroxodicarbonate. The amount of applied charge is given in relation to carbonate. | ||
Waldvogel and Gooßen also explored a Weitz–Scheffer-type epoxidation of unsaturated electron-deficient carbon double bonds in enone systems (Scheme 3).45 Here, an ex-cell approach was applied. This is of tremendous importance, since the synthesis of 1 is conducted at very high current densities which would fragment organic molecules in an unselective manner. First, highly concentrated 1 was synthesized according to an established protocol. After determination of the concentration, a defined amount of 1 solution was added to the enone in ethanol and the mixture was stirred at room temperature until completion of the reaction (1–3 h). The protocol was successfully applied to 12 substrates in yields between 50% and 96%. The protocol is compatible with sensitive quinones, as well as sterically demanding cyclic ketones. In contrast to the previous protocol, highly concentrated peroxodicarbonate eliminates the need for a catalyst (Scheme 3).
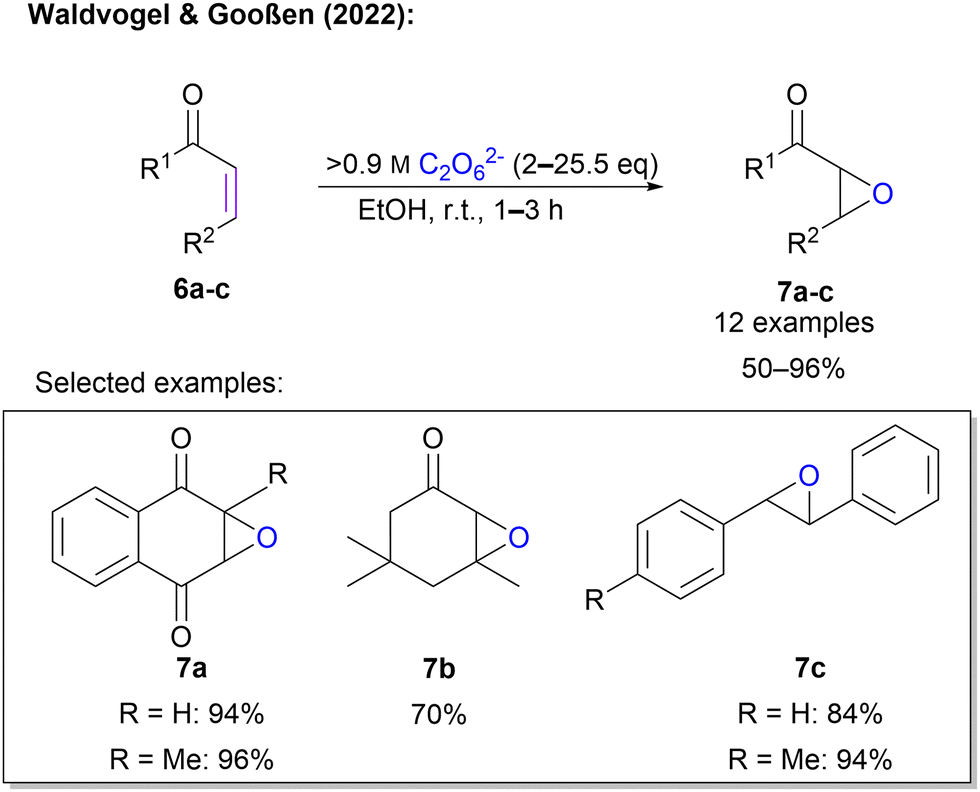 | ||
| Scheme 3 Epoxidation of unsaturated α,β-enones with 1. | ||
Recently, Waldvogel and co-workers expanded the scope of carbon oxygenations by introducing hydroxylation protocols. Through treatment of aromatic boronic acids with highly concentrated 1 in ethanol phenols are obtained.65 It is noteworthy that in the majority of 22 examples, 1.1 eq. of 1 was sufficient to ensure full conversion and a high yield of up to 97%. Moreover, a plethora of functional groups, including various halo substituents, trifluoromethyl, esters, thioethers, nitriles, silyl groups and heterocycles, was tolerated, underlining the high synthetic utility of this protocol. Scalability was demonstrated on a multi-gram scale without a variation in yield. Simple downstream processing was ensured through purification by short-path distillation. In addition to boronic acids, the protocol is also applicable to alkenes. Here, a hydroboration–oxygenation sequence was performed in a one-pot fashion. Even challenging substrates, e.g. norbornene and terpenes, were compatible with the approach and overall satisfying yields between 39 and 76% were found for these 8 examples (Scheme 4).
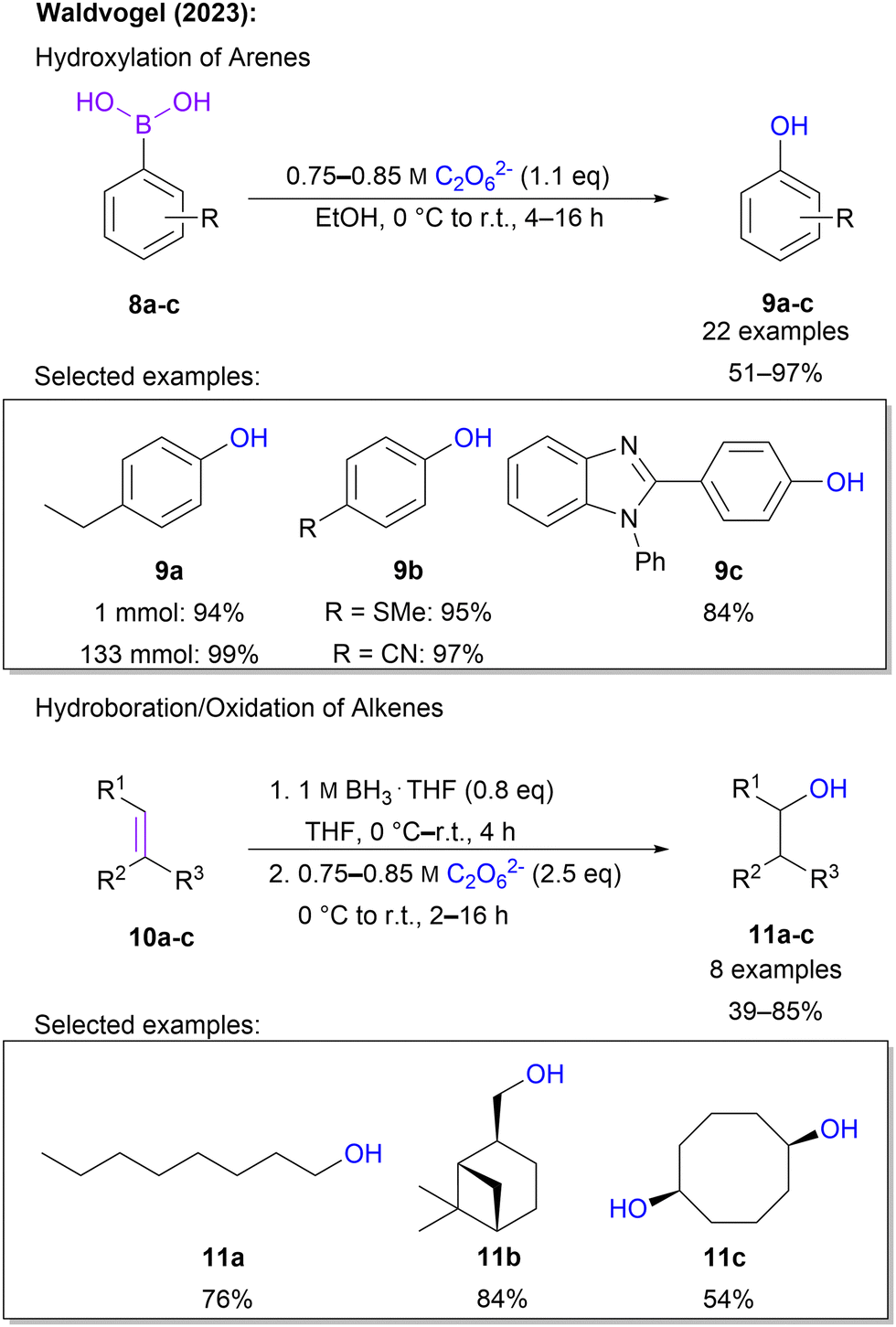 | ||
| Scheme 4 Hydroxylation of organoboron compounds with 1. | ||
Another hydroxylation approach was introduced in 2024 by Waldvogel and co-workers,66 where hydroxybenzaldehydes served as starting materials. This is in particular attractive, since such substrates can be sourced from biogenic feedstock.67,68 In a Dakin-type reaction sequence, these hydroxybenzaldehydes react with 1 to form hydroquinones and catechols. For this application 1 solution fulfils a dual role as it acts as an oxidant and the required base. Again, the reaction was performed using an ex-cell approach. Interestingly, the actual Dakin reaction step was performed in the peroxodicarbonate solution circumventing the use of an additional organic solvent. A broad scope of 20 substrates was successfully tested and a diverse set of hydroquinone and catechol was obtained in yields between 53 and 97% in swift reaction times of 20 minutes (Scheme 5). Noteworthily, the reaction with 1 is very quick and the intermediate aryl formate is hydrolysed after the peroxide is converted preventing over-oxidation to quinones.
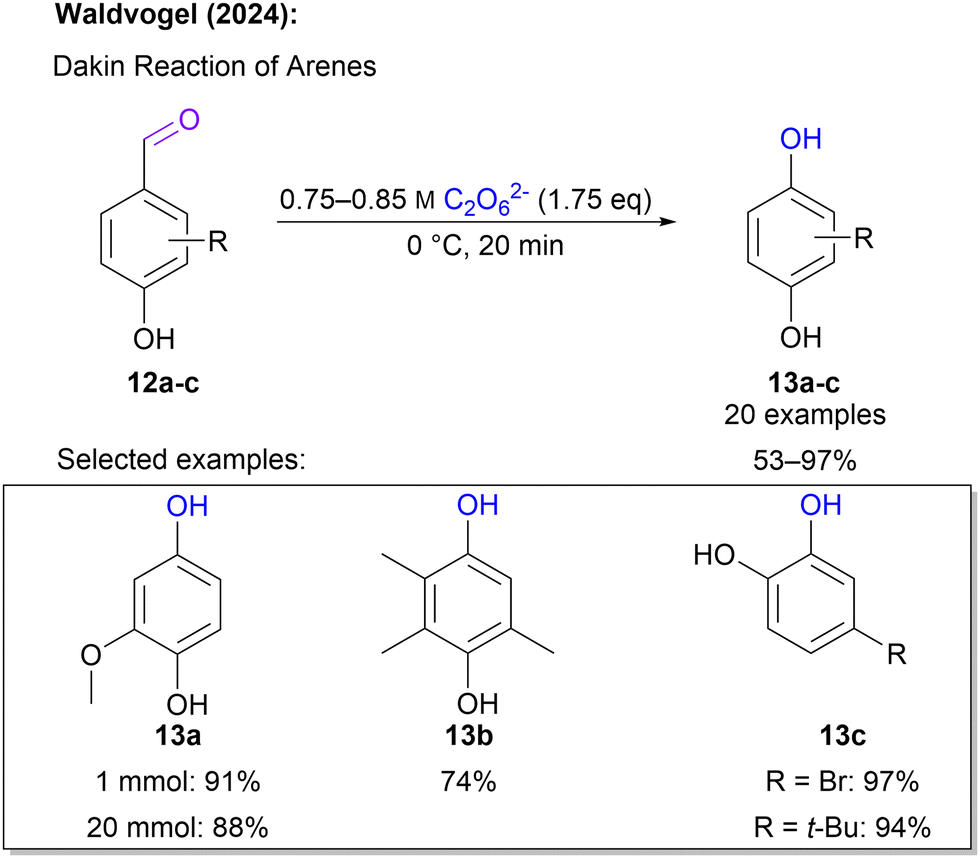 | ||
| Scheme 5 Dakin reaction with 1. | ||
Gooßen and co-workers established the N-oxidation of tertiary amines using a 0.25 M 1 solution. It was found that addition of 0.25 eq. of ethylenediaminetetraacetic acid (EDTA) and 2,2,2-trifluoroacetophenone (TFAP) was crucial to obtain high yields of 97%. The roles of EDTA and TFAP are attributed to the promoted oxygen transfer of EDTA and the formation of an activated oxygen-transfer intermediate from TFAP. Subsequently, the scope of 16 substrates with up to 98% yield was established (Scheme 6). It should be noted that these protocols are limited to aliphatic and benzylic tertiary amines which are sufficiently water-soluble. This can be explained by the high salt concentration of 1 solution.83 By replacing the low concentration peroxodicarbonate solution by a highly concentrated solution, above 0.9 M, N-oxidation was achieved in excellent yields without any additives.45 Moreover, non-water-soluble compounds were successfully converted to the corresponding N-oxide using ethanol as a co-solvent (Scheme 6).
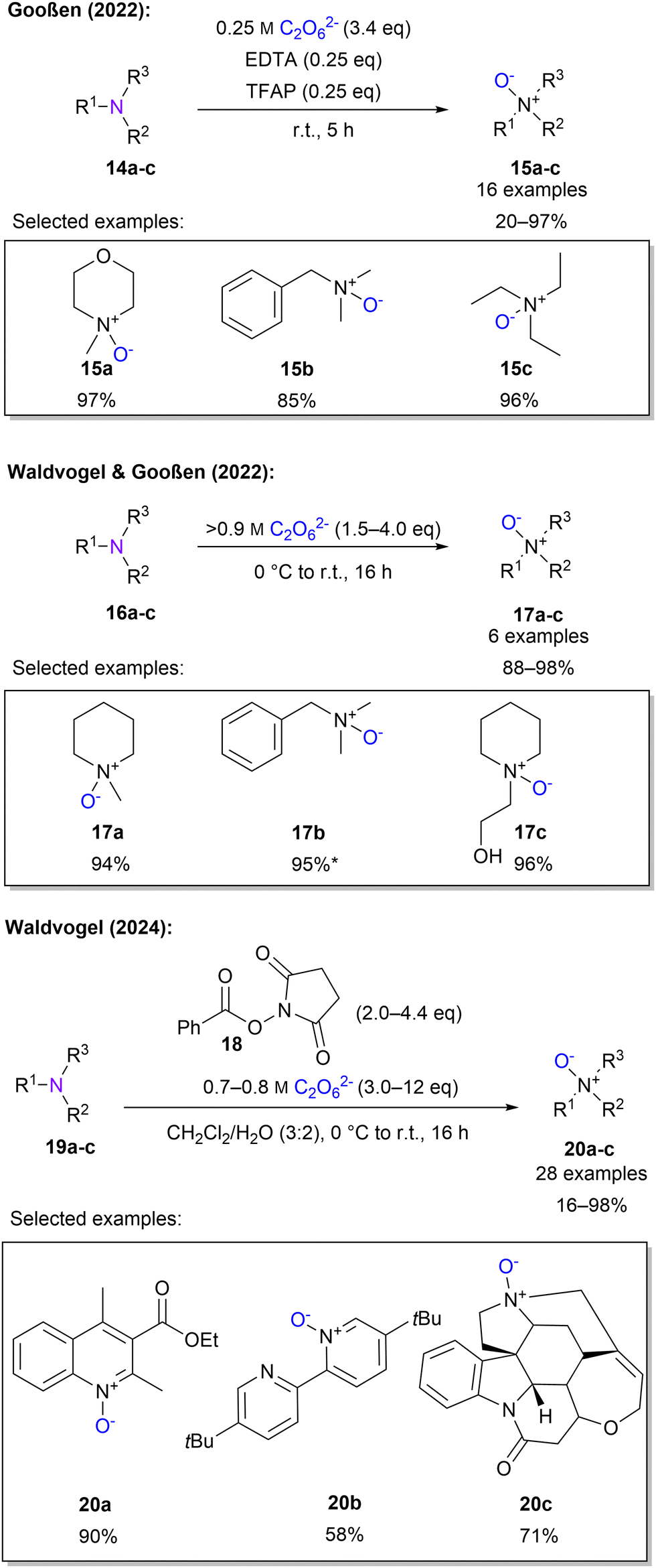 | ||
| Scheme 6 N-Oxidation with peroxodicarbonate. * Ethanol is used as a co-solvent. | ||
To include aromatic, non-water-soluble amines, Waldvogel and co-workers explored a new protocol, by choosing a biphasic mixture of water and dichloromethane and introducing a new set of activators derived from commercially available bleach activators.84 After screening several activators phenyloxy succinimide (POSI) 18 gave the best results (Scheme 6). The authors stated that by introducing 1 to 18, a peracid is formed, which oxidises the amines. It is further explained that there are two important influences on the activator. First, the basicity of the released leaving group and second, the oxidising strength of the peracid. Of particular importance here is the ex-cell approach for generating highly concentrated 1. The protocol, using POSI as an activator and high peroxodicarbonate concentrations, was then successfully applied to 28 examples containing quinolines, pyridines and naturally occurring compounds for late-stage functionalisation.
Waldvogel and Gooßen also investigated the oxidation of sulphur moieties.45 By adjusting the pH of the percarbonate solution, the outcome of sulfide oxidation could be controlled to sulfoxides or sulfones, with a pH of 11 or 12.5, respectively (Scheme 7). In this manner, 6 examples with yields between 80–98% were demonstrated. Furthermore, Waldvogel and co-workers explored the synthesis of sulfoximines from sulfilimines.85 Applying electrical current to sulfilimines did not afford any sulfoximines. However, by using highly concentrated peroxodicarbonate with over 0.8 M in combination with a small amount of acetonitrile, sulfoximines were obtained in high yields with a selection of 5 examples (Scheme 7). Interestingly, a low concentration of peroxodicarbonate requires more equivalents.
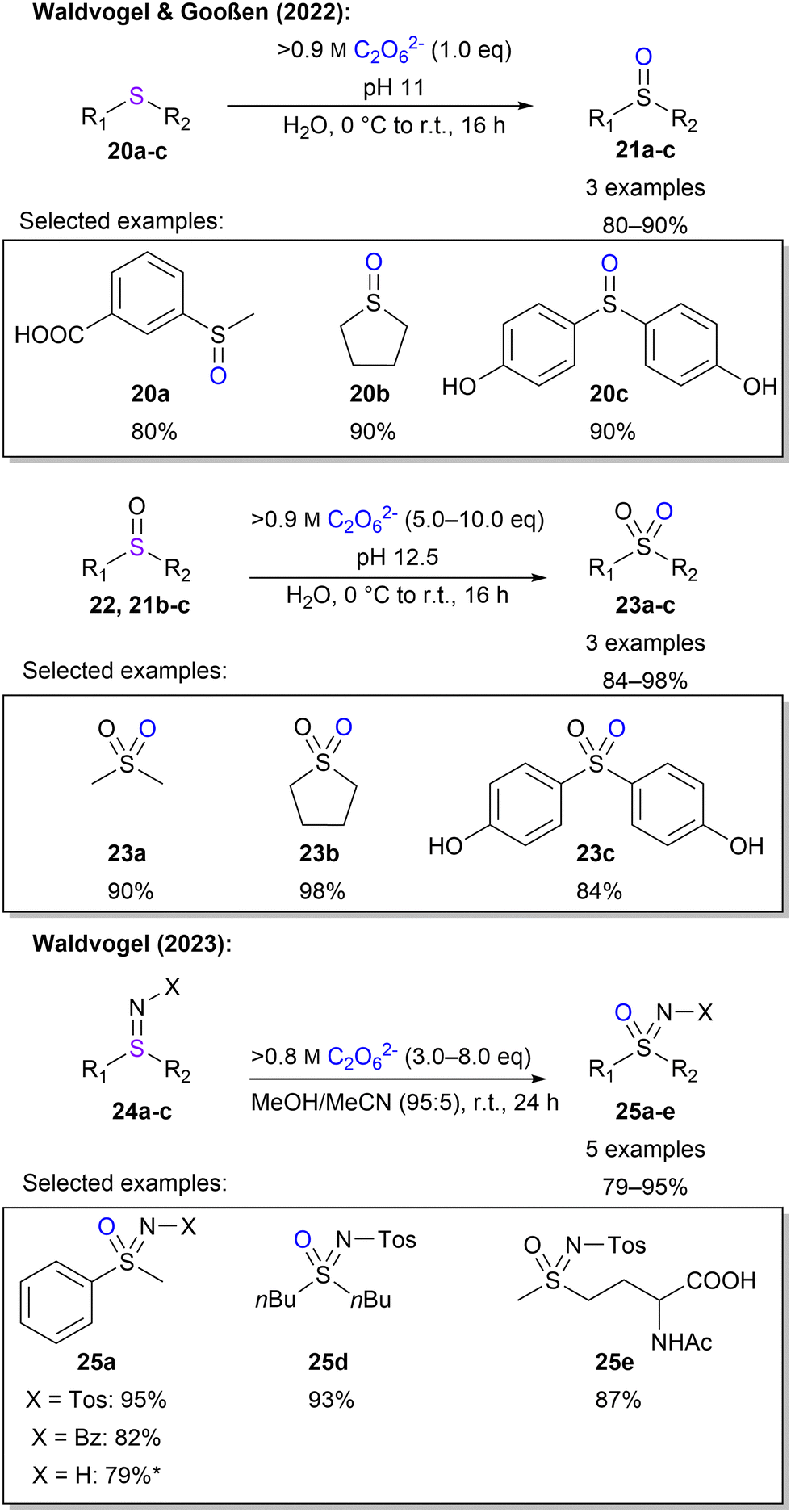 | ||
| Scheme 7 S-oxidation with 1. * 25a (X = H) is formed from 24a with X = C(O)CF3, which is removed after oxidation by methanolysis. | ||
Waldvogel and co-workers published a protocol using 1 to treat different types of lignin and achieved depending on lignin between 5 and 6.2 wt% vanillin and up to 2.3 wt% acetovanillone as a by-product.48,97 Therefore, they added a 0.18 M 1 solution to a solution of lignin dissolved in caustic soda and treated it in a pressured vessel at temperatures of over 100 °C. It is noteworthy that the solution of 1 is first combined with the alkaline lignin solution at temperatures below 100 °C, followed by post-treatment at a strongly elevated temperature. Noteworthily, the remaining carbonate in the residual stream represents the makeup chemical for the pulping plant, enabling technical implementation.98,99
Oxidation of Kraft lignin towards vanillin has also been demonstrated on a pilot-scale at technology readiness level 6 (Fig. 5). The ex-cell continuous production of 1 was combined with a thermal depolymerisation plug flow reactor for kraft lignin conversion. This plant was operated for more than 1200 hours. A final optimisation of the process resulted in a vanillin yield of 8 wt%. Specific focus was put on a model comprising steady-state operation and transient behaviour of the peroxodicarbonate reactor.100 An accompanying study on techno-economic profitability identified the high dilution rate and the electrochemical production of 1 as the major bottlenecks of the current process.48,49
 | ||
| Fig. 5 Technical reactor setup for the production of peroxodicarbonate on a pilot-scale. PSU – power supply unit, HX cooler – heat exchanger. | ||
The bleaching activity of 1 has been studied using indigo carmine, a food colorant, as a test substrate. While 15% hydrogen peroxide did not show any bleaching activity, 1 and percarbonate discoloured the solution. Interestingly, 1 bleached significantly faster and more efficiently than percarbonate.33 A similar effect on bleaching activity was reported for bleaching black tea. Furthermore, lignosulfonate was bleached using an in-cell circular flow setup. However, as a mixture of carbonates and lignosulfonate is electrolysed, it is not clear if 1 is involved in the decolourisation step. In an ex-cell recyclable flow setup, different types of wood veneer and pulp as well as cardboard were bleached. In all cases, a clear bleaching effect was observed. The physical properties and structure of the wood veneers did not change significantly and 1 had a low penetration depth. Noteworthily, the surface polarity of peroxodicarbonate treated wood increased significantly.44
Shellac is obtained by processing Lac, a natural resin excreted by lac insects on trees in Asia, mostly India and Thailand. It is used in a plethora of applications, including food coatings, wood varnish and pharmaceutical coatings. Due to the presence of lac dyes, the natural colour of shellac ranges from yellow to dark brown. However, for most applications, this colour is undesirable and is removed by oxidative bleaching with sodium hypochlorite. Aside from the safety issues and environmental impact of this process, the bleached shellac suffers from severe bleaching damage and chlorination of double bonds. This results in a short shelf-life and fast polymerization of bleached shellac as well as consumption of toxic organochlorides in food and pharmaceutical applications.101 As mentioned earlier, oxygen-based bleach detergents need elevated temperatures or relatively expensive activators, leading to severe bleaching damage or economically unfeasible processes. Recently, a novel bleaching process using 1 at room temperature was developed using acetonitrile as an inexpensive and atom-economical activator. Furthermore, the ionic strength of the peroxodicarbonate buffer was used to facilitate the dewaxing of seedlac. This process has great economic and ecologic potential to replace the current bleaching with sodium hypochlorite.102
Similar to bleaching, degradation of organic pollutants or disinfection of wastewater has been accomplished by the addition of strong oxidizers. Electrochemical technologies have been developed for sustainable wastewater treatment.103 Direct electrooxidation of pollutants at the anode suffers from incomplete decontamination because of mass transport limitations.104 Indirect electrochemical water disinfection is based on the electrochemical production of disinfecting substances from electrolytes present in wastewater, mostly chlorides.1051 has been suggested as an alternative to chloride-deficient water. However, as the natural concentration of carbonate in water is too low to efficiently obtain 1, disinfection of bacteria turned out to be incomplete.106
Conclusions
Peroxodicarbonate as a green platform oxidizer has experienced a renaissance in the past two decades. This can be attributed to an improved accessibility of the boron-doped diamond anodes. The disruptive electrochemical generation of 1 was achieved by two major developments: first, optimized heat dissipation in flow electrolysers. Second, the mixed cation approach that enabled higher carbonate concentrations in the aqueous electrolyte. Peroxodicarbonate solutions have been generated on a pilot-scale. This peroxide has unique features and is safe for handling, since upon standing under ambient conditions the peroxide will slowly decompose. With these highly concentrated peroxodicarbonate solutions in hand, many applications can be achieved in a superior way, wherein peroxide and alkaline bases are required. In typical N- or S-oxidation peroxodicarbonate can be employed. If the salty nature of aqueous 1 impedes direct conversion, activators or catalysts must be used to obtain an optimal yield. In the Dakin-type transformation and deborolative hydroxylation reaction, almost quantitative yields were achieved and robust scalability in the decagram range was proven. The aqueous peroxodicarbonate is an ideal reagent that can be implemented in pulping factories since the decomposition products represent the chemical makeup of these plants. Besides the selective degradation of kraft lignin to vanillin on lab and technical scales, aqueous peroxodicarbonate can be used for bleaching wood and shellac. For the latter the salty nature is even beneficial since separation of the waxes is facilitated. The use of 1 is currently emerging and many more applications will come.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
S. R. W. designed the review together with T. R., N. S., F. S., T. H., and B. W. contributed and prepared elaborated graphics. All authors wrote and edited the manuscript. S. R. W. supervised the project. All authors agreed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under FOR 2982/2-UNODE (WA 1276/23-2). This paper is supported by the European Union's Horizon 2020 research and innovation programme under grant agreement no. 820735 (project LIBERATE) and no. 101006612 (project EBIO). Open Access funding provided by the Max Planck Society.References
- R. A. Bellezoni, A. P. Adeogun, M. X. Paes and J. A. P. de Oliveira, J. Clean. Prod., 2022, 381, 135126 CrossRef CAS.
- K. Solaun and E. Cerdá, Renewable Sustainable Energy Rev., 2019, 116, 109415 CrossRef.
- J. Hart and F. Pomponi, Circ. Econ. Sustainability, 2021, 1, 127–141 CrossRef PubMed.
- P. Bradu, A. Biswas, C. Nair, S. Sreevalsakumar, M. Patil, S. Kannampuzha, A. G. Mukherjee, U. R. Wanjari, K. Renu, B. Vellingiri and A. V. Gopalakrishnan, Environ. Sci. Pollut. Res. Int., 2023, 30, 124488–124519 CrossRef PubMed.
- J. O. Imoniana, W. L. Silva, L. Reginato, V. Slomski and V. G. Slomski, Sustainability, 2021, 13, 218 CrossRef.
- A. M. Oliveira, R. R. Beswick and Y. Yan, Curr. Opin. Chem. Eng., 2021, 33, 100701 CrossRef.
- M. Ostadi, K. G. Paso, S. Rodriguez-Fabia, L. E. Øi, F. Manenti and M. Hillestad, Energies, 2020, 13, 4859 CrossRef CAS.
- D. M. Morales, D. Jambrec, M. A. Kazakova, M. Braun, N. Sikdar, A. Koul, A. C. Brix, S. Seisel, C. Andronescu and W. Schuhmann, ACS Catal., 2022, 12, 982–992 CrossRef CAS.
- D. Qu, S. He, L. Chen, Y. Ye, Q. Ge, H. Cong, N. Jiang and Y. Ha, Front. Chem., 2022, 10, 1055865 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- R. D. Little, J. Org. Chem., 2020, 85, 13375–13390 CrossRef CAS PubMed.
- R. D. Little and K. D. Moeller, Chem. Rev., 2018, 118, 4483–4484 CrossRef CAS PubMed.
- S. B. Beil, D. Pollok and S. R. Waldvogel, Angew. Chem., Int. Ed., 2021, 60, 14750–14759 CrossRef CAS PubMed.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed.
- J. Seidler, J. Strugatchi, T. Gärtner and S. R. Waldvogel, MRS Energy Sustain., 2020, 7, E42 CrossRef.
- S. Arndt, D. Weis, K. Donsbach and S. R. Waldvogel, Angew. Chem., Int. Ed., 2020, 59, 8036–8041 CrossRef CAS PubMed.
- S. Arndt, P. J. Kohlpaintner, K. Donsbach and S. R. Waldvogel, Org. Process Res. Dev., 2022, 26, 2564–2613 CrossRef CAS.
- J. Klein, K. Alt and S. R. Waldvogel, Adv. Sustainable Syst., 2022, 6, 2100391 CrossRef CAS.
- S. Arndt, B. Grill, H. Schwab, G. Steinkellner, U. Pogorevčnik, D. Weis, A. M. Nauth, K. Gruber, T. Opatz, K. Donsbach, S. R. Waldvogel and M. Winkler, Green Chem., 2021, 23, 388–395 RSC.
- C. M. Kisukuri, R. J.-R. Bednarz, C. Kampf, S. Arndt and S. R. Waldvogel, ChemSusChem, 2022, 15, e202200874 CrossRef CAS PubMed.
- K. C. de Freitas Araújo, D. R. da Silva, E. V. dos Santos, H. Varela and C. A. Martínez-Huitle, J. Electroanal. Chem., 2020, 860, 113927 CrossRef.
- J. Zhu, K. K. Hii and K. Hellgardt, ACS Sustainable Chem. Eng., 2016, 4, 2027–2036 CrossRef CAS.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS PubMed.
- J. Börgel, L. Tanwar, F. Berger and T. Ritter, J. Am. Chem. Soc., 2018, 140, 16026–16031 CrossRef PubMed.
- L. Tanwar, J. Börgel and T. Ritter, J. Am. Chem. Soc., 2019, 141, 17983–17988 CrossRef CAS PubMed.
- S. Miao, S. He, Y. Xue, H. Nian, J. Wang, P. Zhang, F. Zhu and W. Wen, Z. Phys. Chem., 2018, 232, 1741–1753 CrossRef CAS.
- E. J. Constam and A. von Hansen, Z. Elektrochem., 1896, 3, 137–144 CrossRef.
- E. H. Riesenfeld and W. Mau, Ber. Dtsch. Chem. Ges., 1911, 44, 3589–3595 CrossRef.
- E. H. Riesenfeld and B. Reinhold, Ber. Dtsch. Chem. Ges., 1909, 42, 4377–4383 CrossRef.
- R. E. Dinnebier, S. Vensky, P. W. Stephens and M. Jansen, Angew. Chem., Int. Ed., 2002, 41, 1922–1924 CrossRef CAS PubMed.
- C. P. Chardon, T. Matthée, R. Neuber, M. Fryda and C. Comninellis, ChemistrySelect, 2017, 2, 1037–1040 CrossRef CAS.
- M. S. Saha, T. Furuta and Y. Nishiki, Electrochem. Solid-State Lett., 2003, 6, D5 CrossRef CAS.
- Y. Einaga, J. S. Foord and G. M. Swain, MRS Bull., 2014, 39, 525–532 CrossRef CAS.
- S. J. Cobb, Z. J. Ayres and J. V. Macpherson, Annu. Rev. Anal. Chem., 2018, 11, 463–484 CrossRef CAS PubMed.
- S. Lips and S. R. Waldvogel, ChemElectroChem, 2019, 6, 1649–1660 CrossRef CAS.
- J. V. Macpherson, Phys. Chem. Chem. Phys., 2015, 17, 2935–2949 RSC.
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- X. Shi, S. Siahrostami, G.-L. Li, Y. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. Zheng and J. K. Nørskov, Nat. Commun., 2017, 8, 701 CrossRef PubMed.
- S. Velazquez-Peña, C. Sáez, P. Cañizares, I. Linares-Hernández, V. Martínez-Miranda, C. Barrera-Díaz and M. A. Rodrigo, J. Chem. Eng., 2013, 230, 272–278 CrossRef.
- J. Zhang and C. W. Oloman, J. Appl. Electrochem., 2005, 35, 945–953 CrossRef CAS.
- E. J. Ruiz-Ruiz, Y. Meas, R. Ortega-Borges and J. L. Jurado Baizabal, Surf. Engin. Appl. Electrochem., 2014, 50, 478–484 CrossRef.
- A. Ziogas, J. Belda, H.-J. Kost, J. Magomajew, R. A. Sperling and P. Wernig, Curr. Res. Green Sustainable Chem., 2022, 5, 100341 CrossRef CAS.
- A.-K. Seitz, P. J. Kohlpaintner, T. van Lingen, M. Dyga, F. Sprang, M. Zirbes, S. R. Waldvogel and L. J. Gooßen, Angew. Chem., Int. Ed., 2022, 61, e202117563 CrossRef CAS PubMed.
- J. Du, Z. Chen, C. Chen and T. J. Meyer, J. Am. Chem. Soc., 2015, 137, 3193–3196 CrossRef CAS PubMed.
- C. M. Hamm, S. Schneider, S. Hild, R. Neuber, T. Matthée, J. Krümberg, M. Stöckl, K.-M. Mangold and J. Kintrup, Front. Catal., 2024, 4, 1323322 CrossRef.
- M. Zirbes, T. Graßl, R. Neuber and S. R. Waldvogel, Angew. Chem., Int. Ed., 2023, 62, e202219217 CrossRef CAS PubMed.
- Á. Cabeza Sánchez, O. Trygve Berglihn, E. Ottaviano, T. Rücker, T. Pettersen, B. Wittgens, A. Aliko, L. Gálvez and M. López, Open Res. Europe, 2024, 4, 5 Search PubMed.
- E. J. Ruiz, R. Ortega-Borges, J. L. Jurado, T. W. Chapman and Y. Meas, Electrochem. Solid-State Lett., 2009, 12, E1 CrossRef CAS.
- M. S. Saha, T. Furuta and Y. Nishiki, Electrochem. Commun., 2004, 6, 201–204 CrossRef CAS.
- S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed.
- C. Gütz, A. Stenglein and S. R. Waldvogel, Org. Process Res. Dev., 2017, 21, 771–778 CrossRef.
- R. J.-R. Bednarz, A. S. Gold, J. Hammes, D. F. Rohrmann, S. Natalello, M. Mann, F. Weinelt, C. Brauer and S. R. Waldvogel, Org. Process Res. Dev., 2024, 28, 1529–1538 CrossRef CAS.
- B. Gleede, M. Selt, R. Franke and S. R. Waldvogel, Chemistry, 2021, 27, 8252–8263 CrossRef CAS PubMed.
- B. Gleede, M. Selt, C. Gütz, A. Stenglein and S. R. Waldvogel, Org. Process Res. Dev., 2020, 24, 1916–1926 CrossRef CAS.
- J. Gorzynski Smith, Synthesis, 1984, 629–656 CrossRef CAS.
- C. Lauret, Tetrahedron: Asymmetry, 2001, 12, 2359–2383 CrossRef CAS.
- C. Schneider, Synthesis, 2006, 3919–3944 CrossRef CAS.
- B. Das and K. Damodar, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar, and S. K. Chattopadhyay, Wiley-VCH, Weinheim, 2011, 3, pp. 63–95 Search PubMed.
- M. I. Childers, J. M. Longo, N. J. van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- X. Xie, Z. Huo, E. Jang and R. Tong, Commun. Chem., 2023, 6, 202 CrossRef CAS PubMed.
- E. J. Vandenberg, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 525–567 CrossRef CAS.
- P. C. B. Page, F. Marken, C. Williamson, Y. Chan, B. R. Buckley and D. Bethell, Adv. Synth. Catal., 2008, 350, 1149–1154 CrossRef CAS.
- P. J. Kohlpaintner, L. Marquart, L. J. Gooßen and S. R. Waldvogel, Eur. J. Org. Chem., 2023, e202300220 CrossRef CAS.
- F. Sprang, N. Schupp, P. Kohlpaintner, L. J. Gooßen and S. R. Waldvogel, Green Chem., 2024, 26, 5862–5868 RSC.
- V. E. Tarabanko and N. Tarabanko, Int. J. Mol. Sci., 2017, 18, 2421 CrossRef PubMed.
- N. Zhou, W. P. D. W. Thilakarathna, Q. S. He and H. P. V. Rupasinghe, Front. Energy Res., 2022, 9, 758744 CrossRef.
- K. Holmberg, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2019, pp. 1–56 Search PubMed.
- D. P. Rice, Toxicol. Appl. Pharmacol., 1977, 39, 377–389 CrossRef CAS PubMed.
- K. Singha, I. Habib and M. Hossain, ChemistrySelect, 2022, 7, e202203537 CrossRef CAS.
- A. Albini, Synthesis, 1993, 263–277 CrossRef CAS.
- O. V. Larionov, Heterocyclic N-Oxides, Springer International Publishing, Cham, 2017, vol. 53 Search PubMed.
- ed. D. Enders and E. Schaumann, Category 5, Compounds with One Saturated Carbon Heteroatom Bond, Georg Thieme Verlag, Stuttgart, 2009 Search PubMed.
- A. Lin, X. Su, D. She, K. Qiu, Q. He and Y. Liu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1008, 65–73 CrossRef CAS PubMed.
- B. Kunze, G. Höfle and H. Reichenbach, J. Antibiot., 1987, 40, 258–265 CrossRef CAS PubMed.
- J. Balzarini, M. Stevens, E. de Clercq, D. Schols and C. Pannecouque, J. Antimicrob. Chemother., 2005, 55, 135–138 CrossRef CAS PubMed.
- A. M. Mfuh and O. V. Larionov, Curr. Med. Chem., 2015, 22, 2819–2857 CrossRef CAS PubMed.
- D. Bernier, U. K. Wefelscheid and S. Woodward, Org. Prep. Proced. Int., 2009, 41, 173–210 CrossRef CAS.
- C. Liu, J. Luo, L. Xu and Z. Huo, ARKIVOC, 2013, 2013, 154–174 Search PubMed.
- V. Boekelheide and W. L. Lehn, J. Org. Chem., 1961, 26, 428–430 CrossRef CAS.
- A. H. Beckett, J. W. Gorrod and P. Jenner, J. Pharm. Pharmacol., 1971, 23, 55S–61S CrossRef CAS PubMed.
- A.-K. Seitz, T. van Lingen, M. Dyga, P. J. Kohlpaintner, S. R. Waldvogel and L. J. Gooßen, Synlett, 2022, 1527–1531 CAS.
- P. J. Kohlpaintner, N. Schupp, N. Ehlenz, L. Marquart, L. J. Gooßen and S. R. Waldvogel, Org. Lett., 2024, 26, 1607–1611 CrossRef CAS PubMed.
- M. Klein, D. L. Troglauer and S. R. Waldvogel, JACS Au, 2023, 3, 575–583 CrossRef CAS PubMed.
- C. Mai, A. Majcherczyk and A. Hüttermann, Enzyme Microb. Technol., 2000, 27, 167–175 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- M. Breiner, M. Zirbes and S. R. Waldvogel, Green Chem., 2021, 23, 6449–6455 RSC.
- M. Zirbes and S. R. Waldvogel, Curr. Opin. Green Sustainable chem., 2018, 14, 19–25 CrossRef.
- I. Bosque, G. Magallanes, M. Rigoulet, M. D. Kärkäs and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef CAS PubMed.
- M. Zirbes, L. L. Quadri, M. Breiner, A. Stenglein, A. Bomm, W. Schade and S. R. Waldvogel, ACS Sustainable Chem. Eng., 2020, 8, 7300–7307 CrossRef CAS.
- M. Zirbes, D. Schmitt, N. Beiser, D. Pitton, T. Hoffmann and S. R. Waldvogel, ChemElectroChem, 2019, 6, 155–161 CrossRef CAS.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- J. Klein and S. R. Waldvogel, ChemSusChem, 2023, 16, e202202300 CrossRef CAS PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- S. R. Waldvogel, M. Zirbes, R. Neuber and T. Matthée, Process for the oxidation of carbon-containing organic compounds with electrochemically produced oxidizing agents and arrangement for carrying out the process. PCT Int. Appl., WO 2020099350 A1 20200522, 2020 Search PubMed.
- D. Schmitt, N. Beiser, C. Regenbrecht, M. Zirbes and S. R. Waldvogel, Sep. Purif. Technol., 2017, 181, 8–17 CrossRef CAS.
- D. Schmitt, C. Regenbrecht, M. Schubert, D. Schollmeyer and S. R. Waldvogel, Holzforschung, 2017, 71, 35–41 CrossRef CAS.
- T. Rücker, T. Pettersen, H. Graute, B. Wittgens, T. Graßl and S. R. Waldvogel, ACS Sustainable Chem. Eng., 2024, sc-2024-02898u Search PubMed.
- K. Li, B. Tang, W. Zhang, Z. Shi, X. Tu, K. Li, J. Xu, J. Ma, L. Liu and H. Zhang, ACS Omega, 2020, 5, 22551–22559 CrossRef CAS PubMed.
- T. Horsten and S. R. Waldvogel, RSC Sustainable, 2024 10.1039/D4SU00228H.
- C. A. Martínez-Huitle and E. Brillas, Appl. Catal., B, 2009, 87, 105–145 CrossRef.
- I. Sirés, E. Brillas, M. A. Oturan, M. A. Rodrigo and M. Panizza, Environ. Sci. Pollut. Res. Int., 2014, 21, 8336–8367 CrossRef PubMed.
- A. Kraft, Int. J. Electrochem. Sci., 2007, 2, 355–385 CrossRef CAS.
- T. Furuta, H. Tanaka, Y. Nishiki, L. Pupunat, W. Haenni and P. Rychen, Diamond Relat. Mater., 2004, 13, 2016–2019 CrossRef CAS.
- S. Mavrikis, S. C. Perry, P. K. Leung, L. Wang and C. Ponce de León, ACS Sustainable Chem. Eng., 2021, 9, 76–91 CrossRef CAS.
- K. Fuku and K. Sayama, Chem. Commun., 2016, 52, 5406–5409 RSC.
- Y. Miyase, S. Iguchi, Y. Miseki, T. Gunji and K. Sayama, J. Electrochem. Soc., 2019, 166, H644–H649 CrossRef CAS.
- K. Fuku, Y. Miyase, Y. Miseki, T. Gunji and K. Sayama, ChemistrySelect, 2016, 1, 5721–5726 CrossRef CAS.
- L. Fan, X. Bai, C. Xia, X. Zhang, X. Zhao, Y. Xia, Z.-Y. Wu, Y. Lu, Y. Liu and H. Wang, Nat. Commun., 2022, 13, 2668 CrossRef CAS PubMed.
- Y. Miyase, Y. Miseki, T. Gunji and K. Sayama, ChemElectroChem, 2020, 7, 2448–2455 CrossRef CAS.
- F. Kuttassery, A. Sebastian, S. Mathew, H. Tachibana and H. Inoue, ChemSusChem, 2019, 12, 1939–1948 CrossRef CAS PubMed.
- S. Mavrikis, M. Göltz, S. Rosiwal, L. Wang and C. Ponce de León, ChemSusChem, 2022, 15, e202102137 CrossRef CAS PubMed.
- H. Bemana and N. Kornienko, iScience, 2024, 27, 109482 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |