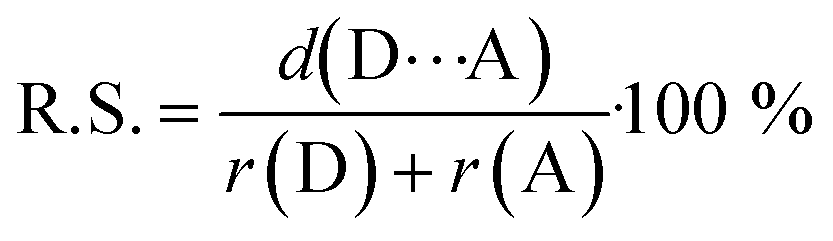DOI:
10.1039/D4CE00557K
(Paper)
CrystEngComm, 2024,
26, 4137-4145
Evaluation of halogen bonding proclivity of oxazole derivatives carrying multiple acceptor sites in cocrystals with perfluorinated iodobenzenes†
Received
3rd June 2024
, Accepted 9th July 2024
First published on 10th July 2024
Abstract
In order to study the competition between halogen bond acceptor sites of multifunctional N,O,X-based halogen bond acceptors (X = S, O, N or π aromatic ring) we have synthesized a family of oxazole derivatives and cocrystallized them with selected iodoperfluorinated benzenes as halogen bond donors. Out of 28 combinations, 19 experiments yielded crystals suitable for single-crystal X-ray diffraction. Structural analysis revealed that in all of the obtained cocrystals the most prominent supramolecular interaction is the I⋯Noxazole halogen bond with relative shortening values of up to 18%, comparable to I⋯Npyridine halogen bond shortening values. The acceptors are ditopic in 10 cocrystals and form additional I⋯N, I⋯O or I⋯π halogen bonds. A majority of cocrystals feature one donor molecule per one acceptor molecule. In order to rank the acceptor sites and establish how the electrostatic potential differences impact the supramolecular landscape around these molecules, the values of molecular electrostatic potentials (MEPs) were calculated on their optimized geometries. These calculations were in agreement with experimental observations, since the best (most negative MEP value) acceptor binding site in the series of used oxazoles is the oxazole nitrogen atom. Depending on the difference between MEP values of the oxazole oxygen atom and the peripheral functionalities, additional halogen bonds could potentially be formed with one of these acceptor sites, leading to the formation of two different types of halogen-bonded supramolecular chains.
1. Introduction
Since the utilization of halogen bonding1 in supramolecular chemistry and especially in the field of crystal engineering in the late 1990s,2,3 there has been an enduring trend of growth in the volume of research directed towards its application in property-driven design of molecular solids.4 This interest is not surprising given that these scientific endeavours have unveiled a diverse array of applications across a wide field within supramolecular and material science. Photochemically active materials,5–7 multicomponent solids of pharmacologically active compounds,8–10 explosive stabilizers11,12 and molecular recognition13–16 exemplify merely a fraction of the manifold applications wherein halogen bonding has exhibited its role in the field of supramolecular systems. Although research on the use of halogen bonding in crystal engineering has recently broadened to include a wider range of different types of acceptor species,4,17–22 by searching the Cambridge Structural Database (CSD),23 one can find around 3104 deposited data sets featuring the N⋯X–D (where X = Br or I, D = C, N, Br or I) interaction that is consistent with the IUPAC definition of halogen bond.1 Somewhat more represented are structures containing O⋯X–D halogen bonds with oxygen as the acceptor (7540 data sets) but less represented are structures containing S⋯X–D halogen bonds (943 data sets). A subset of the above mentioned data corresponds to multicomponent crystals containing perhalogenated benzenes as classic halogen bond donors4,24–26 (PHB), C6Y5X (Y = F, Cl, Br, I; X = Cl, Br, I), with 1627 data sets. The best studied and most reliable acceptors in this data set are nitrogen-containing species. Currently, the N⋯XPHB halogen bond is present in 770 data sets and the most commonly used halogen bond acceptors are pyridyl nitrogen atoms, with 526 data sets for the Npyridyl⋯XPHB motif.27–29 Furthermore, the O⋯XPHB halogen bond is present in 368 data sets. Of those, 130 data sets correspond to structures containing the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯XPHB motif.30–32 Many of these data have been published in reports over the past decade dealing with supramolecular hierarchy of halogen (and hydrogen bonds)30,33–36 and research on the competition/cooperation of acceptor species located on molecules with multiple acceptor sites (i.e. N,O-, S,O-, N,S,O-, N,N,O-, N,O,O-based acceptors etc.).37–45 Recently, Aakeröy and co-workers prepared a family of 13 cocrystals of compounds carrying both nitrogen- and oxygen-based acceptor sites together with a hydrogen bond donor species (–NH–) in order to explore the structural competition between hydrogen- and halogen bonding.37 They established that in these cocrystals the interaction between halogen bond donor and acceptor molecules was either to a Npyridyl (81%), Ocarbonyl (15%) or Cπ (4%). Multifunctional acceptor systems were recently also studied by Shimizu and co-workers.35 They have examined a family of tetrafluorodiiodobenzene cocrystals with N,N,O,O-based acceptors carrying two hydrogen bond donor species (–NH–), and established that the oxalamide self-assembly CO⋯H–N hydrogen bonding motif proved to be strongly directional and consistent, appearing in all 12 cocrystals, which, coupled with an additional I⋯N halogen bonding motif, afforded well-defined supramolecular architectures. Our recent research into halogen-bonded systems in which there is an absence of conventional hydrogen bond donor species led to a systematic study of S,O- and S,N,O-based acceptors in cocrystals with perhalogenated aromates.44 Out of 18 combinations, only 7 (39%) yielded cocrystals, although with a high occurrence of the targeted I⋯S halogen bonding motif (71% of all cocrystals), the I⋯Omorpholine (100% of the imine cocrystals) as well as the I⋯Nimine motif (100%). Furthermore, we also explored the potential of oxygen and nitrogen atoms of morpholine and piperazine fragments which are peripherally located on N,O,O- or N,N,O-based acceptors.45 The molecular electrostatic potential surfaces (MEPs) of four acceptors were evaluated, which allowed for a charge-based ranking. In all cocrystals, halogen bonds are formed with either the morpholinyl oxygen atom or the terminal piperazine nitrogen atom with additional I⋯Ocarbonyl halogen bonding.
O⋯XPHB motif.30–32 Many of these data have been published in reports over the past decade dealing with supramolecular hierarchy of halogen (and hydrogen bonds)30,33–36 and research on the competition/cooperation of acceptor species located on molecules with multiple acceptor sites (i.e. N,O-, S,O-, N,S,O-, N,N,O-, N,O,O-based acceptors etc.).37–45 Recently, Aakeröy and co-workers prepared a family of 13 cocrystals of compounds carrying both nitrogen- and oxygen-based acceptor sites together with a hydrogen bond donor species (–NH–) in order to explore the structural competition between hydrogen- and halogen bonding.37 They established that in these cocrystals the interaction between halogen bond donor and acceptor molecules was either to a Npyridyl (81%), Ocarbonyl (15%) or Cπ (4%). Multifunctional acceptor systems were recently also studied by Shimizu and co-workers.35 They have examined a family of tetrafluorodiiodobenzene cocrystals with N,N,O,O-based acceptors carrying two hydrogen bond donor species (–NH–), and established that the oxalamide self-assembly CO⋯H–N hydrogen bonding motif proved to be strongly directional and consistent, appearing in all 12 cocrystals, which, coupled with an additional I⋯N halogen bonding motif, afforded well-defined supramolecular architectures. Our recent research into halogen-bonded systems in which there is an absence of conventional hydrogen bond donor species led to a systematic study of S,O- and S,N,O-based acceptors in cocrystals with perhalogenated aromates.44 Out of 18 combinations, only 7 (39%) yielded cocrystals, although with a high occurrence of the targeted I⋯S halogen bonding motif (71% of all cocrystals), the I⋯Omorpholine (100% of the imine cocrystals) as well as the I⋯Nimine motif (100%). Furthermore, we also explored the potential of oxygen and nitrogen atoms of morpholine and piperazine fragments which are peripherally located on N,O,O- or N,N,O-based acceptors.45 The molecular electrostatic potential surfaces (MEPs) of four acceptors were evaluated, which allowed for a charge-based ranking. In all cocrystals, halogen bonds are formed with either the morpholinyl oxygen atom or the terminal piperazine nitrogen atom with additional I⋯Ocarbonyl halogen bonding.
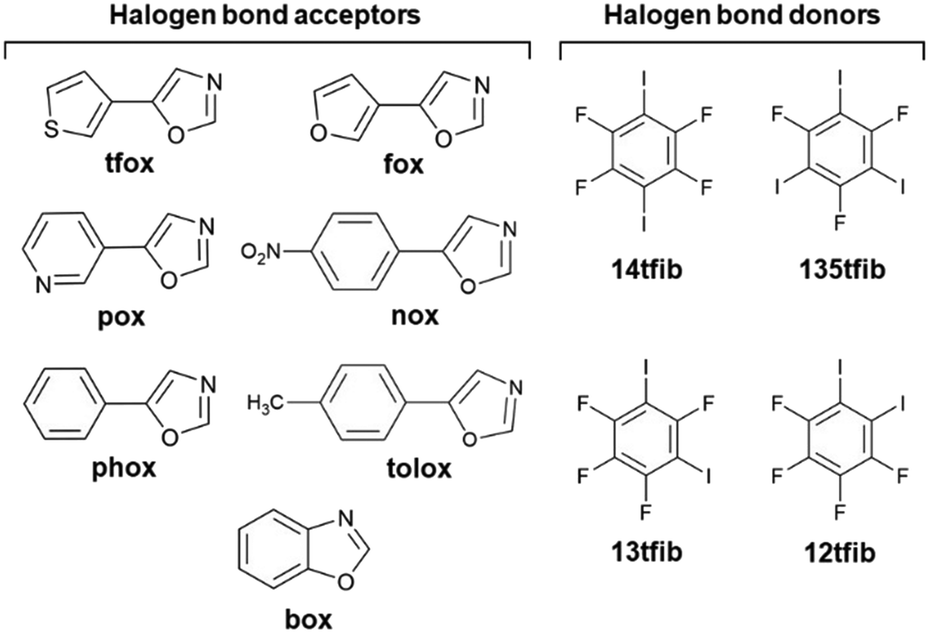
Following our previous research, in this work we decided to explore acceptors carrying nitrogen and oxygen atoms relatively close together in a cyclic fragment. For this purpose, we chose oxazole derivatives which contain a functional group that can be a halogen bond acceptor (S, O, N or π aromatic ring) in addition to the oxazole fragment on one side. Oxazoles46 are a class of heterocyclic aromatic compounds composed of an oxygen, a nitrogen and three sp2 hybridized carbon atoms, and are known for their diverse biological activities47,48 and potential applications in cancer treatment49,50 that make them an important class of compounds for further research. There are around 700 data sets in the Cambridge Structural Database corresponding to systems containing the oxazole fragment, but only four of them feature Br/I⋯Noxazole halogen bonding containing bromo- and iodoperfluorinated benzenes, mainly focusing on luminescence properties.51,52 For this research six acceptors have been prepared by van Leusen's oxazole synthesis from corresponding aromatic aldehydes:53 5-(thiophen-3-yl)oxazole (tfox), 5-(furan-3-yl)oxazole (fox), 5-(pyridin-3-yl)oxazole (pox), 5-(p-nitrophenyl)oxazole (nox), 5-(p-tolyl)oxazole (tolox) and 5-phenyloxazole (phox), Scheme 1. Alongside with the commercially available benzo[d]oxazole (box), all seven acceptor molecules have been cocrystallized with selected perfluorinated iodobenzenes: 1,2-diiodotetrafluorobenzene (12tfib), 1,3-diiodotetrafluorobenzene (13tfib), 1,4-diiodotetrafluorobenzene (14tfib) and 1,3,5-trifluoro-2,4,6-triiodobenzene (135tfib). In order to rank the acceptor sites in target molecules, values of molecular electrostatic potentials were calculated on their optimized geometries.
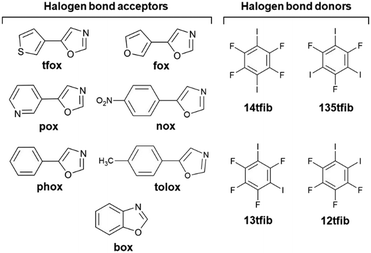 |
| | Scheme 1 Molecular structures of halogen bond acceptors and donors used in this study. | |
2. Experimental methods
2.1. Synthesis of acceptors
Acceptors (with the exception of box which was used as purchased) were prepared by the conversion of appropriate aromatic aldehydes into 5-substituted oxazoles according to the reported general procedure for van Leusen's reaction.53 All reagents were used as purchased, without purification.
General procedure.
In a 250 mL round-bottomed flask equipped with a reflux condenser, the corresponding aldehyde (10.0 mmol) was mixed with equimolar amounts of p-toluenesulfonylmethyl isocyanide (TosMIC) (10.0 mmol, 2.112 g) and potassium carbonate (10.0 mmol, 2.419 g). The reaction mixture was dissolved in methanol (44.8 mL), and was then heated, stirred and refluxed at 70 °C for 3 hours. After cooling to room temperature, distilled water (22.4 mL) was added and the mixture was stirred for a further 10 minutes at the room temperature. Methanol from the resulting solution was removed by rotary evaporation. The residual liquid was transferred to a separatory funnel, and then extracted with methyl tert-butyl ether (3 × 12 mL). The combined organic layers were washed with water (5 mL) and a saturated solution of sodium chloride. The solution was then dried over anhydrous sodium sulfate and filtered. The resultant solution containing the product was used for cocrystallization experiments (as aliquots) without purification and yield determination (see ESI† for details).
2.2. Synthesis of single crystal cocrystals
Aliquots (50–300 μL) of the methyl tert-butyl ether solution obtained by synthesis were transferred into crystallization vessels containing perfluorinated iodobenzenes dissolved in an appropriate solvent or mixture of solvents. Crystallization vessels were left undisturbed either at normal laboratory conditions (20–25 °C, 40–60% RH) or at 4 °C for a couple of days until the formation of single crystals suitable for XRD analysis. The box reactant was first melted and then dissolved in a solvent or a solvent mixture together with perfluorinated iodobenzenes (see ESI† for details).
2.3. Thermal analysis
DSC measurements were performed on a Mettler-Toledo DSC823e instrument. The bulk samples obtained from crystallization experiments were placed in sealed aluminium pans (40 μL) with a pinhole made in the top cover, and heated in flowing nitrogen (150 mL min−1) from −10 °C or 25 °C to 300 °C at a rate of 10 °C min−1. Data collection and analysis were performed using the program package STARe Software 17.00.54
2.4. Powder X-ray diffraction experiments (PXRD)
PXRD experiments were performed on a Malvern PANalytical Aeris X-ray diffractometer with CuKα1 (1.54056 Å) radiation at 15 mA and 40 kV in Bragg–Brentano geometry. The scattered intensities were measured with a line (1D) detector. The angular range was from 5 to 40° (2θ) with an interpolated step size of 0.00543322°. Data analysis was performed using the program Data Viewer.55
2.5. Single-crystal X-ray diffraction experiments (SCXRD)
Crystal and molecular structures of the prepared cocrystals were determined by single crystal X-ray diffraction. Unit cell parameters and refinement data are listed in ESI.† Diffraction measurements were made on a Rigaku Synergy XtaLAB X-ray diffractometer with graphite-monochromated MoKα (λ = 0.71073 Å) radiation. The data sets were collected using the ω scan mode over the 2θ range up to 64°. The CrysAlisPro program package56 was employed for data collection, cell refinement, and data reduction. The structures were solved by direct methods and refined using the SHELXS, SHELXT, and SHELXL programs, respectively.57,58 Structural refinement was performed on F2 using all data. Hydrogen atoms were placed in calculated positions and treated as riding on their parent atoms. Calculations were performed using the WINGX or Olex2 crystallographic suites of programs.59,60 The molecular structures of compounds and their molecular packing projections were prepared using Mercury.61
2.6. Computational details
All calculations were performed using the Gaussian 16 software package.62 Geometry optimizations of oxazole molecules, halogen-bonded dimers and trimers as well as MEP calculations were performed using M062X/def2-tzvp level of theory,63 with ultrafine integration grid (99 radial shells and 590 points per shell). The default Gaussian convergence criteria were used. The same level of theory was used to calculate the binding energies on optimized geometries, employing the Boys–Bernardi counterpoise scheme to account for basis set superposition error.64 Harmonic frequency calculations were performed on the optimized geometries to ensure the success of each geometry optimization. The figures were prepared using GaussView 5.1.65
3. Results and discussion
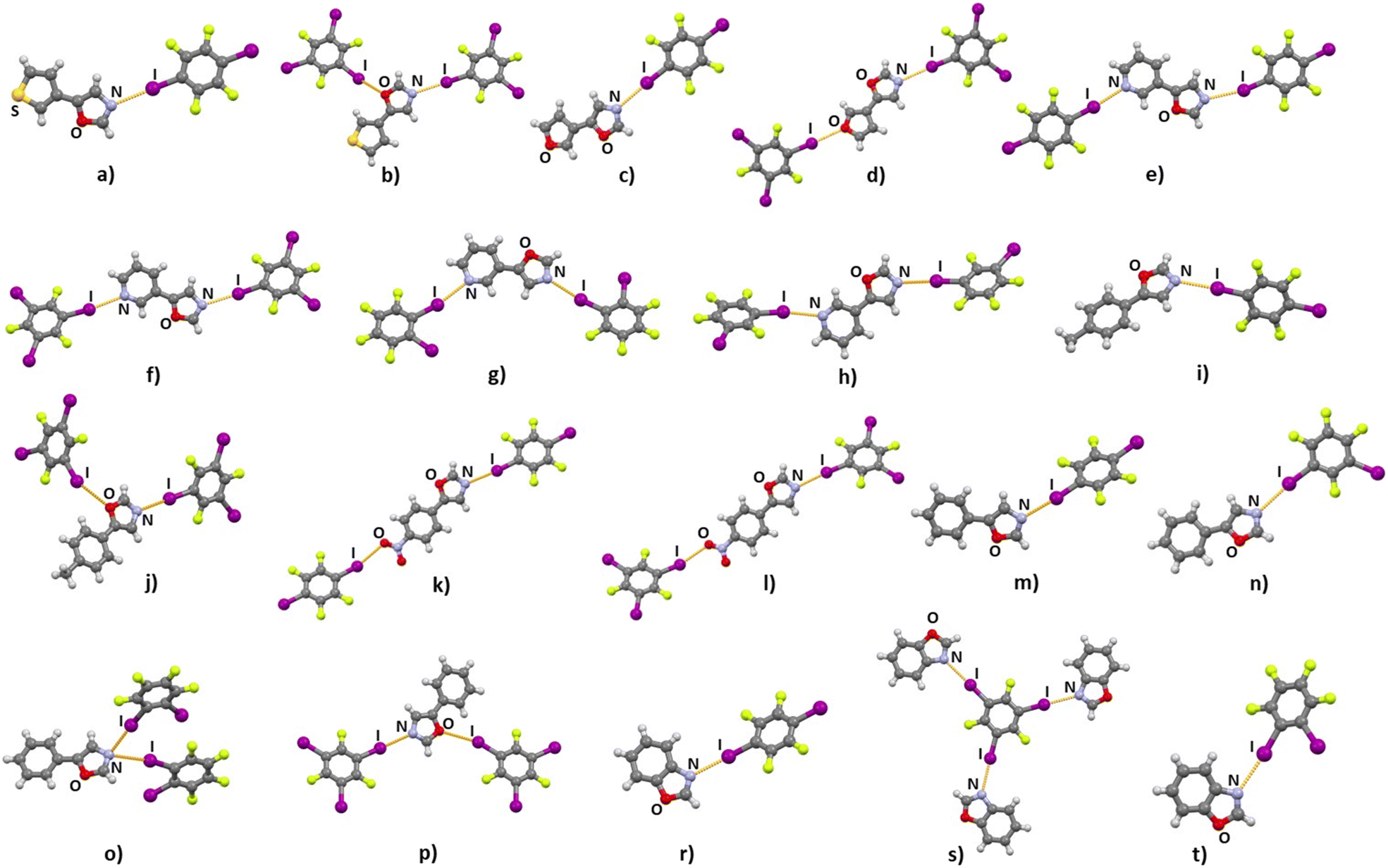
Out of 28 combinations, 19 resulted in cocrystals that were then suitable for SCXRD structural analysis (Fig. 1). Preparation of the cocrystals from solution has been a considerable challenge, since most of the prepared acceptors were obtained in the form of a viscous oil or as low melting point solids. Cocrystallization of halogen bond donors with aliquots of solutions containing the prepared acceptors enabled the preparation of single crystals. This approach, however, was not favourable for the preparation of pure crystal bulk, resulting in solids whose PXRD patterns do not correspond to those simulated from SCXRD data, and/or whose DSC curves contain multiple or irregular peaks (see Table S2 in ESI† and Fig. S22–S52). Therefore, according to PXRD and DSC data we have obtained a pure crystal bulk for only four cocrystals, (tfox)2(14tfib), (tfox)(135tfib), (phox)(14tfib) and (phox)(135tfib). In the small set of reliable DSC data we observed that the melting points for these cocrystals are lower than for pure donors, that tfox cocrystals have lower melting points than phox cocrystals with the same donor molecule, and that 135tfib cocrystals have a higher melting point than 14tfib ones. Crystal structure determination has revealed that, as anticipated, the main supramolecular interaction in all obtained cocrystals is the halogen bond with distances, angles and relative shortening values‡ that follow established trends of dependency on the acceptor moiety. A majority of cocrystals feature one donor molecule per one acceptor molecule. Exceptions are (tfox)2(14tfib), (pox)(12tfib)2 (phox)(12tfib)2 and (box)3(135tfib). The most consistent trend is the formation of a halogen bond between a donor iodine and the oxazole nitrogen atom as an acceptor, regardless of additional acceptor or donor species (Table 1). This bond is present in all (100%) cocrystals. These halogen bonds have high values of relative shortening, up to 18.7%, and averaging at 15.9%, comparable to the values found between iodine and pyridine nitrogen atoms (17.6%). Halogen bonding with the oxazole oxygen atom is considerably less frequent, being present in only 3 cocrystals containing 135tfib due to its geometrical disposition. Contrary to expectations based on recent emerging insights into the proclivity of the sulfur atom as an acceptor and, more specifically, the thiophenyl moiety,44 in cocrystals containing tfox neither halogen nor chalcogen bonding involving the sulfur atom was observed. All halogen bonds in cocrystals are monocentric with the exception of the (phox)(12tfib)2, where trimers are formed via (I⋯)2Noxazole halogen bond in which the oxazole nitrogen atom is a bifurcated acceptor. As expected, all four acceptors with a pyridine moiety participate in a halogen bond with short I⋯Npyridine contacts. Consistency was also achieved with the acceptor nitro group in cocrystals (nox)(14tfib) and (nox)(135tfib) with relative shortening values of I⋯Onitro of 12.1% and 15.4% respectively. These I⋯Onitro halogen bonds are somewhat shorter than those reported previously in similar cocrystals.31,67,68 The I⋯Ofuryl halogen bond was observed in one of the two cocrystals with the furyl moiety.
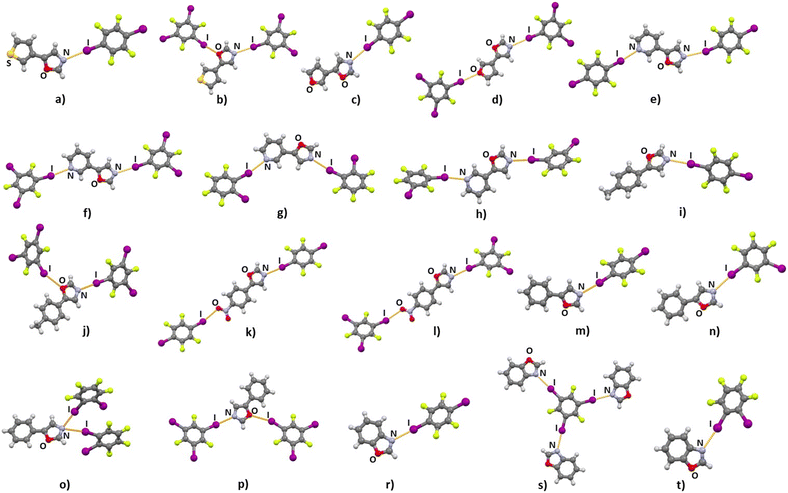 |
| | Fig. 1 Partial crystal structure of cocrystals: a) (tfox)2(14tfib); b) (tfox)(135tfib); c) (fox)(14tfib); d) (fox)(135tfib); e) (pox)(14tfib); f) (pox)(135tfib); g) (pox)(12tfib)2; h) (pox)(13tfib); i) (tolox)(14tfib); j) (tolox)(135tfib); k) (nox)(14tfib); l) (nox)(135tfib); m) (phox)(14tfib); n) (phox)(13tfib); o) (phox)(12tfib)2; p) (phox)(135tfib); r) (box)(14tfib); s) (box)3(135tfib) and t) (box)(12tfib). | |
Table 1 Halogen bond lengths (d), angles (∠) and relative shortening values (R.S.) of D⋯A distances in the herein presented cocrystals
| Cocrystal |
D⋯A |
Acceptor specie |
d(D⋯A)/Å |
R.S./% |
∠(C–D⋯A)/° |
| (tfox)2(14tfib) |
I1⋯N1 |
Oxazole nitrogen |
2.983 |
15.5% |
174.8 |
| (tfox)(135tfib) |
I1⋯N1 |
Oxazole nitrogen |
2.957 |
16.2% |
179.7 |
| I3⋯O1 |
Oxazole oxygen |
3.316 |
5.3% |
157.9 |
| I2⋯I1 |
XB donor |
3.906 |
1.4% |
162.8 |
| (fox)(14tfib) |
I1⋯N1 |
Oxazole nitrogen |
2.934 |
16.9% |
175.9 |
| (fox)(135tfib) |
I1⋯N1 |
Oxazole nitrogen |
2.996 |
15.1% |
175.9 |
| I2⋯O1 |
Furyl oxygen |
2.981 |
14.8% |
176.4 |
| (pox)(14tfib) |
I1⋯N1 |
Pyridyl nitrogen |
2.862 |
18.9% |
174.4 |
| I2⋯N2 |
Oxazole nitrogen |
2.989 |
15.3% |
168.8 |
| (pox)(135tfib) |
I1⋯N1 |
Pyridyl nitrogen |
2.909 |
17.6% |
173.2 |
| I3⋯N2 |
Oxazole nitrogen |
2.895 |
18.0% |
176.8 |
| I2⋯I1 |
XB donor |
3.877 |
2.1% |
154.7 |
| (pox)(12tfib)2 |
I1⋯N1 |
Pyridyl nitrogen |
2.949 |
16.5% |
169.5 |
| I3⋯N2 |
Oxazole nitrogen |
2.974 |
15.8% |
172.2 |
| I4⋯I3 |
XB donor |
3.927 |
0.8% |
151.0 |
| (pox)(13tfib) |
I1⋯N1 |
Pyridyl nitrogen |
2.911 |
17.5% |
170.9 |
| I2⋯N2 |
Oxazole nitrogen |
2.936 |
16.8% |
176.7 |
| (tolox)(14tfib) |
I1⋯N1 |
Oxazole nitrogen |
2.910 |
17.6% |
177.8 |
| (tolox)(135tfib) |
I1⋯N1 |
Oxazole nitrogen |
2.868 |
18.8% |
176.7 |
| I2⋯O1 |
Oxazole oxygen |
3.270 |
6.6% |
157.9 |
| (nox)(14tfib) |
I2⋯N2 |
Oxazole nitrogen |
2.963 |
16.1% |
175.3 |
| I1⋯O2 |
Nitro oxygen |
3.077 |
12.1% |
167.3 |
| (nox)(135tfib) |
I1⋯N2 |
Oxazole nitrogen |
2.925 |
17.1% |
176.1 |
| I2⋯O1 |
Nitro oxygen |
2.962 |
15.4% |
167.6 |
| (phox)(14tfib) |
I1⋯N1 |
Oxazole nitrogen |
2.919 |
17.3% |
176.0 |
| (phox)(135tfib) |
I1⋯N1 |
Oxazole nitrogen |
2.952 |
16.4% |
179.3 |
| I3⋯O1 |
Oxazole oxygen |
3.339 |
4.6% |
157.1 |
| (phox)(12tfib)2 |
I1⋯N1 |
Oxazole nitrogen |
3.082 |
12.7% |
175.5 |
| I3⋯N1 |
Oxazole nitrogen |
3.257 |
7.7% |
170.2 |
| (phox)(13tfib) |
I1⋯N1 |
Oxazole nitrogen |
3.016 |
14.6% |
174.2 |
| (box)(14tfib) |
I1⋯N1 |
Oxazole nitrogen |
2.910 |
17.6% |
176.8 |
| I2⋯π |
π aromatic system |
3.561 |
3.2% |
162.4 |
| (box)3(135tfib) |
I1⋯N1 |
Oxazole nitrogen |
2.978 |
15.6% |
178.6 |
| I3⋯N3 |
Oxazole nitrogen |
2.908 |
17.6% |
178.4 |
| I2⋯N2 |
Oxazole nitrogen |
2.943 |
16.6% |
178.2 |
| (box)(12tfib) |
I1⋯N1 |
Oxazole nitrogen |
2.984 |
15.5% |
173.8 |
| I3⋯N2 |
Oxazole nitrogen |
3.003 |
14.9% |
175.6 |
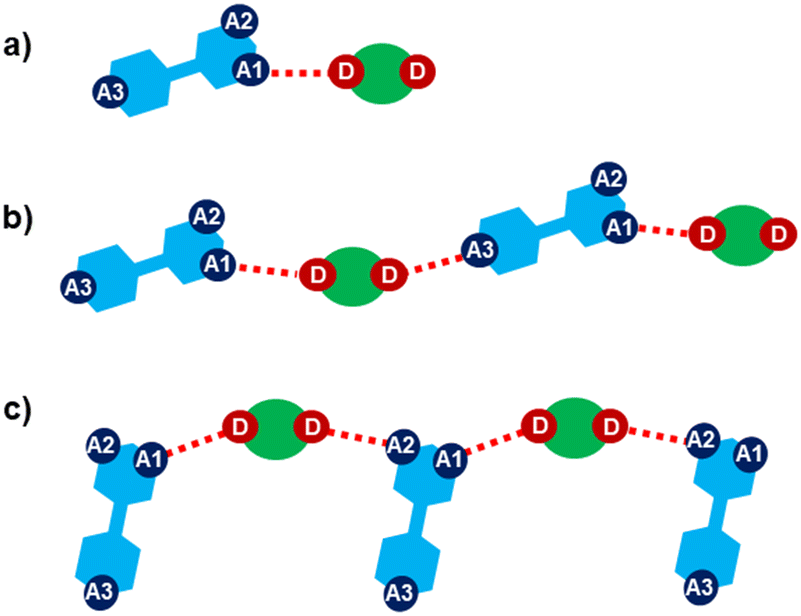
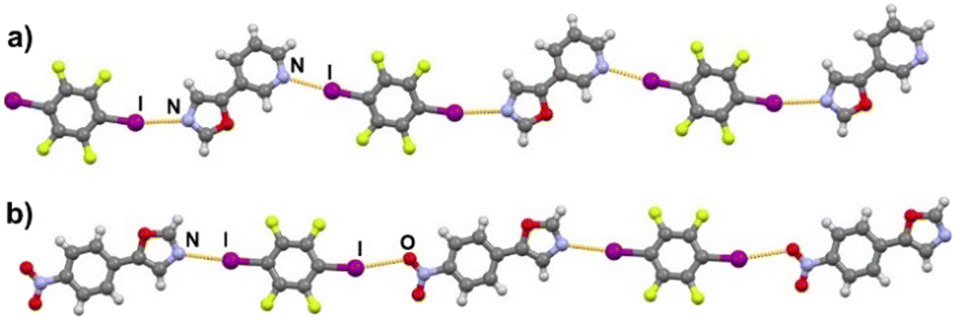
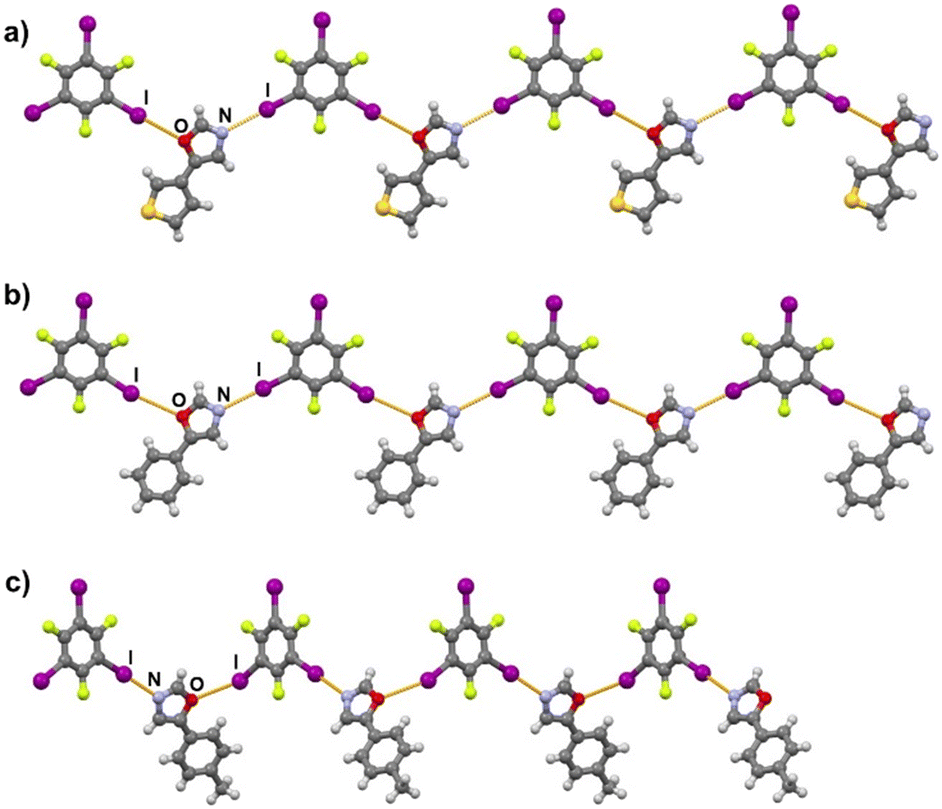
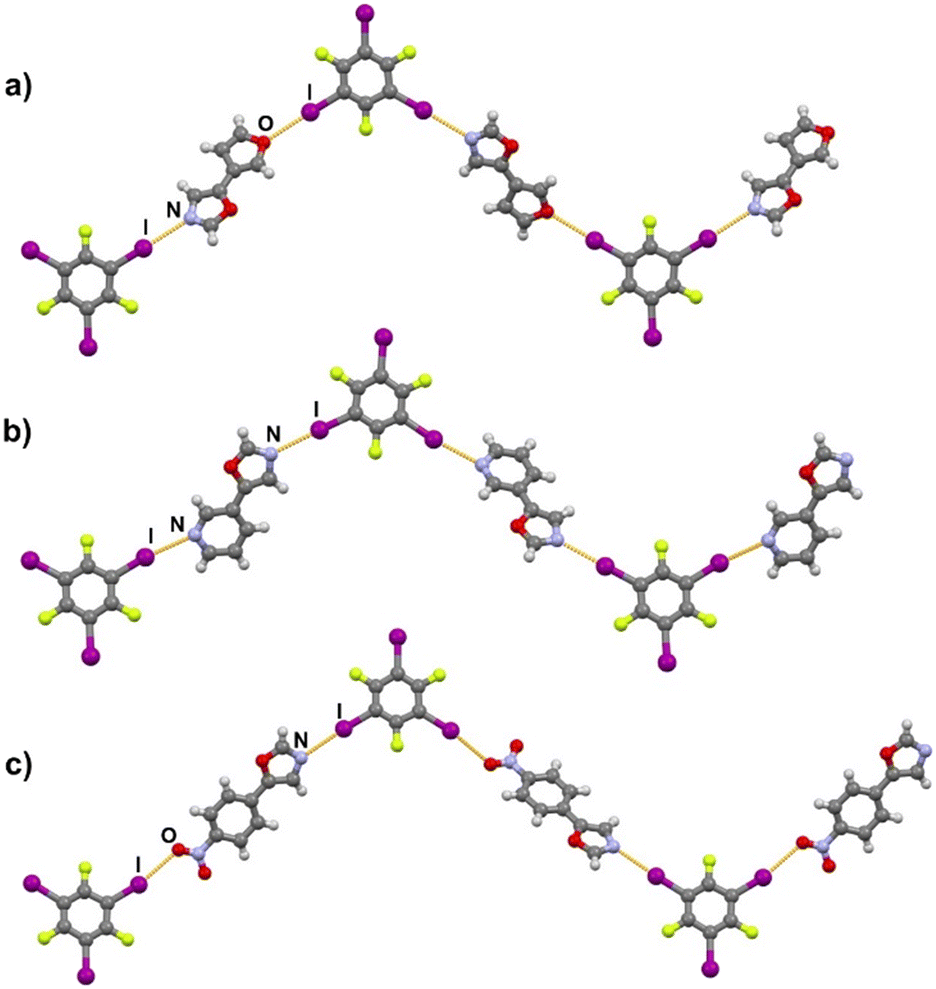
Halogen bonding motifs are fairly diverse from one cocrystal to another, but it is possible to divide them into three main categories: discrete supramolecular complexes, chains interconnected through both oxazole nitrogen and an acceptor moiety originating from the aldehyde, and chains interconnected through both the oxazole nitrogen and oxygen atoms bridged by donor molecules (Fig. 2). Discrete halogen bonded supramolecular complexes (Fig. 2a) are formed in (tfox)2(14tfib), (fox)(14tfib), (pox)(12tfib)2, (pox)(13tfib), (tolox)(14tfib), (phox)(14tfib), (phox)(12tfib), (phox)(13tfib), (box)(12tfib) and (box)3(135tfib) while halogen-bonded chains (Fig. 2b and c) are formed in the remaining cocrystals. It can be observed that 14tfib tends to make discrete complexes in cases where acceptor sites on the aldehyde-originating fragment are absent (phox, tolox) or if the acceptor site on that fragment is somewhat weaker (tfox, fox). However, if the acceptor molecules have strong acceptor sites on both ends (pox, nox), cocrystals with the same donor molecule feature 1D chains as shown in Fig. 2b and 3. This trend is also present to an extent in the (box)(14tfib) cocrystal, where the donor iodine atom forms a halogen bond only with the oxazole nitrogen atom. Rather than stopping at the formation of discrete complexes like in phox and tolox cocrystals, zig-zag halogen-bonded chains are formed through additional I⋯π halogen bonds. In cocrystals containing 135tfib, all systems exhibit the formation of halogen-bonded chains due to the favourable structural arrangement of donor atoms which can link both oxazole acceptor sites and interconnect them into chains even in systems containing phox and tolox that only have acceptor sites on the oxazole fragment. Chains in 135tfib cocrystals can therefore be divided into two groups: linear chains in which 135tfib molecules bridge oxazole molecules through the available oxazole fragment oxygen and nitrogen atoms (Fig. 2c and 4), and zig-zag chains that are formed when there are favourable acceptor sites on both the oxazole and aldehyde fragments (Fig. 2b and 5).
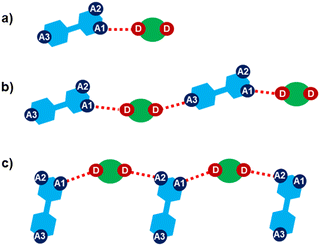 |
| | Fig. 2 Schemes of the three main groups of observed motifs: a) discrete supramolecular complexes; b) chains interconnected through the oxazole nitrogen atom (A1) and the additional acceptor moiety originating from the aldehyde part (A3); c) chains interconnected trough oxazole nitrogen (A1) and oxygen atoms (A2) bridged by donor molecules (D). | |
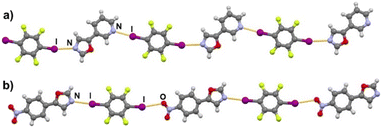 |
| | Fig. 3 Halogen-bonded chains in cocrystals of a) (pox)(14tfib) and b) (nox)(14tfib). | |
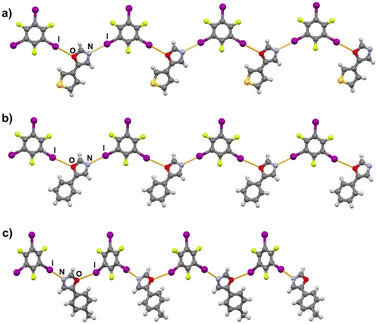 |
| | Fig. 4 Halogen-bonded chains in a) (tfox)(135tfib), b) (phox)(135tfib) and c) (tolox)(135tfib). | |
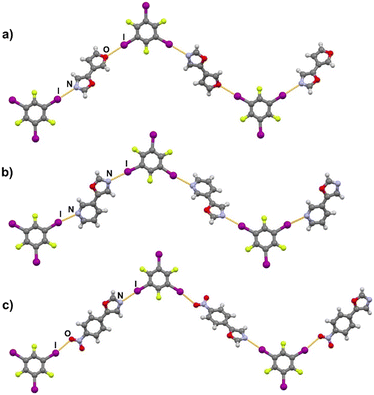 |
| | Fig. 5 Zig-zag halogen-bonded chains in a) (fox)(135tfib), b) (pox)(135tfib) and c) (nox)(135tfib). | |
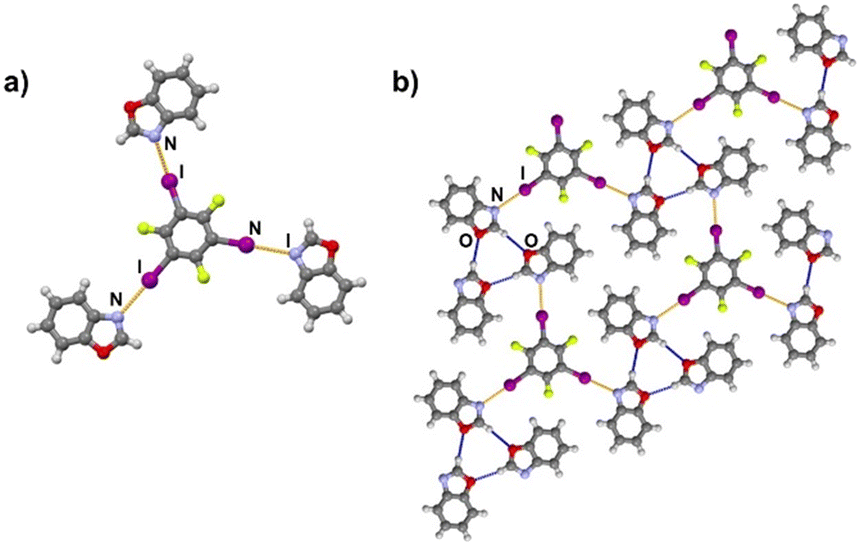
It is noteworthy to compare tolox- and phox-containing cocrystals with 135tfib (Fig. 4b and c). Although they are nearly identical in structure (the only difference is one methyl group relatively far from the acceptor sites), they do not form the same supramolecular motif. In the (tolox)(135tfib) cocrystal the molecules form a directional (linear), but highly branched V-shaped chain (1D) that runs parallel to the c-crystallographic axis. In the (phox)(135tfib) cocrystal, the chain is almost planar and each molecule is symmetrically equivalent. In case of (box)3(135tfib) which differs from other observed stoichiometries, each molecule of 135tfib is tritopic, participating in halogen bonding with three box molecules, is surrounded by a total of six acceptor molecules, and the discrete motifs are connected into a 2D layer through C–H⋯Ooxazole hydrogen bonding (Fig. 6). Further connection into 3D frameworks in all systems is attained via C–H⋯F or weak interhalogen contacts.
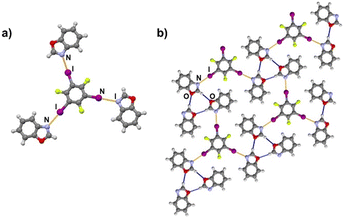 |
| | Fig. 6 a) Discrete complex, and b) hydrogen (blue) and halogen bonds (orange) in a 2D layer in the (box)3(135tfib) cocrystal. | |
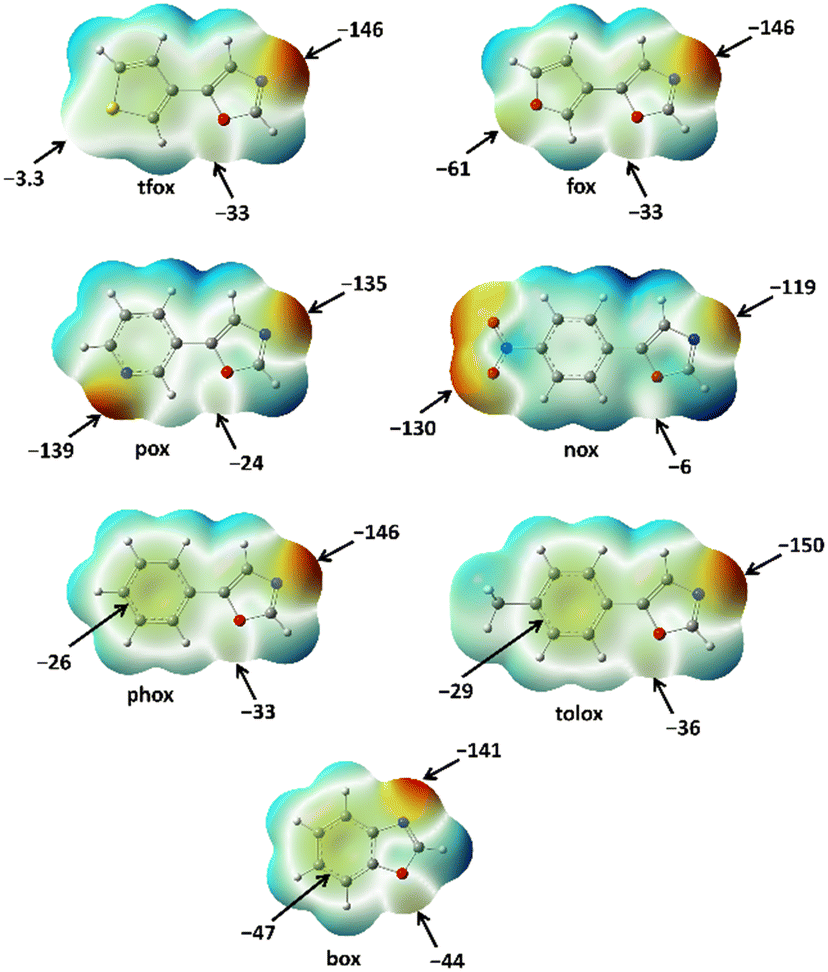
According to the calculated MEP values (Fig. 7) it follows that the oxazole nitrogen (A1) is, on average, the best binding site in the series of used oxazoles, which is in accordance with the trends observed in the crystal structures of the prepared cocrystals. Such a regularity has not been observed for oxazole oxygen atoms (A2) and peripheral binding sites (A3), whose relative acceptor strength depends on the functional groups present in the corresponding oxazole molecule. For the purpose of quantitatively ranking these two acceptor sites by strength, a ΔMEP scale has been defined that corresponds to the difference of MEPs on the peripheral group and that on the oxazole oxygen atom (ΔMEP = MEPA3 − MEPA2). According to the obtained ΔMEP values, the 7 used oxazoles can be divided into two groups: those with ΔMEP < 0 kJ mol−1 e−1 and those with ΔMEP > 0 kJ mol−1 e−1. In tfox, tolox and phox the acceptor site A2 is more negative than A3 which leads to a positive ΔMEP value, and consequently, in corresponding cocrystals an additional halogen bond was formed between a donor atom and the A2 acceptor (oxazole oxygen). On the other hand, acceptors with negative ΔMEP values, nox, pox, fox and box, form a total of 7 cocrystals (with different donors) in which an additional halogen bond with the peripheral acceptor group (I⋯A3) has been formed. The MEP values of the nitro and pyridyl fragment (on the periphery of nox and pox molecules, respectively) are more negative than MEP values of the oxazole nitrogen in the same molecules. Hence, it is not surprising that nox and pox are selectively connected into halogen-bonded chains through two sorts of halogen bonds, including the two strongest acceptor sites in molecules (I⋯A1 and I⋯A3). Furthermore, ΔMEP values for box and fox are significantly higher (less negative) than those for nox and pox, which indicate relatively poor acceptor properties of the furyl oxygen and π-system functionalities compared to the nitro and pyridyl functionalities. Interestingly, the I⋯π and I⋯Ofuryl halogen bonds formed in box and fox cocrystals are still stronger (14.9 kJ mol−1 and 11.6 kJ mol−1) than hypothetical I⋯Ooxazole halogen bonds (10.1 kJ mol−1), which unequivocally explains the observed I⋯A3 motif in the crystal structures of box and fox cocrystals.
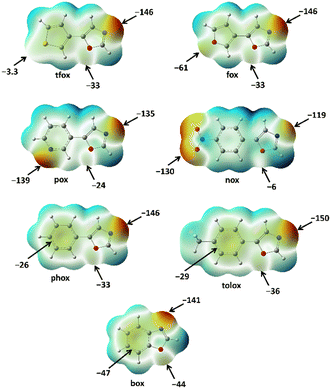 |
| | Fig. 7 Molecular electrostatic potentials of the oxazole molecules used in present study, mapped on isodensity surface (0.001 a.u.). All values are in kJ mol−1 e−1. | |
In addition to the described cocrystals, 5 out of 7 used oxazoles (tfox, tolox, phox, box and fox) form a total of 10 cocrystals in which the molecules are connected into discrete molecular complexes. By calculating the difference between the MEPs on the oxazole nitrogen atoms and the average MEP value of all 7 oxazole nitrogen atoms (−140 kJ mol−1 e−1), it can be observed that the abovementioned five oxazoles have more negative MEP values than average, while the MEPs on the remaining two (nox and pox) are slightly more positive. These differences are also reflected in the I⋯Noxazole halogen bond energies, which are (on average) lower for nox and pox (21.8 kJ mol−1) than for the other five oxazoles (23.9 kJ mol−1). All this supports the statement that the presence of two strong halogen bond acceptors in nox and pox structures leads to the formation of more stable halogen-bonded chains in which both strong acceptors participate in halogen bonding. On the other hand, the formation of the I⋯Noxazole halogen bond with tfox, tolox, phox, box and fox is somewhat more favorable than in the case of nox and pox, and its formation can more easily lead to the formation of cocrystals with discrete molecular complexes, as was also observed in the crystal structures.
4. Conclusions
To conclude, we have prepared and structurally characterized nineteen novel cocrystals of 5-substituted oxazoles with perfluorinated halogen bond donors. As anticipated, the dominant supramolecular interaction in each of the obtained systems is halogen bonding. The oxazole fragment, or more precisely the oxazole ring nitrogen atom turned out to be a reliable acceptor site, as confirmed both computationally and experimentally, participating in halogen bonding in all (100%) of the herein presented cocrystals. Relative shortening values of these contacts are 16% on average, and are comparable to those in halogen bonding between iodine and pyridine nitrogen atoms. In contrast, the oxazole ring oxygen atom selectively partakes in interactions within only three cocrystals, and is confined to systems with favourable steric and geometric dispositions. This is due to the fact that the MEP value on the oxazole oxygen atom is less negative than peripheral functionalities in most oxazole molecules used in this study. Additionally, the acceptor potential for halogen bonding was reconfirmed for both nitro and pyridyl functional groups in acceptors featuring these functionalities. The coexistence of I⋯Noxazole with I⋯Onitro and I⋯Npyridyl halogen bonds shows cooperativities of those interactions in connecting into chains and other supramolecular architectures. Each of the prepared cocrystals features unique geometric dispositions and different topicities of donor atoms and three main categories of structural motifs were observed. This study provides an insight into the reliability of oxazole fragments for halogen bonding. These results could be useful for the design and synthesis of oxazole-containing and similar building blocks potentially enhancing their utility in crystal engineering and supramolecular chemistry applications.
Data availability
The data supporting this article (experimental procedures, DSC curves, PXRD patterns MEP calculations, crystal data and ORTEP representations of obtained structures) have been included as part of the ESI.† Crystallographic data for 21 compounds has been deposited at the Cambridge Crystallographic Data Centre under deposition codes 2358783–2358803.
Author contributions
Investigation: SC and RS performed synthesis of the acceptors and the cocrystallization experiments. Conceptualization: VN, RS and DC conceived and designed the study and formulated research aims. Formal analysis: VN and NBT performed single-crystal X-ray diffraction measurements, thermal analysis and the associated data processing. NB performed MEP calculations. Supervision: the study was supervised by VN and DC. Visualisation: DC, RS and NB prepared schemes and figures. Writing: the manuscript draft was written by RS with contributions from all authors. Reviews and editing were done by VN and DC. DC ensured funding acquisition and project management.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
This research was funded by the Croatian Science Foundation under the research project IP-2019-04-1868. Additionally, we acknowledge support of the project CIuK cofinanced through the European Regional Development Fund-Competitiveness and Cohesion Operational Programme (Grant KK.01.1.1.02.0016).
Notes and references
- G. R. Desiraju, P. Shing Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- V. Amico, S. V. Meille, E. Corradi, M. T. Messina and G. Resnati, J. Am. Chem. Soc., 1998, 120, 8261–8262 CrossRef CAS.
- J. B. Davey, A. C. Legon and E. R. Waclawik, Phys. Chem. Chem. Phys., 1999, 1, 3097–3101 RSC.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- J.-C. Christopherson, F. Topić, C. J. Barrett and T. Friščić, Cryst. Growth Des., 2018, 18, 1245–1259 CrossRef CAS.
- I.-R. Jeon, C. Mathonière, R. Clérac, M. Rouzières, O. Jeannin, E. Trzop, E. Collet and M. Fourmigué, Chem. Commun., 2017, 53, 10283–10286 RSC.
- W. Dai, X. Niu, X. Wu, Y. Ren, Y. Zhang, G. Li, H. Su, Y. Lei, J. Xiao, J. Shi, B. Tong, Z. Cai and Y. Dong, Angew. Chem., Int. Ed., 2022, 61, e202200236 CrossRef CAS PubMed.
- C. Li, E. A. Keene, C. Ortiz-de León and L. R. MacGillivray, Supramol. Chem., 2021, 33, 687–692 CrossRef CAS.
- M. Baldrighi, G. Cavallo, M. R. Chierotti, R. Gobetto, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Mol. Pharmaceutics, 2013, 10, 1760–1772 CrossRef CAS PubMed.
- D. Choquesillo-Lazarte, V. Nemec and D. Cinčić, CrystEngComm, 2017, 19, 5293–5299 RSC.
- K. B. Landenberger, O. Bolton and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 5074–5079 CrossRef CAS PubMed.
- N. Şen, J. Mol. Struct., 2022, 1254, 132381 CrossRef.
- G. Berger, P. Frangville and F. Meyer, Chem. Commun., 2020, 56, 4970–4981 RSC.
- A. Frontera and A. Bauzá, J. Chem. Theory Comput., 2020, 16, 4744–4752 CrossRef CAS PubMed.
- S. Koppireddi, C. Z. Liu, H. Wang, D. W. Zhang and Z. T. Li, CrystEngComm, 2019, 21, 2626–2630 RSC.
- P. Metrangolo, Y. Carcenac, M. Lahtinen, T. Pilati, K. Rissanen, A. Vij and G. Resnati, Science, 2009, 323, 1461–1464 CrossRef CAS PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- L. Happonen, J. M. Rautiainen and A. Valkonen, Cryst. Growth Des., 2021, 21, 3409–3419 CrossRef CAS.
- K. Lisac, F. Topić, M. Arhangelskis, S. Cepić, P. A. Julien, C. W. Nickels, A. J. Morris, T. Friščić and D. Cinčić, Nat. Commun., 2019, 10, 61 CrossRef CAS PubMed.
- F. Meyer and P. Dubois, CrystEngComm, 2013, 15, 3058–3071 RSC.
- V. Nemec, K. Lisac, N. Bedeković, L. Fotović, V. Stilinović and D. Cinčić, CrystEngComm, 2021, 23, 3063–3083 RSC.
- J. Vainauskas, T. H. Borchers, M. Arhangelskis, L. J. McCormick McPherson, T. S. Spilfogel, E. Hamzehpoor, F. Topić, S. J. Coles, D. F. Perepichka, C. J. Barrett and T. Friščić, Chem. Sci., 2023, 14, 13031–13041 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- P. Metrangolo and G. Resnati, Chem. – Eur. J., 2001, 7, 2511–2519 CrossRef CAS PubMed.
- M. Fourmigué and P. Batail, Chem. Rev., 2004, 104, 5379–5418 CrossRef PubMed.
- N. Bedeković, V. Stilinović, T. Friščić and D. Cinčić, New J. Chem., 2018, 42, 10584–10591 RSC.
- M. Saccone, G. Cavallo, P. Metrangolo, A. Pace, I. Pibiri, T. Pilati, G. Resnati and G. Terraneo, CrystEngComm, 2013, 15, 3102–3105 RSC.
- C. B. Aakeröy, T. K. Wijethunga, M. A. Haj, J. Desper and C. Moore, CrystEngComm, 2014, 16, 7218–7225 RSC.
- A. Carletta, M. Zbačnik, M. Vitković, N. Tumanov, V. Stilinović, J. Wouters and D. Cinčić, CrystEngComm, 2018, 20, 5332–5339 RSC.
- M. Zbačnik, M. Vitković, V. Vulić, I. Nogalo and D. Cinčić, Cryst. Growth Des., 2016, 16, 6381–6389 CrossRef.
- V. Nemec and D. Cinčić, CrystEngComm, 2016, 18, 7425–7429 RSC.
- V. Nemec, L. Fotović, T. Vitasović and D. Cinčić, CrystEngComm, 2019, 21, 3251–3255 RSC.
- C. B. Aakeröy, P. D. Chopade, C. Ganser and J. Desper, Chem. Commun., 2011, 47, 4688–4690 RSC.
- C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem. – Eur. J., 2013, 19, 16240–16247 CrossRef PubMed.
- B. A. DeHaven, A. L. Chen, E. A. Shimizu, S. R. Salpage, M. D. Smith and L. S. Shimizu, Cryst. Growth Des., 2019, 19, 5776–5783 CrossRef CAS.
- J. C. Gamekkanda, A. S. Sinha, J. Desper, M. Đaković and C. B. Aakeröy, New J. Chem., 2018, 42, 10539–10547 RSC.
- A. M. Abeysekera, B. B. Averkiev, A. S. Sinha, P. Le Magueres and C. B. Aakeröy, ChemPlusChem, 2021, 86, 1049–1057 CrossRef CAS PubMed.
- B. A. DeHaven, A. L. Chen, E. A. Shimizu, S. R. Salpage, M. D. Smith and L. S. Shimizu, Supramol. Chem., 2018, 30, 315–327 CrossRef CAS.
- K. N. Shunje, B. B. Averkiev and C. B. Aakeröy, Molecules, 2022, 27, 3685 CrossRef CAS PubMed.
- C. J. Setter, J. J. Whittaker, A. J. Brock, K. S. Athukorala Arachchige, J. C. McMurtrie, J. K. Clegg and M. C. Pfrunder, CrystEngComm, 2020, 22, 1687–1690 RSC.
- J. J. Brown, A. J. Brock, M. C. Pfrunder, J. P. Sarju, A. Z. Perry, A. C. Whitwood, D. W. Bruce, J. C. McMurtrie and J. K. Clegg, Aust. J. Chem., 2017, 70, 594–600 CrossRef CAS.
- E. Bosch, G. M. Ferrence, C. J. Powell, D. K. Unruh, H. R. Krueger Jr. and R. H. Groeneman, CrystEngComm, 2022, 24, 3841–3845 RSC.
- B. Ji, W. Wang, D. Deng and Y. Zhang, Cryst. Growth Des., 2011, 11, 3622–3628 CrossRef CAS.
- V. Nemec and D. Cinčić, Cryst. Growth Des., 2022, 22, 5796–5801 CrossRef CAS PubMed.
- R. Sušanj, V. Nemec, N. Bedeković and D. Cinčić, Cryst. Growth Des., 2022, 22, 5135–5142 CrossRef PubMed.
- I. J. Turchi and M. J. S. Dewar, Chem. Rev., 1975, 75, 389–437 CrossRef CAS.
- Z. Yan, A. Liu, M. Huang, M. Liu, H. Pei, L. Huang, H. Yi, W. Liu and A. Hu, Eur. J. Med. Chem., 2018, 149, 170–181 CrossRef CAS PubMed.
- T. Maekawa, N. Sakai, H. Tawada, K. Murase, M. Hazama, Y. Sugiyama and Y. Momose, Chem. Pharm. Bull., 2003, 51, 565–573 CrossRef CAS PubMed.
- N. D. Tangellamudi, S. B. Shinde, V. Pooladanda, C. Godugu and S. Balasubramanian, Bioorg. Med. Chem. Lett., 2018, 28, 3639–3647 CrossRef CAS PubMed.
- X. Yan, J. Wen, L. Zhou, L. Fan, X. Wang and Z. Xu, Curr. Top. Med. Chem., 2020, 20, 1916–1937 CrossRef CAS PubMed.
- G. Fan, X. Yang, R. Liang, J. Zhao, S. Li and D. Yan, CrystEngComm, 2016, 18, 240–249 RSC.
- D. Yan, H. Yang, Q. Meng, H. Lin and M. Wei, Adv. Funct. Mater., 2014, 24, 587–594 CrossRef CAS.
- A. M. van Leusen, Chem. Pharm. Bull., 1979, 27, 793–796 CrossRef.
-
STARe Evaluation Software Version 17.00, Mettler-Toledo GmbH, 2022 Search PubMed.
-
Data Viewer Version 1.9a, PANalytical B.V., Almelo, The Netherlands, 2018 Search PubMed.
-
CrysAlis Pro software, Version 171.41.93a, Rigaku Oxford Diffraction, Gemini CCD system, 2020 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
-
R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 5.1, Semichem Inc., Shawnee, KS, USA, 2008 Search PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- H. Jain, D. Sutradhar, S. Roy and G. R. Desiraju, Angew. Chem., Int. Ed., 2021, 60, 12841–12846 CrossRef CAS PubMed.
- V. K. Seiler, N. Tumanov, K. Robeyns, B. Champagne, J. Wouters and T. Leyssens, Cryst. Growth Des., 2020, 20, 608–616 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2358783–2358803. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00557k |
‡ Relative shortening values (R.S.) describe percentage of shortening of the contacts between donor and acceptor atom relatively to the literature values of the van der Waals radii.66 R.S. were calculated by following formula:
where d(D⋯A) stands for crystallographically measured contact length as r(D) and r(A) are values of van der Waals radii of donor atom and acceptor atom, respectively. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Nikola
Bedeković
,
Nikola
Bedeković