
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper nanoclusters: emerging photoredox catalysts for organic bond formations
Arunachalam
Sagadevan†
,
Kathiravan
Murugesan†
,
Osman M.
Bakr
 and
Magnus
Rueping
*
and
Magnus
Rueping
*
KAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: magnus.rueping@kaust.edu.sa
First published on 12th November 2024
Abstract
Advancements in fine chemical synthesis and drug discovery continuously demand the development of new and more efficient catalytic systems. In this regard, numerous transition metal-based catalysts have been developed and successfully applied in industrial processes. However, the need for innovative catalyst systems to further enhance the efficiency of chemical transformations and industrial applications persists. Metal nanoclusters (NCs) represent a distinct class of ultra-small nanoparticles (<3 nm) characterized by a precise number of metal atoms coordinated with a defined number of ligands. This structure confers abundant unsaturated active sites and unique electronic and optical properties, setting them apart from conventional nanoparticles or bulk metals. The well-defined structure and monodisperse nature of NCs make them particularly attractive for catalytic applications. Among these, copper-based nanoclusters have emerged as versatile and sustainable catalysts for challenging organic bond-forming reactions. Their unique properties, including natural abundance, accessible oxidation states, diverse ligand architectures, and strong photophysical characteristics, contribute to their growing prominence in this field. In this review, we discuss the photocatalytic activities of Cu-based nanoclusters, focusing on their applications in cross-coupling reactions (C–C and C–N), click reactions, multicomponent couplings, and oxidation reactions.
1. Introduction
In contemporary organic synthesis, a central objective is to develop efficient catalytic methods that advance drug discovery, facilitate fine chemical synthesis, and support the sustainable production of industrial building blocks.1–3 Catalysts with precisely defined multiple active sites, coupled with recyclability, offer significant advantages in terms of cost-effectiveness and environmental sustainability.1,4,5 Due to their larger surface area and complex electronic structures, metal nanoparticles (MNPs) may demonstrate superior catalytic performance, exhibiting enhanced activity and selectivity compared to traditional homogenous metal catalyst.1,4,6,7Nanoparticle systems, however, present significant challenges in the design and targeted synthesis, with surface-active sites that are difficult to customize. The exact bonding details of surface molecules often remain unclear due to the lack of detailed structural characterization.7–9 As a result, there is a need to develop new catalysts that combine high atom utilization to enhance catalytic activity, with well-defined structures that enable detailed mechanistic understanding. Achieving this balance is both highly desirable and inherently challenging.
In this context, metal nanoclusters (NCs) represent a unique class of nanomaterials. These clusters consist of a precise number of metal atoms stabilized by a specific number of ligands, resulting in well-defined, accessible structures.8–10 NCs are typically represented as [Mm(L)n]q (where m, n, and q correspond to the total number of metal atoms, ligands, and charge state, respectively).10
The ultra-small size of these clusters (<3 nm) provides a larger specific surface area and enhances catalytic activity.8,9,11 Ligand-protected, atomically precise, high-purity nanoclusters (NCs) can be synthesized via wet chemical methods.8 The properties of these clusters can be finely tuned by adjusting the number of metal atoms and the structure of the ligands, with even a single metal atom substitution significantly impacting the cluster's characteristics.9–12 It is widely recognized that the size of the clusters can vary substantially depending on the specific reaction conditions and the initial concentrations of the reactants.12 These size variations, in turn, lead to changes in the physicochemical properties of the clusters.10 Quantum confinement effects are a key factor in this phenomenon, causing the transformation of continuous conduction and valence bands into discrete energy levels, particularly in clusters with dimensions smaller than 3 nm.9,10,12 As a result, nanoclusters (NCs) with precisely defined compositions can exhibit distinctive electronic transitions in the UV-vis spectrum, which are dependent on the size of the cluster's core.13 The UV-vis absorption spectrum serves as a unique optical fingerprint, enabling both qualitative and quantitative analysis of a specific cluster. Notably, recent advancements in this field highlight the potential of these nanostructures as promising materials for a wide range of applications. A such NCs recently appeared as new catalysts for organic reactions including oxidations, reductions, C–C cross-couplings, A3-couplings, and others.9,14–19
Despite their efficiency, a significant drawback of most nanoclusters is that they are composed of noble metals, which are expensive and scarce, thereby limiting their sustainable applicability.9 Due to their abundance in the Earth's crust, cost-effectiveness, versatile ligand chemistry, and adjustable oxidation states, copper nanoclusters (CuNCs) have emerged as promising and sustainable catalysts for various bond-forming reactions. The catalytic applications of CuNCs have initially been relatively limited for specific reactions (e.g., Click reactions, hydrogenations, and other catalytic processes processes)15,20–27 primarily due to the insufficient availability and stability. However, recent advancements in synthesis have enabled the successful crystallization of a significant number of stable core–shell CuNCs.22,28–30 In 2019 the Bakr research group reported the crystal structure of an atomically precise thiol-protected high-nuclearity core–shell Cu61 NC, [Cu61(StBu)26S6Cl6H14] that can be synthesized in large quantities following a simple synthetic procedure.31
The broad-range absorption in the visible light region, coupled with the richer photoluminescence properties (longer lifetimes) and well-defined structure of high-nuclearity core–shell CuNCs,13,31–37 has inspired organic chemists to explore the catalytic activities of these materials in photoredox reactions.
This feature article provides an overview of the photocatalytic capabilities of copper-based nanoclusters for organic bond formations. Additionally, it outlines prospective directions for catalytic applications based on their precisely determined single-crystal and surface structures (see Fig. 1).
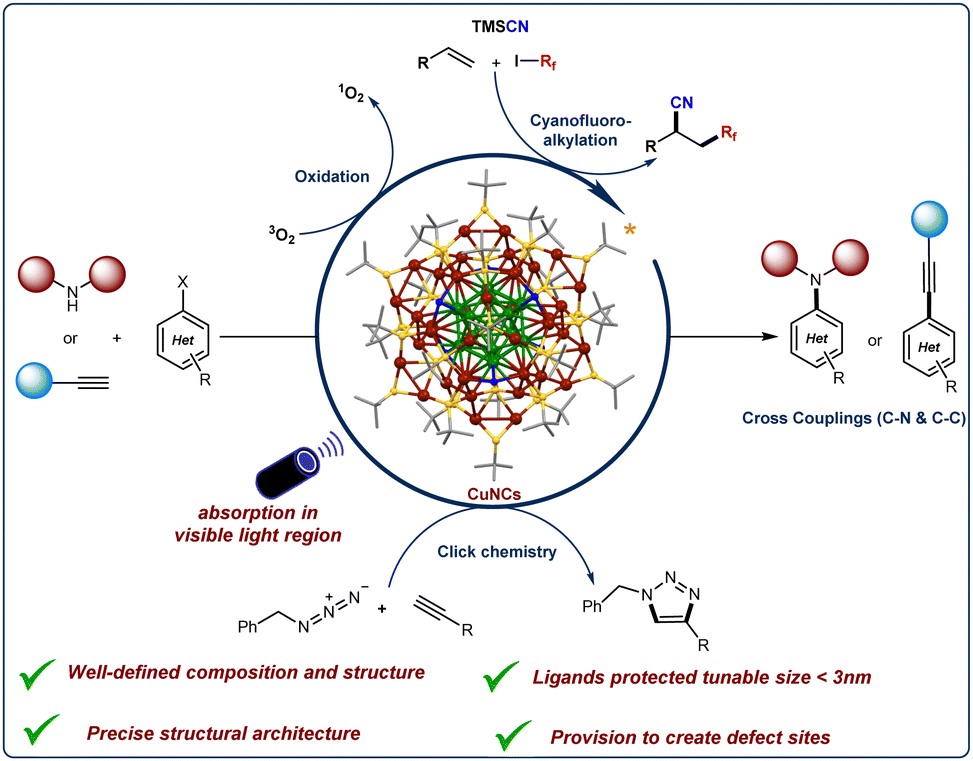 | ||
| Fig. 1 Photoredox copper nanoclusters catalyzed organic bond-forming reactions. | ||
2. Cross-coupling reactions
Cross-coupling reactions are among the most important processes for constructing complex organic molecules.38 In this article, we summarize the C–N and C–C cross-couplings catalyzed by atomically precise Cu-based nanoclusters under photoinduced conditions.2.1. C–N amination of aryl chlorides
In synthetic chemistry, the C–N arylation with nitrogen nucleophiles is a fundamentally significant reaction, widely utilized across various fields including agro- and pharmaceutical chemistry and the development of organic functional materials.38–41Given the importance, numerous robust methodologies have been developed for constructing C–N bonds, including Ullmann–Goldberg couplings,42–44 Buchwald–Hartwig aminations,38 and Chan–Evans–Lam aminations.45 Although copper has been employed as an abundant and inexpensive catalyst in Ullmann–Goldberg coupling, this process typically requires high reaction temperatures and activated electrophiles (e.g., aryl bromides/iodides). In response the groups of Buchwald,46,47 Taillefer,44 and Ma43,48,49 developed copper catalysts with diamine-based ligands to broaden the substrate scope for C–N coupling reactions, yet high temperatures were still necessary.42–44 Fu and Peter's research group reported ultraviolet (UV) light-activated copper-catalyzed Ullmann C–N arylations of heteroatom nucleophiles.50–53 Notably, with a judicious choice of ligand, it became possible to conduct copper-catalyzed C–N cross-couplings under visible light rather than traditional UV light.54–58 More recently, hybrid Cu2O nanocrystals have been employed for C–N cross-couplings via a photoinduced process.59,60 However, while these are considered heterogeneous catalysts, the reaction mechanism remains poorly understood,9 and their use has been limited to aryl iodides/bromide substrates.
As a result, there is a need to develop more effective and precisely structured catalysts for C–N cross-coupling reactions, particularly for the activation of cost-effective aryl chloride coupling agents. Additionally, a comprehensive understanding of catalytic mechanisms and the correlations between catalyst structure and properties would be necessary for advancing more efficient C–N bond forming reactions. Therefore, atomically precise copper nanocluster represents an optimal candidate for catalyzing C–N bond-forming reactions.
In 2022, our research group, in collaboration with the Bakr lab, made a pioneering discovery by demonstrating that the thiol-protected core–shell copper nanocluster [Cu61(StBu)26S6Cl6H14] (Cu61) is an effective photocatalyst for the N-arylation of heterocyclic nucleophiles with unactivated aryl chlorides at room temperature (Fig. 2).61
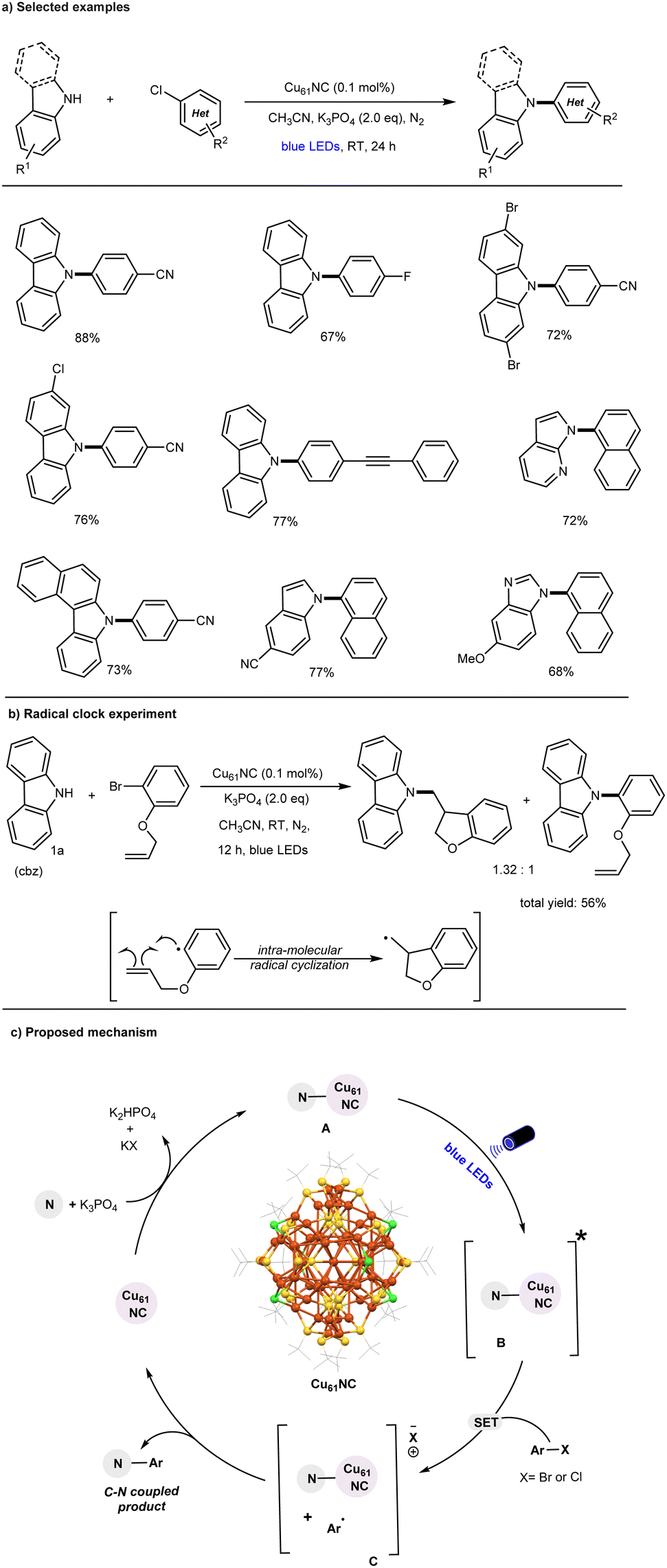 | ||
| Fig. 2 Visible-light activated Cu61 NC catalyzed C–N amination of aryl chlorides. | ||
The initial study established that the combination of Cu61, a base, and visible light is essential for achieving the desired C–N coupling reaction, with yields reaching up to 88%. It was demonstrated that inorganic Cu salts (such as CuCl or CuCl2) are less efficient, primarily due to their ineffectiveness to act as photocatalysts in the visible light region as compared to the Cu61 cluster catalyzed reaction.51,53,54,57
The C–N bond formation catalyzed by the cluster exhibits a broad substrate scope, including typically unreactive aryl(hetero)chlorides. Additionally, various heterocyclic N-nucleophiles were found to be compatible reaction partners, as illustrated in Fig. 2a Notably, this cluster-based approach marks a significant improvement over previously reported thermal copper-catalyzed aryl chloride amination methods.43,62–65 The Cu61 catalysis can be performed under mild reaction conditions (i.e., room temperature) and accommodates a broad range of substrates, making it suitable for widespread application. Mechanistic control experiments, including radical clock experiments (Fig. 2b), competitive reactions, and radical trapping experiments, along with detailed spectroscopic studies, suggest that a facile single-electron transfer (SET) mechanism is operative in the CuNC-catalyzed C–N bond formation of aryl chlorides, in contrast to the traditional two-electron process.
The proposed mechanism is illustrated in Fig. 2c. The in situ-formed Cu61 NC-amine complex (A) is promoted to its photoexcited state (B) upon blue-light irradiation. In this excited state, the cluster complex (B) undergoes a facile single-electron transfer (SET) process with aryl chlorides, generating an aryl radical and an intermediate oxidized Cu61 NC–Nu complex (C). The highly reactive aryl radical subsequently undergoes a radical attack on the surface of complex C, leading to the C–N arylation product through either a radical-radical coupling pathway or a reductive elimination step. Overall, this successful transformation highlights the potential of CuNCs as emerging photocatalysts for challenging organic bond-forming reactions and provides valuable insights into the role of their core–shell structural architecture under photoinduced conditions.
2.2. Sonogashira C–C cross-coupling
The Sonogashira reaction is widely recognized as a straightforward and efficient method for the synthesis of alkyne derivatives. It is commonly used in the production of natural products, medicinal chemistry, pharmaceutical manufacturing, and the development of functional materials.66–68 Given the importance, an array of catalytic methods have been established for such cross-couplings, including (a) conventional Pd/Cu-catalytic methods with amine bases,69,70 (b) Cu-free Pd-catalytic methods,71–73 (c) Pd-free Fe-, Ni-catalysis,74–76 (d) Pd-free, copper catalysis,77–80 and (e) MNPs (including Pd- or Cu-NPs catalysis).81–85 Recently, photocatalytic Cu nanoparticles (Cu-NPs) have also been shown to catalyze Sonogashira couplings under a CO2 atmosphere.86 The required CO2 promotes the formation of an active catalyst surface-bound Cu(I)-phenylacetylide complex via the formation of inorganic carbonate (CuCO3), with a working principle similar to that of conventional Cu catalysts.78 However, modeling nanoparticle systems remains challenging due to their complex atomic surface structures, and the suitability of these ambiguous structures for catalytic applications may be subject to scrutiny.9 In previously reported copper-catalyzed Sonogashira reactions, significant to high amounts of Glaser homocoupling by-products are often formed.79,80,85,86 Mechanistically, the polymeric nature of the copper-acetylide intermediate plays a key role in the formation of Glaser homocoupling by-products during the copper-catalyzed Sonogashira reaction.87–89Addressing these challenges, the development of a novel copper-catalyzed system with enhanced selectivity for C–C coupled products, while minimizing the formation of undesired homocoupled alkyne by-products, remains a key objective. In this context, the Rueping and Bakr groups recently highlighted the unique characteristics of [Cu28H10(C7H7S)18(TPP)3] (Cu28) nanoclusters (NCs), which demonstrate high selectivity as catalysts for Sonogashira C–C coupling reactions under visible light irradiation (Fig. 3).90 Control experiments have revealed that light, base, and Cu28 are essential for the observed reaction. The defective Cu28 nanocluster is compatible with a broad range of substrates, including various aryl iodides and terminal alkynes, delivering Sonogashira C–C coupled products in good to excellent yields (up to 84%). Notably, the presence of defects in Cu28 play a crucial role in achieving high selectivity in the Sonogashira C–C coupling reaction, effectively preventing the problematic formation of undesired homo-coupling alkyne by-products. In contrast, traditional copper catalytic systems, such as CuCl, Cu(CH3CN)4BF4, or CuNPs, exhibit lower yields and inferior selectivity in comparison. Additionally, the study explored a larger cluster family, Cu61 NCs, in the same reaction. However, these clusters proved to be less efficient compared to the defective Cu28 catalyst. A series of control experiments demonstrated that the defective Cu28 outperformed traditional copper catalysts. Notably, after the completion of the reaction, the recovered Cu28 catalyst remained unchanged, highlighting its remarkable stability. A set of mechanistic control experiments support the formation of Ar-radical via SET process between the photoexcited state of Cu28 and aryl iodides. The proposed mechanism is shown in Fig. 3b. Upon blue-light irradiation of ground-state Cu28, its long-lived photoexcited state is generated that participates in a SET process with aryl iodides (Ered = −1.7 to −2.3 V vs. SCE)91,92 to generate the Ar-radical C and oxidized form of Cu28B. The highly reactive Ar-radical attacks the C–C triple bond of the alkynes to produce vinylic-type carbon-centered radical intermediate D, and a subsequent back SET process to Cu28+ (B) regenerates the ground state Cu28 photocatalyst. Finally, base-promoted deprotonation of vinyl cation E leads to form desired Sonogashira C–C coupled products with high selectivity. In contrast to conventional inorganic copper-catalyst79,85,86 and Cu(I)-acetylide cluster,87,89Cu28 may not form a CuNC-phenylacetylide-intermediate (supported by various control experiments). Thus, the defective Cu28 can act as a putative photocatalyst that involves the SET process with aryl iodides to generate the Ar-radical and subsequently attacks the C–C triple bond, thus preventing the formation of the homocoupling byproduct.
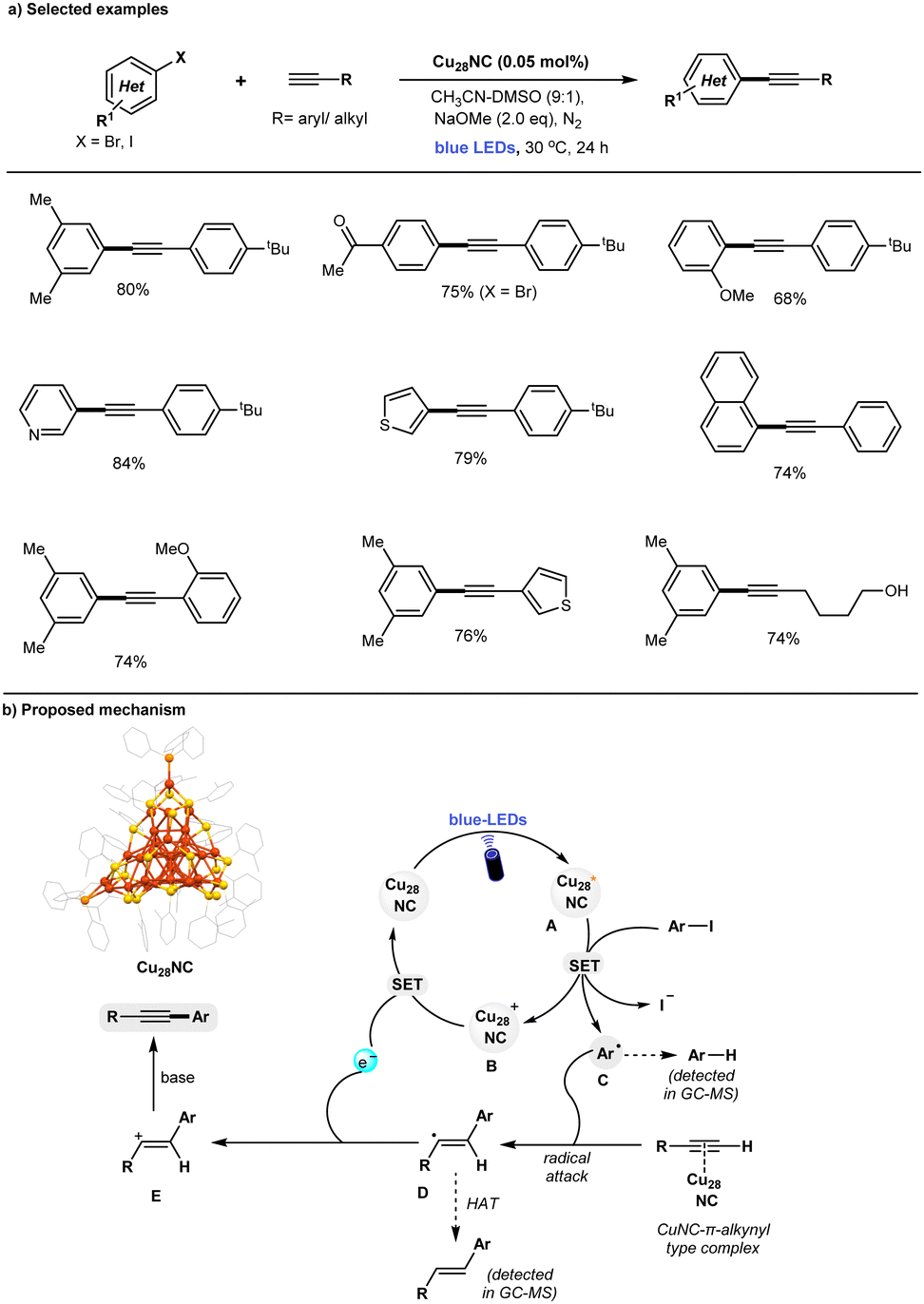 | ||
| Fig. 3 Defects Cu28 NC catalyzed Sonogashira C–C coupling reactions under photoinduced conditions. | ||
Recently, the Negishi group successfully synthesized the luminescent, hydride-free [Cu7(SC5H9)7(PPh3)3] (Cu7 NC) and explored its catalytic application in the Sonogashira cross-coupling reaction under photoinduced conditions (Fig. 4).93 Control experiments revealed that the exclusion of Cu7 NC, light, or base resulted in no reaction. Notably, the Cu7 NC catalyst can be recovered, as confirmed by UV-vis absorption spectra, and remains intact even after 10 reaction cycles.
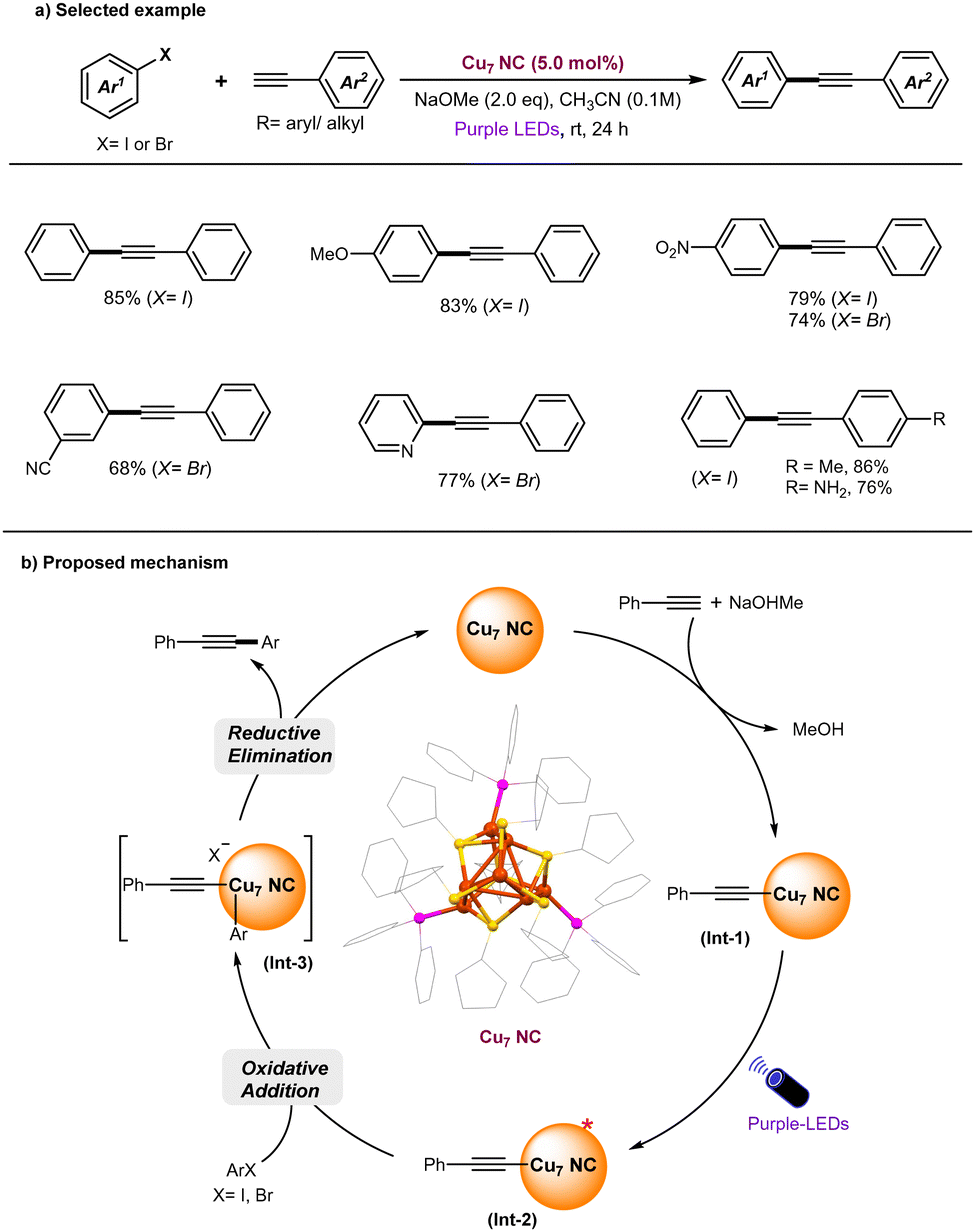 | ||
| Fig. 4 Luminescent Cu7 NC catalyzed Sonogashira C–C coupling reactions under photoinduced conditions. | ||
Under optimized reaction conditions, a broad range of aryl halides (X = I, Br) were successfully coupled with various aryl alkynes, yielding the C–C coupled Sonogashira products in excellent yields and with high selectivity (Fig. 4a). Based on experimental results, supported by theoretical studies, the reaction mechanism is proposed to follow a polar pathway rather than a radical-based mechanism. The proposed mechanism is illustrated in Fig. 4b. Initially, Cu7 nanoclusters (NCs) interact with phenylacetylene in the presence of a base, forming a Cu-phenylacetylide intermediate (Int-1). Upon photoexcitation, the ground-state Int-I is excited to generate a photoexcited state (Int-2), which subsequently undergoes oxidative addition with aryl halides, leading to the formation of Int-III. Finally, a reductive elimination step from Int-III results in the formation of the C–C coupled products and the regeneration of the original Cu7 NCs. It was identified that in the current Cu7 NC-catalyzed Sonogashira C–C coupling reaction, the oxidative addition step is rate-determining, closely aligning with the mechanism observed in thermally mediated Cu-catalyzed Sonogashira reactions78,80 Alternatively, the photoexcited state of Int-2 may undergo a SET process with ArX to generate an aryl radical and the oxidized form of Int-2. The highly reactive aryl radical is set to attack the oxidized form of Int-2 to deliver desired C–C coupled products and allows re-generation of original Cu7 NC. Indeed, inorganic copper(I) phenylacetylides were shown to undergo facile intersystem crossing,94–98 therefore, a radical process cannot be ruled out.
3. Click reactions
The copper(I)-catalyzed [3 + 2] azide–alkyne cycloaddition (CuAAC) stands out as a versatile reaction in the field of “click chemistry for the construction of 1,4-triazoles.99–102 In 2002, the research group of Meldal103 and Sharpless104 disclosed CuAAC reactions, which have had a wide variety of applications in synthetic chemistry,105–107 polymers and material science,108,109 and bio-conjugation chemistry.110–112The significance of click chemistry was underscored by the Nobel Prize in Chemistry awarded to Bertozzi, Meldal, and Sharpless.113 Traditionally, the click reaction is catalyzed by air-sensitive, unstable CuI complexes or by stable CuII salts in the presence of a chemical reducing agent, such as sodium ascorbate, under thermally mediated conditions.114,115 More recently, a photo-click reaction has been reported,116–118 where the active CuI catalyst is generated in situ via the photoreduction of a stable CuII complex. However, light-induced click reactions offer greater benefits compared to traditional click reactions performed in the absence of light (dark conditions). For instance, photo-click reactions can be triggered at precise moments and locations, making them a potent tool for chemical synthesis, bio-orthogonal conjugation, and customized material production.117,119
Hence, there is a strong desire for the creation of a novel and precisely structured copper-based photocatalyst for click reactions. Recently, atomically precise Cu-nanoclusters e.g., (Cu20(PhCC)12(OAc)6) have been shown as a modular catalyst for CuAAC reaction,22,120,121 however, this catalytic system is still operative under thermal conditions rather than a photoinduced process. In the most recent developments, the Rueping and Bakr groups collaboratively published findings on the production of atomically precise copper hydride nanoclusters, [Cu58H20PET36(PPh3)4]2+ (Cu58; PET: phenylethanethiolate; PPh3: triphenylphosphine).122 One surface copper atom of Cu58 is chemically removed during the recrystallization process, which results in a defective analogous nanocluster namely, [Cu57H20PET36(PPh3)4]+ (Cu57) (Fig. 5a). Indeed, the single-atom manipulation on stable copper hydride nanocluster (Cu58) could drastically alter their reactivity of the nanocluster. Catalytic explorations revealed that the defective Cu57 nanocluster acted as an efficient photocatalyst, delivering the click product in 97% yield, whereas the parent copper nanocluster (Cu58) produced only 77% yield after 1.5 hours of visible-light irradiation (Fig. 5b). Cu57 and Cu58 were found to exhibit good stability during photoirradiation under blue LEDs in acetonitrile. Control experiments, such as conducting the reaction in the dark or using conventional copper catalysts (e.g., CuCl), showed significantly lower efficiency. Mechanistic control studies revealed that this photocatalytic click reaction follows a radical pathway, as opposed to the traditional two-electron process. The superior catalytic performance of Cu57 compared to Cu58 is attributed to the creation of an active catalytic site in Cu57 due to the removal of a surface copper atom. This insight could inspire further exploration of single-atom chemistry in metal nanoclusters and enable researchers to fine-tune their properties for catalytic applications in organic reactions.
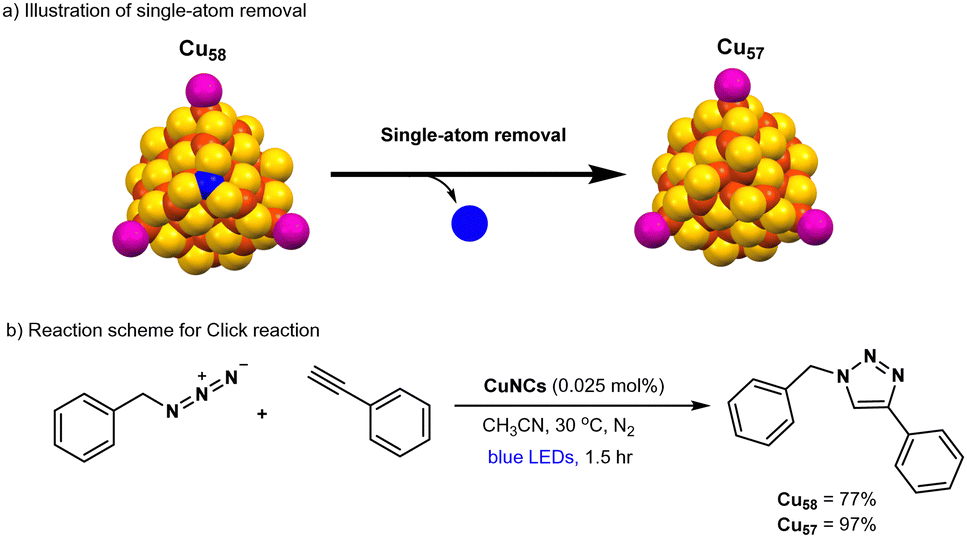 | ||
| Fig. 5 Isostructural Cu57 NC & Cu58 NC catalyzed Click reactions. | ||
In relation to Cu-cluster-catalyzed photo-click reactions, Jiang and co-workers recently synthesized the dimeric anionic supramolecular cluster [Cu8I14]6− (HUBU-1), which has been demonstrated to be an effective photocatalyst for both the oxidative coupling of benzylamine and the azide–alkyne cycloaddition reaction.120
4. Multicomponent coupling
Recently, He and co-workers synthesized and characterized [Cu40H17(2,4-DMBT)24](PPh4) (denoted as Cu40-H NC) which displays enhanced photophysical properties along with excellent air and moisture stability.123 The photoexcitation of this structurally well-defined anionic Cu40-H NC exhibits remarkable NIR-II (1000–1700 nm) emissive properties.Moreover, the Cu40-H NC photocatalyst demonstrated high efficiency in a three-component coupling reaction involving alkenes, fluoroalkyl iodides, and trimethylsilyl cyanide, yielding the desired coupled products in excellent yields with high turnover numbers (TON) (Fig. 6a). Based on a series of control experiments, the proposed reaction mechanism is illustrated in Fig. 6b. Upon irradiation with blue LEDs, the anionic Cu40-H NC is excited to its photoexcited state (Int-A), followed by a single electron transfer (SET) process with fluoroalkyl iodides, generating a reactive alkyl radical (R˙) and a cluster intermediate (Int-B). The highly reactive alkyl radical readily attacks the terminal alkene, generating a secondary alkyl radical. In the next step, cluster intermediate B reacts with TMSCN to form an anionic cyanide-ligated cluster intermediate (Int-C), which subsequently reacts with the secondary alkyl radical to afford the C–C coupling product and regenerate the original anionic Cu40-H NC. Overall, the NIR-II emissive anionic Cu40-H NC has been demonstrated as a highly efficient photoredox catalyst for the cyanofluoroalkylation of alkenes with a broad range of substrates under blue-LED irradiation.
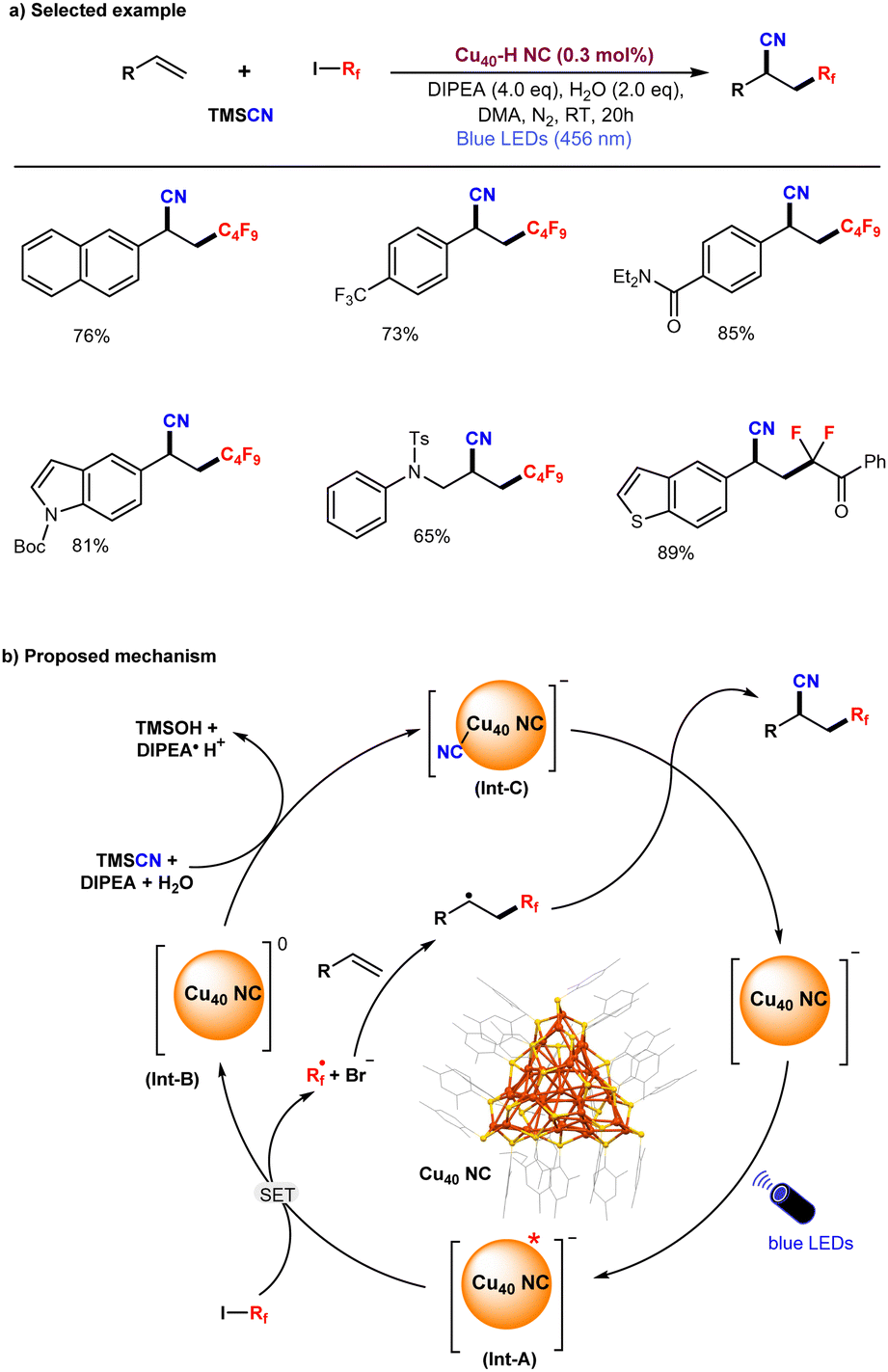 | ||
| Fig. 6 NIR-II emissive anionic Cu40 NC photoredox catalyzed cyanofluoroalkylation of alkenes. | ||
5. Photooxidation by singlet oxygen
Singlet oxygen (1O2) is known to be a key reactive oxygen species (ROS) and plays an indispensable role in photocatalytic organic reactions (e.g., Ene reactions)124 and photodynamic therapy.125 Indeed, metal nanoclusters (MNCs) are shown as powerful photosensitizers for 1O2-mediated photocatalytic organic reactions via an energy transfer process with molecular oxygen (3O2).126 In this context, Zhang et al. recently reported the successful preparation of two quasi-structurally isomeric Cu13 NCs (denoted as Cu13a and Cu13b) and demonstrated their effectiveness in the photocatalytic oxidation of sulfides (Fig. 7a).127 These isomeric structures of Cu13 nanoclusters are protected by CZ-PrAH (9-(prop-2-yn-1-yl)-9H-carbazole) and H4TC4A (p-tert-butylthiacalix[4]arene) ligands. The formula of these NCs is [Cu13Na(CZ-PrA)6(TC4A)2(CH3OH)]·CH3OH·CH2-Cl2·CH3COCH3 and [Cu13Na2(CZ-PrA)6(TC4A)2Cl(CH3OH)2]. The two Cu13 nanoclusters (NCs) exhibit structural similarities; however, the primary distinction lies in the attachment of a chloride anion to the metallic framework, influenced by solvent-induced effects during the crystallization process.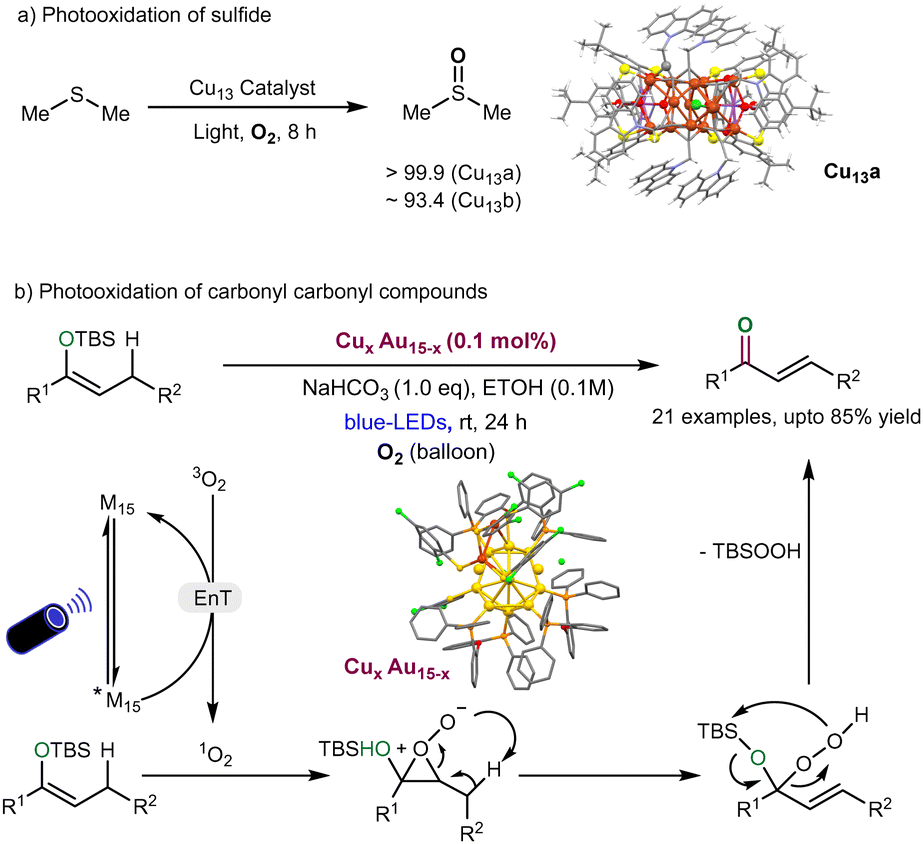 | ||
| Fig. 7 (a) Quasi-structurally isomeric Cu13 NCs catalysed photocatalytic sulfoxidation of sulfides. (b) Photo-oxidation of carbonyl compound derivatives by CuxAu15−x NCs. | ||
Upon photoexcitation, the chloride anion engages in a charge transfer process with the metal core, a process more efficient in the Cu13a structure compared to Cu13b. This charge transfer event significantly alters the electronic band structure of Cu13a, leading to a higher production of singlet oxygen (1O2) rather than superoxide radical anion (O2˙−), compared to Cu13b. As a result, the Cu13a structure demonstrates superior photocatalytic performance in the selective oxidation of dimethyl sulfide, with selectivity exceeding 99.9%. The series of trapping experiments such as DMPO (5,5-dimethyl-1-pyrroline-N-oxide) and TEMPO (2,2,6,6-tetramethylpiperidine-N-oxyl), and DFT study indicates that Cu13a is an effective for selective photocatalytic oxidation of dimethyl sulfide. Overall, this work reveals that the attachment of chloride ions to the surface of Cu nanoclusters (CuNCs) can create more active sites to produce singlet oxygen (1O2), specifically targeting selective photocatalytic oxidation reactions. This discovery significantly expands the potential for various chemical reactions, opening new possibilities for advanced catalytic applications. Very recently, Zhu and coworkers128 synthesized and characterized the homogold Au15 and Au–alloy clusters such as AgAu15 and CuxAu15−x, trimetallic AgxCuyAu15−x−y, and tetramettalic Pt1AgxCuyAu15−x−y. In the evaluation of the photophysical properties of Au–alloy nanoclusters, the phosphorescence of CuxAu15−x nanocluster displayed efficient singlet oxygen generation for photooxidation of carbonyl compounds to produce α,β-unsaturated ketones under blue-LEDs irradiation (Fig. 7b). The reaction pathway follows the energy transfer process and is followed by singlet oxygen generation, enabling photooxidation of carbonyl derivatives.
Recently, our group successfully designed ligand-engineered, atomically precise copper nanoclusters, including Cu13(Nap)3(PPh3)7H10 (Cu13Nap) and Cu13(DCBT)3(PPh3)7H10 (Cu13DCBT) and demonstrated their photocatalytic application in decarboxylative oxidation reactions (Fig. 8).129 Notably, Cu13Nap, stabilized by naphthalene thiolate ligands, achieved a remarkable 90% yield in the decarboxylative oxidation, significantly outperforming the isostructural Cu13(DCBT)3(PPh3)7H10 (Cu13DCBT), which gave a 28% yield. This enhanced performance is attributed to ligand engineering, which modulates the electronic structure, enhances visible light absorption, and facilitates energy transfer mechanisms during the reaction. In contrast to conventional decarboxylative oxidation reactions, which typically follow a single-electron transfer (SET) pathway, the mechanism here is primarily driven by energy transfer, as confirmed by density functional theory (DFT) calculations and experimental evidence. This study highlights the profound impact of ligand modification on copper nanoclusters (Cu NCs), opening new avenues for developing more sustainable and efficient photocatalytic systems. Furthermore, the catalytic system exhibits broad substrate compatibility and excellent tolerance for various functional groups. Particularly, the successful decarboxylation of fully aliphatic acids and the late-stage modification of drug molecules, as shown in examples 11–17, underscores the potential of this catalyst to address key challenges in drug discovery.
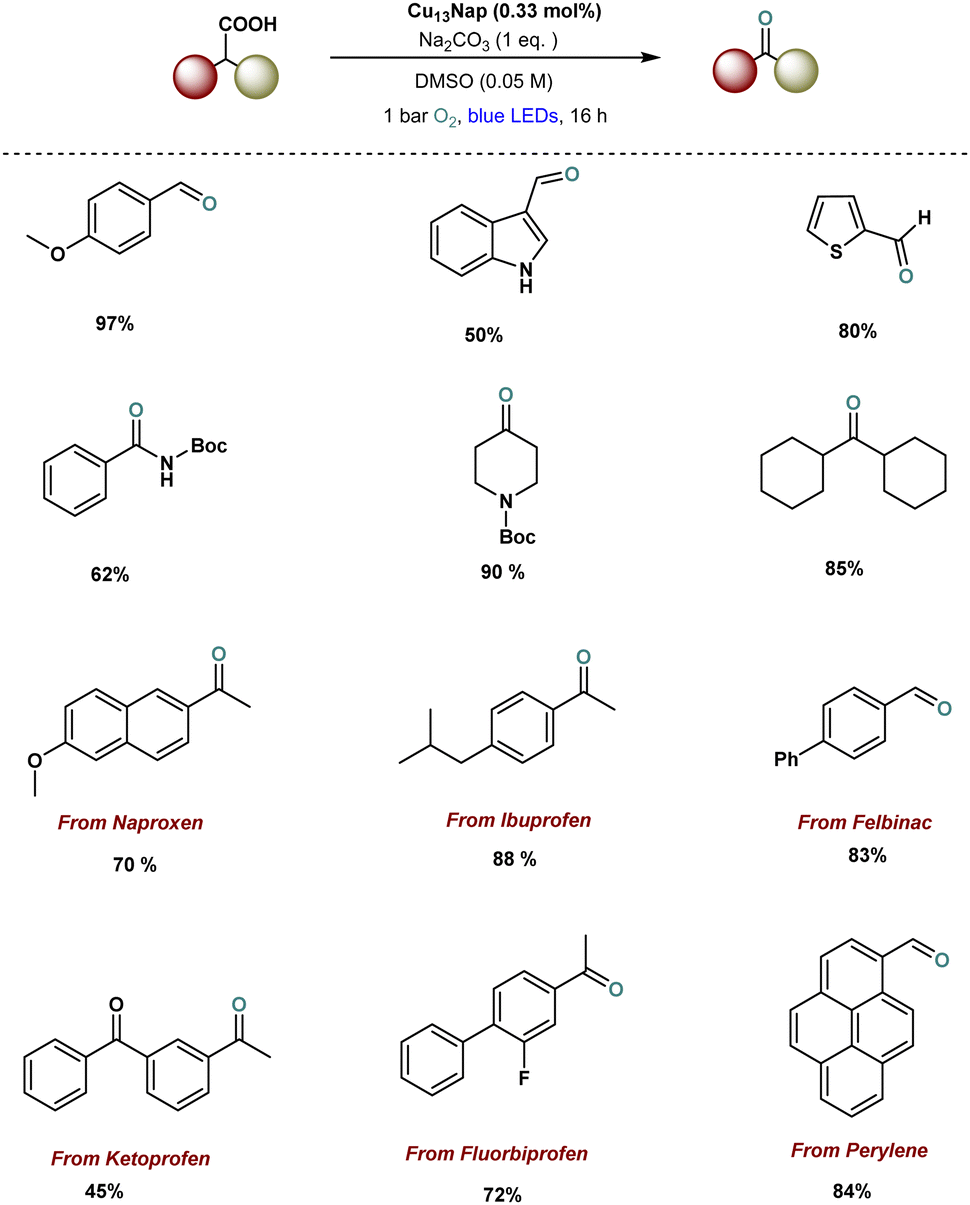 | ||
| Fig. 8 Ligand-engineered Cu13 NCs catalysed photocatalytic decarboxylative oxidation reaction. | ||
6. Conclusions and outlook
In light of the growing applications of modern photochemistry in drug discovery and biological synthesis, the need for the development and refinement of highly efficient catalysts has become increasingly important. This review highlights the potential of atomically precise copper nanoclusters (CuNCs) as innovative photocatalysts for a diverse range of bond-forming reactions, including C–C and C–N couplings, copper-catalyzed azide–alkyne cycloaddition (CuAAC), multicomponent couplings, and oxidation reactions. These findings suggest that CuNCs can play a transformative role in catalysis, extending beyond their conventional use as copper-based catalysts, and offering new possibilities for advancements in chemical synthesis. Unlike noble metal nanoclusters, the synthesis of copper nanoclusters (CuNCs) is significantly more challenging due to their lower half-reduction potential, which raises concerns about their stability in catalytic applications. However, recent studies have shown that the use of carefully selected thiol and phosphorus-containing ligands enables the successful synthesis of stable CuNCs in larger quantities, making them suitable for catalyzing organic reactions. Despite this progress, examining the structural stability of CuNCs when subjected to photoinduced conditions during catalytic reactions is important. To date the structures of Cu7, Cu28, Cu58, and Cu57 have shown to remain stable under blue LED irradiation.In comparison to homogeneous systems, heterogeneous materials are more appropriate for industrial applications due to their stability and recyclability. Therefore, there is a strong demand to develop stable, recyclable and precisely structured CuNCs. Furthermore, the development of chiral CuNCs for enantioselective reactions presents a promising area of interest. However, this endeavour is particularly challenging, as the chiral ligands must fulfil dual roles: stabilizing the clusters while also inducing high enantioselectivity.
In conclusion, the exploration of Cu-based nanocluster (CuNC) catalysis offers promising opportunities for advancing unique bond-forming reactions. We hope that this review will inspire further research into Cu-based cluster catalysis, leading to novel approaches in organic synthesis and expanding the role of CuNCs in catalysis.
Conflicts of interest
The authors declare no competing financial interest.Notes and references
- L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825–1837 CrossRef CAS PubMed.
- I. Ghosh, N. Shlapakov, T. A. Karl, J. Düker, M. Nikitin, J. V. Burykina, V. P. Ananikov and B. König, Nature, 2023, 619, 87–93 CAS.
- C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS.
- N. K. Ojha, G. V. Zyryanov, A. Majee, V. N. Charushin, O. N. Chupakhin and S. Santra, Coord. Chem. Rev., 2017, 353, 1–57 CrossRef CAS.
- B. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
- M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- R.-W. Huang, J. Yin, C. Dong, A. Ghosh, M. J. Alhilaly, X. Dong, M. N. Hedhili, E. Abou-Hamad, B. Alamer, S. Nematulloev, Y. Han, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2020, 142, 8696–8705 CrossRef PubMed.
- T. Higaki, Y. Li, S. Zhao, Q. Li, S. Li, X.-S. Du, S. Yang, J. Chai and R. Jin, Angew. Chem., Int. Ed., 2019, 58, 8291–8302 CrossRef CAS.
- C. Sun, N. Mammen, S. Kaappa, P. Yuan, G. Deng, C. Zhao, J. Yan, S. Malola, K. Honkala, H. Häkkinen, B. K. Teo and N. Zheng, ACS Nano, 2019, 13, 5975–5986 CrossRef CAS PubMed.
- J. Hu, Y.-M. Li, B. Zhang, X. Kang and M. Zhu, Inorg. Chem. Front., 2024, 11, 4974–5003 RSC.
- J. Wang, J. Cai, K.-X. Ren, L. Liu, S.-J. Zheng, Z.-Y. Wang and S.-Q. Zang, Sci. Adv., 2024, 10, eadn7556 CrossRef CAS.
- H. Guo, Y. Chen, Y.-Z. Han, Q. Wu, L. Wang, Q. Xu, R. Huo, X. Gong, J. Sun, Q. Tang and H. Shen, Chem. Mater., 2024, 36, 7243–7251 CrossRef CAS.
- M. S. Mathew, G. Krishnan, A. A. Mathews, K. Sunil, L. Mathew, R. Antoine and S. Thomas, Nanomaterials, 2023, 13, 1874 CrossRef CAS PubMed.
- B. Mondal, K. Basu, R. Jana, P. Mondal, B. Hansda, A. Datta and A. Banerjee, ACS Appl. Nano Mater., 2022, 5, 7932–7943 CrossRef CAS.
- S. Lee, M. S. Bootharaju, G. Deng, S. Malola, W. Baek, H. Häkkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2020, 142, 13974–13981 CrossRef CAS PubMed.
- A. W. Cook, Z. R. Jones, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2018, 140, 394–400 CrossRef CAS.
- G. Yang, Y. Xie, Y. Wang, Y. Tang, L. L. Chng, F. Jiang, F. Du, X. Zhou, J. Y. Ying and X. Yuan, Nano Res., 2023, 16, 1748–1754 CrossRef CAS.
- C.-Y. Liu, T.-Y. Liu, Z.-J. Guan, S. Wang, Y.-Y. Dong, F. Hu, D.-E. Jiang and Q.-M. Wang, CCS Chem., 2024, 6, 1581–1590 CrossRef CAS.
- P. Kumar and M. Nemiwal, Chem. – Asian J., 2024, 19, e202400062 CrossRef CAS PubMed.
- B. Yan, X. You, X. Tang, J. Sun, Q. Xu, L. Wang, Z.-J. Guan, F. Li and H. Shen, Chem. Mater., 2024, 36, 1004–1012 CrossRef CAS.
- L. Dong, L. Yu, X. Sun, X. Tang, X. You, J. Tang, Z.-A. Nan, D. Cao, Y. Jia, S. Li, F. Li, S. Guo and H. Shen, Inorg. Chem. Front., 2023, 10, 5745–5751 RSC.
- T.-A. D. Nguyen, Z. R. Jones, B. R. Goldsmith, W. R. Buratto, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2015, 137, 13319–13324 CrossRef CAS PubMed.
- T.-A. D. Nguyen, Z. R. Jones, D. F. Leto, G. Wu, S. L. Scott and T. W. Hayton, Chem. Mater., 2016, 28, 8385–8390 CrossRef CAS.
- P. Yuan, R. Chen, X. Zhang, F. Chen, J. Yan, C. Sun, D. Ou, J. Peng, S. Lin, Z. Tang, B. K. Teo, L.-S. Zheng and N. Zheng, Angew. Chem., Int. Ed., 2019, 58, 835–839 CrossRef CAS PubMed.
- A. Ghosh, R.-W. Huang, B. Alamer, E. Abou-Hamad, M. N. Hedhili, O. F. Mohammed and O. M. Bakr, ACS Mater. Lett., 2019, 1, 297–302 CrossRef CAS.
- C. Dong, R.-W. Huang, C. Chen, J. Chen, S. Nematulloev, X. Guo, A. Ghosh, B. Alamer, M. N. Hedhili, T. T. Isimjan, Y. Han, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2021, 143, 11026–11035 CrossRef CAS.
- S. Biswas, S. Das and Y. Negishi, Nanoscale Horiz., 2023, 8, 1509–1522 RSC.
- N. Li, B. Li, K. Murugesan, A. Sagadevan and M. Rueping, ACS Catal., 2024, 11974–11989, DOI:10.1021/acscatal.4c03238.
- A. Ghosh, A. Sagadevan, K. Murugesan, S. A. F. Nastase, B. Maity, M. Bodiuzzaman, A. Shkurenko, M. N. Hedhili, J. Yin, O. F. Mohammed, M. Eddaoudi, L. Cavallo, M. Rueping and O. M. Bakr, Mater. Horiz., 2024, 11, 2494–2505 RSC.
- B. Alamer, A. Sagadevan, M. Bodiuzzaman, K. Murugesan, S. Alsharif, R.-W. Huang, A. Ghosh, M. H. Naveen, C. Dong, S. Nematulloev, J. Yin, A. Shkurenko, M. Abulikemu, X. Dong, Y. Han, M. Eddaoudi, M. Rueping and O. M. Bakr, J. Am. Chem. Soc., 2024, 146, 16295–16305 CrossRef CAS PubMed.
- A. Jana, S. Duary, A. Das, A. R. Kini, S. Acharya, J. Machacek, B. Pathak, T. Base and T. Pradeep, Chem. Sci., 2024, 15, 13741–13752, 10.1039/D4SC01566E.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS.
- G. Ćirić-Marjanović, Synth. Met., 2013, 177, 1–47 CrossRef.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS.
- H.-J. Knölker, The alkaloids: chemistry and biology, Academic Press, 2011, vol. 70 Search PubMed.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS PubMed.
- F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed.
- J. X. Qiao and P. Y. S. Lam, Synthesis, 2011, 829–856 CrossRef CAS.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13–31 RSC.
- S.-T. Kim, M. J. Strauss, A. Cabré and S. L. Buchwald, J. Am. Chem. Soc., 2023, 145, 6966–6975 CrossRef CAS PubMed.
- S. Bhunia, S. De and D. Ma, Org. Lett., 2022, 24, 1253–1257 CrossRef CAS PubMed.
- Q. Yang, Y. Zhao and D. Ma, Org. Process Res. Dev., 2022, 26, 1690–1750 CrossRef CAS.
- A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, eaav9713 CrossRef PubMed.
- S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Science, 2012, 338, 647–651 CrossRef CAS.
- A. C. Bissember, R. J. Lundgren, S. E. Creutz, J. C. Peters and G. C. Fu, Angew. Chem., Int. Ed., 2013, 52, 5129–5133 CrossRef CAS.
- D. T. Ziegler, J. Choi, J. M. Muñoz-Molina, A. C. Bissember, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 13107–13112 CrossRef CAS PubMed.
- Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681–684 CrossRef CAS PubMed.
- C. D. Matier, J. Schwaben, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 17707–17710 CrossRef CAS PubMed.
- C. Chen, J. C. Peters and G. C. Fu, Nature, 2021, 596, 250–256 CrossRef CAS PubMed.
- J. M. Ahn, T. S. Ratani, K. I. Hannoun, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 12716–12723 CrossRef CAS PubMed.
- H. Cho, H. Suematsu, P. H. Oyala, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2022, 144, 4550–4558 CrossRef CAS.
- G. Singh, M. Kumar and V. Bhalla, Green Chem., 2018, 20, 5346–5357 RSC.
- Z. Xu, W. Wang, Y. Xu, S. Song, J. Yu, W. Song, H. Xu, H. Fu and Z. Li, ACS Catal., 2023, 13, 13920–13930 CrossRef CAS.
- A. Sagadevan, A. Ghosh, P. Maity, O. F. Mohammed, O. M. Bakr and M. Rueping, J. Am. Chem. Soc., 2022, 144, 12052–12061 CrossRef CAS.
- W. Liu, J. Xu, X. Chen, F. Zhang, Z. Xu, D. Wang, Y. He, X. Xia, X. Zhang and Y. Liang, Org. Lett., 2020, 22, 7486–7490 CrossRef CAS.
- S. Xia, L. Gan, K. Wang, Z. Li and D. Ma, J. Am. Chem. Soc., 2016, 138, 13493–13496 CrossRef CAS PubMed.
- W. Zhou, M. Fan, J. Yin, Y. Jiang and D. Ma, J. Am. Chem. Soc., 2015, 137, 11942–11945 CrossRef CAS PubMed.
- Z. Chen and D. Ma, Org. Lett., 2019, 21, 6874–6878 CrossRef CAS.
- C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed.
- O. Mongin, L. Porrès, L. Moreaux, J. Mertz and M. Blanchard-Desce, Org. Lett., 2002, 4, 719–722 CrossRef CAS PubMed.
- I. Paterson, R. D. M. Davies and R. Marquez, Angew. Chem., Int. Ed., 2001, 40, 603–607 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed.
- K. C. Dissanayake, P. O. Ebukuyo, Y. J. Dhahir, K. Wheeler and H. He, Chem. Commun., 2019, 4973–4976 RSC.
- M. Gazvoda, M. Virant, B. Pinter and J. Košmrlj, Nat. Commun., 2018, 9, 4814 CrossRef.
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- D.-L. Zhu, R. Xu, Q. Wu, H.-Y. Li, J.-P. Lang and H.-X. Li, J. Org. Chem., 2020, 85, 9201–9212 CrossRef CAS.
- M. Carril, A. Correa and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 4862–4865 CrossRef CAS.
- F. M. Moghaddam, G. Tavakoli and H. R. Rezvani, Catal. Commun., 2015, 60, 82–87 CrossRef CAS.
- Y. Liu, V. Blanchard, G. Danoun, Z. Zhang, A. Tlili, W. Zhang, F. Monnier, A. Van Der Lee, J. Mao and M. Taillefer, ChemistrySelect, 2017, 2, 11599–11602 CrossRef CAS.
- F. Monnier, F. Turtaut, L. Duroure and M. Taillefer, Org. Lett., 2008, 10, 3203–3206 CrossRef CAS PubMed.
- A. Sagadevan and K. C. Hwang, Adv. Synth. Catal., 2012, 354, 3421–3427 CrossRef CAS.
- K. V. Arundhathi, P. Vaishnavi, T. Aneeja and G. Anilkumar, RSC Adv., 2023, 13, 4823–4834 RSC.
- S. Diyarbakir, H. Can and Ö. Metin, ACS Appl. Mater. Interfaces, 2015, 7, 3199–3206 CrossRef CAS.
- B. Wang, X. Guo, G. Jin and X. Guo, Catal. Commun., 2017, 98, 81–84 CrossRef CAS.
- A. Elhage, A. E. Lanterna and J. C. Scaiano, ACS Sustainable Chem. Eng., 2018, 6, 1717–1722 CrossRef CAS.
- G. S. More, A. K. Kar and R. Srivastava, Inorg. Chem., 2022, 61, 19010–19021 CrossRef CAS.
- P. Singh and A. C. Shaikh, Chem. Commun., 2023, 59, 11615–11630 RSC.
- M. Shanmugam, A. Sagadevan, V. P. Charpe, V. K. K. Pampana and K. C. Hwang, ChemSusChem, 2020, 13, 287–292 CrossRef CAS.
- S. Zhang and L. Zhao, Nat. Commun., 2019, 10, 4848 CrossRef PubMed.
- A. Bakhoda, O. E. Okoromoba, C. Greene, M. R. Boroujeni, J. A. Bertke and T. H. Warren, J. Am. Chem. Soc., 2020, 142, 18483–18490 CrossRef CAS PubMed.
- S. Zhang and L. Zhao, Nat. Commun., 2023, 14, 6741 CrossRef CAS PubMed.
- S. Nematulloev, A. Sagadevan, B. Alamer, A. Shkurenko, R. Huang, J. Yin, C. Dong, P. Yuan, K. E. Yorov, A. A. Karluk, W. J. Mir, B. E. Hasanov, M. Nejib Hedhili, N. M. Halappa, M. Eddaoudi, O. F. Mohammed, M. Rueping and O. M. Bakr, Angew. Chem., Int. Ed., 2023, 62, e202303572 CrossRef CAS.
- R. J. Enemærke, T. B. Christensen, H. Jensen and K. Daasbjerg, J. Chem. Soc., Perkin Trans. 2, 2001, 1620–1630, 10.1039/B102835A.
- M. Mansour, R. Giacovazzi, A. Ouali, M. Taillefer and A. Jutand, Chem. Commun., 2008, 6051–6053, 10.1039/B814364A.
- S. Biswas, A. Pal, M. K. Jena, S. Hossain, J. Sakai, S. Das, B. Sahoo, B. Pathak and Y. Negishi, J. Am. Chem. Soc., 2024, 146, 20937–20944 CrossRef CAS PubMed.
- M. Majek and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2013, 52, 5919–5921 CrossRef CAS PubMed.
- V. W.-W. Yam, K. Kam-Wing Lo and K. Man-Chung Wong, J. Organomet. Chem., 1999, 578, 3–30 CrossRef CAS.
- A. Sagadevan, A. Ragupathi and K. C. Hwang, Angew. Chem., Int. Ed., 2015, 54, 13896–13901 CrossRef CAS.
- A. Sagadevan, V. P. Charpe, A. Ragupathi and K. C. Hwang, J. Am. Chem. Soc., 2017, 139, 2896–2899 CrossRef CAS.
- A. Sagadevan, A. Ragupathi, C.-C. Lin, J. R. Hwu and K. C. Hwang, Green Chem., 2015, 17, 1113–1119 RSC.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- R. Ramkumar and P. Anbarasan, Cu-Catalyzed Click Reactions in Copper Catalysis in Organic Synthesis, 2020, pp. 177–207 DOI:10.1002/9783527826445.ch9.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- X. Wang, B. Huang, X. Liu and P. Zhan, Drug Discovery Today, 2016, 21, 118–132 CrossRef CAS PubMed.
- J. D. Megiatto, Jr. and D. I. Schuster, J. Am. Chem. Soc., 2008, 130, 12872–12873 CrossRef.
- J. W. Lee, J. H. Kim, B.-K. Kim, W. S. Shin and S.-H. Jin, Tetrahedron, 2006, 62, 894–900 CrossRef CAS.
- D. Döhler, P. Michael and W. H. Binder, Acc. Chem. Res., 2017, 50, 2610–2620 CrossRef PubMed.
- J. Chen, J. Wang, K. Li, Y. Wang, M. Gruebele, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2019, 141, 9693–9700 CrossRef CAS PubMed.
- C. Yang, J. P. Flynn and J. Niu, Angew. Chem., Int. Ed., 2018, 57, 16194–16199 CrossRef CAS PubMed.
- G. Oyman Eyrilmez, S. Doran, E. Murtezi, B. Demir, D. Odaci Demirkol, H. Coskunol, S. Timur and Y. Yagci, Macromol. Biosci., 2015, 15, 1233–1241 CrossRef CAS PubMed.
- The Nobel Prize in Chemistry, 2022, https://www.nobelprize.org/prizes/chemistry/2022/popular-information/.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS.
- K. Min, H. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 1789–1791 CrossRef CAS.
- G. S. Kumar and Q. Lin, Chem. Rev., 2021, 121, 6991–7031 CrossRef CAS PubMed.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS.
- M. A. Tasdelen and Y. Yagci, Tetrahedron Lett., 2010, 51, 6945–6947 CrossRef CAS.
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256–259 CrossRef CAS.
- Y.-Q. Xiao, P. Shang, X.-Y. Chen, X.-Q. Pu, K.-W. Jiang and X.-F. Jiang, Chem. Mater., 2024, 36, 2027–2038 CrossRef CAS.
- T. Yang, J. Jia, L. Xiong, S. Jin and M. Zhu, Chem. Commun., 2024, 60, 9614–9617, 10.1039/D4CC03288H.
- C. Dong, R.-W. Huang, A. Sagadevan, P. Yuan, L. Gutiérrez-Arzaluz, A. Ghosh, S. Nematulloev, B. Alamer, O. F. Mohammed, I. Hussain, M. Rueping and O. M. Bakr, Angew. Chem., Int. Ed., 2023, 62, e202307140 CrossRef CAS PubMed.
- L.-J. Liu, M.-M. Zhang, Z. Deng, L.-L. Yan, Y. Lin, D. L. Phillips, V. W.-W. Yam and J. He, Nat. Commun., 2024, 15, 4688 CrossRef CAS PubMed.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed.
- A. W. Girotti, J. Lipid Res., 1998, 39, 1529–1542 CrossRef CAS.
- M. Cao, R. Pang, Q.-Y. Wang, Z. Han, Z.-Y. Wang, X.-Y. Dong, S.-F. Li, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2019, 141, 14505–14509 CrossRef CAS PubMed.
- C. Zhang, Z. Wang, W.-D. Si, L. Wang, J.-M. Dou, Z.-Y. Gao, C.-H. Tung and D. Sun, ACS Nano, 2022, 16, 9598–9607 CrossRef CAS.
- C. Zhu, Z.-L. Chen, H. Li, L. Lu, X. Kang, J. Xuan and M. Zhu, J. Am. Chem. Soc., 2024, 146, 23212–23220 CrossRef CAS.
- M. Bodiuzzaman, K. Murugesan, P. Yuan, B. Maity, A. Sagadevan, N. Malenahalli, H. S. Wang, P. Maity, M. F. Alotaibi, D.-E. Jiang, M. Abulikemu, O. F. Mohammed, L. Cavallo, M. Rueping and O. M. Bakr, J. Am. Chem. Soc., 2024, 146, 26994–27005 CrossRef CAS PubMed.
Footnote |
| † A. S. and K. M. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |