
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitro group and K+-based secondary building units for the self-assembly of 3D coordination polymers built on dinuclear dianionic helicate connectors†
Raúl
Mendoza-Báez
,
Alan
Molina-Renteria
 and
Juan
Olguín
*
and
Juan
Olguín
*
Departamento de Química, Centro de Investigación y de Estudios Avanzados del IPN (Cinvestav), Avenida IPN 2508, Col. San Pedro Zacatenco, Ciudad de México 07360, Mexico. E-mail: jolguin@cinvestav.mx
First published on 5th April 2024
Abstract
Two 3D coordination polymers based on dinuclear dianionic helicates, K2[MII2(L)3] M = MnII (1) and NiII (2), were obtained by slow liquid–liquid diffusion of solutions of 2-hydroxy-5-nitrobenzaldehyde, 4,4′-methylenedianiline, KOtBu and the metal salt at room temperature. The resulting dianionic triply stranded helicates self-assemble into novel 3D coordination polymers, in which the helicate units act as connectors. The assembly is promoted by the phenolate bridging ability and the coordination of the pre-organized NO2 groups to K–(O–N–O)4–K nodes, resulting in robust paramagnetic materials. SC-XRD shows that the metal centers are located 11.257(2) and 11.264(1) Å apart. The Mn–L bond lengths correspond to the high-spin (HS, S = 5/2) state for 1. The magnetic behavior for both complexes shows weak magnetic exchange, as expected due to the spin carrier separation, and the confirmation of the HS-sate for the MnII centers. For complex 1, an antiferromagnetic exchange is detected, whereas for complex 2, a mixture of intramolecular antiferromagnetic and intermolecular ferromagnetic exchanges are detected. Thus, we demonstrated that nitro groups can be used for the stabilization of anionic complexes because coordination to K+ results in stable intricate architectures.
Introduction
Triply stranded dimetallic helicates1 have received considerable attention as they can offer interesting physicochemical properties such as chirality,2 act as hosts for molecules,3 magnetism,4 and luminescence.5The ligand design is fundamental for obtaining helicate complexes; therefore, two approaches can be followed: a designer approach in which a polydentate multinucleated ligand can be synthesized (investing serious time and effort) or a self-assembly approach, in which in situ formation of the ligand and the helicate complex occurs, due to the right match of subcomponents, e.g., ligands bite angle, metal ion geometry, and angle between coordination pockets. Therefore, the latter approach is commonly used due to mild reaction conditions, and the possibility of using commercially available motifs.
Imine-based ligands are some of the choices for the self-assembly of dinuclear helicates because the formation of the ligand is metal-directed by simply mixing a diamine, containing both NH2 at an angle smaller than 180° using a flexible linker and two equivalents of a monoaldehyde; the latter should contain, at least, a donor moiety. Most of the imine-based helicates are derived from N-heterocycle monoaldehydes, although there are very few examples known based on cathecolatealdehyde-6 and, to a much less extent, hydroxybenzaldehyde-synthons.7,8 These phenolate groups are known to act as bridging ligands, with a M1–OPh–M2 motif, forming discrete or polynuclear homo- and hetero-metallic complexes.9
However, the use of helicates as metalloligands is not common,10 and even fewer examples are found for the construction of more complex 1D-7,11,12 or 3D structures.13 The ligands in this type of structure contain secondary assembly instruction sites that permit the 1D or 3D growth; to date, only pyridine or carboxylate groups have been used. In fact, the majority of metalloligands that are used for the synthesis of intricate designer heterometallic architectures such as metal–organic frameworks (MOF), coordination networks and polymers, contain N or O-functional groups for secondary assembly instruction,14i.e., the interconnections with the second metal synthon are formed using N-heterocycles, amides, amines, imines, and/or carboxylate groups. Due to the robustness and coordination versatility of the latter group, it has been widely used for the generation of such architectures.15
Thus, we have decided to explore the formation of dinuclear helicates based on 2-hydroxy-5-nitrobenzaldehyde and 4,4′-methylenedianiline as the diamine moiety. The resulting imine ligand (H2L) comprises two bidentate (N and O donors) coordination pockets. Thus, upon deprotonation of the OH groups, three ligand strands could wrap two metal centers, resulting in a N3O3 coordination sphere (Fig. 1). The latter coordination sphere could impart interesting magnetic properties for Mn complexes, e.g., spin crossover (SCO).16 Moreover, the ligand contains nitro groups that can be used as secondary assembly instruction because it has been observed that this group can coordinate alkali and alkaline earth metal counter ions (see ESI†) in anionic complexes.
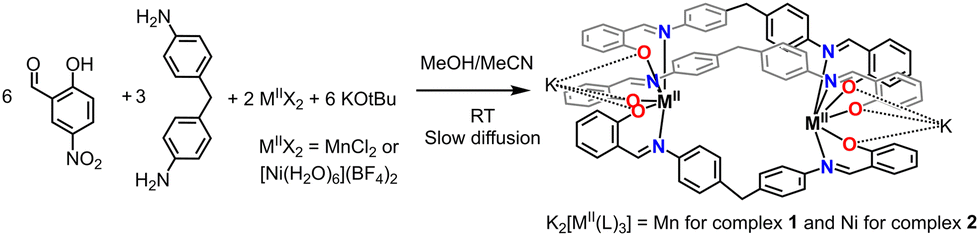 | ||
| Fig. 1 Synthesis of triply stranded dianionic dimetallic helicates K2[M2(L)3], M = Mn2+ and Ni2+, under mild reaction conditions. | ||
Experimental
General
All reactions were carried out in an argon atmosphere using standard Schlenk techniques. The solvents were degassed just before use. Elemental analyses were performed on a Thermo-Finnigan Flash 1112 elemental analyzer at the Chemistry Department CINVESTAV. Infrared (IR) spectra were obtained on a Varian 640-IR spectrometer using a diamond attenuated total reflectance (ATR) method. The spectral data were obtained in a spectral window of 4000–550 cm−1. X-ray diffraction measurements were collected on a Bruker D8 VENTURE diffractometer (PHOTON 100 detector), using a multilayer mirror monochromator and MoKα source (λ = 0.71073 Å).Synthesis
Results
Synthesis and characterization
The attempted architecture was a neutral helicate, where the metal centers would have a 3+ oxidation state. In many cases, coordination of phenolate ligands to low oxidation state metal ions results in metal oxidation. Therefore, the synthesis of the helicates (see Experimental section) was attempted by employing MnCl2 under inert conditions, followed by crystallization in the air for spontaneous oxidation of the metal center. The synthesis of the complexes was achieved by two methods, one in which all the components were mixed in a refluxing solvent resulting in the precipitation of solids, which were insoluble in most of the common solvents, except for DMF or DMSO that partially dissolved the complexes. Therefore, a second method was used, a liquid–liquid slow diffusion method resulted in crystals suitable for single-crystal X-ray diffractometry (SCXRD, see below). Surprisingly, SCXRD showed the formula K2[MnII2(L)3]·MeOH (1), which is a triply-stranded dianionic dimetallic helicate in which the metal ion is found in a +2 oxidation state. To prove that the dianionic helicates were stable, [NiII(H2O)6](BF4)2 was used as the metal source under similar conditions, obtaining K2[NiII2(L)3]·MeCN (2) according to SCXRD, thus confirming the dianionic nature of the helicates.Both complexes were characterized as solids by IR and microanalysis. It is possible to observe in the IR spectra the characteristic υ for the NO2 group at 1593 and 1296 cm−1 for 1 and 1595 and 1292 cm−1 for 2. The imine υN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C is found at 1614 cm−1 for both complexes. Due to the lack of solubility, it was not possible to further characterize the complexes by other techniques.
C is found at 1614 cm−1 for both complexes. Due to the lack of solubility, it was not possible to further characterize the complexes by other techniques.
Single crystal X-ray diffractometry
SCXRD measurements were performed at RT for 1 and 2. Crystal data and refinement parameters are given in Table 1, while Table 2 shows some selected structural parameters. Both metal complexes are isostructural, crystallizing in the C2/c space group, Fig. 2 and S1.† The asymmetric unit consists of one half helicate dianion, one MeOH molecule disordered over two positions for 1, but one half MeCN solvent molecule for 2, and one K+; the rest of the helicate, the second K+ and the solvent molecule are generated by a C2 rotation axis. The N3O3 coordination sphere for the only symmetry-independent metal center, for both complexes, is formed by three Nimine and three Ophenolate in a fac mode, in a distorted octahedral geometry. Complex 1 exhibits a greater degree of distortion compared to 2, based on the values for the ∑Oh parameter17,18 (63.75° and 46.67°, respectively, Table 2 and S3†).| Complex | 1 | 2 |
|---|---|---|
| Empirical formula | C83H62K2Mn2N12O20 | C83H57K2N13Ni2O18 |
| Formula weight | 1735.52 | 1720.03 |
| Temperature | 298(2) K | 296(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å |
| Crystal system | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c |
| Unit cell dimensions | a = 16.080(3) Å | a = 15.7610(11) Å |
| α = 90° | α = 90° | |
| b = 14.472(3) Å | b = 14.5644(9) Å | |
| β = 100.611(8)° | β = 97.921(2)° | |
| c = 37.326(8) Å | c = 36.558(3) Å | |
| γ = 90° | γ = 90° | |
| Volume | 8537(3) Å3 | 8311.7(10) Å3 |
| Z | 4 | 4 |
| Density (calculated) | 1.350 Mg m−3 | 1.375 Mg m−3 |
| Absorption coefficient | 0.469 mm−1 | 0.629 mm−1 |
| F(000) | 3568 | 3536 |
| Crystal size | 0.350 × 0.260 × 0.100 mm3 | 0.384 × 0.217 × 0.056 mm3 |
| Theta range for data collection | 2.221 to 27.195° | 2.250 to 25.026° |
| Index ranges | −20 ≤ h ≤ 20, −18 ≤ k ≤ 18, −47 ≤ l ≤ 47 | −18 ≤ h ≤ 18, −17 ≤ k ≤ 17, −42 ≤ l ≤ 43 |
| Reflections collected | 126![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 744 744 |
204178 |
| Independent reflections | 9469 [R(int) = 0.1708] | 7322 [R(int) = 0.1132] |
| Completeness to theta = 25.242° | 99.70% | 99.60% |
| Absorption correction | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max. and min. transmission | 0.7455 and 0.4820 | 0.7452 and 0.6704 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 9469/36/558 | 7322/0/551 |
| Goodness-of-fit on F2 | 1.026 | 1.066 |
| Final R indices [I > 2sigma(I)] | R 1 = 0.0737, wR2 = 0.1871 | R 1 = 0.0642, wR2 = 0.1671 |
| R indices (all data) | R 1 = 0.1797, wR2 = 0.2480 | R 1 = 0.1076, wR2 = 0.1879 |
| Largest diff. peak and hole | 0.851 and −0.356 e Å−3 | 0.795 and −0.267 e Å−3 |
| Complex | 1 | 2 |
|---|---|---|
| a The octahedral distortion parameter, ∑Oh, is the sum of the absolute deviation values of the twelve cis angles to 90°. ∑Oh = 0 for a perfect octahedron. b X is a methanol O-atom for complex 1, while an acetonitrile N-atom for complex 2. | ||
| M–N [Å] | 2.293(4) | 2.124(3) |
| M–O [Å] | 2.116(3) | 2.040(3) |
| M–M [Å] | 11.257(2) | 11.264(1) |
| K–Ophenolate [Å] | 2.829(3) | 2.803(3) |
| K–Onitro [Å] | 2.951(5) | 2.983(5) |
| K–Xsolventb [Å] | 3.999(7) | 3.448(19) |
| cis O–M–O [°] | 87.07(14) | 85.56(12) |
| cis N–M–N [°] | 99.19(13) | 97.44(13) |
| cis O–M–N [°] | 86.67(13) | 88.16(13) |
| trans O–M–N [°] | 168.68(13) | 171.19(12) |
| ∑Oha | 63.75(46) | 46.67(43) |
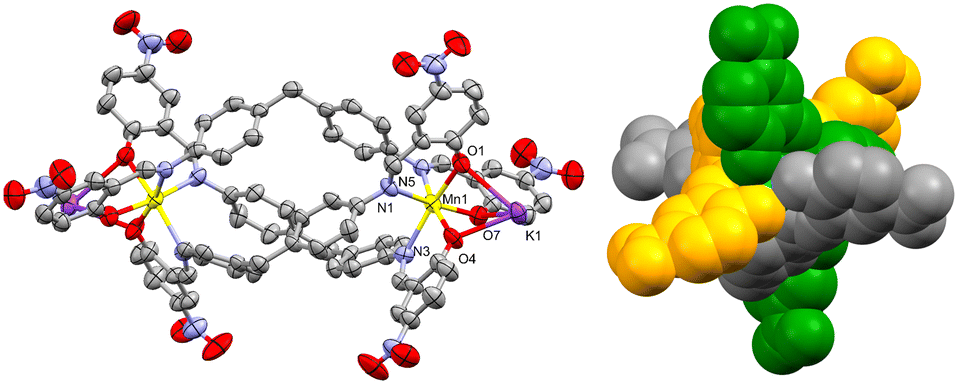 | ||
| Fig. 2 Left: perspective view of 1, 40% probability level ellipsoids. Solvent molecules and H-atoms are omitted. Right: side view of 2 in a space-fill model, each ligand strand is colored differently to highlight the wrapping of the ligands around the metal centers. | ||
The three Ophenolate groups are found in the external face away from the helicate cavity, which permits the O to act as a bridging ligand to a K+. The latter is found in a nine-coordination sphere comprising three Ophenolate and six O coming from four NO2 groups of neighboring complexes, similar to the acetate paddle wheel structures. All the NO2 groups act as μ1,1 and μ1,3-bridging ligands connecting to a symmetry-generated K+ possessing the same coordination environment, promoting the formation of dinuclear potassium units (Fig. 3) interconnecting the dinuclear helicates in three dimensions (Fig. 4).
 | ||
| Fig. 3 Numbering scheme of the symmetry-independent atoms and coordination sphere of the two K+ metal centers interconnected by μ1-μ1,3-bridging NO2 ligands. Dangling oxygen atoms correspond to Ophenolate moieties. The rest of the dinuclear unit, solvent molecule, H-atoms are omitted. | ||
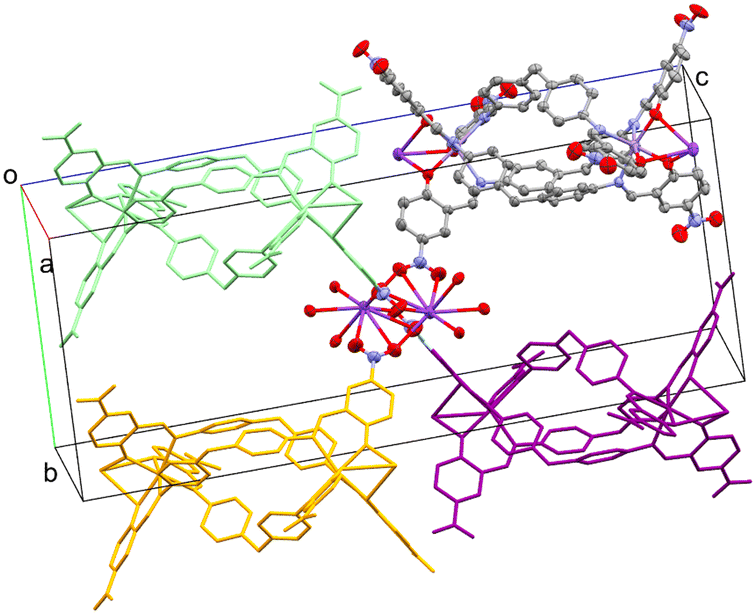 | ||
| Fig. 4 Perspective view of the three-dimensional growth of (1), promoted by the NO2 groups and K+. The dinuclear K unit is drawn with ellipsoids at 30% probability level. The coordinated NO2 to the K dinuclear node come from different helicate units, highlighted in different colors and in a capped stick style. Solvent molecules and H-atoms are omitted for the sake of clarity. | ||
Mn–Ophenolate (2.116(3) Å) and Mn–Nimine (2.293(4) Å) bond lengths of complex 1 agree with the values reported for high spin (S = 5/2) six-coordinate MnII complexes with salicylamide-based ligands (2.073–2.196 Å and 2.185–2.273 Å, respectively).19–22 However, these examples correspond to complexes with discrete (0-D) or one-dimensional arrangements and, in most cases complexes that contain co-ligands/anions/solvent molecules to complete the coordination sphere. In this sense, 1 is a novel example of a 3D homoleptic MnII coordination polymer. For complex 2, the Ni–Ophenolate (2.040(3) Å) and Ni–Nimine (2.124(3) Å) bond lengths also agree with values previously reported for analogous complexes (1.978–2.083 Å and 1.988–2.064 Å, respectively).28–31 In Fig. S2,† the ligands used in the examples mentioned above are shown. In both cases, the M–Nimine length is slightly higher than the range found, possibly due to the effects of the internal triple-stranded structure. This relatively long bond is unusual, as examples of helicates or mesocates of MnII and NiII with Schiff base-derived ligands show shorter M–Nimine lengths compared to 1 and 2.23–26 Only one example showed a Ni–Nimine bond length (2.179 Å), even higher than that reported in complex 2.27 Moreover, both complexes 1 and 2 have the peculiarity of containing the MII metal centers in a dinuclear anionic species, which had not been reported (as far as we know) for MnII and NiII complexes with salicylimine-based-ligands.
Although the K+ center is nine-coordinated, KO9, the solvent molecules also interact with it in a non-covalent manner. In 1, the K–OMeOH distance is 3.999(7) Å (Fig. S3a†), which is close to the previously reported K–OMeOH distances in a highly-coordinated potassium complex (3.913 Å).28 However, this OMeOH cannot be considered as part of the K+ coordination sphere. A search in the CSD database (update June 2023) shows at least 158 crystal structures with ten-coordinated potassium, KO10, where the K–O distances range from 2.574 to 3.389 Å (see histogram in Fig. S4†), suggesting that the K–OMeOH interaction in 1 is weak. Furthermore, there is an H-bond between the methanol –OH group (H donor) and the Ophenolate (H acceptor) atom (Fig. S5†), which could favor the orientation of OMeOH towards K+. Complex 2 shows a weak K–NMeCN interaction, with a distance of 3.448(19) Å (Fig. S3b†). Crystal structures that present ten-coordinated potassium atoms in an N1O9 coordination sphere are rare since only seven examples were found (based on the CSD database, updated in June 2023), of which only one structure presents MeCN as a ligand.29 K–N bond lengths are found in a range of 3.008 to 3.160 Å, which are smaller than the value found for 2. This suggests, as in 1, a weak K–NMeCN interaction.
Thus, the K atom is found in a distorted nine-coordinate sphere. The average K–Ophenolate bond length is 2.828(3) and 2.803(3) Å for 1 and 2, respectively, Table 2, which are notably higher than the average MnII/NiII–Ophenolate of 2.116(3) and 2.040(3) Å, respectively, due to a more covalent interaction between the latter atoms compared to K–Ophenolate. The MnII–K and NiII–K heterometallic intramolecular distances are 3.555(1) and 3.564(1) Å, while the homometallic (M–M) distances are 11.257(2) and 11.264(1) Å, respectively.
Interestingly, the shortest M–M distance is not the intramolecular one, i.e., within a helicate molecular unit. Fig. S6–S8† show the five shortest M–M distances, in a range between 10.73 and 13.05 Å (Table S4†). The shortest M–M distance is the one parallel to the ab plane, 10.817(2) Å for 1 and 10.730(1) Å for 2, while the longest is the one parallel to the ac plane, 13.050(2) Å for 1 and 12.535(1) Å for 2.
As mentioned above, dinuclear K units act as nodes between adjacent metal helicates due to K–Onitro bonds. Each coordinated NO2 group comes from a different helicate unit, as shown in Fig. 4. This allows the growth of the structure through the three axes, forming the 3D network (Fig. S9†). The phenolate oxygen atoms coordinated to the K+ center form O–K–O angles of approximately 59–63°, while the angles formed by the Onitro are ca. 40°. These O–K–O angle values are close to those previously reported in similar complex structures involving metalloligands.28,30 The 3D topology for both complexes is a pcu; 6/4/c1; sqc1, in a cluster representation, according to TopCryst.31 The CSD occurrence of the pcu; 6/4/c1; sqc1 topology in a cluster representation (all nodes and single node) is 3541 structures.
There is another non-covalent interaction that promotes the 3D growth of the crystal structure, that is, the intermolecular π–π stacking between two aromatic rings of terminal nitrophenolate groups, as shown in Fig. S10.† This interaction is an off-centered parallel stacking with an interplanar angle of 0.0°. There is an interplanar normal distance of approximately 3.51–3.53 Å and a horizontal displacement (offset) of 2.30–2.39 Å (Fig. S11 and S12†). Both normal and offset distances agree with the range of typical distances reported in other π–π stacking systems.32,33 These π–π interactions, together with the coordinated K atoms, promote the structural growth of zigzag-type chains along the c axis, Fig. S13.† Furthermore, intramolecular C–H/π interactions occur in complexes 1 and 2, where a C(sp2)–H of a benzene ring from diphenylmethane moiety is directed towards the benzene center of the adjacent ligand strand (Fig. S14 and S15†). Both aromatic rings of a single diphenylmethane moiety act as the C–H/π acceptor, while the remaining two ligands act as the donors. The C–H/π distances are 3.640 and 3.503 Å for complexes 1 and 2, respectively. The interplanar angles are close to 90° (87.27° and 88.89°), and the C–H– π α angle ranges from 167.46° to 168.80°, both parameters indicate a quasi-perpendicular interaction (Fig. S16†).
Magnetic and N2 adsorption properties
The magnetic properties of crystalline samples for both complexes 1 and 2 were obtained in the 5–300 K temperature range under a 0.1 T external magnetic field, Fig. 5. In the case of complex 1, the room temperature χMT = 9.30 cm3 mol−1 K is in good agreement for two non-interacting Mn2+ in the high-spin state (S = 5/2), spin only χMT value of 8.75 cm3 mol−1 K. The value slightly decreases upon cooling reaching 8.24 cm3 mol−1 K at 50 K. The best fit for the data using PHI,34Ĥex = −2JS1·S2, indicates a negligible antiferromagnetic coupling J = −0.0029 cm−1, g = 1.954, TIP = 414 × 10−6 cm3 mol−1, monomeric impurity of 0.07 and a residual error R = 0.92.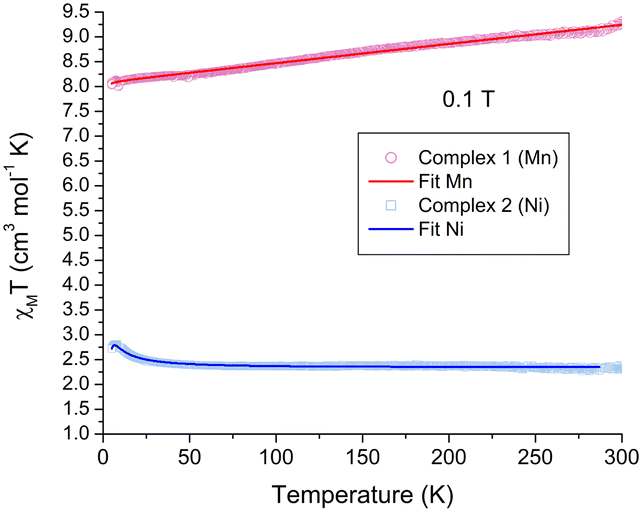 | ||
| Fig. 5 χ M T vs. T magnetic behavior for complexes 1 and 2. The solid lines represent the best fit for both complexes. | ||
In the case of the complex 2, χMT = 2.37 cm3 mol−1 K at RT, which is slightly higher than the expected spin-only value for two S = 1 systems, 2.00 cm3 mol−1 K, and is maintained relatively constant down to 50 K, χMT = 2.40 cm3 mol−1 K. Below this temperature, an increase is observed reaching a maximum at 7.2 K of χMT = 2.80 cm3 mol−1 K, on cooling, this value decreases again. The latter behavior indicates two types of exchange interactions, as seen for other dinuclear Ni complexes.35 The best fit using PHI37 affords an antiferromagnetic intramolecular interaction between Ni centers J = −0.92 cm−1, a ferromagnetic intermolecular interaction zJ = 0.44 cm−1, g = 2.15, a ZFS D parameter of 2.7 cm−1, TIP = 94.8 × 10−6 cm3 mol−1 and R = 0.38, which are in good agreement for dinuclear Ni complexes containing O-based ligands, previously reported.36 The small exchange coupling values for both systems are consistent with distant metal centers found in the helicate units.
In addition, the total pore volume and specific surface area of complex 1 were measured through N2 adsorption isotherms at 77 K. The BET-surface area value found is 0.1915 m2 g−1, with a negligible total pore volume (1 × 10−4 cm3 g−1). The results indicate that no N2 adsorption occurs in complex 1, and a similar behavior would be expected for complex 2.
Discussion
Dinuclear anionic triply stranded helicates are rare species. In fact, to the best of our knowledge, there are only a few examples: polymeric Ti4+ or V4+ helicates37,38 based on catecholate-imine ligands, 2D coordination polymers of Ni2+,39,40 containing pyridine-carboxamide or polyoxamate-type ligands, and two coordination polymers based on Fe3+ and catecholamide or salicylic acid–carboxamide ligand.41,42 In some of these compounds, the helicates act as metalloligands to alkali metal cations, Fig. 6, and the polymeric growth occurs thanks to the O-donors, coming from the catecholate, amide and/or carboxylate groups, that coordinate to an alkali metal counterion. In our case, the ligand is a salicyl-imine derivative, which provides an unusual N3O3 donor set that stabilizes the metal ions in the low oxidation state (M2+). In addition, it is important to emphasize that the nitro group functions as a secondary assembly instruction, coordinating to the K+ nodes and none of the reported structures contain nitro groups, see Fig. 6. Thus, our structures are a new class of anionic [M2L3]2− helicates, which, in general, have been little explored.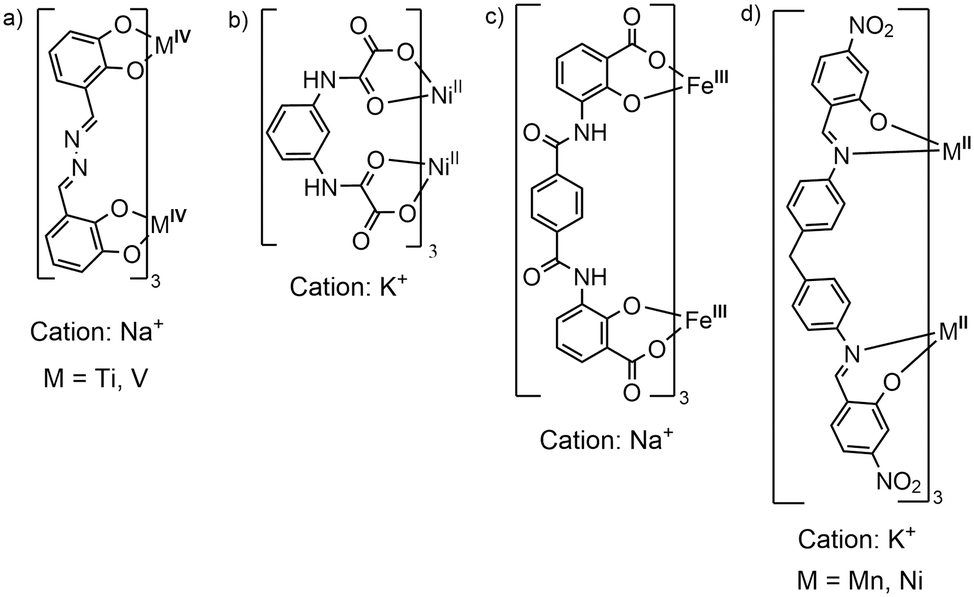 | ||
| Fig. 6 a–c) Structures reported in the literature of triple-stranded anionic dinuclear helicates that form coordination polymers mediated by alkali metal cations. d) This work. | ||
The coordination of nitro group to alkali or alkaline earth metal ions and some other transition metal centers has been previously observed. A search in the CSD database (update June 2023) shows at least 88 crystal structures of aromatic nitro groups coordinated to metal ions. However, it is important to mention that only six out of the 88 structures are based on anionic metalloligands, in which the nitro groups coordinate to the countercation, and from those only four self-assemble in polymeric coordination structures, the rest of the structures correspond to deprotonated ligands counterbalanced by the metal cation. The metal cations observed in the 88 structures correspond to alkali (77), alkaline-earth (8) and transition metals (5), forming discrete (0D), 1D, 2D and 3D structures (Table S5†). The analysis shows that the nitro groups are responsible for the n-dimensional growth (n = 1, 2, 3) of these structures, behaving as μ,1-μ1,3and -μ31 bridging ligands (Fig. S17†). None of the discrete (0-D) structures present the NO2 as a bridging ligand, but only in a mono- and bidentate mode. It is observed that alkali metal ions dominate the list, with the K atom being the most frequent (34 structures), mainly based in homometallic 3D networks. However, as previously mentioned, the presence of metalloligands in this type of nitro-mediated 3D-coordination polymers is very unusual, with few examples similar to those reported in this work.28,43,44 Furthermore, most structures contain less than four nitro groups per metal cation (67 structures), therefore, a high NO2/[M+] ratio is relatively uncommon (21 structures).
Conclusions
We have shown that it is possible to synthesize, under ambient conditions, triply stranded dinuclear helicates, which act as connectors for the self-assembly construction of intricate 3D structures, by harnessing the potential of nitro groups as ligands for K+. The pre-organization and the spatial disposition of the NO2 attached to the aromatic groups, coming from the helicate, permits the use of the latter as connectors, which to the best of our knowledge is an unseen type of building units for the 3D structures. Moreover, the use of NO2 groups as ligands to K+ (and other metal cations) could be used for the stabilization of anionic complexes, without the need to use expensive crown ether type of ligands for the cation, and thus permitting the crystallization and stabilization of the pair by choosing nitro-containing synthons, which are commercially available. The resulting compounds are robust and insoluble in most common solvents, which could be used for the development of novel materials. We are investigating the incorporation of other metal ions to produce interesting magnetic materials, for example, Fe2+ and Co2+ for SCO. In addition, we are exploring other cations as connectors to try to improve the porosity of this type of material.Author contributions
R. Mendoza-Báez and A. Molina-Renteria: investigation, methodology. R. Mendoza-Báez and J. Olguín: formal analysis, data curation, writing – original draft. J. Olguín: conceptualization, funding acquisition, supervision, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Cinvestav for financial support, Dr. Agustin Conde-Gallardo (Departamento de Física, Cinvestav) for access to the PPMS and measurements, Conahcyt for a PhD scholarship to R. M.-B., Marco A. Leyva Ramírez for X-ray diffractometry, to Naytzé Ortiz Pastrana for microanalysis, and Dr. Omar Solorza for access to surface area analyzer.References
- C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005–2062 CrossRef CAS PubMed.
- H. Miyake and H. Tsukube, Chem. Soc. Rev., 2012, 41, 6977–6991 RSC.
- J. Jiao, J. Dong, Y. Li and Y. Cui, Angew. Chem., 2021, 133, 16704–16711 CrossRef.
- L. A. Barrios, R. Diego, M. Darawsheh, J. I. Martínez, O. Roubeau and G. Aromí, Chem. Commun., 2022, 58, 5375–5378 RSC.
- F. Stomeo, C. Lincheneau, J. P. Leonard, J. E. O'Brien, R. D. Peacock, C. P. McCoy and T. Gunnlaugsson, J. Am. Chem. Soc., 2009, 131, 9636–9637 CrossRef CAS PubMed.
- M. Albrecht, I. Janser and R. Fröhlich, Chem. Commun., 2005, 157–165 RSC.
- X. Xi, Y. Fang, T. Dong and Y. Cui, Angew. Chem., Int. Ed., 2011, 50, 1154–1158 CrossRef CAS PubMed.
- S. Fernández-Fariña, I. Velo-Heleno, M. Martínez-Calvo, M. Maneiro, R. Pedrido and A. M. González-Noya, Int. J. Mol. Sci., 2023, 24, 8654 CrossRef PubMed.
- M. Andruh, Dalton Trans., 2015, 44, 16633–16653 RSC.
- M. Albrecht, X. Chen and D. Van Craen, Chem. – Eur. J., 2019, 25, 4265–4273 CrossRef CAS PubMed.
- H. Yoo, J. Lee, P. Kang and M.-G. Choi, Dalton Trans., 2015, 44, 14213–14216 RSC.
- P. Cucos, L. Sorace, C. Maxim, S. Shova, D. Patroi, A. Caneschi and M. Andruh, Inorg. Chem., 2017, 56, 11668–11675 CrossRef CAS PubMed.
- Y. Liu, X. Xi, C. Ye, T. Gong, Z. Yang and Y. Cui, Angew. Chem., Int. Ed., 2014, 53, 13821–13825 CrossRef CAS PubMed.
- G. Kumar and R. Gupta, Chem. Soc. Rev., 2013, 42, 9403–9453 RSC.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- J. Olguín, Coord. Chem. Rev., 2020, 407, 213148 CrossRef.
- M. G. B. Drew, C. J. Harding, V. McKee, G. G. Morgan and J. Nelson, J. Chem. Soc., Chem. Commun., 1995, 1035–1038 RSC.
- F. A. Deeney, C. J. Harding, G. G. Morgan, V. McKee, J. Nelson, S. J. Teat and W. Clegg, J. Chem. Soc., Dalton Trans., 1998, 1837–1844 RSC.
- M. Mikuriya, K. Nakadera and T. Tokii, Inorg. Chim. Acta, 1992, 194, 129–131 CrossRef CAS.
- M. Mikuriya, T. Fujii, S. Kamisawa, Y. Kawasaki, T. Tokii and H. Oshio, Chem. Lett., 1990, 19, 1181–1184 CrossRef.
- B. Mabad, P. Cassoux, J. P. Tuchagues and D. N. Hendrickson, Inorg. Chem., 1986, 25, 1420–1431 CrossRef CAS.
- J. Adhikary, A. Chakraborty, S. Dasgupta, S. K. Chattopadhyay, R. Kruszynski, A. Trzesowska-Kruszynska, S. Stepanović, M. Gruden-Pavlović, M. Swart and D. Das, Dalton Trans., 2016, 45, 12409–12422 RSC.
- J. R. Pioquinto-Mendoza, J. A. Rosas-Ortiz, R. Reyes-Martínez, P. Conelly-Espinosa, R. A. Toscano, J. M. Germán-Acacio, A. Avila-Sorrosa, O. Baldovino-Pantaleón and D. Morales-Morales, Inorg. Chim. Acta, 2015, 438, 146–152 CrossRef CAS.
- S. D. Reid, C. Wilson, C. I. De Matteis and J. B. Love, Eur. J. Inorg. Chem., 2007, 2007, 5286–5293 CrossRef.
- J. Sanmartín, A. M. García-Deibe, M. R. Bermejo, F. Novio, D. Navarro and M. Fondo, Eur. J. Inorg. Chem., 2003, 2003, 3905–3913 CrossRef.
- Z. Xu, L. K. Thompson, D. O. Miller, H. J. Clase, J. A. K. Howard and A. E. Goeta, Inorg. Chem., 1998, 37, 3620–3627 CrossRef CAS PubMed.
- J. Mayans, M. Font-Bardia, L. Di Bari, L. Arrico, F. Zinna, G. Pescitelli and A. Escuer, Chem. – Eur. J., 2018, 24, 7653–7663 CrossRef CAS PubMed.
- G. W. Bates, P. A. Gale, M. E. Light, M. I. Ogden and C. N. Warriner, Dalton Trans., 2008, 4106–4112 RSC.
- T. Han and C.-F. Chen, J. Org. Chem., 2008, 73, 7735–7742 CrossRef CAS PubMed.
- V. K. Bhardwaj, Dalton Trans., 2015, 44, 8801–8804 RSC.
- A. P. Shevchenko, A. A. Shabalin, I. Y. Karpukhin and V. A. Blatov, Sci. Technol. Adv. Mater.: Methods, 2022, 2, 250–265 Search PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- D. P. Malenov and S. D. Zarić, Coord. Chem. Rev., 2020, 419, 213338 CrossRef CAS.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- A. Patra, L. M. Carrella, P. Vojtíšek, E. Rentschler and S. C. Manna, Polyhedron, 2022, 226, 116098 CrossRef CAS.
- K. Matelková, R. Boča, L'. Dlháň, R. Herchel, J. Moncol, I. Svoboda and A. Mašlejová, Polyhedron, 2015, 95, 45–53 CrossRef.
- M. Albrecht, I. Janser, H. Houjou and R. Fröhlich, Chem. – Eur. J., 2004, 10, 2839–2850 CrossRef CAS PubMed.
- M. Albrecht, S. Kamptmann and R. Fröhlich, Polyhedron, 2003, 22, 643–647 CrossRef CAS.
- M. A. Palacios, A. Rodríguez-Diéguez, A. Sironi, J. M. Herrera, A. J. Mota, J. Cano and E. Colacio, Dalton Trans., 2009, 8538–8547 RSC.
- C. A. Simosono, R. M. R. da Silva, N. R. De Campos, M. A. R. Silva, A. C. Doriguetto, L. S. Flores, C. C. Correa, T. R. G. Simões, A. K. S. M. Valdo, F. T. Martins, F. Garcia, G. P. Guedes, B. R. L. Galvão, J. Cancino-Bernardi, R. D. dos Reis, H. O. Stumpf, D. D. Justino, P. F. R. Ortega, W. D. do Pim, M. Julve and M. V. Marinho, Molecules, 2023, 28, 2086 CrossRef CAS PubMed.
- J.-L. Do, H. M. Titi, L. A. Cuccia and T. Friščić, Chem. Commun., 2021, 57, 5143–5146 RSC.
- E. J. Enemark and T. D. P. Stack, Inorg. Chem., 1996, 35, 2719–2720 CrossRef CAS.
- S. Samanta, S. Mukhopadhyay, D. Mandal, R. J. Butcher and M. Chaudhury, Inorg. Chem., 2003, 42, 6284–6293 CrossRef CAS PubMed.
- W. Zhang and J. Zhao, Inorg. Chem. Commun., 2006, 9, 397–399 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic and characterization data. CCDC 2326580 and 2326581. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00255e |
| This journal is © The Royal Society of Chemistry 2024 |