
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ XPS investigation of the SEI formed on LGPS and LAGP with metallic lithium†
Yi
Liang
 a,
Matthew
Burton
ab,
Ben
Jagger
a,
Hua
Guo
ab,
Johannes
Ihli
a and
Mauro
Pasta
*ab
a,
Matthew
Burton
ab,
Ben
Jagger
a,
Hua
Guo
ab,
Johannes
Ihli
a and
Mauro
Pasta
*ab
aDepartment of Materials, University of Oxford, Parks Road, Oxford OX1 3PH, UK. E-mail: mauro.pasta@materials.ox.ac.uk
bThe Faraday Institution, Quad One, Becquerel Avenue, Harwell Campus, Didcot OX11 0RA, UK
First published on 11th October 2024
Abstract
In situ X-ray photoelectron spectroscopy (XPS) techniques have proven to be powerful tools for the characterisation of the solid electrolyte interphase (SEI) formed between the anode and solid electrolyte (SE) in solid-state batteries. XPS offers access to time and operational condition-resolved information on the SEI's chemical composition in the absence of destructive sample preparation. Here we present a Virtual Electrode Plating XPS (VEP-XPS) investigation of the composition and stability of the SEI formed between lithium metal and two different solid electrolytes: Li10GeP2S12 (LGPS) and Li1.5Al0.5Ge1.5(PO4)3 (LAGP). LAGP shows slower SEI formation kinetics, as proven by the emergence of a metallic lithium signal, while LGPS exhibits rapid SEI growth that prevents metallic lithium from plating. We attribute these observations to the SEI composition, distribution and physical properties of secondary decomposition products and in particular to the mixed ion-electron conductive Li3P which can be observed in LGPS and not in LAGP.
Solid-state batteries (SSBs) employing a solid electrolyte (SE) and a lithium metal anode (LMA) have been regarded as the ‘holy grail’ of energy storage due to expectations of enhanced safety and higher energy density compared to conventional lithium-ion batteries (LIBs).1–3 The compatibility of the LMA with the SE is key to the realisation of stable, high energy density Li metal SSBs.4 The solid electrolyte interphase (SEI) formed between the SE and LMA plays a crucial role in this regard. Formed as a result of interfacial reactions, this nano-composite consisting of crystalline and amorphous reaction products determines battery performance. The ideal SEI exhibits high ionic conductivity and low electronic conductivity to facilitate lithium ion transport while limiting the extent of electrochemical reactions.5–7 Investigation of the SEI, in terms of composition, spatial extent, and reaction pathways, should therefore be a key part of the SSB battery development and optimisation.8–10 While the number of high ionic conductivity SEs investigated has steadily increased over the last two decades,11–14 the characterisation of SEIs has remained challenging on account of their thinness (often a few nanometres) and compositional heterogeneity.6,15
Virtual electrode plating X-ray photoelectron spectroscopy (VEP-XPS) is an emerging technique that allows time- and operational condition-resolved characterisation of the SEI formed in the first charge. This method replicates the Li plating process within the XPS chamber, allowing for detailed characterisation about the SEI formation dynamics. The in situ Li sputtering technique allows the characterisation of the composition of the formed SEI as soon as the LMA is brought into contact with SEs. These techniques could eliminate the need for destructive and potentially invasive sample preparation steps in many cases.16–19 Either via virtual electrode plating or Ar-ion sputtering (Fig. 1a and Fig. S1, ESI† respectively), a thin film of Li is gradually deposited onto the surface of a SE under ultra-high vacuum (UHV) conditions. Iterative XPS measurements during this process enable the formation and evolution of the SEI as the deposited Li reacts with the SE to be investigated.20–23 Using VEP-XPS and in situ Li sputtering techniques, we report on the composition, chemical stability, and electrochemical stability of the SEIs formed between Li metal and two types of SE: Li10GeP2S12 (LGPS) and NASICON-type Li1.5Al0.5Ge1.5(PO4)3 (LAGP). With an ionic conductivity of up to 12 mS cm−1, LGPS has been proposed as an attractive SE, yet has failed to find practical implementation in light of previously reported unstable SEI growth during battery operation. LAGP has also been heavily investigated as a SE due to its high air stability and promising ionic conductivity,24–27 but SEI growth has also been limiting its application.28
 | ||
| Fig. 1 Virtual Electrode Plating X-ray Photoelectron Spectroscopy (VEP-XPS). (a) Schematic of the VEP-XPS experiment illustrating the mechanism of operation, whereby the electron neutraliser provides an electron beam and generates a bias across the sample stack which facilitates Li-ion migration towards the top surface; (b) experiment workflow: XPS measurements and e−-beam plating are periodically alternated; (c) illustration of SEI growth and composition as a function of passed charge; chemical phases are determined from deconvoluted XPS spectra. | ||
The pristine structures of these SEs both contain Ge4+, which is susceptible to reduction to Ge0 and alloying to form LixGe when in contact with the LMA,21,22,25,29 which is an undesirable electronic conductive phase in the SEI. How this affects the stability of the SEI depends on the evolution of the other SEI components, necessitating a study of the whole degradation process.
XPS experiments probing the stability of LAGP at the interface with Li metal revealed reduced reaction kinetics compared to the sulphide LGPS electrolyte. The SEI formed between LGPS and Li metal displays an apparent continuous growth behaviour due to the presence of the electronic conductive and ionic conductive Ge(Li) and Li3P phases.23,30 These experiments highlight the importance of the chemical composition of the SEI, and their electronic and ionic properties. This work also introduces a potential approach for quantifying the SEI formation kinetics, which could improve our understanding of the SEI formation process.
The VEP-XPS technique works by utilizing the electron neutraliser gun. The generated electron beam is directed to the sample surface, which provides a bias across the sample stack. This causes a driving force for the migration of Li+ through to the SE surface, where the Li+ is reduced. This provides a source of Li at the surface to react with the SE, causing the formation, growth, and evolution of an SEI. These SEI formation processes were probed by alternating between XPS spectra acquisition and e−-beam (Li) plating. In view of the relatively recent development of VEP-XPS,22,23 we performed an in situ Li sputtering (see ESI†) XPS experiment to cross-validate observations. This is summarised in Fig. S2 and S3 (ESI†).
Firstly, to confirm that the observed electrolyte decomposition is not caused by the applied electron beam, ‘’VEP-like’’ experiments were performed on both LGPS and LAGP SEs without a Li metal source (the Li foil) on the underside of the SE (Fig. S4, ESI†). During these experiments, no change in the SE composition was detected, confirming that the observed changes in chemical information on the surface are due to the electrochemical reaction between the electrolyte and the plated Li metal.
Fig. 2 provides a comparison of the XPS spectra collected for (a) LGPS and (b) LAGP during the VEP experiment. The evolution of the core spectra for the main elements (from left to right Li, Ge, P and Al/S, respectively) is shown as a function of Li “plating” time (O is shown in Fig. S5, ESI†). For the pristine (labelled as 0 min) LGPS and LAGP, we observe a single Li environment in the Li 1s spectra centred at 55.4 eV and 55.5 eV, respectively. A similar observation is made for the Ge 3d region, with a single peak at approximately 30.5 eV and 32.5 eV, consistent with values reported by He et al. and Yu et al. for the Ge4+ environment.29,31 The difference in Ge binding energy (BE) between LGPS and LAGP is due to the fact that the Ge atom is bonded to six more electronegative oxygen (O) atoms in LAGP, forming a GeO6 octahedron, whereas GeS4 is present in LGPS.32 Correspondingly, the phosphorus atom is found in the PS43− or PO43− structure, as visible in the P 2p spectra. The Al 2p and S 2p spectra show the bonding environments of AlO6 octahedra (in LAGP) and PS43− (in LGPS), respectively.
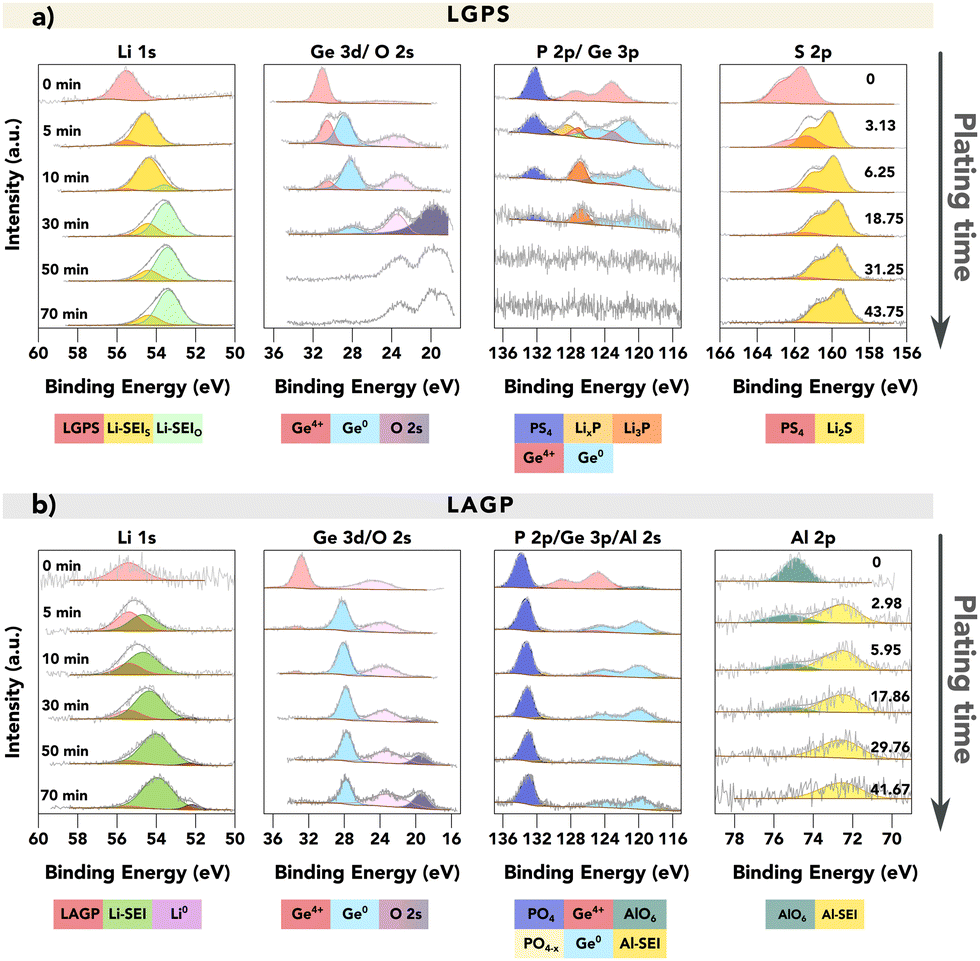 | ||
| Fig. 2 VEP-XPS investigations of LGPS and LAGP (available enlarged as Fig. S6 and non-normalised in Fig. S7, ESI†). The plots show time series of XPS core spectra acquired during VEP-XPS experiments examining the SEI formation on top of (a) LGPS and (b) LAGP. The total Li plating time (e−-beam exposure time) is given in the first column, and the passed charge qA (μA h cm−2), normalised by area, is labelled in the last column. Acquired spectra (grey) are shown along with linear combination fitting results. The individual spectral components are coloured; a key is provided below each group of spectra. For LGPS SE, two Li 1s SEI peaks are labelled in two colours with the subscripts donating the main non-Li contributing element, as their close BE values make it difficult to deconvolute the peaks accurately. The peak intensities were normalised for each acquired core spectrum to improve the visibility of minor spectral contributions; the raw peak intensity variations are provided in the ESI.† The applied beam current was 20 μA for both samples. | ||
After 5 minutes of in situ Li plating, the onset of SEI formation is detectable, with decomposition products visible in both LGPS and LAGP spectra. The Li 1s region shows the formation of a second, signal-dominating Li environment of reduced oxidation state, which is assigned as “Li-SEI” for both LGPS and LAGP. The Ge 3d spectral regions also change, revealing the formation of a secondary Ge environment of reduced oxidation state (the electronically-conductive Ge metal phase) in both LGPS and LAGP at about 28 eV.
The differences between the SEIs on LGPS and LAGP are visible in the S 2p (LGPS), Al 2p (LAGP), and also P 2p spectral regions. Correlated with the reduction in Ge in the LGPS structure, the P–S structure in the LGPS SE underwent significant degradation, as we observed the substantial formation of Li2S and the reduced phosphide species including LixP and Li3P in line with previous reports about LGPS SEI formation.21,22 In the case of LAGP SE, the formation of a new Al-containing component at the interphase is indicated by the BE shift of the Al 2p peak from 74.5 eV to 72.3 eV. This decrease in BE signifies a change in the Al bonding environment, transitioning from the initial AlO6 octahedra, where the Al atom is bonded to six O atoms, to a new structure. XPS measurements on a piece of aluminium foil were performed to verify if the new Al-SEI phase is in its metallic form. The result, illustrated in Fig. S8 (ESI†), shows that the Al 2p peak for metallic Al has a BE of 71.6 eV, which is approximately 0.7 eV lower than the BE of the Al-SEI peak. This difference suggests that the Al atoms in the Al-SEI phase are bonded to fewer O atoms, potentially forming LiAlO2 where the Al atom is bonded with four O atoms in the AlO4 tetrahedral structure, which possesses promising stability against Li metal.33–35 Further investigations are required to identify this Al-containing decomposition product. However, the peak intensity of Al 2p is rather low, as an indication of the low atomic concentration of the Al-SEI phase in the SEI.
The peak in the P 2p spectra of LAGP exhibits a relatively stable BE of around 133.2 eV up to this point and it remains detectable during continued plating, suggesting that the P atoms maintain their PO4 tetrahedral structure.31,36 The implication of this is that Li3PO4 is a major component of the LAGP SEI, in contrast to the LGPS SEI in which Li2S predominates. This also indicates that the strong bonding strength of P–O bonds helps the PO4 tetrahedral structure to remain intact. With continuous Li plating, the phosphorus species would be reduced to Li3P, but interestingly this is not seen in LAGP SE. SEI growth continues on both electrolyte materials as more Li is plated.
By the 30th minute of the in situ Li plating, the spectral signatures of pristine LGPS and LAGP are almost completely replaced by SEI-associated spectra, since the XPS-probed volume now consists mostly of SEI material. Here, the metallic Li (Li0) peak was only observed for LAGP SE, as an indication of the formation of a more passivating SEI layer for LAGP SE.
As Li plating continues up to 70 minutes, differences in the stabilities of the SEIs formed on LGPS and LAGP are revealed. These differences are most visible in the Li 1s and Ge 3d spectral regions, in two ways.
Firstly, while the SEI of LAGP contains a significant fraction of electronically conductive Ge0 in the SEI throughout, the fraction of this phase in the LGPS SEI decreases with plating time, which agrees with a previous report.22 The intensity attenuating of the Ge signal as well as the P signal may result from the formation of Li2O on the surface, which obscures the signal from the underneath layer. The formation of Li2O is likely due to the reaction between the formed Li2S and the residual air inside the XPS chamber. This is evident in Fig. S9 (ESI†), which shows that a pellet of pure Li2S continuously forms Li2O on its surface.
Secondly, for both LGPS and LAGP, we observe a gradual shift in the Li-SEI peak position towards a lower BE with increased plating time. The emergence of a Li0 peak would signify that the reaction rate between Li and the underlying SEI layer is slower than the time needed to collect the Li 1s spectra (∼30 minutes). In contrast, the absence of a Li0 peak would denote that the plated Li continues to react with the underlying SE faster than the time required to collect the Li 1s spectra. The latter would result in faster SEI growth and thus a thicker SEI layer. In this work, the formation of metallic Li was only observed on LAGP SE, which is indicative of a more passivating SEI, compared to LGPS. For LGPS, no spectroscopic signature of metallic Li could be detected throughout the experiment as shown in Fig. 2, which we attribute to the fast reaction kinetics between LGPS SE and lithium and the formation of a more ionically or electronically conductive SEI layer.
Janek et al. suggested that the SEI growth relies on the availability of both electrons and ions and is therefore limited by the slowest of either ionic or electronic transport.37 One possible explanation for the different observed behavior of the two electrolytes could be found in percolation theory,38 which predicts that a percolation threshold greater than 15 vol% is required for a continuous pathway to exist.
Assuming the following decomposition reactions:21,33
| Li1.5Al0.5Ge1.5(PO4)3 + 6Li → 0.5LiAlO2 + Li4P2O7 + Li3PO4 + 1.5Ge |
| Li10GeP2S12 + 20Li → 12Li2S + 2Li3P + Ge |
In contrast, the only ionically and electronically conductive phase within the initial LAGP SEI nanostructure is Ge0, occupying less than 13 vol%. As a result, the LAGP SE shows restrained reaction kinetics against Li metal compared to LGPS. This concept is schematically depicted in Fig. 3. Additional information about SEI nanostructure and exact composition would be required to assess if the growth is limited by electrons or ions.
 | ||
| Fig. 3 Schematic of the multi-component SEI layer formed on top of LGPS and LAGP SEs. | ||
Previous studies reporting an unstable SEI were performed at the LAGP-Li interface during cycling within a confined cell setup where the build-up of strain and mechanical degradation could facilitate the SE decomposition.26,39 In contrast, here, this experimental setup removes the effects of pressure, allowing solely the chemical properties of the SEI to be probed to help gain a better understanding of degradation mechanisms. It is therefore necessary to further investigate SEI stability in this system.
As an attempt to study the SEI evolution kinetics, continuous XPS measurements were conducted on the surface of the LAGP SE after a strong Li0 peak was observed as shown in Fig. 4, without any further Li plating. In principle, the SEI formation kinetics can be extracted from the Li0 peak attenuation rate, and the increasing rate of other characteristic peaks from different SEI components. In the case of LAGP SE, Ge, P and O spectra are available for this quantification process. As shown in Fig. 4, Ge stays in its metallic form (evidenced by both Ge 3d and Ge 3p spectra). The BE of the P 2p-PO4 peak stays constant, suggesting stability of the PO4 structure against Li metal. Both Ge and P signals show a slight decrease in peak intensity over time.
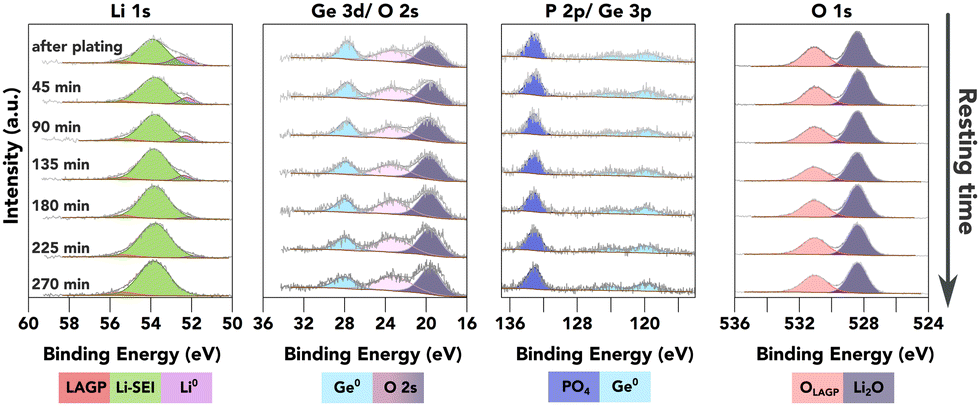 | ||
| Fig. 4 XPS investigation of the further reaction of plated Li metal with the underneath layer (available enlarged as Fig. S11, ESI†). The plots show a time series of XPS core spectra acquired after a strong Li0 peak was observed on top of LAGP. The resting time is given in the first column. | ||
There are two possible degradation pathways for the plated Li metal on the surface: it can be oxidised by the residual air inside the XPS chamber, or react with the underneath layer to form SEI. The oxidation rate of metallic Li inside the XPS chamber was investigated by continuous XPS measurements on a piece of sputter-cleaned Li foil as shown in Fig. S10 (ESI†), which demonstrates that over 50 at% of the metallic Li was oxidised within the first 50 minutes, resulting in the formation of Li2O and LiOH. However, the O 1s peak from LiOH, with a BE of approximately 530.7 eV, is less than 0.4 eV away from the O 1s-PO4 peak. This proximity can introduce errors to the quantification process, thus reducing the accuracy of reaction kinetics estimations. The proximity of these two peaks characteristic of two possible pathways mean that no quantification of the reaction kinetics could reliably be conducted due to the errors introduced into the quantification process.
Further, no significant differences in overall SEI composition and stability could be detected between the results of the VEP-XPS experiment reported above and those of the equivalent in situ Li sputtering XPS experiments (Fig. S2 and S3, ESI†).
In summary, virtual electrode plating and in-situ Li sputtering XPS experiments performed on LGPS and LAGP SEs provided consistent results in terms of SEI composition and growth. LAGP shows slower reaction kinetics, as proven by the emergence of a Li0 peak, while LGPS exhibits rapid SEI growth that prevents metallic lithium from plating. The absence of applied pressure in a VEP-XPS experiment, allows us to attribute these observations to the SEI composition, distribution and physical properties of secondary decomposition products and in particular to the mixed ion-electron conductive Li3P which can be observed in LGPS and not in LAGP. Additionally, the in situ XPS technique provides the potential to investigate SEI formation kinetics by continuously monitoring the surface information after the Li0 is observed. However, to accurately quantify the reaction kinetics, an ultra-high vacuum level (much lower than 10−7 Pa) is required to minimise the reaction between the Li metal and the chamber environment.
This work was supported by the Faraday Institution SOLBAT project (grant number FIRG056) and Henry Royce Institute (through UK Engineering and Physical Science Research Council grant EP/R010145/1). M. P. is grateful for the support of Nissan Motor Co. Ltd, Japan. B. J. is grateful for the support of the Clarendon Fund Scholarships.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- X.-B. Cheng, et al. , Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- D. Lin, et al. , Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS.
- J. Lu, et al. , Electrochem. Energy Rev., 2018, 1, 35–53 CrossRef CAS.
- J. Janek, et al. , Nat. Energy, 2016, 1, 16141 CrossRef.
- A. Banerjee, et al. , Chem. Rev., 2020, 120, 6878–6933 CrossRef CAS PubMed.
- S. A. Pervez, et al. , ACS Appl. Mater. Interfaces, 2019, 11, 22029–22050 CrossRef CAS PubMed.
- S. Wenzel, et al. , Solid State Ionics, 2015, 278, 98–105 CrossRef CAS.
- W. D. Richards, et al. , Chem. Mater., 2016, 28, 266–273 CrossRef CAS.
- Y.-Y. Sun, et al. , J. Mater. Chem. A, 2021, 9, 9481–9505 RSC.
- H.-D. Lim, et al. , Energy Storage Mater., 2020, 25, 224–250 CrossRef.
- J. Janek, et al. , Nat. Energy, 2023, 8, 230–240 CrossRef.
- M. Uitz, et al. , J. Electroceram., 2017, 38, 142–156 CrossRef CAS.
- J. C. Bachman, et al. , Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- Y. Lee, et al. , ACS Energy Lett., 2021, 171–179 Search PubMed.
- M. B. Dixit, et al. , Energy Environ. Sci., 2021, 14, 4672–4711 RSC.
- F. Walther, et al. , Chem. Mater., 2019, 31, 3745–3755 CrossRef CAS.
- Z. Wang, et al. , Nano Lett., 2016, 16, 3760–3767 CrossRef CAS.
- S. K. Otto, et al. , Adv. Mater. Interfaces, 2022, 9, 2102387 CrossRef CAS.
- C. Ma, et al. , Nano Lett., 2016, 16, 7030–7036 CrossRef CAS PubMed.
- S. Wenzel, et al. , Solid State Ionics, 2018, 318, 102–112 CrossRef CAS.
- S. Wenzel, et al. , Chem. Mater., 2016, 28, 2400–2407 CrossRef CAS.
- A. L. Davis, et al. , J. Electrochem. Soc., 2021, 168, 070557 CrossRef.
- S. Narayanan, et al. , Nat. Commun., 2022, 13, 7237 CrossRef CAS.
- A. Paolella, et al. , Adv. Energy Mater., 2020, 10, 2001497 CrossRef CAS.
- L. He, et al. , ACS Appl. Mater. Interfaces, 2019, 11, 20895–20904 CrossRef CAS PubMed.
- J. Tippens, et al. , ACS Energy Lett., 2019, 4, 1475–1483 CrossRef CAS.
- Z. Tong, et al. , ACS Appl. Energy Mater., 2022, 5, 11694–11704 CrossRef CAS.
- J. A. Lewis, et al. , ACS Energy Lett., 2019, 4, 591–599 CrossRef CAS.
- Q. Yu, et al. , ACS Appl. Mater. Interfaces, 2019, 11, 9911–9918 CrossRef CAS PubMed.
- J. Li, et al. , SmartMat, 2023, 4, e1200 CrossRef CAS.
- L. He, et al. , Chem. Mater., 2021, 33, 6841–6852 CrossRef CAS.
- S. P. Ong, et al. , Energy Environ. Sci., 2013, 6, 148–156 RSC.
- Y. Zhu, et al. , ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS.
- Q. Zhao, et al. , SusMat, 2022, 2, 104–112 CrossRef CAS.
- Y. Wu, et al. , Electrochim. Acta, 2017, 235, 463–470 CrossRef CAS.
- M. Weiss, et al. , ACS Appl. Mater. Interfaces, 2018, 10, 10935–10944 CrossRef CAS.
- C. D. Alt, et al. , Joule, 2024, 8, 1–22 CrossRef.
- H. Scher, et al. , J. Chem. Phys., 1970, 53, 3759–3761 CrossRef CAS.
- B. Ozdogru, et al. , ACS Appl. Energy Mater., 2022, 5, 2655–2662 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: [DETAILS]. See DOI: https://doi.org/10.1039/d4cc04462b |
| This journal is © The Royal Society of Chemistry 2024 |