
First-principles studies on the electronic and photocatalytic water splitting properties of surface functionalized Y2C-based MXenes†
Sheng-Yi
Zhang
,
Ni-Ping
Shi
,
Chuan-Kui
Wang
* and
Guang-Ping
Zhang
 *
*
Shandong Key Laboratory of Medical Physics and Image Processing & Shandong Provincial Engineering and Technical Center of Light Manipulations, School of Physics and Electronics, Shandong Normal University, Jinan 250358, China. E-mail: zhangguangping@sdnu.edu.cn; ckwang@sdnu.edu.cn
First published on 29th November 2023
Abstract
Recently, MXenes, an emerging family of two-dimensional (2D) materials, have attracted increasing interest for photocatalytic water splitting due to their various excellent physical and chemical properties, such as large specific surface area, good hydrophilicity, and remarkable light absorption ability. However, the photocatalysts of MXenes with symmetric structures are limited by rapid recombination of photo-generated carriers and the prerequisite of a large band gap no less than 1.23 eV. Differently, Janus MXenes with different surface functional groups facilitate the separation of photo-generated electrons and holes with the help of the intrinsic electric field. And, at the same time, there is no prerequisite for the band gap of Janus MXene photocatalysts as long as they possess appropriate band edge positions. Here, we explored the structural, electronic and photocatalytic water splitting properties of symmetric Y2CT2 and Janus Y2CTT′ MXenes (T, T′ = H, F, Cl, OH) using the density functional theory (DFT) method. Our calculations show that all the investigated Y2CT2 are not suitable photocatalysts for photocatalytic water splitting at all pH values (pH = 0, 7, and 14). In contrast, all the investigated Janus Y2CTT′ MXenes are good water splitting photocatalysts with high optical absorption coefficients and remarkable solar-to-hydrogen (STH) efficiencies larger than 18% at pH = 14. Moreover, the STH efficiencies are larger than 18% even at all investigated pH values for Y2CHCl (18.5–22.6%), Y2 CFCl (∼18.7%), and Y2 C(OH)Cl (∼19.4%). Based on the first-principles calculations, we here for the first time propose an easy strategy to design Janus MXene photocatalyst candidates with possible high STH efficiency according to the electronic properties of their symmetric counterparts. Our study is helpful for the future design of Janus MXenes and more generally Janus 2D photocatalysts for water splitting with high STH efficiency.
1 Introduction
With the rapid development of human society, we are facing increasingly serious energy and environmental problems. Researchers are devoted to finding renewable clean energy to replace traditional fossil fuels. Due to the high energy density and harmless combustion products (H2O), hydrogen has been considered as the ultimate solution to current energy and environmental crises. However, producing hydrogen is not an easy task, and many researchers have developed many kinds of hydrogen production methods such as methanol thermochemical conversion and electrolysis of water.1 Among all these hydrogen production methods, photocatalytic water splitting is definitely an ideal way to obtain hydrogen economically, environmentally friendly and safely. In general, a good photocatalyst for water splitting should meet the following three conditions. (1) Photo-generated electron and hole carriers should be spatially separated efficiently. (2) It should have appropriate band alignments with the redox potentials of H+/H2 and H2O/O2, namely, the potential of its conduction band minimum (CBM) should be higher (more negative) than the hydrogen reduction potential (0 V vs. standard hydrogen electrode (SHE)), while the potential of its valence band maximum (VBM) should be lower (more positive) than the water oxide potential (1.23 V vs. the SHE). This means that the band gap should be larger than 1.23 eV for structurally symmetric photocatalysts. (3) It should have good sunlight absorption characteristics to fully utilize the solar energy. Since the discovery of TiO2 photocatalysts, many other semiconductors have been studied for photocatalytic water splitting.2–4 But these photocatalysts have disadvantages of inefficient carrier separation and low sunlight absorption.5 Therefore, searching efficient photocatalysts for photocatalytic water splitting has long been a vital task in producing hydrogen.In 2004, graphene was successfully prepared in experiments, which opened a new era of two-dimensional (2D) materials.6 Since then, plenty of 2D materials have been newly discovered and prepared;7–12 among which 2D transition metal carbides, nitrides and carbonitrides (MXene) have attracted widespread attention because of their large specific surface area, variety of compositions and structures and other remarkable properties.13,14 MXenes can be formulated as Mn+1XnTx, in which M denotes transition metals (e.g. Sc, Ti, V, Cr, Mn, Y, Zr, Nb, Mo, Hf, Ta, W, or their combination), X denotes C or/and N, and T denotes surface functional groups (such as –H, –F, –O, –OH, and –Cl).15,16 So far, hundreds of MXenes have been experimentally synthesized or theoretically predicted with diverse applications, such as energy storage devices,17,18 gas sensors,19 supercapacitors,20 electromagnetic interference shielding21,22 and photocatalysts.23–26
In 2019, the stacked sheets of Y2CF2 were successfully synthesized by Druffel et al.27 In the next year, Maeda et al. succeeded in reducing the protons into hydrogen using the stacked sheets of Y2CF2.28 According to the two studies mentioned above, although Y2CF2 has a band gap of about 1.90 eV and its CBM potential is more negative than the hydrogen reduction potential, the VBM potential of Y2CF2 is more negative than the water oxidation potential, which forbids the process of water oxidation (H2O to O2). This means the electrons excited to conduction bands have sufficient energy to reduce protons H+ to H2 but the energy of holes that remained in valence bands is not low enough for them to accept electrons from H2O to produce O2. Therefore, other reductants must be added to enable the redox reaction process to proceed, which will bring unnecessary cost and other products besides H2 and O2. On the other hand, photo-excited carriers in symmetric Y2CF2 can easily recombine and the relatively large band gap also limits the utilization of sunlight, resulting in low photocatalytic efficiency of water splitting.
In general, assuming that both light absorbance and quantum efficiency can reach 100% and considering that the appropriate band gap is about 2.0 eV for conventional water splitting photocatalysts, then the theoretical solar-to-hydrogen (STH) efficiency (a parameter often used to evaluate the conversion rate from solar energy to hydrogen) for conventional water splitting photocatalysts is at most ca. 18%.29 In fact, the experimentally reported STH efficiencies by far rarely exceed a criterion of 10% for practical applications.30–34 Therefore, exploring photocatalysts for photocatalytic water splitting with high STH efficiency is of great importance. For this aim, Yang et al. suggested asymmetric 2D materials as a new class of photocatalysts for photocatalytic water splitting as long as the potential of the VBM contributed from one surface is lower than the oxidation potential of H2O to O2 and the potential of the CBM contributed from the other surface is higher than the reduction potential of H+ to H2.35 This proposed mechanism of photocatalytic water splitting is very similar to that in Z-scheme heterostructures which has been verified experimentally.36–38 They have revealed that the intrinsic electric field (IEF) in structurally asymmetric 2D materials facilitates the carrier separation; meanwhile, it is no longer a prerequisite for water-splitting photocatalysts to have a band gap larger than 1.23 eV due to different band edge positions for two surfaces. Recently, lots of 2D Janus materials have been predicted to possess high photocatalytic efficiency for water splitting.29,39–43 For example, Fu et al. theoretically investigated the characteristics of 2D M2X3 (M = Al, Ga, In; X = S, Se, Te) for photocatalytic water splitting.29 All the studied systems are promising photocatalysts for photocatalytic water splitting and moreover, the calculated STH efficiencies of Al2Te3, Ga2Se3, Ga2Te3 (with 3% strain), In2Se3, and In2Te3 are lager than 18%. Similarly, Janus MXenes with different functional groups on two surfaces are also expected to be good candidates for water-splitting photocatalysts.44,45 For example, Zhang et al. theoretically reported that Sc2C(OH)Cl, Sc2CFCl, and Sc2CHCl are promising photocatalysts for photocatalytic water splitting.44
Though Janus MXenes have the advantages of efficient separation of light-generated electrons and hole carriers and no prerequisite for the band gaps to be good photocatalysts for photocatalytic water splitting, they should still have appropriate band alignments with the redox potentials of water and good sunlight absorption characteristics. Therefore, it is still not a trivial task to search such Janus MXenes satisfying the above two prerequisites. In order to search more Janus MXenes as excellent water splitting photocatalysts with high STH and more importantly to explore the underlying design strategy for Janus MXene photocatalysts for photocatalytic water splitting, here we studied the structural, electronic and photocatalytic properties of Y2C-based MXenes Y2CT2 and Y2CTT′ (T, T′ = H, F, Cl, OH) by using the density functional theory (DFT) method. Our first-principles calculations reveal that though all the investigated Y2CT2 are not suitable photocatalysts for photocatalytic water splitting at all investigated pH values (pH = 0, 7, and 14), all the Janus Y2CTT′ MXenes are good water splitting photocatalysts at pH = 14 with STH efficiencies of larger than 18%. Furthermore, the STH efficiencies of Y2CHCl, Y2CFCl, and Y2C(OH)Cl are larger than 18% at all pH values. Finally, we propose an easy strategy to design Janus MXene photocatalysts with possible high STH efficiency by simply inspecting the band edge positions of their symmetric counterparts.
2 Computational details
All the calculations in this work were based on density functional theory (DFT) implemented in the Vienna ab initio simulation package (VASP).46,47 The projector-augmented wave (PAW) pseudopotentials were employed to describe the interactions between valence electrons and cores.48 The valence electron density was expanded in the plane wave basis set with an energy cutoff of 500 eV. As sketched by the red dashed lines in Fig. 1, the primitive cell was used to perform the calculations. The periodic boundary condition was used within the plane of 2D MXenes. For the direction perpendicular to the 2D MXene plane, a lattice parameter of 30 Å was used (i.e. a vacuum of about 23–25 Å). And, at the same time, a dipole correction was applied during the calculations for all Janus MXenes. For the structural relaxation, the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) was chosen as the exchange–correlation functional,49 and a criterion of 0.01 eV Å−1 for the residual force on each atom was set. On the other hand, the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional was used to calculate the electronic and optical properties. The k-point meshes of 11 × 11 × 1 and 15 × 15 × 1 generated using the Monkhorst–Pack scheme were used for structural optimizations and electronic self-consistent calculations, respectively. For the dynamic stability calculations, 3 × 3 × 1 supercells were constructed based on the optimized primitive cells and the force constants were calculated by density functional perturbation theory (DFPT) using the PHONOPY code,50,51 for which a k-point sampling of 5 × 5 × 1 was used instead. | ||
| Fig. 1 Top and side views of structural models for (a) Y2CT2 MXenes and (b) Y2CTT′ Janus MXenes. The primitive cells are indicated by the red dashed lines. | ||
3 Results and discussion
3.1 Geometric structures
According to previous studies,52–55 the surface functional groups prefer to locate at the hollow sites of the Y2C MXene. There are two kinds of hollow sites in the Y2C MXene. One is above the hollow of three neighboring C atoms of the middle C atomic layer, aligned with a Y atom of either Y atomic layers (denoted as hollow site A). The other is above the hollow of three neighboring Y atoms of either Y atomic layers, aligned with a C atom of the middle C atomic layer (denoted as hollow site B). When the two surfaces of the Y2 C MXene are functionalized with the same functional group (i.e., Y2CT2 MXenes), as seen from Fig. 1(a), there are three possible structure models for this case. For Model-1, the functional groups at each surface locate at the A hollow sites. For Model-2, the functional groups locate at the B hollow sites. For Model-3, the functional groups locate at the A hollow sites on one surface but at the B hollow sites on the other surface. Obviously, Model-1 and Model-2 are structurally symmetric while Model-3 is structurally asymmetric. Therefore, when it turns to the cases of Janus functionalized MXenes, two different structural models will derive from the above asymmetric Model-3 by exchanging the functional groups on the two surfaces, which are respectively denoted as Model-3 and Model-4 as shown in Fig. 1(b).To figure out which structural model is energy preferable, we calculated the total energies of considered structural models for each Y2CT2/Y2CTT′ MXene, which are listed in Table 1. It can be clearly seen from Table 1 that for both Y2CT2 and Y2CTT′ MXenes, Model-1 is the most stable structure with the lowest energy, which is in good agreement with previously reported Sc2C and Ti2C MXenes.44,56 To further confirm the dynamic stability of Model-1, phonon dispersion of all investigated MXenes with the Model-1 structure has been inspected by using the density functional perturbation theory (DFPT) implemented in the PHONOPY code. As seen from Fig. 2 for Y2CH2 and Y2CFCl (refer to Fig. S1 of the ESI† for the other 8 systems), there are only few imaginary frequencies near the Γ point, which are of acoustic nature8,57 and expected to be eliminated by using a larger supercell in the phonon dispersion analysis (but accompanying a dramatic increase in computation).58 At this point, it can be concluded that all the investigated systems with the Model-1 structure are also dynamically stable. Therefore, we used the optimized Model-1 structure for the subsequent analyses. The optimized lattice parameters for all investigated systems are given in Table 2. Besides, we also optimized the structure of stacked sheets of Y2CF2, which was experimentally synthesized recently.27,28 The calculated lattice constant of stacked sheets of Y2CF2 is 3.63 Å, which agrees well with an experimental value of 3.66 Å.27,28 This suggests that the DFT-GGA method is good enough for geometry optimization. From Table 2, we can see that the lattice constant of the Y2CF2 monolayer is 3.57 Å, which is very close to the value of stacked sheets. Also, it can be seen that the lattice constant is closely related to the surface functional groups. More specifically, as shown in Table 2, MXenes with the –Cl functional group have relative larger lattice constants. In contrast, the lattice constants for those without the –Cl functional group are relatively smaller and they share a common value of around 3.58 ± 0.1 Å. This can be attributed to the fact that the size of –H, –F and –(OH) groups are much smaller than that of the –Cl group.
| Systems | Model-1 | Model-2 | Model-3 | Model-4 |
|---|---|---|---|---|
| Y2CH2 | 0 | 0.627 | 0.294 | — |
| Y2CF2 | 0 | 1.045 | 0.507 | — |
| Y2CCl2 | 0 | 0.627 | 0.308 | — |
| Y2C(OH)2 | 0 | 0.607 | 0.305 | — |
| Y2CHF | 0 | 0.894 | 0.495 | 0.309 |
| Y2CHCl | 0 | 0.651 | 0.256 | 0.366 |
| Y2C(OH)H | 0 | 0.637 | 0.294 | 0.322 |
| Y2CFCl | 0 | 0.844 | 0.551 | 0.261 |
| Y2C(OH)F | 0 | 0.838 | 0.301 | 0.559 |
| Y2C(OH)Cl | 0 | 0.623 | 0.335 | 0.267 |
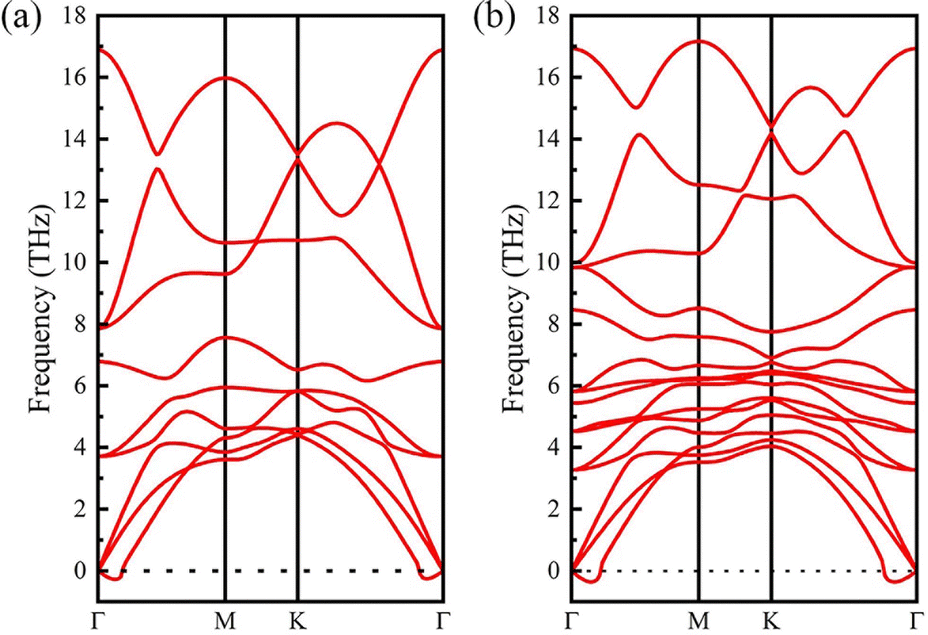 | ||
| Fig. 2 Phonon dispersion spectra of (a) Y2CH2 and (b) Y2CFCl. | ||
| Systems | a (Å) | E gap (eV) | Direct/indirect gap | Φ (eV) | ΔΦ (eV) | E IEF (V Å−1) | |
|---|---|---|---|---|---|---|---|
| Y2CH2 | 3.59 | 1.663 | I | 4.478 | 0 | 0 | |
| Y2CF2 | 3.57 | 1.952 | I | 4.615 | 0 | 0 | |
| Y2CCl2 | 3.71 | 1.630 | I | 6.223 | 0 | 0 | |
| Y2C(OH)2 | 3.58 | 0.684 | D | 1.427 | 0 | 0 | |
| Y2CHF | 3.58 | 1.793 | I | 4.780(H) | 4.710(F) | 0.070 | 0.014 |
| Y2CHCl | 3.65 | 1.578 | I | 4.636(H) | 6.186(Cl) | 1.550 | 0.281 |
| Y2C(OH)H | 3.58 | 1.098 | I | 1.809(OH) | 4.729(H) | 2.920 | 0.481 |
| Y2CFCl | 3.64 | 1.747 | I | 4.452(F) | 6.292(Cl) | 1.840 | 0.327 |
| Y2C(OH)F | 3.58 | 1.127 | D | 2.122(OH) | 4.402(F) | 2.270 | 0.367 |
| Y2C(OH)Cl | 3.64 | 0.896 | D | 1.689(OH) | 6.099(Cl) | 4.410 | 0.654 |
3.2 Electronic properties
It has been known that the electronic density of states (DOS) and band gaps of MXenes can be modified by the surface functional groups so that their electronic properties can be tuned in a large range from dielectrics to semiconductors, semimetals, and metals.14 Here, we examined the electronic properties of Y2CT2 and Y2CTT′ MXenes. From Table 2, one can see that all the investigated Y2C-based MXenes are semiconductors. It should be mentioned that for stacked sheets of Y2CF2 our calculations from the HSE06 functional give a more reliable band gap (1.987 eV) than the pure GGA functional (1.138 eV) in comparison to an experimental observed value of 1.90 eV.28 This is why we used the HSE06 functional to calculate the electronic and optical properties of Y2C-based MXenes in our study. The band gap of Y2C-based MXenes ranges from 0.684 to 1.952 eV, which closely depends on the surface functional groups. Roughly speaking, the –(OH) functional group tends to lower the band gap while the –F functional group is in favor of increasing the band gap. By this, Y2CF2 has the biggest band gap of 1.952 eV, while Y2 C(OH)2 has the smallest one of 0.684 eV. This can also be confirmed by the band structures shown in Fig. 3 (also refer to Fig. S2 of the ESI†) as well as the density of states (DOS) in Fig. S3 of the ESI.† The conduction bands are mainly donated by 4d orbitals of Y atoms while the valence bands are dominated by Y 4d and C 2p orbitals for the investigated Y2 C-based MXenes. For MXenes with the –(OH) functional group, the CBM contributed from 2p orbitals of the O atom in the –(OH) group at the Γ point can be seen, which significantly reduces the band gaps and hence Y2C(OH)2, Y2C(OH)Cl, and Y2C(OH)F are changed from indirect band semiconductors to direct ones. | ||
| Fig. 3 Band structures of (a) Y2CF2, (b) Y2C(OH)2, (c) Y2CHF, and (d) Y2 C(OH)Cl. The dashed lines indicate the Fermi level EF, which is put at the middle of the band gap and is set to zero. | ||
As one of the basic physical characteristics, the work function Φ also plays an important role in the electronic properties, which is closely related to the surface functional groups for MXenes.56 Therefore, we calculated Φ for each investigated MXene, which is defined as the energy difference between EF and vacuum level (electrostatic potential far away from the surface). As seen in Table 2, the calculated Φ values for Y2CH2, Y2CF2, Y2CCl2, and Y2C(OH)2 are 4.478 eV, 4.615 eV, 6.223 eV, and 1.427 eV, respectively. It can be seen that surfaces with the –Cl functional group have the highest Φ value. In contrast, surfaces with the –(OH) functional group have the lowest Φ value. The calculated very low work function of Y2C(OH)2 agrees with previous theoretical and experimental reports for other OH-terminated MXenes.56,59,60 This is because the change in work functions of functionalized MXenes has a positive linear correlation with the change in surface dipole moments, while the intrinsic dipole moment of the polar –(OH) group makes a large and negative contribution to the total surface dipole moments.56 When it turns to Y2CTT′ Janus MXenes, each individual surface has its own Φ. As shown in Fig. 4 (also see Table 2), there is a work function difference ΔΦ for each Y2CTT′ Janus MXene, which in turn contributes to an IEF perpendicular to the plane of the Janus MXenes. The intensity of IEF (EIEF) relates to ΔΦ by the following equation:35
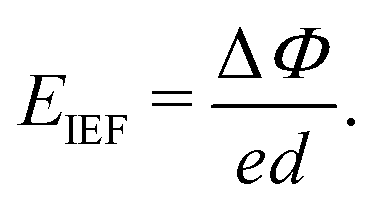 | (1) |
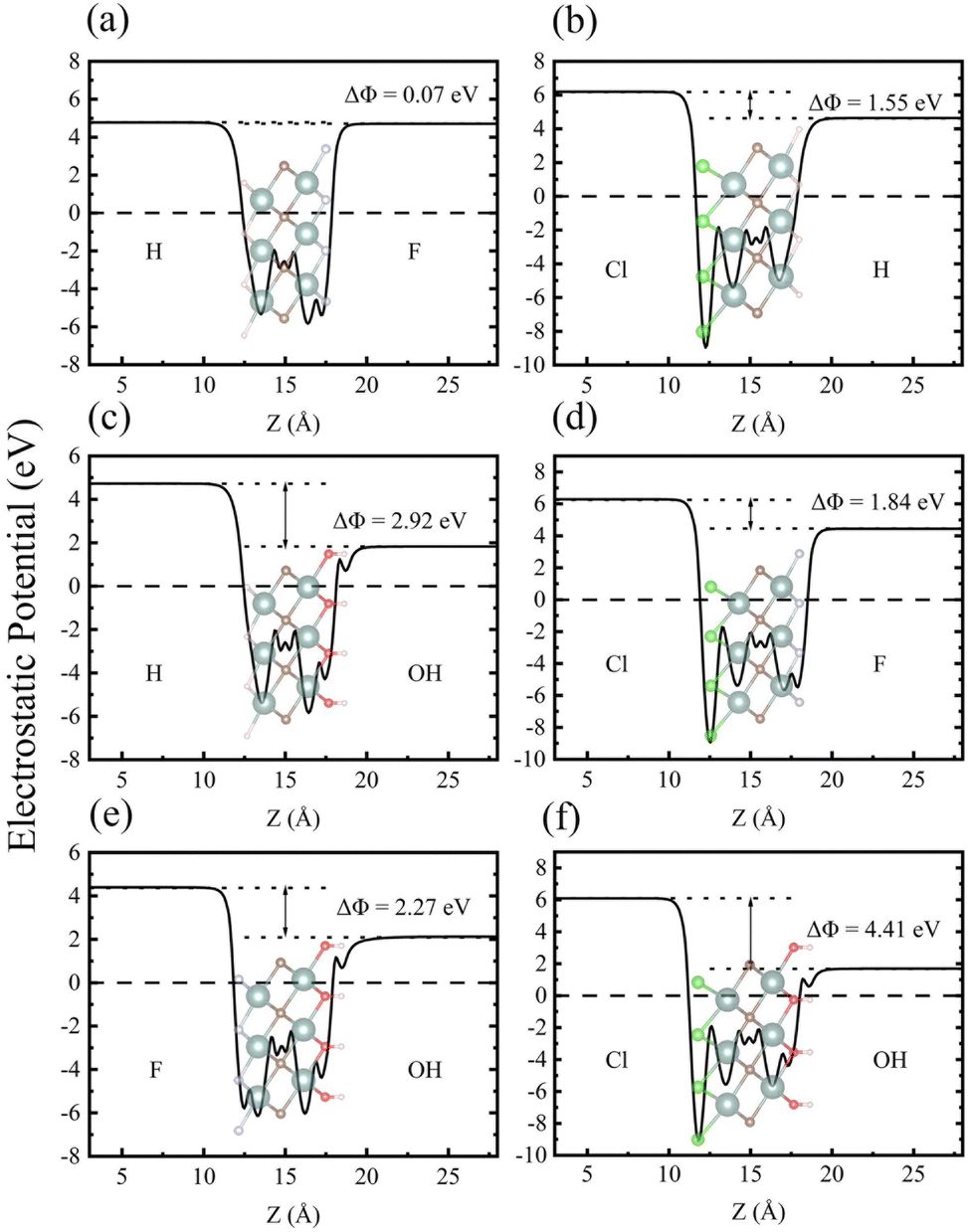 | ||
| Fig. 4 The calculated electrostatic potential energy differences of Janus MXenes: (a) Y2CHF, (b) Y2CHCl, (c) Y2C(OH)H, (d) Y2CFCl, (e) Y2C(OH)F, and (f) Y2(OH)Cl. The dotted and dashed lines represent the vacuum level of each sides and the Fermi level which is set to zero, respectively. | ||
Therefore, it is expected that Janus MXenes with the –(OH) group on one side and the –Cl group on the other side (i.e., Y2C(OH)Cl) will have the largest ΔΦ and hence the strongest EIEF, which can be easily confirmed from Table 2 and Fig. 4. The IEF in Janus MXenes facilitates the spatial separation of photo-excited electrons and holes onto different functional groups of two surfaces, which can be verified in Fig. S4 in the ESI.† The spatial separation of photo-excited electrons and holes is in favor of the photocatalytic water splitting on the surfaces.
3.3 Photocatalytic properties
For photocatalytic water splitting, the key processes are reduction of H+ to H2 and oxidation of H2O to O2. For these two processes to proceed, it needs electrons with high energy and holes with low energy. Because electrons with high energy show strong reducibility and can easily reduce H+ to H2, while holes with low energy have strong oxidability and can readily oxidize H2 O to O2. The reduction potential of H+ to H2 is 0 V vs. the SHE and oxidation potential of H2 O to O2 is 1.23 V vs. SHE, which align with the band energy values of −4.44 and −5.67 eV vs. absolute vacuum scale (AVS), respectively. Therefore, a photocatalyst candidate for water splitting should simultaneously have a CBM of higher than −4.44 eV and a VBM of lower than −5.67 eV. Here, the absolute band edge positions vs. AVS were calculated to study the redox ability of Y2CT2 and Janus Y2CTT′ MXenes. | ||
| Fig. 5 The calculated band edge positions of (a) Y2CH2, (b) Y2CF2, (c) Y2CCl2, (d) Y2C(OH)2, (e) Y2CHF, (f) Y2CHCl, (g) Y2C(OH)H, (h) Y2CFCl, (i) Y2C(OH)F, and (j) Y2C(OH)Cl vs. AVS (left) and SHE (right). The red, purple, and blue dash dot (dash) lines represent the reduction (oxidation) potential of water at pH = 0, 7, and 14, respectively. | ||
We further checked the photocatalytic properties for other pH values (here taking pH = 7 and pH = 14 as examples). As is known, the redox potentials (EP) of water change with the pH value, which follows the Nernst equation.61
| EP(pH) = EP(pH = 0) − 0.0591 × pH | (2) |
As shown in Fig. 5, though pH changes the redox potentials (EP) of water, the relative relationships of their alignments with the VBMs and CBMs of Y2CT2 MXenes are not fundamentally altered. Therefore, the four structurally symmetric Y2CT2 MXenes still cannot act as photocatalysts for water splitting at pH = 7 and 14.
Though the structurally symmetric Y2CT2 MXenes are not suitable photocatalysts for water splitting at all pH values, for Y2CH2 (noting that the VBM of Y2CH2 is merely 0.02 eV lower than the water oxidation potential at pH = 14, which is believed not large enough to efficiently drive the water oxidation), Y2CF2, and Y2C(OH)2, their electrons in conduction bands have relatively high energies, showing very large over-potentials, especially Y2C(OH)2. This will make the process of hydrogen evolution much faster. On the other hand, Y2CCl2 has a very positive valence band potential, which makes the process of oxygen evolution much faster instead. Therefore, with the addition of suitable reactants, Y2CT2 MXenes can be utilized to individually produce hydrogen or oxygen.
A Janus MXene can be simply considered as a combination of its two symmetric counterparts. And if the potential of the VBM for one counterpart is lower than the oxidation potential of H2O to O2 and the potential of the CBM for the other counterpart is higher than the reduction potential of H+ to H2, then this Janus MXene is more likely to have appropriate alignments of its band edge positions with the redox potentials of water. According to this simple strategy for finding photocatalyst candidates, we predicted that Y2CHCl, Y2CFCl, and Y2C(OH)Cl are promising photocatalyst candidates for water splitting at pH = 0, which can be easily verified in Fig. 5. Even though the band gap of Y2C(OH)Cl is only 0.896 eV, the redox over-potentials are still large demonstrating a fast rate of the photocatalytic water splitting reaction.
Since Y2CH2, Y2CF2, and Y2C(OH)2 belong to the same kind symmetric MXene with only the potential of the CBM higher than the reduction potential of H+ to H2 at pH = 0, the hybridizations of them do not provide effective photocatalyst candidates for water splitting. However, according to the Nernst equation (eqn (2)), the increase of the pH value can move the water redox potentials towards a negative direction, which makes their hybrid Janus MXenes possible photocatalyst candidates of water splitting. For example, at pH = 7, except for Y2CHCl, Y2CFCl, and Y2C(OH)Cl, two additional Janus MXenes Y2CHF and Y2C(OH)H can act as photocatalysts for water splitting. When the pH value increases to 14, all the investigated Janus MXenes become photocatalysts for water splitting.
To explore the energy conversion, we calculated the upper limits of STH efficiency for all investigated Janus MXenes at pH = 0, 7 and 14, which are listed in Table 3 and Tables S1, S2 of the ESI,† respectively (refer to the calculation method of STH efficiency in the ESI†). We can see from Table 3 that the corrected STH efficiencies at pH = 0 for Y2CHCl, Y2CFCl, and Y2C(OH)Cl are 22.6%, 18.7%, and 19.4%, respectively, all of which are lager than a theoretical limit of about 18% for structurally symmetric photocatalysts. It is worth mentioning that the corrected STH efficiencies of Y2CHCl, Y2CFCl, and Y2C(OH)Cl remain nearly unchanged and are always larger than 18% when the pH value is changed from 0 to 14 due to their large over-potentials for hydrogen and oxygen evolution reactions, illustrating their stable photocatalytic abilities. The corrected STH efficiencies of Y2CHF and Y2C(OH)H at pH = 7 are only 10.1% and 12.2% (refer to Table S1 of the ESI†), respectively, due to their small over-potentials of oxygen evolution. However, when the pH value increases to 14, the corrected STH efficiencies for all the investigated Janus MXenes are larger than 18% (refer to Table S2 of the ESI†).
| Systems | χ(H2) | χ(O2) | η abs | η cu | η STH | η′STH |
|---|---|---|---|---|---|---|
| Y2CHF | 1.02 | −0.39 | — | — | — | — |
| Y2CHCl | 0.83 | 1.06 | 58.6 | 53.9 | 31.6 | 22.6 |
| Y2C(OH)H | 3.13 | −0.34 | — | — | — | — |
| Y2CFCl | 1.15 | 1.2 | 48.7 | 53.2 | 25.9 | 18.7 |
| Y2C(OH)F | 2.88 | −0.71 | — | — | — | — |
| Y2C(OH)Cl | 3.17 | 0.91 | 88.4 | 71.7 | 63.4 | 19.4 |
As stated before, a good photocatalyst should also have a good sunlight absorption characteristic so that the solar energy can be utilized at most. To further check the ability of solar energy utilization, the optical absorption coefficients α(ω) of all investigated Janus Y2CTT′ MXenes are calculated and shown in Fig. 6 according to the following equation.62
 | (3) |
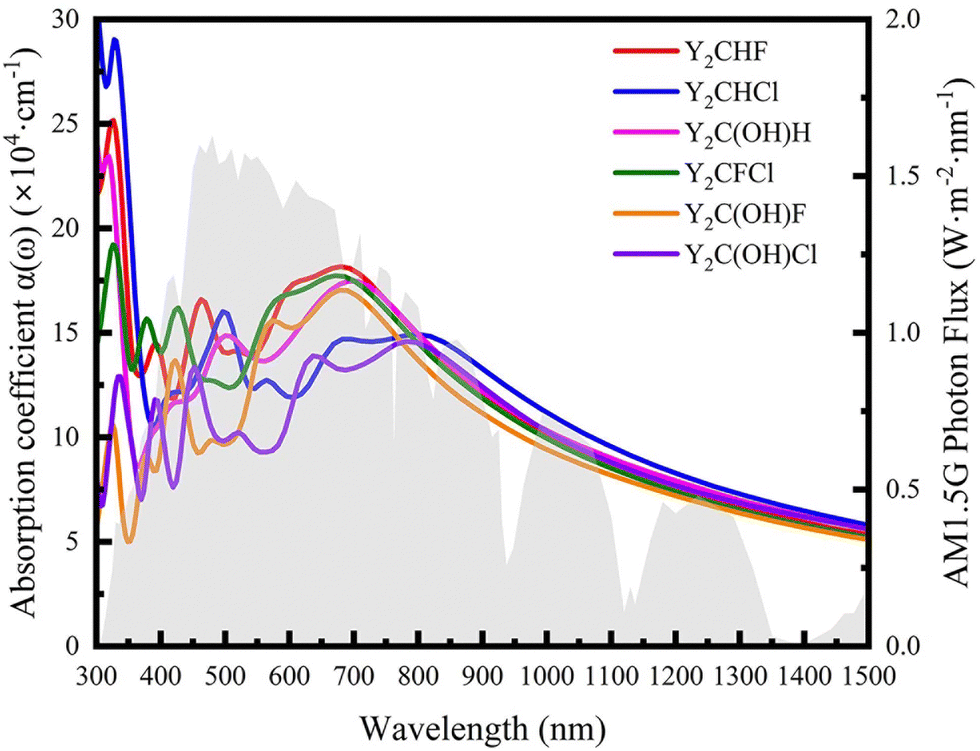 | ||
| Fig. 6 The calculated absorption rates of Janus Y2CTT′, and the gray shadow background is the solar spectral irradiance as Air Mass 1.5G. | ||
4 Conclusions
In summary, based on the first-principles calculations, the structural, electronic and photocatalytic properties of surface functionalized Y2CT2 and Janus Y2CTT′ MXenes (T, T′ = H, F, Cl, OH) have been systematically studied. The lattice constant, band gap, work-function, and photocatalytic properties of Y2C-based MXenes are closely related to the surface functional groups. The –Cl group increases the lattice constant of Y2C-based MXenes in comparison with the –H, –F or –(OH) group. For photocatalytic water splitting, all the four structurally symmetric Y2CT2 MXenes are not suitable photocatalysts for water splitting at all pH values (pH = 0, 7, and 14). In contrast, all the investigated Janus Y2CTT′ MXenes can act as good photocatalysts for water splitting since they have high absorption coefficients and meanwhile have significant STH efficiencies of larger than 18% at pH = 14. Furthermore, Y2CHCl, Y2CFCl, and Y2CFCl are predicted to be good photocatalysts for water splitting with STH efficiencies larger than 18% at all investigated pH values. Finally, for the first time, we propose an easy strategy to design Janus MXene candidates for photocatalysts with possible high STH efficiency by preliminarily inspecting the band structures and work functions of their symmetric counterparts. This work is helpful for the future design of Janus MXenes and more generally Janus 2D photocatalysts for photocatalytic water splitting with high STH efficiency.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the financial support from the National Natural Science Foundation of China (Grant No. 22173052 and 21933002).References
- G. Garcia, E. Arriola, W.-H. Chen and M. D. De Luna, Energy, 2021, 217, 119384 CrossRef CAS.
- T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrovic, D. Volbers, R. Wyrwich, M. Döblinger, A. S. Susha, A. L. Rogach, F. Jäckel, J. K. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013–1018 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- B. Lalmi, H. Oughaddou, H. Enriquez, A. Kara, S. Vizzini, B. Ealet and B. Aufray, Appl. Phys. Lett., 2010, 97, 223109 CrossRef.
- S. Cahangirov, M. Topsakal, E. Aktürk, H. Sahin and S. Ciraci, Phys. Rev. Lett., 2009, 102, 236804 CrossRef CAS.
- M. E. Dávila, L. Xian, S. Cahangirov, A. Rubio and G. Le Lay, New J. Phys., 2014, 16, 095002 CrossRef.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, abf1581 CrossRef.
- Z. Fu, N. Wang, D. Legut, C. Si, Q. Zhang, S. Du, T. C. Germann, J. S. Francisco and R. Zhang, Chem. Rev., 2019, 119, 11980–12031 CrossRef PubMed.
- C. Zhan, W. Sun, Y. Xie, D.-E. Jiang and P. R. C. Kent, ACS Appl. Mater. Interfaces, 2019, 11, 24885–24905 CrossRef.
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef PubMed.
- Y.-H. Liu, C.-Y. Wang, S.-L. Yang, F.-F. Cao and H. Ye, J. Energy Chem., 2022, 66, 429–439 CrossRef.
- X.-f Yu, Y.-c Li, J.-b Cheng, Z.-b Liu, Q.-z Li, W.-z Li, X. Yang and B. Xiao, ACS Appl. Mater. Interfaces, 2015, 7, 13707–13713 CrossRef.
- Y. Xia, T. S. Mathis, M.-Q. Zhao, B. Anasori, A. Dang, Z. Zhou, H. Cho, Y. Gogotsi and S. Yang, Nature, 2018, 557, 409–412 CrossRef CAS PubMed.
- F. Shahzad, M. Alhabeb, C. B. Hatter, B. Anasori, S. M. Hong, C. M. Koo and Y. Gogotsi, Science, 2016, 353, 1137–1140 CrossRef CAS PubMed.
- A. Iqbal, P. Sambyal and C. M. Koo, Adv. Funct. Mater., 2020, 30, 2000883 CrossRef CAS.
- Z. Guo, J. Zhou, L. Zhu and Z. Sun, J. Mater. Chem. A, 2016, 4, 11446–11452 RSC.
- H. Zhang, G. Yang, X. Zuo, H. Tang, Q. Yang and G. Li, J. Mater. Chem. A, 2016, 4, 12913–12920 RSC.
- C.-F. Fu, X. Li, Q. Luo and J. Yang, J. Mater. Chem. A, 2017, 5, 24972–24980 RSC.
- I. Ihsanullah, J. Environ. Chem. Eng., 2022, 10, 107381 CrossRef CAS.
- D. L. Druffel, M. G. Lanetti, J. D. Sundberg, J. T. Pawlik, M. S. Stark, C. L. Donley, L. M. McRae, K. M. Scott and S. C. Warren, Chem. Mater., 2019, 31, 9788–9796 CrossRef CAS.
- K. Maeda, H. Wakayama, Y. Washio, A. Ishikawa, M. Okazaki, H. Nakata and S. Matsuishi, J. Phys. Chem. C, 2020, 124, 14640–14645 CrossRef CAS.
- C.-F. Fu, J. Sun, Q. Luo, X. Li, W. Hu and J. Yang, Nano Lett., 2018, 18, 6312–6317 CrossRef CAS.
- C. R. Cox, J. Z. Lee, D. G. Nocera and T. Buonassisi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14057–14061 CrossRef CAS.
- L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng, Q. Yu, X. Cai, J. Zhao, Z. Ren, H. Fang, F. Robles-Hernandez, S. Baldelli and J. Bao, Nat. Nanotechnol., 2014, 9, 69–73 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- X. Li, Z. Li and J. Yang, Phys. Rev. Lett., 2014, 112, 018301 CrossRef PubMed.
- X. Wu, Y. Hu, Y. Wang, Y. Zhou, Z. Han, X. Jin and G. Chen, Appl. Surf. Sci., 2019, 464, 108–114 CrossRef CAS.
- B.-J. Ng, L. K. Putri, X. Y. Kong, Y. W. Teh, P. Pasbakhsh and S.-P. Chai, Adv. Sci., 2020, 7, 1903171 CrossRef CAS PubMed.
- L. Guo, H. Huang, L. Mei, M. Li and Y. Zhang, Mater. Chem. Front., 2021, 5, 2484–2505 RSC.
- Y. Liu, Y. Wan, B. Li, C. Yang, X. Lv and Y. Shi, J. Mater. Chem. A, 2023, 11, 21713–21720 RSC.
- Z. Gao, X. He, W. Li, Y. He and K. Xiong, Appl. Surf. Sci., 2023, 639, 158146 CrossRef CAS.
- N. Ghobadi, A. Rezavand, S. Soleimani-Amiri and S. Gholami Rudi, Appl. Surf. Sci., 2023, 639, 158278 CrossRef CAS.
- G. Nan, W. Zhang, X. Yan, X. Qin, S. Wu, R. Tang, M.-X. Tang, L. Hu, L. Liu, S. Wang, Y. Feng and W. Yi, Phys. Chem. Chem. Phys., 2023, 25, 24594–24602 RSC.
- Y. Zhao, B. Zhang and J. Lin, Phys. Chem. Chem. Phys., 2023, 25, 26666–26678 RSC.
- Y. Zhang, B. Sa, N. Miao, J. Zhou and Z. Sun, J. Mater. Chem. A, 2021, 9, 10882–10892 RSC.
- K. Xiong, Z. Cheng, J. Liu, P.-F. Liu and Z. Zi, RSC Adv., 2023, 13, 7972–7979 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- X. Gonze and C. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10355–10368 CrossRef.
- M. Khazaei, M. Arai, T. Sasaki, C.-Y. Chung, N. S. Venkataramanan, M. Estili, Y. Sakka and Y. Kawazoe, Adv. Funct. Mater., 2013, 23, 2185–2192 CrossRef CAS.
- Y. Lee, S. B. Cho and Y.-C. Chung, ACS Appl. Mater. Interfaces, 2014, 6, 14724–14728 CrossRef CAS PubMed.
- Y. Lee, Y. Hwang, S. B. Cho and Y.-C. Chung, Phys. Chem. Chem. Phys., 2014, 16, 26273–26278 RSC.
- J. Yang, X. Luo, S. Zhang and L. Chen, Phys. Chem. Chem. Phys., 2016, 18, 12914–12919 RSC.
- M. Khazaei, M. Arai, T. Sasaki, A. Ranjbar, Y. Liang and S. Yunoki, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 075411 CrossRef.
- H. Sahin, S. Cahangirov, M. Topsakal, E. Bekaroglu, E. Akturk, R. T. Senger and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 155453 CrossRef.
- J. He, X. Li, P. Lyu and P. Nachtigall, Nanoscale, 2017, 9, 2246–2252 RSC.
- X. Bai, X.-H. Zha, Y. Qiao, N. Qiu, Y. Zhang, K. Luo, J. He, Q. Li, Q. Huang, J. S. Francisco, C.-T. Lin and S. Du, Nanoscale, 2020, 12, 3795–3802 RSC.
- C. Peng, T. Zhou, P. Wei, H. Ai, B. Zhou, H. Pan, W. Xu, J. Jia, K. Zhang, H. Wang and H. Yu, Chem. Eng. J., 2022, 439, 135685 CrossRef CAS.
- V. Chakrapani, J. C. Angus, A. B. Anderson, S. D. Wolter, B. R. Stoner and G. U. Sumanasekera, Science, 2007, 318, 1424–1430 CrossRef CAS PubMed.
- J. Zhang, X. Tang, M. Chen, D. Ma and L. Ju, Inorg. Chem., 2022, 61, 17353–17361 CrossRef CAS.
- C. A. Gueymard, D. Myers and K. Emery, Sol. Energy, 2002, 73, 443–467 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04191c |
| This journal is © the Owner Societies 2024 |