
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancements in π-conjugated polymers: harnessing cycloalkyl straps for high-performance π-conjugated materials
Charles
Ochonma
 ,
Victor S.
Francis
,
Sayan Kumar
Biswas
and
Nagarjuna
Gavvalapalli
*
,
Victor S.
Francis
,
Sayan Kumar
Biswas
and
Nagarjuna
Gavvalapalli
*
Department of Chemistry and Institute for Soft Matter Synthesis and Metrology, Georgetown University, 37th and O St, NW, Washington DC, USA. E-mail: ng554@georgetown.edu
First published on 4th November 2024
Abstract
Pendant alkyl chains are widely used to successfully obtain a wide variety of soluble linear 1D π-conjugated polymers. Over the past several decades, a wide variety of π-conjugated polymers have been synthesized to realize the desired properties and improve the performance of organic electronic devices. However, this strategy is not suitable for generating soluble 2D-π-conjugated materials, including ladder polymers, nanoribbons, and 2D-π-conjugated polymers, due to strong van der Waals interactions between the ribbons and sheets. The drive to synthesize higher dimensional polymers and to enhance polymers' properties has spurred the exploration of a novel direction in materials chemistry—the synthesis of unconventional monomers and polymers. The Gavvalapalli research group has developed and used cycloalkyl straps containing aryl building blocks for the synthesis of conjugated polymers. These cycloalkyl straps, positioned either above or below the π-conjugation plane, have been shown to directly control the π–π interactions between the polymer chains. We have demonstrated that π-face masking cycloalkyl straps hinder interchain π–π interactions. The first part of this review article highlights the use of cycloalkyl straps for the synthesis of higher dimensional π-conjugated polymers. In this section, we discuss the synthesis of 2D-H-mers, dispersible hyperbranched π-conjugated polymers, and conjugated porous polymers without the pendant solubilizing chains. The second part of the feature article highlights how the cycloalkyl straps can be used to gain control over polymer–acceptor interactions, including the interaction strength and the location of the acceptor along the polymer backbone. We conclude the article with the future outlook on cycloalkyl strap-containing building blocks in the world of conjugated polymers.
 Charles Ochonma | Charles Ochonma was born in Lagos, Nigeria. He received his undergraduate bachelor's degree in industrial chemistry from the prestigious Federal University of Technology, Akure, Nigeria (FUTA) as well as an MSc in Chemistry from Georgetown University (GU), USA. He has vast industrial experience in the auto lubricant industry and crude oil exploration. He is pursuing his PhD in the Gavvalapalli group at GU, where he is currently investigating the development of novel semiconductor polymers as well as stimuli-responsive hydrogels and organogels with target applications in medicine, electronics and CO2 capture. |
 Victor S. Francis | Victor Stanley Francis is a PhD candidate in Prof. Gavvalapalli's research group at Georgetown University's Department of Chemistry. With a master's in chemistry from Georgetown and a First-Class Bachelor's in Industrial Chemistry from the University of Jos, Nigeria, Francis specializes in functional polymer development and structural characterization. His undergraduate thesis delved into the impact of oxidation on the physicochemical properties of starch. Present research focuses on n-type doped azaacene polymers and polymer hydrogels for energy and bioelectronic applications. He has been recognized with prestigious awards like the ISMSM Graduate Fellowship and has contributed to several publications in the field of chemistry. |
 Sayan Kumar Biswas | Sayan was born in India and received his MSc from the Indian Institute of Technology Patna in 2023. He is currently pursuing his PhD under the supervision of Prof. Nagarjuna Gavvalapalli, working on the synthesis of higher dimensional pi-conjugated materials for optical, electronic, and optoelectronic applications and synthesis of polymer hydrogels for CO2 capture and release experiments. |
 Nagarjuna Gavvalapalli | Prof. Nagarjuna Gavvalapalli, professor in the Department of Chemistry at Georgetown University, leads research on the design, synthesis, and study of polymers. His work aims to uncover critical macromolecular parameters necessary for developing materials that address global challenges in energy and the environment. His group focuses on two key areas: the creation of novel building blocks for next-generation π-conjugated polymers used in electronic and bioelectronic applications and the development of tough, elastomeric, and resilient polymer networks for various applications. He is a recipient of the NSF CAREER and ACS-PMSE Young Investigator awards. |
Introduction
π-Conjugated polymers have emerged as captivating protagonists in materials science, showing promising electronic, optical, and structural properties.1–3 The interplay between synthetic organic chemistry and materials science has become an indispensable driving force, leading to the development of a wide variety of π-conjugated polymers with desired properties and performance.4,5 Higher dimensional π-conjugated organic materials are a burgeoning class of materials characterized by extended π-electron delocalization.6–13 Delocalization of π-electrons in more than one-dimension provides intriguing optical and electronic properties to higher dimensional π-conjugated polymers including ladder polymers,14 nanoribbons,15 and 2D-π-conjugated polymers16 compared to 1D π-conjugated polymers. However, controlling polymer growth beyond one dimension and synthesizing solution-processable higher dimensional π-conjugated polymers have been persistent challenges in materials science due to strong interchain van der Waals and π–π interactions in these materials. The conventional alkyl pendant chains, which effectively render soluble 1D-π-conjugated polymers, do not directly shield the π-faces of repeat units, hence failing to address the strong interchain π–π interactions encountered in higher dimensional π-conjugated materials.17,18 We have also investigated the control of polymer–acceptor interactions using cycloalkyl straps by tuning the interaction strength and the location of the acceptor along the polymer backbone (Fig. 1). | ||
| Fig. 1 Cycloalkyl strapped aryl building blocks developed by the Gavvalapalli group to access soluble higher dimensional π-conjugated materials and to control polymer–dopant interactions. | ||
Previous work
Recognizing the importance of hindering interchain interactions, in 1994, Schluter and co-workers developed building blocks whose π-face is masked. They used alkyl-strapped ansa-monomers for polymerization (Fig. 2).19 The alkyl strap masks the π-face of the repeat unit on one side, either above or below. Fortunately, alkyl straps are present randomly on either side of the π-face upon polymerization due to a lack of control over the orientation of the repeat unit during the polymerization. Masking the π-face of the polymer backbone and disrupting interchain (π–π) interactions indeed helped to generate ladder oligomers and polymer P1 without pendant solubilizing chains (Fig. 2). Gel permeation chromatography (GPC) results revealed its number average molecular weight (Mn) and polydispersity index Đ to be 7–14 kDa and 2.5–2.8, respectively. The alkyl straps successfully suppressed the chain aggregation and rendered soluble oligomers of Mn ranging from 2–5 kDa. However, their results revealed a chain length-dependent solubility limit for their polymers, where higher molecular weight fractions of both linear and strapped polymers are insoluble in organic solvents. The presence of a single alkyl strap for a larger aryl monomer unit is not sufficient to completely hinder interchain interactions and render soluble high-molecular-weight polymers. Nonetheless, this work presents the possibility of generating ladder polymers using ansa-monomers and provides a foundation for devising straps to mask the π-surface. | ||
| Fig. 2 Chemical structure of a typical ansa polymer (left). Alkyl-strapped ladder polymer P1 (right) reported by Schluter and coworkers.19 | ||
Since then, remarkable efforts have been made to develop different approaches to obtain different strapped aryl monomers, advancing the development of novel π-conjugated polymers for diverse applications. A few groups over the years including Bronstein, Sugiyasu, and Takeuchi have developed π-conjugated polymers that contain both straps and pendant solubilizing chains.20–22 The straps in P2 and P3 (Fig. 3) were used to disrupt the interchain π–π interactions and to gain access to the desired properties associated with hindering strong interchain π–π interactions, such as an increase in photoluminescence quantum yield, thermoformability, and enhanced sensory response towards analytes (vide infra). The pendant chains helped to render the soluble and solution-processable polymers.
 | ||
| Fig. 3 Chemical structures of strapped polymers containing both straps and pendant solubilizing chains reported by (left) Sugiyasu and coworkers (P2), reproduced with permission from ref. 22. Copyright American Chemical Society (2010); (right) Bronstein and coworkers (P3), reproduced with permission from ref. 20. Copyright American Chemical Society (2018). | ||
While alkyl straps reduce interchain interactions, they alone cannot render soluble, high-molecular-weight conjugated polymers. All the high-molecular-weight strapped conjugated polymers reported (vide infra) have pendant solubilizing chains to make them soluble. Building blocks that do not require pendant chains yet still produce soluble conjugated polymers, which will enable the elusive solution-state synthesis of higher dimensional π-conjugated materials.23
Our approach
Cycloalkyl straps to gain access to the high-molecular-weight linear π-conjugated polymers without pendant solubilizing chains
Realizing the significant gap in knowledge and recognizing the potential of strapping, the Gavvalapalli group was motivated to design and develop straps that would result in high-molecular-weight π-conjugated polymers that are soluble without pendant solubilizing chains.18 Once such straps are identified, they would be more appropriate for the controlled synthesis of higher dimensional π-conjugated materials. A critical aspect of the design involves the implementation of cycloalkyl straps as opposed to linear alkyl chains (Fig. 4). | ||
| Fig. 4 Advantages of cycloalkyl strapped aryl building blocks over conventional pendant solubilizing chains. | ||
The cycloalkyl straps effectively conceal the π-face of the repeating unit and hinder interchain interactions. More importantly, the strength of the π-face masking ability offered by the cycloalkyl straps can be adjusted by changing the specific cycloalkyl group.
The synthesis of the strapped monomers (6 and 12) used for the polymers (P4 and P5) is shown in Fig. 5. The 1,4-diethynyl cyclohexanocyclophane monomer (6) was synthesized from commercially available 1,4-cyclohexyl dimethanethiol (3) in a few steps. First, 1,4-cyclohexyl dimethanol (1) was converted into 1,4-cyclohexyl dimethanethiol (3). This intermediate was then reacted with 2,5-dibromo-p-xylylene dibromide under dilute conditions to produce 1,4-dibromo cyclohexanocyclophane (4). Sonogashira coupling of compound 4 with TMS-acetylene, followed by desilylation, yielded the desired 1,4-diethynyl cyclohexanocyclophane monomer (6). Similarly, the 1,4-diethynyl adamantanocyclophane monomer (12) was synthesized from 1,3-adamantane dimethanol (7) using protocols similar to those described above. The formation of the cyclophane is a key step in both synthetic schemes. To minimize undesired side products, such as linear chain structures, the reactants were slowly added dropwise over 24 hours into a reaction flask containing a large excess of solvent. The cyclophane formation results in a mixture of stereoisomers (a racemic mixture) due to planar chirality. The purified mixture of stereoisomers was used for polymerization without enantioseparation, resulting in atactic strapped polymers. The atactic nature of the polymer improves its solubility in organic solvents. The sulfur-containing linkers are prone to oxidation, forming sulfoxide and eventually sulfone under oxidizing conditions. However, no oxidized cyclophanes were observed in our studies, as the synthetic procedures did not involve strong oxidizing agents.
 | ||
| Fig. 5 Synthesis of cycloalkyl strapped monomers. Reaction conditions: (a) thioacetic acid, DIAD, PPh3, THF; (b) LiAlH4, THF; (c) 2,5-dibromo-p-xylylene dibromide, KOH, C6H6, EtOH; (d) (i) TMS-acetylene, Pd(PPh3)4, CuI, toluene, triethylamine; (e) TBAF, THF; (g) TMS-acetylene, Pd(PPh3)4, CuI, piperidine. | ||
There are two key reasons for selecting a thioether bridge between the alkyl strap and the conjugated main chain. First, the synthesis of adamantanocyclophane small molecules using dithiol chemistry was already known and reported by Vogtel in 1993.24 The reported reaction conditions were easily adaptable, yielding monomers 6 and 12 in decent quantities. Although we successfully synthesized adamantanocyclophane with an ether bridge (using oxygen instead of sulfur), the yields were low (<10%). Second, the thioether group can be selectively cleaved, allowing for the removal of the insulating cycloalkyl straps after film processing, if desired, to enhance interchain interactions between polymer chains.
Using single-crystal X-ray spectroscopy, the height of the straps (distance from the plane of the phenyl ring to the farthest atom on the cycloalkyl group) of the different cycloalkyl groups was determined: 7.22 Å for an adamantyl cycloalkyl group in P5 and 5.90 Å for the cyclohexyl group in P4 (Fig. 6). The cycloalkyl straps rendered higher molecular weight polymers that were soluble (Mn = 24 kDa) compared to ansa polymers with linear alkyl straps (Fig. 1) (Mn < 5 kDa) as reported by Scherf and coworkers,25 or pendant solubilizing chains (P6) (Mn = 16 kDa). Even though none of the cycloalkyl-strapped polymers have pendant solubilizing chains, all of them are soluble in typical organic solvents. Also, the solubility limit for cycloalkyl strap-containing conjugated polymers in conventional organic solvents like chloroform is higher than with linear pendant solubilizing chains. There is a clear correlation between the height of the straps and the solubility of the polymer. The polymer solubility limit in chloroform follows the order: P6 (16 kDa) < P4 (11 kDa) < P5 (15 kDa), which confirms that the height of the cycloalkyl unit indeed influences its solubility. We further evaluated the impact of dissimilar heights on the solubility limits by synthesizing a copolymer P7 having both adamantyl and cycloalkyl strapped monomer units randomly arranged. Results showed a three-fold increase in the solubility limit of the copolymer compared to the adamantyl homopolymer of similar molecular weight. This enhanced solubility limit is due to the more pronounced ruggedness of the π-surface of the copolymer with respect to the homopolymers.
 | ||
| Fig. 6 Chemical structures of polymers P4–P7 along with their Mn, solubility limit, and Ksv. The heights of the straps from the phenyl plane as deduced by single-crystal X-ray spectroscopy are shown. Reproduced with permission from ref. 18 under a Creative Commons Attribution-Non-commercial 3.0 Unported Licence. Royal Society of Chemistry (2019). | ||
PL quenching experiments further elucidated how the cycloalkyl strap monomer height influences the interaction between the conjugated backbone and small molecule acceptors. Using TCNQ (tetracyanoquinodimethane) as an acceptor, the Stern–Volmer quenching constant (Ksv) decreased with increasing monomer height, following the order: P6 > P4 > P7 > P5, with an order of magnitude difference between P6 with the pendant chains and P4 with the cycloalkyl straps. This drastic change in Ksv, despite all polymers having similar conjugated backbones, is because the height of the cycloalkyl straps governs how close the TCNQ quencher can approach the conjugated polymer backbone.
The cycloalkyl strap design was initially developed to reduce interchain π–π interactions between polymer chains, thus making 1D and higher dimensional π-conjugated materials more soluble. The random orientation (either up or down) along the π-face of the polymer backbone prevents incoming polymer chains from approaching the backbone closely enough (∼3.5 Å) to form strong π–π stacking interactions. However, this restriction does not apply to small molecules, which can still access the opposite face of the strapped repeat units. Despite this, both simulations and experimental observations indicate that the interaction strength with small molecules is weaker. Density functional theory (DFT) simulations on a trimer (containing both strapped and non-strapped units) interacting with an acceptor show that the highest binding energy is obtained when the acceptor resides on the non-strapped repeat unit, compared to that when it resides on the opposite face of the strapped monomers (vide infra).26 Also, the Ksv trend observed in PL studies where it decreases as the size of the strap increases suggests that the strapped repeat units freely rotate around the polymer axis, forming a molecular sheath around the polymer backbone, which hinders polymer interaction with small molecules (acceptors).
Although the cycloalkyl straps insulate only one π-face of the repeat unit, their random orientation and the free rotation of the repeat units effectively shield the polymer backbone, reducing interactions with both other polymer chains and small molecules. Note that, for π-conjugated polymers with a molecular weight below 20 kDa, the likelihood of the polymer folding onto itself is minimal. Therefore, intrachain interactions were not considered in the discussion. Since adamantyl straps render high-molecular-weight soluble π-conjugated polymers without pendant solubilizing chains, the Gavvalapalli group has employed these straps to synthesize higher dimensional π-conjugated materials.
Molecular design of 2D-π-conjugated materials
Synthesis of 2D-π-conjugated oligomers: the non-slanted H-mers
As a starting point in our journey into the higher dimensional π-conjugated materials world, we synthesized and studied 2D-π-conjugated oligomers. The synthesis and study of structurally well-defined, monodisperse π-conjugated oligomers of diverse shapes that are conjugated in 2-dimensions provide insights into the impact of molecular shape on the extension of π-conjugation in 2-dimensions.27–30 Also, nanosized 2D-π-conjugated oligomers lend an opportunity to realize unique physical and electronic properties that are unattainable with linearly conjugated oligomers. Toward this, we have developed non-slanted H-mers31 that are soluble without pendant solubilizing chains (Fig. 7).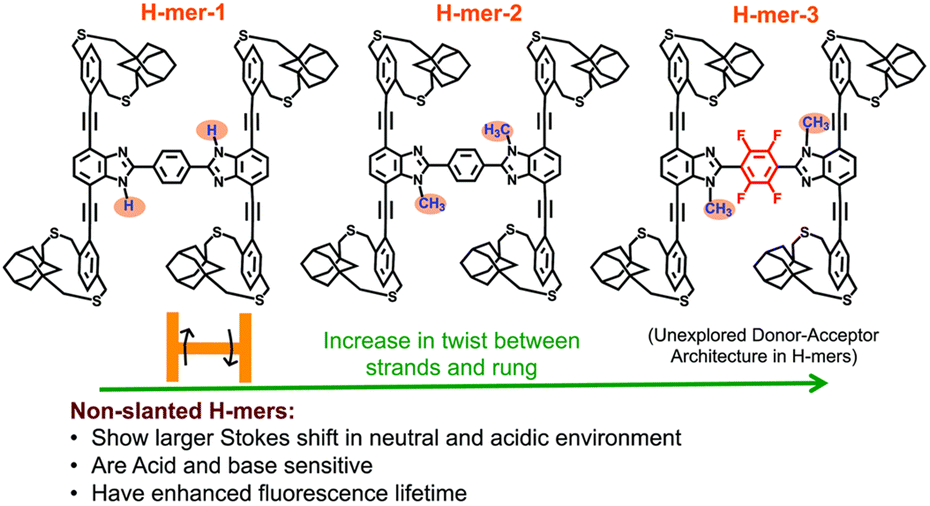 | ||
| Fig. 7 Chemical structures of non-slanted H-mers. Reproduced with permission from ref. 31 under a Creative Commons Attribution-Non-commercial 3.0 Unported Licence. Royal Society of Chemistry (2020). | ||
Non-slanted H-mers electronically differ from the slanted H-mers developed by the Zhao group.27 In both the H-mers, two strands are connected via a rung but differ in terms of the delocalization of π-electrons. In the slanted H-mers, the rungs are directly grafted onto strands, and therefore only half of the strands are in conjugation with each other, resulting in a conjugation path that resembles the letter Z, whereas in the non-slanted H-mers, the two strands are connected to the rung via a fused imidazole-based heterocycle, which involves the whole strand in the π-conjugation rather than just half of the strand, resulting in extended charge and exciton delocalization. Thus, in H-mers, a rung functions as a bridge between the strands, playing a crucial role in extending the π-conjugation across them.
We synthesized three H-mers to understand the impact of the twist between the strands and rungs as well as the electronic nature of the rung on the H-mer optical, sensing, and photophysical properties. To obtain soluble H-mers, we employed adamantyl straps reported by our group18 since they were shown to impart solubility to polymers without the need for pendant solubilizing chains. The benzimidazole (fused imidazole) containing H-mers are synthesized in one step via a dialdehyde and diamine condensation reaction. All the H-mers were readily soluble without heating in chloroform and dichloromethane. UV-vis absorption and emission spectra of all three H-mers were recorded in neutral, acidic, and basic environments. Structural changes and pH have a minimal impact on the UV-vis absorption spectra of H-mers. Interestingly, H-mer-3 with an electron-deficient rung and twist displayed a larger Stokes shift compared to other H-mers. The larger Stokes shift in the H-mer is attributed to efficient intramolecular charge transfer in the excited state from the strands to the electron-deficient rung. The red shift in the emission spectra upon protonation is significantly higher compared to T-mers that have analogous absorption and emission spectra in a neutral state.32 Notably, the excited state lifetime of H-mer-3 is at least five times longer than that of H-mer-1 and H-mer-2, underscoring the significance of the electronic nature of the rung. The photoluminescence of H-mer-3 is quenched by an electron acceptor such as tetracyanoethylene (TCNE). The non-slanted H-mers represent the repeat units of 2D-π-conjugated materials.33,34 Therefore, in addition to extended π-conjugation between the strands, these H-mers are also appropriate to grow into polymeric structures due to the presence of mirror-plane symmetry compared to the slanted H-mers.
Synthesis of strapped π-conjugated hyperbranched polymers
The synthesis of soluble 2D-oligomers without the pendant solubilizing chains strengthened our hypothesis and enabled us to develop higher dimensional polymers without pendant solubilizing chains. Toward this, we synthesized hyperbranched strapped-π-conjugated polymers,35 strapped polyazomethines, without pendant solubilizing chains. We synthesized strapped polyazomethines by harnessing the power of strapped building blocks and dynamic imine chemistry (Fig. 8). For this, an adamantyl-strapped monomer was selected as the aryl-strapped building block due to its demonstrated efficiency in hindering interchain interactions compared to the cyclohexyl-strapped monomer.18,36 Dynamic imine37 chemistry was used because of its reversible nature and has been successfully used to create elusive yet significant materials, such as macrocycles and covalent organic frameworks.38,39 More importantly, the imine reaction facilitates the extension of π-conjugation between the building blocks.40,41 Imiane condensation polymerization of a strapped aryl dialdehyde monomer (A2) with a trifunctional aryl amine unit (B3) generated solution-dispersible and processable hyperbranched polyazomethines (HB-PAMs) that were dispersible in chloroform without the need for pendant solubilizing chains. The straps prevent the extensive aggregation of polymer chains, leading to the formation of a dispersible hyperbranched polymer. | ||
| Fig. 8 (a) Synthesis of HB-PAM-1 discussed in this study. (b) AFM image of HB-PAM-1. Reproduced with permission from ref. 35 under a Creative Commons Attribution-Non-commercial 3.0 Unported Licence. Royal Society of Chemistry (2024). | ||
The polymer's topology and architecture were inferred from its degree of branching (DB). Polymers with a DB between 0.35 and 0.65 are classified as hyperbranched polymers.35 The topological structure of a polymer with a high DB is thought to be similar to dendrimer analogues, whereas a low DB suggests that the structure is more like a linear polymer. HB-PAM-1's DB was determined from NMR using the formula DB = (T + D)/(T + D + L), which was proposed by Frechet and coworkers.42 HB-PAM-1's DB is found to be 0.46, which indicates a hyperbranched architecture.
By meticulously controlling key polymerization conditions, such as the catalyst, monomer concentration, and solvent, we managed to control the growth and morphology of the resulting HB-PAM particles. A noteworthy discovery about the morphology of the nanoparticles is that they all have a pancake-like morphology (Fig. 8b), which means that their diameter is greater than their height. The diameter of the HB-PAM nanoparticles ranges from 60 to 300 nm, while their height varies from 6 to 60 nm. The pancake morphology is observed because the polymer architecture is unable to support its hyper-branched structure, causing it to collapse onto itself. This pancake morphology was also found in dendrimers, which are regarded as analogues of defect-free hyperbranched polymers, as well as in hyperbranched polymers. The higher surface area to volume ratio of HB-PAMs resulting from the pancake morphology is advantageous for effective interaction with small molecule dopants for electronic and optoelectronic applications.
Synthesis of strapped π-conjugated porous polymers
Access to soluble hyperbranched polymers through the cycloalkyl strapped building blocks encouraged us to use the strapped building blocks to generate π-conjugated porous polymers (CPP) and study the impact of the strapped building blocks on the CPP network growth and solubility (Fig. 9). | ||
| Fig. 9 Cartoon representation of how straps reduce interchain π–π stacking interactions in higher dimensional π-conjugated porous polymers. | ||
Although there has been some progress in creating solution-processable 2D π-conjugated organic materials by reducing interchain interactions via either varying inter-arylene torsion angles and/or pendant solubilizing chains,43,44 techniques that directly target the interchain π–π stacking interactions at the molecular level are still desperately needed to control the growth and processability of 2D π-conjugated organic materials.45–49
Strapped tris(arylethynyl)phosphane porous polymer networks (S-CPP-1–3) (Fig. 10) were synthesized by reacting phosphorous trichloride knots (A3) with adamantane cyclophane diacetylene struts (B2) in the presence of a Ni(II) catalyst in a mixture of toluene and triethylamine. PCl3 was selected as the trifunctional A3 monomer for two reasons. Phosphorus extends the π-conjugation across the network, unlike the meta-conjugated 1,3,5-phenylene trifunctional core. Also, tri-, di-, and mono-reacted phosphorus with aryl acetylenes result in crosslinked (dendritic), linear, and terminal groups respectively, and are relatively easy to differentiate and identify using 31P-NMR compared to a completely carbon-based network (Fig. 11). To tune the network crosslinking density, the molar ratio of the knot was systematically increased from 0.66 to 1.33 with respect to the strut. The knot-to-strut ratio was varied to control the degree of branching and to demonstrate that the straps provide synthetic control over the growth of the CPP. A non-strapped CPP (NS-CPP) was synthesized for comparison following a similar synthetic protocol.
 | ||
| Fig. 10 Chemical structures of the series of strapped CPPs (S-CPP-1–3) and a non-strapped CPP (NS-CPP). Reproduced with permission from ref. 50 under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence. Royal Society of Chemistry (2023). | ||
 | ||
| Fig. 11 (a) Synthesis of a strapped CPP. (b) Cartoon representation of the CPP network; purple, yellow and grey balls represent dendritic, linear, and terminal groups. Reproduced with permission from ref. 50 under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence. Royal Society of Chemistry (2023). | ||
As anticipated, S-CPPs were dispersible in typical solvents used to dissolve conjugated polymers, including chloroform and o-dichlorobenzene (o-DCB). Dendritic groups represent the crosslinking centers in the CPP and are useful to extend the delocalization of π-electrons throughout the network. The percentage of dendritic groups in S-CPP-1 (38%) is almost twice that of the non-strapped CPP (NS-CPP: 21%), even though both of them have similar knot-to-strut ratios. The larger percentage of dendritic groups in S-CPP-1 suggests that it has a higher crosslinking density than NS-CPP. This was attributed to the increase in the network's residence time in the reaction mixture, which allowed higher crosslinking density. DLS studies indicate that the straps also generate a swollen network (Table 1). Thus, the straps reduce interchain π–π interactions, enhance the network residence time, and generate a swollen network. This allows the monomer to diffuse into the network and react.
Similar to S-CPP-1, S-CPP-2 and S-CPP-3 were synthesized by increasing the knot ratio from 0.66 to 1 and 1.33. Both these reaction mixtures contain more knots than the required stoichiometry of reactive functional groups. The percentage of dendritic groups in S-CPP-2 and S-CPP-3 is 24% and 39%, respectively. When the knot ratio rises in non-conjugated hyperbranched polymers, the degree of branching decreases.45 However, there are no reports on how the degree of branching (crosslinking density) of π-conjugated polymer porous networks varies as the knot ratio increases. In this work, the percentage of dendritic groups did not show a clear trend as the knot ratio increased. This could be due to the change in growth kinetics associated with the change in swellability and morphology of the network, which alters the percentage of monomer conversion, thus pushing the crosslinking density away from the non-conjugated polymers trend line.
Dispersibility limits of the CPPs were determined to see if the strapped adamantyl groups decrease interchain interactions, stabilize particles, and prevent S-CPPs from aggregating and undergoing sedimentation. S-CPPs showed dispersibility limits that were 8–16 times higher than NS-CPP. The dispersibility limit of S-CPP-1 is 16 times greater than that of control NS-CPP with the same knot-to-strut ratio. The presence of adamantyl straps accounts for the S-CPP-1's increased dispersibility limit when compared to NS-CPP. The straps shield the π-face of the aryl repeat unit and hinder interparticle π–π interactions against the formation of larger aggregates and sedimentation. Within the S-CPP series, the dispersibility limit of S-CPP-1–3 reduces as the knot-to-strut ratio rises. The percentage of dendritic groups is similar for S-CPP-1 and S-CPP-3; thus, S-CPP-1's higher dispersibility limit is due to its smaller network size. The S-CPP-2 network's dispersibility limit lies between S-CPP-1 and S-CPP-3, and the pattern is consistent with the network size.
To understand the impact of the knot-to-strut ratio on the efficiency of chemical doping, the dopant was titrated into the CPP dispersion, and the change in absorbance of the F4TCNQ˙− peak (873 nm peak) was plotted against the weight percentage of the dopant (Fig. 12). The chemical doping efficiency of S-CPPs is 4 to 18 times higher than that of NS-CPP, with S-CPP-1 being 18 times more efficient than NS-CPP. Within the strapped series, as the knot ratio increased, doping efficiency decreased; larger particles showed lower doping efficiency. This highlights the importance of controlling the growth of CPPs and controlling the degree of branching. Compared to poly(phenyleneethynylene) CPP, the conductivity values of the produced CPPs are approximately three orders of greater magnitude greater. The main distinction between the poly(phenyleneethynylene) CPP and the CPPs studied in this work is that the struts in the former are connected via meta-phenylene linkages, which do not extend the conjugation between struts, while the struts in the latter are connected via phosphorus, which is known to extend the conjugation across the network as shown by Lucht and Gates.51 Since NS-CPP and S-CPP-1–3 have comparable conductivities, straps do not impede interchain charge transport.
 | ||
| Fig. 12 (a) Dispersibility limit of CPPs in CHCl3 (1 mg mL−1 dispersions of S-CPP-1 and NS-CPP are shown for comparison in the inset); (b) UV-vis absorption spectra of CPPs in chloroform; (c) increase in the absorbance of F4TCNQ˙− upon increasing the loading of F4TCNQ in S-CPP-1 solution (legend: weight% of F4TCNQ with respect to S-CPP-1); (d) change in absorbance at 873 nm (which corresponds to the absorbance of F4TCNQ˙−) as the weight% of F4TCNQ is increased in solution for S-CPP-1–3 and NS-CPP, doping efficiency of CPPs increases as the porous polymer network solvodynamic size decreases. Reproduced with permission from ref. 50 under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence. Royal Society of Chemistry (2023). | ||
The novel π-face masking straps effectively prevent interchain π–π interactions by directly shielding the π-face of the repeat unit and increasing the network's residence time in the reaction mixture compared to the conventional linear pendant alkyl chains. By reducing interchain π–π interactions during network growth, these straps create a swollen network that facilitates monomer diffusion and reaction, further giving control over the crosslinking density and network size.50 The strapped networks outperform the non-strapped ones in terms of crosslinking density, processability, and percentage of charge transfer. Thus, the use of straps is a powerful strategy to realize control over CPP growth and particle size.
Controlling π-conjugated polymer donor–acceptor interactions using straps
As mentioned above, a few groups have used strapped monomers along with pendant solubilizing chains to control interchain interactions and to gain access to the properties associated with hindering strong interchain π–π interactions, such as increased photoluminescence quantum yield, thermoformability, and enhanced sensory response towards analytes.17,20–22 The Bronstein group synthesized a series of doubly strapped π-conjugated polymers based on diketopyrrolopyrrole (DPP) or alkylene-encapsulated anthracene that show excellent properties due to π-face masking.20,21 The DPP monomer unit is sheathed by its side chains using a covalent encapsulation approach, thereby suppressing π-stacking interactions between the backbones of the polymer. The encapsulation effectively impedes interchain interactions, as evidenced by the high fluorescence quantum yields recorded in solution and solid states.52 The Takeuchi and Sugiyasu groups have developed a unique self-threading polythiophene insulated by its own cyclic side chains.22 The doubly strapped monomer building block provides full insulation of the π-face of the resulting polymer. The fully strapped homopolymer exhibited superior photoluminescence (PL) quantum yield in solution and solid states. More interestingly, the polymers were excellently thermoformable, typical of conventional plastics, which is rare for π-conjugated polymers.Recent works by our research group and others show that the molecular sheathing of a π-conjugated polymer backbone controls the photoinduced charge transfer from the polymer to the acceptor.53,54 The photoluminescence quenching of a donor polymer by an acceptor is one of the key steps in charge generation in organic electronics. For example, organic thermoelectrics involve a molecular doping process wherein the charge transfer from the polymer to the dopant results in ionized species that determine the electrical conductivity and thermoelectric performance of the material.55 Unlike small molecules, oligomers and polymers have several available sites for the dopant to bind along their backbone.56–61 Control over the polymer–dopant interactions, including the interaction strength, spatial location, and dopant concentration, all of which determine the thermoelectric performance, is highly desired.
Previous work
The recent increased interest towards organic thermoelectrics has spurred new research directions aimed at unmasking the intricacies of molecular doping in organic semiconductors. The importance of controlling the dopant binding location along the polymer backbone is demonstrated by Kim and coworkers using a conventional (no straps) donor–acceptor polymer.62 They have shown that for a donor–acceptor copolymer, the dopant binding strength varies depending on its location on the backbone, i.e., the repeat unit with which it is interacting. They observed that for the 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4-TCNQ) doped isoindoloindole-based donor–acceptor conjugated polymer, the dopant binding strength depends on the binding location.The Smith group showed that the strapped building blocks are useful in enhancing the PL emission quenching rate.59 They used sterically encumbered m-terphenyls as straps. The oxacyclophane canopy in the polymer was responsible for defined molecular clefts and free spaces around the π-conjugated backbone, which facilitated the entry of analytes. The polymers were then used for molecular sensing of small molecules like nitro-substituted organics, recording up to a 100-fold emission quenching rate enhancement in response to analytes. However, so far, there are no design rules to control the polymer–acceptor interactions at the molecular level for a better understanding of charge generation and transport and the thermoelectric performance of organic π-conjugated polymers.
Our approach
These precedents motivated us to investigate the potential to control the binding strength and location of the dopant by synthesizing a series of oligomers containing three covalently linked phenyl units. These trimers: PCP (phenyl–cyclophane–phenyl), CPC (cyclophane–phenyl–cyclophane), and PPP (phenyl–phenyl–phenyl) have a strapped unit in the middle, strapped at both edges, and no straps, respectively (Fig. 13).26 In addition to the PPP control trimer, PLP (phenyl–linear–phenyl) and LPL (linear–phenyl–linear) trimers that are analogous to PCP and CPC, respectively, but have linear substituents, were also synthesized. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexation of the five donor trimers with the dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) acceptor (dopant) was studied in solution to demonstrate that the acceptor strongly interacts with the non-strapped units of the strapped trimers (PCP and CPC), and it is possible to control the location of the acceptor in a strapped–non-strapped copolymer.
:1 complexation of the five donor trimers with the dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) acceptor (dopant) was studied in solution to demonstrate that the acceptor strongly interacts with the non-strapped units of the strapped trimers (PCP and CPC), and it is possible to control the location of the acceptor in a strapped–non-strapped copolymer.
 | ||
| Fig. 13 The percentage configuration of DDQ residing on each of the trimer is determined using DFT calculations and is shown on the arrow. DDQ complex with (left) a non-strapped donor (PPP); (middle) middle repeat unit strapped donor (PCP); and (right) terminal repeat unit strapped donor (CPC). The green cylinder around the cyclophane indicates the insulating sheath due to rotation of the cyclophane unit. Blue and red color arrows indicate the most and least accessible locations for DDQ along the trimer backbone. Bottom left and right structures show PLP–DDQ and LPL–DDQ complexes respectively. Reproduced with permission from ref. 26 under a Creative Commons Attribution-Non-commercial 3.0 Unported Licence. Royal Society of Chemistry (2022). | ||
The density functional theory (DFT)-determined binding energy of DDQ with CPC and PCP is similar but higher than that with PPP. DDQ showed a 25% higher preference to interact with the middle-repeat unit of the fully non-strapped trimer (PPP). On the linearly substituted controls, DDQ residing on the middle phenyl unit had the highest percentage configuration of ca. 64% and 96% for PLP and LPL, respectively, whereas in the case of strapped trimers (CPC and PCP), the DDQ prefers to reside on the non-strapped repeat units irrespective of their location by at least 25% more. The highest percentage configuration (ca. 74%) for the two strapped trimers was the geometry where DDQ resides on the non-strapped phenyl unit. Despite the cycloalkyl straps only masking one of the π-faces of the phenyl unit, the free rotation generates a cylindrical insulating sheath around the unit, thus restricting the approach of the dopant to the π-face.
UV-vis spectroscopy was used to determine the percentage of dopant ionization in the donor–acceptor complex (Fig. 14). The DDQ concentration of the radical anion, which is formed upon complexation with the donors, increased by 15% and 59% in the case of PCP and CPC trimers compared to the PPP donor. On the other hand, the linear substituted LPL and PLP had a lower percentage (smaller by 15–20%) of DDQ ionization with respect to the PPP complex. Thus, the strapped units provide control over the dopant location and binding energies as well as provide higher dopant ionization. The ability to control donor–acceptor interactions will have a significant impact on the development of organic electronics that use donor and acceptor π-conjugated materials, including light-emitting diodes, solar cells, and thermoelectrics.64
 | ||
| Fig. 14 Normalized UV-vis absorption spectra of (a) trimers and DDQ in chloroform, (b) charge transfer peak shown for clarity, (c) charge-transfer complexes in tetrahydrofuran and (d) non-normalized spectra of PLP–DDQ and LPL–DDQ complexes in tetrahydrofuran. Reproduced with permission from ref. 26 under a Creative Commons Attribution-Non-commercial 3.0 Unported Licence. Royal Society of Chemistry (2022). | ||
 | ||
| Fig. 15 Chemical structure of the P8 polymer containing both adamantyl strapped and p-CP comonomers. Randomly positioned p-CP units along the insulated polymer backbone enhance the photoinduced charge transfer to acceptor molecules resulting in fluorescence quenching. Reproduced with permission from ref. 65 Copyright American Chemical Society (2021). | ||
To understand the specific role of the p-CP units, we synthesized four different random copolymers P8 of adamantyl strapped monomers (a-CP) with an increasing fraction of p-CP monomers from 5–30% (Mn between 11–14 kDa).65 As more p-CP units are incorporated into the adamantanocyclophane polymer, the copolymer's solubility decreases due to the π–π interactions between the phenyl units of the dithia[3.3]paracyclophanes and the π-conjugated polymer backbone. Hence, solubility was the primary reason for limiting the p-CP comonomer mol% to a maximum of 30% and restricting the copolymer's molecular weight to below 15 kDa.
There was a marked difference in the UV-vis absorbance of peaks less than 300 nm and above 450 nm (Fig. 16). The increase in absorbance between 455 and 500 nm as the percentage of the p-CP monomer increases is attributed to the intramolecular charge transfer between the a-CP and p-CP units. The fluorescence of the copolymers was quenched more effectively by the tetracyanoquinodimethane (TCNQ) acceptor with an increasing percentage of p-CP. P8 with 30% p-CP showed a Ksv that is approximately 4.5 times higher than that of the a-CP homopolymer with 0% p-CP. The p-CP units are smaller in size compared to the a-CP units. Hence, the observed enhancement in Ksv is due to the reduced steric hindrance from the p-CP unit compared to the a-CP unit, which allows TCNQ to easily access the polymer backbone and quench the emission.
 | ||
| Fig. 16 (a) Chemical structures, (b) UV-vis absorption spectra, and (c) emission spectra of P5, P7, and P8. Reproduced with permission from ref. 65 Copyright American Chemical Society (2021). | ||
Also, the through-space transannular π–π interactions in p-CP are a plausible reason for enhanced quenching. To investigate the contribution of the transannular π–π interactions to the observed changes in the PL quantum yield, we synthesized a control copolymer (P7) with equimolar amounts of adamantyl and cyclohexyl groups. The cyclohexyl group is the saturated version of the phenyl ring and does not provide transannular π–π interactions. The cyclohexyl strap has the same height as the p-CP; hence, the observed difference in the optical properties between P7 and P8 can be attributed to the through-space-conjugated phenyl ring transannular interaction in P8. The Ksv of P8 is 3.4 times higher than that of P7, which highlights the role of through-space (transannular) interaction in enabling the acceptor interaction with the polymer backbone.
 | ||
| Fig. 17 Copolymers (bottom) with a mixture of strapped and non-strapped units direct acceptors to strongly interact with the non-strapped units because the π-face is less hindered. The random distribution of non-strapped units increases non-neighbor acceptor binding events and results in higher PL quenching. Reproduced with permission from ref. 53 Copyright American Chemical Society (2023). | ||
A series of adamantyl strapped copolymers (P8–P12) having 20 mol% (feed ratio) randomly incorporated non-strapped repeat units were synthesized (Fig. 18a).53 Control polymers P5 and P6 with fully strapped and fully non-strapped repeat units were also synthesized. By employing the freeze–thaw–run approach, copolymers of approximately similar molecular weights were generated to eliminate any discrepancies that may arise due to molecular weight differences. 1H-NMR confirms that all copolymers contain 16–20 mol% of the non-strapped aryl units. The UV-vis absorption (Fig. 18b) maxima of all polymers were close, between 418 and 423 nm, which indicates that the substituents on the non-strapped units have no significant impact on the energy band gap of the copolymers (P8, P9, and P10). Also, similar emission maxima for the copolymers suggested that the conjugated segment length where exciton migration occurs upon excitation is approximately the same for all copolymers.
 | ||
| Fig. 18 (a) Chemical structures; (b) UV-vis absorption spectra and (c) emission spectra of P5, P6, and P8–P12 (n = ∼20% in the copolymers). Reproduced with permission from ref. 53. Copyright American Chemical Society (2023). | ||
To investigate the impact of a mixture of strapped and non-strapped units on the polymer–TCNQ interactions, photoinduced PL quenching studies were conducted on the polymers in the presence of the dopant (Table 2). The Ksv of all the copolymers was higher than that of the P5 homopolymer, which indicates easy access of the polymer backbone to the dopant. More importantly, the Ksv of one of the strapped copolymer P9 is higher than those of the homopolymers and co-polymers studied in this work including the conventional pendant chain polymer P10. Concentration-dependent (high vs. low conc.) studies showed that Ksv of P9 increased 75 times moving from lower to higher concentrations, while there was only a 11-fold increase for P6. In the case of P6, as the concentration of TCNQ increases, the newly added TCNQ molecules preferentially bind next to the already existing polymer–TCNQ complexes (neighboring group effect), as it is more energetically favored.50 Hence, the contribution of newly added TCNQ molecules toward the polymer PL quenching is low. In the case of strapped copolymers, non-strapped units are the preferred binding sites for TCNQ molecules due to the lack of straps and they are randomly distributed along the polymer backbone. The TCNQ molecules bind to energetically preferred non-strapped units, which are randomly distributed throughout the chain and avoid neighboring strapped units because of the lower binding energy. The strapped copolymer P9 showed a higher Ksv than the conventional polymer P6 because the TCNQ molecules interact with the polymer backbone randomly throughout the polymer chain, leading to efficient PL quenching. Thus, the random mixing of strapped and non-strapped units in conjugated donor copolymers is an effective strategy to enhance the strength of the polymer–acceptor interaction as well as the ability to direct the location of the acceptor along the polymer backbone.
| Polymer | Nonlinear Ksv (× 103) (M−1) | Linear Ksv (× 103) | K sv2/Ksv1 | |
|---|---|---|---|---|
| K sv1 (M−1) | K sv2 (M−1) | |||
| P9 | 225 ± 20 | 28 ± 0.3 | 2092 ± 370 | 75 |
| P10 | 30 ± 07 | 14 ± 1 | 335 ± 20 | 23 |
| P8 | 44 ± 3 | 20 ± 0·1 | 246 ± 13 | 12 |
| P11 | 96 ± 0.5 | 19 ± 1.6 | 1360 ± 74 | 70 |
| P12 | 369 ± 7 | 242 ± 15 | 15716 ± 1600 | 65 |
| P5 | 13 ± 1 | 8 ± 0.1 | 17 ± 1 3 | 2 |
| P6 | 110 ± 10 | 33 | 362 | 11 |
Applications of strapped π-conjugated polymers
Encapsulating the π-face of conjugated polymer repeat units has significant advantages, like the manipulation of polymer solubility, intra- and inter-chain charge transport, and sensory responses amongst others.17 A common question surrounding the incorporation of straps is their effects on conductivity and bulk charge transfer efficiency for use in electronic devices. We have earlier shown that the conductivity of a strapped CPP (S-CPP) is similar to that of a non-strapped CPP (NS-CPP), suggesting that the straps do not significantly hinder charge transport in CPPs.50 Takeuchi and coworkers developed a doubly strapped molecular wire (with straps on both sides of the repeat unit) by designing a self-threading polythiophene.12 Incorporating double straps reportedly enhanced the effective conjugation length of the backbone, resulting in an intrinsic hole mobility of 0.9 cm2 V−1 s−1 and a bulk conductivity of 2 × 10−3 S cm−1. However, this observed conductivity is two orders of magnitude lower than that of the corresponding control polythiophene, which is attributed to lower interchain charge transfer. In contrast, the minimal impact on the conductivity of straps on our S-CPPs could be due to the fact that only one of the π-faces of the repeat unit is masked by the straps, unlike in the doubly strapped polythiophene, where both faces are insulated. However, to fully understand the influence of straps on interchain charge transport, more detailed studies on conductivity and charge mobility in strapped polymers and networks are needed.Several recent research has leveraged the incorporation of straps to tune the bulk properties of materials. We have used molecular straps to control CPP growth, chemical doping efficiency, and conductivity.50 Takeuchi and Sugiyasu demonstrated that incorporating doubly strapped conjugated polymers made them thermoformable, similar to conventional plastics—overcoming a common limitation of conjugated polymers.67 The Smith group introduced a steric canopy using sterically encumbered m-terphenyls as straps to create a conjugated sensory system for detecting nitroaromatics.68 The McCulloch group innovatively utilized ethylene glycol straps to induce ion sensory capabilities by exploiting the complexation of sodium and potassium ions with the ethylene glycol straps.69 The Bronstein group reported that their doubly strapped π-conjugated polymers, based on DPP, exhibited enhanced photoluminescence quantum yield (PLQY) in thin films due to the suppression of π-stacking interactions between polymer backbones. The straps were employed to structurally control the degree of energetic disorder in the polymers.20,21 The Ikeda group synthesized a conjugated polyrotaxane with a thiophene backbone encapsulated by the electron-deficient tetracationic receptor cyclobis(paraquat-p-phenylene) (CBPQT4+). The migration of counterions from the solvent into the polythiophene backbone made this polymer effective as an electrochromic material.70 Anderson and coworkers explored the impact of encapsulation on charge transport dynamics using poly(para-phenylenevinylene) (PPV)-based polyrotaxanes encapsulated with cyclodextrins (CDs).71 They fabricated a single-layer polymer LED device that exhibited optimal electroluminescence performance despite the encapsulation.
Future outlook
Controlled synthesis of π-conjugated materials beyond 1-dimension is still in its infancy. At present, there are no well-established design rules and strategies to overcome interchain π–π interactions in higher dimensional π-conjugated materials, including CPPs, nanoribbons, 2D-π-conjugated polymers, and covalent organic frameworks. Pendant alkyl chains, which are widely used to successfully obtain soluble linear 1D-π-conjugated polymers, do not overcome strong interchain π–π interactions in higher dimensional π-conjugated materials because they do not directly mask the π-face of polymer chains. This is the key reason for the lack of bulk solution phase synthesis, single-layer synthesis, and/or synthetic control of these materials. The π-face masking straps, especially the cycloalkyl straps, that are positioned either above or below the π-conjugation plane have been shown to directly control the π–π interactions between the polymer chains. We have shown that π-face masking cycloalkyl straps mask the π-surface, hinder interchain π–π interactions, and render high-molecular-weight soluble 1D-conjugated polymers without pendant solubilizing chains. The synthesized strapped polymers are soluble in several organic solvents commonly used for solubilizing π-conjugated polymers, including THF, CHCl3, DMF, and chlorobenzene, as highlighted in our report.18 Introducing polar or ionic straps could enable the development of strapped polymers that are soluble in solvents across a wider range of polarities. One example is the work of McCulloch and coworkers, where ethylene glycol-strapped repeat units were used alongside pendant solubilizing chains.68 However, the authors did not comment on the solubility of these polymers in polar solvents. We plan to explore some of these approaches in future studies. It is also worth noting that the obtained polydispersity values in this work are typical for π-conjugated polymers. However, the specific effect of polydispersity on the physical properties of strapped polymers has not yet been investigated. More importantly, π-face masking cycloalkyl straps will also help to systematically tune the interchain π–π interactions and provide control over the growth of higher dimensional π-conjugated materials. We have demonstrated the synthesis of soluble 2D-H-mers, dispersible hyperbranched π-conjugated polymers, and conjugated porous polymers without the pendant solubilizing chains. Cycloalkyl straps helped to attain a higher degree of branching in the CPPs compared to the control non-strapped CPP due to hindered interchain interactions and a swollen network in the former, which highlights that the cycloalkyl straps provide control over the CPP growth, branching density, and solubility. However, the cycloalkyl strapped-CPPs’ degree of branching did not increase systematically as the knot feed ratio decreased due to enhanced interchain interactions. In order to completely gain control over the growth and degree of branching, there is a need to efficiently hinder interchain interactions. Using a mixture of straps of different heights, the solubility limit of linear conjugated copolymers was increased by three times compared to the corresponding homopolymers. Thus, employing straps of different heights is expected to efficiently frustrate the interchain interactions, swell the network, enhance network growth time, and realize control over the network growth and crosslinking density for a given range of crosslinker ratios. The use of cycloalkyl straps for the synthesis of nanoribbons, nanoladders, and covalent organic frameworks will enable bulk solution phase synthesis of these materials. In addition, the use of cycloalkyl straps will also provide individually dispersed polymers/sheets of these materials, which has been a longstanding challenge for the higher dimensional materials and will push the frontiers of molecular electronics.We have also shown that cycloalkyl straps can be used to control the donor–acceptor interactions. DFT simulations and experimental data on the cycloalkyl strapped trimer-acceptor systems show that the location of the acceptor can be controlled and, moreover, the percentage of charge transfer can be enhanced more than that of conventional pendant chain trimers. An analogous observation was made in the mixture of strapped-non-strapped random copolymers as well. The Ksv, which is a measure of photoinduced charge transfer from the polymer to the acceptor, of a cycloalkyl strapped–non-strapped random copolymer was higher than that of a conventional pendant chain containing control polymer due to the minimized neighboring group effect and random distribution of the acceptors across the polymer backbone. To completely gain control over the spatial location of the acceptor along the polymer backbone, sequence-controlled copolymers containing strapped and non-strapped building blocks should be synthesized and complexed with acceptors. The well-designed sequence-controlled polymers will help to unearth the molecular level insights into the impact of the spatial location of the acceptor as well as the concentration of acceptors along the polymer backbone on charge generation, transport, and thermoelectric performance of the conjugated polymers.
Author contributions
All the authors contributed to the draft writing.Data availability
No primary research results, software, or codes have been included and no new data have been generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Science Foundation CAREER Grant (NSF-1944184) and Georgetown University. V. S. F. acknowledges the Georgetown University-Institute for the Soft Matter Synthesis and Metrology Graduate Student Fellowship. N. G. would like to thank all the current and former members of the Gavvalapalli group who have contributed to the work discussed in this feature article. We acknowledge the use of a large language model for assistance with English language editing and grammar refinement of this manuscript.Notes and references
- Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 RSC.
- K. M. Coakley and M. D. McGehee, Chem. Mater., 2004, 16, 4533–4542 CrossRef.
- S. B. Mdluli, M. E. Ramoroka, S. T. Yussuf, K. D. Modibane, V. S. John-Denk and E. I. Iwuoha, Polymers, 2022, 14, 716 CrossRef PubMed.
- U. Mehmood, A. Al-Ahmed and I. A. Hussein, Renewable Sustainable Energy Rev., 2016, 57, 550–561 CrossRef.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef.
- C. S. Hartley, E. L. Elliott and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 4512–4513 CrossRef PubMed.
- I. C. Y. Hou, Y. Hu, A. Narita and K. Müllen, Polym. J., 2017, 50, 3–20 CrossRef.
- D. K. Fu, B. Xu and T. M. Swager, Tetrahedron, 1997, 53, 15487–15494 CrossRef.
- J. S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef PubMed.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012 RSC.
- D. Taylor, S. J. Dalgarno, Z. Xu and F. Vilela, Chem. Soc. Rev., 2020, 49, 3981–4042 RSC.
- M. G. M. Bai, H. V. Babu, V. Lakshmi and M. R. Rao, Mater. Chem. Front., 2021, 5, 2506–2551 RSC.
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- J. Lee, A. J. Kalin, T. Yuan, M. Al-Hashimi and L. Fang, Chem. Sci., 2017, 8, 2503–2521 RSC.
- A. Narita, X. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, H. Yang, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen, A. H. R. Koch, G. Fytas, O. Ivasenko, B. Li, K. S. Mali, T. Balandina, S. Mahesh, S. De Feyter and K. Müllen, Nat. Chem., 2013, 6, 126–132 CrossRef PubMed.
- D. Barpuzary, K. Kim and M. J. Park, ACS Nano, 2019, 13, 3953–3963 CrossRef CAS PubMed.
- J. Royakkers and H. Bronstein, Macromolecules, 2021, 54, 1083–1094 CrossRef CAS.
- S. Chaudhuri, M. Mohanan, A. V. Willems, J. A. Bertke and N. Gavvalapalli, Chem. Sci., 2019, 10, 5976–5982 RSC.
- A.-D. Schlüter, M. Löffler and V. Enkelmann, Nature, 1994, 368, 831–834 CrossRef.
- A. Leventis, J. Royakkers, A. G. Rapidis, N. Goodeal, M. K. Corpinot, J. M. Frost, D.-K. Bučar, M. O. Blunt, F. Cacialli and H. Bronstein, J. Am. Chem. Soc., 2018, 140, 1622–1626 CrossRef CAS PubMed.
- D. G. Congrave, B. H. Drummond, V. Gray, A. D. Bond, A. Rao, R. H. Friend and H. Bronstein, Polym. Chem., 2021, 12, 1830–1836 RSC.
- K. Sugiyasu, Y. Honsho, R. M. Harrison, A. Sato, T. Yasuda, S. Seki and M. Takeuchi, J. Am. Chem. Soc., 2010, 132, 14754–14756 CrossRef CAS PubMed.
- S. D. Diwakara, W. S. Y. Ong, Y. H. Wijesundara, R. L. Gearhart, F. C. Herbert, S. G. Fisher, G. T. McCandless, S. B. Alahakoon, J. J. Gassensmith, S. C. Dodani and R. A. Smaldone, J. Am. Chem. Soc., 2022, 144, 2468–2473 CrossRef CAS PubMed.
- R. Lemmerz, M. Nieger and F. Vögtle, J. Chem. Soc., Chem. Commun., 1993, 1168–1170 RSC.
- R. Fiesel, J. Huber, U. Apel, V. Enkelmann, R. Hentschke, U. Scherf and K. Cabrera, Macromol. Chem. Phys., 1997, 198, 2623–2650 CrossRef CAS.
- J. L. Sartucci, A. Maity, M. Mohanan, J. Bertke, M. Kertesz and N. Gavvalapalli, Org. Biomol. Chem., 2022, 20, 375–386 RSC.
- N. Zhou, L. Wang, D. W. Thompson and Y. Zhao, Tetrahedron, 2011, 67, 125–143 CrossRef CAS.
- B. A. G. Hammer and K. Müllen, Chem. Rev., 2015, 116, 2103–2140 CrossRef PubMed.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453–465 CrossRef CAS PubMed.
- D. Hertel, U. Scherf and H. Bässler, Adv. Mater., 1998, 10, 1119–1122 CrossRef CAS.
- M. K. Das, F. Hameed, R. Lillis and N. Gavvalapalli, Mater. Adv., 2020, 1, 2917–2925 RSC.
- T. Inouchi, T. Nakashima, M. Toba and T. Kawai, Chem. – Asian J., 2011, 6, 3020–3027 CrossRef CAS PubMed.
- S.-L. Cai, K. Zhang, J.-B. Tan, S. Wang, S.-R. Zheng, J. Fan, Y. Yu, W.-G. Zhang and Y. Liu, ACS Macro Lett., 2016, 5, 1348–1352 CrossRef CAS PubMed.
- W. Liu, X. Luo, Y. Bao, Y. P. Liu, G.-H. Ning, I. Abdelwahab, L. Li, C. T. Nai, Z. G. Hu, D. Zhao, B. Liu, S. Y. Quek and K. P. Loh, Nat. Chem., 2017, 9, 563–570 CrossRef CAS PubMed.
- M. Mohanan, X. Zhang and N. Gavvalapalli, Polym. Chem., 2024, 15, 422–430 RSC.
- M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028–1064 CrossRef CAS PubMed.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003 RSC.
- T. Ide, D. Takeuchi and K. Osakada, Chem. Commun., 2012, 48, 278–280 RSC.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef PubMed.
- A. Bolduc, A. Al Ouahabi, C. Mallet and W. G. Skene, J. Org. Chem., 2013, 78, 9258–9269 CrossRef CAS PubMed.
- A. Bolduc, S. Dufresne and W. G. Skene, J. Mater. Chem., 2012, 22, 5053 RSC.
- C. J. Hawker, R. Lee and J. M. J. Frechet, J. Am. Chem. Soc., 1991, 113, 4583–4588 CrossRef CAS.
- S. Bandyopadhyay, P. Pallavi, A. G. Anil and A. Patra, Polym. Chem., 2015, 6, 3775–3780 RSC.
- G. Cheng, T. Hasell, A. Trewin, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 12727–12731 CrossRef CAS PubMed.
- S. R. Bheemireddy, M. P. Hautzinger, T. Li, B. Lee and K. N. Plunkett, J. Am. Chem. Soc., 2017, 139, 5801–5807 CrossRef CAS PubMed.
- S. R. Bheemireddy, M. P. Hautzinger, T. Li, B. Lee and K. N. Plunkett, J. Am. Chem. Soc., 2017, 139, 5801–5807 CrossRef CAS PubMed.
- L. Ma, S. Wang, X. Feng and B. Wang, Chin. Chem. Lett., 2016, 27, 1383–1394 CrossRef CAS.
- L. Yang and D.-C. Wei, Chin. Chem. Lett., 2016, 27, 1395–1404 CrossRef CAS.
- W. Yang, A. Lucotti, M. Tommasini and W. A. Chalifoux, J. Am. Chem. Soc., 2016, 138, 9137–9144 CrossRef CAS PubMed.
- M. Mohanan, H. Ahmad, P. Ajayan, P. K. Pandey, B. M. Calvert, X. Zhang, F. Chen, S. J. Kim, S. Kundu and N. Gavvalapalli, Chem. Sci., 2023, 14, 5510–5518 RSC.
- B. W. Rawe, M. R. Scott, C. M. Brown, H. K. MacKenzie and D. P. Gates, Macromolecules, 2017, 50, 8916–8927 CrossRef CAS.
- D. G. Congrave, B. H. Drummond, V. Gray, A. D. Bond, A. Rao, R. H. Friend and H. Bronstein, Polym. Chem., 2021, 12, 1830–1836 RSC.
- F. Hameed, M. Mohanan, N. Ibrahim, C. Ochonma, J. Rodríguez-López and N. Gavvalapalli, Macromolecules, 2023, 56, 3421–3429 CrossRef CAS PubMed.
- B. P. Morgan, R. J. Gilliard Jr., R. S. Loungani and R. C. Smith, Macromol. Rapid Commun., 2009, 30, 1399–1405 CrossRef CAS PubMed.
- A. Mityashin, Y. Olivier, T. Van Regemorter, C. Rolin, S. Verlaak, N. G. Martinelli, D. Beljonne, J. Cornil, J. Genoe and P. Heremans, Adv. Mater., 2012, 24, 1535–1539 CrossRef CAS PubMed.
- C. L. Anderson, T. Zhang, M. Qi, Z. Chen, C. Yang, S. J. Teat, N. S. Settineri, E. A. Dailing, A. Garzón-Ruiz, A. Navarro, Y. Lv and Y. Liu, J. Am. Chem. Soc., 2023, 145, 5474–5485 CrossRef CAS PubMed.
- M. Goel and M. Thelakkat, Macromolecules, 2020, 53, 3632–3642 CrossRef CAS.
- C. Wang, C. Li, E. R. C. Rutledge, S. Che, J. Lee, A. J. Kalin, C. Zhang, H.-C. Zhou, Z.-H. Guo and L. Fang, J. Mater. Chem. A, 2020, 8, 15891–15899 RSC.
- W. Wu, S. Yu, D. S. Forbes, H. Jiang, M. Ahmed and J. Mei, J. Am. Chem. Soc., 2023, 146, 578–585 CrossRef PubMed.
- E. Peterson, R. L. Maust, R. Jasti, M. Kertesz and J. D. Tovar, J. Am. Chem. Soc., 2022, 144, 4611–4622 CrossRef CAS PubMed.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed.
- S. E. Yoon, Y. Kang, S. Y. Noh, J. Park, S. Y. Lee, J. Park, D. W. Lee, D. R. Whang, T. Kim, G.-H. Kim, H. Seo, B.-G. Kim and J. H. Kim, ACS Appl. Mater. Interfaces, 2019, 12, 1151–1158 CrossRef PubMed.
- K. Susuki and M. N. Rasband, Curr. Opin. Cell Biol., 2008, 20, 616–623 CrossRef CAS PubMed.
- K. S. Mayer, D. J. Adams, N. Eedugurala, M. M. Lockart, P. Mahalingavelar, L. Huang, L. A. Galuska, E. R. King, X. Gu, M. K. Bowman and J. D. Azoulay, Cell Rep. Phys. Sci., 2021, 2, 100467 CrossRef CAS.
- R. Lillis, M. R. Thomas, M. Mohanan and N. Gavvalapalli, Macromolecules, 2021, 54, 3112–3119 CrossRef.
- Y. Morisaki and Y. Chujo, Polym. Chem., 2011, 2, 1249 RSC.
- C. Pan, K. Sugiyasu, Y. Wakayama, A. Sato and M. Takeuchi, Angew. Chem., Int. Ed., 2013, 52, 10775–10779 CrossRef PubMed.
- R. C. Smith, Macromol. Rapid Commun., 2009, 30, 2067–2078 CrossRef PubMed.
- A. Giovannitti, C. B. Nielsen, J. Rivnay, M. Kirkus, D. J. Harkin, A. J. P. White, H. Sirringhaus, G. G. Malliaras and I. McCulloch, Adv. Funct. Mater., 2015, 26, 514–523 CrossRef.
- T. Ikeda and M. Higuchi, Langmuir, 2011, 27, 4184–4189 CrossRef PubMed.
- F. Cacialli, J. S. Wilson, J. J. Michels, C. Daniel, C. Silva, R. H. Friend, N. Severin, P. Samorì, J. P. Rabe, M. J. O’Connell, P. N. Taylor and H. L. Anderson, Nat. Mater., 2002, 1, 160–164 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |