
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinspired hydrogels: polymeric designs towards artificial photosynthesis
Reina
Hagiwara
a,
Ryo
Yoshida
 b and
Kosuke
Okeyoshi
*a
b and
Kosuke
Okeyoshi
*a
aGraduate School of Advanced Science and Technology, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. E-mail: okeyoshi@jaist.ac.jp
bDepartment of Materials Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 1st November 2024
Abstract
Aquatic environments host various living organisms with active molecular systems, such as the enzymes in the thylakoid membrane that realise photosynthesis. Various challenges in achieving artificial photosynthesis, such as water splitting, have been studied using both inorganic and organic molecules. However, several problems persist, including diffusion-limited reactions and multiple redox reactions in the liquid phase. In this Feature Article, we discuss the significant challenges in using polymer networks as active mediators for photoinduced water splitting. In the creation of artificial chloroplasts, polymer networks offer various advantages, such as stable dispersions of multiple types of functional molecules and close molecular arrangements. To incorporate these features, stepwise synthesis and integration can be utilized during the hierarchical construction of polymer networks. The constituent molecules such as ruthenium complex and platinum nanoparticles in the photoinduced electron transfer circuits are closely arranged to smoothly operate forward reactions by polymer networks. The quantum efficiency of photoinduced H2 generation in gel systems is considerably higher than that of conventional solution systems. Additionally, a thermoresponsive poly(N-isopropylacrylamide) (PNIPAAm) network of microgels can be used to integrate catalytic nanoparticles into the inside by using the electrostatic interaction and the mesh size changes. By focusing on the redox changes of copolymerised molecules that induce swelling/shrinking at a constant temperature, active electron transfer can be precisely achieved using the coil–globule transition of the PNIPAAm having viologen. This article highlights the potential of polymer networks to develop strategies for active electron transfer and energy conversion systems similar to those found in living organisms.
 Reina Hagiwara | Reina Hagiwara received her BA in Science and Technology from Kochi University (Japan) in 2022. She then joined JAIST and obtained an MS in materials science in 2024. She is currently pursuing a PhD at JAIST, where she works on soft matter and polymer science in the laboratory of Prof. Kosuke Okeyoshi. |
 Ryo Yoshida | Ryo Yoshida is a professor at The University of Tokyo. He received a PhD degree in 1993 from Waseda University, Japan. He was a research associate at Tokyo Women's Medical University in 1993, a researcher at the National Institute of Materials and Chemical Research in 1994–1997, and an assistant professor at the University of Tsukuba in 1997–2000. In 2001, he moved to The University of Tokyo as an associate professor and became a full professor in 2012. He got The Award of the Society of Polymer Science, Japan (SPSJ) 2019, etc. |
 Kosuke Okeyoshi | Kosuke Okeyoshi is an Associate Professor at JAIST (Japan). He received his PhD in 2010 from The University of Tokyo (Japan). With the support of the Japan Society for the Promotion of Science fellowships DC1, PD, and Research Abroad, he subsequently worked at The University of Tokyo, RIKEN (Japan), and Harvard University (U.S.A.). He started working at JAIST in 2014 with a focus on bioinspired soft materials. He has received several awards, including The Commendation for Science and Technology from the Minister of Education, Culture, Sports, Science, and Technology. |
Introduction
The phase transitions and functions of polymer gels have been studied extensively.1,2 Hydrogel is defined as a three-dimensional polymer network having crosslinking points, which absorbs water and swells. As they act as open systems capable of transforming energy or substances, many types of bioinspired gels have been developed.3 In addition to simple polymer gels4–9 that respond to various stimuli (temperature, pH, light, solvent composition, ionic strength, electric field, etc.), information-converting polymer gels, such as chemo-mechanical gels10 and molecular-recognition gels,11 have been fabricated. These gels convert chemical substrates from their surroundings into mechanical energy, resulting in a change in volume. Because of their “soft and wet” properties, which are similar to those of living organisms, polymer networks are expected to have applications in biomaterials and regenerative medicine.12–21 Recent gel science provides advanced technologies such as a variety of crosslinking methods and molecular arrangements, allowing for the design of tough hydrogels or self-healing hydrogels. By using the structural and rheological advantages of the polymer networks, we can expect artificial cartilage, etc. as like materials working in living systems. The network also provides nanometer-scale compartmentalized spaces, if the multiple kinds of molecules are immobilized inside.Focusing on high-order functions in living organisms, the metabolism of energy and substances is broadly classified into two categories: catabolism and anabolism. Catabolism includes processes such as cellular respiration, where ATP is used to generate mechanical energy. Anabolism is exemplified by photosynthesis in plants, which uses carbon dioxide to produce glucose. Although anabolism-inspired gels have been designed, catabolism-inspired gels have not yet been developed. Using recent advances in gel science and technology, it is possible to design polymer networks that mediate electron transfer, similar to the multiple redox reactions occurring on the thylakoid membranes of chloroplasts.
Photosynthesis in chloroplasts is primarily achieved by photosystems PS I and PS II, as shown in Fig. 1A. PS I sensitises and fixes carbon dioxide through electron transfer using the reduced species nicotinamide adenine dinucleotide phosphate. PS II mainly sensitises and generates oxygen using four electrons through chlorophyll and manganese clusters. The total photosynthetic reaction can be expressed as follows:
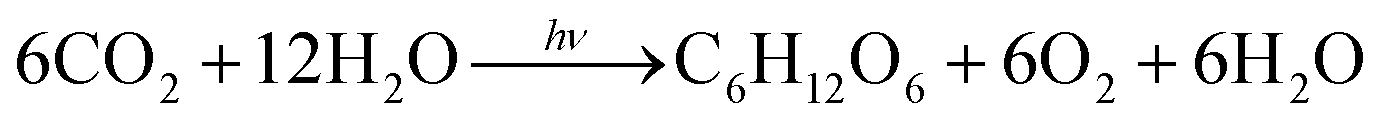 | (1) |
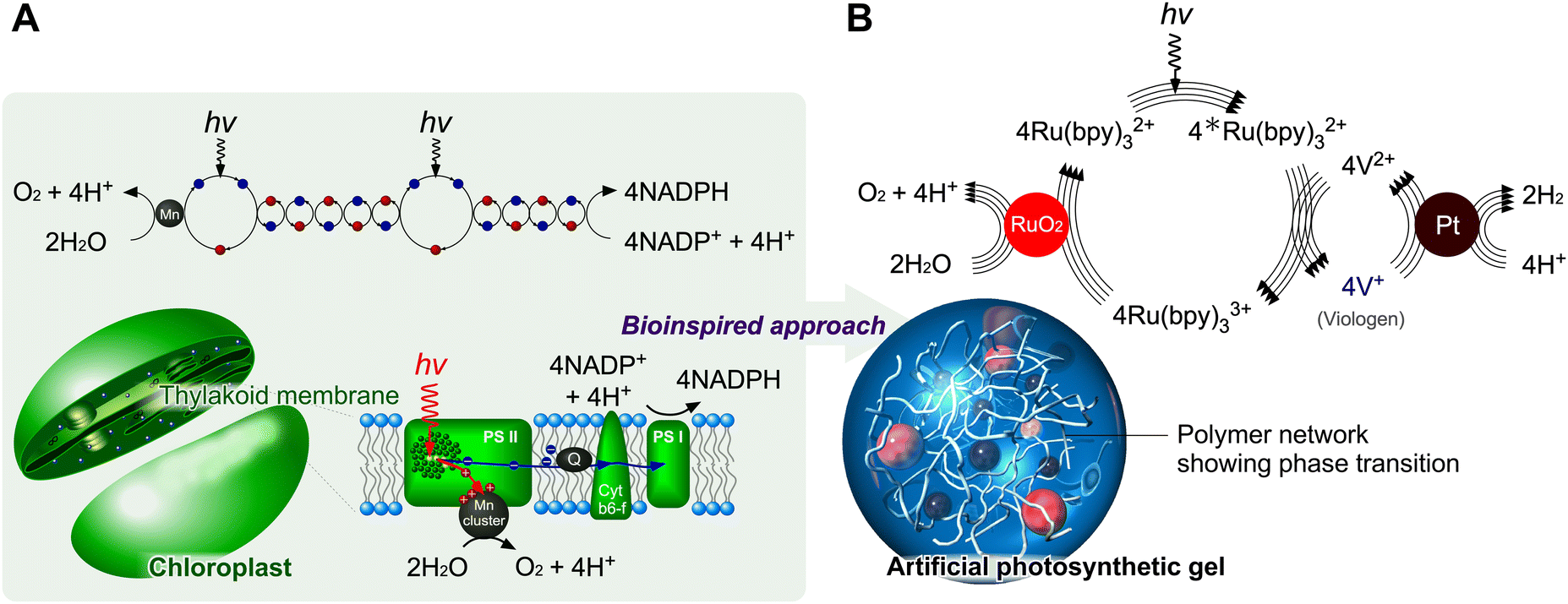 | ||
| Fig. 1 Design of photoinduced electron transfers in polymer networks inspired by chloroplasts. (A) Mechanism of photosynthesis. (B) Design of artificial photosynthetic gels. | ||
Here, the membrane does not participate in the photoreaction but provides reaction fields. If proteins such as photosystems were randomly mixed in the liquid phase without the lipid molecules forming the membrane, it would be difficult to achieve the photosynthesis reaction shown in eqn (1). Theoretically, the maximum energy efficiency for CO2 fixation through the Calvin cycle is 34%. However, under normal growth conditions, the actual performance of plants is significantly lower, reaching only up to 1% of this theoretical value, even in high-performing plants such as sugarcane.22 This is because biochemical reactions in plants and environmental conditions limit photosynthetic performance.
Inspired by photosynthesis, various inorganic and organic molecules have been used for photocatalytic water splitting. The total reaction for water splitting, with the simultaneous transfer of four electrons, is expressed as follows:
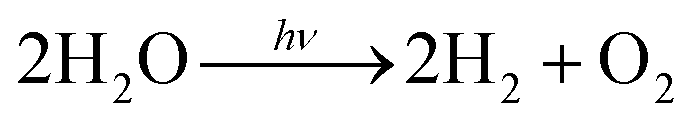 | (2) |
This Feature Article discusses recent strategies that use polymer networks for achieving artificial photosynthesis. Utilising a polymer network as a mediator for photoinduced water splitting addresses key challenges in solution systems, such as the inhibition of self-aggregation. Polymer networks containing solvents also provide nanometre-scale compartmentalised spaces if multiple types of molecules are immobilised (Fig. 1B). The immobilisation/mobilisation of each molecule could be easily designed by the copolymerization and each catalytic nanoparticle could be easily caught in the polymer network. This spatial situation allows close molecular arrangements to accelerate forward reactions. Furthermore, by using a thermoresponsive polymer, poly(N-isopropylacrylamide) (PNIPAAm), it is possible to control swollen/shrunken states and mesh sizes in the polymer gels in the normal temperature range. PNIPAAm has a lower critical solution temperature around 30 °C and the copolymers having a monomer showing two states such as red/ox and cis/trans, obtain hydrophilic/hydrophobic states at constant temperature. The polymer conformational changes allow for controlling the macromolecular distance in the network and for the theoretical design of electron transfers based on Marcus theory.51,52 PNIPAAm is one of the examples for the design of a stimuli-responsive polymer network. The advantage of PNIPAAm, a secondary amide, is the stable chemical structure even during the photoinduced reactions, in contrast to primary amides such as acrylamide. We could expect drastic conformational changes reversibly for the electron transfer in the circuit. Besides, the synthesis of acrylamide type is easy to design on the side chain with keeping the stimuli-responsive behaviors originated from the main PNIPAAm chain. Meanwhile, when the need for optimization of the transition temperature arises, other thermoresponsive polymers would be useful.
Gel systems for photoinduced O2 generation
Design of polymer networks
Electron-transfer systems for visible-light-induced O2 generation have been designed using several strategies.24,53–66 An electron transfer circuit is generally composed of an O2-generating catalyst, a sensitizer, and an oxidant. Molecular arrangement is a key strategy for achieving complete artificial photosynthesis and driving multiple forward reactions. In this study, O2-generating gel systems containing RuO2 as the catalyst and Ru(bpy)32+ as the sensitiser were constructed (Fig. 2).67 An important aspect of this photoexcited reaction is the simultaneous transfer of four electrons to generate an O2 molecule.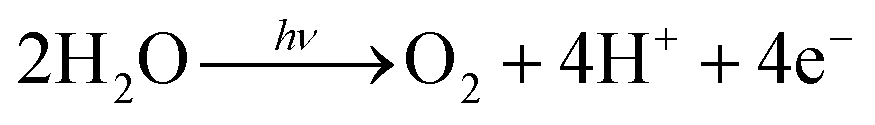 | (3) |
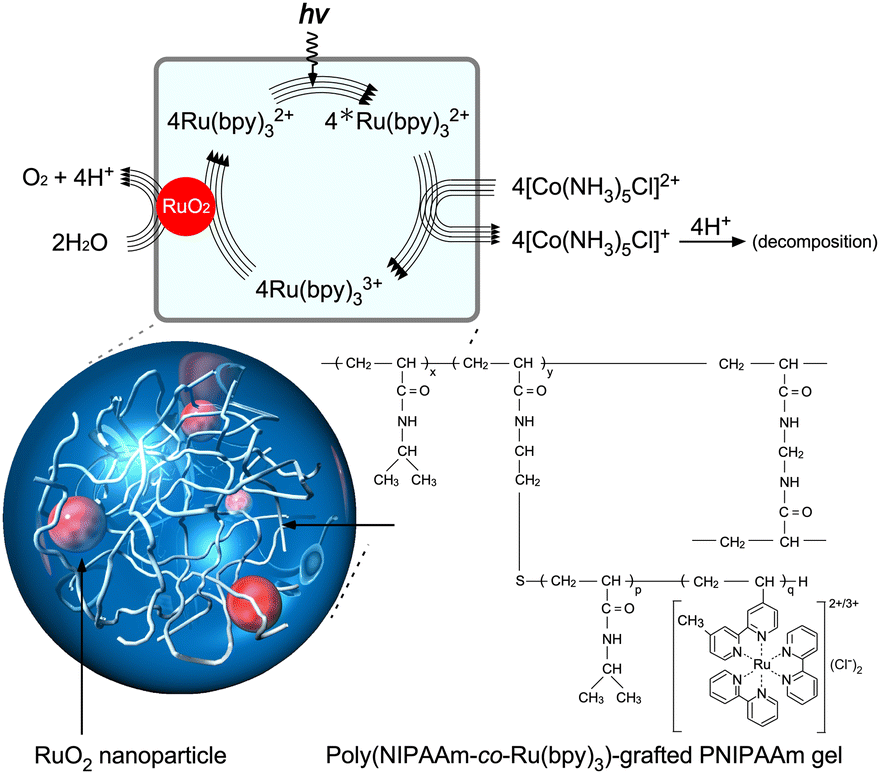 | ||
| Fig. 2 Photoinduced O2-generating system using a poly(NIPAAm-co-Ru(bpy)3)-grafted PNIPAAm gel containing RuO2 nanoparticles (NPs). Reproduced from ref. 67 with permission from Wiley-VCH, Copyright 2010. | ||
Therefore, the interactions between RuO2 and Ru(bpy)33+ are smooth. To arrange molecules closely, the nanometre-scale structures of the polymer network are crucial. The networks can be constructed in a stepwise manner to inhibit self-aggregation, which reduces forward electron transfer. In addition, polymer networks provide stable dispersions of nanoparticles and facilitate close molecular arrangements among heterofunctional groups through electrostatic interactions.
When this system was prepared without polymer networks, an increase in Ru(bpy)32+ concentration led to the aggregation of SDS-RuO2 nanoparticles (NPs) at high salt concentrations. Polymer networks are useful for preventing aggregation and providing a stable, close molecular arrangement. As shown in Fig. 3A, poly(NIPAAm-co-Ru(bpy)3) (PNR) with a double bond at the terminal end group was prepared as a macromonomer. This double bond is useful for further polymerisation. As shown in Fig. 3B, during the mixing of the NPs and polymer in water, the cationic Ru(bpy)32+ units immobilised in the polymer approach the anionic surface of the NP via electrostatic interactions, and the polymer spontaneously surrounds the NPs. The mixture exhibits a stable dispersion because of the steric effect of the polymer. Subsequently, by polymerising NIPAAm as the main monomer, N,N′-methylenebisacrylamide (BIS) as a crosslinker, a PNR-grafted PNIPAAm gel containing RuO2 NPs can be prepared. This stepwise arrangement produces different units: RuO2 NP and Ru(bpy)32+ coexist closely via electrostatic interactions and also exist separately because of physical restrictions. In fact, by irradiating visible light to the gel system, O2 gas is generated continuously. This indicates that electrons from the water molecules were simultaneously transferred to RuO2 and Ru(bpy)33+ in the polymer network for the forward reaction, which is O2 generation.
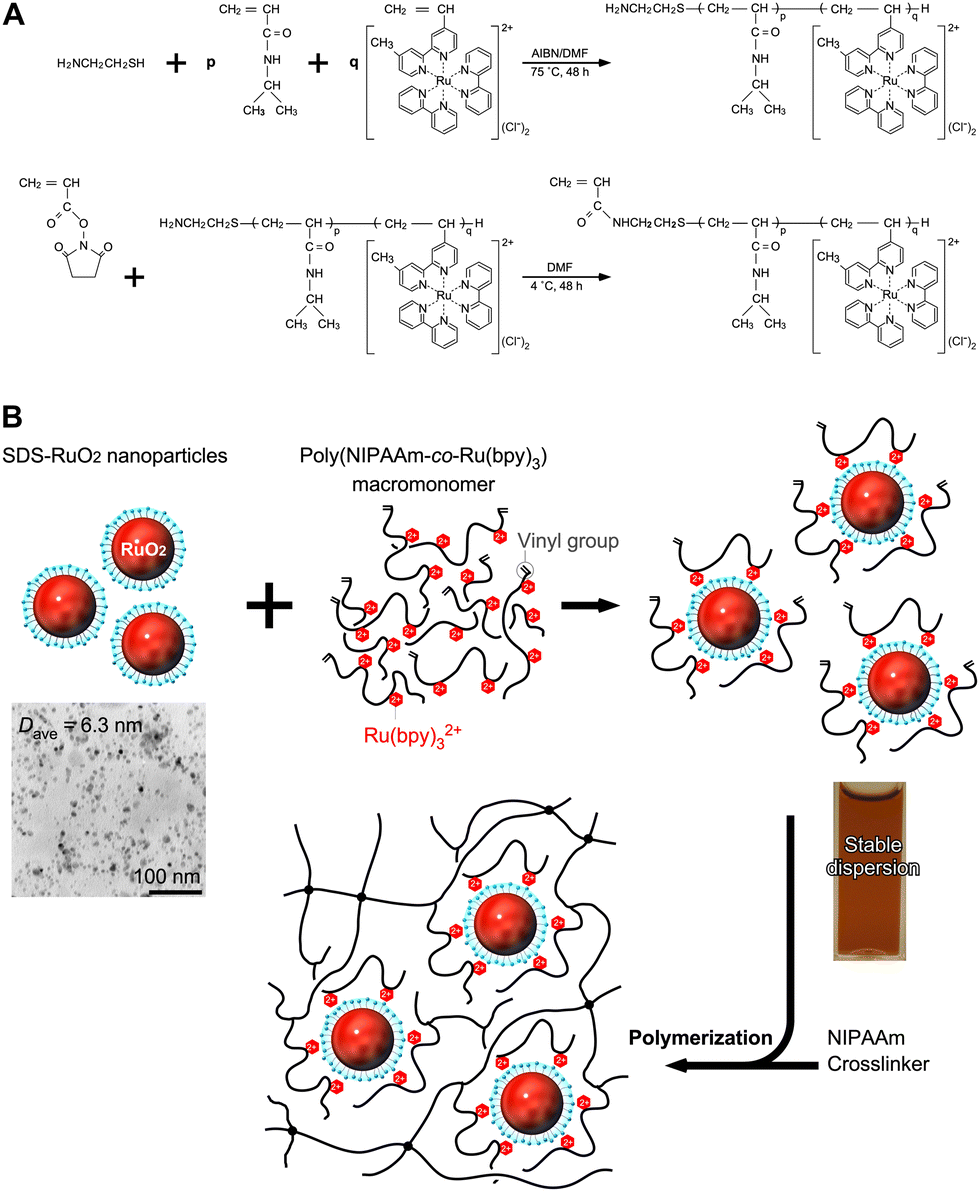 | ||
| Fig. 3 (A) Synthetic procedure for the poly(NIPAAm-co-Ru(bpy)3) (PNR) macromonomer using telomerization and amide condensation. (B) Synthetic procedure for the PNR-grafted PNIPAAm gel containing RuO2 NPs by radical copolymerization using a crosslinker. Reproduced from ref. 67 with permission from Wiley-VCH, Copyright 2010. | ||
Gel systems for photoinduced H2 generation
Electron pathways using a polymer network
Many researchers have attempted to construct photoinduced H2-generating systems using the sacrificial reducer ethylenediaminetetraacetic acid (EDTA). When visible light and water are supplied to this system, the reactions can be evaluated by eliminating the diffusion-limited process of O2 generation. The molecules necessary for H2 generation can be immobilised on the polymer network. To achieve efficient H2 generation, this network provides a reaction field for a two-electron transfer circuit (Fig. 4A).68,69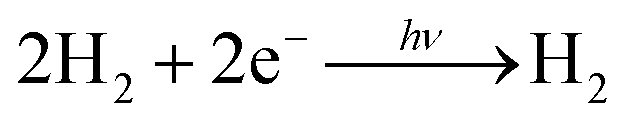 | (4) |
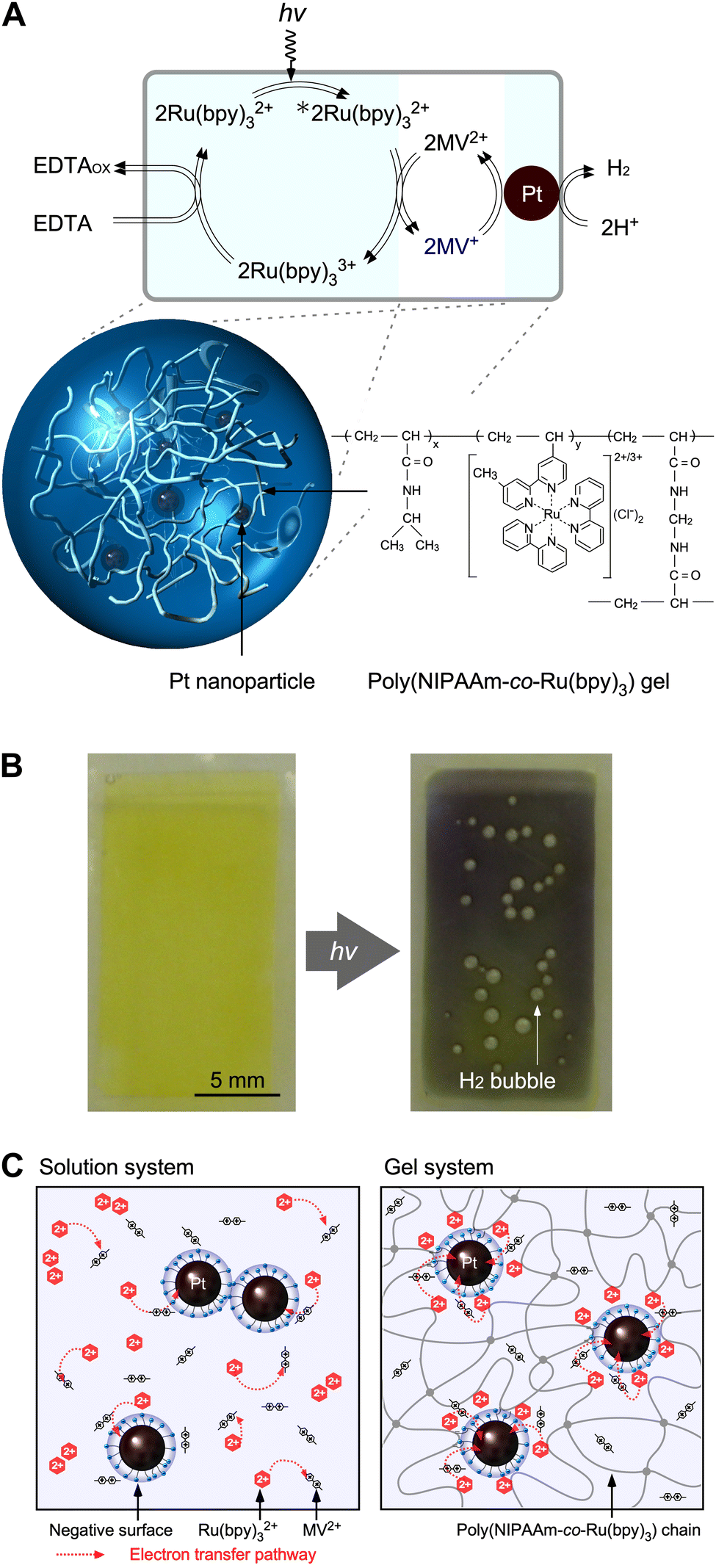 | ||
| Fig. 4 (A) Photoinduced H2-generating system using PNR gel containing Pt NPs. (B) Color change and H2 bubble generation in the gel system under visible-light irradiation. (C) Schematic illustration of electron transfer in conventional solution and gel systems. Reproduced from ref. 68 and 69 with permission from the Royal Society of Chemistry, Copyright 2009. | ||
Under visible light irradiation, electron transfer happens in the gel during generating hydrogen. In gel systems, the necessary components are not dispersed, as in conventional solution systems, where colloidal nanoparticles become unstable because of the high salt concentration and side reactions easily occur. Catalytic Pt nanoparticles (Pt NPs) are trapped in the polymer network, and the Ru(bpy)32+ photosensitizer can be copolymerised.
Fig. 4B shows the photoinduced H2 generation from a PNR gel containing Pt NPs in an aqueous solution of EDTA and methylviologen (MV2+) as an electron acceptor. Before irradiation, the gel suspension was yellow due to the presence of Ru(bpy)32+. After irradiation, the colour of the gel changed from yellow to blue, indicating the generation of the reduced state of MV (MV+), and H2 bubbles were clearly observed. A comparison between the conventional solution system and gel system will help understand the role of the crosslinked polymer network (Fig. 4C). At higher Ru(bpy)32+ concentrations, the H2 generation rate in the solution system initially peaked and then decreased significantly. This was due to the concentration quenching of Ru(bpy)32+ and back reactions.
Conversely, the H2-generation rates in the gel systems increased with increasing Ru(bpy)32+ concentrations. In the polymer network, a higher number of copolymerised Ru(bpy)32+ species localize around the Pt NPs without self-aggregation. As a result of this stability, the generated MV+ species provide electrons to the Pt NPs for H2 generation. In addition, because of the electrostatic interactions between the cationic molecules and anionic surface of the Pt NPs dispersed by the surfactant, electron pathways are constructed at multiple points, and the circuit operates smoothly. Therefore, hydrogen generation in gel systems is significantly enhanced compared to conventional solution systems.
Effect of polymers on quantum efficiency
The microenvironments of several polymers for Ru(bpy)32+ molecules were clarified by comparing their fluorescence properties in the following radiative process: *Ru(bpy)32+ → Ru(bpy)32+ + hν′.70,71 The fluorescence intensities of two types of non-crosslinked polymers, namely poly(acrylamide-co-Ru(bpy)3) and PNR, were approximately two times larger than that of the Ru(bpy)32+ solution (Fig. 5A and B). While Ru(bpy)32+ in solution easily loses the received energy owing to quenching by water molecules,33 this quenching is suppressed in polymer solutions. This is because the Ru(bpy)32+ molecules copolymerise, which induces steric hindrance between them. Moreover, the intensities of the gel suspensions were slightly higher than those of the polymer solution, suggesting that the cross-linked polymer network limits the moving radius of the Ru(bpy)32+ molecules and suppresses self-quenching. The effect of copolymerisation was also confirmed by blue-shifted fluorescence due to rigidochromism.72–75 The rigidity from the isopropyl group creates a more hydrophobic microenvironment around Ru(bpy)32+, which suppresses nonradiative processes such as intermolecular rotation. Because of reduced energy loss, more excited electrons are transmitted by the forward reactions. Thus, by immobilising a photosensitizer in a polymer chain with a hydrophobic group, the polymer exhibits an efficient radiative process.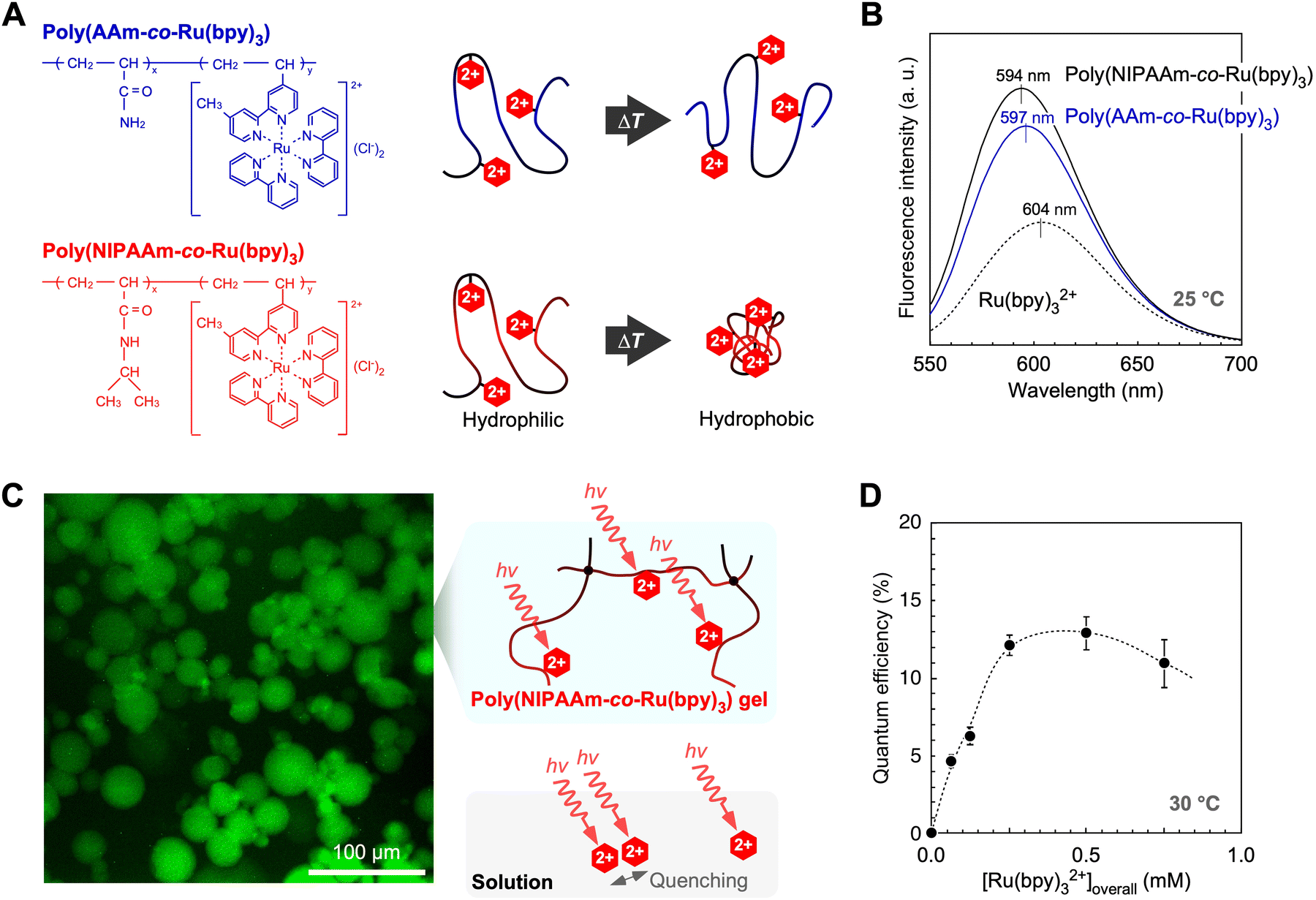 | ||
| Fig. 5 (A) Chemical structures of PNR and poly(AAm-co-Ru(bpy)3) and schematics of their linear chains. (B) Effect of copolymerization on fluorescence properties. (C) Fluorescence microscopy image of PNR gel particles containing Pt NPs and a schematic of the polymer network. (D) Overall quantum efficiency of PNR gel systems for photoinduced H2 generation. Reproduced from ref. 70 and 71 with permission from the Royal Society of Chemistry (Copyright 2011) and the American Chemical Society (Copyright 2016). | ||
By using a PNR network with crosslinking points, the overall quantum efficiency for photoinduced H2 generation was higher than that of conventional solution systems. The efficiency for the H2-generation, Φoverall, was calculated using the following equation:
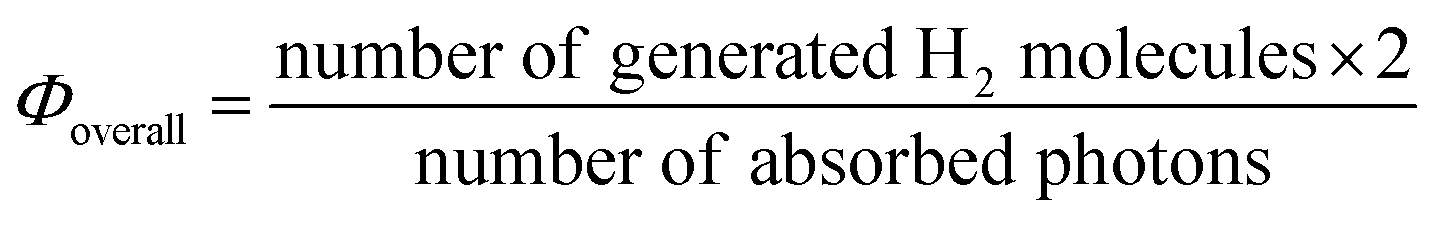 | (5) |
As the amount of gel increased, the quantum efficiency increased to more than 10% and attained its maximum value at approximately 13% (Fig. 5D). The quantum efficiency of the solution system was ∼3.5% at most. A drastic increase in efficiency was achieved by immobilising Ru(bpy)32+ and Pt NPs in the polymer network. This increase can be attributed to the network design for photoexcitation and electron transfer in the close molecular arrangement between Ru(bpy)32+ and the Pt NPs.
Temperature control of the polymer network
The effect of thermoresponsive polymer networks on photochemical reactions was demonstrated using a gel system composed of a PNR gel containing Pt NPs. Similar to conventional PNIPAAm gels, drastic changes in volume and transmittance were observed at the phase transition near 30 °C (Fig. 6A). The volume phase transition considerably affects H2 generation during light irradiation (Fig. 6B). Below the volume phase transition temperature (VPTT), the H2 generation rate, rH2, increased as the temperature increased, according to the Arrhenius equation.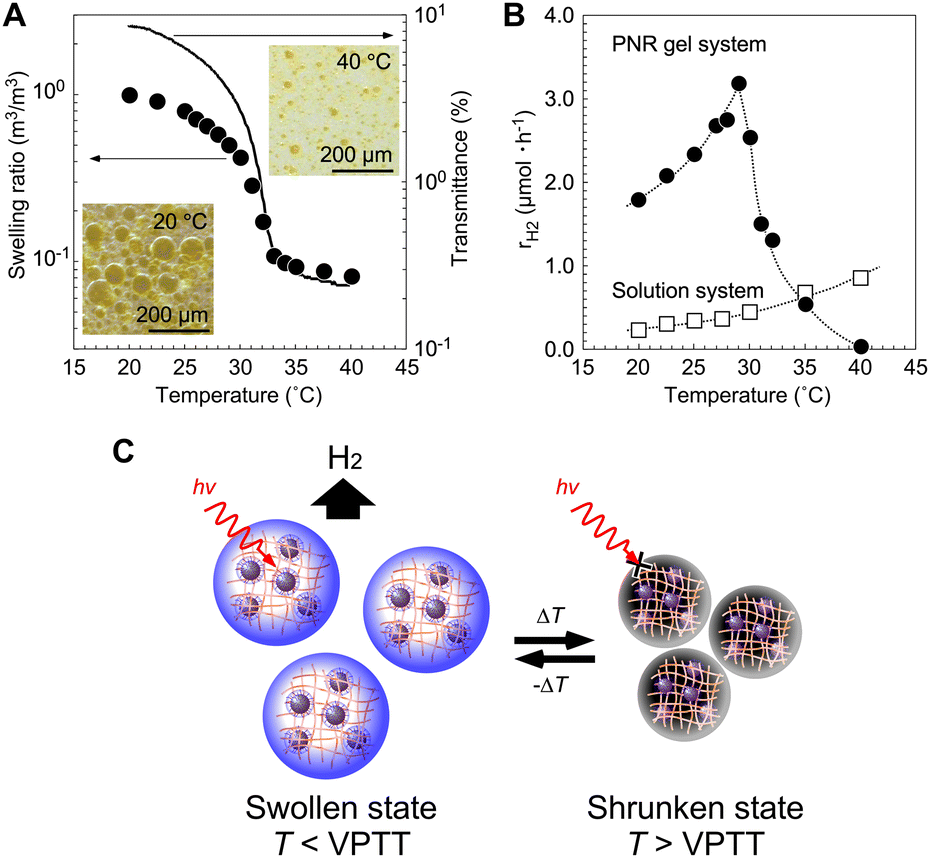 | ||
| Fig. 6 (A) Equilibrium swelling ratio of the PNR gel containing Pt NPs in a solution of EDTA and MV, and the transmittance of the gel-particle suspension. (B) Temperature dependence of H2-generating rates in the PNR gel and solution systems. (C) Temperature control of the H2-generating gel system using a thermoresponsive polymer network for ON–OFF switching. Reproduced from ref. 69 with permission from the Royal Society of Chemistry, Copyright 2009. | ||
Considering this, the decreases in the swelling ratio and the distances between the molecules in the network appeared to be larger than the decrease in transmittance. Above the VPTT, rH2 decreased significantly because of the drastic decrease in light transmission due to gel shrinkage, solution discharge from the gel, and suppressed electron transfer. Thus, an optimal temperature is necessary for photoinduced H2 generation in gel systems. Using a thermoresponsive polymer network, the ON–OFF control of photoinduced H2 generation is possible. This is caused by the reversibility of H2 generation in the network under stepwise temperature changes above and below the VPTT.
Nano-integration
Based on photochemical reactions in polymer networks using microgels prepared by precipitation polymerization, H2-nanogenerators have been proposed (Fig. 7A). Microgels with diameters smaller than the wavelength of visible light can also be useful in designing photonic crystals as optical waveguides. Photonic crystals composed of microgels can enhance photon absorption owing to the light-confinement effect. As the first step, a photoinduced H2-nanogenerator was designed by integrating a photosensitizer and a catalytic nanoparticle with a close molecular arrangement in the network.76,77 Electrostatic interactions among the molecules and shrinking of the thermoresponsive PNIPAAm network were utilised. Positively charged PNR microgels and negatively charged Pt NPs protected with anionic surfactants were prepared. By mixing them in the swollen state of the gel, the Pt NPs in the surrounding solution can be introduced into the interior via electrostatic interactions. When the temperature was increased while avoiding collapse, the network shrank to physically immobilise the Pt NPs in the interior network. The TEM images in Fig. 7B show the Pt NP-immobilised-PNR microgels, with Pt NPs clearly visible and having diameters of 2–3 nm within the network. This result clearly indicates that the two hetero-nanomaterials were successfully integrated. The Pt NPs remained in the gel, even as the gel network shrank with increasing ionic strength. By controlling the crosslinker contents (Fig. 7C), the optimum cross-linking density for the immobilisation of NPs and H2 generation was clarified. Considering the diameter of the Pt NPs (1–2 nm), the network mesh size should be significantly larger than that in the swollen state.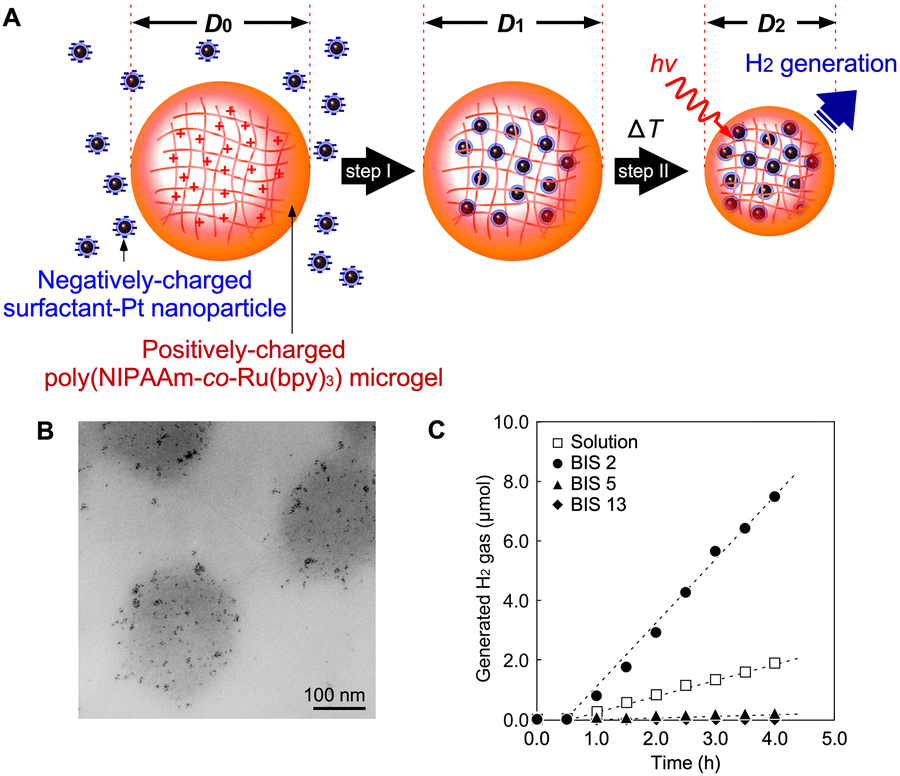 | ||
| Fig. 7 (A) Nano-integration of Pt NPs and PNR microgels. The positively charged PNR network absorbs the negatively charged Pt NPs (step I), which are immobilized in the thermoresponsive network by shrinking (step II). (B) TEM images of PNR microgel containing Pt NPs. (C) H2-generation at 25 °C under light irradiation. Numbers placed after BIS in the graph indicate initial concentrations of the crosslinker during polymerization. Reproduced from ref. 76 and 77 with permissions from Wiley-VCH (Copyright 2011) and the American Chemical Society (Copyright 2012). | ||
Electron transfer in the polymer network
To prepare an all-in-one gel, a viologen unit was introduced into the network of a poly(NIPAAm-co-Ru(bpy)3-co-viologen) gel containing Pt NPs (Fig. 8A). Under visible-light irradiation, the colour of the gel changed from orange to blue because of the reduced state of the viologen (Fig. 8B), suggesting that electrons were transferred from Ru(bpy)32+ to the viologen in the polymer network. Simultaneously, H2 gas was continuously generated (Fig. 8C), suggesting that electrons were transferred effectively among the three components in the gel. However, the H2 generation rate in this system was a thousand of times lower than that of PNR gel systems using free MV (see Fig. 4). This is mainly because Ru(bpy)32+ and the viologen are copolymerised randomly in PNIPAAm-based chains, and most viologens are placed far from the Pt NPs. Therefore, they cannot approach or transfer electrons to Pt. Hence, Ru(bpy)32+ and viologens should be independently immobilised in the polymer network using other molecular strategies. Recent molecular technologies for viologens have expanded their potential using organic architectures, such as covalent organic networks, for applications including water splitting, piezochromic materials, and hydrochromic materials.78–84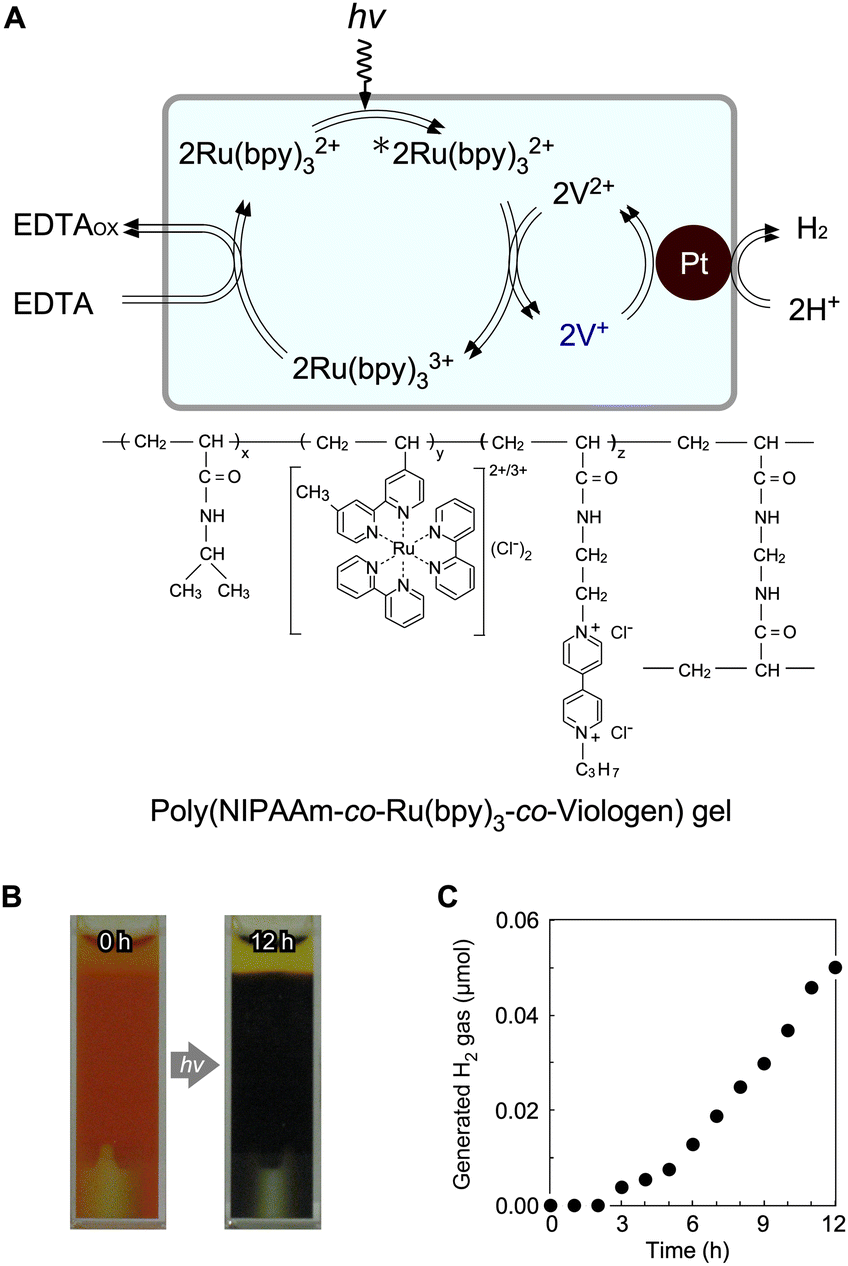 | ||
| Fig. 8 (A) Photoinduced H2-generating system composed of a poly(NIPAAm-co-Ru(bpy)3-co-viologen) gel containing Pt NPs. (B) Images of the gel suspension before and after light irradiation. (C) H2-generation in the gel system. Reproduced from ref. 68 with permission from the Royal Society of Chemistry, Copyright 2009. | ||
Utilization of polymeric coil–globule transitions
To solve these problems for effective distance between viologen and Pt NP, a polymeric system utilising poly(NIPAAm-co-viologen) (PNV) was designed (Fig. 9A).85 Here, the coil–globule transitions of the polymer with reversible hydrophilic/hydrophobic changes accelerated the cyclic electron transfers for H2 generation. Unlike conventional solution systems without polymers, PNV systems have the following advantages: (i) conformational changes in the PNV molecule are caused by redox changes (Fig. 9B); (ii) multiple types of molecules are closely arranged by electrostatic interactions, including positively charged PNV and negatively charged Pt NPs, which are dispersed by an anionic surfactant; (iii) Pt NPs are stably dispersed, even at high salt concentrations, by the surrounding polymer chains. In this PNV system, when the excited photoexcited sensitizer, *Ru(bpy)32+, donates an electron to the viologen in PNV, the reduced viologen (V+) causes shrinkage to approach a hydrophobic space on the surface of the Pt NPs. Subsequently, H2 is efficiently generated using the accumulated electrons on the Pt NPs and nearby protons (Fig. 9C). After providing electrons to the Pt NPs, PNV expands again, and the circuit works continuously.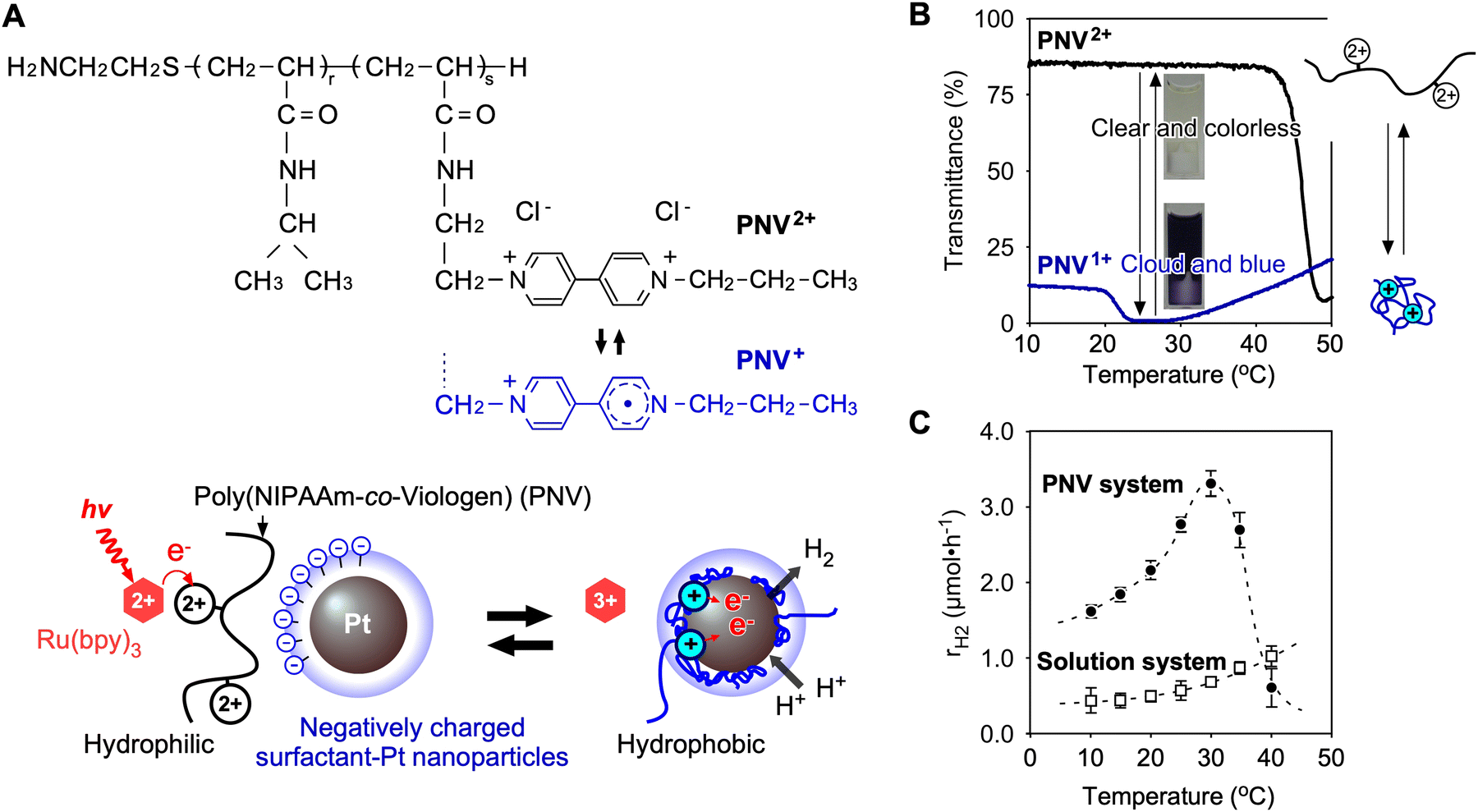 | ||
| Fig. 9 (A) Chemical structure and redox states of poly(NIPAAm-co-viologen) (PNV) and mechanism of electron transfer driven by the coil–globule transitions of PNV while approaching the Pt NPs. (B) Temperature dependence of the transmittance of PNV2+/+ solutions. (C) Temperature dependence of H2 generation rates in the PNV system and solution system. Reproduced from ref. 85 with permission from Wiley-VCH, Copyright 2019. | ||
Precise design for active electron transfer
An advantage of the PNV system is its capacity to enhance cyclic electron transfer through polymeric coil–globule transitions. However, this system also has certain disadvantages. When the viologen in a free PNV molecule is in a reduced state and far from the Pt NPs, it shrinks and readily flocculates with neighbouring PNV molecules. Therefore, the electrons in PNV are difficult to transfer during forward reactions. To overcome this limitation, a polymeric system with a precise molecular arrangement is required to remove free PNV. One strategy involves the conjugation of the polymer onto the surface of the Pt NPs. This allows for active electron transfer and the development of a novel catalytic system. As shown in Fig. 10A and B, a copolymer-conjugated nanocatalytic system for active electron transfer using a ternary random copolymer, poly(NIPAAm-co-acrylamide-co-viologen) (PNAV), was designed.86 Using telomerization, the composition of the monomers and length of the polymer chain are controllable to place each component at an effective distance. PNAV2+/+ exhibited different critical transition temperatures, and the coil–globule transitions during redox changes at constant temperature were useful (Fig. 10C). In the second step, PNAV was conjugated to the surface of the Pt NPs using copolymerised acrylamide (AAm). Thus, the PNAV-conjugated Pt NPs were designed for active electron transport and H2 generation. When the copolymerised viologen undergoes redox changes, the polymer exhibits coil–globule transitions, cyclically extending and shrinking as it absorbs photoenergy and generates H2 (Fig. 10D). Compared to the PNV system, the PNAV system was precisely organized on a single-nanometre scale for electron transfer. According to Marcus theory, electron transfer occurs when the distance between an electron donor and acceptor is less than ∼2 nm.51,52 In the PNAV system, when a photoexcited sensitiser, such as *Ru(bpy)32+, provides an electron to the viologen, the reduced viologen triggers immediate shrinkage of the polymer, causing it to approach the surface of the Pt NPs for H2 generation. Subsequently, the polymer expands again, enabling the electronic transfer circuit to continue. During viologen redox cycles, the polymer chain acts as a distance regulator and an electron transporter. By controlling the compositions in the polymer, the search for the quantum efficiency would be expected as like that of the PNV system showing the optimum temperature range for H2 generation.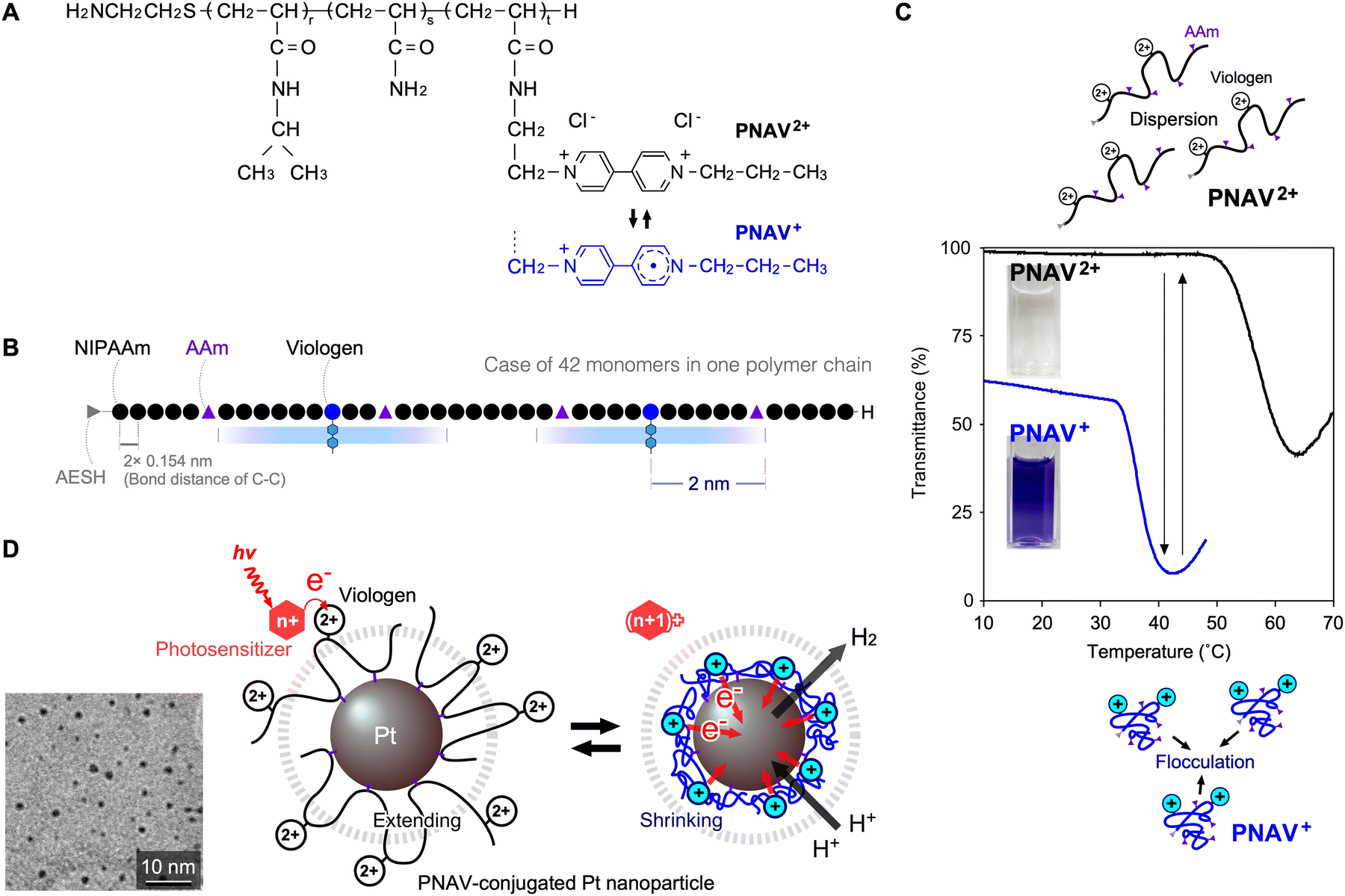 | ||
| Fig. 10 (A) Chemical structure and redox states of poly(NIPAAm-co-AAm-co-viologen) (PNAV). (B) Possible schematic of PNAV, estimated by experimental properties. (C) Phase transition of PNAV solutions in redox states contingent on temperature. (D) Schematics of the photoinduced H2 generation system through electron transfer driven by coil–globule transitions of PNAV-conjugated on Pt NP. The dot circle means the effective distance from the Pt NP surface. Reproduced from ref. 86 with permission from Copyright 2024, Royal Society of Chemistry. | ||
Conclusions and outlook
Bioinspired materials with energy conversion systems are crucial for constructing a sustainable society. Artificial photosynthetic hydrogels have been designed and partially constructed by arranging multiple functional molecules in polymer networks (Fig. 11). This article outlines design strategies for molecular arrangements involving photoinduced electron transfer, with a focus on O2 and H2 generation. To achieve photoinduced water splitting, it is necessary to hierarchically construct multiple functional molecules such as photosensitizers and catalytic nanoparticles. Polymer networks mediate reaction fields, similar to the thylakoid membrane. The next challenge would be integrating the active electron transfers that happen not only on the path such as viologen and Pt NPs, but also among multiple paths including photosensitizer and O2-generating catalysts in the compartmentalised nanospace. Because the gel systems featured in this article were prepared by versatile functional molecules and versatile polymerization methods, it would be of huge potential to develop the networks using supramolecules or special synthesis methods. Considering the recent advances in molecular and polymer technologies, a more evolved network is expected to be designed. It is possible to immobilise photosensitizers onto polymer networks not only as monomers or macromonomers67,87 but also as crosslinkers.88 Furthermore, immobilisation on natural supramolecules, such as tubulin/microtubules, is possible.71,89,90 These molecular techniques will help achieve ideal molecular systems for artificial photosynthesis. This initiative may facilitate the development of a hydrogen energy-based society that uses sunlight and water as sustainable energy sources. | ||
| Fig. 11 Illustration of artificial photosynthetic gels that generate H2 and O2 in an environment containing sunlight and water. | ||
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a grant-in-Aid for Challenging Research (Exploratory) (Grant No. JP21K18998) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT).Notes and references
- P. J. Flory, Principle of Polymer Chemistry, Cornell University Press, Ithaca, New York, 1953 Search PubMed.
- T. Tanaka, Phys. Rev. Lett., 1978, 40, 820 CrossRef CAS.
- T. Tanaka, From Gels to Life, University of Tokyo Press, Tokyo, Japan, 2004 Search PubMed.
- T. Tanaka, D. Fillmore, S. T. Sun, I. Nishio, G. Swislow and A. Shah, Phys. Rev. Lett., 1980, 45, 1636 CrossRef CAS.
- I. Ohmine and T. Tanaka, J. Chem. Phys., 1982, 11, 5725 CrossRef.
- T. Tanaka, I. Nishio, S. T. Sun and S. Ueno-Nishio, Science, 1982, 218, 467–469 CrossRef CAS PubMed.
- Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1984, 81, 6379 CrossRef.
- S. Katayama, Y. Hirokawa and T. Tanaka, Macromolecules, 1984, 17, 2641–2643 CrossRef CAS.
- A. Suzuki and T. Tanaka, Nature, 1990, 346, 345–347 CrossRef CAS.
- E. Kokufuta, Y.-Q. Zhang and T. Tanaka, Nature, 1991, 351, 302–304 CrossRef.
- E. Kokufuta and T. Tanaka, Macromolecules, 1991, 24, 1605–1607 CrossRef CAS.
- Y. Osada and J. P. Gong, Adv. Mater., 1998, 10, 827–837 CrossRef CAS.
- T. Shimizu, M. Yamato, Y. Isoi, T. Akutsu, T. Setomaru, K. Abe, A. Kikuchi, M. Umezu and T. Okano, Circ. Res., 2002, 90, e40–e48 CrossRef CAS PubMed.
- R. Yoshida, Adv. Mater., 2010, 22, 3463–3483 CrossRef CAS PubMed.
- A. Patwa, A. Thiéry, F. Lombard, M. K. S. Lilley, C. Boisset, J.-F. Bramard, J.-Y. Bottero and P. Barthélémy, Sci. Rep., 2015, 5, 11387 CrossRef PubMed.
- K. Matsumoto, N. Sakikawa and T. Miyata, Nat. Commun., 2018, 9, 2315 CrossRef PubMed.
- P. Siqueira, É. Siqueira, A. E. De Lima, G. Siqueira, A. D. Pinzón-Garcia, A. P. Lopes, M. E. Cortés Segura, A. Isaac, F. V. Pereira and V. R. Botaro, Nanomaterials, 2019, 9, 78 CrossRef PubMed.
- C. G. Han, X. Qian, Q. Li, B. Deng, Y. Zhu, Z. Han, W. Zhang, W. Wang, S.-P. Feng, G. Chen and W. Liu, Science, 2020, 368, 1091–1098 CrossRef CAS PubMed.
- S. Ishikawa, Y. Iwanaga, T. Uneyama, X. Li, H. Hojo, I. Fujinaga, T. Katashima, T. Saito, Y. Okada, U. Chung, N. Sakumichi and T. Sakai, Nat. Mater., 2023, 22, 1564–1570 CrossRef CAS PubMed.
- C. Tangsombun and D. K. Smith, J. Am. Chem. Soc., 2023, 145, 24061–24070 CrossRef CAS PubMed.
- A. Al Kindi, N. S. Courelli, K. Ogbonna, J. M. Urueña, A. L. Chau and A. A. Pitenis, Langmuir, 2024, 40, 9926–9933 CrossRef CAS PubMed.
- G. S. Singhal, G. Renger, S. K. Sopory, K.-D. Irrgang and G. Govindjee, Concepts in Photobiology, Springer Dordrecht, New Delhi, 1999 Search PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Kalyanasundaram and M. Grätzel, Angew. Chem., Int. Ed. Engl., 1979, 18, 701–702 CrossRef.
- W. E. Ford, J. W. Otvos and M. Calvin, Nature, 1978, 274, 507–508 CrossRef CAS PubMed.
- K. Takuma, T. Sakamoto, T. Nagamura and T. Matsuo, J. Phys. Chem., 1981, 85, 619–621 CrossRef CAS.
- M. Grätzel, Acc. Chem. Res., 1981, 14, 376–384 CrossRef.
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163–170 CrossRef CAS.
- M. R. Wasielewski, Chem. Rev., 1992, 435–461 CrossRef CAS.
- E. Amouyal, Sol. Energy Mater. Sol. Cells, 1995, 38, 249–276 CrossRef CAS.
- M. Sykora, K. A. Maxwell, J. M. DeSimone and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7687–7691 CrossRef CAS PubMed.
- M. Kaneko, Prog. Polym. Sci., 2001, 26, 1101–1137 CrossRef CAS.
- H. Inoue, S. Punyu, Y. Shimada and S. Takagi, Pure Appl. Chem., 2005, 77, 1019–1033 CrossRef CAS.
- K. Yamauchi, S. Masaoka and K. Sakai, J. Am. Chem. Soc., 2009, 131, 8404–8406 CrossRef CAS PubMed.
- B. Happ, A. Winter, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 2222–2255 RSC.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- H. Zhou, P. Li, J. Liu, Z. Chen, L. Liu, D. Dontsova, R. Yan, T. Fan, D. Zhang and J. Ye, Nano Energy, 2016, 25, 128–135 CrossRef CAS.
- H. Lu, W. Chen, S. Li, J. Sun, K. Du, Q. Xia and F. Feng, ACS Appl. Mater. Interfaces, 2017, 9(12), 10355–10359 CrossRef CAS PubMed.
- S. Naya, T. Kume, R. Akashi, M. Fujishima and H. Tada, J. Am. Chem. Soc., 2018, 140, 1251–1254 CrossRef CAS PubMed.
- K. Yamamoto, A. Call and K. Sakai, Chem. – Eur. J., 2018, 24, 16620–16629 CrossRef CAS PubMed.
- X. Ji, J. Wang, L. Mei, W. Tao, A. Barrett, Z. Su, S. Wang, G. Ma, J. Shi and S. Zhang, Adv. Funct. Mater., 2018, 28, 1705083 CrossRef.
- G. F. Luo, Y. Biniuri, W.-H. Chen, E. Neumann, M. Fadeev, H.-B. Marjault, A. Bedi, O. Gidron, R. Nechushtai, D. Stone, T. Happe and I. Willner, Nano Lett., 2019, 19(9), 6621–6628 CrossRef CAS PubMed.
- Y. Li, T. Kong and S. Shen, Small, 2019, 15(32), 1900772 CrossRef PubMed.
- X. Fang, L. Meng, A. Prominski, E. N. Schaumann, M. Seebald and B. Tian, Chem. Soc. Rev., 2020, 49, 7978–8035 RSC.
- Y. Wang, X. Shang, J. Shen, Z. Zhang, D. Wang, J. Lin, J. C. S. Wu, X. Fu, X. Wang and C. Li, Nat. Commun., 2020, 11, 3043 CrossRef CAS PubMed.
- T. Çeper, D. Costabel, D. Kowalczyk, K. Peneva and F. H. Schacher, ACS Appl. Polym. Mater., 2023, 5(8), 6493–6503 CrossRef.
- D. L. Jiang, C. K. Choi, K. Honda, W. S. Li, T. Yuzawa and T. Aida, J. Am. Chem. Soc., 2004, 126, 12084–12089 CrossRef CAS PubMed.
- H. Imahori, H. Yamada, D. M. Guldi, Y. Endo, A. Shimomura, S. Kundu, K. Yamada, T. Okada, Y. Sakata and S. Fukuzumi, Angew. Chem., Int. Ed., 2002, 41, 2344–2347 CrossRef CAS PubMed.
- S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74–77 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CrossRef CAS.
- C. Creutz and N. Sutin, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 2858–2862 CrossRef CAS PubMed.
- K. Kalyanasundaram, O. Mićić, E. Pramauro and M. Grätzel, Helv. Chim. Acta, 1979, 62, 2432–2441 CrossRef CAS.
- J. Kiwi and M. Grätzel, Nature, 1979, 281, 657–658 CrossRef CAS.
- J. Kiwi, J. Chem. Soc., Faraday Trans., 1982, 78, 339–345 RSC.
- M. Yagi, S. Tokita, I. Ogino and M. Kaneko, J. Chem. Soc., Faraday Trans., 1996, 92, 2457–2461 RSC.
- I. Ogino, K. Nagoshi, M. Yagi and M. Kaneko, J. Chem. Soc., Faraday Trans., 1996, 92, 3431–3434 RSC.
- L. Sun, H. Berglund, R. Davydov, T. Norrby, L. Hammarstrolm, P. Korall, A. Börje, C. Philouze, K. Berg, A. Tran, M. Andersson, G. Stenhagen, J. Mårtensson, M. Almgren, S. Styring and B. Åkermark, J. Am. Chem. Soc., 1997, 119, 6996–7004 CrossRef CAS.
- T. Abe, Y. Tamada, H. Shiroishi, M. Nukaga and M. Kaneko, J. Mol. Catal. A: Chem., 1999, 144, 389–395 CrossRef CAS.
- K. C. Pillai, A. S. Kumar and J. M. Zen, J. Mol. Catal. A: Chem., 2000, 160, 277–285 CrossRef CAS.
- M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21–36 CrossRef CAS PubMed.
- G. C. Dismukes, V. V. Klimov, S. V. Baranov, J. DasGupta and A. Tyryshkin, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2170–2175 CrossRef CAS PubMed.
- G. C. Dismukes, Science, 2001, 292, 447–448 CrossRef CAS PubMed.
- M. Yagi, K. V. Wolf, P. J. Baesjou, S. L. Bernasek and G. C. Dismukes, Angew. Chem., Int. Ed., 2001, 113, 3009–3012 CrossRef.
- R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spicca, Angew. Chem., Int. Ed., 2008, 47, 7335–7338 CrossRef CAS PubMed.
- K. Okeyoshi and R. Yoshida, Adv. Funct. Mater., 2010, 20, 708–714 CrossRef CAS.
- K. Okeyoshi and R. Yoshida, Soft Matter, 2009, 5, 4118–4123 RSC.
- K. Okeyoshi and R. Yoshida, Chem. Commun., 2009, 6400–6402 RSC.
- K. Okeyoshi and R. Yoshida, Chem. Commun., 2011, 47, 1527–1529 RSC.
- K. Okeyoshi, R. Kawamura, R. Yoshida and Y. Osada, Langmuir, 2016, 32, 626–631 CrossRef CAS PubMed.
- M. Ogawa, M. Inagaki, N. Kodama, K. Kuroda and C. Kato, J. Phys. Chem., 1993, 97, 3819–3823 CrossRef CAS.
- M. Ogawa and K. Kuroda, Chem. Rev., 1995, 95, 399–438 CrossRef CAS.
- P. J. Giordano and M. S. Wrighton, J. Am. Chem. Soc., 1979, 101, 2888–2897 CrossRef CAS.
- J. McKiernan, J.-C. Pouxviel, B. Dunn and J. I. Zink, J. Phys. Chem., 1989, 93, 2129–2133 CrossRef CAS.
- K. Okeyoshi, D. Suzuki, A. Kishimura and R. Yoshida, Small, 2011, 7, 311–315 CrossRef CAS PubMed.
- K. Okeyoshi, D. Suzuki and R. Yoshida, Langmuir, 2012, 28, 1539–1544 CrossRef CAS PubMed.
- C. Liu, J. Tang, H. M. Chen, B. Liu and P. Yang, Nano Lett., 2013, 13, 2989–2992 CrossRef CAS PubMed.
- Q. Sui, X.-T. Ren, Y.-X. Dai, K. Wang, W.-T. Li, T. Gong, J.-J. Fang, B. Zou, E.-Q. Gao and L. Wang, Chem. Sci., 2017, 8, 2758–2768 RSC.
- B. Wang, H. Tahara and T. Sagara, ACS Appl. Mater. Interfaces, 2018, 10, 36415–36424 CrossRef CAS PubMed.
- G. F. Luo, Y. Biniuri, W.-H. Chen, E. Neumann, M. Fadeev, H.-B. Marjault, A. Bedi, O. Gidron, R. Nechushtai, D. Stone, T. Happe and I. Willner, Nano Lett., 2019, 19, 6621–6628 CrossRef CAS PubMed.
- T. Škorjanc, D. Shetty, M. A. Olson and A. Trabolsi, ACS Appl. Mater. Interfaces, 2019, 11, 6705–6716 CrossRef PubMed.
- Z. Mi, T. Zhou, W. Weng, J. Unruangsri, K. Hu, W. Yang, C. Wang, K. A. I. Zhang and J. Guo, Angew. Chem., Int. Ed., 2021, 60, 9642–9649 CrossRef CAS PubMed.
- Y. Luo, J.-P. Liu, L.-K. Li and S.-Q. Zang, Inorg. Chem., 2023, 62, 14385–14392 CrossRef CAS PubMed.
- K. Okeyoshi and R. Yoshida, Angew. Chem., Int. Ed., 2019, 58, 7304–7307 CrossRef CAS PubMed.
- R. Hagiwara, S. Nishimura and K. Okeyoshi, Chem. Commun., 2024, 60, 280–283 RSC.
- R. Mitsunaga, K. Okeyoshi and R. Yoshida, Chem. Commun., 2013, 49, 4935–4937 RSC.
- M. Aizenberg, K. Okeyoshi and J. Aizenberg, Adv. Funct. Mater., 2018, 28, 170425 CrossRef.
- K. Okeyoshi, R. Kawamura, R. Yoshida and Y. Osada, J. Mater. Chem. B, 2014, 2, 41–45 RSC.
- K. Okeyoshi, R. Kawamura, R. Yoshida and Y. Osada, Chem. Commun., 2015, 51, 11607–11610 RSC.
| This journal is © The Royal Society of Chemistry 2024 |