
Theoretical design of durable and strong polycarbonates against photodegradation†
Xiao
Huang
a,
Yuuichi
Orimoto
b and
Yuriko
Aoki
 *b
*b
aDepartment of Interdisciplinary Engineering Sciences, Chemistry and Materials Science, Interdisciplinary Graduate School of Engineering Sciences, Kyushu University, 6-1 Kasuga Park, Fukuoka 816-8580, Japan
bDepartment of Material Sciences, Faculty of Engineering Sciences, Kyushu University, 6-1 Kasuga Park, Fukuoka 816-8580, Japan. E-mail: aoki.yuriko.397@m.kyushu-u.ac.jp
First published on 30th November 2023
Abstract
The photodegradation mechanism of polycarbonate (PC) was investigated by quantum chemistry, and a novel antidegradation molecular design using substituents was proposed. It was demonstrated that electron-withdrawing substituents in the phenyl moiety controlled bond alternation, leading to inhibition of the O–C bond cleavage in the carbonate moiety. These results provide a promising alternative for durable PC synthesis.
Bisphenol-A polycarbonate (BPA PC) is a widely used material in our daily life owing to its high transparency, high toughness, and other good performances.1 It loses its physical properties notably if it is overexposed to sunlight or ultraviolet (UV) light.2,3 To improve PC performance, a common strategy in experiment4–12 is to modify its chemical structure. Replacing PC phenyl hydrogen atoms with electron-donating or -withdrawing groups would make it possible to achieve the above goal by suppressing PC photodegradation.
A few computational mechanistic studies have focused on the photodegradation of PC13 and aromatic esters.14,15 Recently, our group reported the mechanism of PhO–COO bond scission using a simplified PC model bisphenol-A hydrogen carbonate (BPAHC, Fig. 1) based on quantum chemical methods.13 As shown in Fig. 1, two transitions are involved in the O–C cleavage:  and
and 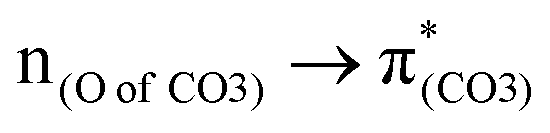 . The former leads to C–C elongations by strengthening the C–C out-of-phase overlap within the phenyl groups and enhances the two C
. The former leads to C–C elongations by strengthening the C–C out-of-phase overlap within the phenyl groups and enhances the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds by C–C in-phase overlap. The two CC bonds induce the formation of the PhO double bond owing to geometric constraints, facilitating quinoid-like structure formation and breaking the PhO–COO bond. The latter further enhances PhO–COO elongation via the reinforced out-of-phase overlap. These two transitions based on the ground state (GS) S0 geometry jointly cleave the PhO–COO bond in the excited state (ES) S13 geometry.
C double bonds by C–C in-phase overlap. The two CC bonds induce the formation of the PhO double bond owing to geometric constraints, facilitating quinoid-like structure formation and breaking the PhO–COO bond. The latter further enhances PhO–COO elongation via the reinforced out-of-phase overlap. These two transitions based on the ground state (GS) S0 geometry jointly cleave the PhO–COO bond in the excited state (ES) S13 geometry.
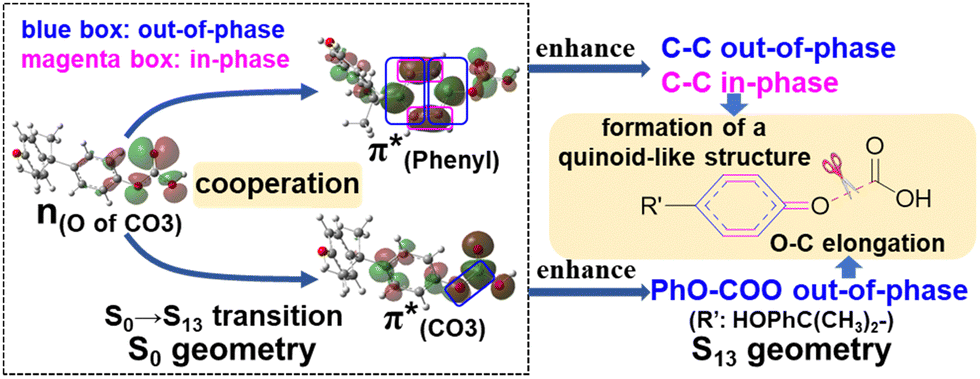 | ||
| Fig. 1 The PhO–COO bond cleavage of BPAHC. | ||
Given the lack of theoretical studies on the photodegradation of PC, our goal was to provide a detailed investigation of the mechanism affected by substituents, with the aim of designing light-resistant PC materials. Therefore, based on our previous study,13 we applied the density functional theory (DFT) and time-dependent DFT (TDDFT16) methods to study the substituent effect on the photodegradation of two PC models with electron-donating (–NH2) and -withdrawing (–NO2) groups as phenyl substituents.
The GS and ES geometries of m(NH2)-BPAHC and m(NO2)-BPAHC (Fig. 2) were fully optimized using B3LYP17–19/6-31G(d) and TD-B3LYP/6-31G(d), respectively, as B3LYP can generate reasonable results (Fig. S1–S5 and Tables S1–S3, ESI†) for these organic systems.20–22 The S0 and Sn geometries in this study are the optimized geometries of the singlet GS and singlet nth ES, respectively. The computational details are provided in the ESI.†
 | ||
| Fig. 2 Molecular structures of the BPAHC and substituted BPAHC at the –C(CH3)2-meta positions (denoted as m(R)-BPAHC, R = NH2, NO2) (bold green: atomic number). | ||
To assess the substituent effect on the carbonate O–C bond, Fig. 3 shows the transition behaviors of the models studied. As reported in our previous study on BPAHC,13 the electronic transition (corresponding to S0 → S13 excitation in the current study) attributed to 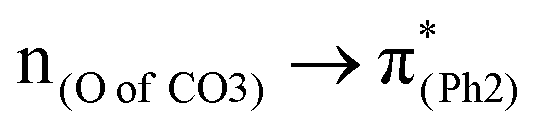 and
and 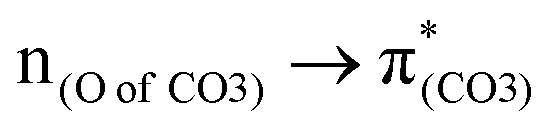 plays a key role in breaking the PhO–COO bond. Hence, S0 → S21 (m(NH2)-BPAHC) and S0 → S40 (m(NO2)-BPAHC) transitions are discussed because their major contributions also focus on the excitations to
plays a key role in breaking the PhO–COO bond. Hence, S0 → S21 (m(NH2)-BPAHC) and S0 → S40 (m(NO2)-BPAHC) transitions are discussed because their major contributions also focus on the excitations to 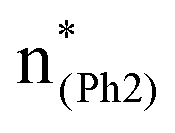 and
and 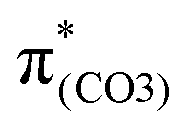 as in the S0 → S13 transition (Fig. 3). Relative to the oscillator strength of 0.0294 in the S0 → S13 transition, those of S0 → S21 and S0 → S40 transitions have similar values (0.0364 and 0.0272, respectively), implying a similar impact on the absorption spectra. The absorption spectra and parameters of the vertical excitation are shown in Fig. S6 and Table S4 (ESI†), respectively.
as in the S0 → S13 transition (Fig. 3). Relative to the oscillator strength of 0.0294 in the S0 → S13 transition, those of S0 → S21 and S0 → S40 transitions have similar values (0.0364 and 0.0272, respectively), implying a similar impact on the absorption spectra. The absorption spectra and parameters of the vertical excitation are shown in Fig. S6 and Table S4 (ESI†), respectively.
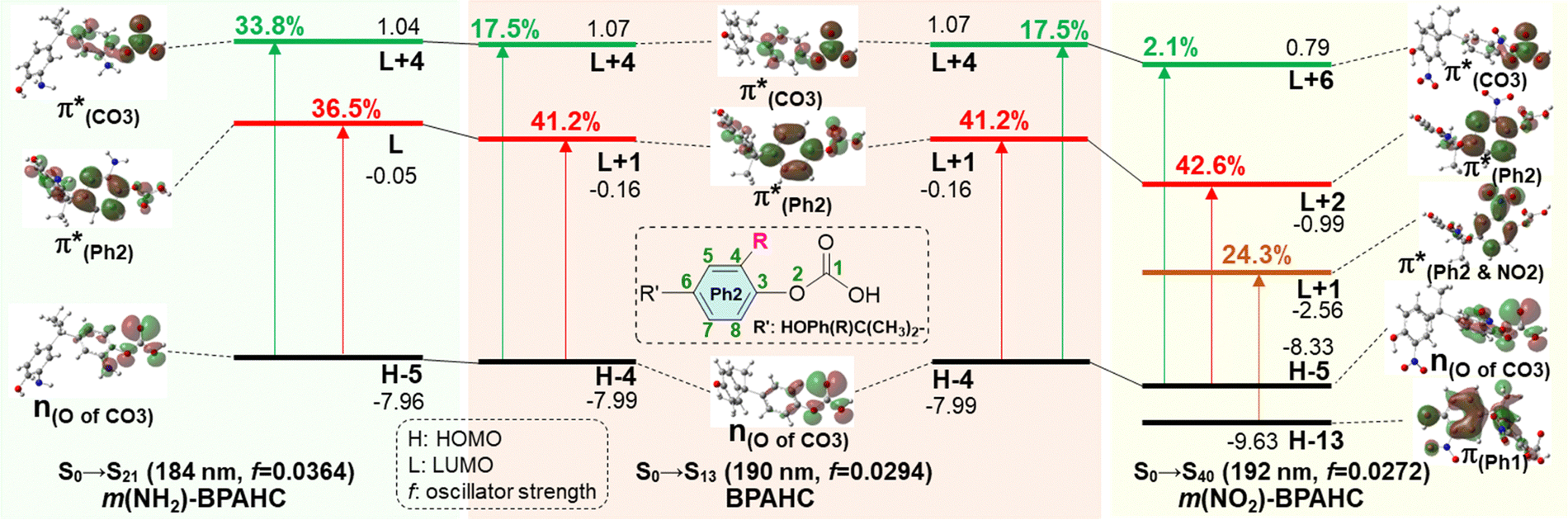 | ||
| Fig. 3 Calculated molecular orbitals (MOs) (isovalue = 0.03) involved in vertical excitation of BPAHC, m(NH2)-BPAHC, and m(NO2)-BPAHC in the S0 geometry (percentage values: transition contribution, black color values: orbital energy in eV). | ||
The effect of substituents on the O–C bond was determined by examining the variation in the transition contributions compared to those of BPAHC. As shown in Fig. 3 (left), the large 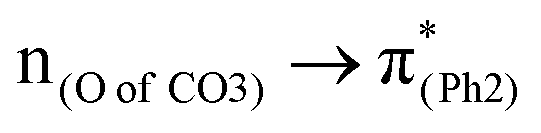 contribution (41.2 → 36.5%) maintained and the remarkable increased
contribution (41.2 → 36.5%) maintained and the remarkable increased 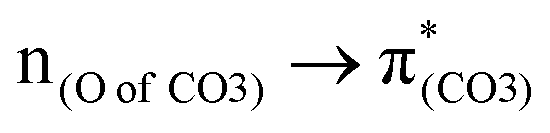 contribution (17.5 → 33.8%) indicate the promotion of O2–C1 bond scission under the –NH2 effect. As shown in Fig. 3 (right), the contributions excited to Ph2 π* orbitals remain substantial (41.2 → 42.6%), while those excited to
contribution (17.5 → 33.8%) indicate the promotion of O2–C1 bond scission under the –NH2 effect. As shown in Fig. 3 (right), the contributions excited to Ph2 π* orbitals remain substantial (41.2 → 42.6%), while those excited to 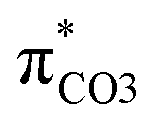 orbitals are nearly eliminated (17.5 → 2.1%), implying the suppression of O2–C1 bond cleavage influenced by the –NO2 group.
orbitals are nearly eliminated (17.5 → 2.1%), implying the suppression of O2–C1 bond cleavage influenced by the –NO2 group.
To further examine the impact of substituents on the O–C bond, the GS and ES geometries of the models studied were investigated (Fig. 4 and Fig. S7, ESI†). Upon excitation, the S13 geometry (Fig. 4, center) of BPAHC with a distorted Ph2 group relative to S0 geometry is prone to be a quinoid-like structure along the C6–C3–O2 line with C4C5, C7C8, and C3O2 bonds, finally cleaving the O2–C1 bond.
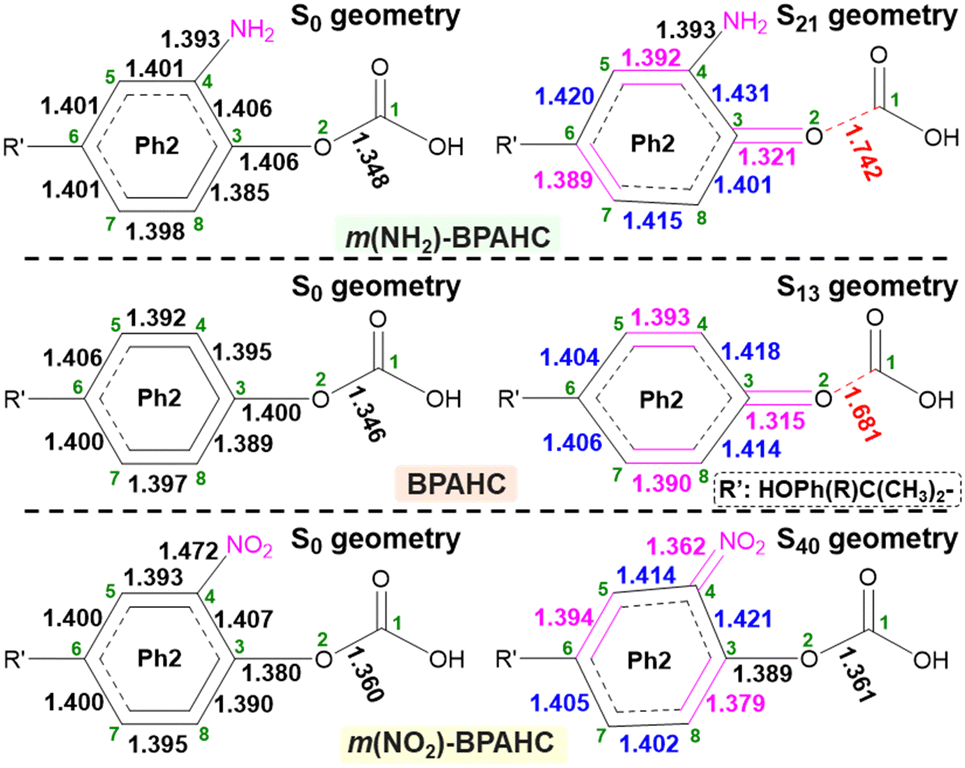 | ||
| Fig. 4 GS and ES geometries of BPAHC, m(NH2)-BPAHC, and m(NO2)-BPAHC, including their main bond lengths (Å). | ||
Similarly, upon excitation of 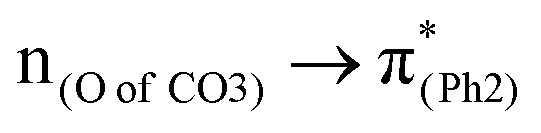 , the S21 geometry (Fig. 4, top) of m(NH2)-BPAHC tends to have a different quinoid-like structure with C4C5, C6C7, and C3O2 bonds from that of BPAHC. This might be because the in-phase interaction on C7C8 in BPAHC is destroyed on C7–C8 at the L
, the S21 geometry (Fig. 4, top) of m(NH2)-BPAHC tends to have a different quinoid-like structure with C4C5, C6C7, and C3O2 bonds from that of BPAHC. This might be because the in-phase interaction on C7C8 in BPAHC is destroyed on C7–C8 at the L 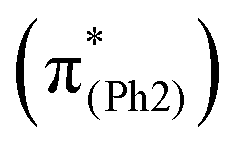 orbital due to the phase-node along the C7–C4–NH2 line (Fig. 3, left) in m(NH2)-BPAHC. The single-bond nature of C4–NH2 maintained in the S21 geometry relative to the S0 geometry enables the bond alternation along the O2–C3–C4–C5 line, leading to the C3O2 double bond, finally breaking the O2–C1 bond, which is the same behavior observed in the S13 geometry of BPAHC.13
orbital due to the phase-node along the C7–C4–NH2 line (Fig. 3, left) in m(NH2)-BPAHC. The single-bond nature of C4–NH2 maintained in the S21 geometry relative to the S0 geometry enables the bond alternation along the O2–C3–C4–C5 line, leading to the C3O2 double bond, finally breaking the O2–C1 bond, which is the same behavior observed in the S13 geometry of BPAHC.13
Distinct from the results of BPAHC,13 for m(NO2)-BPAHC (Fig. 4, bottom), the O2–C1 bond maintains the single-bond nature in the S40 geometry, as in S0 geometry, implying that it is not easily broken by excitation under the –NO2 effect. The C4–NO2 bond (1.362 Å) in the S40 geometry is much shorter than that (1.472 Å) in the S0 geometry, revealing C4NO2 bond formation in the S40 geometry. Relative to the S13 geometry of BPAHC, the S40 geometry tends to be a new quinoid-like structure with C3C8, C5C6, and C4NO2 bonds upon excitation. It can be considered that the in-phase interaction on C7C8 in BPAHC is destroyed on C7–C8 at the L + 2 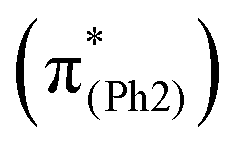 orbital due to –NO2 substitution (Fig. 3, right) as in m(NH2)-BPAHC. The C4NO2 bond formation allows bond alternation along the C7–C4–NO2 line, maintaining the single-bond nature of C3–O2, and finally suppressing the O2–C1 bond scission. For the ES geometry of m(NO2)-BPAHC, there may be another possible alternated structure with C3O2 and C7C8 bonds that was not obtained via calculations (Fig. S8, ESI†). However, such an ES structure similar to S40 geometry may not exist in BPAHC and m(NH2)-BPAHC. As no substituent is located at the –C(CH3)2-meta positions in BPAHC, and the C4–NH2 bond maintains the single-bond nature in the S21 geometry of m(NH2)-BPAHC. To exclude the possibility of such an ES structure in m(NO2)-BPAHC, Fig. S9 (ESI†) shows geometric comparisons of the different starting structures
orbital due to –NO2 substitution (Fig. 3, right) as in m(NH2)-BPAHC. The C4NO2 bond formation allows bond alternation along the C7–C4–NO2 line, maintaining the single-bond nature of C3–O2, and finally suppressing the O2–C1 bond scission. For the ES geometry of m(NO2)-BPAHC, there may be another possible alternated structure with C3O2 and C7C8 bonds that was not obtained via calculations (Fig. S8, ESI†). However, such an ES structure similar to S40 geometry may not exist in BPAHC and m(NH2)-BPAHC. As no substituent is located at the –C(CH3)2-meta positions in BPAHC, and the C4–NH2 bond maintains the single-bond nature in the S21 geometry of m(NH2)-BPAHC. To exclude the possibility of such an ES structure in m(NO2)-BPAHC, Fig. S9 (ESI†) shows geometric comparisons of the different starting structures 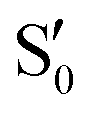 and
and 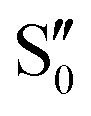 . The results indicate that this ES structure is not favored, even for different starting structures.
. The results indicate that this ES structure is not favored, even for different starting structures.
To explore the influence of substituents on the above geometric changes, Fig. 5 displays MO comparisons in the S0 geometry for the models studied. In BPAHC (Fig. 5, center) by excitation to 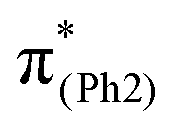 , the C3–C4, C3–C8, C5–C6, and C6–C7 bonds are elongated by enhancing the out-of-phase overlaps (blue boxes of
, the C3–C4, C3–C8, C5–C6, and C6–C7 bonds are elongated by enhancing the out-of-phase overlaps (blue boxes of 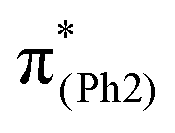 ). These C–C elongations lead to a distorted Ph2 in the S13 geometry relative to the S0 geometry, with the in-phase C4C5 and C7C8 bonds enhanced by C–C in-phase overlaps (magenta boxes of
). These C–C elongations lead to a distorted Ph2 in the S13 geometry relative to the S0 geometry, with the in-phase C4C5 and C7C8 bonds enhanced by C–C in-phase overlaps (magenta boxes of 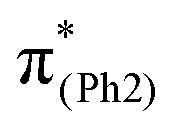 ), forming a quinoid-like structure in the S13 geometry. Upon excitation to
), forming a quinoid-like structure in the S13 geometry. Upon excitation to 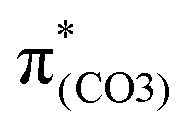 , the reinforced O2–C1 out-of-phase overlap (blue box of
, the reinforced O2–C1 out-of-phase overlap (blue box of 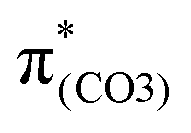 ) resulted in O2–C1 bond elongation. These two excitations cause O2–C1 bond breakage in the S13 geometry.
) resulted in O2–C1 bond elongation. These two excitations cause O2–C1 bond breakage in the S13 geometry.
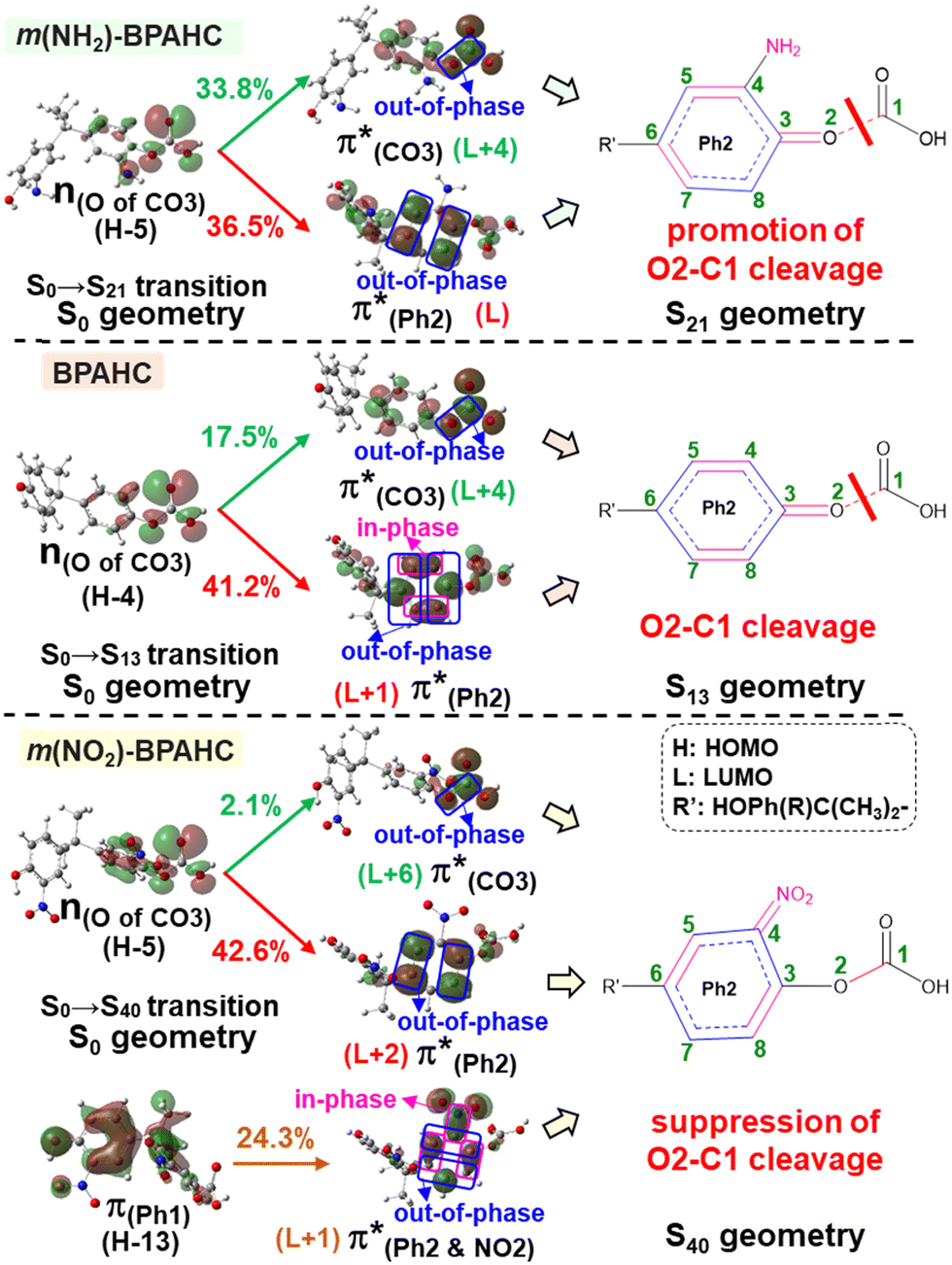 | ||
| Fig. 5 Transition of BPAHC, m(NH2)-BPAHC, and m(NO2)-BPAHC in S0 geometry, and corresponding ES geometries (percentage values: transition contribution). | ||
In m(NH2)-BPAHC, the O2–C1 bond cleavage in the S21 geometry relative to the S0 geometry, occurs as in BPAHC. As shown in Fig. 5 (top), the C3–C8 and C5–C6 bonds are stretched owing to the enhanced out-of-phase overlaps (blue boxes of 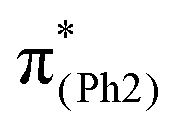 ) upon excitation to
) upon excitation to 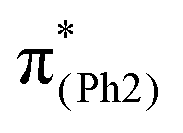 . These two C–C elongations result in a distorted Ph2 group in the S21 geometry relative to S0 geometry, forming C4C5 and C6C7 bonds due to geometric constraints. Hence, another quinoid-like structure different from that of BPAHC is formed in the S21 geometry, breaking the O2–C1 bonds. Upon excitation to
. These two C–C elongations result in a distorted Ph2 group in the S21 geometry relative to S0 geometry, forming C4C5 and C6C7 bonds due to geometric constraints. Hence, another quinoid-like structure different from that of BPAHC is formed in the S21 geometry, breaking the O2–C1 bonds. Upon excitation to 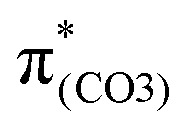 , the O2–C1 elongation occurs because of the enhanced out-of-phase overlap (blue box of
, the O2–C1 elongation occurs because of the enhanced out-of-phase overlap (blue box of 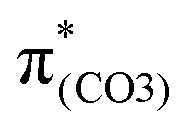 ). These two transitions cause O2–C1 bond scission in the S21 geometry as in BPAHC. Besides, the presence of a stronger driving force elongates the O2–C1 bond owing to a higher transition contribution in m(NH2)-BPAHC upon excitation to
). These two transitions cause O2–C1 bond scission in the S21 geometry as in BPAHC. Besides, the presence of a stronger driving force elongates the O2–C1 bond owing to a higher transition contribution in m(NH2)-BPAHC upon excitation to 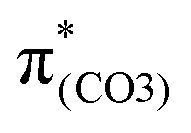 (33.8%) relative to that of BPAHC (17.5%), implying that the –NH2 group can promote O2–C1 bond scission.
(33.8%) relative to that of BPAHC (17.5%), implying that the –NH2 group can promote O2–C1 bond scission.
Conversely, in m(NO2)-BPAHC, the O2–C1 bond maintains a single-bond nature in the S40 geometry as in the S0 geometry. There are two factors to explain this situation (Fig. 5, bottom): (1) the enhanced C4–NO2 in-phase overlap (magenta box of 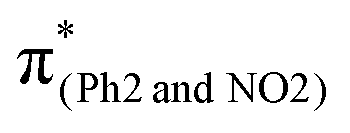 ), and (2) the out-of-phase overlap at C3–C8 and C5–C6 (blue boxes of
), and (2) the out-of-phase overlap at C3–C8 and C5–C6 (blue boxes of 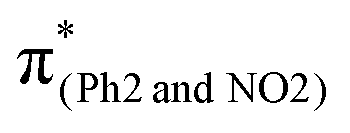 ). The former leads to much shorter C4NO2 formation as well as shorter C3–C8 and C5–C6, while the latter is longer in all lengths between carbons in the phenyl ring. Factor (2) has a larger weight than (1) by excited state calculations. Nevertheless, the effect of (2) does not appear in excited geometries. From factors (1) and (2), it is suggested that the resulting C3C8 and C5C6 imply a very strong geometric effect of bond alternation caused by (1) C4NO2 compared to the out-of-phase effect in (2). Finally, a new quinoid-like structure of the S40 geometry along the C7–C4–NO2 line is strongly affected by the very short C4NO2, suppressing O2–C1 bond cleavage under the –NO2 effect.
). The former leads to much shorter C4NO2 formation as well as shorter C3–C8 and C5–C6, while the latter is longer in all lengths between carbons in the phenyl ring. Factor (2) has a larger weight than (1) by excited state calculations. Nevertheless, the effect of (2) does not appear in excited geometries. From factors (1) and (2), it is suggested that the resulting C3C8 and C5C6 imply a very strong geometric effect of bond alternation caused by (1) C4NO2 compared to the out-of-phase effect in (2). Finally, a new quinoid-like structure of the S40 geometry along the C7–C4–NO2 line is strongly affected by the very short C4NO2, suppressing O2–C1 bond cleavage under the –NO2 effect.
To analyze the O–C bond scission affected by substituents, Fig. 6 shows the potential energy surfaces (PESs) obtained by scanning the O2–C1 bond length and the C4–C3–O2–C1 dihedral angle for the models studied, and Fig. S10–S12 (ESI†) display the other three views of the PESs. In Fig. 6 (center) and Fig. S10 (ESI†), the points f1, e1, and g1 correspond to the local minima on the S13 PES of BPAHC.13 The point f1 corresponds to the S13 geometry where the O2–C1 bond is broken. It has a lower energy of about 2.9 kcal mol−1 relative to point b1 (the ES state via vertical excitation from a1 (S0 geometry)). The energy barrier (a1 → f1) for breaking the O2–C1 bond is about 18.1 kcal mol−1, implying that this barrier can be easily overcome to break the O2–C1 bond. Points e1 and g1 are the intersections between the S0 and S13 PESs, and the recombination (e1 → a1) or separation (e1 → h1 or g1 → h1) of the two radicals (PhO˙ and ˙COOH) may occur on the S0 PES from these two points. Besides, possible intersystem crossing exists from the excited singlet to the triplet via a crossing point (Fig. S13, ESI†).
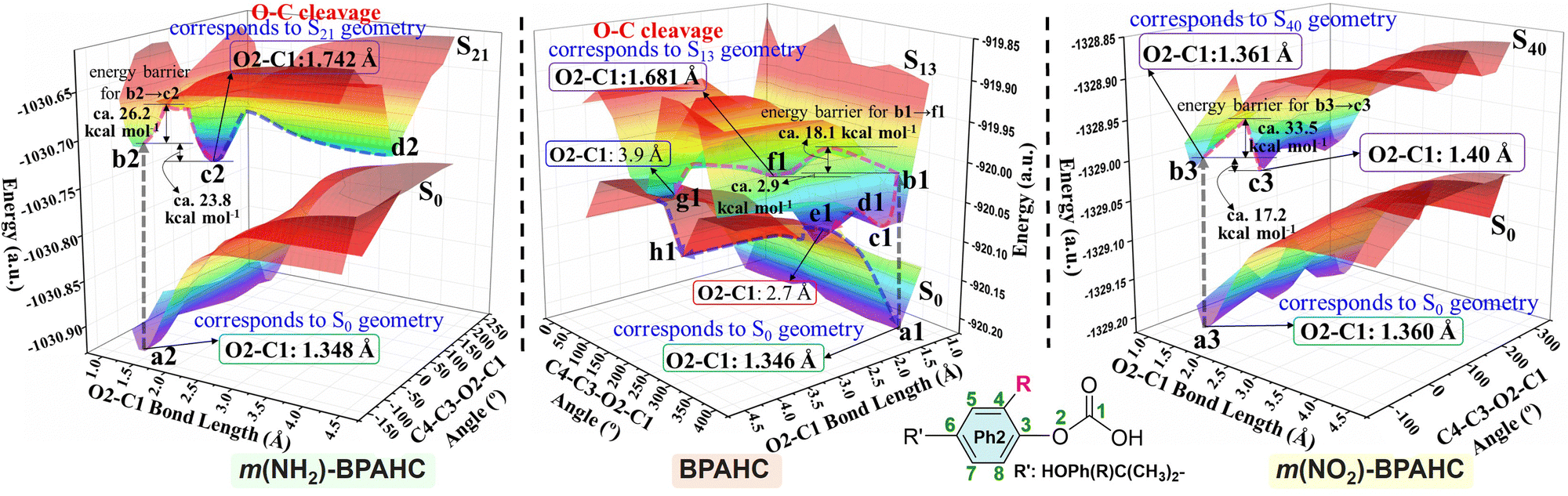 | ||
| Fig. 6 PESs of the GS and ES states for BPAHC, m(NH2)-BPAHC, and m(NO2)-BPAHC as functions of O2–C1 bond lengths and C4–C3–O2–C1 dihedral angles. | ||
On the PESs of m(NH2)-BPAHC, shown in Fig. 6 (left) and Fig. S11 (ESI†), the O2–C1 bond lengths at points a2 and c2 are 1.348 Å for the S0 geometry and 1.742 Å for the S21 geometry. The O2–C1 bond is cleaved on the S21 PES with an energy barrier of about 26.2 kcal mol−1 (b2 → c2), which is slightly higher than that in BPAHC. It is difficult to judge the substituent effect on O2–C1 bond scission from this barrier via current coarse grid calculations. Relative to the energy at point f1 (−2.9 kcal mol−1) of BPAHC, the energy of the local minimum point c2 is much lower (−23.8 kcal mol−1) than that at point b2, revealing that it is more likely to reach point c2. In contrast to the PES result of BPAHC, there is no intersection between the S0 and S21 PESs in the current calculation, implying that recombining the broken O2⋯C1 pathway is blocked to some extent and the O2–C1 bond is more easily cleaved. This indicates that the –NH2 group can promote O2–C1 bond scission due to the much lower energy of the local minimum point c2 than that of BPAHC and the blockade to some degree of the damaged O2⋯C1 bond recombination.
For m(NO2)-BPAHC, as shown in Fig. 6 (right) and Fig. S12 (ESI†), points a3 and b3 correspond to S0 and S40 geometries, respectively. Unlike BPAHC, O2–C1 bond cleavage does not occur in m(NO2)-BPAHC because the O2–C1 bond lengths are similar in S0 (1.360 Å) and S40 (1.361 Å) geometries. On the S40 PES, a local minimum point c3 has a lower energy (−17.2 kcal mol−1) relative to point b3. The energy barrier (b3 → c3) is about 33.5 kcal mol−1, which is much higher than that of BPAHC, indicating that it is difficult to overcome this barrier to reach point c3. Besides, relative to BPAHC, it is difficult to recombine the broken O2⋯C1 bond as there is no intersection between S0 and S40 PESs. Hence, point b3 may be a better location than c3 (still 1.40 Å) on the S40 PES, maintaining the single-bond nature of O2–C1. This indicates that –NO2 on the phenyl rings can suppress O2–C1 bond breakage.
In a word, the excitation to the phenyl π* orbital is responsible for forming the quinoid-like structure and the carbonate π* orbital is responsible for elongating the carbonate O–C bond. These two excitations conjointly cleave the O–C bond in the ES geometry. Relative to the results of BPAHC, the maintained high 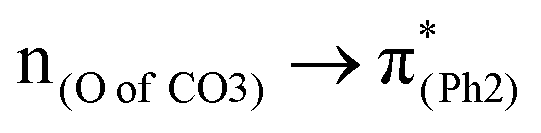 transition facilitates quinoid-like structure formation with the PhO bond, and the enhanced
transition facilitates quinoid-like structure formation with the PhO bond, and the enhanced 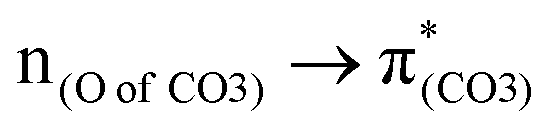 transition facilitates O–C bond extension, finally promoting O–C bond cleavage in m(NH2)-BPAHC. This promotion is further confirmed by the fact that the broken O⋯C bond recombination is somewhat hindered because there is no intersection between the GS and ES PESs. It is difficult to overcome the much higher energy barrier of the S40 geometry with the single-bond nature of O–C than that of BPAHC, and the energy minimum at the S40 energy surface is still short, maintaining a bonding with 1.40 Å. Even if it is broken, it is difficult to recombine the O⋯C bond because of the separation between the S0 and S40 PESs. These results provide a comprehensive awareness and feasible direction for designing UV-resistant PC material.
transition facilitates O–C bond extension, finally promoting O–C bond cleavage in m(NH2)-BPAHC. This promotion is further confirmed by the fact that the broken O⋯C bond recombination is somewhat hindered because there is no intersection between the GS and ES PESs. It is difficult to overcome the much higher energy barrier of the S40 geometry with the single-bond nature of O–C than that of BPAHC, and the energy minimum at the S40 energy surface is still short, maintaining a bonding with 1.40 Å. Even if it is broken, it is difficult to recombine the O⋯C bond because of the separation between the S0 and S40 PESs. These results provide a comprehensive awareness and feasible direction for designing UV-resistant PC material.
This work was financially supported by a Grant-in-Aid for JSPS Fellows DC1 (KAKENHI: 202321990) from the Japan Society for the Promotion of Science (JSPS), the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) (KAKENHI: JP23245005, JP16KT0059, JP25810103, JP15KT0146, JP16K08321, and JP20H00588), and the Japan Science and Technology Agency (JST), CREST. The computations were carried out using Linux systems in our research group and high-performance computing systems at the Research Institute for Information Technology at Kyushu University.
Conflicts of interest
There are no conflicts to declare.References
- A. Rivaton, Polym. Degrad. Stabil., 1995, 49, 163–179 CrossRef.
- A. Ram, O. Zilber and S. Kenig, Polym. Eng. Sci., 1985, 25, 535–540 CrossRef.
- H. Liu, M. Zhou, Y. Zhou, S. Wang, G. Li, L. Jiang and Y. Dan, Polym. Degrad. Stabil., 2014, 105, 218–236 CrossRef.
- A. Factor and M. L. Chu, Polym. Degrad. Stabil., 1980, 2, 203–223 CrossRef.
- A. Factor, W. V. Ligon and R. J. May, Macromolecules, 1987, 20, 2461–2468 CrossRef.
- D.-J. Liaw and P. Chang, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 2453–2460 CrossRef.
- A. Factor and P. T. Engen, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2231–2236 CrossRef CAS.
- A. Factor and J. C. Lynch, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 3413–3422 CrossRef CAS.
- A. Rivaton and J. Lemaire, Polym. Degrad. Stabil., 1988, 23, 51–73 CrossRef.
- G. M. Coppinger and E. R. Bell, J. Phys. Chem., 1966, 70, 3479–3489 CrossRef CAS.
- D.-J. Liaw and P. Chang, Polymer, 1996, 37, 2857–2863 CrossRef CAS.
- A. Rivaton, B. Mailhot, J. Soulestin, H. Varghese and J.-L. Gardette, Eur. Polym. J., 2002, 38, 1349–1363 CrossRef CAS.
- X. Huang, Y. Orimoto and Y. Aoki, J. Phys. Chem. A, 2021, 125, 6662–6673 CrossRef CAS PubMed.
- S. Grimme, Chem. Phys., 1992, 163, 313–330 CrossRef CAS.
- J. M. Toldo, M. Barbatti and P. F. B. Gonçalves, Phys. Chem. Chem. Phys., 2017, 19, 19103–19108 RSC.
- E. K. U. Gross and W. Kohn, Adv. Quantum Chem., 1990, 21, 255–291 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- Y. Orimoto, K. Ishimoto and Y. Aoki, J. Phys. Chem. C, 2018, 122, 4546–4556 CrossRef.
- J. Xu, A. Takai, Y. Kobayashi and M. Takeuchi, Chem. Commun., 2013, 49, 8447–8449 RSC.
- A. Francés-Monerris, M. Lineros-Rosa, M. A. Miranda, V. Lhiaubet-Vallet and A. Monari, Chem. Commun., 2020, 56, 4404–4407 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03533f |
| This journal is © the Owner Societies 2024 |