First-principles prediction of the thermal conductivity of two configurations of difluorinated graphene monolayer
Received
11th October 2023
, Accepted 23rd November 2023
First published on 25th November 2023
Abstract
Lattice thermal conductivity (κL) plays a crucial role in the thermal management of electronic devices. In this study, we systematically investigate the thermal transport properties of monolayer fluorinated graphene using a combination of machine learning-based interatomic potentials and the phonon Boltzmann transport equation. At a temperature of 300 K, we find that the κL values for chair-configured fluorinated graphene monolayers are 184.24 W m−1 K−1 in the zigzag direction and 205.57 W m−1 K−1 in the armchair direction. For the boat configuration, the κL values are 120.45 W m−1 K−1 and 64.26 W m−1 K−1 in the respective directions. The disparities in κL between these two configurations predominantly stem from differences in phonon relaxation times, which can be elucidated by examining the Grüneisen parameters representing the degree of anharmonicity. A more in-depth analysis of bond strengths, as assessed by the crystal orbital Hamiltonian population, reveals that the stronger in-plane CC bonds in chair-configured fluorinated graphene monolayers are the primary contributors to the observed variations in anharmonicity.
1. Introduction
The discovery of graphene by Novoselov et al. in 20041 has sparked substantial interest in two-dimensional (2D) materials.2–6 The outstanding thermal, mechanical, magnetic, and electronic properties of graphene make it a promising candidate for nano-electronic devices.7–11 However, its high electron mobility, stemming from the absence of an intrinsic bandgap,12,13 hampers its progress and application in electronic fields, such as high-performance transistors.14,15 Consequently, there has been a significant research emphasis on introducing a bandgap.16,17 The most common approach is to open the bandgap through physical methods while preserving the atomic structure, such as interactions with substrates, physical adsorption, and strain. Woods et al. constructed a heterostructure consisting of graphene and hexagonal boron nitride (hBN), enabling graphene to adjust its periodicity to match that of hBN.18 The observed small bandgap can be attributed to the breaking of sublattice symmetry in graphene, as domain walls would divide matched lattices and accumulate generated strain. Papagno et al. investigated the effect of Na adsorption on graphene with and without an epitaxial Ir cluster superlattice on Ir (111).19 The results indicate that co-adsorption of Na onto an Ir cluster superlattice results in a significant bandgap of 740 meV. Pereira et al. analyzed the electronic structure of graphene with an observed bandgap under uniaxial tensile strain.20 They pointed out that a high strain of 20% deformation is required to induce this phenomenon, attributed to the merging of two inequivalent Dirac points. Nevertheless, many other researchers have focused on chemical engineering approaches, often involving the destruction of the pristine lattice to achieve flexibility and scalable production.15 One possible approach is to introduce defects; Wang et al. synthesized nanoporous graphene with variable bandgaps, where the large bandgap arises from the confinement of π-electrons due to pore generation and the non-planar structure.21 Lee et al. proposed the design of inverse nanoporous graphene/graphene heterobilayers with electric-field-tunable bandgaps.22 Chemical modification is another feasible strategy,23 involving the introduction of oxygen, fluorine, or hydrogen atoms to create new covalent bonds, altering the hybridization of carbon atoms from sp2 to sp3 and thus opening the bandgap.24–26
Fluorination emerges as the most promising method among chemical modifications due to its superior stability when compared to hydrogenated or oxidized graphene.13,27 Fluorinated graphene (FG) has garnered significant attention in both synthesis and application since its initial preparation by Geim et al.13 and Otyepka et al.28 It has been reported to possess a bandgap of 3.5 eV,29 or even as high as 7.4 eV,30 attributed to the introduction of CF bonds. Consequently, extensive research on fluorinated graphene, encompassing both experimental and theoretical investigations, has been undertaken. Due to its outstanding mechanical properties, FG exhibits significant potential as an ultrathin lubrication protective film. Matsumura et al. conducted a study on the tribological properties of both monolayer and multilayer FG treated with fluorine plasma, revealing that fluorination improved the durability of both types of FG.31 In multilayer FG, the friction coefficient decreased from 0.20 to 0.15, while in monolayer FG, it increased from 0.21 to 0.27. Fluorine, being the most electronegative element, induces changes in the electron distribution of graphene, leading to alterations in its optical and electronic characteristics. Jeon et al. produced monolayer FG through direct chemical fluorination of pristine graphene and discovered its luminescence across a broad spectrum, including the UV and visible light regions, exhibiting both excitonic and direct optical absorption and emission features.32 This suggests its potential for applications in optoelectronics and energy harvesting. Ho et al. delved into the dielectric properties of FG and identified its suitability as a gate dielectric in graphene-based field-effect transistors.16 Furthermore, FG displays promise in electrochemical applications. Wu et al. proposed that incorporating FG into ether-based electrolytes could enhance the discharge capacity and improve the stability of Li-metal in Li–O2 batteries.33 Additionally, Feng et al.24 in 2016 and Chen et al.25 in 2022 provided comprehensive summaries of the properties and applications of FG.
Thermal properties, e.g., lattice thermal conductivity (κL), are also of paramount importance for various applications. In the field of thermoelectric materials, researchers strive to identify materials with improved conversion efficiency, necessitating a reduction in κL.6,34–39 Simultaneously, researchers in the field of thermal management concentrate on enhancing κL to efficiently transfer heat from heat sources to the surrounding environment.40–44 Consequently, gaining a comprehensive understanding of the thermal transport properties of novel materials is essential, as it can provide valuable insights for tuning κL. However, there has been relatively limited research conducted on the thermal properties of FG. Huang et al. investigated the lattice thermal conductivity κL of FG using molecular dynamics (MD) simulations.45 It was observed that the introduction of fluorine atoms led to a decrease in κL, and as the fluorine content varied from 0% to 100%, κL exhibited a U-shaped trend due to variations in phonon scattering. Cepellotti et al. delved into the phonon transport and heat conductivity of several 2D materials using density-functional perturbation theory, suggesting that the room-temperature κL of FG is approximately 10−2 orders of magnitude.46 However, an experimental study in 2017 demonstrated that the κL of single-layer FG is around 80 W m−1 K−1, significantly lower than the prediction by Cepellotti et al.47 In contrast, Vu et al. prepared exfoliated FG films with variable thickness, showcasing a superior in-plane κL of 242 W m−1 K−1 at a thickness of 10 μm, with κL varying from 88 to 242 W m−1 K−1 as the thickness changed.48 These discrepancies highlight a significant knowledge gap regarding the thermal transport properties of FG.
In this study, we systematically investigated the κL of two distinct configurations of fluorinated graphene monolayer (FGM), i.e., the chair and boat configurations. We employed a machine learning interatomic potential (MLIP) approach49–52 in conjunction with the phonon Boltzmann transport equation (PBTE) method.53 Our findings reveal high κL values for both configurations, along with varying degrees of anisotropy. Notably, the chair configuration FGM (CFGM) exhibits significantly higher κL compared to the boat configuration FGM (BFGM). At approximately 300 K, κL values are measured at 184.24 W m−1 K−1 and 205.57 W m−1 K−1 for CFGM, and 120.45 W m−1 K−1 and 64.26 W m−1 K−1 for BFGM, along the zigzag and armchair directions, respectively. Further analysis, including an examination of phonon properties and crystal orbital Hamiltonian population (COHP), indicates the presence of stronger in-plane CC bonds in CFGM. These stronger bonds contribute to reduced anharmonicity, smaller Grüneisen parameters, longer phonon relaxation times, and superior κL in CFGM. Our findings offer valuable insights into the phonon transport properties of FGM, which can be instrumental in informing thermal management strategies for FGM in electronic devices.
2. Computational details
First-principles calculations, including structure optimization and ab initio molecular dynamics (AIMD) simulations, are conducted using the Vienna ab initio simulation package54 (VASP). The interaction among atoms is described by the projector augmented wave (PAW) pseudopotential,55 and generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional is employed to portray the exchange–correlation function for structure relaxation.56 A cutoff energy of 550 eV is adopted for the plane-wave basis set and a 6 × 3 × 1 Monkhorst–Pack k-mesh is implemented to sample the Brillouin zones of the FG unit cell. Structure relaxation is carried out until convergence is achieved, with forces on individual atoms reduced to 10−5 eV Å−1 whilst the energy is less than 10−6 eV. A 400 fs AIMD simulation ranging from 50 K to 800 K, using a time step of 1 fs and a supercell of 5 × 5 × 1, is conducted to obtain the trajectory of atoms for training the moment tensor potential (MTP) within the MLIP package. This allows us to calculate the second-order interatomic force constants (2nd IFCs) and third-order IFCs (3rd IFCs) with the MTP, thus circumventing the substantial computational cost of density functional theory (DFT), as previously demonstrated in our earlier work.57,58 A supercell of 5 × 5 × 1 is adopted to calculate 2nd IFCs and 3rd IFCs, which are subsequently employed in the iterative solution of the PBTE using the ShengBTE package.59 To ensure accuracy, we employ a dense phonon q-grid of 44 × 33 × 1 for CFGM and 60 × 50 × 1 for BFGM following convergence tests. This allows us to ultimately access the κL. The equation is expressed as follows:| | 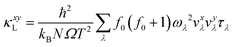 | (1) |
where x and y are the cartesian directions, and kB, N, Ω, and T are the Boltzmann constant, number of phonon wave vectors, individual unit cell volume and absolute temperature, respectively. λ denotes the phonon vibrational mode (i.e., qν that consists of wave vector q and phonon polarization ν), while ωλ, τλ and νxλ are the phonon frequency, relaxation time and group velocity along cartesian direction x of the phonon mode λ, respectively. ħ and f0 are the Planck constant and Bose–Einstein distribution function.
3. Results and discussion
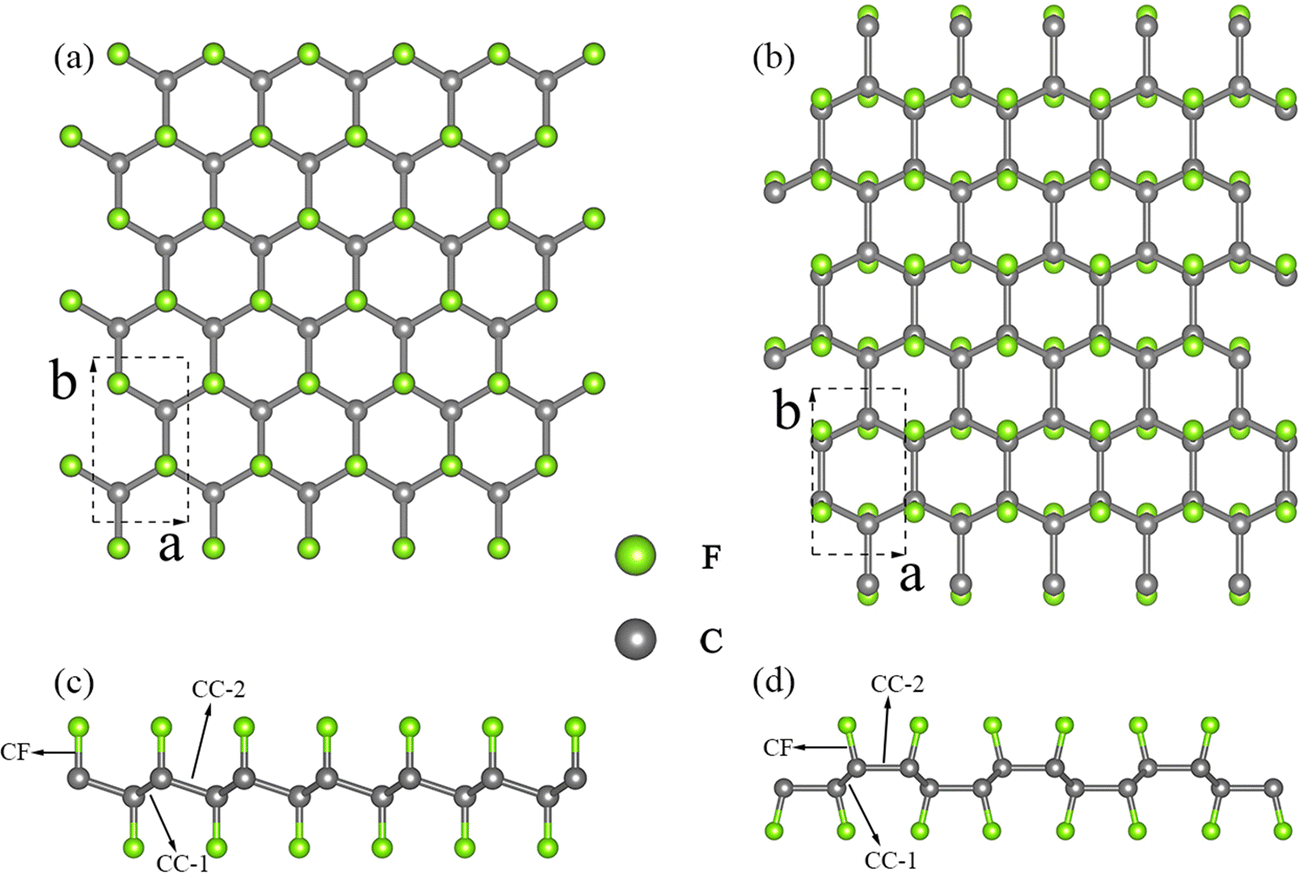
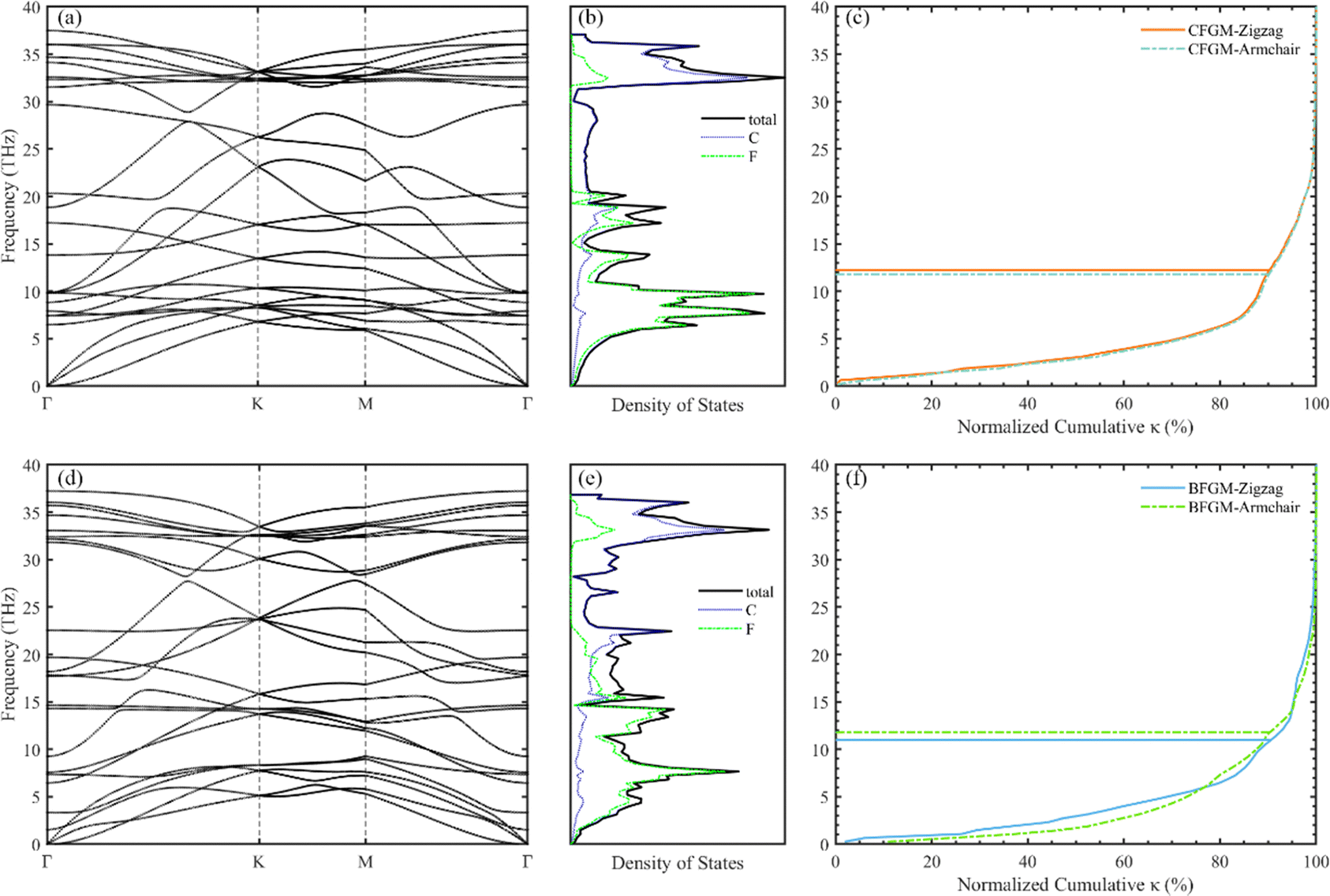
We conducted an investigation into two distinct configurations of FGM, wherein fluorine atoms form covalent bonds with carbon atoms on both sides. This bonding arrangement leads to the transformation of sp2 hybridized carbon atoms into sp3 hybridization, resulting in the creation of folds within the graphene monolayer. These two configurations, as illustrated in Fig. 1, are commonly referred to as the “chair” and “boat” configurations, based on the arrangement of the CF bonds. As aforementioned, there is limited available information regarding the phonon transport properties of FGM, and there may be discrepancies between these two configurations. Therefore, we chose to study the two configurations to gain a comprehensive understanding. To facilitate comparison, both structures were described using an orthogonal lattice containing 4 carbon atoms and 4 fluorine atoms. The lattice parameters after relaxation for CFGM are a = 2.606 Å, b = 4.512 Å, while for BFGM, they are a = 2.580 Å, b = 4.609 Å. These values are in good agreement with those reported in previous literature.27 To further confirm the stability and elucidate the phonon transport characteristics of the two configurations, we calculated the phonon spectra using the Phonopy code60 with the 2nd IFCs derived from the MTP. The results are displayed in Fig. 2(a) and (d). The absence of imaginary frequencies in the phonon spectrum indicates the dynamic stability of both configurations. The phonon frequency ranges from 0 to 37.5 THz for CFGM and 0 to 37.2 THz for BFGM, with no apparent band gap observed in the phonon spectrum due to the similar mass of the constituent elements. However, it is noteworthy that BFGM exhibits stronger intersections between acoustic modes and between acoustic modes and optical modes at low frequencies compared to CFGM. This observation demonstrates stronger acoustic–acoustic coupling and acoustic–optical coupling between low-frequency phonons in BFGM, creating more scattering channels for low-frequency phonons, such as acoustic + acoustic → optical. In addition, we present the projected density of states (PDOS) for both configurations in Fig. 2(b) and (e). From these plots, we can observe that the F atoms primarily contribute to the low-frequency modes. Both configurations exhibit extended states, implying larger mean free paths (MFP), longer phonon relaxation times, and consequently a substantial κL.61 It is widely recognized that low-frequency modes play a significant role in determining the κL of 2D materials, with the ZA (out-of-plane acoustic) mode, for instance, contributing as much as 75% of the κL in graphene.62 To assess this contribution, we calculated the normalized cumulative κL for both configurations at 300 K, as depicted in Fig. 2(c) and (f). In the following sections, we will refer to the unit cell vectors a and b as the zigzag and armchair directions, respectively. The horizontal lines in the figures represent the frequencies at which the contribution reaches 90%, underscoring the dominant role of low-frequency modes in κL. Notably, phonons with frequencies below 13 THz in CFGM and below 12 THz in BFGM collectively contribute 90% to κL. Therefore, our analysis will primarily focus on phonons with frequencies below 15 THz.
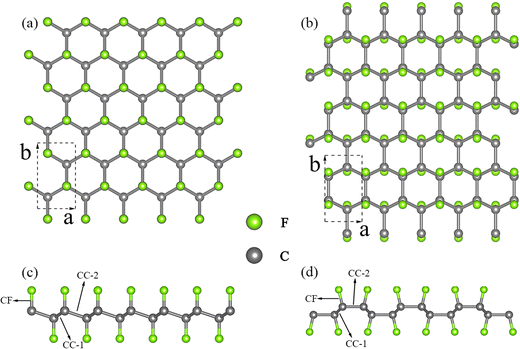 |
| | Fig. 1 Schematic diagrams of two different configurations of fluorinated graphene monolayer: (a) (001) plane, (c) (100) plane of chair configuration, (b) (001) plane, and (d) (100) plane of boat configuration. The dashed boxes show the unit cell. | |
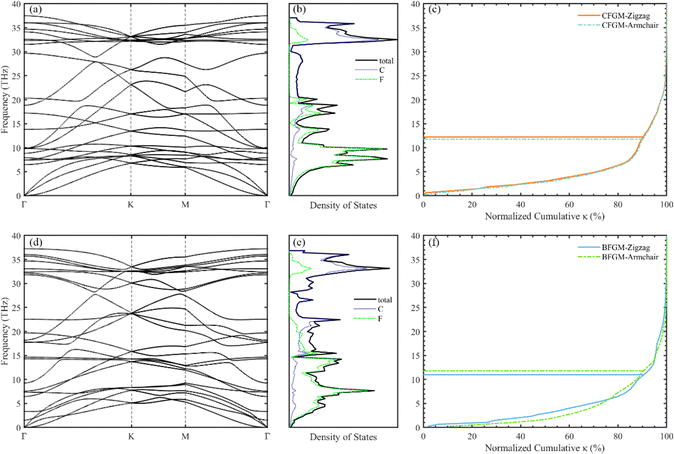 |
| | Fig. 2 Phonon spectrum, PDOS, normalized cumulative directional κL with respect to the frequency of CFGM (a)–(c) and BFGM (d)–(f) at 300 K. | |
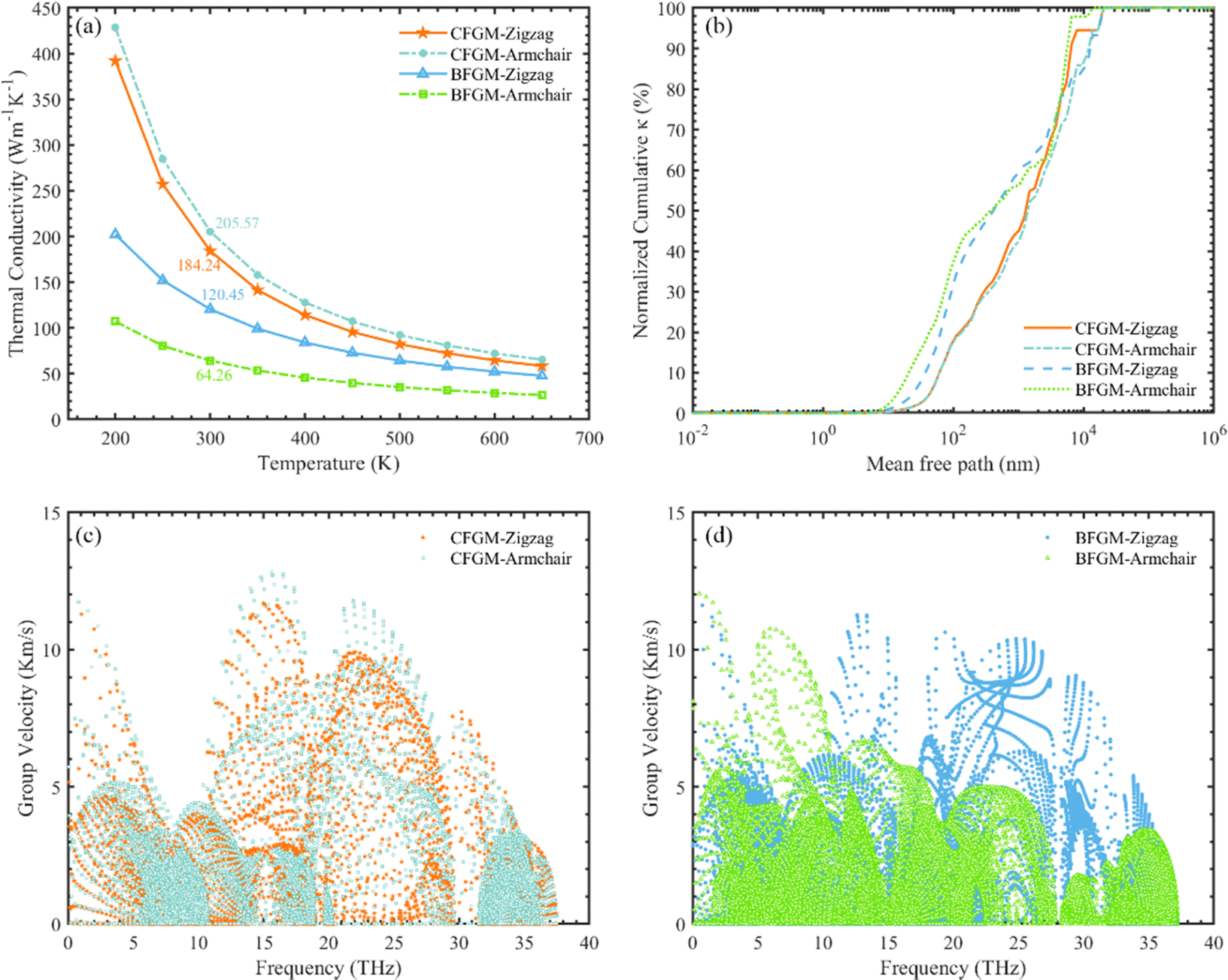
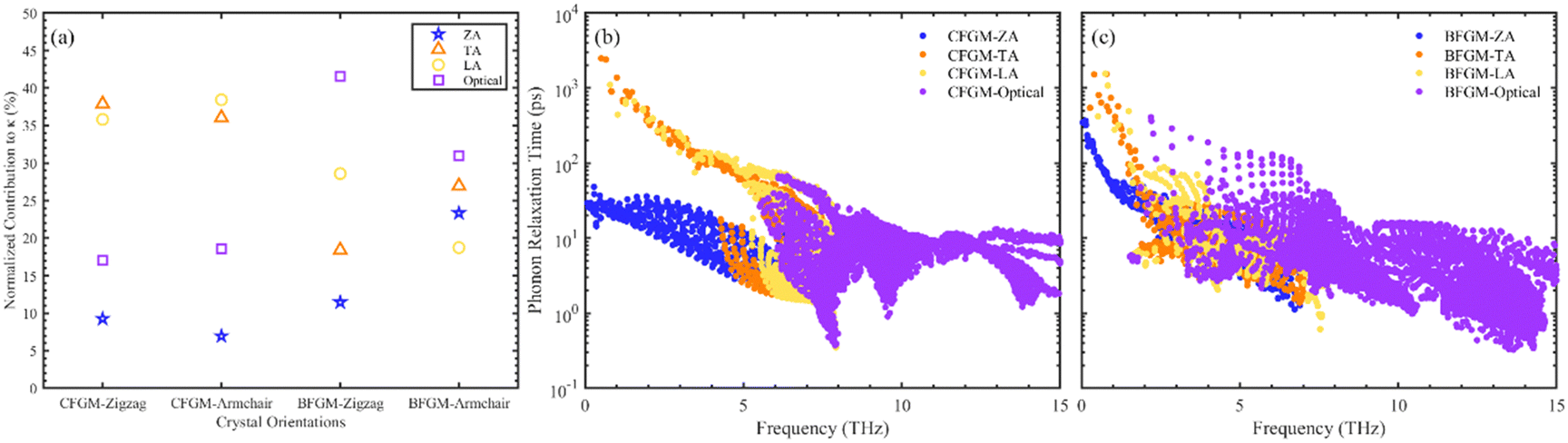
The in-plane κL of CFGM and BFGM as a function of temperature is presented in Fig. 3(a), i.e., κzigzag and κarmchair, as the κL along the out-of-plane direction is not meaningful. As the temperature increases, κL generally decreases, approximately following an inverse relationship due to stronger lattice vibrations and enhanced Umklapp processes. Interestingly, κL exhibits a certain degree of anisotropy and configuration dependence. For CFGM, κarmchair is slightly higher than κzigzag across the entire temperature range, while the opposite is true for BFGM. At room temperature, the κzigzag and κarmchair of CFGM are measured at 184.24 W m−1 K−1 and 205.57 W m−1 K−1, respectively. However, for BFGM, the κzigzag and κarmchair are 120.45 W m−1 K−1 and 64.26 W m−1 K−1, representing only two-thirds and one-third of the values in CFGM, respectively. These values suggest a significant difference between the two configurations. To elucidate the primary causes of anisotropy and discrepancies in thermal conductivity, we delved deeper into the phonon transport properties of both configurations. As per eqn (1), higher phonon group velocity (v) and longer phonon relaxation time (τ) contribute to higher κL. We examined the normalized cumulative κL as a function of MFP and directional phonon group velocity at 300 K, as illustrated in Fig. 3(b–d). From Fig. 3(b), it is evident that the differences between the zigzag and armchair directions are negligible for both configurations. Phonons with longer relaxation times generally exhibit larger MFP, which would manifest as a downward shift in the curve. Therefore, it appears that relaxation time may not be the primary cause of the anisotropy in κL. An analysis of directional group velocity reveals that, for CFGM, varmchair is slightly higher than vzigzag across the entire frequency range. Conversely, for BFGM, vzigzag is approximately twice varmchair. These findings are consistent with the observed κL values, suggesting that the directional differences in group velocity contribute significantly to the anisotropy in κL. Furthermore, we redirected our focus towards phonon relaxation time, given that phonons with longer MFP in CFGM contribute more significantly than those in BFGM. As previously noted, low-frequency phonons exert a more substantial influence compared to their high-frequency counterparts. Consequently, we have depicted the percentage contribution of low-frequency phonon modes to κL in all crystal orientations at 300 K, as shown in Fig. 4(a). Unlike graphene, ZA modes make minimal contributions to κL, owing to the folds generated by CF bonds that restrict out-of-plane vibrations. In CFGM, in-plane transverse and longitudinal acoustic (TA and LA) modes dominate, collectively contributing more than 70% to the total κL. Conversely, in BFGM, optical modes assume greater importance due to the pronounced phonon–phonon scattering between acoustic and low-frequency optical modes, as reflected in the phonon spectrum in Fig. 2(d). Fig. 4(b) and (c) present the phonon relaxation time for different phonon modes as a function of frequency at 300 K. Overall, the relaxation time of ZA and optical modes for both configurations is comparable, while that of TA and LA modes in CFGM is nearly an order of magnitude higher than that in BFGM. This suggests that the longer relaxation time primarily accounts for the higher κL in CFGM.
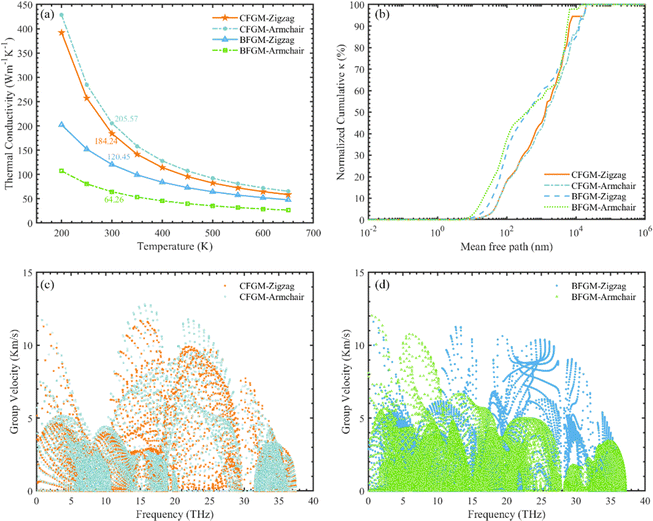 |
| | Fig. 3 (a) Lattice thermal conductivity of CFGM and BFGM as a function of temperature ranging from 200 to 650 K. (b) Normalized cumulative directional κL in dependency of MFP at 300 K. Phonon group velocities versus frequency of (c) CFGM and (d) BFGM at 300 K. | |
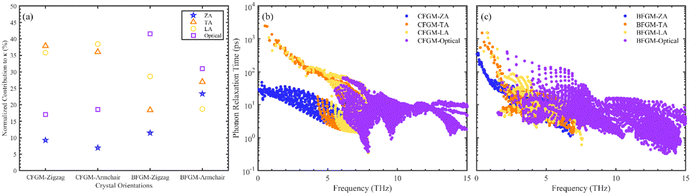 |
| | Fig. 4 (a) Percentage contribution from low-frequency phonon modes to κL in all crystal orientations. Phonon relaxation time with respect to frequency of (b) CFGM and (c) BFGM. | |
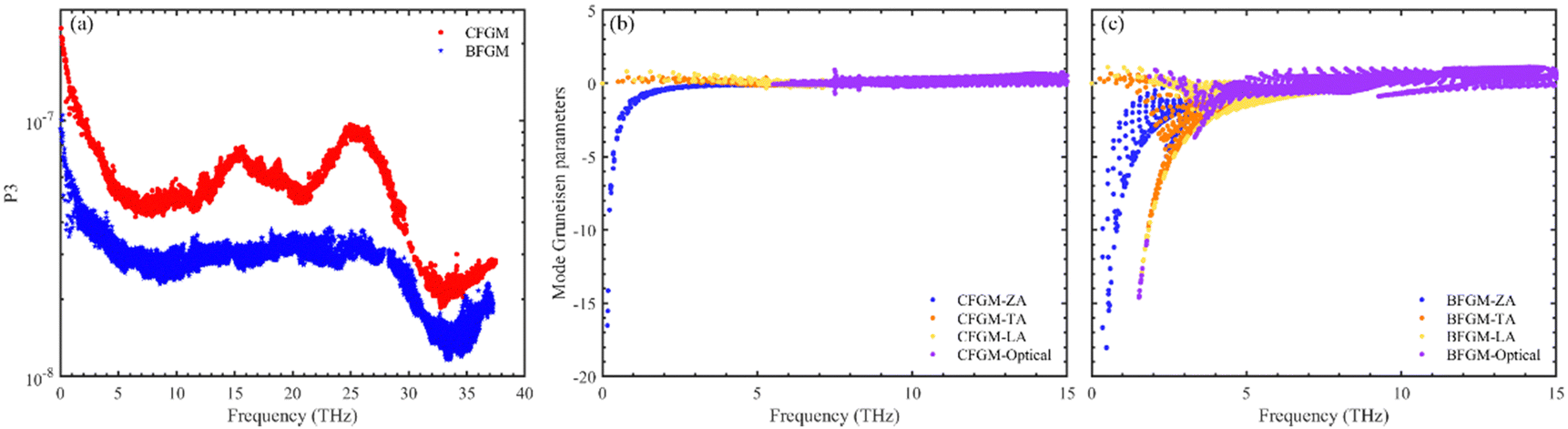
To further clarify the origin of differences in phonon relaxation time and the underlying mechanisms, we present the phase space P3 and mode Grüneisen parameters γ at 300 K in Fig. 5. P3 measures the phase space available for three-phonon processes across all phonon modes, representing the number of available scattering channels.63 Mode Grüneisen parameters offer insights into anharmonic interactions and are directly derived from 3rd IFCs.59,64 In Fig. 5(a), we illustrate the frequency-dependent P3 of both configurations. Notably, P3 for CFGM is slightly larger than that for BFGM across the entire frequency range, indicating a greater number of scattering channels in CFGM and, consequently, a lower relaxation time. However, the frequency-dependent mode Grüneisen parameters shown in Fig. 5(b) and (c) exhibit an opposite trend. It is important to highlight that γ for BFGM is significantly larger than that for CFGM, especially for TA and LA modes, suggesting much stronger anharmonicity in BFGM. Taking into account the combined impact of P3 and Grüneisen parameters, we can attribute the differences in relaxation time to the various anharmonicity. Furthermore, several formulas have been proposed to describe the relationship between κL and Grüneisen parameters, such as the widely accepted Slack model65,66 expressed as follows:
| | 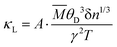 | (2) |
where
A is a collection of physical constants,
δ3 is the volume of each atom,
n is the number of atoms in the primitive unit cell,
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif)
is the average mass of an atom in the crystal,
γ is the acoustic mode Grüneisen parameter, and
θD is the Debye temperature of acoustic modes given by
| | 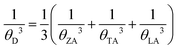 | (3) |
and
θλ of acoustic phonon mode
λ (
λ = ZA, TA, LA) is defined as
| | 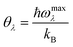 | (4) |
where
ωmaxλ is the maximum frequency of phonon mode
λ. In accordance with
eqn (3) and (4), the calculated Debye temperatures are presented in
Table 1. Consequently, it becomes evident that the differential Grüneisen parameters and Debye temperature play a pivotal role in determining
κL, as outlined in
eqn (2).
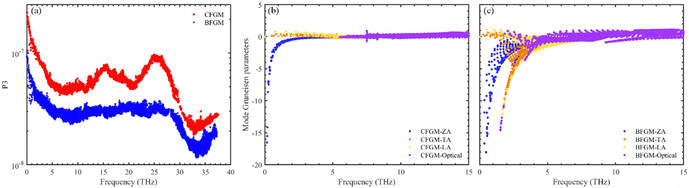 |
| | Fig. 5 (a) Frequency-dependent phase space of both configurations; frequency-dependent mode Grüneisen parameters of (b) CFGM and (c) BFGM. | |
Table 1 Bond lengths d, -ICOHP values and Debye temperatures ΘD of the two configurations
| Configuration bond |
Chair |
Boat |
| CF |
CC-1 |
CC-2 |
CF |
CC-1 |
CC-2 |
|
d/Å |
1.383 |
1.581 |
1.581 |
1.377 |
1.571 |
1.664 |
| -ICOHP |
11.04 |
8.81 |
8.73 |
11.17 |
9.02 |
7.54 |
|
Θ
D/K |
358.5 |
351.5 |
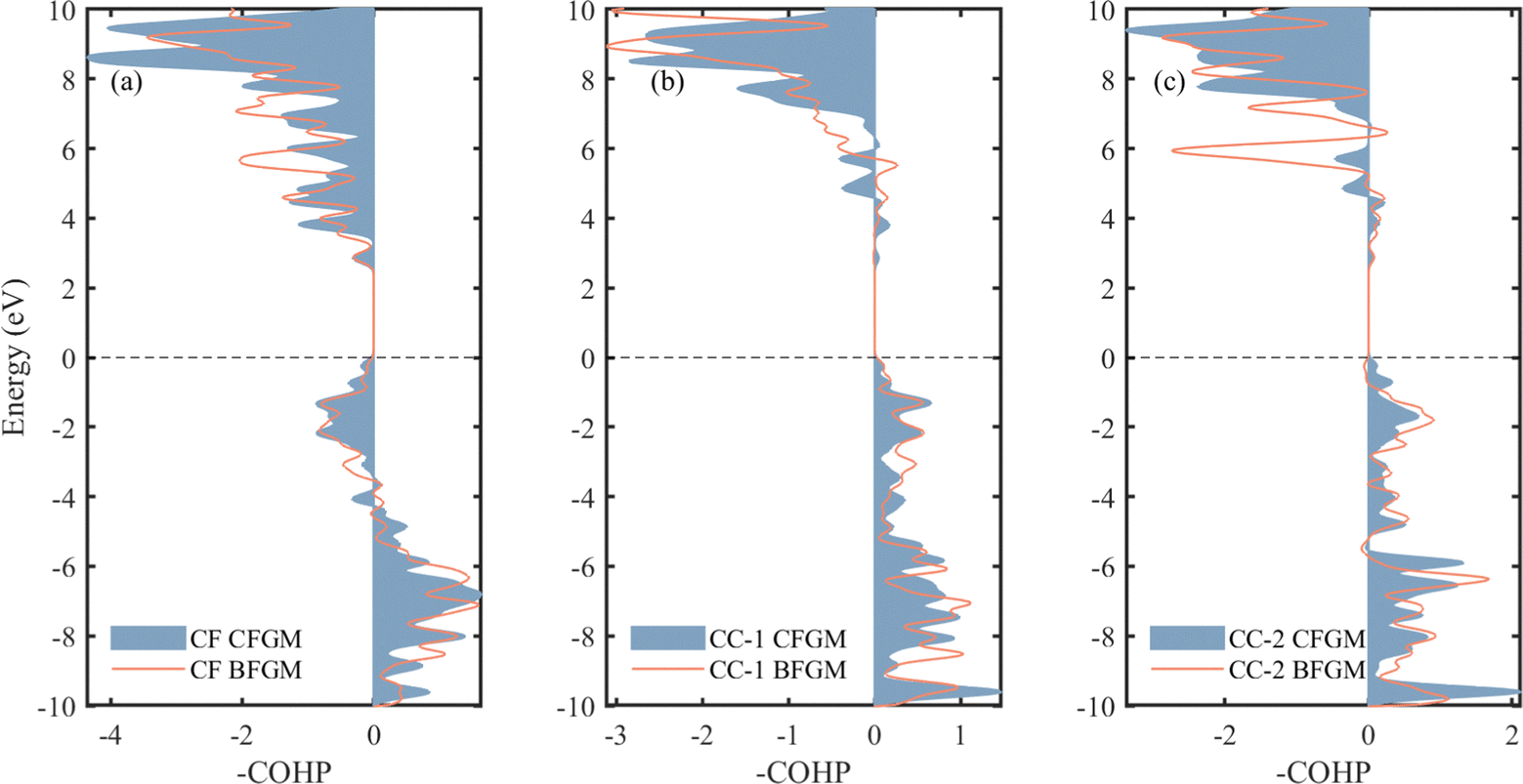
In the case of layered materials, an increase in in-plane bonding strength will enhance in-plane κL while maintaining out-of-plane bonding constants.67 Furthermore, variations in bond strength can influence anharmonicity and harmonic properties.68,69 Thus, it is imperative to investigate the bonding conditions. In each configuration, we have identified three types of chemical bonds, as marked in Fig. 1, categorized as CF, CC-1, and CC-2, respectively. To gain a deeper understanding of bonding strength, we conducted a COHP analysis, as depicted in Fig. 6. COHP is a quantitative method that provides information about the bonding characteristics between specific pairs of atoms below the Fermi level.70 A negative COHP value indicates a bonding state that stabilizes the system, whereas a positive value signifies an anti-bonding state associated with higher system energy. Here, we present the results in terms of –COHP for improved comprehension and comparison. Below the Fermi level, CF pairs exhibit an anti-bonding state in the energy range from −4 to 0 eV, while the other regions and entire CC pairs are in a bonding state. It is noteworthy that the CF curve and area for the two configurations show minimal differences. However, in CFGM, the CC pairs display a higher peak at approximately −9.6 eV, indicating a larger value of integrated COHP (ICOHP) and stronger bonding. Consequently, we have compiled the corresponding -ICOHP values in Table 1. These values are obtained by integrating the -COHP below the Fermi level, providing a measure and evaluation of the total bond strength. As expected, the values for CF and CC-1 pairs are similar, while in the case of CC-2 pairs, the value for CFGM is approximately 15% larger than that in BFGM. These results indicate that the in-plane CC bonds in CFGM are generally stronger than those in BFGM, while the out-of-plane CF bonds exhibit less variation. This trend is consistent with the bond lengths presented in Table 1, where a shorter length signifies greater bond strength. The comparable strength of CF bonds and the increased strength of CC bonds in CFGM suggest a higher ability for atoms to restore, which reduces anharmonicity. This, in turn, explains the smaller Grüneisen parameters and longer relaxation time observed in CFGM.
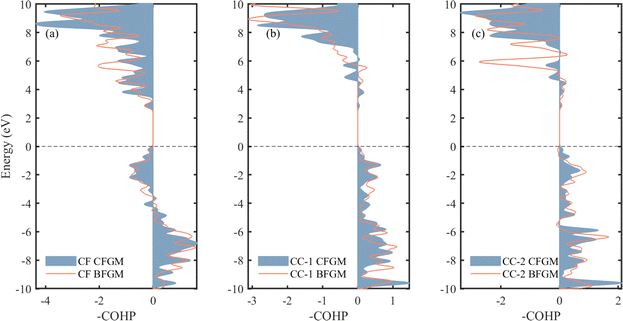 |
| | Fig. 6 -COHP of (a) CF, (b) CC-1 and (c) CC-2 pairs for two different configurations of fluorinated graphene monolayer. | |
It is worth highlighting that both configurations exhibit superior κL at room temperature when compared to a plethora of other 2D materials. Gao et al. developed an empirical model for 2D materials fitted as 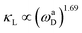 , where ωaD is the largest acoustic phonon frequency.71 For CFGM and BFGM, ωaD is 8.26 and 7.74 THz, respectively, which surpass the values observed in many other 2D materials. This suggests a potentially stronger acoustic phonon vibration, which bodes well for thermal transport and κL. As previously analyzed, FGMs demonstrate a high phonon group velocity and long phonon relaxation time, with relaxation times extending up to 103 ps. These values are comparable to those of well-known high-κL 2D materials such as MoS2 and WS2.72 This is primarily due to the lower amplitude of Grüneisen parameters and weaker anharmonicity. Additionally, CFGM and BFGM exhibit high Debye temperatures, which represent the threshold below which phonon modes become “frozen out”.73 Consequently, higher Debye temperatures imply that fewer phonons are activated at room temperature, leading to reduced phonon population and scattering rates.73–75
, where ωaD is the largest acoustic phonon frequency.71 For CFGM and BFGM, ωaD is 8.26 and 7.74 THz, respectively, which surpass the values observed in many other 2D materials. This suggests a potentially stronger acoustic phonon vibration, which bodes well for thermal transport and κL. As previously analyzed, FGMs demonstrate a high phonon group velocity and long phonon relaxation time, with relaxation times extending up to 103 ps. These values are comparable to those of well-known high-κL 2D materials such as MoS2 and WS2.72 This is primarily due to the lower amplitude of Grüneisen parameters and weaker anharmonicity. Additionally, CFGM and BFGM exhibit high Debye temperatures, which represent the threshold below which phonon modes become “frozen out”.73 Consequently, higher Debye temperatures imply that fewer phonons are activated at room temperature, leading to reduced phonon population and scattering rates.73–75
4. Conclusions
In summary, we have conducted a comprehensive investigation into the thermal transport properties of two distinct configurations of FGM through first-principles calculations, MLIP and the PBTE. This approach offers an efficient strategy for exploring the lattice thermal conductivity of materials with polyatomic unit cells. At room temperature, CFGM exhibits a κzigzag of 184.24 W m−1 K−1 and a κarmchair of 205.57 W m−1 K−1, while BFGM displays a κzigzag of 120.45 W m−1 K−1 and a κarmchair of 64.26 W m−1 K−1. Further analysis confirms that the disparity in thermal conductivity between the two configurations can be attributed to the longer relaxation time in BFGM. As evidenced by COHP analysis and -ICOHP values, the stronger in-plane CC bonds in CFGM enhance the restorative ability of atoms and reduce anharmonicity, resulting in reduced Grüneisen parameters. This work contributes to a deeper understanding of the phonon transport properties of FGMs and their potential application in thermal management for electronic devices.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 12074115), the Science and Technology Innovation Program of Hunan Province (Grant No. 2023RC3176), the Natural Science Foundation of Hunan Province (Grant No. 2023JJ40248), and the Scientific Research Project of Hunan Provincial Education Department (Grant No. 22B0868).
References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- J. Gou, H. Bai, X. Zhang, Y. L. Huang, S. Duan, A. Ariando, S. A. Yang, L. Chen, Y. Lu and A. T. S. Wee, Nature, 2023, 617, 67–72 CrossRef CAS PubMed.
- D. Wang, C. Zhou, A. S. Filatov, W. Cho, F. Lagunas, M. Wang, S. Vaikuntanathan, C. Liu, R. F. Klie and D. V. Talapin, Science, 2023, 379, 1242–1247 CrossRef CAS PubMed.
- X.-K. Chen, J.-L. Tan, M. Pang, Z.-X. Xie, W.-X. Zhou and J. Liu, Appl. Phys. Lett., 2022, 121, 142203 CrossRef CAS.
- K. Wang, W. Zhou, Y. Cheng, M. Zhang, H. Wang and G. Zhang, Nanoscale, 2021, 13, 10882–10890 RSC.
- P.-Z. Jia, J.-P. Xie, X.-K. Chen, Y. Zhang, X. Yu, Y.-J. Zeng, Z.-X. Xie, Y.-X. Deng and W.-X. Zhou, J. Phys.: Condens. Matter, 2023, 35, 073001 CrossRef CAS.
- C. Cai, T. Wang, G. Qu and Z. Feng, Chin. Chem. Lett., 2021, 32, 1293–1298 CrossRef CAS.
- A. F. Carvalho, B. Kulyk, A. J. S. Fernandes, E. Fortunato and F. M. Costa, Adv. Mater., 2022, 34, 2101326 CrossRef CAS.
- L. Lin, L. Fu, K. Zhang, J. Chen, W. Zhang, S. Tang, Y. Du and N. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 39062–39067 CrossRef CAS PubMed.
- E. Riccardi, M. A. Méasson, M. Cazayous, A. Sacuto and Y. Gallais, Phys. Rev. Lett., 2016, 116, 066805 CrossRef CAS PubMed.
- S.-Z. Chen, Y.-X. Deng, X.-H. Cao, W.-X. Zhou, Y.-X. Feng, L.-M. Tang and K.-Q. Chen, J. Mater. Chem. A, 2019, 7, 21976–21984 RSC.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- R. R. Nair, W. Ren, R. Jalil, I. Riaz, V. G. Kravets, L. Britnell, P. Blake, F. Schedin, A. S. Mayorov, S. Yuan, M. I. Katsnelson, H.-M. Cheng, W. Strupinski, L. G. Bulusheva, A. V. Okotrub, I. V. Grigorieva, A. N. Grigorenko, K. S. Novoselov and A. K. Geim, Small, 2010, 6, 2877–2884 CrossRef CAS.
- T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2012, 57, 1061–1105 CrossRef CAS.
- X. Xu, C. Liu, Z. Sun, T. Cao, Z. Zhang, E. Wang, Z. Liu and K. Liu, Chem. Soc. Rev., 2018, 47, 3059–3099 RSC.
- K.-I. Ho, C.-H. Huang, J.-H. Liao, W. Zhang, L.-J. Li, C.-S. Lai and C.-Y. Su, Sci. Rep., 2014, 4, 5893 CrossRef.
- B. Standley, A. Mendez, E. Schmidgall and M. Bockrath, Nano Lett., 2012, 12, 1165–1169 CrossRef.
- C. R. Woods, L. Britnell, A. Eckmann, R. S. Ma, J. C. Lu, H. M. Guo, X. Lin, G. L. Yu, Y. Cao, R. V. Gorbachev, A. V. Kretinin, J. Park, L. A. Ponomarenko, M. I. Katsnelson, Y. N. Gornostyrev, K. Watanabe, T. Taniguchi, C. Casiraghi, H. J. Gao, A. K. Geim and K. S. Novoselov, Nat. Phys., 2014, 10, 451–456 Search PubMed.
- M. Papagno, S. Rusponi, P. M. Sheverdyaeva, S. Vlaic, M. Etzkorn, D. Pacilé, P. Moras, C. Carbone and H. Brune, ACS Nano, 2012, 6, 199–204 CrossRef PubMed.
- V. M. Pereira, A. H. Castro Neto and N. M. R. Peres, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 045401 CrossRef.
- D. Wang, X. Lu, Arramel, M. Yang, J. Wu and A. T. S. Wee, Small, 2021, 17, 2102246 CrossRef CAS.
- B. Lee and J. Kang, Adv. Electron. Mater., 2022, 8, 2200252 CrossRef CAS.
- D. W. Boukhvalov and M. I. Katsnelson, J. Phys.: Condens. Matter, 2009, 21, 344205 CrossRef CAS.
- W. Feng, P. Long, Y. Feng and Y. Li, Adv. Sci., 2016, 3, 1500413 CrossRef PubMed.
- X. Chen, K. Fan, Y. Liu, Y. Li, X. Liu, W. Feng and X. Wang, Adv. Mater., 2022, 34, 2101665 CrossRef CAS.
- J. Carpenter, H. Kim, J. Suarez, A. van der Zande and N. Miljkovic, ACS Appl. Mater. Interfaces, 2023, 15, 2429–2436 CrossRef CAS.
- O. Leenaerts, H. Peelaers, A. D. Hernández-Nieves, B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 195436 CrossRef.
- R. Zbořil, F. Karlický, A. B. Bourlinos, T. A. Steriotis, A. K. Stubos, V. Georgakilas, K. Šafářová, D. Jančík, C. Trapalis and M. Otyepka, Small, 2010, 6, 2885–2891 CrossRef.
- J. C. Charlier, X. Gonze and J. P. Michenaud, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 16162–16168 CrossRef CAS.
- A. Markevich, R. Jones and P. R. Briddon, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 115439 CrossRef.
- K. Matsumura, S. Chiashi, S. Maruyama and J. Choi, Appl. Surf. Sci., 2018, 432, 190–195 CrossRef CAS.
- K. J. Jeon, Z. Lee, E. Pollak, L. Moreschini, A. Bostwick, C. M. Park, R. Mendelsberg, V. Radmilovic, R. Kostecki, T. J. Richardson and E. Rotenberg, ACS Nano, 2011, 5, 1042–1046 CrossRef CAS PubMed.
- X. Wu, X. Wang, Z. Li, L. Chen, S. Zhou, H. Zhang, Y. Qiao, H. Yue, L. Huang and S.-G. Sun, Nano Lett., 2022, 22, 4985–4992 CrossRef CAS PubMed.
- A. Suwardi, J. Cao, L. Hu, F. Wei, J. Wu, Y. Zhao, S. H. Lim, L. Yang, X. Y. Tan, S. W. Chien, Y. Yin, W.-X. Zhou, W. L. Mun Nancy, X. Wang, S. H. Lim, X. Ni, D. Li, Q. Yan, Y. Zheng, G. Zhang and J. Xu, J. Mater. Chem. A, 2020, 8, 18880–18890 RSC.
- C.-W. Wu, X. Ren, G. Xie, W.-X. Zhou, G. Zhang and K.-Q. Chen, Phys. Rev. Appl., 2022, 18, 014053 CrossRef CAS.
- P.-Z. Jia, Z.-X. Xie, Y.-X. Deng, Y. Zhang, L.-M. Tang, W.-X. Zhou and K.-Q. Chen, Appl. Phys. Lett., 2022, 121, 043901 CrossRef CAS.
- X.-K. Chen, E.-M. Zhang, D. Wu and K.-Q. Chen, Phys. Rev. Appl., 2023, 19, 044052 CrossRef CAS.
- Z.-X. Xie, X.-K. Chen, X. Yu, Y.-X. Deng, Y. Zhang, W.-X. Zhou and P.-Z. Jia, Comput. Mater. Sci., 2023, 220, 112041 CrossRef CAS.
- P.-Z. Jia, Z.-X. Mo, L.-Q. Deng, Y. Zhang, X. Yu, Y.-J. Zeng, Y.-X. Deng and Z.-X. Xie, Diamond Relat. Mater., 2024, 141, 110609 CrossRef CAS.
- W.-X. Zhou, Y. Cheng, K.-Q. Chen, G. Xie, T. Wang and G. Zhang, Adv. Funct. Mater., 2020, 30, 1903829 CrossRef CAS.
- C. Xiang, C.-W. Wu, W.-X. Zhou, G. Xie and G. Zhang, Front. Phys., 2021, 17, 13202 CrossRef.
- C.-W. Wu, H. Pan, Y.-J. Zeng, W.-X. Zhou, K.-Q. Chen and G. Zhang, Nanoscale, 2023, 15, 6732–6737 RSC.
- J. Kwon, H. Ma, A. Giri, P. E. Hopkins, N. B. Shustova and Z. Tian, ACS Nano, 2023, 17, 15222–15230 CrossRef CAS PubMed.
- X.-K. Chen, X.-Y. Hu, P. Jia, Z.-X. Xie and J. Liu, Int. J. Mech. Sci., 2021, 206, 106576 CrossRef.
- W. Huang, Q.-X. Pei, Z. Liu and Y.-W. Zhang, Chem. Phys. Lett., 2012, 552, 97–101 CrossRef CAS.
- A. Cepellotti, G. Fugallo, L. Paulatto, M. Lazzeri, F. Mauri and N. Marzari, Nat. Commun., 2015, 6, 6400 CrossRef CAS.
- M. Narasaki, H. Wang, T. Nishiyama, T. Ikuta and K. Takahashi, Appl. Phys. Lett., 2017, 111, 093103 CrossRef.
- M. C. Vu, N. A. Thi Thieu, J.-H. Lim, W.-K. Choi, J. Chan Won, M. A. Islam and S.-R. Kim, Carbon, 2020, 157, 741–749 CrossRef.
- B. Mortazavi, I. S. Novikov, E. V. Podryabinkin, S. Roche, T. Rabczuk, A. V. Shapeev and X. Zhuang, Appl. Mater. Today, 2020, 20, 100685 CrossRef.
- B. Mortazavi, E. V. Podryabinkin, I. S. Novikov, T. Rabczuk, X. Zhuang and A. V. Shapeev, Comput. Phys. Commun., 2021, 258, 107583 CrossRef.
- I. S. Novikov, K. Gubaev, E. V. Podryabinkin and A. V. Shapeev, Mach. Learn.: Sci. Technol., 2021, 2, 025002 Search PubMed.
- Z. Fan, Z. Zeng, C. Zhang, Y. Wang, K. Song, H. Dong, Y. Chen and T. Ala-Nissila, Phys. Rev. B, 2021, 104, 104309 CrossRef CAS.
- J. Garg, N. Bonini, B. Kozinsky and N. Marzari, Phys. Rev. Lett., 2011, 106, 045901 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C.-W. Wu, X. Ren, S.-Y. Li, Y.-J. Zeng, W.-X. Zhou and G. Xie, Appl. Phys. Lett., 2022, 121, 172201 CrossRef CAS.
- A. Chen, H. Tong, C.-W. Wu, G. Xie, Z.-X. Xie, C.-Q. Xiang and W.-X. Zhou, Chin. Phys. B, 2023, 32, 058201 CrossRef.
- W. Li, J. Carrete, N. A. Katcho and N. Mingo, Comput. Phys. Commun., 2014, 185, 1747–1758 CrossRef CAS.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- M. S. Islam, K. Ushida, S. Tanaka, T. Makino and A. Hashimoto, Comput. Mater. Sci., 2014, 94, 35–43 CrossRef CAS.
- L. Lindsay, D. A. Broido and N. Mingo, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 115427 CrossRef.
- L. Lindsay and D. A. Broido, J. Phys.: Condens. Matter, 2008, 20, 165209 CrossRef.
- B. Peng, H. Zhang, H. Shao, Y. Xu, R. Zhang, H. Lu, D. W. Zhang and H. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 20977–20985 CrossRef CAS.
- G. A. Slack, J. Phys. Chem. Solids, 1973, 34, 321–335 CrossRef CAS.
- D. T. Morelli and J. P. Heremans, Appl. Phys. Lett., 2002, 81, 5126–5128 CrossRef CAS.
- Z. Wei, Y. Chen and C. Dames, Appl. Phys. Lett., 2013, 102, 011901 CrossRef.
- S. P. Keshri and S. K. Pati, ACS Appl. Energy Mater., 2022, 5, 13590–13599 CrossRef CAS.
- C. Peng, G. Qin, L. Zhang, G. Zhang, C. Wang, Y. Yan, Y. Wang and M. Hu, J. Phys. D: Appl. Phys., 2018, 51, 315303 CrossRef.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef PubMed.
- Z. Gao, F. Tao and J. Ren, Nanoscale, 2018, 10, 12997–13003 RSC.
- X. Gu and R. Yang, Appl. Phys. Lett., 2014, 105, 131903 CrossRef.
- T. Nakashima and Y. Umakoshi, Philos. Mag. Lett., 1992, 66, 317–321 CrossRef.
- A. Shafique, A. Samad and Y.-H. Shin, Phys. Chem. Chem. Phys., 2017, 19, 20677–20683 RSC.
- C. Hu, L. Zhou, X. Hu, B. Lv and Z. Gao, Appl. Surf. Sci., 2023, 613, 156064 CrossRef.
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Hua
Tong
a,
Cheng-Wei
Wu
a,
Shi-Yi
Li
a,
Pin-Zhen
Jia
a,
Hua
Tong
a,
Cheng-Wei
Wu
a,
Shi-Yi
Li
a,
Pin-Zhen
Jia
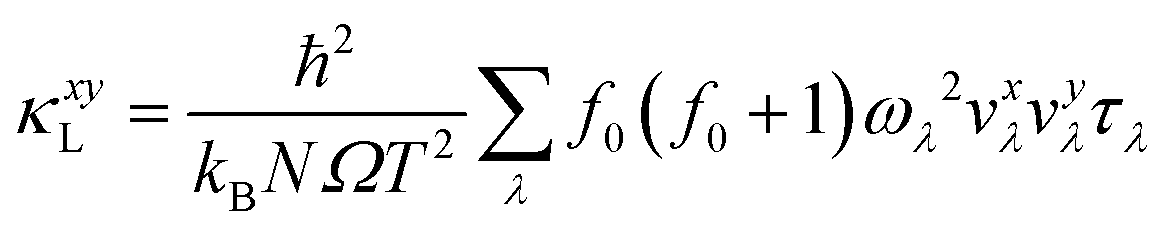
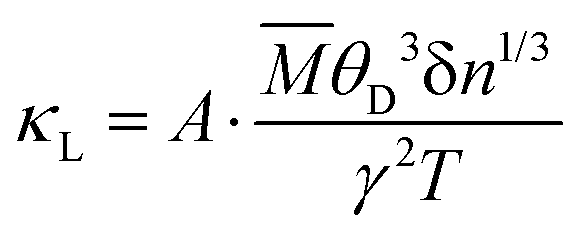
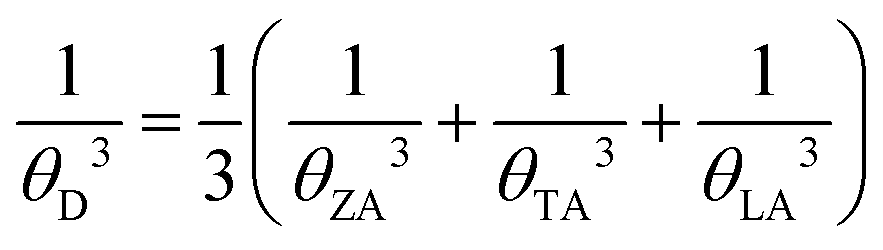
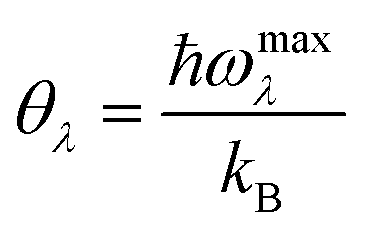
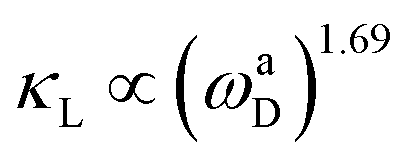 , where ωaD is the largest acoustic phonon frequency.71 For CFGM and BFGM, ωaD is 8.26 and 7.74 THz, respectively, which surpass the values observed in many other 2D materials. This suggests a potentially stronger acoustic phonon vibration, which bodes well for thermal transport and κL. As previously analyzed, FGMs demonstrate a high phonon group velocity and long phonon relaxation time, with relaxation times extending up to 103 ps. These values are comparable to those of well-known high-κL 2D materials such as MoS2 and WS2.72 This is primarily due to the lower amplitude of Grüneisen parameters and weaker anharmonicity. Additionally, CFGM and BFGM exhibit high Debye temperatures, which represent the threshold below which phonon modes become “frozen out”.73 Consequently, higher Debye temperatures imply that fewer phonons are activated at room temperature, leading to reduced phonon population and scattering rates.73–75
, where ωaD is the largest acoustic phonon frequency.71 For CFGM and BFGM, ωaD is 8.26 and 7.74 THz, respectively, which surpass the values observed in many other 2D materials. This suggests a potentially stronger acoustic phonon vibration, which bodes well for thermal transport and κL. As previously analyzed, FGMs demonstrate a high phonon group velocity and long phonon relaxation time, with relaxation times extending up to 103 ps. These values are comparable to those of well-known high-κL 2D materials such as MoS2 and WS2.72 This is primarily due to the lower amplitude of Grüneisen parameters and weaker anharmonicity. Additionally, CFGM and BFGM exhibit high Debye temperatures, which represent the threshold below which phonon modes become “frozen out”.73 Consequently, higher Debye temperatures imply that fewer phonons are activated at room temperature, leading to reduced phonon population and scattering rates.73–75