
First-principles study on the design of nickel based bimetallic catalysts for xylose to xylitol conversion†
Shedrack G.
Akpe
a,
Sun Hee
Choi
c and
Hyung Chul
Ham
 *ab
*ab
aDepartment of Chemistry and Chemical Engineering, Education and Research Center for Smart Energy and Materials, Inha University, Incheon, 22212, Republic of Korea. E-mail: ham.hyungchul@inha.ac.kr
bProgram in Smart Digital Engineering, Inha University, Incheon, 22212, Republic of Korea
cCenter for Hydrogen Fuel Cell Research, Korea Institute of Science and Technology, 5 Hwarang-ro 14-gil, Seongbuk-gu, Seoul 02792, Republic of Korea
First published on 23rd November 2023
Abstract
A significant challenge for effective biomass utilization and upgrading is catalysis. This research paper focuses on the conversion of xylose into xylitol, a valuable chemical used in the pharmaceutical and food industries. The primary objective is to design more efficient and cost-effective catalysts for this conversion process. The study investigates the use of Ni-bimetallic catalysts by employing a first-principles technique. Catalyst models derived from subsets of Ni (111) surfaces with various transition metals (M = Ti, V, Cr, Fe, Co, and Cu) are examined. The catalyst surfaces are screened based on the rate-determining step (RDS) involved in the conversion of xylose to xylitol, with Ni (111) serving as a reference. Electronic structure calculations are used to analyze the activities of the investigated Ni-bimetallic catalysts relative to the RDS. The results show that certain bimetallic surfaces exhibit significantly lower kinetic barriers compared to the Ni (111) surface. The hydrogenation process when investigated using different transition state paths, reveals that hydrogenation commences at the carbon atom of the carbonyl group of xylose after the ring-opening step. Stability segregation tests demonstrate varying behaviors among the screened catalysts, with Ni (111)/Cr/Ni showing greater stability than Ni (111)/Co. This study sheds light on the theoretical design of catalysts for xylose conversion, providing insights for the development of more efficient and active catalysts for industrial applications. The research highlights the significance of theoretical methodologies in tailoring catalyst surfaces to optimize their performance in biomass upgrading.
1. Introduction
Studies on upgrading hemicellulose have mostly focused on the primary sugar, xylan, which accounts for 8–25% of biomass content.1 On the other hand, xylose conversion studies (see Fig. 1) majorly focus on furfural production and its derivatives owing to the value-added chemicals that can be obtained from the latter.2–7 Additionally, an important valued chemical that can be obtained from xylose is xylitol. Xylitol is an alcohol sugar frequently used in the pharmaceutical and food industries as a sucrose alternative due to its sweetness and low-calorie content.8,9 It can also serve as a platform chemical for producing ethylene/propylene glycol,10,11 which are frequently used as a feedstock for generating hydrogen.12,13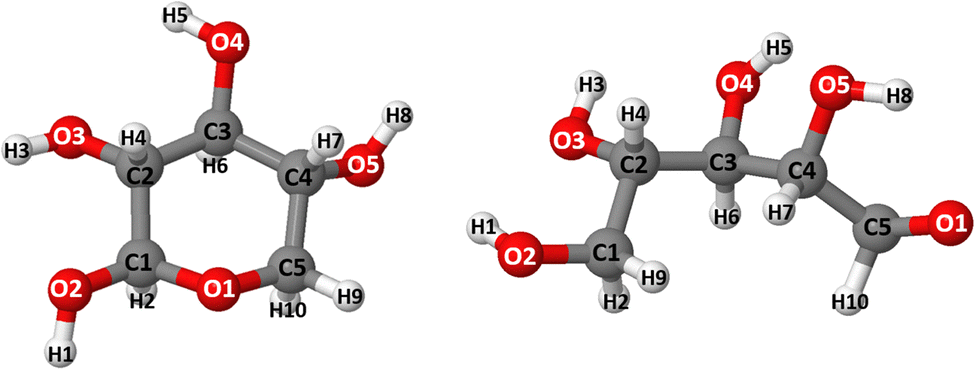 | ||
| Fig. 1 The structural configuration of cyclic (XC) and acyclic (XAC) forms of xylose. The C, O, and H atoms are represented by dark gray, red, and white balls that are numbered in clockwise directions. | ||
A significant challenge for effective biomass utilization and upgrading is catalysis. Several catalysts have been tested for biomass processing,2,9,14–21 but only a few are efficient, time-stable, recyclable, and selective to the target product. For commercial xylitol production via xylose hydrogenation, a RANEY® nickel catalyst is frequently used for this process because it is less expensive and easier to handle. Nonetheless, it has several limitations, including leaching, and organic impurity accumulation on its surface, which often leads to poisoning of active sites, and ultimately faster deactivation.22,23 Catalyst minimization and efficiency can be accomplished by combining active sites (metals), supports, and promoters.24
Utilizing bimetallic catalysts in biomass feedstock refining proves to be a smart approach due to the capacity of metal interaction to modify the catalyst's surface properties.24–27 These modifications have the potential to significantly boost catalytic activity, modify selectivity towards the desired product, and enhance the stability of the catalyst when faced with impurities derived from biomass and more rigorous reaction conditions.24
In our previous study,28 we investigated the activities of single crystal metallic catalysts (Ru, Pt, Pd, Ni, and Rh) in the reductive hydrogenation of cyclic xylose to xylitol. Our findings revealed that direct hydrogenation of cyclic xylose to xylitol is not a feasible process. However, we observed that the minimum energy path (MEP) for this reaction on all examined surfaces involves the initial transformation of xylose from its cyclic conformation to an acyclic form followed by step-wise hydrogenation. Consequently, in the current study, we have excluded the mechanism of directly hydrogenating cyclic xylose. To enhance the performance of nickel (Ni) catalysts employed in the industrial production of xylitol from xylose, our study focused on investigating the activities of Ni-bimetallic catalysts using a first-principles technique. Specifically, we examined Ni-bimetallic catalyst models derived from subsets of Ni (111) surfaces, incorporating surface and subsurface layers of various 3d ordbital transition metals denoted as M (where M = Ti, V, Cr, Fe, Co, and Cu). The surface layer configurations are represented as Ni (111)/M (M = Co, Cu, and Fe), while the subsurface layer configurations are denoted as Ni (111)/M/Ni (M = Ti, V, Cr, Co, and Fe). The purpose of this investigation is to explore how these Ni-bimetallic catalysts would affect the conversion of xylose to xylitol while using Ni (111) as a reference surface. We understand that the real-world applicability of theoretical model structures is a common challenge in the field of catalysis.29 However, theoretical calculations serve as a valuable tool in understanding the fundamental interactions and potential reaction mechanisms at play, which in turn informs the design of more effective catalysts. Our work aims to contribute to this understanding by investigating transition metal overlayer and sublayer structures on Ni (111) catalysts using first-principles calculations. A recent study on the theoretical and experimental study of NiM (111) (M = Fe, Co, Cu, Zn) bimetallic catalysts for the water–gas shift reaction revealed the mechanistic advantage of NiCo (111) compared to Ni (111). The experimental evaluations within the same study also verify the theoretical predictions, showing a good correlation between theoretical and real-world observations.25
From our previous study, we can recall that the RDS for xylose hydrogenation to xylitol on virtually all surfaces involved the transformation of cyclic xylose into its acyclic form. With this knowledge in mind, we conducted a screening process to assess the selected Ni-bimetallic catalysts in terms of their impact on the barrier of the RDS involved in the conversion of xylose to xylitol. As a reference point, we used Ni (111) as the basis for comparison. Also, we employed electronic structure calculations to elucidate the trend in activities of our investigated Ni-bimetallic catalyst relative to the RDS.
During the liquid-phase catalytic hydrogenation of xylose, it is observed that a minor fraction of side products, such as xylulose, are generated through xylose isomerization, subsequently resulting in the production of arabinitol and xylitol.22 Studies have demonstrated that employing low temperatures and high hydrogen pressures can effectively mitigate the formation of these side products.30 Therefore, the primary objective of this study is to prioritize the production of xylitol as the main product of xylose hydrogenation. This study highlights the importance of first-principles techniques in optimizing the catalyst surfaces for enhanced performance in upgrading biomass, particularly for the ring-opening of cyclic xylose.
2. Computational details and methodologies
All density functional theory (DFT) calculations were performed within the Vienna ab initio simulation package (VASP).31 The exchange–correlation interactions are described by the generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE).32 Core electron interactions are modeled using the projector augmented wave (PAW) pseudopotentials, and the electronic states were expanded on a plane wave basis with a kinetic energy cutoff of 400 eV. The Ni-bimetallic surfaces were modeled using a 6 × 4, 3-layer unit cell of bulk nickel as employed in ref. 28, with surface and/or subsurface layers of M (M = Ti, V, Cr, Mn, Fe, Co, and Cu).Furthermore, for both the adsorption studies and reactions occurring on the different surfaces, the irreducible Brillouin-zone integrations were sampled with a 1 × 1 × 1.33,34 For the various supercells, the atoms at the bottom two layers were kept fixed in their bulk structure, whereas the atoms in the top layer, including the adsorbate species, were allowed to relax using the conjugate gradient method. In our investigation, we examined the adsorption of xylose on some specific surface slabs with 4 layers (see Table 2S in the ESI†). For these slabs, the atoms within the bottom two layers remained fixed in their bulk structure, while the atoms in the top two layers, including the adsorbate species, were allowed to relax using the conjugate gradient method. The variation in xylose adsorption energy between the slabs with two different numbers of layers (3 and 4 layers) is minimal, and both slabs exhibit a consistent trend. This observation suggests that a 3-layer slab can yield reliable and accurate results. In addition, to avoid interactions with periodic images, a vacuum distance of ≈20 Å was introduced between repeated slabs along the z-direction. Also, for molecules in the gas phase, the total energies were calculated using a 20 × 20 × 20 simulation box with a gamma k-point mesh. For the electronic and ionic relaxations of slabs and adsorbates, convergence criteria of 1 × 10−6 eV and 0.05 eV Å−1 were set. The transition states (TS*s) and barriers for the elementary steps are located using the climbing-image nudged elastic band (CI-NEB) method35 with six images per calculation created between the initial and final states. The images were relaxed until the forces approached values of <0.1 eV.36 For the xylose hydrogenation pathway, the minimum energy path (MEP) shown in Schemes 1 and 2, already investigated on single crystal surfaces,28 was employed. For all the elementary reactions examined in the current study, reaction energies (ΔE) were calculated from the difference between the energies of the initial and final states. Zero-point energy corrections were ignored in this study, and the adsorption, activation, and reaction energies were calculated using eqn (1)–(3). Furthermore, eqn (4) was used to obtain the d-band centers of the slab surfaces (refer to the ESI† for a comprehensive explanation of the d-band center calculation). Additionally, the segregation energy was obtained as the difference in total energy between the segregated slab and the normal slab (see the ESI† for more information).
| Eads = Etot − Eslab − Especies | (1) |
| Ea = ETS* − Erxt | (2) |
| ΔEr = Eprod − Erxt | (3) |
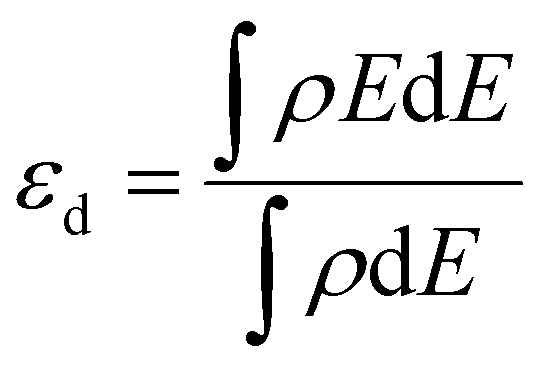 | (4) |
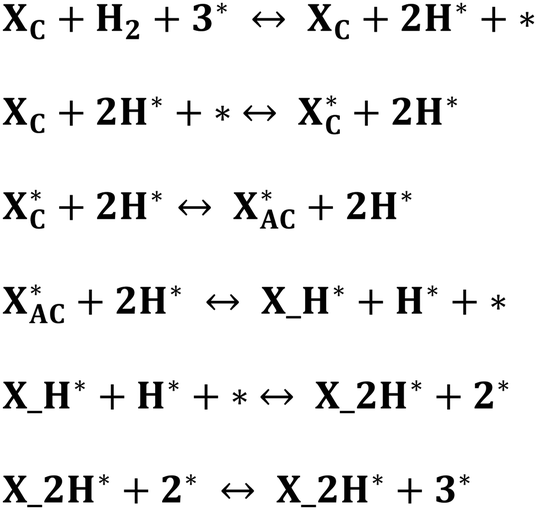 | ||
| Scheme 1 Proposed mechanisms for the reductive hydrogenation of xylose to xylitol. XC and XAC denote cyclic and acyclic xylose, respectively, while X_H denotes the first hydrogenation intermediate and X_2H, the second hydrogenation intermediate (xylitol). X, X_H, or X_2H with * represent the adsorbed species, while the bare surface (s) is represented by * only. | ||
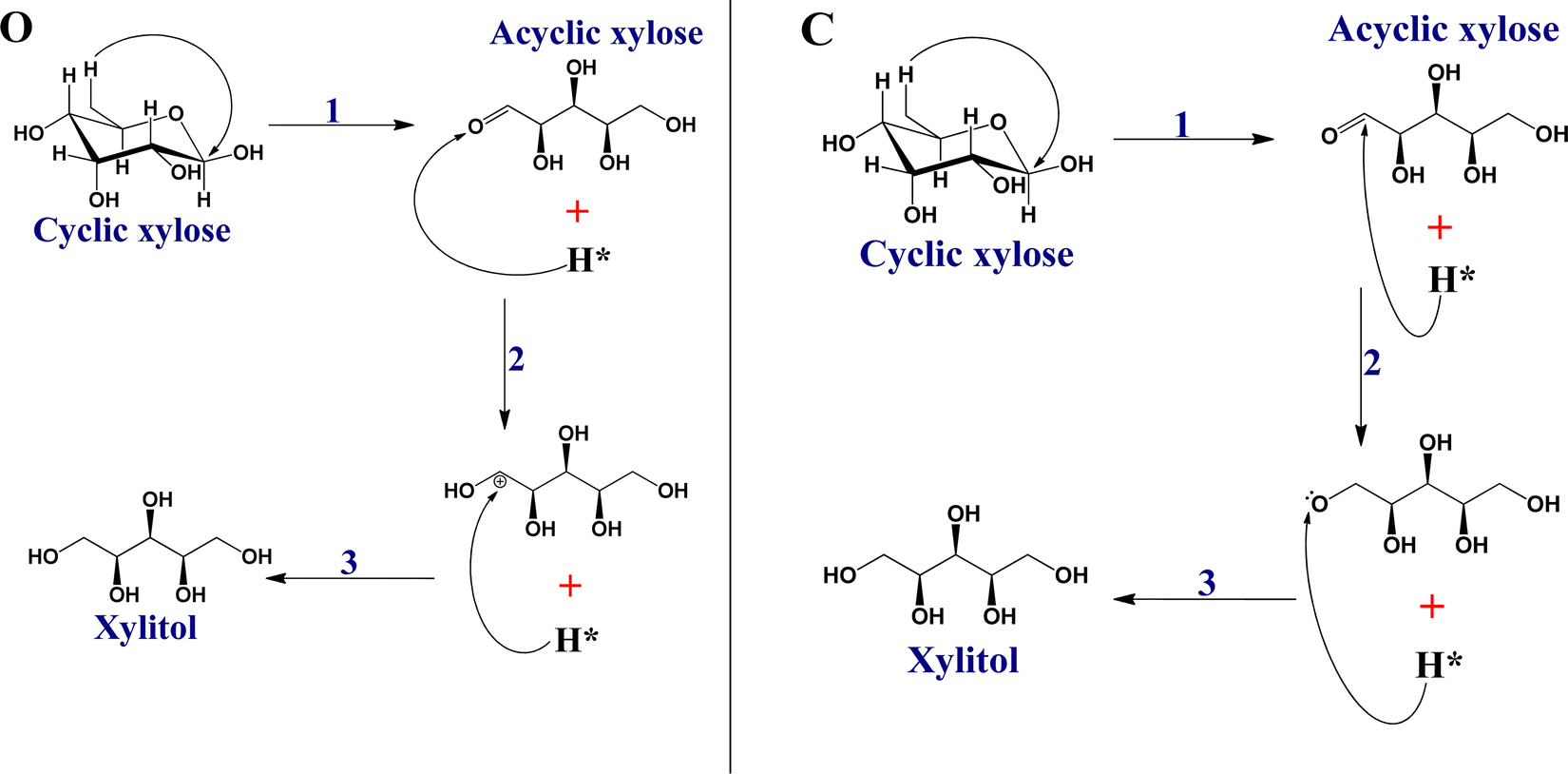 | ||
| Scheme 2 Schematic representations for the proposed mechanisms for the reductive hydrogenation of xylose to xylitol. O and C represent transition state paths (TSP) via different hydrogenation pathways designated as “Hydro_via_O” and “Hydro_via_C,” respectively. For TSP O and C, 1: ring-opening of cyclic to xylose via C–O bond cleavage initiated by the rearrangement of atomic hydrogen; 2 and 3: O–H & C–H and C–H & O–H bond formations, respectively. | ||
The reaction steps are summarized in Schemes 1 and 2, respectively.
3. Results and discussions
3.1. Stability of the Ni-bimetallic slabs, Ni (111)/M and Ni (111)/M/Ni
We have calculated the binding energies (indicated by EB.layer) of the M or Ni surface monolayers in the Ni (111)/M or Ni (111)/M/Ni slab. Here, the EB.layer = E[Ni (111)/M or Ni (111)/M/Ni] − E[Ni (111) or Ni (111)/M] − E[M or Ni] where E[Ni (111)/M or Ni (111)/M/Ni] − E[Ni (111) or Ni (111)/M], and E[M or Ni] represent the total energy of Ni (111)/M or Ni (111)/M/Ni slab, Ni (111) or Ni (111)/M slab and M or Ni monolayer, respectively. As shown in Table 1, the binding energies of the various M and Ni overlayers in the Ni (111)/M or Ni (111)/M/Ni slab are all negative, indicating stronger interaction between M and Ni and in turn high stability of the bimetallic Ni–M slabs.| Surfaces | E B.layer (eV) |
|---|---|
| Ni (111)/Co | −32.96 |
| Ni (111)/Cu | −19.13 |
| Ni (111)/Fe | −32.08 |
| Ni (111)/Ti/Ni | −43.34 |
| Ni (111)/V/Ni | −41.64 |
| Ni (111)/Cr/Ni | −32.71 |
| Ni (111)/Co/Ni | −85.71 |
| Ni (111)/Fe/Ni | −4.02 |
3.2. Screening of Ni-bimetallic surfaces based on cyclic xylose ring-opening barrier
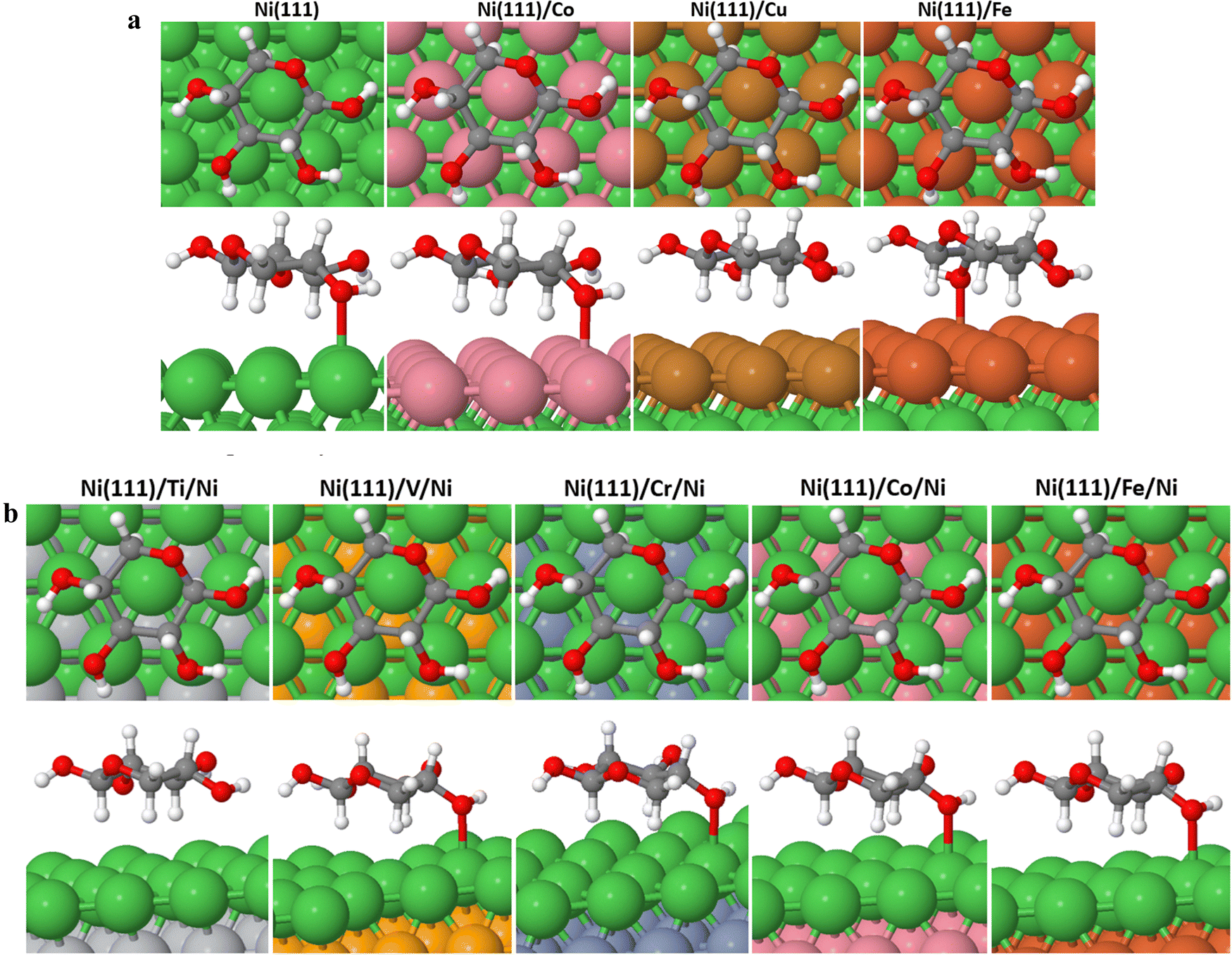 | ||
| Fig. 2 (a) Most stable binding conformations of XC on the Ni (111)/M surfaces showing the front and bottom views, respectively, where M = Ni, Co, Cu, and Fe. (b) Most stable binding conformations of XC on the Ni (111)/M/Ni surfaces showing the front and bottom views, respectively, where M = Ti, V, Cr, Co, and Fe. | ||
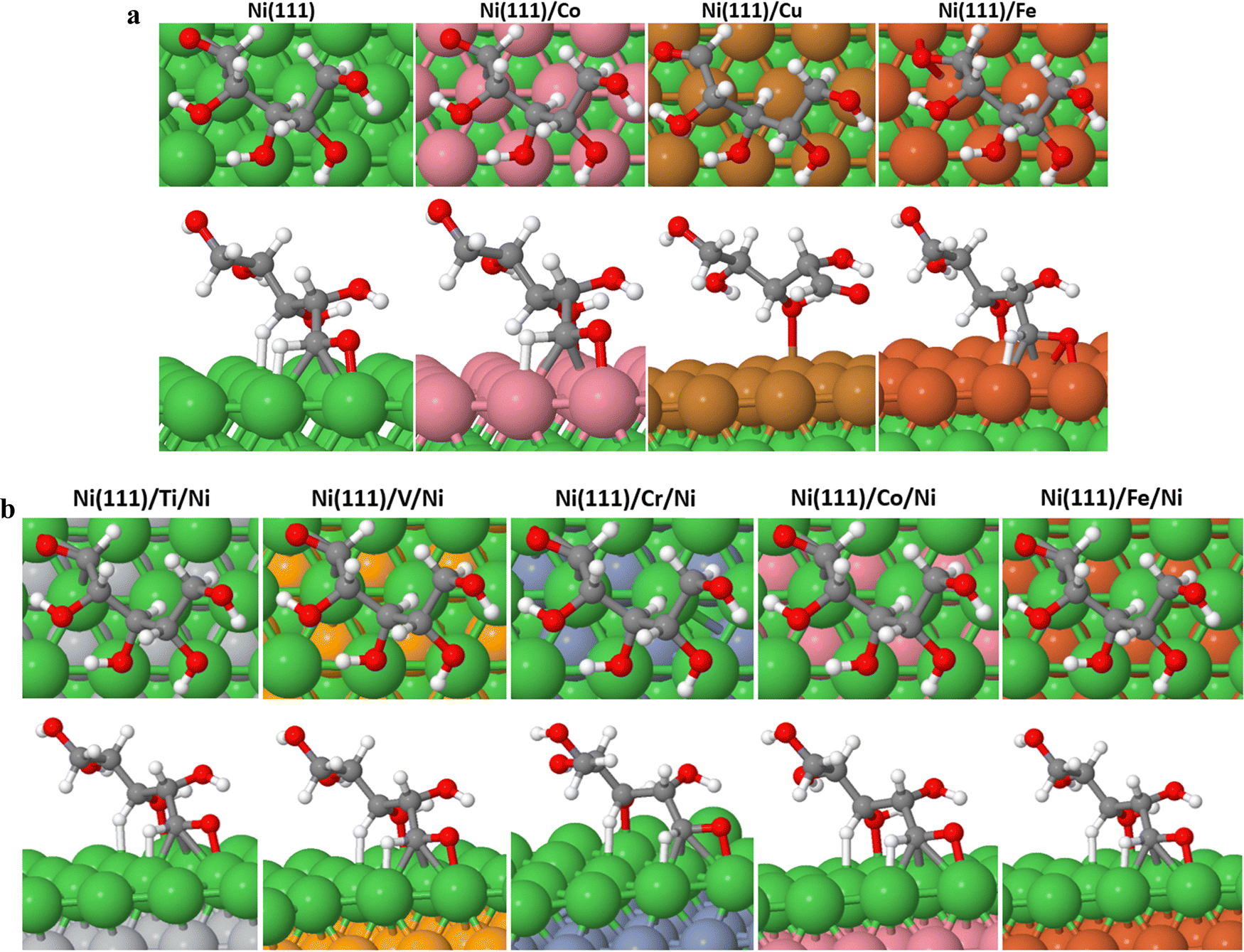 | ||
| Fig. 3 (a) Most stable binding conformations of XAC on the Ni (111)/M surfaces showing the front and bottom views, respectively, where M = Ni, Co, Cu, and Fe. (b) Most stable binding conformations of XAC on the Ni (111)/M/Ni surfaces showing the front and bottom views, respectively, where M = Ti, V, Cr, Co, and Fe. | ||
| Surfaces | E b_Xc (eV) | E b_Xac (eV) |
|---|---|---|
| Ni (111) | −0.32 | −0.28 |
| Ni (111)/Co | −0.33 | −0.37 |
| Ni (111)/Cu | −0.27 | −0.04 |
| Ni (111)/Fe | −0.46 | −0.78 |
| Ni (111)/Ti/Ni | −0.43 | −0.18 |
| Ni (111)/V/Ni | −0.35 | −0.38 |
| Ni (111)/Cr/Ni | −0.35 | −0.52 |
| Ni (111)/Co/Ni | −0.35 | −0.33 |
| Ni (111)/Fe/Ni | −0.34 | −0.15 |
The results presented in Table 2 provide valuable insights into the binding energies of cyclic (XC) and acyclic (XAC) xylose on the investigated surfaces. Starting with the Ni (111) surface, we observe that both XC and XAC exhibit relatively similar binding energies, with XC having a slightly higher binding energy of −0.32 eV compared to XAC at −0.28 eV. This suggests a reasonably stable interaction between the xylose molecules and the Ni (111) surface.
Moving on to the bimetallic surfaces, we can observe variations in the binding energies. Ni (111)/Co shows comparable binding energies for XC and XAC, indicating a relatively stable interaction. However, Ni (111)/Cu stands out with significantly weaker binding energies for both XC (−0.27 eV) and XAC (−0.04 eV), suggesting a poor affinity between xylose and the Ni (111)/Cu surface. Interestingly, Ni (111)/Fe demonstrates the strongest binding energies for both XC (−0.46 eV) and XAC (−0.78 eV) among all the surfaces studied. This indicates a favorable interaction and strong affinity between xylose and the Ni (111)/Fe surface, suggesting its potential as an efficient catalyst for xylose conversion. However, we must understand that too strong or too weak adsorption may result in a sluggish reaction.37
For the other bimetallic surfaces, including Ni (111)/Ti/Ni, Ni (111)/V/Ni, Ni (111)/Cr/Ni, Ni (111)/Co/Ni, and Ni (111)/Fe/Ni, the binding energies of XC and XAC are comparable to or slightly weaker than those observed on the Ni (111) surface, except for Ni (111)/Cr/Ni, where the binding energy of XAC showed significant improvement compared to the Ni (111) surface. This suggests that the presence of the 3d transition metals on the subsurface layer does not significantly alter the binding characteristics of XC on these surfaces.
Overall, the binding energy results provide valuable information about the strength of interaction between (XC and XAC) and the investigated surfaces. These findings can guide the design and optimization of catalyst surfaces for improved xylose conversion in biomass upgrading processes.
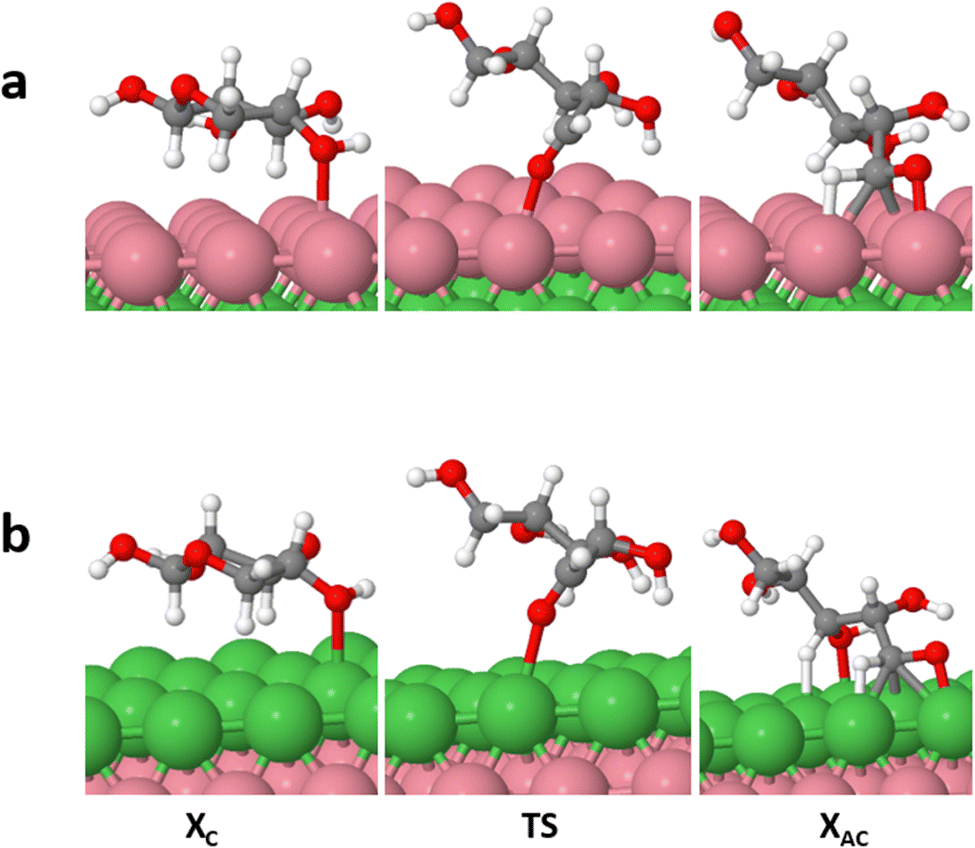 | ||
| Fig. 4 Ring-opening process of cyclic xylose on Ni (111)/Co (a) and Ni (111)/Co/Ni (b) as representative surfaces. TS denotes the transition state obtained from the kinetic process with the CI-NEB method. | ||
| Surfaces | E a (eV) | ΔE (eV) |
|---|---|---|
| Ni (111) | 0.827 | 0.177 |
| Ni (111)/Co | 0.770 | 0.107 |
| Ni (111)/Cu | 0.890 | 0.376 |
| Ni (111)/Fe | 0.816 | −0.178 |
| Ni (111)/Ti/Ni | 0.854 | 0.389 |
| Ni (111)/V/Ni | 0.821 | 0.108 |
| Ni (111)/Cr/Ni | 0.791 | −0.020 |
| Ni (111)/Co/Ni | 0.846 | 0.172 |
| Ni (111)/Fe/Ni | 0.836 | 0.331 |
Table 3 provides important insights into the kinetic barriers associated with the conversion of cyclic xylose to its acyclic form on the investigated surfaces. Starting with Ni (111) as a reference, the kinetic barrier for the transformation process is determined to be 0.827 eV. Among the bimetallic surfaces, Ni (111)/Co and Ni (111)/Cr/Ni exhibit the lowest kinetic barriers of 0.770 eV and 0.791 eV, respectively. This indicates a relatively easier conversion process compared to Ni (111), suggesting that the presence of Co as a second metal on the surface or Cr as a second metal on the subsurface can enhance the catalytic activity for the ring-opening of cyclic xylose.
On the other hand, Ni (111)/Cu shows the highest kinetic barrier of 0.890 eV, indicating a more challenging conversion process compared to the other surfaces. This suggests that the presence of Cu as a second metal may hinder the ring-opening reaction of cyclic xylose.
Furthermore, Ni (111)/Fe, Ni (111)/Ti/Ni, Ni (111)/V/Ni, Ni (111)/Co/Ni, and Ni (111)/Fe/Ni surfaces exhibit kinetic barriers that are comparable to or slightly higher than the Ni (111) surface. This suggests no improvement in catalytic activity compared to Ni (111).
Overall, these results highlight the significance of different surface compositions in influencing the kinetics of cyclic xylose conversion. The lower kinetic barriers observed on specific bimetallic surfaces indicate their enhanced catalytic performance relative to the Ni (111) surface. Furthermore, Fig. 5(a), a subset of Fig. 5, illustrates the correlation between the binding energies (Eb_Xac) of acyclic xylose and the corresponding kinetic barriers (Ea) for its conversion from the cyclic form. On the other hand, Fig. 5(b) showcases the relationship between the reaction energies (ΔH) and the kinetic barriers (Ea) involved in the transformation process of cyclic xylose to its acyclic form. Despite the relatively narrow range of the kinetic barrier values (0.77 eV to 0.89 eV) on all surfaces in the present study, there is a clear linear relationship between the kinetic barrier and both the binding energies of acyclic xylose and the reaction energy associated with its transformation from the cyclic form.
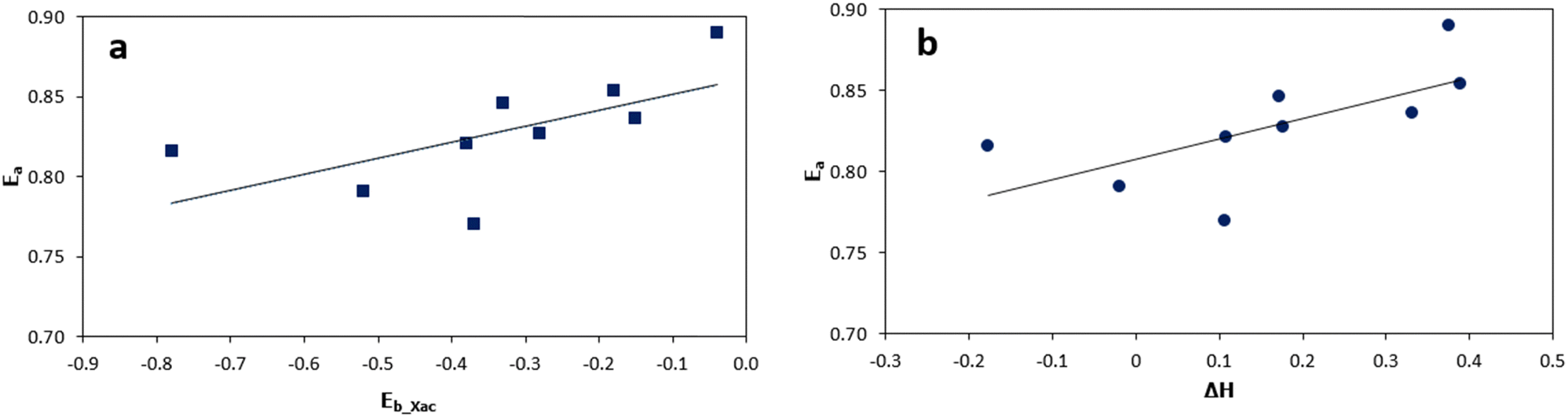 | ||
| Fig. 5 Relationship between ring-opening kinetic barriers and acyclic xylose binding energy (a) and between ring-opening kinetic barriers and reaction energies (b) on all investigated surfaces. | ||
The observed differences in kinetic barriers appear relatively subtle, with values of approximately 0.827 eV for Ni (111), 0.770 eV for Ni (111)/Co, and 0.791 eV for Ni (111)/Cr/Ni. We acknowledge that these differences may not seem substantial on their own. However, it's important to consider these results within the broader context of catalysis under investigation. For example, the kinetic difference in the key reaction step can be better understood by the reaction rate constant. It is generally known that a reaction rate constant can be described using the Arrhenius equation k = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/RT),38 where k is the rate constant of the reaction, A is the prefactor, often interpreted as the frequency of collisions or the frequency of attempts to overcome the energy barrier, Ea is the activation energy of the reaction, R is the universal gas constant, and T is the temperature. If we assume the same value of perfactor A for each of these reactions (which implies that the frequency of attempts to overcome the energy barrier is the same for all three surfaces), the rate constants k for these reactions at a given temperature T will be determined solely by the exponential term of activation energy Ea, rather than the linear perfactor A. The Arrhenius equation shows that a lower activation energy results in a higher rate constant k, implying a faster reaction rate. At a typical reaction temperature of 473 K, we estimate the rate constant difference for the catalysts. We find the about four times higher [Ni (111)/Co (k ≈ 6.42 × 10−9)] and two times higher [Ni (111)/Cr/Ni (k ≈ 3.84 × 10−9)] rate constants than for the Ni (111) case (k ≈ 1.59 × 10−9), indicating that the Ni (111)/Cr/Ni catalyst has a higher reaction rate, followed by Ni (111)/Cr/Ni, and then Ni (111). This analysis based on the Arrhenius equation supports our DFT results that the bimetallic surfaces Ni (111)/Co and Ni (111)/Cr/Ni exhibit lower kinetic barriers and, hence, are likely to exhibit enhanced catalytic activity for the ring-opening of cyclic xylose compared to Ni (111).
exp(−Ea/RT),38 where k is the rate constant of the reaction, A is the prefactor, often interpreted as the frequency of collisions or the frequency of attempts to overcome the energy barrier, Ea is the activation energy of the reaction, R is the universal gas constant, and T is the temperature. If we assume the same value of perfactor A for each of these reactions (which implies that the frequency of attempts to overcome the energy barrier is the same for all three surfaces), the rate constants k for these reactions at a given temperature T will be determined solely by the exponential term of activation energy Ea, rather than the linear perfactor A. The Arrhenius equation shows that a lower activation energy results in a higher rate constant k, implying a faster reaction rate. At a typical reaction temperature of 473 K, we estimate the rate constant difference for the catalysts. We find the about four times higher [Ni (111)/Co (k ≈ 6.42 × 10−9)] and two times higher [Ni (111)/Cr/Ni (k ≈ 3.84 × 10−9)] rate constants than for the Ni (111) case (k ≈ 1.59 × 10−9), indicating that the Ni (111)/Cr/Ni catalyst has a higher reaction rate, followed by Ni (111)/Cr/Ni, and then Ni (111). This analysis based on the Arrhenius equation supports our DFT results that the bimetallic surfaces Ni (111)/Co and Ni (111)/Cr/Ni exhibit lower kinetic barriers and, hence, are likely to exhibit enhanced catalytic activity for the ring-opening of cyclic xylose compared to Ni (111).
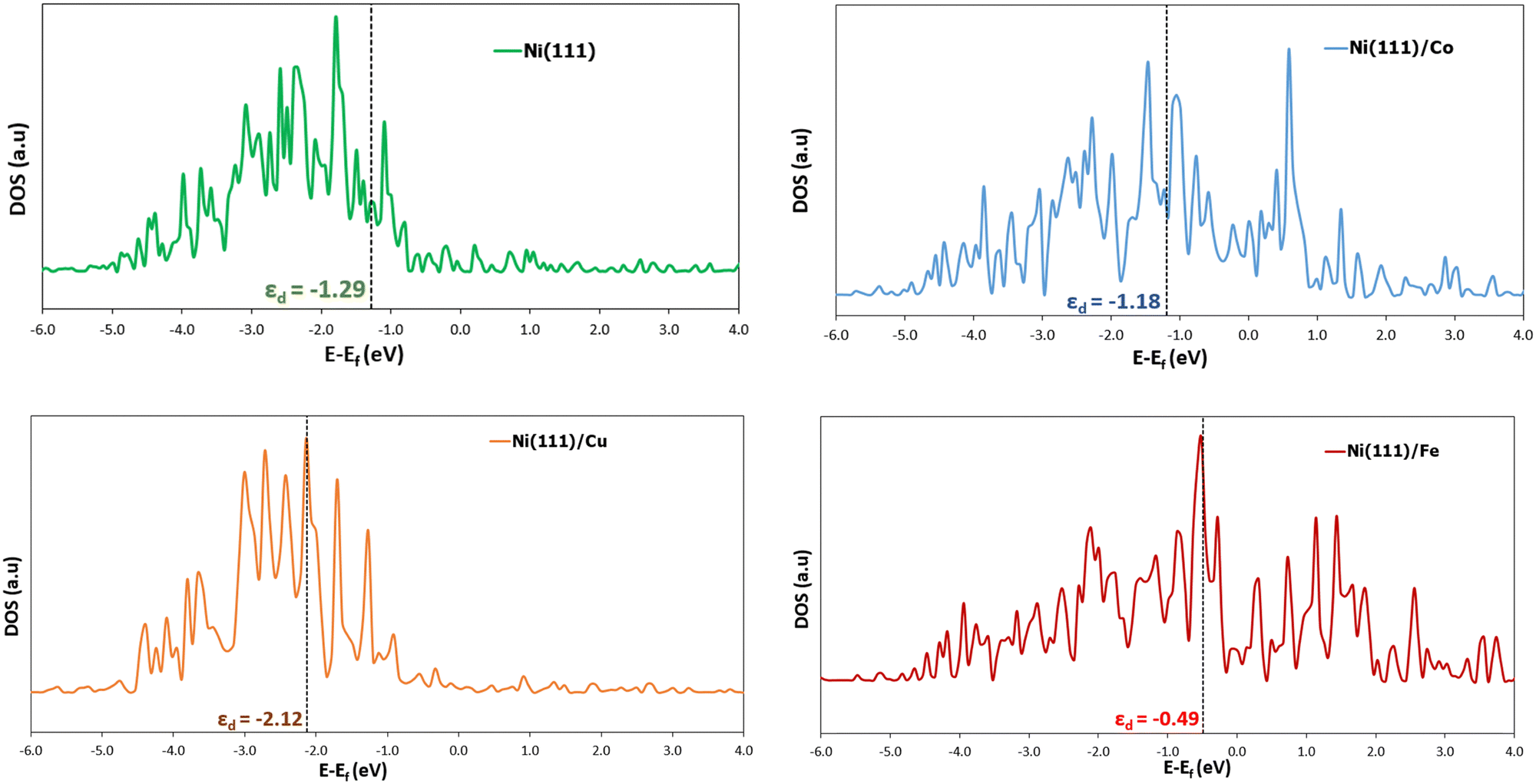 | ||
| Fig. 6 Representation of the partial density of states (DOS) of the d band surface atom(s) of Ni (111), Ni (111)/Co, Ni (111)/Cu, and Ni (111)/Fe, respectively. The 0 eV is the Fermi level position, and the dotted lines denote the d-band centers (εd). Refer to the supplementary page for the partial DOS of the other surfaces (ESI†). | ||
3.3. Comprehensive study of xylose hydrogenation to xylitol
In this section, we conducted a complete investigation of xylose hydrogenation to xylitol on Ni (111), Ni (111)/Co, and Ni (111)/Cr/Ni. These specific Ni-bimetallic surfaces were selected due to their superior performance in the ring-opening process of cyclic xylose (the rate-determining step), as compared to the Ni (111) surface. Recall that this criterion served as the foundation for screening the various surfaces examined in this study.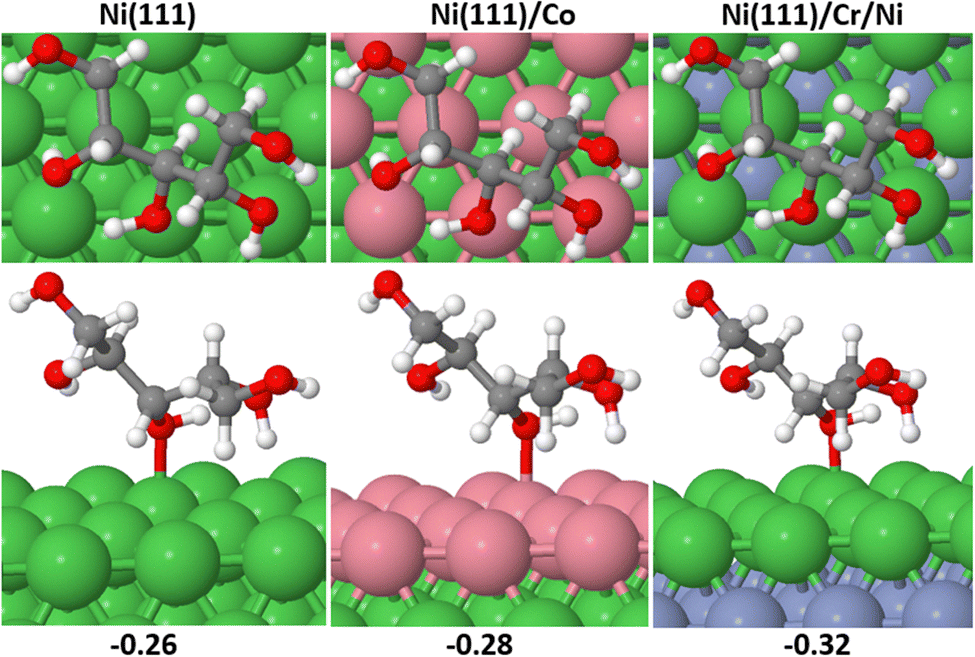 | ||
| Fig. 7 Binding conformations of X_2H on the screened surfaces showing the front and bottom views, respectively. The values represent the adsorption energies in eV. | ||
3.3.2.1. TS path O: acyclic xylose hydrogenation starting at O. In this TS path, hydrogenation begins with co-adsorbed XAC and H, where the adsorbed H directly attacks the O1 atom of XAC (refer to Fig. 1 for XAC numbering), yielding the first hydrogenation intermediate (X_H). On Ni (111), Ni (111)/Co, and Ni (111)/Cr/Ni surfaces, kinetic barriers of 0.84 eV, 0.93 eV, and 0.91 eV, respectively, were observed. The corresponding reaction energies are 0.14 eV, 0.21 eV, and 0.11 eV, respectively. Notably, all surfaces, including Ni (111), indicated an endothermic process. However, the values of the reaction barriers and energies obtained for the bimetallic surfaces do not demonstrate a significant improvement compared to the Ni (111) surface. Additionally, for the second hydrogenation stage, the kinetic barriers encountered to yield the final xylitol product are 0.66, 0.75, and 0.78 with reaction energies of −0.45, −0.33, and −0.35, respectively. Contrary to the first hydrogenation stage, this hydrogenation stage predicts an exothermic process on all surfaces. It should be noted that proximity energy contribution terms, which is the energy required to bring the reacting species from infinite separated distances to co-adsorbed state have been included in both the barriers and reaction energies, respectively.
Fig. 8 shows the intermediate's structures and transition states of the TS path O on the investigated surfaces, while Fig. 9 depicts the energy profile diagram for changes in energy on Ni (111), Ni (111)/Co, and Ni (111)/Cr/Ni, surfaces, respectively.
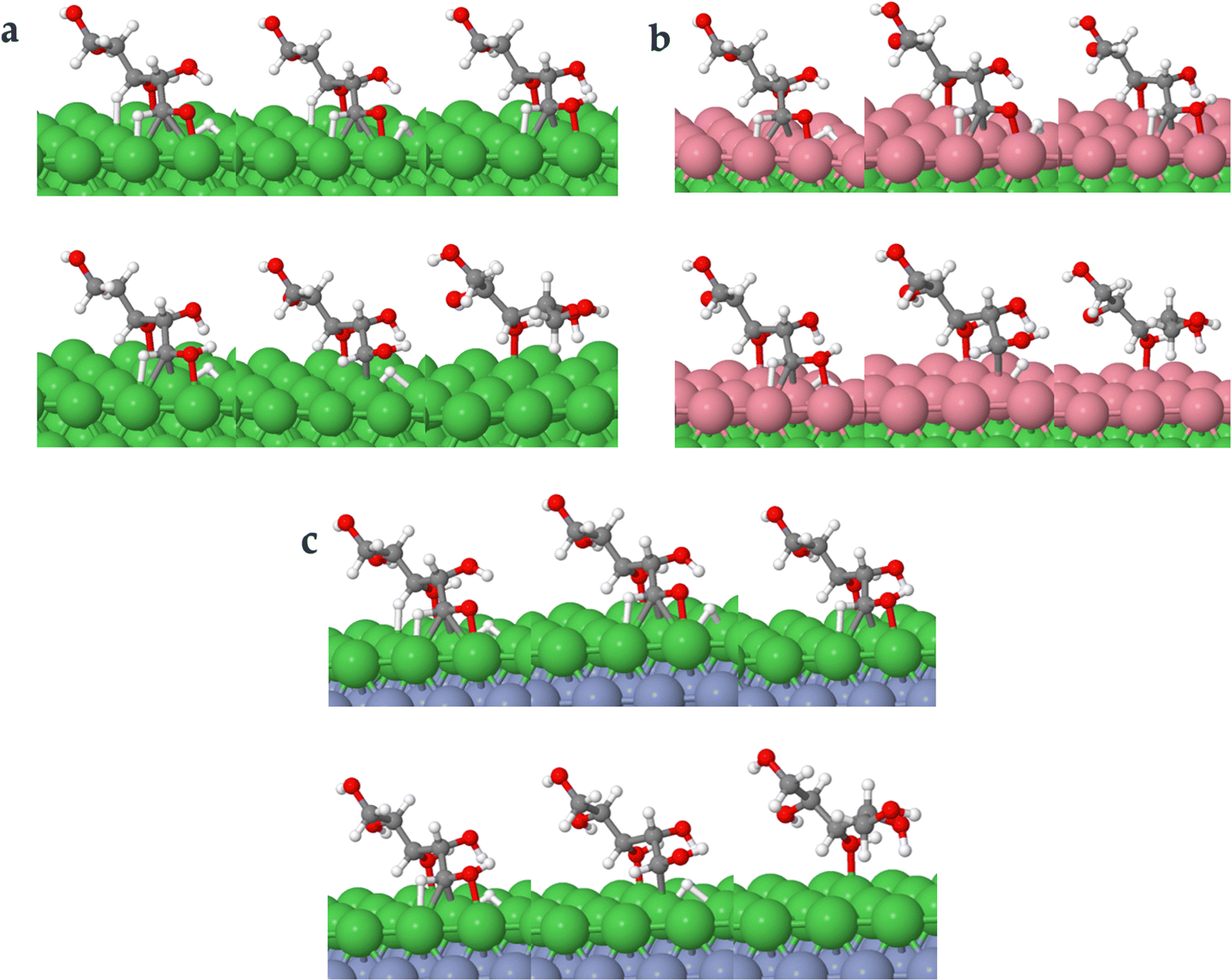 | ||
| Fig. 8 Surface intermediate's structures and transition states along the hydrogenation of acyclic xylose to xylitol via TS path O on Ni (111) (a), Ni (111)/Co (b), and Ni (111)/Cr/Ni (c) surfaces. | ||
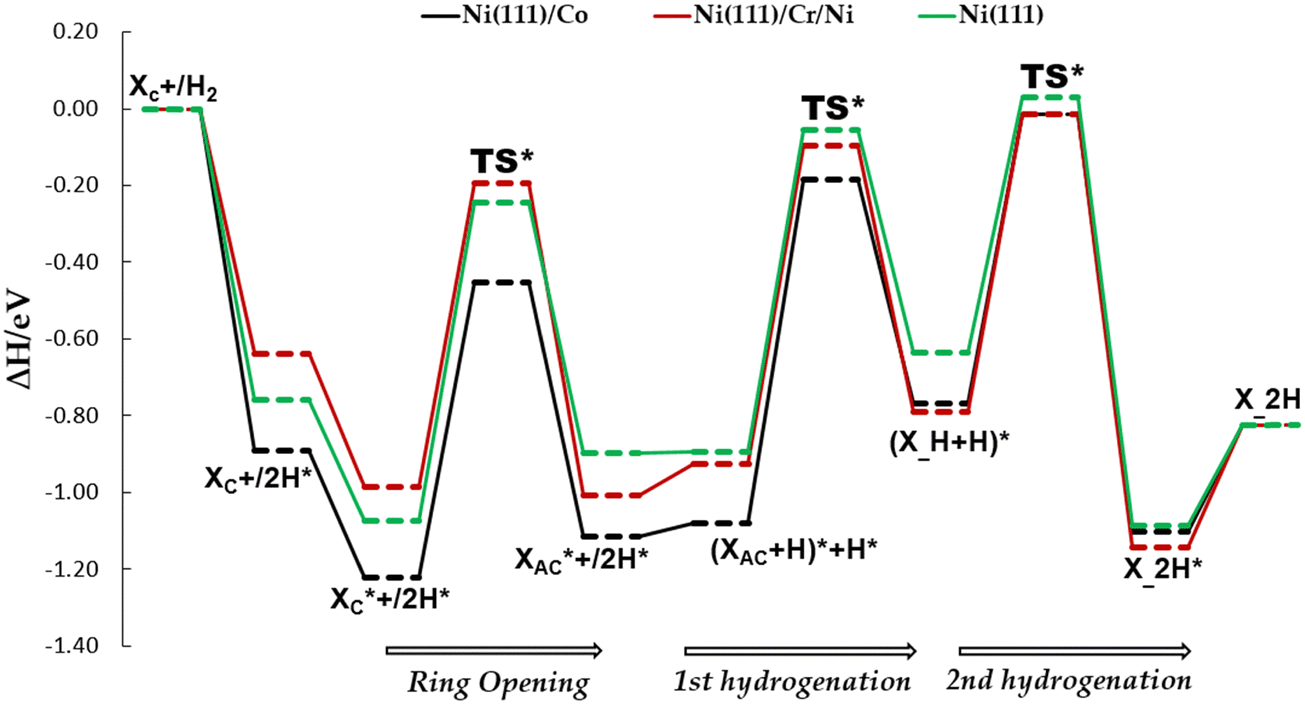 | ||
Fig. 9 Potential energy profile diagrams (PEDs) for the ring-opening of cyclic xylose and the subsequent hydrogenation of acyclic xylose to xylitol via TS path O on Ni (111)/Co, Ni (111)/Cr/Ni, and Ni (111) surfaces. XC and 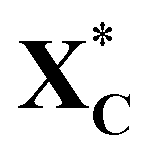 denote gas-phase and adsorbed phase cyclic xylose, while X_H* and X_2H* represent the adsorbed first and second hydrogenation intermediate/product. The reactant species in infinite separation and co-adsorbed states are denoted by A*+/B* and (A + B)*. denote gas-phase and adsorbed phase cyclic xylose, while X_H* and X_2H* represent the adsorbed first and second hydrogenation intermediate/product. The reactant species in infinite separation and co-adsorbed states are denoted by A*+/B* and (A + B)*. | ||
3.3.2.2. TS path B2: XC C–O bond scission to XAC followed by hydrogenation via C. In this TS path, the adsorbed H first attacks the C5 atom of XAC before attacking the O1 atom (refer to Fig. 1 for XAC numbering), yielding the first hydrogenation intermediate (X_H) and subsequently the xylitol product. For the first stage hydrogenation, kinetic barriers of 0.71 eV, 0.55 eV, and 0.43 eV, respectively on Ni (111), Ni (111)/Co, and Ni (111)/Cr/Ni surfaces were encountered. The corresponding reaction energies are 0.20 eV, −0.03 eV, and 0.02 eV, respectively. Contrary to TS path O, the values of the reaction barriers and energies obtained for the bimetallic surfaces demonstrate significant improvement compared to the Ni (111) surface. Subsequently, for the second hydrogenation stage, kinetic barriers of 0.51, 0.54, and 0.73 eV, respectively were observed while the corresponding reaction energies were −0.45, 0.20, and −0.29 eV, respectively. It is noticeable that on the examined surfaces, this TS path showed lower kinetic barriers for both hydrogenation stages compared with the TS path O.
Fig. 10 shows the intermediate's structures and transition states of TS path C on the investigated surfaces, while Fig. 11 depicts the energy profile diagram for changes in energy on Ni (111), Ni (111)/Co, and Ni (111)/Cr/Ni surfaces, respectively.
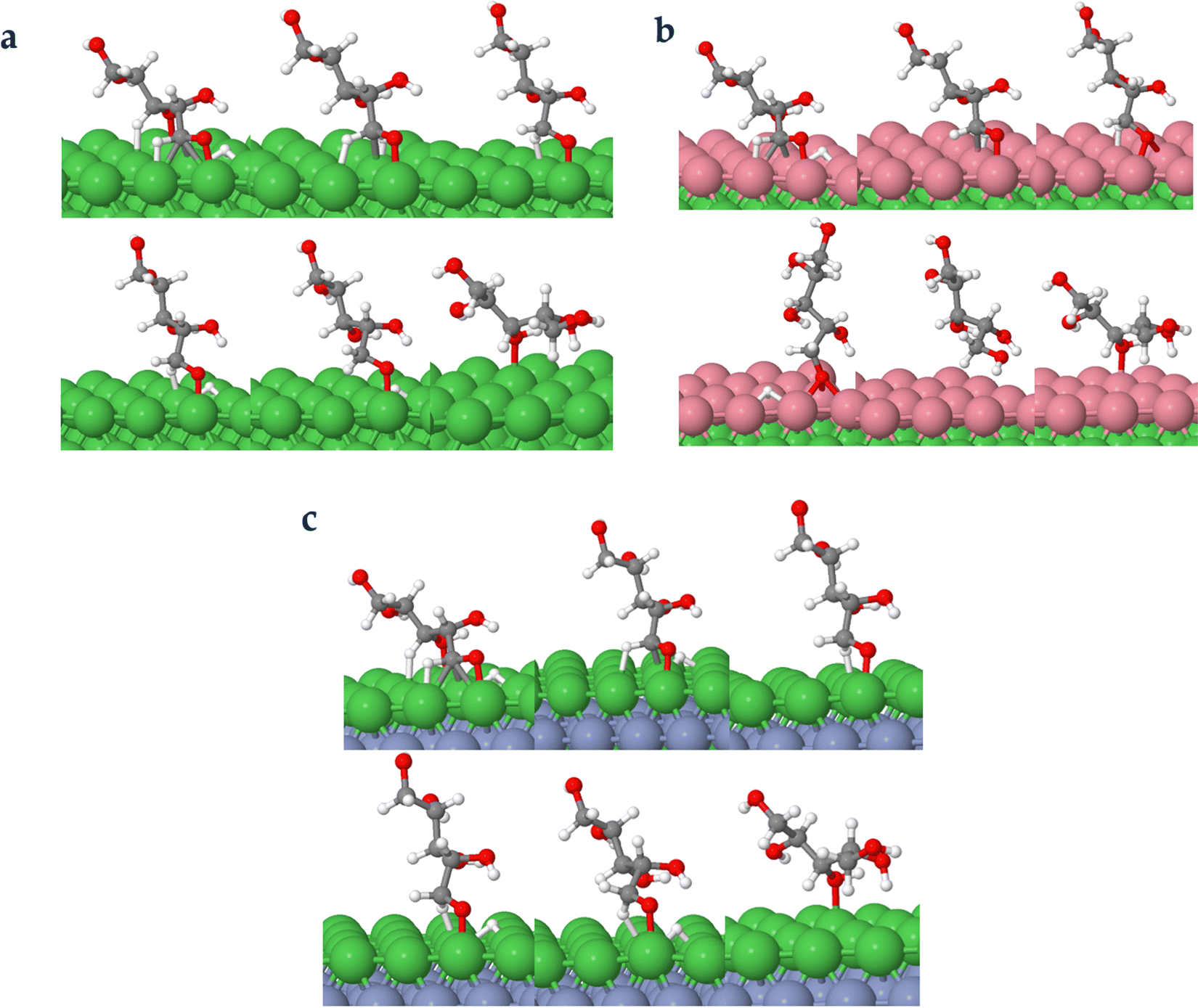 | ||
| Fig. 10 Surface intermediate's structures and transition states along the hydrogenation of acyclic xylose to xylitol via the TS path C on Ni (111) (a), Ni (111)/Co (b), and Ni (111)/Cr/Ni (c) surfaces. | ||
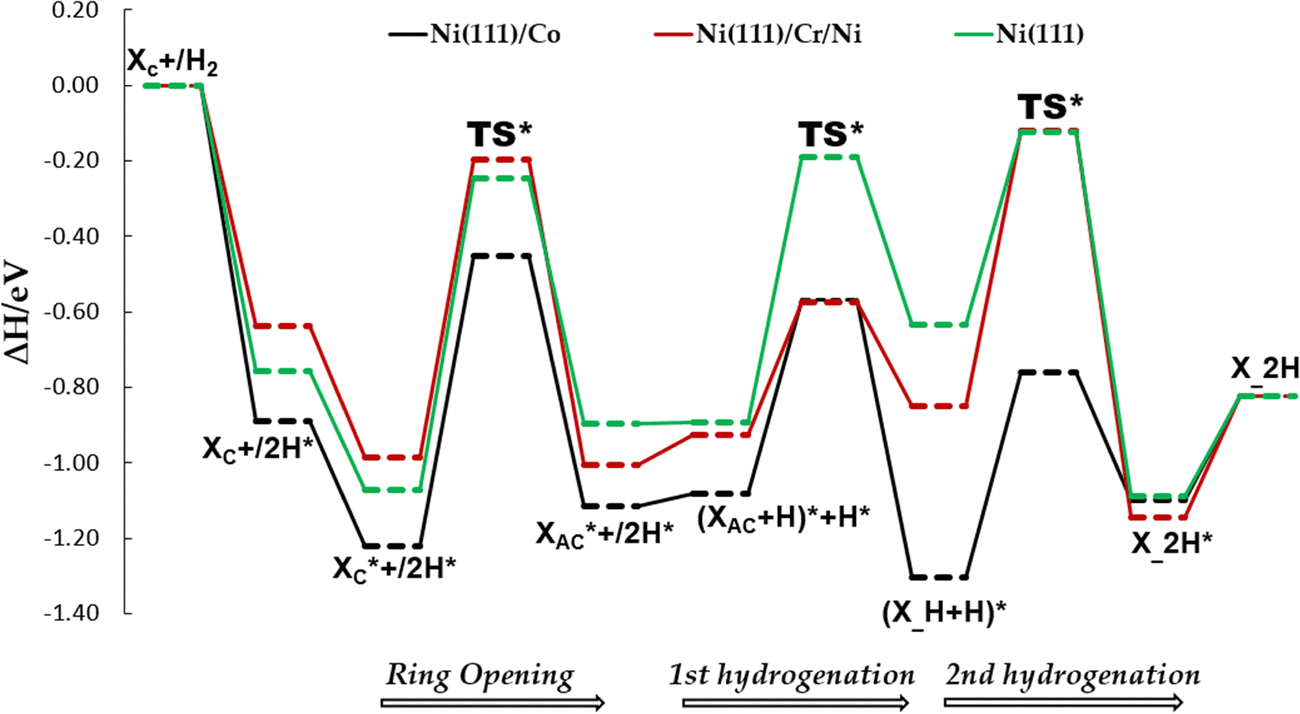 | ||
Fig. 11 Potential energy profile diagrams (PEDs) for the ring-opening of cyclic xylose and the subsequent hydrogenation of acyclic xylose to xylitol via the TS path C on Ni (111)/Co, Ni (111)/Cr/Ni, and Ni (111) surfaces. XC and 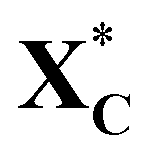 denote gas-phase and adsorbed phase cyclic xylose, while X_H* and X_2H* represent the adsorbed first and second hydrogenation intermediate/product. The reactant species in infinite separation and co-adsorbed states are denoted by A*+/B* and (A + B)*. denote gas-phase and adsorbed phase cyclic xylose, while X_H* and X_2H* represent the adsorbed first and second hydrogenation intermediate/product. The reactant species in infinite separation and co-adsorbed states are denoted by A*+/B* and (A + B)*. | ||
Tables 4 and 5 provide a comprehensive overview of the kinetic barriers and reaction energy requirements for the two investigated TS paths associated with the complete hydrogenation process, which begins with the ring-opening of cyclic xylose and leading to xylitol production.
| Surfaces | E a1 (eV) | ΔE1 (eV) | E a2 (eV) | ΔE2 (eV) | E a3 (eV) | ΔE3 (eV) |
|---|---|---|---|---|---|---|
| Ni (111) | 0.827 | 0.177 | 0.839 | 0.143 | 0.664 | −0.453 |
| Ni (111)/Co | 0.770 | 0.107 | 0.931 | 0.208 | 0.754 | −0.334 |
| Ni (111)/Cr/Ni | 0.791 | −0.020 | 0.911 | 0.111 | 0.776 | −0.354 |
| Surfaces | E a1 (eV) | ΔE1 (eV) | E a2 (eV) | ΔE2 (eV) | E a3 (eV) | ΔE3 (eV) |
|---|---|---|---|---|---|---|
| Ni (111) | 0.827 | 0.177 | 0.705 | 0.199 | 0.510 | −0.452 |
| Ni (111)/Co | 0.770 | 0.107 | 0.545 | −0.032 | 0.544 | 0.204 |
| Ni (111)/Cr/Ni | 0.791 | −0.020 | 0.432 | 0.018 | 0.732 | −0.292 |
Catalyst segregation effects occur when certain elements or components in a catalyst material tend to accumulate or segregate at the catalyst's surface or specific regions during catalytic reactions,41 thereby impacting on the catalyst's performance and reactivity. Segregation tests were conducted on Ni (111)/Co, Ni (111)/Cr/Ni catalysts both in vacuum and under adsorbed XC and XAC conditions. Fig. 12 depicts the segregation scheme for Ni (111)/Co and Ni (111)/Cr/Ni under XAC adsorption conditions, while Table 6 presents the segregation energies in both vacuum and adsorbed states (see the ESI† for calculation procedures). The results from Table 6 indicate that Ni (111)/Co exhibits a high tendency for segregation during the reaction, whereas segregation is not feasible in Ni (111)/Cr/Ni. This suggests that the latter configuration possesses greater stability compared to the former.
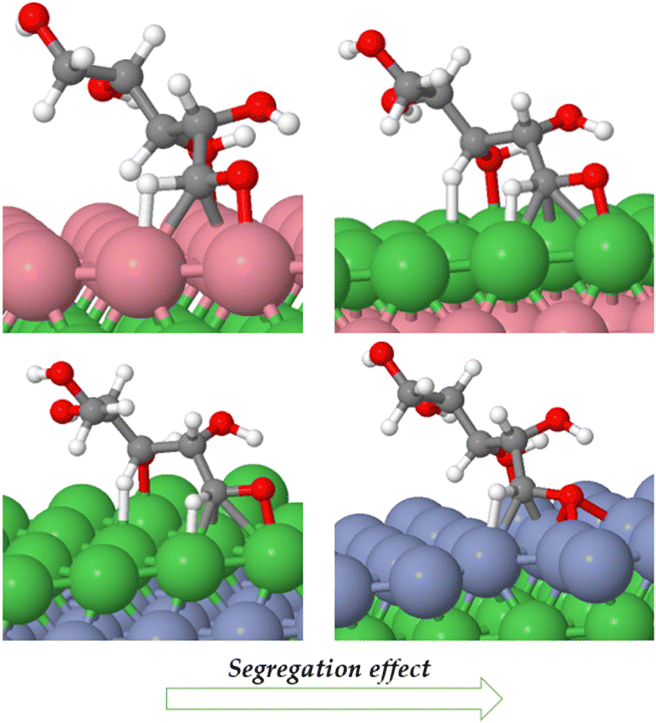 | ||
| Fig. 12 Subsurface to surface segregation of Ni (111)/Co and Ni (111)/Cr/Ni under XAC adsorbed conditions. | ||
| Surfaces | ΔEvac (eV) | ΔEXc (eV) | ΔEXac (eV) |
|---|---|---|---|
| Ni (111)/Co | −3.92 | −3.94 | −3.87 |
| Ni (111)/Cr/Ni | 10.91 | 9.49 | 8.47 |
4. Conclusions
RANEY® nickel is primarily used in commercial xylitol synthesis via xylose hydrogenation due to its availability and low cost. The use of bimetallic catalysts for upgrading biomass feedstock is an excellent technique because the interaction between metals can alter the surface properties of the catalyst. These modifications can significantly enhance the catalytic activity, alter selectivity to the targeted product, and boost catalyst stability in the presence of biomass-derived impurities and harsher reaction conditions.In this study we employed a first-principles technique to design Ni-bimetallic catalysts and conducted xylose conversion into xylitol using the Vienna ab initio simulation package (VASP). Our modeled catalysts were subsets of Ni (111), namely Ni (111)/M (M = Co, Cu, and Fe) and Ni (111)/M/Ni (M = Ti, V, Cr, Co, and Fe), with the Ni (111) surface serving as a reference. First, we screened the catalyst surfaces based on the rate-determining step (RDS), which involves the transformation of cyclic xylose (XC) into its acyclic form (XAC). Our results revealed a linear relationship between the kinetic barrier and the binding energies of acyclic xylose, as well as the reaction energy associated with its transformation from the cyclic form.
Furthermore, among the bimetallic surfaces, Ni (111)/Co and Ni (111)/Cr/Ni exhibited appreciably lower kinetic barriers of 0.770 eV and 0.791 eV, respectively, compared to a kinetic barrier of 0.827 eV for the Ni (111) surface. To that end, we conducted comprehensive hydrogenation studies on these screened surfaces, following two transition state paths (TS path O and TS path C). Our findings showed that for all three examined surfaces, including Ni (111), the TS path C exhibited lower kinetic barriers for the first and second hydrogenation steps compared to TS path O. This implies that after the ring-opening step, which yields acyclic xylose, hydrogenation begins at the carbon atom of the carbonyl group rather than at the oxygen atom.
Finally, segregation tests were conducted on Ni (111)/Co and Ni (111)/Cr/Ni catalysts under both vacuum and adsorbed XC and XAC conditions. Our results demonstrated that while Ni (111)/Co exhibited a highly favorable tendency for segregation during the reaction process, segregation was not feasible in Ni (111)/Cr/Ni. This suggests that the latter configuration possesses greater stability compared to the former. Overall, this study provides insights into the computational design of more cost-effective and active catalysts for the conversion of xylose into xylitol.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Hydrogen Energy Innovation Technology Development Program of the National Research Foundation of Korea (NRF), funded by the Korean government (Ministry of Science and ICT (MSIT)) (NRF-2020M3E6A1043955). This work was also supported by INHA UNIVERSITY Research Grant and by the National Supercomputing Center with supercomputing resources including technical support (TS-2023-RE-0027).References
- J. J. Cheng, Biomass to renewable energy processes, CRC Press: Taylor & Francis, Boca Raton, London, New York, 2010 Search PubMed.
- K. R. Enslow and A. T. Bell, The role of metal halides in enhancing the dehydration of xylose to furfural, ChemCatChem, 2015, 7, 479–489 CrossRef CAS.
- J. Iglesias, J. A. Melero, G. Morales, M. Paniagua and B. Hernández, Dehydration of xylose to furfural in alcohol media in the presence of solid acid catalysts, ChemCatChem, 2016, 8, 2089–2099 CrossRef CAS.
- Z. Qi, Q. Wang, C. Liang, J. Yue, S. Liu, S. Ma, X. Wang, Z. Wang, Z. Li and W. Qi, Highly efficient conversion of xylose to furfural in a Water–MIBK system catalyzed by magnetic carbon-based solid acid, Ind. Eng. Chem. Res., 2020, 59, 17046–17056 CrossRef CAS.
- Y. Liu, C. Ma, C. Huang, Y. Fu and J. Chang, Efficient conversion of xylose into furfural using sulfonic acid-functionalized metal–organic frameworks in a biphasic system, Ind. Eng. Chem. Res., 2018, 57, 16628–16634 CrossRef CAS.
- O. H. Pardo Cuervo, G. P. Romanelli, J. A. Cubillos, H. A. Rojas and J. J. Martínez, Selective Catalytic Dehydration of Xylose to Furfural and Fructose and Glucose to 5-Hydroximethylfurfural (HMF) Using Preyssler Heteropolyacid, ChemistrySelect, 2020, 5, 4186–4193 CrossRef CAS.
- X. Cheng, D. Jiang, X. Hu, B. Barati, Y. Hu, L. Qian, Z. He, S. Wang and H. Li, Mechanism of solvothermal conversion of xylose to furfural in rich-methanol solution: A study based on density functional theory, J. Anal. Appl. Pyrolysis, 2021, 154, 104996, DOI:10.1016/j.jaap.2020.104996.
- E. Subroto, Chemical and Biotechnological Methods for the Production of Xylitol: A Review, Int. J. Emerg. Technol. Eng. Res., 2020, 8, 2508–2512, DOI:10.30534/ijeter/2020/49862020.
- Y. Delgado Arcaño, O. D. Valmaña García, D. Mandelli, W. A. Carvalho and L. A. Magalhães Pontes, Xylitol: A review on the progress and challenges of its production by chemical route, Catal. Today, 2020, 344, 2–14, DOI:10.1016/j.cattod.2018.07.060.
- H. Liu, Z. Huang, C. Xia, Y. Jia, J. Chen and H. Liu, Selective hydrogenolysis of xylitol to ethylene glycol and propylene glycol over silica dispersed copper catalysts prepared by a precipitation–gel method, ChemCatChem, 2014, 6, 2918–2928 CrossRef CAS.
- C. Heisig, C. Glotzbach, S. Schirrmeister and T. Turek, Selective Hydrogenolysis of Biomass-Derived Xylitol to Glycols: Reaction Network and Kinetics, Chem. Eng. Technol., 2021, 44, 761–772 CrossRef CAS.
- X. Zhao, Y. Lü, W. Liao, M. Jin and Z. Suo, Hydrogen production from steam reforming of ethylene glycol over supported nickel catalysts, J. Fuel Chem. Technol., 2015, 43, 581–588, DOI:10.1016/S1872-5813(15)30017-7.
- J. Zhang and N. Xu, Hydrogen Production from Ethylene Glycol Aqueous Phase Reforming over Ni–Al Layered Hydrotalcite-Derived Catalysts, Catalysts, 2020, 10, 54 CrossRef CAS.
- Y. Zhu, K. Kanamori, N. Brun, C.-H. Pélisson, N. Moitra, F. Fajula, V. Hulea, A. Galarneau, K. Takeda and K. Nakanishi, Monolithic acidic catalysts for the dehydration of xylose into furfural, Catal. Commun., 2016, 87, 112–115, DOI:10.1016/j.catcom.2016.09.014.
- V. Krzelj, D. P. Ferrandez and M. F. Neira D’Angelo, Sulfonated foam catalysts for the continuous dehydration of xylose to furfural in biphasic media, Catal. Today, 2021, 365, 274–281, DOI:10.1016/j.cattod.2020.12.009.
- B. Rohini and H. U. Hebbar, Photocatalytic Conversion of Xylose to Xylitol over Copper Doped Zinc Oxide Catalyst, Catal. Lett., 2021, 151, 2583–2594, DOI:10.1007/s10562-020-03499-z.
- Q. Xia, G. Zhang, J. Wang, W. Zhang, M. Liu, Y. Li, B. Yin, C. Yang, J. Shen and X. Jin, Synergistic Bimetallic Pd–Pt/TiO2 Catalysts for Hydrogenolysis of Xylitol with In Situ-Formed H2, Ind. Eng. Chem. Res., 2020, 59, 13879–13891 CrossRef CAS.
- Q. Xia, X. Jin, G. Zhang, M. Liu, J. Wang, Y. Li, T. Fang, J. Ding, D. Zhang and K. Meng, Catalytic deoxygenation of xylitol to renewable chemicals: Advances on catalyst design and mechanistic studies, Chem. Rec., 2021, 21, 133–148 CrossRef CAS PubMed.
- S. J. You, I. G. Baek, Y. T. Kim, K.-E. Jeong, H.-J. Chae, T.-W. Kim, C.-U. Kim, S.-Y. Jeong, T. J. Kim, Y.-M. Chung, S.-H. Oh and E. D. Park, Direct conversion of cellulose into polyols or H2 over Pt/Na(H)-ZSM-5, Korean J. Chem. Eng., 2011, 28, 744–750, DOI:10.1007/s11814-011-0019-3.
- P. Kuchonthara, B. Puttasawat, P. Piumsomboon, L. Mekasut and T. Vitidsant, Catalytic steam reforming of biomass-derived tar for hydrogen production with K2CO3/NiO/γ-Al2O3 catalyst, Korean J. Chem. Eng., 2012, 29, 1525–1530, DOI:10.1007/s11814-012-0027-y.
- M. Yadav, D. K. Mishra and J.-S. Hwang, Catalytic hydrogenation of xylose to xylitol using ruthenium catalyst on NiO modified TiO2 support, Appl. Catal., A, 2012, 425–426, 110–116, DOI:10.1016/j.apcata.2012.03.007.
- M. Yadav, D. K. Mishra and J.-S. Hwang, Catalytic hydrogenation of xylose to xylitol using ruthenium catalyst on NiO modified TiO2 support, Appl. Catal., A, 2012, 425–426, 110–116, DOI:10.1016/j.apcata.2012.03.007.
- H. Xia, L. Zhang, H. Hu, S. Zuo and L. Yang, Efficient Hydrogenation of Xylose and Hemicellulosic Hydrolysate to Xylitol over Ni–Re Bimetallic Nanoparticle Catalyst, Nanomaterials, 2020, 10, 73, DOI:10.3390/nano10010073.
- D. Martin Alonso, S. G. Wettstein and J. A. Dumesic, Bimetallic catalysts for upgrading of biomass to fuels and chemicals, Chem. Soc. Rev., 2012, 41, 8075–8098, 10.1039/C2CS35188A.
- P. Yin, H. Meng, L. Wang, Y. Lai, Y. Jie, J. Yu, W. Liu, X. Zhao, T. Shen, X. Zhang, J. Han, Y. Yang, H. Yan and M. Wei, Theoretical and experimental exploration of NiM(111) (M = Fe, Co, Cu, Zn) bimetallic catalysts for the water-gas shift reaction, J. Mater. Chem. A, 2022, 10, 16610–16619, 10.1039/D2TA00991A.
- M.-Q. Li, Y.-L. Ma, X.-X. Ma, Y.-G. Sun and Z. Song, Insight into the efficient catalytic conversion of biomass to EG and 1,2-PG over W–Ni bimetallic catalyst, RSC Adv., 2018, 8, 10907–10913, 10.1039/c8ra00584b.
- M. Kim, A. Badakhsh, S. G. Akpe, Y. Kim, K.-J. Nam, Y. Kim, H. Jeong, S. W. Nam, H. C. Ham, S. H. Choi and H. Sohn, Highly selective PtCo bimetallic nanoparticles on silica for continuous production of hydrogen from aqueous phase reforming of xylose, Int. J. Hydrogen Energy, 2023, 48, 29162–29177, DOI:10.1016/j.ijhydene.2023.03.458.
- S. G. Akpe, S. Hee Choi and H. Chul Ham, Conversion of cyclic xylose into xylitol on Ru, Pt, Pd, Ni, and Rh catalysts: a density functional theory study, Phys. Chem. Chem. Phys., 2021, 23, 26195–26208, 10.1039/D1CP04660H.
- Y. Kang, O. Cretu, J. Kikkawa, K. Kimoto, H. Nara, A. S. Nugraha, H. Kawamoto, M. Eguchi, T. Liao, Z. Sun, T. Asahi and Y. Yamauchi, Mesoporous multimetallic nanospheres with exposed highly entropic alloy sites, Nat. Commun., 2023, 14, 4182, DOI:10.1038/s41467-023-39157-2.
- J.-P. Mikkola, H. Vainio, T. Salmi, R. Sjöholm, T. Ollonqvist and J. Väyrynen, Deactivation kinetics of Mo-supported RANEY® Ni catalyst in the hydrogenation of xylose to xylitol, Appl. Catal., A, 2000, 196, 143–155, DOI:10.1016/S0926-860X(99)00453-6.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- Y.-Q. Su, Y. Wang, J.-X. Liu, I. A. Filot, K. Alexopoulos, L. Zhang, V. Muravev, B. Zijlstra, D. G. Vlachos and E. J. Hensen, Theoretical approach to predict the stability of supported single-atom catalysts, ACS Catal., 2019, 9, 3289–3297 CrossRef CAS.
- M. Yang, B. Wang, M. Fan and R. Zhang, HCOOH decomposition over the pure and Ag-modified Pd nanoclusters: Insight into the effects of cluster size and composition on the activity and selectivity, Chem. Eng. Sci., 2021, 229, 116016, DOI:10.1016/j.ces.2020.116016.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- S. Li, S. Singh, J. A. Dumesic and M. Mavrikakis, On the nature of active sites for formic acid decomposition on gold catalysts, Catal. Sci. Technol., 2019, 9, 2836–2848, 10.1039/C9CY00410F.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis, J. Catal., 2015, 328, 36–42, DOI:10.1016/j.jcat.2014.12.033.
- K. J. Laidler, A glossary of terms used in chemical kinetics, including reaction dynamics (IUPAC Recommendations 1996), Pure Appl. Chem., 1996, 68, 149–192, DOI:10.1351/pac199668010149.
- B. Hammer and J. K. Nørskov, Theoretical surface science and catalysis—calculations and concepts, Adv. Catal., 2000, 71–129, DOI:10.1016/S0360-0564(02)45013-4.
- S. Jiao, X. Fu and H. Huang, Descriptors for the Evaluation of Electrocatalytic Reactions: d-Band Theory and Beyond, Adv. Funct. Mater., 2022, 32, 2107651, DOI:10.1002/adfm.202107651.
- S. Zafeiratos, S. Piccinin and D. Teschner, Alloys in catalysis: phase separation and surface segregation phenomena in response to the reactive environment, Catal. Sci. Technol., 2012, 2, 1787–1801, 10.1039/C2CY00487A.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03503d |
| This journal is © the Owner Societies 2024 |