
Simulation of exciton spectra in disordered supramolecular polymers: exciton localization in cisoid indolenine squaraine hexamers†
David
Fischermeier
 a,
Arthur
Turkin
b,
Joshua
Selby
b,
Christoph
Lambert
bc and
Roland
Mitrić
*a
a,
Arthur
Turkin
b,
Joshua
Selby
b,
Christoph
Lambert
bc and
Roland
Mitrić
*a
aInstitut für Physikalische und Theoretische Chemie, Julius-Maximilians-Universität Würzburg, Emil-Fischer-Str. 42, 97074 Würzburg, Germany. E-mail: roland.mitric@uni-wuerzburg.de
bInstitut für Organische Chemie, Universitüt Würzburg, Am Hubland, D-97074 Würzburg, Germany. E-mail: christoph.lambert@uni-wuerzburg.de
cCenter for Nanosystems Chemistry, Universität Würzburg, Theodor-Boveri-Weg, D-97074 Würzburg, Germany
First published on 16th November 2023
Abstract
In order to understand the effects of disorder and defects in oligomers and polymers on the localization of excitons, we investigated the spectral properties of the squaraine B hexamer using long range corrected tight-binding TDDFT (lc-TDDFTB) and Frenkel-exciton model based calculations. Employing classical molecular dynamics, the cisoid indolenine squaraine hexamers helix was propagated in DCM and acetone to obtain ensembles of realistic structures, which naturally exhibit considerable disorder. The trajectories together with several model squaraine systems were studied to show the profound effects of disorder in the superstructure and disorder of the local monomer geometry on optical properties like absorption and exciton localization. We further compared lc-TDDFTB and exciton theory derived spectral data to related experimental data on absorption, exciton transfer and localization in squaraine polymers and oligomers.
1 Introduction
Conjugated polymer chains are often considered as highly promising systems for the development of optoelectronic devices and organic solar cells.1–9 For this purpose an understanding of fundamental processes like energy transfer and exciton localization is crucial.10–16 These traits are influenced and can thus be controlled by the structural diversity and flexibility exhibited by supramolecular conjugated polymers.While linear, elongated polymers promote a red-shift of the monomer's main absorption signal, which is typical for J-aggregates,17 folded structures like helices form H-aggregates and lead to a blue-shift. These structures, however, are idealized and limiting cases. In reality a whole spectrum of intermediate structural motifs between both extremes can be found. Aside from the absorption wavelength, these motifs also influence exciton localization and transfer within or between polymer chains.
The most common description of intermolecular or interchromophoric exciton transfer is provided by the Förster theory.9,18–20 According to the exciton model in an ideal and symmetric polymer, the exciton states should be delocalized over the whole system consisting of electronically coupled monomers. Polymers, however, rarely exhibit this kind of ideal geometry and therefore monomeric units will exhibit varying chemical environments due to conformational disorder as well as local structural differences due to vibrational motion. The chemical environment and local geometry of those monomers frequently leads to exciton localization as it was already shown for squaraine dimers21 and other systems.22–26
Since the Frenkel-exciton model views the coupling monomers as independent units, the relative orientation of the monomers fully determines the optical properties of the polymer within the dipole–dipole approximation. Earlier it was shown how this model can serve to accurately simulate optical properties like absorption bands and coherence length.27 While local geometry can easily be modelled as diagonal disorder and off-diagonal disorder can readily account for effects like the relative orientation of the monomers, effects arising from conjugation as well as deconjugation in polymers are distinctly harder to model and parametrize without explicit simulations. Structural motifs at the site of polymeric binding, which break conjugation to a considerable level can affect exciton diffusion and localization to a profound degree and are often referred to as “defects” or “disorders”.3,23,24,28–30
In former studies, it was suggested, that cisoid indolenine squaraine hexamers (SQB) polymers and oligomers (Fig. 1) adopt different superstructures, i.e. helical or unfolded and linear, resulting in different predominant aggregate types depending on the solvent, and thus exhibit widely different absorption behavior, that is, in e.g. acetone a helix with a hypsochromic H-type shift of the low energy absorption bands was observed while in dichloromethane (DCM) a loose random coil was observed with a J-type bathochromic absorption. These polymers were also found to feature multiple absorption sites, which facilitated a transfer of excitons between them during relaxation processes.31,32
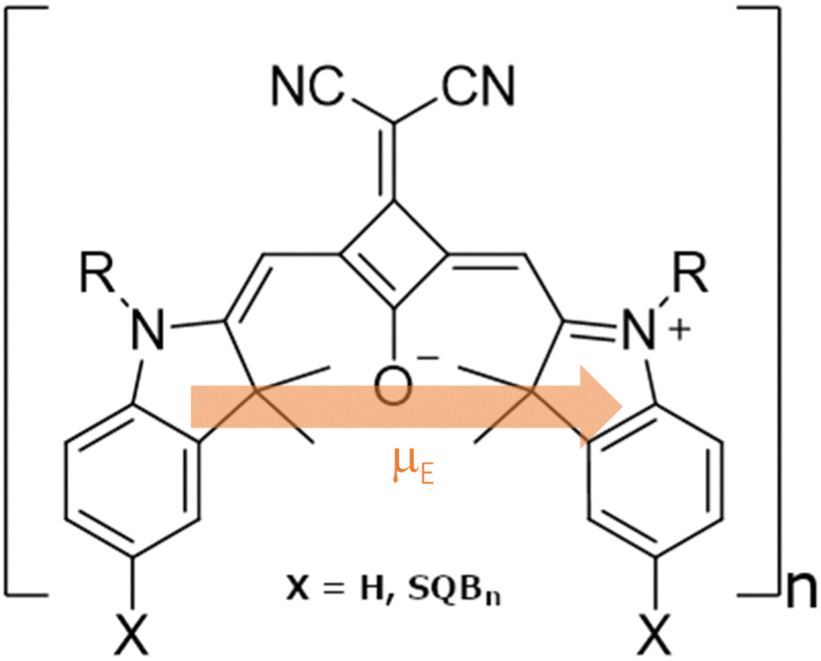 | ||
| Fig. 1 Structural formula of the SQB monomeric unit in SQBn oligomers and polymers. The electronic transition dipole moment (μE) is schematically included. | ||
In this study, we perform classical MD simulations on SQB hexamers to investigate the behavior of an SQB helix in acetone and dichloromethane (DCM). The resulting trajectories were used to generate structural ensembles of multiple geometrically distinct folding states for both solvents at varying timesteps, whereas each ensemble consists of 100 structures spaced 2 ps apart. The optical properties of those structures and several ideal model systems were then examined using lc-TD-DFTB33 and the exciton model. The obtained results were finally compared with experimental data to present a broad picture of the profound effects of static, structural defects and local geometries, which are not accounted for in the traditional exciton model, but lead to an array of interesting absorption properties in squaraine dyes.
2 Computational methods
2.1 Molecular dynamics
All classical MD calculations unless specified otherwise were performed using NAMD2.1234 as well as the latest CHARMM3635 parameters and topologies for solvents and aliphatic tails. These parameters also formed the basis on which the SQB parameters were built. A complete summary of used parameters can be found in the ESI.† The simulations were performed using a 1 fs timestep at 300 K, a pressure of 1 atm and within periodic boundary conditions using Langevin dynamics36 employing the Nosé–Hoover–Langevin piston method.37,38 The nonbonded pairlist distance was set to 14.0 Å with 12.0 Å cutoff distance and a switching distance of 10 Å.The trajectories were obtained using the following methodology: the actual propagation is preceded by an equilibration of the system. For this purpose the system consisting of the polymer and the solvent is first minimized for 5000 steps, then propagated for 2 ns with a fixed polymer within a flexible cell until the boundary box closely encompasses the solvent. Aside from enabling the flexible cell, the equilibration was done using the same options as all other classical MD calculations. This was followed by a 10 ns propagation.
2.2 Parameter development
The bonded parameters (bonds, angles, dihedrals) as well as Coulomb parameters (partial charges) required for the CHARMM36 forcefield employed in NAMD2.12 were obtained using the Force Field Tool Kit (FFTK) plugin39 built into vmd 2.09 software.40 The procedure was performed as described in the official tutorial and documentation. The initial van der Waals (VdW) parameters were derived from molecules similar to parts of SQB; indole for the 1,2-substituted double ring system and actone for the squaric acid, which are readily parametrized in the latest CGENFF36 distribution.41 The FFTK was used in conjunction with GAUSSIAN0942 for the required quantum chemical calculations. For these, DFT was used employing the CAM-B3LYP43 functional and the def2-SVP44,45 basis set. No implicit solvent model was used.The final VdW parameters were obtained in a Monte-Carlo-like fashion using NAMD2.12 employing simulation parameters identical to those specified for the classical MD calculations. The procedure is documented in the ESI.†
2.3 Linear absorption spectra
Linear absorption spectra were calculated using the lc-TDDFTB as implemented in the DFTbaby33 package, which has shown good results for other organic chromophore aggregates by avoiding erroneous charge transfer transitions due to its long-range correction. The lc-TDDFTB employed an active space of 24 occupied and 24 virtual orbitals, with which the first 6 excited singlet states were calculated. Vibrational modes were not taken into account. The spectra were averaged over an ensemble of 100 structures taken from a classical MD trajectory with a time difference of 2 ps between each structure. The spectra obtained for each single structure were broadened using Gaussian functions with a FWHM of 120 cm−1. For the comparison of different ensemble spectra, the intensities are normalized to the highest intensity signal of all spectra. In order to analyze the excitonic states, the fraction of transition density per monomer (FTDM) was calculated from the transition density matrix generated by DFTBaby in a fashion following earlier, similar studies:22,46,47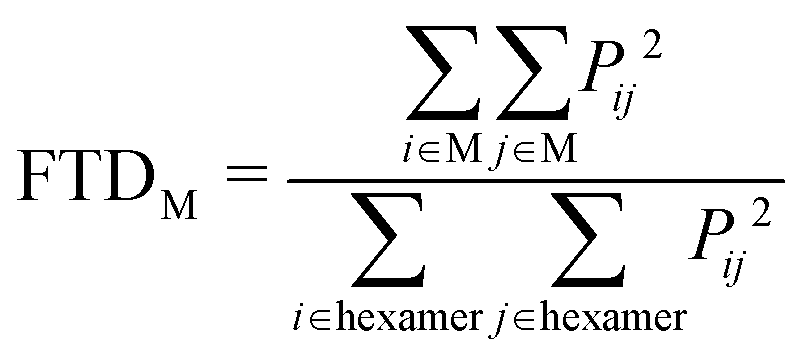 | (1) |
Here Pij denotes the matrix elements of the transition density matrix. Complementary to the full lc-TDDFTB simulations, the exciton model19 was used to simulate linear absorption spectra directly from the monomeric excitation energy calculated by Gaussian TD-DFT (unfortunately DFTBaby at this point does not provide transitions charges) calculations and transition charges representing the transition dipole moment for some selected systems. The eigenvalues were obtained from diagonalizing the exciton Hamiltonian, in which the diagonal elements represent the monomer's excitation energies and the off-diagonal elements represent the electronic coupling between two monomers. We note, that for the hexamer, one should use the total excited state energy of the hexamer, i.e. the excited state energy of the active monomer plus the ground state energy of all other monomers. Since the excited state energy of the active monomer is comprised of the ground state energy and the excitation energy and we assume all monomers to have the same ground state energy (of the optimized SQB monomer), we can ignore all ground state energies and only use the excitation energy.
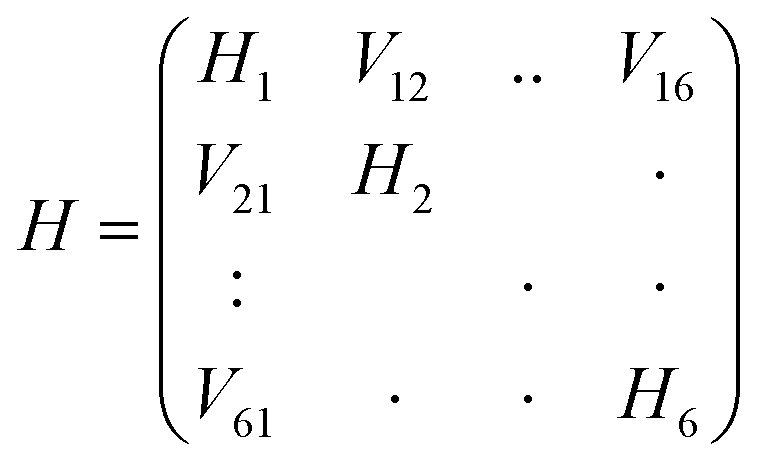 | (2) |
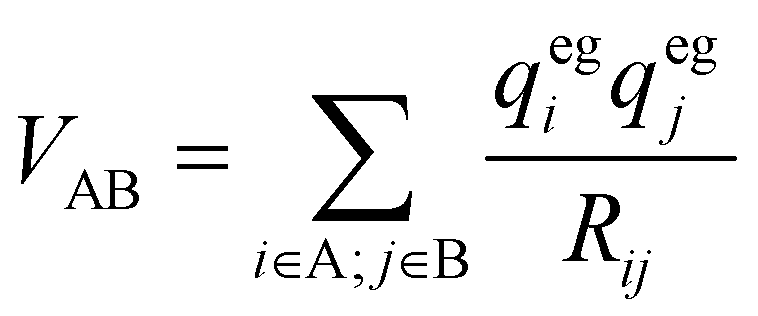 | (3) |
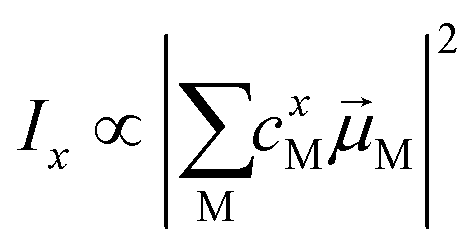 | (4) |
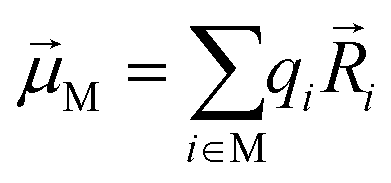 | (5) |
3 Results and discussion
Our MD simulations show that the helical structure of the SQB hexamer with an average sheet distance around 5 Å after equilibration remains folded in acetone over a time interval 10 ns, while it rapidly unfolds in DCM. After 10 ns both structures' total energy seems to have converged, which indicates an equilibrium for both simulations (see ESI†).3.1 Linear absorption spectra of the SQB hexamer in acetone
Since the helix remains stable in acetone, the change in the absorption spectrum is small over the course of 10 ns as seen in Fig. 2a. The main signal exhibits a minor red-shift while losing intensity within the first nanoseconds. In turn the signals below 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1 grow in as the helix opens up slightly. The spectrum does not change noticeably for the remaining simulation time, suggesting, that the helix has equilibrated.
500 cm−1 grow in as the helix opens up slightly. The spectrum does not change noticeably for the remaining simulation time, suggesting, that the helix has equilibrated.
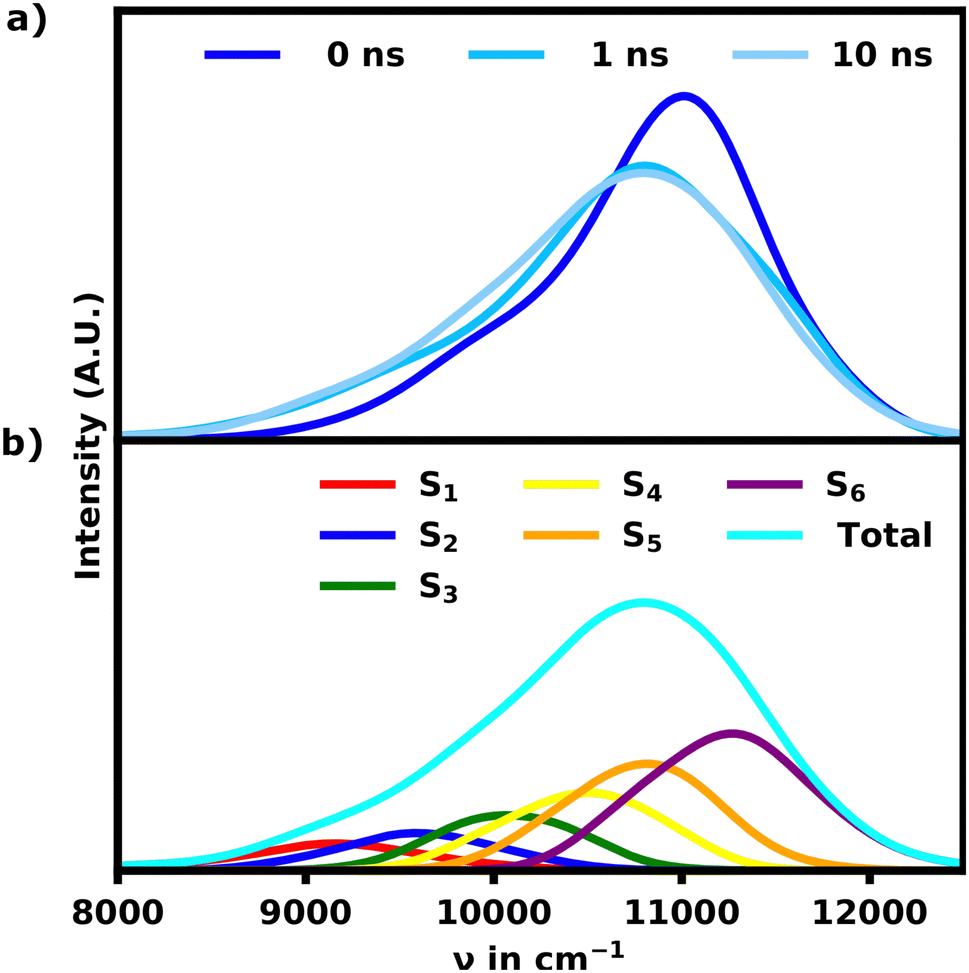 | ||
| Fig. 2 (a) Time evolution of the ensemble absorption spectra for the SQB helix in acetone. (b) Ensemble spectrum of the equilibrated helix after 10 ns split into the S1–6 transitions. | ||
The spectrum of the fully equilibrated helix at 10 ns shows a prominent main peak at 10800 cm−1, which is constituted by the most intense transitions, S5 and S6, but also a prominent shoulder (S1, S2) as seen in Fig. 2b. This shoulder is stretching as low as 9000 cm−1 and overlapping with the main absorption signal for the unfolded structures as shown in Fig. 3a, which shows a comparison of the unfolded and helical SQB6 spectrum. S5 and S6 transitions carrying the highest intensity is expected from a helix, since the H-aggregates formed by the neighbouring sheets result in a main signal blue-shifted to the monomeric HOMO–LUMO transition (10000 cm−1). The prominent red-shifted shoulder signals show, that the linearly bound monomers still promote some J-aggregate like behavior.
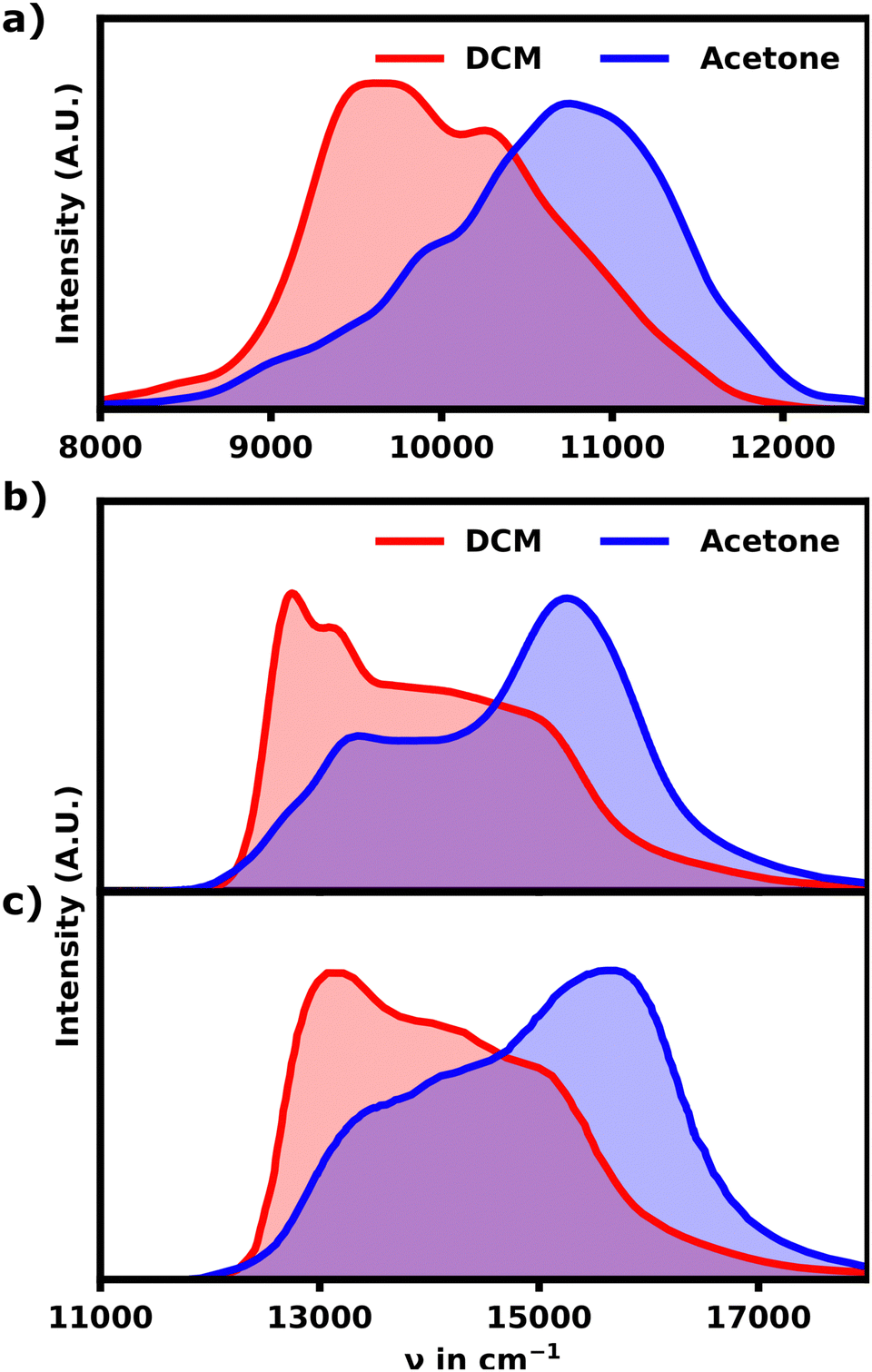 | ||
| Fig. 3 (a) Linear absorption spectra for the DCM and acetone SQB hexamer ensemble after 10 ns calculated with lc-TDDFTB. (b) Exprimental absorption spectra for the hexamers in DCM and acetone. (c) Exprimental absorption spectra for a mixture of higher polymers in DCM and acetone. | ||
The calculated spectrum differs from the experiment in two major ways. First the calculated spectrum exhibits an overall red-shift by roughly 4000 cm−1. Secondly the bandwidth of the signal is much more narrow for the calculated spectrum. Since lc-TDDFTB underestimates the transition dipole moment and transition energy for the monomer (experimental: 10.1 D, lc-TDDFTB: 7.5 D), the width of the band is understated as well. Qualitative, however, the comparison with the experimental spectra for the hexamer and polymer31 shown in Fig. 3b and c is quite favourable.
While the calculated spectrum does reproduce the main signal and relative shoulder positions quite well, the latter appears less pronounced compared to the experiment shown in Fig. 3b and c. From the experimental linear absorption spectrum,31 it appears, that the helix should exhibit a more intense shoulder signal around 9500 cm−1, which constitutes the main absorption region for an unfolded hexamer. This could indicate that the polymer should either exhibit a somewhat linear, loose section and/or higher pitch distances in general. Both of which would weaken the H-aggregate coupling and promote higher absorption intensity at lower wavenumbers relative to the main absorption signal. Overall the agreement between the experimental and theoretical data is still very satisfactory proving that the hexamer in acetone indeed adopts a helical conformation.
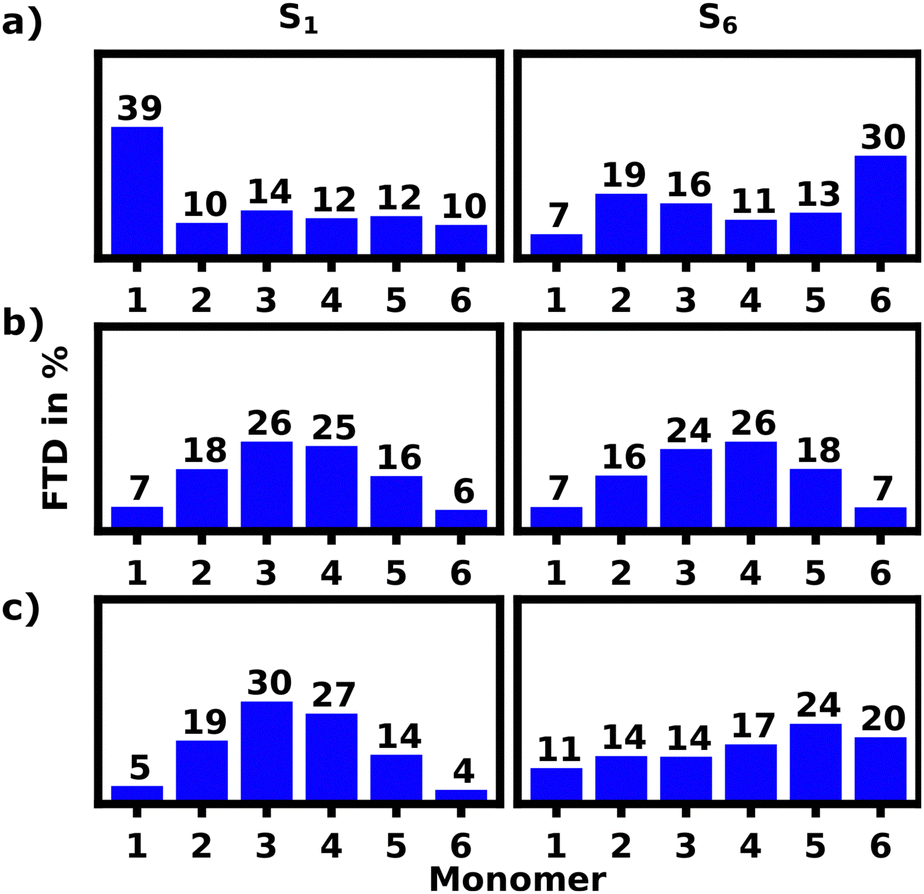
For the S1 transition a clear pattern of exciton localization in the helix is observable with 47% of total FTD on M1 and 18% on M4, giving the two stacked monomers a combined 65% of the total FTD. The monomer with the lowest overall transition density is M5, which is stacked on top of M2 exhibiting very little transition density as well.
Fig. 4a illustrates the H-aggregate pairs and highlights their approximate pitch distances. The pair with the lowest distance and thus strongest coupling, M2 and M5, exhibits the least FTD for the S1 transition. Meanwhile the monomer pair with the lowest coupling, M1 and M4, exhibits by far the highest FTD regarding the S1 transition, especially M1.
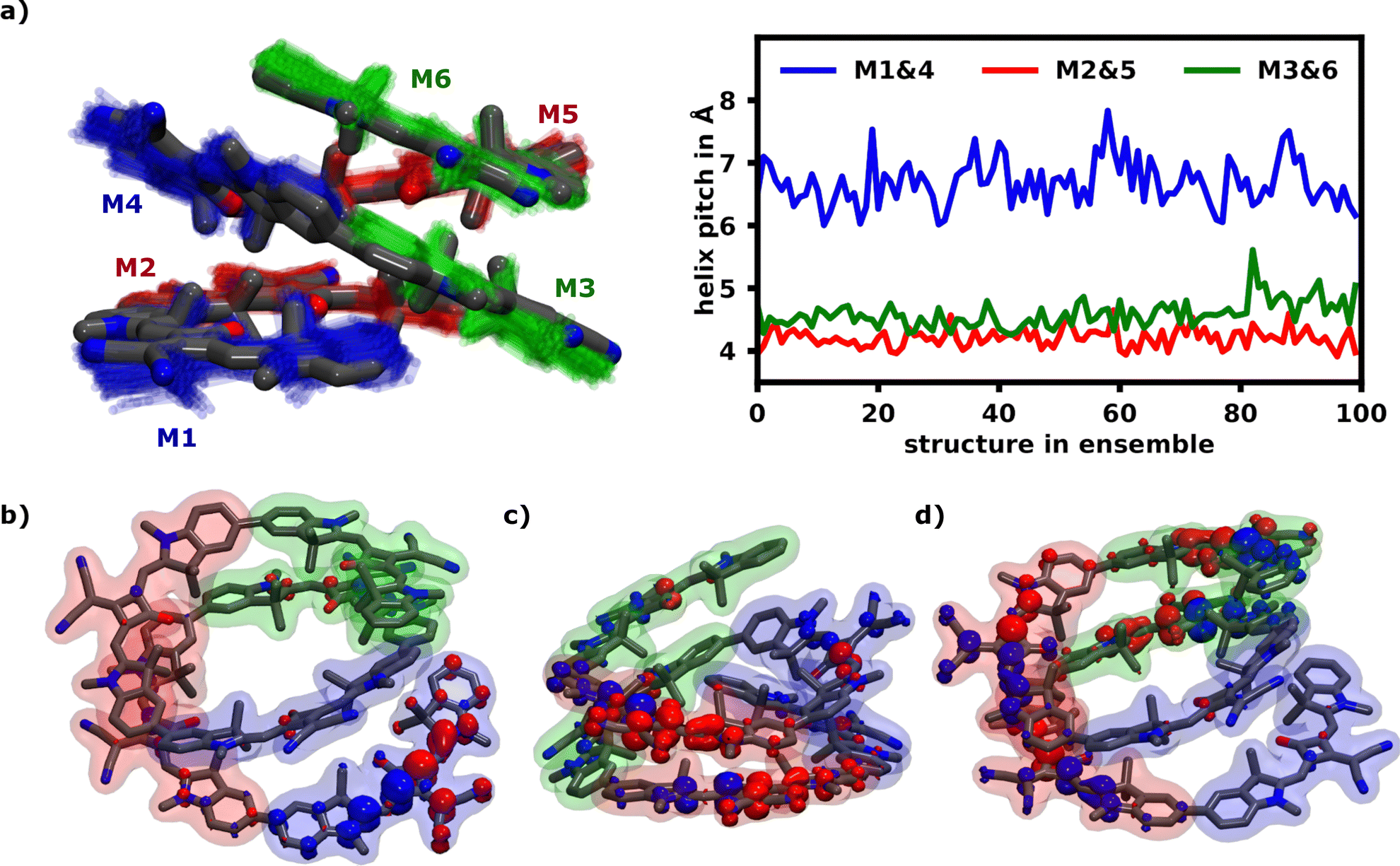 | ||
| Fig. 4 (a) Superimposed structures of the 100 molecule ensemble at 10 ns obtained from the equilibrated helix in acetone (left) and the evolution of approximate pitch distances over all structures (right). The three pairs forming H-aggregates are the bottom end M1 and M4 (blue) forming the weakest aggregate with an average pitch distance of 7 Å. M3 and the top end M6 (green) form the second pair and M2 and M5 (red) present the most strongly coupling H-aggregate with an average pitch distance around 4.3 Å. (b)–(d) Common transition density patterns of the S1 and S6 transition as localization indicator. (b) shows an almost complete localization on M1 for an S1 transition, while (c) shows S6 delocalization over the H-aggretated M2 and M5 and (d) shows strong S6 delocalization over the two H-aggregates M2/M5 and M3/M6. | ||
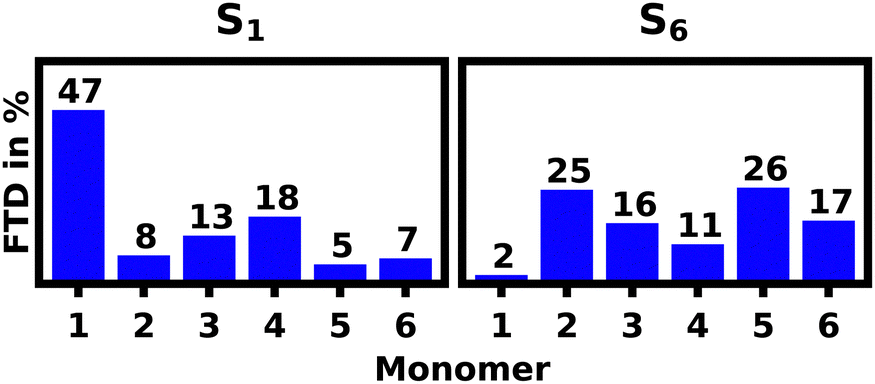 | ||
| Fig. 5 Fractions of transition density (FTD) per monomer over the ensemble of 100 structures for the electronic S1 and S6 transitions of the equilibrated SQB hexamer helix in acetone at 10 ns. | ||
Contrasting this behavior, M2 and M5 exhibit the highest FTD for the S6 transition, while very little FTD is observed for S1.
Three of the most common localization schemes are shown in Fig. 4b–d. The localization schemes exhibiting delocalization over two or more monomers show a clear parallel orientation of transition dipole moments within H-aggregates. These localization patterns demonstrate, how geometrical differences in the helix yield sites with widely different absorption behaviors. For one, as expected, the helix exhibits strong H-aggregate type coupling monomers, which provide a strong absorption signal at higher wavenumbers, which also tend to delocalization due to their strong coupling (Fig. 4c and d). Secondly more loose parts of the helix show absorption signals at wavenumbers lower than the monomeric absorption, which exhibits exciton localization mostly on one monomer (Fig. 4b). Qualitatively this aligns well with the exciton model, where the highest energy excitons should be localized on the most tightly stacked monomers forming an H-aggregate. We have also done some calculations using the exciton model shown in the ESI.†
3.2 Linear absorption spectra of the unfolded SQB hexamer in DCM
In DCM the MD simulations show an unfolding of the helix. In accordance the intensity of the main signal is gradually diminishing and red-shifting as shown in Fig. 6a and d–g. This signal shift continues for 10 ns, albeit at a drastically reduced rate as the helix is already mostly unfolded during the first nanosecond.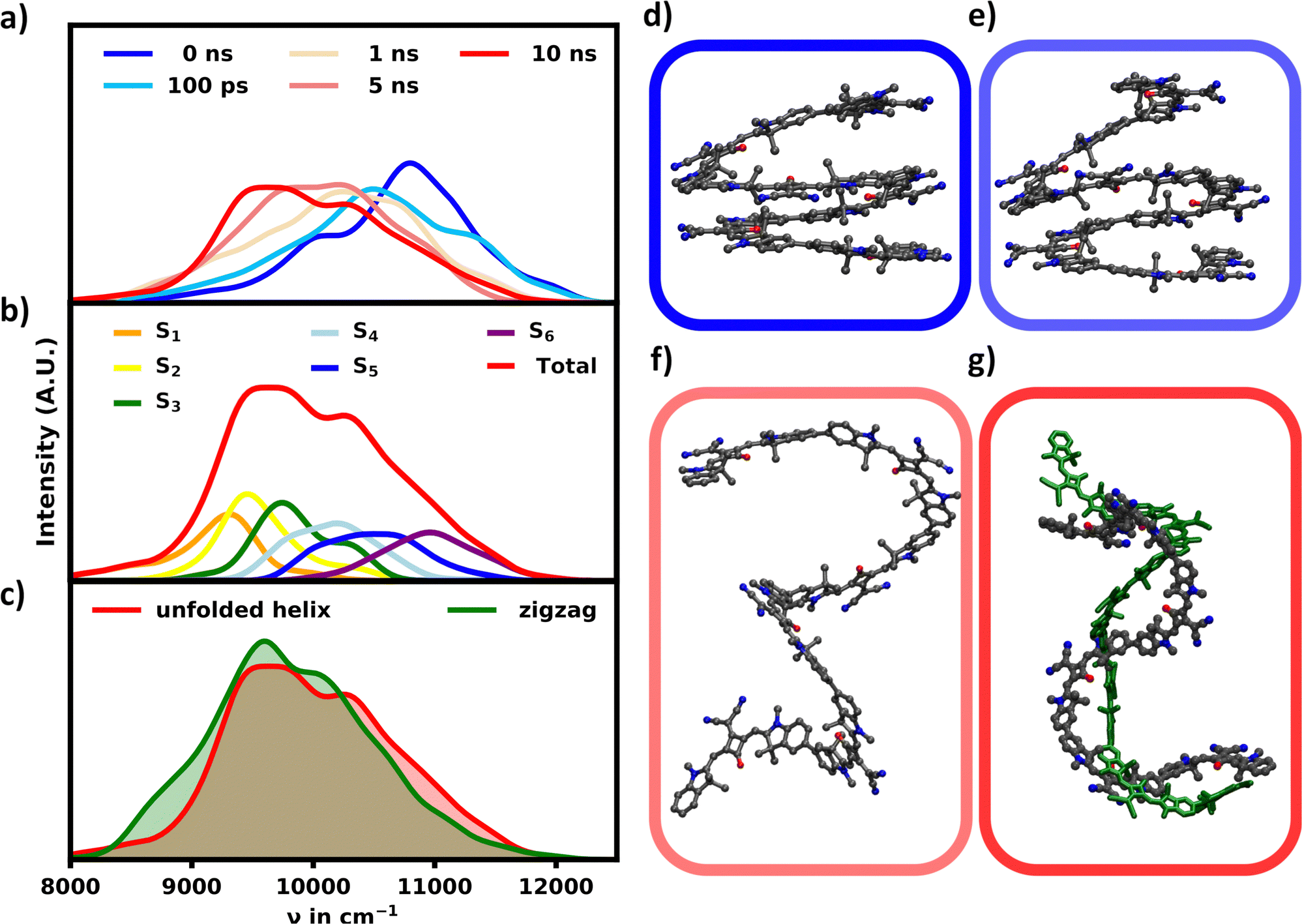 | ||
| Fig. 6 (a) Evolution of the unfolding helix's ensemble absorption spectrum over 10 ns. (b) Absorption spectrum of the unfolded hexamer at 10 ns split into contributions for the S1–6 transitions. (c) Comparison of the equilibrated unfolded helix and equilibrated linear hexamer absorption spectrum. (d)–(f) Unfolded helix structures at 0 ns, 100 ps and 1 ns. (g) Unfolded helix at 10 ns and equilibrated linear hexamer (green). | ||
At 10 ns the spectrum of the unfolded structure starting from the helix (further referred to as “unfolded”) closely resembles the spectrum obtained 10 ns after starting from the fully linear zigzag configuration in DCM (further referred to as “linear”). Thus it is safe to assume that the unfolding process is finished and an equilibrium is reached as the spectra from both simulations converge.
Both simulations also converge regarding the polymer's structure to some extent, as the helix becomes unfolded and more extended, the “linear” structure becomes more twisted in turn. While a difference in structural configuration still remains, the absorption behavior appears nearly identical, which indicates that the amount of conformational space, which is occupied by these unordered, non-helical structures is very large compared to the more ordered and blue-shifted helical structures. The observed change in the absorption spectrum over the course of the unfolding process from the helix to the “unfolded” structure is expected for two reasons. As the helix unfolds, the distance between parallel stacked monomers increases and the polymer loses its H-aggregate character. The transition from the helix to this “unfolded” structure thus decreases the intensity in the blue-shifted regions, while increasing the intensity in the red-shifted regions.
Aside from the expected prominent main signal at low wavenumbers, the unfolded hexamer's cisoid bonded monomers still yield a rather prominent shoulder signal at the main absorption region of the helix. In order to understand the origin of this signal we performed simulations on the SQB dimer to obtain an absorption spectrum within the exciton model as well as lc-TDDFTB, which are illustrated in Fig. 7.
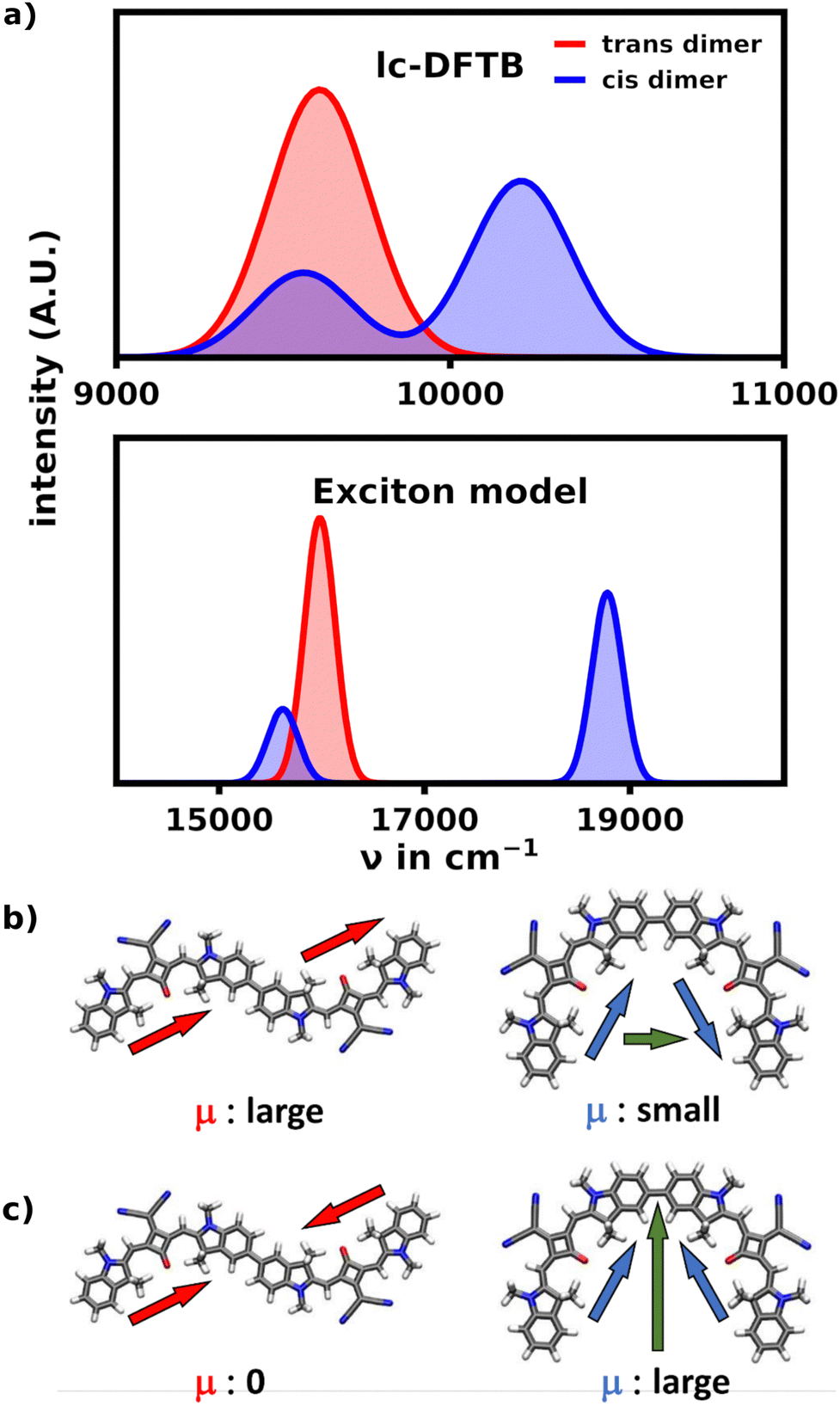 | ||
| Fig. 7 Comparison of the cis and trans dimer. (a) Comparison of the spectrum of the SQB transoid and cisoid dimer calculated with lc-TDDFTB and the exciton model respectively. The transoid dimer only exhibits one signal at low wavenumbers, while the cisoid dimer exhibits a large signal at higher wavenumbers and a smaller signal at the position of the transoid dimer signal. (b) The combination of monomeric transition dipole moments. In the S1 transition the dipole moments are oriented head-to-tail due to the geometric structure, forming a large total transition dipole moment. For the S2 transition both cancel. (c) Contrasting the transoid dimer the S1 dipole orientation only produces a small dipole moment, while the S2 transition yields a larger one. This behavior causes the high wavenumber shoulders of the unfolded hexamer. | ||
The exciton model as well as TD-DFTB describe these ideal cases very well. While the exciton spectrum is heavily blue-shifted due to the blue-shifted monomer energy obtained from TD-DFT (17158 cm−1), they fully align qualitatively in showing the aggregation effects of both dimeric orientations. They show, that in the transoid configuration of the dimer, the S1 transition is the most intense, while the the S2 transition is not visible in the spectrum. Within the exciton model, this is well described by observing the orientation of the monomeric dipole moments. In the trans configuration the dipoles are oriented head-to-tail and thus parallel for the S1 transition, yielding a large total transition dipole moment, while the antiparallel, head-to-head oriented dipole moments cancel for the S2 transition. The cisoid configuration contrasts this behavior, as the head-to-head orientation yields the biggest total transition dipole moment, since the dipoles are no longer (anti-)parallel.
Equilibrated after 10 ns the “unfolded” structure features the S2 transition with the highest intensity, while the other transitions are also quite intense. This is unlike a perfect J-aggregate, which would warrant a most intense S1 transition, with little intensity in the other transitions.
The calculated spectrum shows a lack of intensity (about 30% less) in the region around 11000 cm−1 compared to the experimental spectrum. This behavior is expected, when vibrational transitions are excluded, but could also result from some statistical bias due to the relatively small size in the structural sample. Another explanation is the lack of other possible structural motifs, e.g. the number and/or angle of cisoid conformers. The missing intensity at 11000 cm−1 is even more pronounced in the “linear” ensemble, exhibiting more and more pronounced transoid conformers. In summary though, the overall very good agreement with the experimental spectra suggests, that the “unfolded” hexamer's structural motifs are likely a good average representation of the motifs found in larger polymers unfolded in DCM. While this behavior does not rule out the existence of helical moieties in higher polymers in DCM, it shows the capability of unordered, non-helical structures to exhibit considerable absorption at wavenumbers blueshifted from the monomer typically associated with H-aggregates.
Fig. 8a shows the S1 and S6 localization patterns of the unfolded hexamer in DCM calculated with lc-TDDFTB. 39% of all S1 transition density is localized on M1, while the remaining FTD is evenly split among the other monomers. The S2–5 transitions strongly tend towards delocalization and are not discussed in detail, but are presented in the ESI.† Finally the S6 transitions seem to favour localization on M6 with 30% of total FTD.
 | ||
| Fig. 8 FTD of the S1/6 transition per monomer of the unfolded hexamer in DCM at 10 ns: (a) original ensemble calculated with lc-TDDFTB. (b) Original ensemble calculated with the exciton model. (c) Reconstructed ensemble calculated with lc-TDDFTB. | ||
This strong contrast in absorption behavior of M1 and M6 regarding the S1 and S6 transition can be caused by various effects arising from a difference in geometric features and chemical environments of the two monomers. In the following we will discuss three major factors: (1) the Coulomb coupling, which depends on the relative orientation of the monomers, (2) defects and disorders within the polymeric bond, (3) local geometries affecting the absorption energies. These effects will be discussed in detail, while focusing on the S1 and S6 state and mainly the M1 and M6 monomer.
First, we employed the exciton model to re-calculate the FTD, since this model should at least qualitatively reproduce this localization pattern, if the Coulomb coupling was indeed the determining factor for this localization pattern. Fig. 8b shows, that M1 and M6 behave nearly identical for the S1 and S6 transitions. This contradicts the data calculated by lc-TDDFTB and indicates, that the FTD distribution pattern found using lc-TDDFTB is not a result of pure Coulomb coupling. This is not surprising as the unfolded hexamer still appears to be rather symmetric and thus the Coulomb splitting should not differ much for M1 and M6.
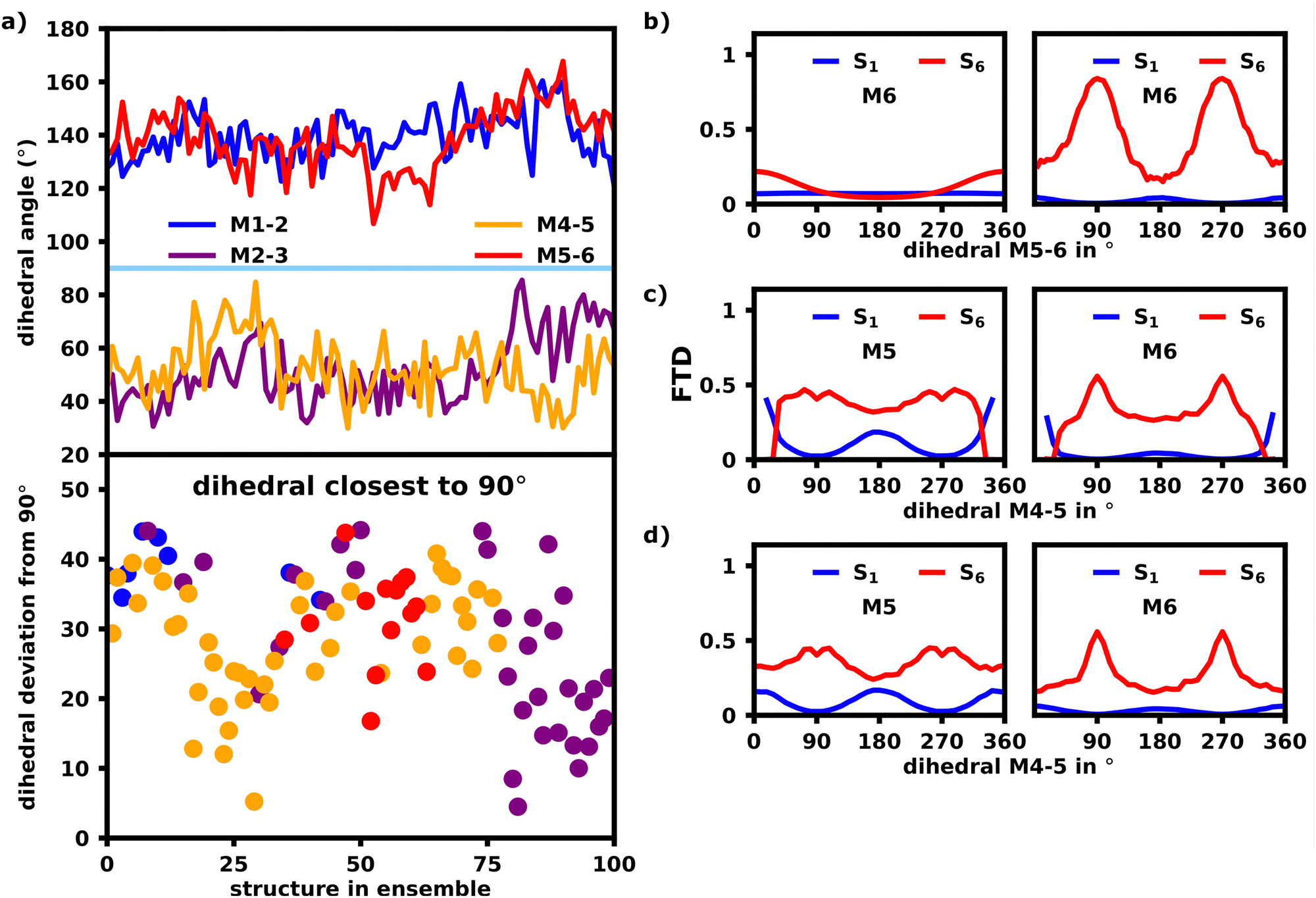
To assess the effects of possible defects on exciton localization, we inspected the dihedral angles of the polymer bonds in Fig. 9a (top) to find, that on average the angles opposite to each other, i.e. M1–M2, M5–M6 and M2–M3, M4–M5, are very similar, but have sizable deviations for each individual structure.
 | ||
| Fig. 9 (a) The top graph shows the polymeric bond dihedrals (M1–M2, M2–M3, etc.) for the ensemble of the unfolded helix at 10 ns. The bottom graph marks the the dihedral, which is the closest to a perpendicular orientation, and its deviation from 90°. At the start the M4–M5 dihedral appears to be the most perpendicular, while the M2–M3 angle appears to be the most perpendicular at the end of the ensemble. (b) S6 FTD dependence for M6 on the M5–M6 dihedral angle calculated with the exciton model (left) and lc-TDDFTB (right). (c) and (d) S6 FTD dependence for M5 and M6 on the M4–M5 dihedral angle calculated with M5/M6 oriented trans (c) and cis (d) with lc-TD-DFTB. | ||
Fig. 9b shows the dependency of the M6 FTD for the S6 transition on the M5–M6 dihedral in an ideal, model hexamer. In this model, the angle between M5 and M6 was rotated by increments of 5° and the FTD distribution for each structure was calculated using lc-TDDFTB (right) as well as the exciton model (left). All other dihedral angles were left fixed at 180° forming a perfect transoid bond.
The exciton model shows, that for an angle of 180°, which constitutes an ideal J-aggregate with trans oriented monomer units, M6 exhibits the lowest FTD, whereas at an angle of 0° and 360°, M6 exhibits an FTD maximum, as it is now cisoid oriented to M5. Since cisoid orientations facilitate higher intensities for blue-shifted states as already shown in Fig. 7, this transition would naturally occur on the cisoid oriented monomers within the exciton model.
The lc-TDDFTB data in Fig. 9b contrasts this behavior heavily. While an angle of 0°, still results in a higher FTD on M6 compared to M1 than an angle of 180°, the maximum FTD on M6 is now established for an angle of 90/270°, which constitutes the point of maximum deconjugation and thus decoupling of the monomers, which is in agreement with earlier literature on deconjugated polymers.23,24,30
Fig. 9c and d also depict the same FTD to angle relation, although this time the dihedral of M4 and M5 is rotated. The dihedral M5–M6 was set to 0° and 180° respectively, while all other dihedrals were left at an angle of 180°. Rotating the M4–M5 dihedral towards 90/270° results in a large FTD on M6, while little FTD on M1 is found for the S6 transition, which shows, that M4–M5 has a very similar effect on exciton localization as the M5–M6 dihedral. If the distribution of FTD is in fact a result of the dihedrals, we would expect, that for the structures within the ensemble, the dihedral M4–M5 or M5–M6 should be closest to 90°. We note, that the interplay of all dihedrals is of course an important factor, but to combat the complexity we will only focus on the most deconjugated polymer bond and assume, that the polymer is chromophorically split at the bond with the most perpendicular angle.
Fig. 9a (bottom) shows which dihedral has the lowest deviation from 90° per structure. We find, that indeed either M4–M5 or M5–M6 is closest to 90° for the first 80 structures, after which M2–M3 exhibits the lowest deviation from 90°. Looking at Fig. 10a, M1 indeed exhibits more S6 FTD in the last 20–25 structures, than the first 75–80, which is what we would expect from the dihedral angles. An opposite, but less clear, trend can be observed for S6 and M6, which shows slightly less FTD in the later structures. To exclude the influence of differing oligomeric bond-lengths as well as the difference in monomeric geometries and resulting excitation energies, we reconstructed the structures in the ensemble from identical optimized monomers. For each structure the five oligomeric bond lengths and dihedrals were measured. Those structures were then reconstructed by connecting six identical, geometrically optimized monomers with the average bondlength and dihedrals of the original structure. The exciton localization within these reconstructed, idealized structures should only be affected by direct orbital coupling as well as coulomb coupling. The original and reconstructed ensemble are structurally very close with an rmsd of 2.06 Å (further referred to as “reconstructed”), as such the coulomb coupling should be nearly identical to the original ensemble.
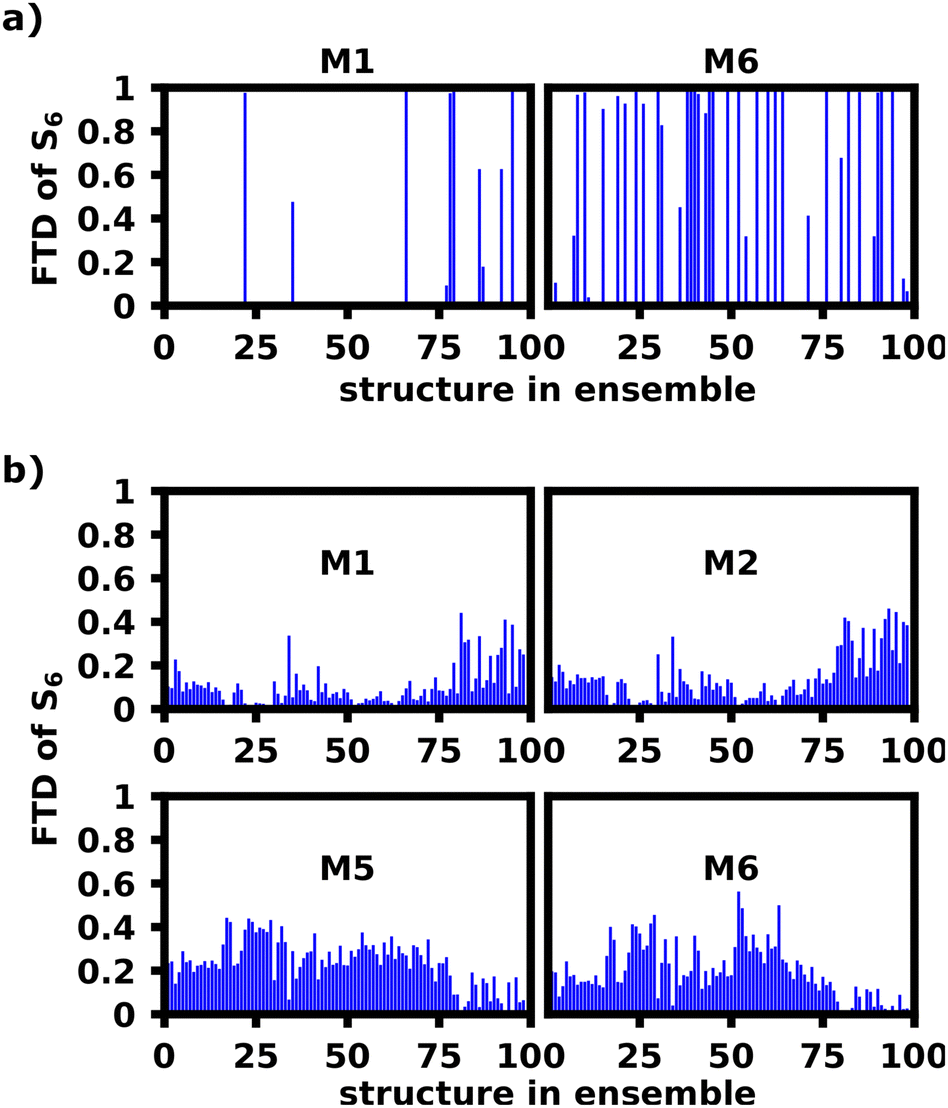 | ||
| Fig. 10 FTD of the S6 transition for M1 and M6 of the unfolded hexamer in DCM at 10 ns calculated with lc-TDDFTB for the original ensemble (a) and reconstructed ensemble (b). | ||
Fig. 8c shows the FTD distribution for the reconstructed ensemble, which exhibits a similar, but less extreme trend for the S6 transition compared to the original ensemble (a). Fig. 10b shows the S6 FTD per monomer and structure, which for the first 75–80 structures shows, that M5 and M6 exhibit a considerably higher FTD than for later structures. The opposite behavior can be seen for M1 and M2, which was expected, since the M2–M3 dihedral is the most perpendicular angle for the last 20 structures, thus decoupling M1 and M2 from the rest of the polymer considerably more than M5 and M6. We also see the highest S6 FTD on M6 between structure 50 and 60, which coincides nicely with the region in which M5–M6 is most perpendicular.
This shows, that the behavior of the dihedrals shown in Fig. 9a can convincingly explain the tendency of the S6 transitions to localize on M6 for the given structures.
Overall, however, there are also strong contrasts between the FTD distribution of the original and reconstructed ensemble. In general the FTD distribution is far more smoothed out and transitions never occur on only one monomer within the reconstructed ensemble, whereas the original had a very high rate of transitions localizing on a single monomer. Secondly, Fig. 9b and c show S1 localization patterns very similar to the exciton model, which are in contradiction to the experiments and the original ensemble calculations in Fig. 9a showing a strong preference for S1.
The monomeric geometries and thus excitation energies constitute one difference for both ensembles and thus we calculated the transition energies for all monomers independently to study the effect of fluctuating monomer geometries on transition energies. For this, the polymer was split at the oligomeric bonds and the monomers were capped with hydrogens, after which their S1 excited state energy and excitation energy was calculated separately using lc-TDDFTB. Analyzing the monomeric transitions in Fig. 11, we see, that the capping monomers M1 and M6 exhibit the lowest excited state energy (and ground state energies). These monomers experience the least strain since they are the most flexible units in the chain. The excitation energies for all monomers are more similar, which should be the governing factor leading to a stabilization of the S1 exciton on the monomers with the lowest excitation energies, i.e. M4 in this case followed by M1, M3 and M6.
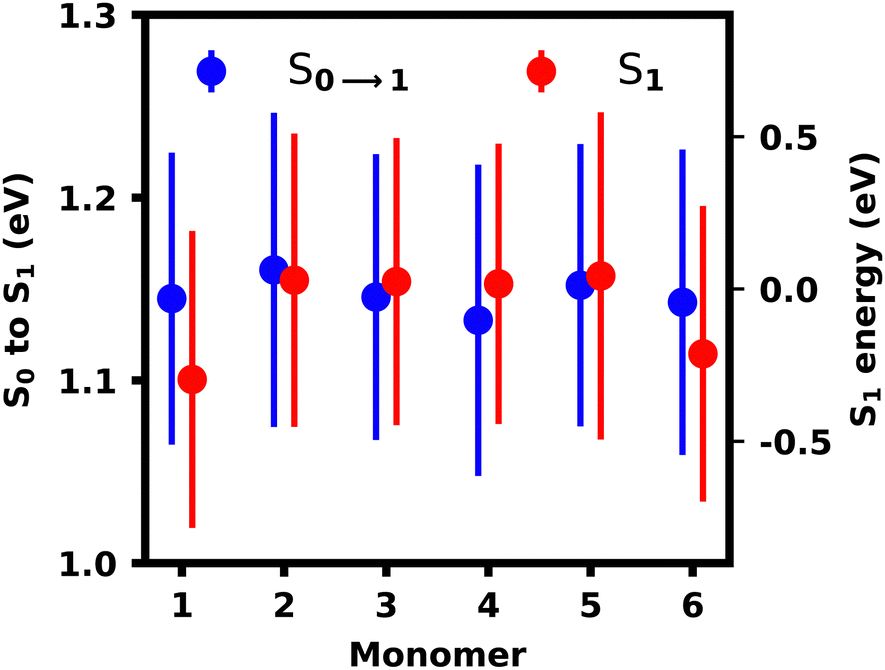 | ||
| Fig. 11 Monomer excitation and excited state energies with root-mean-squared deviation indicated as bars. | ||
Thus we can conclude, that the S1 localization pattern does not purely result from the monomer's geometry affecting the excitation and excited state energy. By sequestering the monomers, we also exclude any sterical or electronic effects, which seem to be governing the exciton localization observed in the lc-TDDFTB calculations either by lowering the excitation energy of M1 or increasing the excitation energies of other monomers.
In the reconstructed ensemble, however, all monomers possess the same transition energies and thus the stabilization of the inner monomers M2–M5 by coupling provides the energetically most stable site, as it was also seen in the exciton model simulations. As the exciton energy of M6 is still lower than for M2 and M5, S6 transitions should not occur on M6 based in monomeric excited state energies alone. M2–M5, however, are better stabilized due to similar monomeric energies and more favourable dihedrals and thus localization of the S6 transition on M6 is favoured, as discussed previously.
In the previous section we showed, that the S1 and S6 transitions tend to localize on opposite parts of the molecule. From these localization sites, possible exciton transfer pathways during internal conversion can be extrapolated. Examining the unfolded structure, it stands to reason, that excitation at the helical structure's main absorption signal around 11000 cm−1 would excite the molecule into the S6 state, which shows a strong localization at one end of the molecule (M6). From the localization pattern of the lowest excited state S1, it appears, that after relaxing to the S1 state, the exciton would be most likely localized at the other end of the polymer (M1). This is in great agreement with experimental transient pump–probe and 2D experiments published previously31 again highlighting the possibility of multiple differently behaving absorption sites in the disordered unfolded structure. For a larger polymer we would expect the behavior of the S6 transition to be rather similar to the hexamer, as high energy transitions tend to occur on the shorter and more isolated parts of a linear polymer chain. Though it stands to reason, that in a large polymer, lower energy excitons would find more conjugated and stable sets of monomers than in a hexamer and behave more similar to what was observed for the reconstructed ensemble.
The helical structure also exhibits a strong tendency towards exciton localization on M1 for S1 transitions. S6 transition often occur on the pair of M2 and M5, but most notably never solely on M1. Excitation of the S6 state should predominantly occur on the M2 and M5 H-aggregate pair and relax to the loose side, presumably M1. This is in so far in agreement with the experiment, in that an exciton transfer from a helical to a more linear (in our case loosely stacked part of the helix) appears possible.
4 Conclusion
We were able to develop parameters for the CHARMM36 force field, which can be applied to study the superstructures of squaraine dye oligomers and polymers in acetone and DCM. The equilibrated ensembles of helical and unfolded structure were used to simulate linear absorption spectra and analyze exciton localization sites. Contrasting earlier theoretical studies, which were based on highly ordered and ideal structures, as well as exciton model simulations presented in this work, our lc-TDDFTB simulations fully account for the disorder effects and while the spectra exhibited a noticeable red-shift, we were able to reproduce not only the main signal's absorption behavior, but also the broad shoulder signals qualitatively. The helix showed two absorption sites, which could clearly be distinguished by their helical pitch. On the loosely stacked part of the helix, which is made up by the M1 and M4 monomeric units, the high average pitch provided a weak H-aggregate coupling leading to stronger absorption signals for low wavenumbers, while the pairs M2 and M5 as well as M3 and M6 provided the strong main absorption signal at higher wavenumbers.For the unfolded structure in DCM the considerable shoulder in the region of the helix's main absorption peak were found to be facilitated by cisoid oriented polymer bonds, which provide a strong absorption region at higher wavenumbers than the trans oriented polymers found in the ideal, linear J-aggregate.
We further provided evidence linking the exciton localization patterns of the high-energy transitions within the unfolded hexamer to a deconjugation of the π-system. These localization patterns compare favourably with earlier experimental studies in terms of possible exciton transfer pathways,31 although we were not able to fully explain the complex interplay of structural motifs and monomeric geometrical distortions for the low energy transitions of the unfolded hexamer in DCM. Yet we demonstrate, that by combining classical MD simulations with quantum mechanical modelling of the absorption spectrum the spectral features and the exciton localization patterns can be revealed in squaraine dye polymers. We showed the importance and interplay of the local monomer geometry, the relative monomer orientation and defects in the conjugated π-system, which are not accounted for by the Frenkel-exciton model.
Conflicts of interest
There are no conflicts to declare.References
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef PubMed.
- G. D. Scholes, Annu. Rev. Phys. Chem., 2003, 54, 57–87 CrossRef CAS PubMed.
- I. Hwang and G. D. Scholes, Chem. Mater., 2011, 23, 610–620 CrossRef CAS.
- E. Hennebicq, G. Pourtois, G. D. Scholes, L. M. Herz, D. M. Russell, C. Silva, S. Setayesh, A. C. Grimsdale, K. Müllen, J.-L. Brédas and D. Beljonne, J. Am. Chem. Soc., 2005, 127, 4744–4762 CrossRef CAS PubMed.
- D. Beljonne, G. Pourtois, C. Silva, E. Hennebicq, L. M. Herz, R. H. Friend, G. D. Scholes, S. Setayesh, K. Mullen and J. L. Bredas, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10982–10987 CrossRef CAS PubMed.
- K. Becker and J. M. Lupton, J. Am. Chem. Soc., 2006, 128, 6468–6479 CrossRef CAS PubMed.
- N. Banerji, J. Mater. Chem. C, 2013, 1, 3052 RSC.
- L. D. Siebbeles and F. C. Grozema, Charge and exciton transport through molecular wires, John Wiley & Sons, 2011 Search PubMed.
- F. Laquai, Y.-S. Park, J.-J. Kim and T. Basché, Macromol. Rapid Commun., 2009, 30, 1203–1231 CrossRef CAS PubMed.
- P. Parkinson, C. Müller, N. Stingelin, M. B. Johnston and L. M. Herz, J. Phys. Chem. Lett., 2010, 1, 2788–2792 CrossRef CAS.
- M. M.-L. Grage, P. W. Wood, A. Ruseckas, T. Pullerits, W. Mitchell, P. L. Burn, I. D. W. Samuel and V. Sundström, J. Chem. Phys., 2003, 118, 7644 CrossRef CAS.
- M.-L. Grage, T. Pullerits, A. Ruseckas, M. Theander, O. Inganäs and V. Sundström, Chem. Phys. Lett., 2001, 339, 96–102 CrossRef CAS.
- T. E. Dykstra, E. Hennebicq, D. Beljonne, J. Gierschner, G. Claudio, E. R. Bittner, J. Knoester and G. D. Scholes, J. Phys. Chem. B, 2009, 113, 656–667 CrossRef CAS PubMed.
- B. J. Schwartz, Nat. Mater., 2008, 7, 427–428 CrossRef CAS PubMed.
- K. Becker, M. Fritzsche, S. Höger and J. M. Lupton, J. Phys. Chem. B, 2008, 112, 4849–4853 CrossRef CAS PubMed.
- M. R. Talipov, A. Boddeda, Q. K. Timerghazin and R. Rathore, J. Phys. Chem. C, 2014, 118, 21400–21408 CrossRef CAS PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- T. Förster, Die Naturwiss., 1946, 33, 166–175 CrossRef.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- M. I. S. Röhr, H. Marciniak, J. Hoche, M. H. Schreck, H. Ceymann, R. Mitric and C. Lambert, J. Phys. Chem. C, 2018, 122, 8082–8093 CrossRef.
- E. Titov, A. Humeniuk and R. Mitrić, Phys. Chem. Chem. Phys., 2018, 20, 25995–26007 RSC.
- T. Nelson, S. Fernandez-Alberti, A. E. Roitberg and S. Tretiak, Phys. Chem. Chem. Phys., 2013, 15, 9245 RSC.
- B. Van Averbeke and D. Beljonne, J. Phys. Chem. A, 2009, 113, 2677–2682 CrossRef CAS PubMed.
- S. Heun, R. F. Mahrt, A. Greiner, U. Lemmer, H. Bassler, D. A. Halliday, D. D. C. Bradley, P. L. Burn and A. B. Holmes, J. Phys.: Condens. Matter, 1993, 5, 247–260 CrossRef CAS.
- J. Singh, E. R. Bittner, D. Beljonne and G. D. Scholes, J. Chem. Phys., 2009, 131, 194905 CrossRef PubMed.
- A. Turkin, P. Malý and C. Lambert, Phys. Chem. Chem. Phys., 2021, 23, 18393–18403 RSC.
- S. Heun, H. Baessler, U. Mueller and K. Muellen, J. Phys. Chem., 1994, 98, 7355–7358 CrossRef CAS.
- U. Rauscher and H. Baessler, Macromolecules, 1990, 23, 398–405 CrossRef CAS.
- J. C. Bolinger, M. C. Traub, J. Brazard, T. Adachi, P. F. Barbara and D. A. Vanden Bout, Acc. Chem. Res., 2012, 45, 1992–2001 CrossRef CAS PubMed.
- C. Lambert, F. Koch, S. F. Völker, A. Schmiedel, M. Holzapfel, A. Humeniuk, M. I. S. Röhr, R. Mitric and T. Brixner, J. Am. Chem. Soc., 2015, 137, 7851–7861 CrossRef CAS PubMed.
- A. Turkin, M. Holzapfel, M. Agarwal, D. Fischermeier, R. Mitric, R. Schweins, F. Gröhn and C. Lambert, Chem. – Eur. J., 2021, 27, 8380–8389 CrossRef CAS PubMed.
- A. Humeniuk and R. Mitrić, Comput. Phys. Commun., 2017, 221, 174–202 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- D. Quigley and M. I. J. Probert, J. Chem. Phys., 2004, 120, 11432–11441 CrossRef CAS PubMed.
- G. J. Martyna, D. J. Tobias and M. L. Klein, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS.
- C. G. Mayne, J. Saam, K. Schulten, E. Tajkhorshid and J. C. Gumbart, J. Comput. Chem., 2013, 34, 2757–2770 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, J. Comput. Chem., 2009, 671–690 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian ∼09 Revision E.01, 2016.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- S. Fernandez-Alberti, D. V. Makhov, S. Tretiak and D. V. Shalashilin, Phys. Chem. Chem. Phys., 2016, 18, 10028–10040 RSC.
- D. Ondarse-Alvarez, S. Kömürlü, A. E. Roitberg, G. Pierdominici-Sottile, S. Tretiak, S. Fernandez-Alberti and V. D. Kleiman, Phys. Chem. Chem. Phys., 2016, 18, 25080–25089 RSC.
- T. Lu and F. Chen, Acta Chim. Sin. (Chin. Ed.), 2011, 69, 2393 CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04557a |
| This journal is © the Owner Societies 2024 |