
PdPt/SrTiO3:Al-catalysed redox-selective photoreduction of unsaturated carboxylic acids using minimal electron-donor and water†
Shogo
Mori
 *a,
Farzaneh
Soleymani Movahed‡
b,
Sha
Xue‡
b,
Yuji
Sakai
b,
Daling
Lu
c,
Takashi
Hisatomi
c,
Kazunari
Domen
cd and
Susumu
Saito
*ab
*a,
Farzaneh
Soleymani Movahed‡
b,
Sha
Xue‡
b,
Yuji
Sakai
b,
Daling
Lu
c,
Takashi
Hisatomi
c,
Kazunari
Domen
cd and
Susumu
Saito
*ab
aIntegrated Research Consortium on Chemical Sciences, Nagoya University, Chikusa, Nagoya, Aichi 464-8602, Japan. E-mail: mori.shogo.n1@f.mail.nagoya-u.ac.jp; saito.susumu.c4@f.mail.nagoya-u.ac.jp
bDepartment of Chemistry, Graduate School of Science, Nagoya University, Chikusa, Nagoya, Aichi 464-8602, Japan
cResearch Initiative for Supra-Materials, Interdisciplinary Cluster for Cutting Edge Research, Shinshu University, Wakasato, Nagano, Nagano 380-8553, Japan
dOffice of University Professors, The University of Tokyo, Yayoi, Bunkyo, Tokyo 113-8656, Japan
First published on 2nd October 2024
Abstract
We developed a semiconductor photocatalyst, Pd–Pt alloy nanoparticle-loaded, Al-doped SrTiO3 (PdPt/STO:Al), for photoreduction of unsaturated carboxylic acids. Due to the cooperative STO:Al surface and Pd–Pt alloy nanoparticles, the catalyst dispersed in water provided highly redox-selective photoreduction against oxidative degradation of starting materials/products and against reductive evolution of H2, where minimal glycolic acid worked as an efficient electron-donating fuel.
Semiconductor photocatalysts (SPs) have shown great potential for the realization of green and sustainable chemical reactions. Representative examples include organic pollutant degradation, and more notably, water splitting, where photo-generated holes (h+s) and electrons (e−s) promote oxidation of water to O2 and reduction of protons (H+s) to H2via surface hydrogen species (H˙ = e− + H+), respectively.1 A vault of modification methods of SPs has been developed, and loading metal nanoparticles on the SP surfaces is one of the most effective strategies to facilitate the generation of the surface H˙ for efficient H2 evolution.2 For instance, Domen, Hisatomi and their co-workers reported a well-designed SP, Rh–Cr–Co-coloaded, Al-doped SrTiO3 (RhCrCo/STO:Al).3
By changing metal nanoparticles on SPs, the reactivity of the surface H˙ can be altered for the reduction of organic compounds, in which various e−-donors have been used instead of explosive H2 gas.4 Although several SPs have been developed for photoreduction, large excess amounts of e−-donors were needed for the quantitative conversion of the starting materials.4–8 This is probably due to competition with oxidative degradation of the organic substrates/products by the h+s,5 and/or with the dissipation of the surface H˙ for undesirable H2 evolution.6 In other words, the SP-promoted photo-oxidation and reduction were barely controlled. Aiming at a green and sustainable organic synthesis, the use of the organic e−-donors must be rationally reduced,9 and also water should be used on purpose more effectively.10
In this work, we newly developed Pd–Pt alloy nanoparticle (ANP)-loaded STO:Al (PdPt/STO:Al) that promotes photoreduction of organic compounds using a minimal11 e−-donor in water (Scheme 1). For instance, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (double) bonds of unsaturated carboxylic acids (CAs), corresponding to 2e−-acceptor, were hydrogenated by using minimal glycolic acid (GA) as a 6e−-donor [CC bond of 1a:GA = 3 (×2e−-acceptor):1 (×6e−-donor) mol/mol]. PdPt/STO:Al provides highly redox-selective photoreduction,12 which is promoted exclusively against the oxidative degradation of organic substrates/products, and against the reductive evolution of H2. Unlike many privileged photo-oxidations of CAs (e.g., decarboxylation) catalysed by TiO2,13–15 photoreduction of CAs predominates in the PdPt/STO:Al system. In addition, CA products are easily isolated by a simple work-up procedure including neutralization with a Brønsted acid and decantation to remove the heterogeneous catalyst. The recovered catalyst was reused for at least four cycles with a minute decrease in the catalytic activity. Besides, PdPt/STO:Al functions under simulated solar light irradiation and in fully aqueous media without organic solvents.16 These features are advantageous in many respects for green and sustainable organic synthesis.
C (double) bonds of unsaturated carboxylic acids (CAs), corresponding to 2e−-acceptor, were hydrogenated by using minimal glycolic acid (GA) as a 6e−-donor [CC bond of 1a:GA = 3 (×2e−-acceptor):1 (×6e−-donor) mol/mol]. PdPt/STO:Al provides highly redox-selective photoreduction,12 which is promoted exclusively against the oxidative degradation of organic substrates/products, and against the reductive evolution of H2. Unlike many privileged photo-oxidations of CAs (e.g., decarboxylation) catalysed by TiO2,13–15 photoreduction of CAs predominates in the PdPt/STO:Al system. In addition, CA products are easily isolated by a simple work-up procedure including neutralization with a Brønsted acid and decantation to remove the heterogeneous catalyst. The recovered catalyst was reused for at least four cycles with a minute decrease in the catalytic activity. Besides, PdPt/STO:Al functions under simulated solar light irradiation and in fully aqueous media without organic solvents.16 These features are advantageous in many respects for green and sustainable organic synthesis.
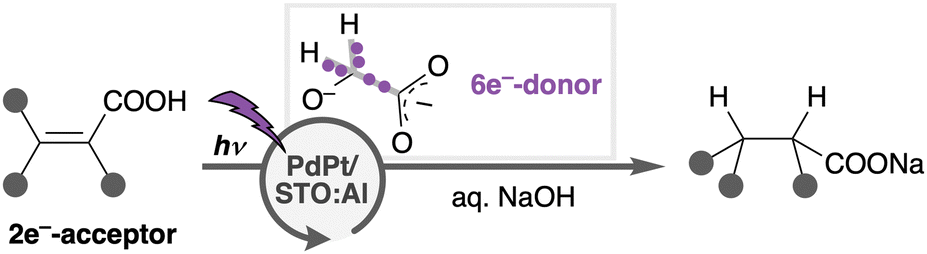 | ||
| Scheme 1 PdPt/STO:Al-catalysed redox-selective photoreduction using minimal glycolic acid (GA) and water. | ||
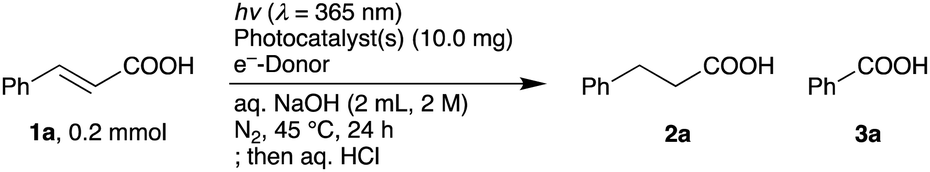
We initially studied a series of reactions of 1a with various SPs under near-UV light irradiation (λ = 365 nm, Fig. S1, ESI†) to synthesize 2a in basic water without organic e−-donors (Table 1). Pristine STO:Al did not give 2a (2a: <1%, entry 1). While Pd/STO:Al (1.5 wt% Pd) and Pt/STO:Al (1.5 wt% Pt) gave small amounts of 2a (ca. 20%, entries 2 and 3), PdPt/STO:Al (1.5 wt% Pd and Pt) promoted the desirable reaction more efficiently (2a: 55%, entry 4). The formation of the Pd–Pt ANPs on STO:Al surfaces was confirmed by scanning transmission electron microscopy (STEM) with energy dispersive X-ray spectroscopy (EDX) (Fig. 1a–d). The EDX line scanning profiles obviously demonstrated that Pd and Pt were randomly mixed in the ANPs (Fig. 1a, e, and f). The high resolution (HR) TEM image showed lattice fringes with d-spacings of 0.220 nm and 0.199 nm, assignable to the (111) and (200) planes of the fcc Pd–Pt ANPs, respectively (Fig. 1g).17,18 When Pd/STO:Al and Pt/STO:Al were used instead of PdPt/STO:Al, a lower amount of 2a was obtained in 26% (entry 5), suggesting that the Pd–Pt ANP formation on the same STO:Al surface is critical. In a control experiment using PdPt/STO without Al-doping, the hydrogenation hardly proceeded (2a: 3%, entry 6). Al-doping would prevent recombination of the photo-generated h+ and e− by diminishing recombination sites in the STO lattice, resulting in enhanced catalytic ability.3 The yield of 2a was further increased when 0.1 mmol of 1a was used (2a: 86%, entry 7). PdPt/STO:Al was also driven by simulated solar light irradiation (2a: 86%, entry 8, Fig. S2, ESI†). Heterogeneous PdPt/STO:Al was easily recovered by centrifugation and decantation after the reaction, and was reused in at least four successive cycles without a significant decrease in the catalytic ability (2a: 81% in the 4th run, entry 9, Fig. S4, ESI†). PdPt/STO:Al recovered after the 4th run was characterized to investigate important factors of the catalytic activity, as will be described later.
|
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst(s) | e−-Donor | Yield of 2a (%) | Yield of 3a (%) |
| a Conditions: 1a (0.2 mmol), hν (λ = 365 nm), photocatalyst(s) (10.0 mg), e−-donor, aq. NaOH (2 mL, 2 M), N2, 45 °C, 24 h. Yields were determined by 1H NMR analysis after neutralization by aq. HCl. b Photocatalysts (5.0 mg each). c 1a (0.1 mmol). d CO2 (50 μmol) was detected. e Simulated solar light, 96 h. f In the 4th run of catalyst recycling. g CO2 (30 μmol) was detected. h 1a (1 mmol), 8 h, hν (λ = 370 nm). i 2e−-donor. j 4e−-donor. k 6e−-donor. | ||||
| 1 | STO:Al | — | <1 | <1 |
| 2 | Pd/STO:Al | — | 19 | <1 |
| 3 | Pt/STO:Al | — | 22 | 2 |
| 4 | PdPt/STO:Al | — | 55 | 4 |
| 5b | Pd/STO:Al and Pt/STO:Al | — | 26 | 3 |
| 6 | PdPt/STO | — | 3 | <1 |
| 7cd | PdPt/STO:Al | — | 86 ± 3 | 5 |
| 8ce | PdPt/STO:Al | — | 86 | 4 |
| 9cf | PdPt/STO:Al | — | 81 | 4 |
| 10c | PdPt/STO:Al | Oxalic acid (0.1 mmol)i | 68 | 3 |
| 11c | PdPt/STO:Al | Glyoxylic acid (0.05 mmol)j | 88 | 3 |
| 12cg | PdPt/STO:Al | GA (0.034 mmol)k | >99 | <1 |
| 13h | PdPt/STO:Al | GA (0.34 mmol)k | 86 | 1 |
|
||||
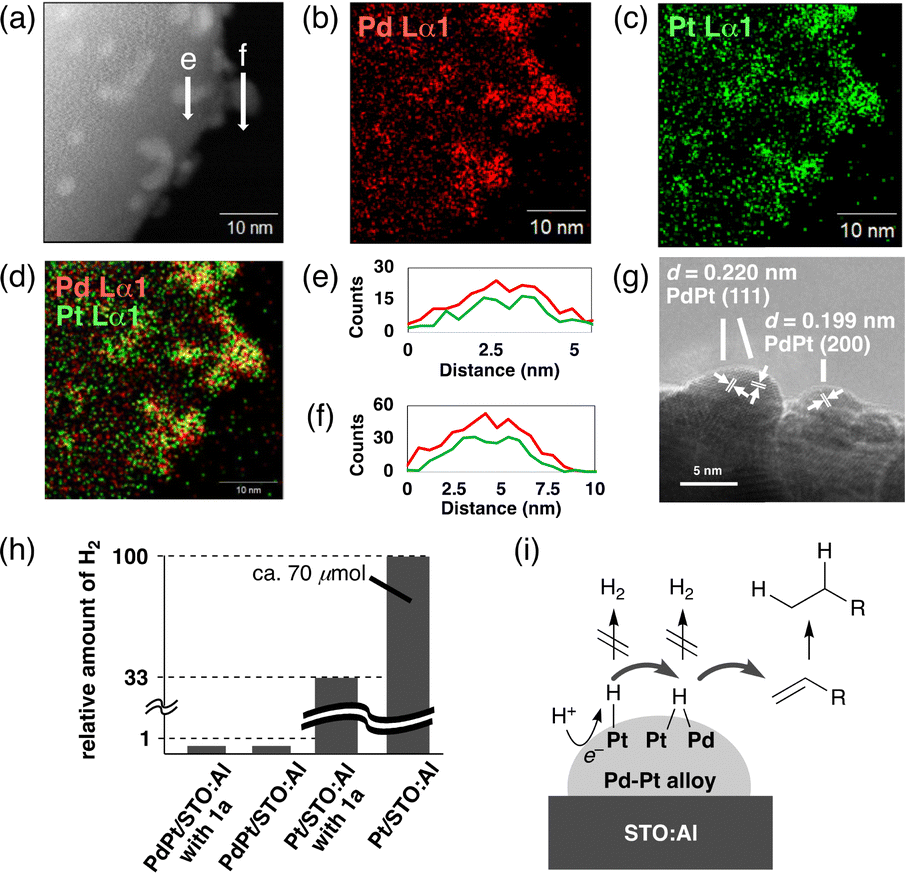 | ||
| Fig. 1 Pd–Pt ANPs on STO:Al (a) STEM image. STEM-EDX mapping images for (b) Pd (Lα1, red), (c) Pt (Lα1, green), and (d) overlay (overlapped points were highlighted in yellow). (e) and (f) EDX line scanning profiles. (g) HR-TEM image. (h) H2 evolution abilities of the photocatalysts. Conditions: hν (λ = 365 nm), photocatalyst (10.0 mg), GA (ca. 0.035 mmol), aq. NaOH (2 mL, 2 M), N2, 45 °C, 24 h. (i) Proposed surface H˙-relay mechanism. Stoichiometry was omitted for simplicity. | ||
Other reaction parameters were optimized, as shown in Tables S1–S3 (ESI†). It should be noted that the reaction was promoted in water without base additives, albeit with low efficiency. The use of NaOH was effective for dissolution of 1a and for the dispersion of PdPt/STO:Al in the reaction mixture, resulting in a better reactivity. Control experiments indicated that light irradiation and the photocatalyst are necessitated and that the protocol is robust against the presence of O2 in air and ions in tap water (Table S4, ESI†).
According to the liquid- and gas-phase analyses for the reaction using PdPt/STO:Al (entry 7), benzoic acid (3a: 5%) and CO2 (50 μmol) were detected, while O2, which is the product of water oxidation, was not generated, indicating that oxidative degradation of 1a and/or 2a occurred and the extracted e−s therefrom were used for the hydrogenation of 1a. When benzaldehyde, seemingly derived by the oxidation of 1a,19 was used instead of 1a, 3a was produced (30%) but CO2 was hardly detected (Scheme S1, ESI†). In a similar reaction with 2a as a starting material, no 3a but CO2 (30 μmol) was detected (Scheme S2, ESI†).13–15 Thus, 1a and 2a would be oxidized mainly to 3a and CO2, respectively (Scheme S3, ESI†).
Inspired by a preceding report on the smooth oxidation of lactic acid with H2 evolution enabled by an interplay of photocatalytic TiO2-surfaces and loaded Pt nanoparticles,20 we envisioned that selective oxidation of α-hydroxycarboxylic acid or its structural analogues, which more strongly bind to the SP surface, would outperform undesirable oxidation of 1a and 2a by a similar synergistic effect of STO:Al surfaces and Pd–Pt ANPs (entries 10–12). Finally, GA was found to be a competent e−-donor: 1a was converted into 2a exclusively when minimal GA was used (2a: >99%, 3a: <1%, CC bond![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GA = 3:1 mol/mol, entry 12).11 At the same time, CO2 (30 μmol) was detected.21 We also conducted a similar reaction using D2O instead of H2O. A post-reaction sample of the D2O phase was analyzed by 1H NMR immediately after solid residues were filtrated, where GA and relevant small organic molecules derived therefrom were undetected (Fig. S5, ESI†). These observations indicate that GA functioned as the efficient 6e−-donor. The D-labeling experiment also suggested that water was the source of two H+s incorporated into the hydrogenated product (Fig. S5 and Table S7, ESI†).22–24 A more scalable reaction (1a, 1 mmol) was successful (2a: 86% entry 13, Fig. S3 and Table S5, ESI†).
:GA = 3:1 mol/mol, entry 12).11 At the same time, CO2 (30 μmol) was detected.21 We also conducted a similar reaction using D2O instead of H2O. A post-reaction sample of the D2O phase was analyzed by 1H NMR immediately after solid residues were filtrated, where GA and relevant small organic molecules derived therefrom were undetected (Fig. S5, ESI†). These observations indicate that GA functioned as the efficient 6e−-donor. The D-labeling experiment also suggested that water was the source of two H+s incorporated into the hydrogenated product (Fig. S5 and Table S7, ESI†).22–24 A more scalable reaction (1a, 1 mmol) was successful (2a: 86% entry 13, Fig. S3 and Table S5, ESI†).
To get further insights into the function of the Pd–Pt ANPs, H2 evolution abilities among photocatalysts were compared (Fig. 1h). The detection of H2 was negligible under the standard conditions using PdPt/STO:Al and 1a. In the absence of 1a, PdPt/STO:Al hardly produced H2 as well. In contrast, Pt/STO:Al produced relatively large amounts of H2 both in the presence and absence of 1a. These observations could be interpreted as follows (Fig. 1i): a photo-excited e− of PdPt/STO:Al accommodated at a Pt site, whose work function is larger than that of Pd,25 would reduce a H+ to a surface H˙ at the Pt site (Pt–H). While the Pt–H is prone to be consumed for H2 evolution, it would also be possible to be transferred to an interface between Pt and Pd in the ANP, which can store a higher concentration of H˙ than the Pt site.26 The formed Pd–H–Pt would be used predominantly for the hydrogenation of the CC bond of 1a that outclasses the undesirable H2 evolution. Similar surface H˙-relay mechanisms involving Pd–Pt ANPs loaded on other SP surfaces were also proposed previously.7,8
Overall, the smooth and selective e− transfer from GA to 1a would be promoted by the cooperative role of the STO:Al surfaces and the Pd–Pt ANPs: GA would be oxidized through the interplay of the STO:Al surfaces and the Pd–Pt ANPs, followed by reduction of 1a through the surface H˙-relay mechanism on the Pd–Pt ANPs.
Under the established standard conditions, a variety of CC– or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C (triple) bonds of CAs were photocatalytically hydrogenated efficiently and selectively (Fig. 2a). trans-Cinnamic acids containing an electron-donating, an electron-withdrawing or a sterically demanding substituent underwent the hydrogenation smoothly (2b–2d: ≥90%). The hydrogenation took place efficiently even when the olefin moiety was not directly π-conjugated with the carboxyl group (2e: 71%). A CA with a CC bond (1f) was doubly hydrogenated to 2a using minimal GA within 24 h [2a: 95%, CC bond:GA = 3 (×4e−-acceptor):2 (×6e−-donor) mol/mol]. Just by shortening the reaction time to 6 h, a partially hydrogenated product with a cis-configuration was obtained selectively (1a′: 70%).
C (triple) bonds of CAs were photocatalytically hydrogenated efficiently and selectively (Fig. 2a). trans-Cinnamic acids containing an electron-donating, an electron-withdrawing or a sterically demanding substituent underwent the hydrogenation smoothly (2b–2d: ≥90%). The hydrogenation took place efficiently even when the olefin moiety was not directly π-conjugated with the carboxyl group (2e: 71%). A CA with a CC bond (1f) was doubly hydrogenated to 2a using minimal GA within 24 h [2a: 95%, CC bond:GA = 3 (×4e−-acceptor):2 (×6e−-donor) mol/mol]. Just by shortening the reaction time to 6 h, a partially hydrogenated product with a cis-configuration was obtained selectively (1a′: 70%).
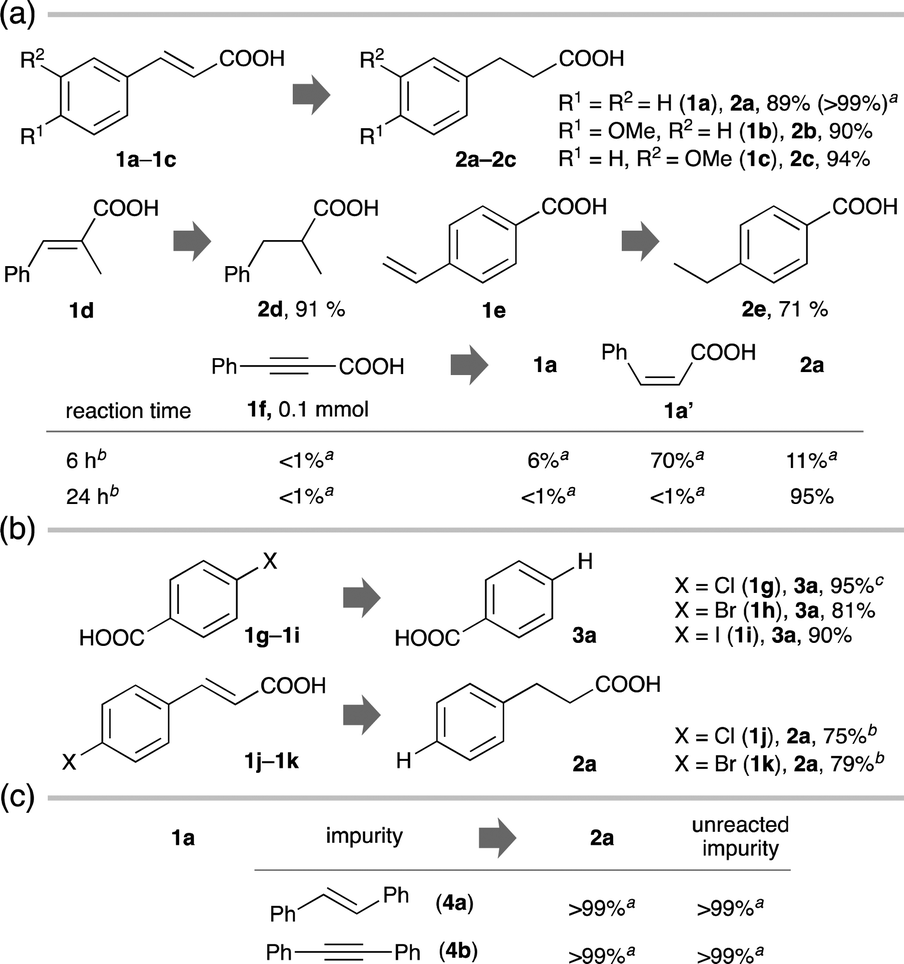 | ||
| Fig. 2 Generality and chemoselectivity. (a) Hydrogenation. (b) Hydrodehalogenation. (c) Chemoselective hydrogenation in the presence of impurities (0.1 mmol). Conditions: CA (1, 0.1 mmol), hν (λ = 365 nm), PdPt/STO:Al (10.0 mg), GA (ca. 0.035 mmol), aq. NaOH (2 mL, 2 M), N2, 45 °C, 24 h. Isolated yield after neutralization by aq. HCl. aDetermined by 1H NMR analysis after neutralization by aq. HCl. bGA (ca. 0.07 mmol). c30 h. | ||
Cl, Br and I-substituted benzoic acids (1g–1i) underwent photocatalytic hydrodehalogenation (substitution of halogens with hydrogen) under the PdPt/STO:Al system (3a: >80%, Fig. 2b). Accordingly, CAs with a CC bond and a C(sp2)–Cl or a C(sp2)–Br bond (1j and 1k) provided uniformly the doubly hydrogenated product (2a: ≥75%).
Emphasis should be placed on the ease of isolation of the products. The desirable products, which were formed cleanly, could be isolated with analytically high purities just by a simple work-up procedure including neutralization by an aqueous solution of HCl and extraction, without column chromatography.
As demonstrated above, CAs soluble in water were competent substrates, while non-polar chemicals with a CC bond (4a) or with a CC bond (4b) remained largely intact (Scheme S4, ESI†). Even in the presence of non-polar 4a or 4b, water-soluble 1a with the carboxyl group underwent the hydrogenation chemoselectively while 4a and 4b were hardly hydrogenated (Fig. 2c).
The newly developed PdPt/STO:Al was characterized by several spectroscopic and microscopic techniques. By inductively coupled plasma-atomic emission spectroscopy (ICP-AES), the metal composition of PdPt/STO:Al was determined to be 1.38 wt% for Pd and 1.27 wt% for Pt. X-ray photoelectron spectroscopy (XPS) showed that freshly prepared PdPt/STO:Al mainly contains Pd0 and Pt0 rather than Pd2+, Pt2+ and Pt4+ (Fig. S6a and d, ESI†). The binding energies for Pd0 3d and Pt0 4f of PdPt/STO:Al were lower than the standard values of bulk Pd0 and Pt0, suggesting electronic interactions of both Pd and Pt with STO:Al. The TEM image of PdPt/STO:Al showed that fine Pd–Pt ANPs were randomly dispersed on all the STO:Al surfaces since the ANPs were loaded by the standard impregnation method (Figs. S7a, ESI†). Other characterization data of PdPt/STO:Al are shown in Fig. S8 and S9 (ESI†).
In the catalyst recycling experiments, the photocatalytic ability was slightly decreased through each run (Fig. S4, ESI†). The possible causes of a gradual deactivation of the catalyst: (i) leaching of the Pd–Pt ANPs from the STO:Al surfaces; (ii) changes of the electronic properties (formal charges) of Pd and/or Pt; and (iii) aggregation of the ANPs were investigated. (i) The ICP-AES analysis demonstrated that Pd and Pt were scarcely leached into the solution even after the 4th cycle (1.35 wt% Pd, 1.24 wt% Pt) compared with the initial state (1.38 wt% Pd, 1.27 wt% Pt). (ii) The XPS analysis of PdPt/STO:Al recovered after the 4th run showed that oxidized Pd2+ and Pt4+ were dominant species rather than Pd0 or Pt0 (Fig. S6b and e, ESI†). Although the oxidation states of Pd and Pt of the recovered PdPt/STO:Al were restored to their initial states by reduction with NaBH4 (Fig. S6c and f, ESI†), the photocatalytic ability could not be refreshed (Table S6, ESI†). These results suggested that the oxidation states of Pd and Pt would not be important for the photocatalytic performance. (iii) According to the XPS analysis, the surface atomic ratios of Pd and Pt relative to Ti were decreased after the 4th run, implying that the fine Pd–Pt ANPs would aggregate (before reaction: Pd/Ti = 0.48, Pt/Ti = 0.34; after the 4th run: Pd/Ti = 0.27, Pt/Ti = 0.30). In the TEM image of the PdPt/STO:Al recovered after the 4th run, most of the fine Pd–Pt ANPs disappeared and relatively large Pd–Pt ANPs were mainly observed (Fig. S7b, ESI†). Therefore, we can conclude that the aggregation of the Pd–Pt ANPs negatively affected the photocatalytic performance.
In summary, the redox-selective reduction of organic compounds was enabled by the newly developed PdPt/STO:Al photocatalyst with minimal GA as the e−-donor in water. Selective e−-transfer from GA to CAs would be enabled by the cooperation of the STO:Al surfaces and the Pd–Pt ANPs. This study demonstrates the great potential of ANP-loaded SPs for clean and selective organic synthesis directed toward sustainable material production.
This work was partially supported by MEXT/JSPS Grant-in-aid for Early-Career Scientists, Specially Promoted Research, Transformative Research Areas (A): Green Catalysis, and International Leading Research, KAKENHI (Grant # 24K17676 to SM, 23H05404, 23H04904, and 22K21346 to SS). This work was also partially supported by JST CREST (Grant # JPMJCR22L2 to SS), Yashima Environment Technology Foundation (to SM), Iketani Science and Technology Foundation (to SM), The Naito Research Grant (to SM), Foundation of Public Interest of Tatematsu (to SM), and the Asahi Glass Foundation (step-up grant to SS). STEM and XPS measurements were performed by Kimitaka Higuchi (Nagoya U) and Michiko Obata (Shinshu U), respectively. We thank Dr Satoshi Muratsugu (Nagoya U) and Dr Satoru Ikemoto (Nagoya U) for insightful discussions.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919 CrossRef CAS PubMed.
- K. Wenderich and G. Mul, Chem. Rev., 2016, 116, 14587 CrossRef CAS PubMed.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411 CrossRef CAS.
- D. Ma, S. Zhai, Y. Wang, A. Liu and C. Chen, Molecules, 2019, 24, 330 CrossRef PubMed.
- A. Kinoshita, K. Nakanishi, R. Yagi, A. Tanaka, K. Hashimoto and H. Kominami, Appl. Catal., A, 2019, 578, 83 CrossRef CAS.
- K. Nakanishi, R. Yagi, K. Imamura, A. Tanaka, K. Hashimoto and H. Kominami, Catal. Sci. Technol., 2018, 8, 139 RSC.
- M. Li, N. Zhang, R. Long, W. Ye, C. Wang and Y. Xiong, Small, 2017, 13, 1604173 CrossRef.
- Y. Shiraishi, Y. Takeda, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, Chem. Commun., 2011, 47, 7863 RSC.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111 RSC.
- M. Yamauchi, H. Saito, T. Sugimoto, S. Mori and S. Saito, Coord. Chem. Rev., 2022, 472, 214773 CrossRef CAS.
- The “minimal” e−s for reduction of a CC bond are two. The number of e−s provided by an organic e−-donor can be counted as 2× the number of C–H and C–C bonds available from an e−-donor. Stable C–O bonds cannot be used as e−-sources. The e−-donor is finally converted into CO2, representing the fully oxidized/oxygenated form of carbon.
- Z. Liu, J. Caner, A. Kudo, H. Naka and S. Saito, Chem. – Eur. J., 2013, 19, 9452 CrossRef CAS.
- Q. Zhu and D. G. Nocera, J. Am. Chem. Soc., 2020, 142, 17913 CrossRef CAS.
- D. W. Manley, R. T. McBurney, P. Miller, R. F. Howe, S. Rhydderch and J. C. Walton, J. Am. Chem. Soc., 2012, 134, 13580 CrossRef CAS PubMed.
- Z. Huang, Z. Zhao, C. Zhang, J. Lu, H. Liu, N. Luo, J. Zhang and F. Wang, Nat. Catal., 2020, 3, 170 CrossRef CAS.
- F. Zhou, Z. Hearne and C.-J. Li, Curr. Opin. Green Sustainable Chem., 2019, 18, 118 CrossRef.
- Q. Yang, L. Shi, B. Yu, J. Xu, C. Wei, Y. Wang and H. Chen, J. Mater. Chem. A, 2019, 7, 18846 RSC.
- H. Dai, T. Zhang, H. Pu, K. Dong, Y. Wang and Y. Deng, Appl. Surf. Sci., 2022, 602, 154318 CrossRef.
- X. Li, Q. Wang, J. Lyu and X. Li, ChemistrySelect, 2021, 6, 9735 CrossRef.
- K. Liu, A. Litke, Y. Su, B. G. Van Campenhout, E. A. Pidko and E. J. M. Hensen, Chem. Commun., 2016, 52, 11634 RSC.
- The experimental value of the detected CO2 (30 μmol) was lower than the theoretical value (ca. 70 μmol). This discrepancy might be because a portion of the generated CO2 was dissolved in the aqueous media.
- C. Liu, Z. Chen, C. Su, X. Zhao, Q. Gao, G.-H. Ning, H. Zhu, W. Tang, K. Leng, W. Fu, B. Tian, X. Peng, J. Li, Q.-H. Xu, W. Zhou and K. P. Loh, Nat. Commun., 2018, 9, 80 CrossRef.
- E. Zhao, W. Zhang, L. Dong, R. Zborǐl and Z. Chen, ACS Catal., 2023, 13, 7557 CrossRef.
- H. Sun, W. Ou, L. Sun, B. Wang and C. Su, Nano Res., 2022, 15, 10292 CrossRef.
- H. B. Michaelson, J. Appl. Phys., 1977, 48, 4729 CrossRef CAS.
- M. Yamauchi, H. Kobayashi and H. Kitagawa, ChemPhysChem, 2009, 10, 2566 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, and characterization of organic compounds and semiconductor photocatalysts. See DOI: https://doi.org/10.1039/d4cc03791j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |