
Allosteric regulation in SARS-CoV-2 spike protein†
Yong
Wei
a,
Amy X.
Chen
b,
Yuewei
Lin
 c,
Tao
Wei
*d and
Baofu
Qiao
*e
c,
Tao
Wei
*d and
Baofu
Qiao
*e
aDepartment of Computer Science, High Point University, High Point, NC 27268, USA
bThomas Jefferson High School for Science and Technology, Alexandria, VA 22312, USA
cComputational Science Initiative, Brookhaven National Laboratory, Upton, NY 11973, USA
dDepartment of Chemical Engineering and Department of Biomedical Engineering, University of South Carolina, Columbia, SC 29208, USA. E-mail: tao.wei@Howard.edu
eDepartment of Natural Sciences, Baruch College, City University of New York, New York, NY 10010, USA. E-mail: baofu.qiao@baruch.cunyu.edu
First published on 2nd February 2024
Abstract
Allosteric regulation is common in protein–protein interactions and is thus promising in drug design. Previous experimental and simulation work supported the presence of allosteric regulation in the SARS-CoV-2 spike protein. Here the route of allosteric regulation in SARS-CoV-2 spike protein is examined by all-atom explicit solvent molecular dynamics simulations, contrastive machine learning, and the Ohm approach. It was found that peptide binding to the polybasic cleavage sites, especially the one at the first subunit of the trimeric spike protein, activates the fluctuation of the spike protein's backbone, which eventually propagates to the receptor-binding domain on the third subunit that binds to ACE2. Remarkably, the allosteric regulation routes starting from the polybasic cleavage sites share a high fraction (39–67%) of the critical amino acids with the routes starting from the nitrogen-terminal domains, suggesting the presence of an allosteric regulation network in the spike protein. Our study paves the way for the rational design of allosteric antibody inhibitors.
1. Introduction
Allosteric regulation refers to the mechanism that an event (e.g., ligand binding) at one place of a protein leads to influences on a remote domain of the protein, such as the local mobility of the distal domain and interactions with another protein.1–6 In addition to the design of drugs that directly bind the active sites of proteins, allosteric regulation provides a new route for drug design.7–9 Nevertheless, our current understanding of allosteric regulation is remarkably limited and its molecular mechanism remains mostly unrevealed due to the complicated folded structures of proteins.10 It thus limits the progress of allosteric regulation-based drug design.Coronavirus disease 2019 (COVID-19), due to infection of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has caused a global pandemic for three years, leading to over 6.9 million deaths and about 0.77 billion confirmed cases worldwide according to the report of the World Health Organization (https://covid19.who.int/). SARS-CoV-2 virus attacks human cells via the binding of its spike protein with the angiotensin-converting enzyme 2 (ACE2) receptor, which is highly expressed on the surface of type II cells.11–13 The coronavirus spike protein, typically known as the spike protein, is a trimeric glycoprotein. It appears on the virus surface as outward-facing 23 nm molecular “spikes”14 and thus plays a key role in binding receptors.15 A spike protein is composed of three subunits, each composed of around 1270 amino acids.16 Therefore, each trimeric spike protein has around 3800 amino acids, where there exists a huge amount of protein–protein interactions, standing for a highly complicated example of folded proteins.
The allosteric regulation of SARS-CoV-2 spike protein has been reported experimentally17–21 and in computer simulations.22 Specifically, Chi, et al.17 found that antibody 4A8, which was isolated from recovered patients, binds to the nitrogen-terminal domains (NTDs) of the spike protein. These NTDs are around 4–8 nm away from the binding interface between the spike's receptor-binding domain (RBD) and human cell receptor ACE2. Another antibody CR3022, also isolated from a recovered patient, was found to target a highly conserved epitope of SARS-CoV-2 (and SARS-CoV in 2013), which is distal from the spike RBD.18 And antibody 47D11 was reported to bind to a non-RBD epitope of the SARS-CoV-2 (and SARS-CoV) spike protein.19 Very recently, Tulsian et al.20 presented extensive studies on the allosteric regulation in the SARS-CoV-2 spike protein upon the binding of nine antibodies (four from their work and five existing ones, including 4A8 and CR3022). Same as 4A8, antibody LSI-CoVA-017 was found to bind to the spike NTD. Impressively, upon the LSI-CoVA-017 binding to NTD, the S1/S2 cleavage site and other distal domains of the spike protein displayed notable changes in the conformational dynamics using the hydrogen–deuterium exchange mass spectrometry (HDX-MS). Conformational changes were also observed in distal sites of NTD and the S2 subunits of the spike protein when the other antibodies were bound to the spike RBD. The allosteric regulation between the S1/S2 cleavage site and RBD was also predicted by Qiao and Olvera de la Cruz22 using all-atom explicit solvent molecular dynamics (MD) simulations and reported by Chen et al.21 using HDX-MS. Note that the influences of glucans on allosteric regulation in the SARS-CoV-2 spike protein were also examined20 as well as the possible allosteric regulation in ACE2.23,24 Taken together, these experimental observations and in silico prediction support that the allosteric regulation in the SARS-CoV-2 spike protein could reach up to a distance of around 4–8 nm between NTD and RBD,17,20 around 10 nm between the S1/S2 cleavage site and RBD,20–22 or more than 10 nm between the S2 subunit and RBD.20
Even though the allosteric regulation in the spike protein has been reported, the mechanism remains elusive. Specifically, the pathway of signal transmission from the allosteric sites (NTD, the S1/S2 cleavage site) to the active site (RBD) is unknown, which is nevertheless required for the rational design of allosteric neutralizing ligands for the SARS-CoV-2 spike protein. Inspired by the recent findings on the allosteric regulation between the S1/S2 cleavage site and RBD20–22 and between NTD and RBD,17,20 here we examined the routes for the allosteric regulation in the SARS-CoV-2 spike protein. All-atom explicit solvent MD simulations were carried out along with the contrastive learning25 and Ohm26 approaches. These methods collectively reveal the route of the allosteric regulation in the spike protein, which will be beneficial for our understanding of the mechanism of allosteric regulations as well as allosteric inhibitor design.
2. Methods
2.1. All-atom explicit solvent MD simulations
All-atom MD simulations were carried out for the spike–ACE2 complex. Each subunit of the spike trimer was composed of 1273 amino acids (M1–T1273) along with 597 amino acids (S19–D615) for ACE2. Three parallel simulations were performed on the spike–ACE2 complex with one tetrapeptide EELE (Glu-Glu-Leu-Glu) which was bound to the polybasic cleavage site (R682RAR685, PCS) on the subunit A (PCS-A). PCS is a part of the S1/S2 cleavage site (residues 672–69520). The initial structure is provided in Fig. S1 (ESI†). Note that although all three subunits of the spike trimer have the same amino acid sequence, they are structurally different in the “Up” conformation when ACE2 binds to the RBD on the subunit C (RBD-C). We examined the structural change of the spike protein that could be activated by the electrostatic binding between the negatively charged tetrapeptide EELE and the positively charged PCS motif.These simulations were performed using the package GROMACS (version 2019.6)27 at the Texas Advanced Computing Center. Like in our previous work,22 the CHARMM 36m potential28 was used, along with the recommended CHARMM TIP3P water model29 with the water structures constrained using the SETTLE algorithm.30
The spike–ACE2 complex structure was downloaded from the Zhang-Server.15 The subunit C of the spike protein was in the “Up” conformation and binding to ACE2. The spike–ACE2 complex was solvated in a water box with a size of 16 nm × 18 nm × 24 nm. A salt concentration of 0.15 M was applied. The system had 692![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 370 atoms in total.
370 atoms in total.
The energy minimization of the whole system was first conducted using the steepest descent algorithm to remove possible close contact between different molecules. Subsequent equilibrations were conducted for one simulation of 1 ps using the canonical ensemble (constant number of particles, volume, and temperature, NVT) and another simulation of 1 ps using the isothermal–isobaric ensemble (constant number of particles, pressure, and temperature, NPT). The velocity-rescale temperature coupling and the Berendsen pressure coupling were applied. Afterward, the solvated system was equilibrated for another 10 ns under the NPT ensemble with the Nosé–Hoover temperature coupling at 298 K and the Parrinello–Rahman barostat at 1.0 bar.31 The integration time step of 2 fs was used with all the hydrogen-involved covalent bonds constrained using the LINCS algorithm.32,33 In the equilibration simulations above, the coordinates of the non-hydrogen atoms of the spike protein trimer, ACE2, and the tetrapeptides were restrained using a force constant of 1000 kJ mol−1 nm−2 to preserve the binding structure. The restraints were then removed in the production simulations. The other parameters were the same as those in the production simulation. Each production simulation was carried out for a duration of 100 ns using the NPT ensemble. The simulation trajectory was saved at a frequency of 10 frames per 1 ns. A total of 1000 snapshots were thus extracted for each system to collect the contact map of the spike protein Cα atoms.
The contact map between all the Cα atoms of the spike protein from each extracted snapshot was calculated using gmx distance, a utility program of GROMACS. The evolution trajectory of the spike protein was represented by a sequence of contact maps. A contact map C was a two-dimensional matrix whose element, C(i, j), was the spatial Euclidean distance between the Cα atoms of the ith and jth amino acids of the spike protein at a particular moment.
It is noteworthy that additional simulations were performed which had three tetrapeptides EELE, each binding to one of the three PCSs on the spike trimer. These simulations suggested the relatively stronger binding affinity between PCS-A and the peptide EELE neighbor. Specifically, only the binding between PCS-A and the associated peptide EELE was stable for the whole simulation duration of 100 ns. In contrast, the peptides bound to PCS-B and PCS-C became dissociated at less than 100 ns: the peptide EELE bound to PCS-B became dissociated at around 10 ns in the first parallel simulation and was stable for 100 ns in the second simulation; the peptide bound to PCS-C became dissociated at around 40 ns and 30 ns in the two parallel simulations. This is qualitatively consistent with our previous observations.22 The observed dissociation of peptides bound to PCS-B and PCS-C also rationalized the simulation duration of 100 ns for protein–peptide interactions here, though it is likely too short for protein–protein interactions of specifically the large-sized spike protein. Moreover, calculations of the RBD–ACE2 binding affinity in the presence of varying number of neutralizing peptide EELE also supported the dominant role of PCS-A in destabilizing the distal RBD–ACE2 binding (Table S1, ESI†). Therefore, in what follows only the simulations with one peptide EELE bound to PCS-A were further analyzed.
2.2. Contact map feature extraction using contrastive learning
Machine learning (ML) has proven its efficacy in comprehending protein structures, even when dealing with unlabeled data. The MD simulation trajectories of protein structures typically lack labels, necessitating an unsupervised approach for interpretation. To address this, data feature extractors, such as autoencoders combined with clustering algorithms, have been employed to identify phases of protein structure changes or fluctuations.34,35 Self-supervised learning stands as another ML category that can yield robust feature representations from unlabeled data for subsequent tasks.36 Backbone models responsible for generating data feature representations are trained through solving “pretext” tasks, encompassing activities like predicting rotations,37 learning inpainting,38 solving jigsaw puzzles,39 and image coloring.40 However, these hand-crafted pretext tasks often rely on ad hoc heuristics, limiting the generality of the data representations.Contrastive learning25 represents a state-of-the-art self-supervised learning algorithm. It is dataset-agnostic and has demonstrated its efficacy across a broad spectrum of applications, including the study of protein structures.41,42 As shown in Fig. 1, the contrastive learning algorithm learns the feature representations of contact maps by maximizing the agreement between a positive pair (![[x with combining tilde]](https://www.rsc.org/images/entities/i_char_0078_0303.gif) i, j) via a loss function, in which i and j are correlated views of the same contact map x, generated by stochastic data augmentations t ∼ T and t′ ∼ T, respectively. The loss function between a positive pair is defined in eqn (1).
i, j) via a loss function, in which i and j are correlated views of the same contact map x, generated by stochastic data augmentations t ∼ T and t′ ∼ T, respectively. The loss function between a positive pair is defined in eqn (1).
 | (1) |
 is cosine similarity of contact maps (u, v). τ is a temperature parameter, which is empirically determined. In Fig. 1, f(·) is the backbone representation encoder. Resnet50 is used for this purpose. g(·) is a projection head, which in this work is a multilayer perceptron with one hidden layer. Both f(·) and g(·) are trained to maximize the agreement between the positive pairs of augmented views of contact maps using the loss function. The dimension of the extracted contact map representation is a 2048 × 1 vector in our work. The augmentation candidate set T are the following that are sequentially and randomly (with a probability of 0.5) applied: random cropping followed by resizing back to the original size, Sobel filtering, random horizontal flipping, and Gaussian blurring. After the contrastive learning model is trained, the projection head is thrown away. The output of the backbone representation encoder is the feature representation of the corresponding contact map. The feature representation vectors of contact maps obtained by the all-atom MD simulations are then grouped via the k-means clustering algorithm to reveal the evolution stages of SARS-CoV-2 spike protein structures in the process of binding to the human cell receptor ACE2.
is cosine similarity of contact maps (u, v). τ is a temperature parameter, which is empirically determined. In Fig. 1, f(·) is the backbone representation encoder. Resnet50 is used for this purpose. g(·) is a projection head, which in this work is a multilayer perceptron with one hidden layer. Both f(·) and g(·) are trained to maximize the agreement between the positive pairs of augmented views of contact maps using the loss function. The dimension of the extracted contact map representation is a 2048 × 1 vector in our work. The augmentation candidate set T are the following that are sequentially and randomly (with a probability of 0.5) applied: random cropping followed by resizing back to the original size, Sobel filtering, random horizontal flipping, and Gaussian blurring. After the contrastive learning model is trained, the projection head is thrown away. The output of the backbone representation encoder is the feature representation of the corresponding contact map. The feature representation vectors of contact maps obtained by the all-atom MD simulations are then grouped via the k-means clustering algorithm to reveal the evolution stages of SARS-CoV-2 spike protein structures in the process of binding to the human cell receptor ACE2.
 | ||
| Fig. 1 The framework of contrastive learning contact map feature extraction and protein structure transition stage detection. (i, j) are considered a positive pair when they represent correlated views of the same contact map x, produced through stochastic data augmentations t ∼ T and t′ ∼ T, respectively. The augmentation sequence T, employed in this study, is executed sequentially and applied randomly with a 0.5 probability, encompassing random cropping followed by resizing back to the original dimensions, Sobel filtering, random horizontal flipping, and Gaussian blurring. The backbone representation encoder, denoted as f(·), specifically employs Resnet50. g(·), a compact projection head, is designed as a multilayer perceptron (MLP) featuring a single hidden layer. Both f(·) and g(·) are trained with the primary objective of maximizing agreement among positive pairs of augmented contact map views using the loss function outlined in eqn (1). The feature extractor, obtained via contrastive learning, processes the contact maps. Subsequently, the k-means clustering algorithm is used to group the series of contact maps into stages representing structural transitions. | ||
2.3. Ohm for allosteric regulation pathway
Based on the contact matrix of proteins, Wang, et al. very recently proposed the Ohm method.26 In the Ohm method, a perturbation propagation algorithm was developed, which was a repeated stochastic process of perturbation propagation on a network of interacting amino acids in a protein. Therefore, Ohm specializes in characterizing the allosteric regulation of proteins by examining the propagation of protein structure perturbation. To predict the allosteric regulation pathway, Ohm relies exclusively on the protein structure, making it computationally efficient. Ohm was found to be able to successfully map allosteric networks for a database of 20 proteins for which the allosteric sites were experimentally known. Wang, et al. further developed an automated web server (https://dokhlab.med.psu.edu/ohm/) for mapping, visualizing, and characterizing allosteric communication networks.Here, the Ohm server was employed. By specifying the start of the allosteric pathway (PCS and NTD, see Table S2, ESI†) and the end which is the RBD on the subunit C (residues L455–Y505), Ohm reports all the critical amino acids on the allosteric route.
Note that antibody 4A8 primarily binds to two motifs of NTD3 and NTD5 of the spike protein.17 After examining the amino acids on the NTD3 and NTD5 epitopes, we found that the NTD3 epitope (L141GVYYHK147NNK150SWMESE156) is similar to PCS. Specifically, the central fragment K147NNK150 is positively charged and exposed, akin to PCS. It might be promising to design NTD3-targeting, negatively charged neutralizing peptides that could destabilize the spike–ACE2 binding given the fact that the spike protein and ACE2 are both negatively charged.22 Therefore, in the present work we are focusing on the NTD3 motif when identifying the allosteric regulation routes from NTD to the RBD on the subunit C.
3. Results and discussion
We first carried out all-atom explicit solvent MD simulations on the spike–ACE2 complex. One tetrapeptide EELE was initially associated with the PCS on the subunit A of the spike trimer. Note that the subunit C was in the “Up” conformation and formed direct binding with ACE2. Illustrated in Fig. 2 is the final structure of one simulation. The parallel simulations supported the stable binding between PCS-A and the EELE peptide. | ||
| Fig. 2 Molecular structure of the spike–ACE2 complex in the presence of the neutralizing tetrapeptides EELE. (a) Front view and (b) back view. The three subunits of the trimeric spike protein are colored in ice blue/cyan/orange for the subunits A/B/C, respectively. The spike protein's PCS, NTD, and RBD are colored in blue, dark blue, and orange, respectively. The ACE2 receptor and the tetrapeptide EELE are colored in silver and red, respectively. | ||
We hypothesize that the strong electrostatic attractions between the positively charged PCS motif (R682RAR685) and the negatively charged EELE tetrapeptides trigger a local structural fluctuation that might eventually lead to a global conformational adaptation of the spike protein. In this work, we are primarily examining the route from the tetrapeptide EELE-triggered local structural fluctuation to the distal influence in destabilizing the RBD–ACE2 binding.
3.1. Stages of the spike protein structure transition obtained via contrastive learning and clustering
Each atomistic simulation ran 100 ns, where 1000 frames were saved at the frequency of 0.1 ns per frame. The contact map of the spike protein Cα atoms was consequently generated as a function of the simulation time. See the Methods section for the details. Contrastive ML and clustering were subsequently performed for the obtained contact maps, and the characteristic structures and stages were determined accordingly.Fig. 3 shows the results of the contrastive ML analysis of one of the three parallel MD trajectories of spike protein structure transition in the process of protein–ACE2 binding. The corresponding contrastive ML analyses for the other two parallel MD trajectories are provided in Fig. S2 (ESI†). A sequence of 1000 contact maps of the spike protein was generated based on the atomistic MD trajectory. Feature vectors of the contact maps are extracted by the backbone feature extractor of the contrastive learning model, which is a deep resnet50 model43 in this work. The contrastive learning model is trained by maximizing the agreement of positive pairs (augmented views of the same contact maps). The details of contrastive learning are discussed in the Methods section. After the contrastive model is trained, the feature extractor is utilized to generate feature vectors of the contact maps. A feature vector in this work is a 2048 × 1 vector. These contact map feature vectors are then grouped using the k-means algorithm to find the stages of the protein structure transition. To find the optimal number of clusters k, cluster numbers ranging from 1 to 15 were tried, and the elbow method and the average silhouette scores method were utilized (Fig. 3(a) and (b)) to determine the optimal number of clusters. Both the elbow and silhouette score methods indicate an optimal number of clusters of k = 10. The contact map that is the closest to the centroid of a cluster is used as the representative of the state. As shown in Fig. 3(c), these contact map IDs are 26, 91, 222, 351, 467, 567, 644, 729, 860, and 954 (occurred at 2.6 ns, 9.1 ns, 22.2 ns, 35.1 ns, 46.7 ns, 56.7 ns, 64.4 ns, 72.9 ns, 86.0 ns, and 95.4 ns, respectively).
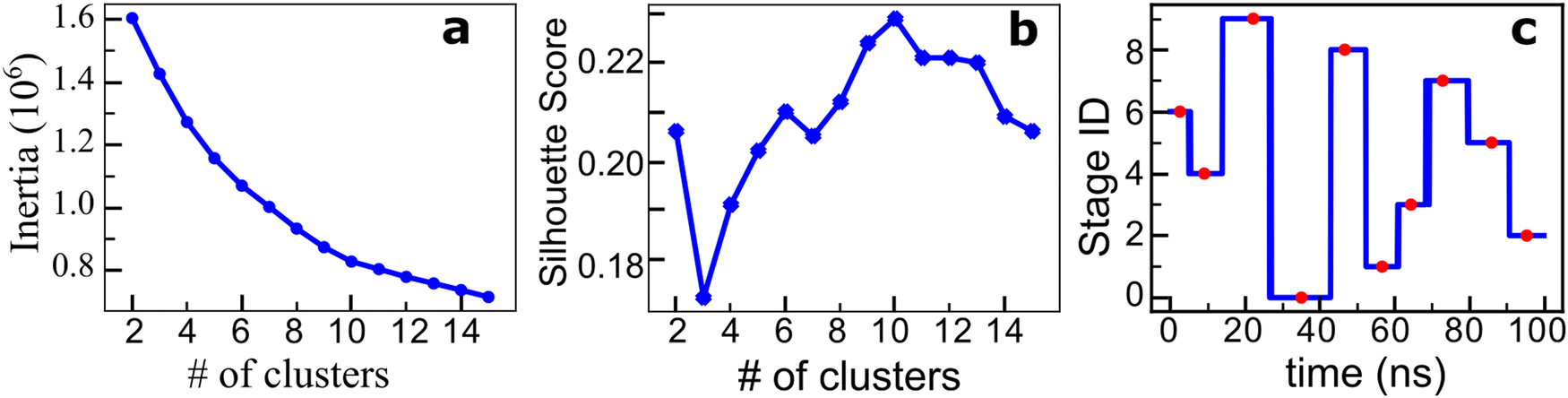 | ||
| Fig. 3 SARS-CoV-2 spike protein structure transition analysis using contrastive learning and k-means clustering. (a) Elbow method using inertia. (b) The average silhouette score with different numbers of clusters. Both criteria indicate that k = 10 is the optimal number of clusters. (c) Ten clustered stages of spike protein structure transition in the process of the spike–ACE2 binding, in chronological order. The red dots are the corresponding positions of the contact map that are closest to the centroid of each cluster. | ||
3.2. Structural fluctuations in the spike structure in the stages from contrastive ML
As provided in the contrastive ML learning and clustering (Fig. 3(c)), a total of 10 stages were identified in the spike–ACE2 complex in the presence of the tetrapeptides EELE. Accordingly, we calculated the root-mean-square fluctuation (RMSF) of the spike protein Cα atoms for all the ten stages. The calculated RMSFs are presented in Fig. 4. | ||
| Fig. 4 Structural fluctuations of the spike protein in the presence of a tetrapeptide EELE obtained from the atomistic MD simulation. (a) Subunit A, (b) subunit B, and (c) subunit C. The Cα RMSFs were calculated for the 10 stages derived from the contrastive ML. The RMSFs are shifted by (n − 1) × 0.5 nm for the display, where n = 1–10 stands for the corresponding stage ID provided on the right of panel (c). | ||
As demonstrated in Fig. 4(a), PCS-A displays the strongest structural fluctuation in the first stage which is ascribed to the tetrapeptide EELE binding, which became gradually weakened over the simulation time. The RBD on the subunit C, which directly binds ACE2, displayed elevated structural fluctuation from stage 2 till the end of the simulation (Fig. 4(c)). In contrast with the remarkable structural fluctuation of PCS-A, it is much weaker for the PCSs (R682RAR685) on the subunits B and C (Fig. 4(b) and (c)), indicating their negligible impacts.
The residues close to the N-terminal (residue numbers less than 300) display relatively larger fluctuations for all three subunits. This motif is actually the N-terminal domain (NTD), which is known to bind antibodies 4A817 and LSI-CoVA-017.20 The calculated RMSFs thus support the intrinsically flexible feature of NTD, which is desired for structural fluctuation and propagation. The relatively large fluctuations on the C-terminal residues (residues 1000–1273), which are located on the S2 subunits, are ascribed to the partially unstructured features and are thus ignored here.
3.3. Pathway of the allosteric regulation from PCS to RBD, and from NTD to RBD
As illustrated in Fig. 4, the contrastive ML supports the correlated structural fluctuation between PCS-A and RBD on the subunit C. However, the detailed route is still missing. In this regard, the Ohm approach26 is employed to predict the allosteric regulation pathway.We examined the allosteric regulation route from all three PCSs to the RBD on the subunit C. As illustrated in Fig. 5, the backbone atoms of the critical residues are highlighted to direct the pathway. Impressively, the allosteric regulation route is found to propagate across different subunits. For instance, the route starting with PCS-A propagates starting from subunit A to subunit C, to subunit B, and eventually to subunit C (Fig. 5(a)), indicating the nonlinear nature of allosteric regulation.
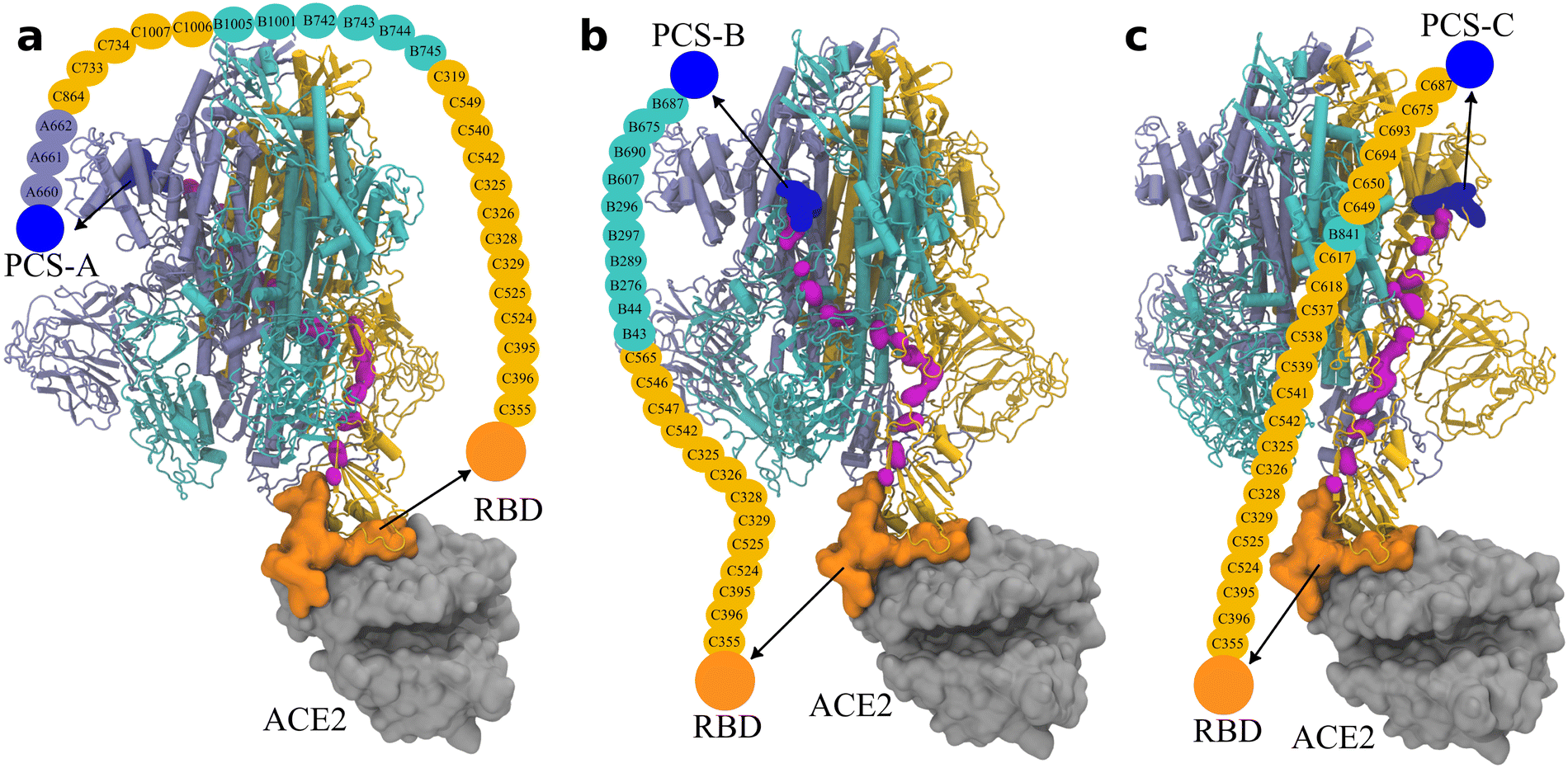 | ||
| Fig. 5 Pathway of the allosteric regulation in the spike protein obtained via Ohm from (a) PCS-A, (b) PCS-B, and (c) PCS-C, to the RBD on subunit C. The three subunits of the trimeric spike protein are colored in ice blue/cyan/orange for the subunits A/B/C, respectively. PCSs are colored in blue and the RBD on subunit C in orange, which is in direct contact with ACE2 (in silver). The backbone atoms on the connecting amino acids are indicated by magenta; the names of the connecting amino acids are provided in the illustration. The corresponding rotation movies are provided as Movie S1 (ESI†). | ||
Moreover, the protein backbone-labeled route is shown to be discontinuous at a couple of sites. See also the Movies (ESI†). It supports that in addition to the backbone atoms, the side chains of the critical residues are also involved in the structural propagation, which is absent in the contrastive ML (Fig. 3) and the RMSF calculations (Fig. 4). That said, for a complete understanding of the allosteric regulation route, different approaches are collectively desired.
Owing to the long distance of approximately 10 nm from the PCSs to the RBD, there exist 27 amino acids on the allosteric regulation routes from PCS-A to RBD, and 23 amino acids from the other two PCSs to RBD.
Given the significant role of NTD observed in the experiments17,20 and our RMSF calculations (Fig. 4), we also examined the route of the allosteric regulation from NTD to RBD (Fig. 6). Even though NTD-A is physically closer (∼9.1 nm) to the RBD–ACE2 binding interface than PCS-A to the interface (∼10.6 nm), there exist more critical residues than that for PCS-A (33 vs. 27), indicating the indirect feature of allosteric regulation. Surprisingly, the route from NTD-A to RBD shares a large number (i.e., 18) of critical residues with the route from PCS-A to RBD. This accounts for 54.5% (18/33) for the NTD-A–RBD route and 66.7% (18/27) for the PCS-A–RBD route, respectively. Similarly, the route starting with NTD-B shares 63.6% (14/22) critical residues with the route starting with PCS-B (14/23 = 60.9%), and the route from NTD-C shares 39.1% (9/23) critical residues with the one from PCS-A (9/23 = 39.1%). It thus indicates that the routes from the PCSs and the NTDs are likely correlated, that is, the presence of an allosteric regulation network in the spike protein (Movie S3, ESI†).
 | ||
| Fig. 6 Pathway of the allosteric regulation in the spike protein obtained via Ohm from (a) NTD-A, (b) NTD-B, and (c) NTD-C, to the RBD on subunit C. NTDs are colored in dark blue. The other color codes are the same as those in Fig. 5. The corresponding rotation movies are provided as Movie S2 (ESI†). | ||
4. Conclusions
We demonstrate the route of allosteric effects in the spike protein of SARS-CoV-2. The EELE tetrapeptides prefer binding to the polybasic cleavage site on the first subunit of the trimeric spike protein. The fluctuation of the spike protein was activated upon the binding of the EELE tetrapeptide. The structural fluctuation is found to propagate across different subunits, and amino acid side chains are also contributing to the propagation. Impressively, we found that the routes from the PCSs to RBD share a large number of the critical amino acids, ranging from 39% up to 67%, with the corresponding routes from the NTDs to RBD. It thus suggests the presence of an allosteric regulation network in the SARS-CoV-2 spike protein and likely in other proteins.In summary, by coupling contrastive learning-based contact map feature extraction, all-atom explicit solvent MD simulations, and Ohm, we have revealed the route of allosteric regulation in the spike protein of SARS-CoV-2. Impressively, the NTDs are found to share the majority of route of allosteric regulations with the PCSs. This work thus sheds insights into the fundamental understanding of allosteric regulations in protein–protein interactions as well as into the rational design of allosteric drugs.
Author contributions
Y. W. developed machine learning software, carried out machine learning simulations, analyzed data and machine learning results, and wrote the manuscript. A. C. performed atomistic simulations, analyzed data, and wrote the manuscript. Y. L. designed the machine learning approach and analyzed machine learning results. B. Q. performed the Ohm study. T. W. and B. Q. designed the project, contributed to the atomistic simulations and data analysis, and wrote the manuscript.Conflicts of interest
The authors have no conflicts to disclose.Acknowledgements
Y. W. is grateful for the computational resources offered by the National Science Foundation (#1548562) through its Extreme Science and Engineering Discovery Environment (XSEDE) at Pittsburgh Supercomputing Center (PSC). A. C. and B. Q. acknowledge the Texas Advanced Computing Center (TACC), The University of Texas at Austin for the computational resources. T. W. and B. Q. thank the support from the National Science Foundation awards (#: 2118099 to T. W. and #: 2328095 to B. Q.).Notes and references
- H. N. Motlagh, J. O. Wrabl, J. Li and V. J. Hilser, Nature, 2014, 508, 331–339 CrossRef CAS PubMed.
- N. M. Goodey and S. J. Benkovic, Nat. Chem. Biol., 2008, 4, 474–482 CrossRef CAS PubMed.
- J. Kuriyan and D. Eisenberg, Nature, 2007, 450, 983–990 CrossRef CAS PubMed.
- J. Xie and L. Lai, Curr. Opin. Struct. Biol., 2020, 62, 158–165 CrossRef CAS PubMed.
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS PubMed.
- T. Zhang, T. Wei, Y. Han, H. Ma, M. Samieegohar, P.-W. Chen, I. Lian and Y.-H. Lo, ACS Cent. Sci., 2016, 2, 834–842 CrossRef CAS PubMed.
- E. Guarnera and I. N. Berezovsky, Curr. Opin. Struct. Biol., 2020, 62, 149–157 CrossRef CAS.
- A. F. Abdel-Magid, ACS Med. Chem. Lett., 2015, 6, 104–107 CrossRef PubMed.
- R. Nussinov and C.-J. Tsai, Cell, 2013, 153, 293–305 CrossRef CAS PubMed.
- A. J. Faure, J. Domingo, J. M. Schmiedel, C. Hidalgo-Carcedo, G. Diss and B. Lehner, Nature, 2022, 604, 175–183 CrossRef CAS PubMed.
- J. Machhi, J. Herskovitz, A. M. Senan, D. Dutta, B. Nath, M. D. Oleynikov, W. R. Blomberg, D. D. Meigs, M. Hasan and M. Patel, J. Neuroimmune Pharmacol., 2020, 15, 359–386 CrossRef.
- Y. Y. Zuo, W. E. Uspal and T. Wei, ACS Nano, 2020, 14, 16502–16524 CrossRef CAS PubMed.
- Y. J. Hou, K. Okuda, C. E. Edwards, D. R. Martinez, T. Asakura, K. H. Dinnon III, T. Kato, R. E. Lee, B. L. Yount and T. M. Mascenik, Cell, 2020, 182, 429–446.e414 CrossRef CAS.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C.-L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS.
- C. Zhang, W. Zheng, X. Huang, E. W. Bell, X. Zhou and Y. Zhang, J. Proteome Res., 2020, 19, 1351–1360 CrossRef CAS PubMed.
- A. C. Walls, Y.-J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 181, 281–292.e286 CrossRef CAS PubMed.
- X. Chi, R. Yan, J. Zhang, G. Zhang, Y. Zhang, M. Hao, Z. Zhang, P. Fan, Y. Dong, Y. Yang, Z. Chen, Y. Guo, J. Zhang, Y. Li, X. Song, Y. Chen, L. Xia, L. Fu, L. Hou, J. Xu, C. Yu, J. Li, Q. Zhou and W. Chen, Science, 2020, 369, 650–655 CrossRef CAS PubMed.
- M. Yuan, N. C. Wu, X. Zhu, C.-C. D. Lee, R. T. Y. So, H. Lv, C. K. P. Mok and I. A. Wilson, Science, 2020, 368, 630–633 CrossRef CAS PubMed.
- C. Wang, W. Li, D. Drabek, N. M. A. Okba, R. van Haperen, A. D. M. E. Osterhaus, F. J. M. van Kuppeveld, B. L. Haagmans, F. Grosveld and B.-J. Bosch, Nat. Commun., 2020, 11, 2251 CrossRef CAS PubMed.
- N. K. Tulsian, R. V. Palur, X. Qian, Y. Gu, B. Shunmuganathan, F. Samsudin, Y. H. Wong, J. Lin, K. Purushotorman, M. M. Kozma, B. Wang, J. Lescar, C.-I. Wang, R. K. Gupta, P. J. Bond and P. A. MacAry, Nat. Commun., 2023, 14, 6967 CrossRef CAS PubMed.
- C. Chen, R. Zhu, E. A. Hodge, M. A. Díaz-Salinas, A. Nguyen, J. B. Munro and K. K. Lee, ACS Infect. Dis., 2023, 9, 1180–1189 CrossRef CAS PubMed.
- B. Qiao and M. Olvera de la Cruz, ACS Nano, 2020, 14, 10616–10623 CrossRef CAS PubMed.
- K. Dutta, ACS Pharmacol. Transl. Sci., 2022, 5, 179–182 CrossRef CAS PubMed.
- M. L. Mugnai and D. Thirumalai, J. Chem. Phys., 2023, 158, 215102 CrossRef CAS PubMed.
- T. Chen, S. Kornblith, M. Norouzi and G. Hinton, Presented in part at the Proceedings of the 37th International Conference on Machine Learning, 2020 Search PubMed.
- J. Wang, A. Jain, L. R. McDonald, C. Gambogi, A. L. Lee and N. V. Dokholyan, Nat. Commun., 2020, 11, 3862 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmuller and A. D. MacKerell Jr, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- D. Bhowmik, S. Gao, M. T. Young and A. Ramanathan, BMC Bioinf., 2018, 19, 47–58 CrossRef PubMed.
- J. Chen, E. Xu, Y. Wei, M. Chen, T. Wei and S. Zheng, Langmuir, 2022, 38, 10817–10825 CrossRef CAS PubMed.
- A. Kolesnikov, X. Zhai and L. Beyer, arXiv, 2019, arXiv:1901.09005, DOI:10.48550/arXiv.1901.09005.
- S. Gidaris, P. Singh and N. Komodakis, arXiv, 2018, arXiv: 1803.07728 DOI:10.48550/arXiv.1803.07728.
- D. Pathak, P. Krahenbuhl, J. Donahue, T. Darrell and A. A. Efros, arXiv, 2016, arXiv: 1604.07379, DOI:10.48550/arXiv.1604.07379.
- M. Noroozi and P. Favaro, Presented in part at the European conference on computer vision, 2016 Search PubMed.
- R. Zhang, P. Isola and A. A. Efros, Presented in part at the European conference on computer vision, 2016 Search PubMed.
- P. C. T. Souza, S. Thallmair, S. J. Marrink and R. Mera-Adasme, J. Phys. Chem. Lett., 2019, 10, 7740–7744 CrossRef CAS PubMed.
- P. Hermosilla and T. Ropinski, arXiv, 2022, arXiv: 2205.15675, DOI:10.48550/arXiv.2205.15675.
- K. He, X. Zhang, S. Ren and J. Sun, Presented in part at the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures, table, and movies. See DOI: https://doi.org/10.1039/d4cp00106k |
| This journal is © the Owner Societies 2024 |