
Conformational preferences of modified nucleobases in RNA aptamers and their effect on Förster resonant energy transfer†
David
Fischermeier
 a,
Christian
Steinmetzger
b,
Claudia
Höbartner
b and
Roland
Mitrić
*a
a,
Christian
Steinmetzger
b,
Claudia
Höbartner
b and
Roland
Mitrić
*a
aInstitut für Physikalische und Theoretische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: roland.mitric@uni-wuerzburg.de
bInstitut für Organische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 24th November 2023
Abstract
Förster resonant energy transfer (FRET) can be utilized in the study of tertiary structures of RNA aptamers, which bind specific fluorophoric ligands to form a fluorogenic aptamer complex. By introducing the emissive nucleobase analog 4-cyanoindole into the fluorogenic Chili RNA aptamer a FRET pair was established. The interpretation of studies aiming to investigate those tertiary structures using FRET, however, relies on prior knowledge about conformational properties of the nucleobase, which govern exciton transfer capabilities. Herein we employed classical molecular dynamics combined with Förster exciton theory to elucidate the preferred orientation relative to proximate bases and the influence on exciton transfer efficiency in multiple substitution sites. We did this by comparing the chromophoric distances emergent from MD simulations with experimental FRET data based on structural data of the native aptamer. We present the outlined methodology as a means to reliably evaluate future nucleobase analogue candidates in terms of their structural behavior and emergent exciton transfer properties as exemplified in the study of the preferred orientation of 4-cyanoindole in the Chili RNA aptamer.
1 Introduction
Fluorogenic RNA aptamers have been successfully applied as genetically encodable tags for RNA imaging.1–4 These tags are used as components of biosensors for metabolite detection5–9 as well a basis for responsive nucleic acid architectures.10,11 Several varieties of assembling a FRET pair to achieve those functionalities exist12–14 and recently the binding of small organic molecules to form a FRET pair with bases of the Chili aptamer was achieved.15–17 Those small, organic molecules are not fluorescent when free in solution but become emissive in the bound state due to reduced non-radiative decay of the excited state. Examples can be found in the families of Spinach,18 Broccoli,19 Corn20 and Chili,15 which bind derivatives of HBI dyes related to the chromophore of green fluorescent proteins (GFP), while the Mango and Peach family bind derivatives of the generic cyanine dye Thiazole Orange.21–24The exploration of FRET between nucleobase-modified RNA and bound ligands is a new addition to fluorogenic aptamers that was demonstrated for the Chili RNA aptamer, which activates fluorescence of DMHBI+ and DMHBO+. The universal nucleobase analogue 4-cyanoindole (CI) was identified as a suitable FRET partner of the Chili ligands and used to map their binding site within the RNA aptamer.17,25 However, quantitative interpretation of the results was hampered by the lack of information on the relative orientation of CI and the ligand.
Recently, the structure of the Chili aptamer in complex with two cationic ligands, DMHBI+ and DMHBO+,15,16 was solved by X-ray crystallography26 (Fig. 1). The RNA forms a central two-tiered G-quadruplex as the ligand-binding element, which is anchored by a basal stem and an apical stem-loop. A long-range base pair helps to establish a preorganized proton transfer pathway to N7 of G15, which enables efficient ligand-to-RNA excited state proton transfer (ESPT) in less than 130 fs and causes fluorescence emission with a large Stokes shift. Site-specific modification of Chili with the universal fluorescent ribonucleoside analog r4CI was used to engineer a supramolecular base-to-ligand FRET system. By incorporating r4CI at eight positions (G6, A11, G17, U23, U34, G36, G45, G46) based on previously reported Tb3+ probing and mutagenesis experiments,15,16 the exciton transfer efficiency was mapped for the different domains of the Chili aptamer earlier experimentally.17
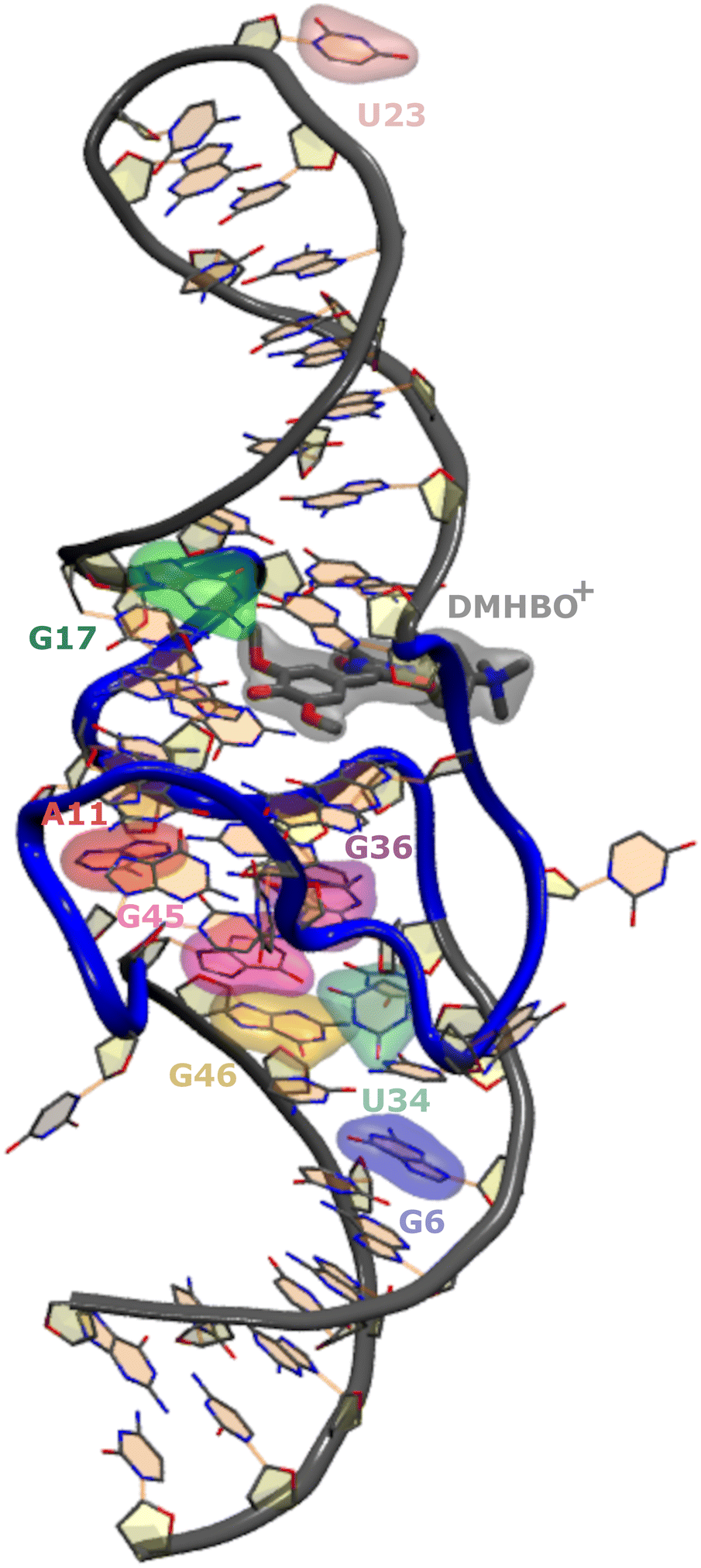 | ||
| Fig. 1 Crystal structure of the Chili RNA aptamer complex. The quadruplex (backbone highlighted in blue) at the center serves as a binding site for DMHBO+. The investigated sites for base substitution with r4CI are highlighted. | ||
The goal of this study is assessing the effect of those substitutions on the structural integrity of the wild-type aptamer as well as the orientational preferences of r4CI. The orientation of FRET paired bases is vital to the exciton transfer process and thus the interpretation of experimental FRET data. Due to a lack of knowledge regarding the orientation of the fluorescent base one often assumes isotropic behavior, which can lead to errors of orders of magnitude when trying to assess the pair distance from FRET experiments as already shown for DNA analogues.27,28 This orientation may deviate from the orientation of the unsubstituted base in the native aptamer's crystal structure due to differences in binding affinities between original base and substitute. Insight into the orientations of the base can be gained from MD simulations. While this study focuses heavily on the Chili aptamer complex, the outlined methodology is not restricted to this field of research and is widely usable in any context where FRET experiments are utilized to gain insights into the structure of large systems.
Using classical MD simulations we were able to gain some insights into the influence of the newly introduced base on pairing, stacking and the general structure of the immediate neighbouring bases. We were also able to analyze the preferred orientations of the substituted bases and their effect on the exciton transfer and quenching. To this end, these classical MD simulations were combined with quantum chemical TD-DFT calculations to theoretically study the exciton transfer based on the classical Förster model.
The outlined methodology can help scan a plethora of different bases and select for stability of the tertiary structure as well as the desired orientation patterns in synthetic nucleoside strands, which allows for a faster and more accurate interpretation of future experimental studies and selection of modification sites and bases. As the employed method only relies on a wild-type crystal structure, the often prohibitively arduous – or in case of in vivo FRET studies impossible – process of isolating and crystallizing a multitude of mutations can be partially or fully replaced by these simulations.
2 Computational methods
Classical MD simulations were performed using the NAMD 2.12 simulation package.29,30 The required bonded parameters for cyanoindole were derived from the parameters of indole and the cyano group from the CHARMM36 cgenff parameters.31 Bonded parameters for DMHBO+ were calculated using the FFTK package provided with VMD 1.9.432 in conjunction with GAUSSIAN16.33 Atomic charges were calculated using the R-ESP routine in nwchem.34 The calculated parameters are outlined in ESI.† The remaining parameters for the canonical bases are already provided by CHARMM36.35–37 The MD simulations were conducted using a 1 fs timestep at 300 K and 1 atm within a periodic water box (initially 80 Å cube) neutralized with KCl maintained by Langevin damping of 1 ps−1. Each trajectory was equilibrated starting with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 step optimization followed by 0.1 ns of propagation of hydrogens and solvent molecules within a flexible periodic cell. The equilibration was concluded by another 10000 step optimization and 0.1 ns of propagation with all atoms in a fixed cell. Long range electrostatics were handled by the implemented PME method. As the parameters for the chromophores were not as rigorously tested as standard nucleobase parameters, we opted to forgo the RATTLE38 or SHAKE39 treatment. The production runs consisted of 10 ns of propagation. According to the Förster Model, the orientation factor, which is a measure for exciton transfer, (κ) was calculated every 10 fs according to:
000 step optimization followed by 0.1 ns of propagation of hydrogens and solvent molecules within a flexible periodic cell. The equilibration was concluded by another 10000 step optimization and 0.1 ns of propagation with all atoms in a fixed cell. Long range electrostatics were handled by the implemented PME method. As the parameters for the chromophores were not as rigorously tested as standard nucleobase parameters, we opted to forgo the RATTLE38 or SHAKE39 treatment. The production runs consisted of 10 ns of propagation. According to the Förster Model, the orientation factor, which is a measure for exciton transfer, (κ) was calculated every 10 fs according to:κ = ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) A·D − 3(A· A·D − 3(A·![[R with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0052_20d1.gif) )(D·), )(D·), | (1) |
| κ = cosθD–A − 3cosθA–RcosθD–R, | (2) |
A and D are the normalized donor (CI) and acceptor (DMHBO+) transition dipole moment vectors and is the normalized chromophores' distance vector. θD–A, θD–R and θA–R give the angles between the vectors as shown in Fig. 2. Instead of calculating the transition dipole moments directly for every frame, the transition dipole vectors can be constructed from the atomic transition charges of the S1 transition of optimized donor and acceptor structures. For this, the donor and acceptor were optimized and the atomic transition charges were derived via Multiwfn40,41 based on GAUSSIAN16 TD-DFT calculations. All quantum chemical calculations including the parametrization were performed at the CAM-B3LYP42/def2-SVP43,44 D3 corrected45 level of DFT without any PCM. The Förster radius46,47R0 was calculated according to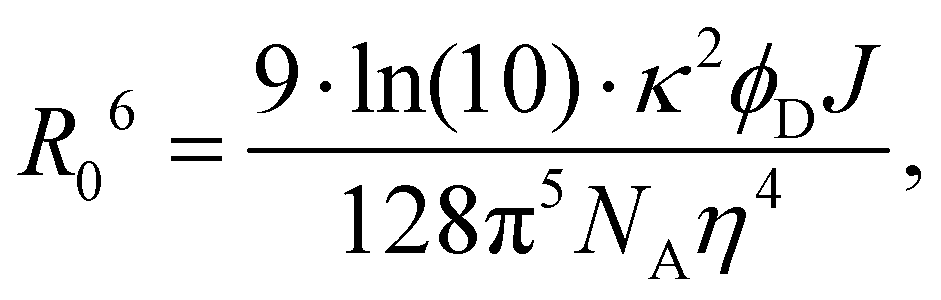 | (3) |
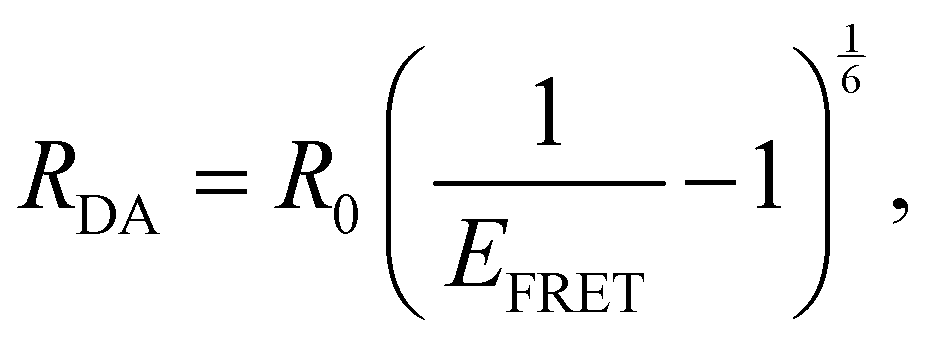 | (4) |
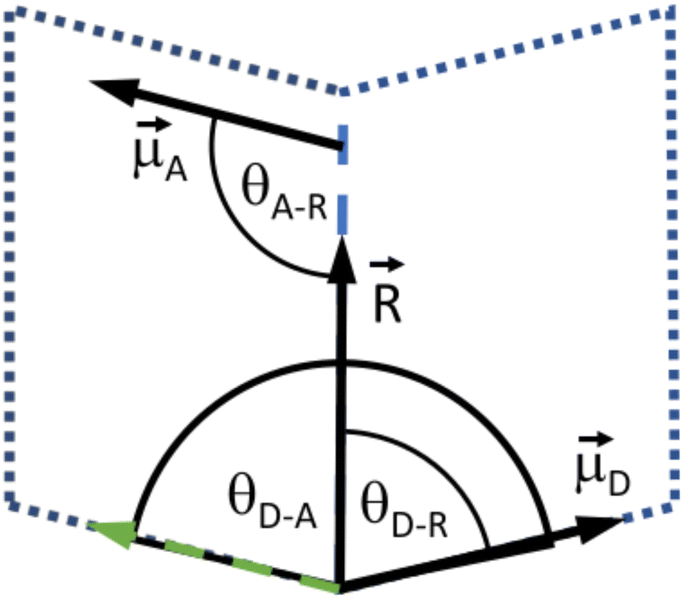 | ||
| Fig. 2 Depiction of the angular relation between the transition dipole moment (D and A) and distance vectors (). | ||
To assess the stability of the system the root mean squared deviation (RMSD) of the atomic positions for parts and the entirety of the system have been calculated according to
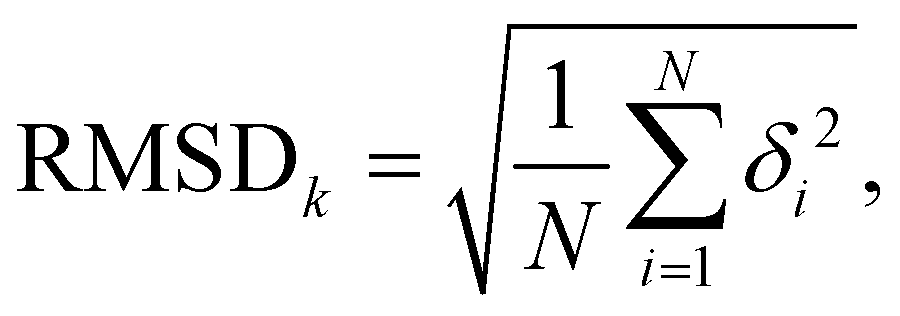 | (5) |
3 Results and discussion
We compared the theoretically obtained vertical excitation energies of the chromophores' S0 → S1 transitions with experimental spectral data. The donor's (CI) S1 transition was calculated to be 4.60 eV, which is in reasonable agreement with the experimental value of 4.09 eV,49 while the acceptor's (DMHBO+) excitation energy was calculated to be 3.07 eV corresponding well to the 2.78 eV observed experimentally.50To evaluate the the orientation factor κ, the orientations of the correct transition dipole moments are paramount. Fig. 3 shows the orientation of the transition dipole moments of both chromophores' and experimental data for comparison. For CI the transition dipole calculated by TD-DFT is in excellent agreement with experimental data.49 While the DMHBO+ transition dipole moment has not been determined yet, several similar species are already studied, which show similar angles (70–100°) between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and the transition dipole moment50 suggesting that both transition dipole vectors are accurately simulated. The FRET donor r4CI was individually introduced into the wild-type Chili RNA aptamer at multiple sites (G6, A11, G17, U23, U34, G36, G45, G46). However, not all orientations of these bases, e.g. stacked, unstacked and different rotational degrees of freedom, are guaranteed to be visited within a single, limited trajectory due to the energy barriers involved likely to lead to trapping a replica in a local minimum. Thus for every substitution several starting orientations were propagated. Unless not permitted by the position of the original base (e.g. sterical constraints) these starting orientations consisted of two varieties with r4CI occupying the intrahelical space of the original base, one of which features a 180° rotation (I1,2). Furthermore several orientations with extrahelical r4CI pointing outwards of the complex (E1–3, see ESI.† Fig. S1–S35) were also simulated.
O bond and the transition dipole moment50 suggesting that both transition dipole vectors are accurately simulated. The FRET donor r4CI was individually introduced into the wild-type Chili RNA aptamer at multiple sites (G6, A11, G17, U23, U34, G36, G45, G46). However, not all orientations of these bases, e.g. stacked, unstacked and different rotational degrees of freedom, are guaranteed to be visited within a single, limited trajectory due to the energy barriers involved likely to lead to trapping a replica in a local minimum. Thus for every substitution several starting orientations were propagated. Unless not permitted by the position of the original base (e.g. sterical constraints) these starting orientations consisted of two varieties with r4CI occupying the intrahelical space of the original base, one of which features a 180° rotation (I1,2). Furthermore several orientations with extrahelical r4CI pointing outwards of the complex (E1–3, see ESI.† Fig. S1–S35) were also simulated.
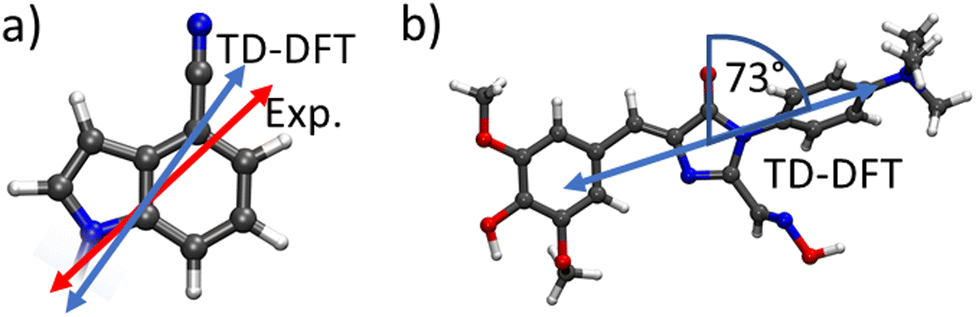 | ||
| Fig. 3 (a) Comparison of the experimentally and theoretically determined transition dipole orientations for CI. (b) Depiction of the calculated transition dipole moment for DMHBO+. The angle between the CO bond and the transition dipole moment was found to be 73°. | ||
To assess the movement and stability of the system the root mean squared deviation (RMSD) of the atomic positions was calculated for the structures within the trajectories for the complete systems as well as the chromophores as shown in Fig. 4. The U23 species was chosen as a good baseline example, since U23 is located at the aptamer's loop and as such can't pair or stack as bases in the helix/tetraloop could and as such can reasonably only take a single orientation, where the cyanoindole can freely rotate around the glycosidic bond (see ESI.† Fig. S36–S43 for RMSD data of the remaining species). To exclude the influence of rotational and translational motion of the complete system, the structures of the complete system were aligned with each other along the backbone of the aptamer. The U23 E1 modified system is equilibrated within roughly 1 ns, after which the RMSD of atomic positions of the system lies between 3 and 5 Å relative to the starting orientation. The atomic positions of DMHBO+ fluctuate roughly around 2 Å when aligning the complete system. An illustration of the positional changes of the complete system and DMHBO+ is shown in Fig. 5. This positional rigidity implies a strong interaction between DMHBO+ and the aptamer. r4CI shows the highest positional flexibility with an RMSD between 2 and 7 Å. In the same way the rotation and translation of the system were excluded by aligning its structures, the change in the local geometry of the chromophores can be characterized by aligning the chromophore's structures. CI shows nearly no change in geometry, while DMHBO+ also shows very little change in structure along the trajectory. This shows, that no rotation of the rings is observed for DMHBO+. The RMSD data for r4CI and the aligned CI show, that CI is very rigid as expected, but can rotate rather freely around the glycosidic bond for the U23 species. This is also true for all sterically unrestricted orientations.
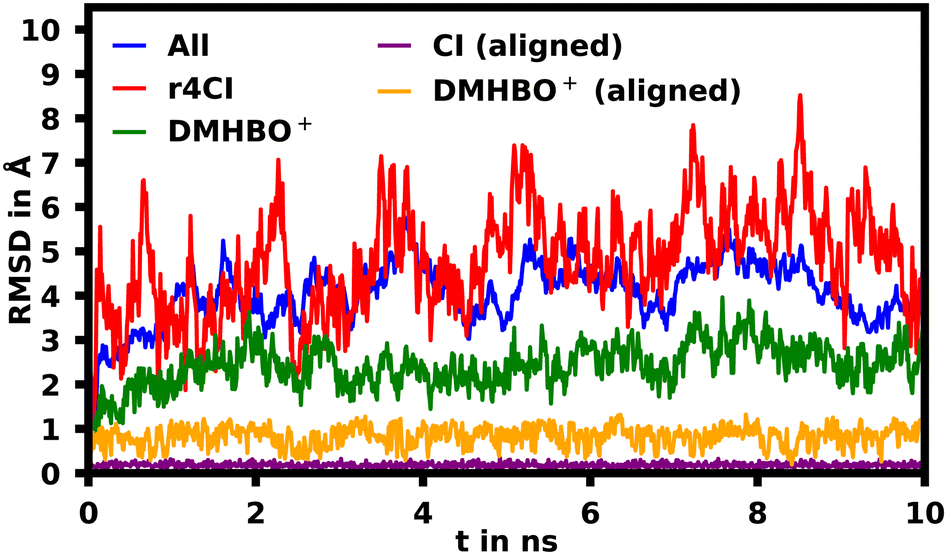 | ||
| Fig. 4 Root mean squared deviation (RMSD) of the atomic positions in Å of the complete backbone aligned aptamer DMHBO+ (blue) complex, r4CI (red) and DMHBO+ (green) for the U23 substitution. The RMSD for the self-aligned DMHBO+ and CI is shown in orange and purple. | ||
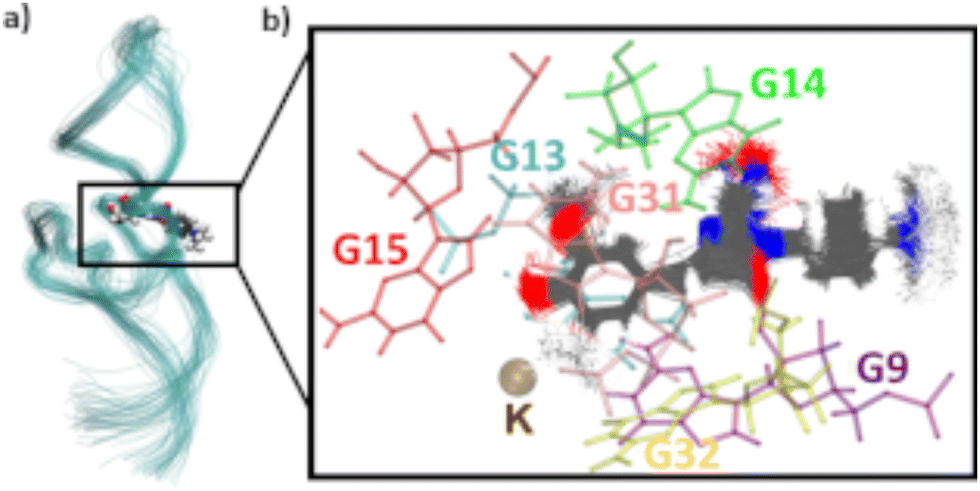 | ||
| Fig. 5 (a) Superimposed tube representation of the RNA backbone over 10 ns. The frames were taken in increments of 0.2 ns. (b) Superimposed frames of DMHBO+ in increments of 0.01 ns as well as the crystal structure positions of the surrounding bases of the binding pocket. DMHBO+ remains bound to the pocket without large rotations. | ||
From MD simulations the chromophore distance RDA (MD) is readily available for each orientation of all substituted species. Using the exciton model this chromophoric distance (RDA (Exc.)) can also be calculated with eqn (4) from κ, which is obtainable from eqn (2). If the chromophoric distance RDA (Exc.) based on experimental data and the orientation factor is sufficiently congruent with RDA (MD), it suggests that the orientation and chromophoric distance observed during the MD simulation is indeed dominant in solution. Fig. 6 shows a comparison of the donor–acceptor distance, RDA (MD), over the course of the MD simulation as well as RDA (Exc.), which was obtained from experimental data17 shown in Table 1 in conjunction with the MD simulations. We show the data for the trajectories, which yield the best agreement for the chromophoric distances as well as other noteworthy behavior affecting those. A table with all values for all trajectories can be found in the ESI.† We note here, that for all species multiple starting orientations give good agreements for the RDA values as they cross over and converge geometrically over the course of the trajectory, which is also shown and discussed in the ESI.†
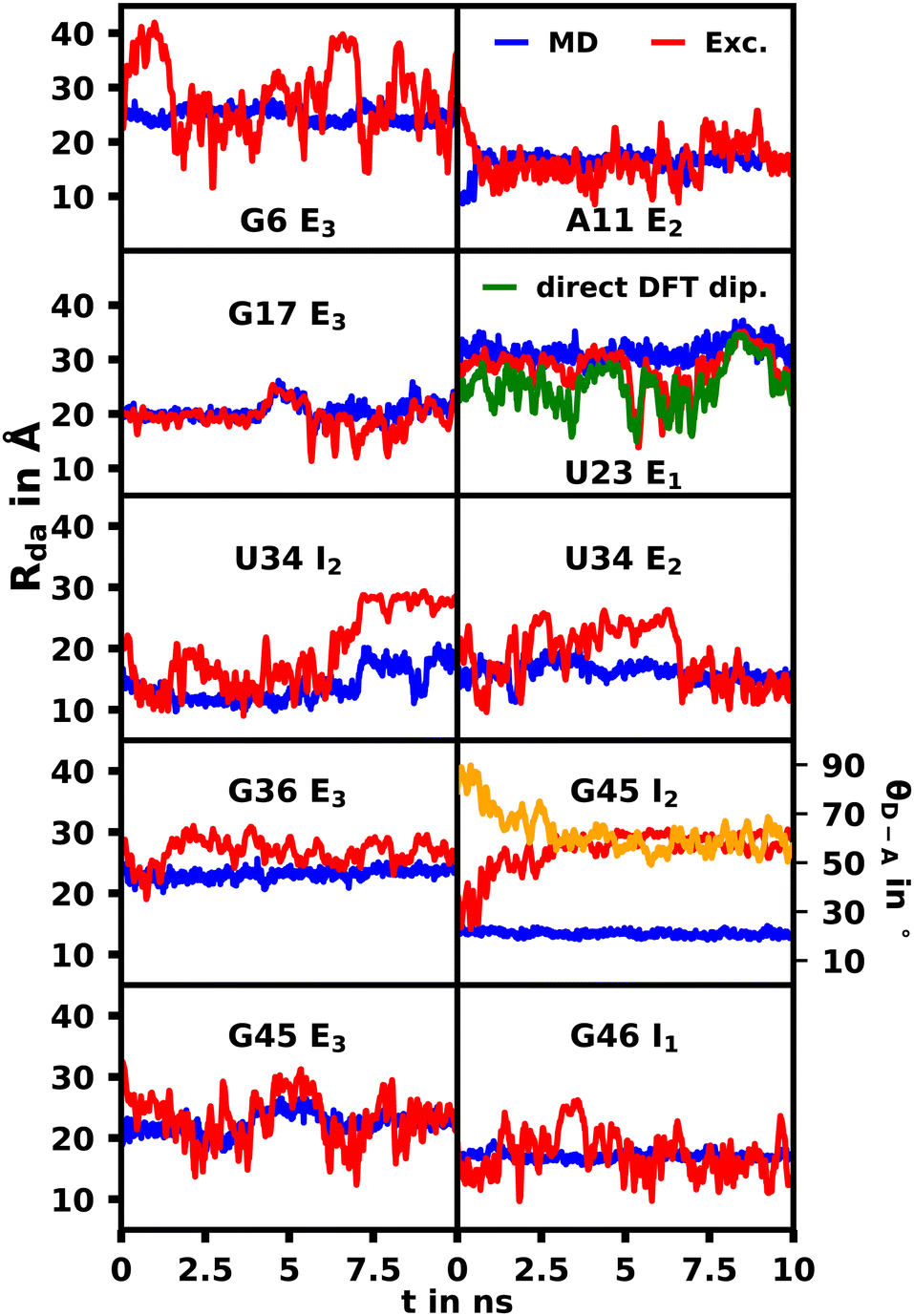 | ||
| Fig. 6 Comparison of the donor–acceptor distance, RDA, over the course of the MD simulation (MD, blue) as well as calculated based on the exciton model (exc.,red) and experimental values shown in Table 1. For visibility the 10 value running average for the exciton model is shown due to the high fluctuations. For G45 I2 the angle between the donor and acceptor transition dipoles (θD–A) is also displayed in yellow. | ||
| Species | Quantum yield | E τ | J | R 0 (Å) | κ 2 | R DA (Å) | |
|---|---|---|---|---|---|---|---|
| MD | Exc. | ||||||
| a 1014 l nm4 cm−1 mol−1. | |||||||
| G6 E3 | 0.48 | 0.36 | 4.40 | 29.7 | 0.13 | 25.0 | 28.2 |
| A11 E2 | 0.25 | 0.67 | 4.90 | 22.5 | 0.09 | 18.1 | 20.0 |
| G17 E3 | 0.07 | 0.86 | 4.10 | 21.0 | 0.65 | 20.6 | 19.4 |
| U23 E1 | 0.49 | 0.80 | 4.80 | 35.8 | 0.50 | 31.5 | 28.1 |
| U34 E2 | 0.15 | 0.70 | 4.50 | 22.0 | 0.15 | 21.1 | 19.5 |
| G36 E3 | 0.28 | 0.82 | 4.40 | 24.0 | 0.95 | 22.0 | 28.2 |
| G45 E3 | 0.10 | 0.37 | 4.60 | 15.4 | 0.15 | 22.5 | 23.2 |
| G46 I1 | 0.28 | 0.62 | 4.80 | 18.9 | 0.03 | 17.1 | 18.3 |
Overall we were able to identify at least one trajectory for every mutation, which produced chromophoric distances very similar to the distances calculated with the exciton model based on the MD and experimental spectroscopic data. A selection of those will be discussed herein, while the datasets for all simulations are shown in the ESI† (Fig. S44–S50). Some external orientations for each species did cross over into each other at some points, for example G6 E2/E3. The G46 mutation was the only species, for which the orientation located inside the helix (G46 I1) produced the best agreement. This confirms the conclusions found by thermal melting studies17 suggesting that substituted sites possess more conformational freedom compared to their unsubstituted variant and that the modified aptamer's r4CI exhibits a significant unstacked population. The I2 orientation of U34 also produced very good agreement for the majority of the trajectory until the orientation changes substantially due to movement in the RNA backbone, which leads to a divergence between the RDA values. This suggests that its dominant orientation might be facing inward or an equilibrium of I2 and E2. The G45 I2 orientation also produces two close RDA curves initially, but diverges rapidly due to multiple subtle geometry changes in the aptamer complex, which result in a steady change of θD–A effecting the change in RDA. This, however, could indicate that r4CI in the G45 species might also be in an inwards facing orientation or in an equilibrium of G45 I2 and G45 E3. Notably the U34, G45 and G46 moieties are very close in proximity at the very bottom of the quadruplex. Thus it seems that this region, where the helix opens up to transition into the quadruplex, allows r4CI to pair with neighbouring bases possibly due to the lack of sterical constraint.
The G17 E3 and U23 E1 species exhibit orientation factors close to the isotropic limit of 2/3. In Fig. 7 we show that θD–A, which should show high fluctuations for isotropic behavior, is nearly constant for U23 E1 and in most other trajectories. Although r4CI exhibits considerable movement when located outside of the helix as seen in Fig. 4, the most freely accessible movement for any unpaired r4CI nucleotide is the rotation around the glycosidic bond. Fig. 3 shows the almost parallel alignment of r4CI's transition dipole moment with the glycosidic bond, which results in the invariance of θD–A in regards to this rotation. Thus the chromophores' relative transition dipole orientation is mostly constant and the value for the orientation factor is not a result of isotropic behavior, but rather a rather rigid orientation resulting in the same value as the isotropic limit.
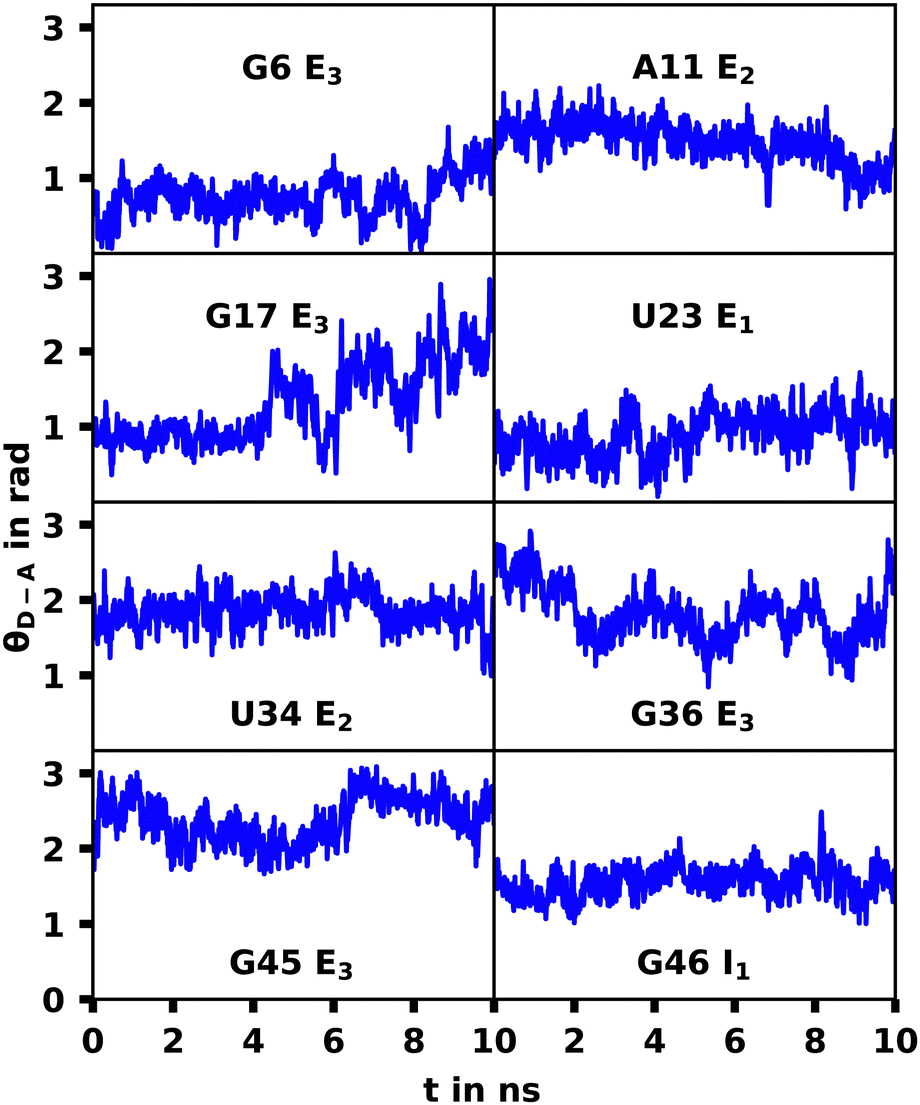 | ||
| Fig. 7 θ D–A over the course the selected MD trajectories. The constant behavior shows a rigid orientation of the transition dipole moment. | ||
For the G17 E3 species the angular relation between the transition dipole moments shows different behavior with a large change at roughly 5 ns suggesting a more flexible environment. In fact the same trend can be observed in Fig. 6, where the agreement of both RDA values is very close before 5 ns with discrepancies arising afterwards. This shift in behavior was found to be a result of a sort of “flipping” motion in the RNA backbone at the G17 position (see Fig. 8). Whereas r4CI “leans” on the helix at the beginning of the simulation, it unlatches from that position and the backbone rotates r4CI further away from the helix tilting the loop portion of the complex during the motion. Following this motion the congruence in RDA fluctuates, but still shows very good agreement at times. This could suggest, that the flipping motion might represent real behavior of the aptamer complex. Some other sites like G6 and G45, where the backbone seems less rigid, also undergo frequent, albeit mostly minor changes in orientation, which can, however, result in large spikes for the calculated RDA.
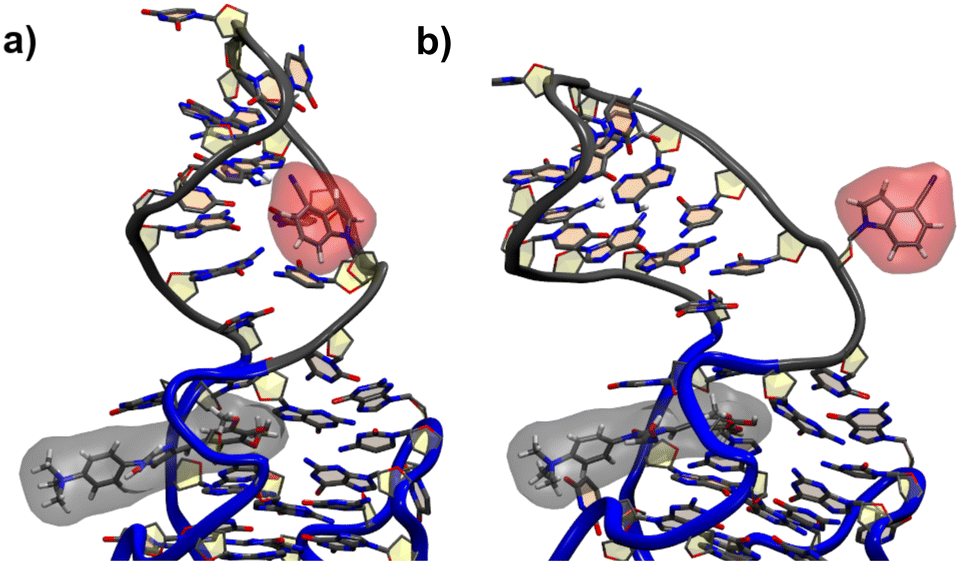 | ||
| Fig. 8 The initial leaning orientation of r4CI (red) in the G17 position starting from the G17 E3 orientation is illustrated in (a), while (b) shows r4CI facing away from the complex. | ||
While we have shown that the chromophores exhibit a rather rigid geometry due to their extensive aromatic, planar rings, these geometric changes might still affect the transition dipole vectors, which would affect the orientation factors and thus fail to produce accurate values for RDA. To assess whether our method of constructing the transition dipole vectors from transition charges is sufficiently accurate, the transition dipole vectors of both chromophores were directly calculated using TD-DFT in GAUSSIAN16 along the trajectory of the U23 species. Furthermore the chromophore distance was calculated subsequently based on those vectors as seen in Fig. 6.
The average chromophoric distances of U23 with 25.2 Å for DFT and 28.1 Å for the exciton method show a good agreement and both curves appear similar in shape especially after the first 4 ns, showing, that both react very similarly to changes in relative orientations as seen in Fig. 6. This shows that the dipole moments constructed using atomic transition charges are in good agreement with the transition dipole moments obtained directly from DFT and thus the exciton model derived transition dipole vectors and orientation factors should give an accurate description of the exciton transfer process.
4 Conclusion
We have performed extensive MD simulations on variants of the native Chili RNA aptamer to probe the effect of introducing r4CI into the system on stability and exciton transfer. The tertiary structure of the DMHBO+ binding aptamer remained stable for all modifications and did not affect DMHBO+ binding noticeably according to our simulations. TD-DFT was able to calculate the transition dipole vectors accurately and in conjunction with experimental data, we were able to find the dominant orientations of r4CI in all modified species. Based on these orientations, we conclude that r4CI prefers to be localized outside of the helix in most cases suggesting the existence of a considerable unstacked population. This orientation of the donor also explains why the exciton is resistant to quenching by neighbouring, stacked guanines as experiments have found and thus long-lived enough to participate in the FRET process. However, we also note, that the basal region of the quadruplex around the U34, G45 and G46 moieties, where the helix starts to open up in transition to the quadruplex seems to also allow for an inward facing orientation of r4CI for said moieties.Even localized outside of the aptamer complex the angular relation of the r4CI and DMHBO+ is similarly constant as it is in an orientation facing inwards to the complex. This suggests that the isotropic limit of κ2 = 2/3 should not be assumed when working with r4CI as nucleoside analog.
Studying which characteristics allow for and enforce intrahelical, well defined extrahelical orientations or enable free movement could prove important to finding suitable sites for base substitution in the future.
The method employed was shown to be comparatively accurate in obtaining transition dipoles and orientation factors to full DFT at a fraction of the computational cost and produced results widely in agreement with prior experimental data. Obtaining crystals of modified biomacromolecules is often virtually impossible due to the prohibitive effort required especially while maintaining the precise tertiary structure present in solution. The outlined methodology could be widely employed to gain very detailed insight into the topology of a variety of artificial nucleobases and their influence on the tertiary structural stability as well as any other chromophoric biomolecules used in FRET applications. Provided a crystal structure for the unmodified system alongside FRET data is provided, both FRET chromophores can be studied to obtain properties like pairing, stacking or true isotropic behavior.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council (ERC/No 682586 to CH and ERC/No 646737 to RM).Notes and references
- M. You and S. R. Jaffrey, Annu. Rev. Biophys., 2015, 44, 187–206 CrossRef CAS PubMed.
- E. V. Dolgosheina and P. J. Unrau, Wiley Interdiscip. Rev.: RNA, 2016, 7, 843–851 CrossRef CAS PubMed.
- R. J. Trachman and A. R. Ferré-D'Amaré, Quarterly Rev. Biophys., 2019, 52, e8 CrossRef PubMed.
- S. Neubacher and S. Hennig, Angew. Chem., Int. Ed., 2019, 58, 1266–1279 CrossRef CAS PubMed.
- N. Mukkayyan, R. Poon, P. N. Sander, L.-Y. Lai, Z. Zubair-Nizami, M. C. Hammond and S. S. Chatterjee, ACS Omega, 2022, 7, 32749–32753 CrossRef CAS PubMed.
- S. R. Jaffrey, Advances in Pharmacology, Elsevier, 2018, vol. 82, pp. 187–203 Search PubMed.
- R. Wu, A. P. K. K. Karunanayake Mudiyanselage, F. Shafiei, B. Zhao, Y. Bagheri, Q. Yu, K. McAuliffe, K. Ren and M. You, Angew. Chem., Int. Ed., 2019, 58, 18271–18275 CrossRef CAS PubMed.
- M. G. Costales and M. D. Disney, Cell Chem. Biol., 2019, 26, 463–465 CrossRef CAS PubMed.
- M. You, J. L. Litke, R. Wu and S. R. Jaffrey, Cell Chem. Biol., 2019, 26, 471–481.e3 CrossRef CAS PubMed.
- A. Chopra, S. Sagredo, G. Grossi, E. Andersen and F. Simmel, Nanomaterials, 2019, 9, 507 CrossRef CAS PubMed.
- V. Goldsworthy, G. LaForce, S. Abels and E. Khisamutdinov, Nanomaterials, 2018, 8, 984 CrossRef PubMed.
- M. D. E. Jepsen, S. M. Sparvath, T. B. Nielsen, A. H. Langvad, G. Grossi, K. V. Gothelf and E. S. Andersen, Nat. Commun., 2018, 9, 18 CrossRef PubMed.
- R. J. Trachman, R. Cojocaru, D. Wu, G. Piszczek, M. Ryckelynck, P. J. Unrau and A. R. Ferré-DAmaré, Structure, 2020, 28, 776–785.e3 CrossRef CAS PubMed.
- S. C. Jeng, R. J. Trachman, F. Weissenboeck, L. Truong, K. A. Link, M. D. Jepsen, J. R. Knutson, E. S. Andersen, A. R. Ferré-D'Amaré and P. J. Unrau, RNA, 2021, 27, 433–444 CrossRef CAS PubMed.
- C. Steinmetzger, N. Palanisamy, K. R. Gore and C. Höbartner, Chem. – Eur. J., 2019, 25, 1931–1935 CrossRef CAS PubMed.
- C. Steinmetzger, I. Bessi and A.-K. Lenz, Nucleic Acids Res., 2019, 47, 13 CrossRef PubMed.
- C. Steinmetzger, C. Bäuerlein and C. Höbartner, Angew. Chem., Int. Ed., 2020, 59, 6760–6764 CrossRef CAS PubMed.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed.
- G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 16299–16308 CrossRef CAS PubMed.
- K. D. Warner, L. Sjekloća, W. Song, G. S. Filonov, S. R. Jaffrey and A. R. Ferré-D'Amaré, Nat. Chem. Biol., 2017, 13, 1195–1201 CrossRef CAS PubMed.
- E. V. Dolgosheina, S. C. Y. Jeng, S. S. S. Panchapakesan, R. Cojocaru, P. S. K. Chen, P. D. Wilson, N. Hawkins, P. A. Wiggins and P. J. Unrau, ACS Chem. Biol., 2014, 9, 2412–2420 CrossRef CAS PubMed.
- A. Autour, S. C. Y. Jeng, A. D. Cawte, A. Abdolahzadeh, A. Galli, S. S. S. Panchapakesan, D. Rueda, M. Ryckelynck and P. J. Unrau, Nat. Commun., 2018, 9, 656 CrossRef PubMed.
- R. J. Trachman, A. Autour, S. C. Y. Jeng, A. Abdolahzadeh, A. Andreoni, R. Cojocaru, R. Garipov, E. V. Dolgosheina, J. R. Knutson, M. Ryckelynck, P. J. Unrau and A. R. Ferré-DAmaré, Nat. Chem. Biol., 2019, 15, 472–479 CrossRef PubMed.
- K. Y. Kong, S. C. Jeng, B. Rayyan and P. J. Unrau, RNA, 2021, 27, 604–615 CrossRef CAS PubMed.
- K. T. Passow and D. A. Harki, Org. Lett., 2018, 20, 4310–4313 CrossRef CAS PubMed.
- M. Mieczkowski, C. Steinmetzger, I. Bessi, A.-K. Lenz, A. Schmiedel, M. Holzapfel, C. Lambert, V. Pena and C. Höbartner, Nat. Commun., 2021, 12, 3549 CrossRef CAS PubMed.
- K. Börjesson, S. Preus, A. H. El-Sagheer, T. Brown, B. Albinsson and L. M. Wilhelmsson, J. Am. Chem. Soc., 2009, 131, 4288–4293 CrossRef PubMed.
- A. Iqbal, S. Arslan, B. Okumus, T. J. Wilson, G. Giraud, D. G. Norman, T. Ha and D. M. J. Lilley, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11176–11181 CrossRef CAS PubMed.
- D. Quigley and M. I. J. Probert, J. Chem. Phys., 2004, 120, 11432–11441 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, J. Comput. Chem., 2009, 671–690 Search PubMed.
- C. G. Mayne, J. Saam, K. Schulten, E. Tajkhorshid and J. C. Gumbart, J. Comput. Chem., 2013, 34, 2757–2770 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian’16 Revision C.01, 2016 Search PubMed.
- E. Aprà, E. J. Bylaska, W. A. de Jong, N. Govind, K. Kowalski, T. P. Straatsma, M. Valiev, H. J. J. van Dam, Y. Alexeev, J. Anchell, V. Anisimov, F. W. Aquino, R. Atta-Fynn, J. Autschbach, N. P. Bauman, J. C. Becca, D. E. Bernholdt, K. Bhaskaran-Nair, S. Bogatko, P. Borowski, J. Boschen, J. Brabec, A. Bruner, E. Cauët, Y. Chen, G. N. Chuev, C. J. Cramer, J. Daily, M. J. O. Deegan, T. H. Dunning, M. Dupuis, K. G. Dyall, G. I. Fann, S. A. Fischer, A. Fonari, H. Früchtl, L. Gagliardi, J. Garza, N. Gawande, S. Ghosh, K. Glaesemann, A. W. Götz, J. Hammond, V. Helms, E. D. Hermes, K. Hirao, S. Hirata, M. Jacquelin, L. Jensen, B. G. Johnson, H. Jónsson, R. A. Kendall, M. Klemm, R. Kobayashi, V. Konkov, S. Krishnamoorthy, M. Krishnan, Z. Lin, R. D. Lins, R. J. Littlefield, A. J. Logsdail, K. Lopata, W. Ma, A. V. Marenich, J. Martin del Campo, D. Mejia-Rodriguez, J. E. Moore, J. M. Mullin, T. Nakajima, D. R. Nascimento, J. A. Nichols, P. J. Nichols, J. Nieplocha, A. Otero-de-la Roza, B. Palmer, A. Panyala, T. Pirojsirikul, B. Peng, R. Peverati, J. Pittner, L. Pollack, R. M. Richard, P. Sadayappan, G. C. Schatz, W. A. Shelton, D. W. Silverstein, D. M. A. Smith, T. A. Soares, D. Song, M. Swart, H. L. Taylor, G. S. Thomas, V. Tipparaju, D. G. Truhlar, K. Tsemekhman, T. Van Voorhis,. Vázquez-Mayagoitia, P. Verma, O. Villa, A. Vishnu, K. D. Vogiatzis, D. Wang, J. H. Weare, M. J. Williamson, T. L. Windus, K. Wolinski, A. T. Wong, Q. Wu, C. Yang, Q. Yu, M. Zacharias, Z. Zhang, Y. Zhao and R. J. Harrison, J. Chem. Phys., 2020, 152, 184102 CrossRef PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- B. R. Brooks, C. L. Brooks, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- H. C. Andersen, J. Comput. Phys., 1983, 52, 24–34 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- T. Lu and F. Chen, Acta Chim. Sin. – Chinese Edition, 2011, 69, 2393 CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- B. W. van der Meer, FRET – Förster Resonance Energy Transfer, John Wiley & Sons, Ltd, 2013, pp. 23–62 Search PubMed.
- R. M. Clegg, Fret and Flim Techniques, Elsevier, 2009, vol. 33, pp. 1–57 Search PubMed.
- W. Kabsch, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1978, 34, 827–828 CrossRef.
- M.-L. Hebestreit, M. Schneider, H. Lartian, V. Betz, M. Heinrich, M. Lindic, M. Y. Choi and M. Schmitt, Phys. Chem. Chem. Phys., 2019, 21, 14766–14774 RSC.
- T. Ansbacher, H. K. Srivastava, T. Stein, R. Baer, M. Merkx and A. Shurki, Phys. Chem. Chem. Phys., 2012, 14, 4109 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04704k |
| This journal is © the Owner Societies 2024 |