
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron–vibrational renormalization in fullerenes through ab initio and machine learning methods†‡
Pablo
García-Risueño
 *a,
Eva
Armengol
b,
Àngel
García-Cerdaña
b,
Juan María
García-Lastra
c and
David
Carrasco-Busturia
*de
*a,
Eva
Armengol
b,
Àngel
García-Cerdaña
b,
Juan María
García-Lastra
c and
David
Carrasco-Busturia
*de
aIndependent scholar, Barcelona, Spain. E-mail: risueno@unizar.es
bArtificial Intelligence Research Institute, (IIIA, CSIC) Carrer de Can Planes, s/n, Campus UAB, 08193 Bellaterra, Catalonia, Spain
cDepartment of Energy Conversion and Storage, Technical University of Denmark, 2800 Kgs. Lyngby, Denmark
dDTU Chemistry, Technical University of Denmark, DK-2800 Kgs. Lyngby, Denmark
eDivision of Theoretical Chemistry and Biology, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: davidcdb@kth.se
First published on 18th June 2024
Abstract
The effect of nuclear vibrations on the electronic eigenvalues and the HOMO–LUMO gap is known for several kinds of carbon-based materials, like diamond, diamondoids, carbon nanoclusters, carbon nanotubes and others, like hydrogen-terminated oligoynes and polyyne. However, it has not been widely analysed in another remarkable kind which presents both theoretical and technological interest: fullerenes. In this article we present the study of the HOMO, LUMO and gap renormalizations due to zero-point motion of a relatively large number (163) of fullerenes and fullerene derivatives. We have calculated this renormalization using density-functional theory with the frozen-phonon method, finding that it is non-negligible (above 0.1 eV) for systems with relevant technological applications in photovoltaics and that the strength of the renormalization increases with the size of the gap. In addition, we have applied machine learning methods for classification and regression of the renormalizations, finding that they can be approximately predicted using the output of a computationally cheap ground state calculation. Our conclusions are supported by recent research in other systems.
I. Introduction
Fullerenes1–4 have been an object of great interest since the discovery of the first one, the buckminsterfullerene in the 1980s, which was awarded with a Nobel Prize. In recent times there has been an increasing research effort in fullerenes due to their properties and applications. These include superconductivity, ferromagnetism and many potential biomedical utilities.4–10 In addition, fullerenes have acquired a prominent role in photovoltaics.11–17 For example, fullerene derivatives are suitable for the elaboration of photovoltaic cells and photodetectors.18 In the last decades the market has been dominated by inorganic, silicon-based solar cells; these, however, present marked drawbacks, like their cost, weight, lack of flexibility and high fabrication-related environmental and energetic costs.11 In contrast, these inconveniences are solved to a great extent in organic solar cells which include fullerenes: their preparation is cheaper and less polluting, they can be light, flexible, semitransparent and suitable for large-area devices.11,14,17,19 In perovskite-based solar cells, the addition of fullerenes can double the power conversion efficiency.12 These properties have boosted a strong interest in fullerene-based photovoltaic materials. In them, fullerenes can take the role of acceptors, taking electrons in their unoccupied states, which enables a photocurrent which is a source of electrical energy.11,12 The interplay between a donor polymer and a fullerene acceptor leads to an increased performance of the polymer solar cells compared with inorganic cells.The electron–vibrational interaction can have a remarkable impact on properties,20,21 like the eigenvalues of the HOMO and LUMO and the HOMO–LUMO gap, which are central for many applications, such as the mentioned photovoltaics.22,23 The LUMO level itself is a key quantity for the performance of solar cells, and the difference between LUMO levels of the donor and acceptor can be of the same order of magnitude (hundreds of meV)11 as the typical renormalizations due to phonons of carbon-based materials; the effect of nuclear motion on electronic properties is strong in bulk diamond,24,25 diamondoids26,27 and graphene.28,29 Hence, the calculation of the impact of electron–vibrational interaction on electronic eigenvalues must be taken into account for an appropriate analysis of relevant properties of fullerenes. Despite the fact that high-quality works on the electron–phonon interaction in fullerenes have been performed,30 there is a lack, to the best of our knowledge, of an extensive analysis of it in a wide number of fullerenes, which makes it possible to extract a general conclusion on the strength of such interaction in these systems. In this article we attempt to perform such an analysis. We present the HOMO–LUMO gap renormalization of several fullerenes, as well as fullerene derivatives with applications in photovoltaics.18,19,31 These renormalizations were calculated in the density-functional theory (DFT) framework.32 Despite the fact that more expensive methods, like GW,33,34 provide higher accuracy,25,30 DFT is expected to provide reasonable results in the calculation of quantities derived from the electron–vibrational interaction.27,35 Moreover, recent research proved that gap renormalizations calculated with GGA-PBE are similar to those of B3LYP for small molecular carbon compounds.26
During the past years, machine learning36–38 (ML) has emerged as an incredibly powerful tool for a variety of applications. Since ML is especially suitable for identification of patterns, some of these applications include medical diagnoses, language processing and generation (chatbots), computer vision, automatic driving or internet search engines, among many others. The complexity of quantum mechanics, where numerous particles interact in intricate manners, makes the suitability of ML for forecasting of features of quantum systems far from granted, for an unaffordable amount of data might be necessary for appropriate training.39,40 Nonetheless, in recent times numerous authors have proved that ML is indeed suitable also in a quantum mechanical context. For example, ML has achieved impressive results in biophysics, where the application of deep neural networks has found a solution to the problem of protein folding,41 which had been unresolved for over 50 years. Other remarkable applications of ML to problems of physics include materials discovery,38,39,42 accelerating molecular dynamics or promptly finding solutions for equations in a variety of formalisms, including the Schrödinger equation (directly), reduced density matrix theory,43 Green's functions or tight-binding Hamiltonians.38,44,45 Through the usage of data from DFT, which is probably the most widely employed theory for discovery of materials, ML has been applied to several purposes, like approximating density functionals46–49 or determining properties of the system, like structures,50,51 excitation and atomization energies,52,53 catalytic activities,54 and many other quantities like band gap or electron affinity.55 This article continues the latter line, presenting ML forecasts for renormalization of electronic eigenvalues due to the interaction between electrons and phonons. Some authors have already used machine learning in the context of electron–phonon theory, e.g. for efficient computation of potential energy surfaces.56 Others have suggested its use for efficient material discovery57 or materials characterization.58 Recently, ML (neural networks, in particular) has also been applied to calculate energy levels renormalized due to the electron–phonon interaction using Holstein Hamiltonians in a Heisenberg chain59 and many-body perturbation (Allen–Heine–Cardona) theory in diamond.60 Moreover ref. 61 employed an approach very similar to the one that we present in this paper. They also evaluated the renormalization of electronic energies due to electron–phonon interaction through ab initio methods in less than two hundred (133) systems and used machine learning methods for forecasting them without further explicit electron–phonon calculations. The primary differences between the analysis presented in ref. 61 (whose research project is fully independent and had no communication with ours) and our current study lie in both the analyzed systems and the methods employed for calculating renormalizations. While the former investigates 2D materials using the special displacement method, our work focuses on fullerenes and utilizes the frozen phonon approach (with corrections for the effects due to crossings and anticrossings26). Recent research hints that the special displacement method might yield not fully accurate predictions for band gap renormalizations in some cases, like C214N and C510N62 using moderately sized supercells.
The present article is structured as follows. In Section II we present the input data employed in our calculations; in Section III we present the methods used to perform these calculations. Section III.A presents our procedures for performing the calculations of renormalization of electronic eigenvalues due to electron–phonon interactions. In Section III.B we present the machine-learning methods employed for efficient forecasting of these renormalizations, with its subsections describing the methods for regression using random forests (Section III.B.1) and the methods for calculations of classification using decision trees (Section III.B.2). Our results are presented in Section IV, which presents the calculated renormalizations (Section IV.A) and the results of ML-based forecasts of them (Section IV.B). Finally, in Section V we briefly outline the conclusions of this work.
II. Data
Our analysis takes atomic positions of fullerenes as the starting point. Many of them were generated through geometric means, which means that not all them correspond to synthesized and isolated molecules. Synthesis of fullerenes has so far proved to be a tricky task, and researchers continue to search for better ways for it. The easiest-to-produce fullerenes are63 C60-Ih (buckminsterfullerene, footballene) and C70-D5h (rugbyballene), yet others are also attainable. For example, plasma synthesis contains a mixture of fullerenes, with C60 being dominant.64 Several research works state that fullerenes of many different sizes have been produced (e.g. Cn for all even numbers between 30 and 8465). Many fullerenes with over 70 carbon atoms (higher fullerenes) have been synthesized in the past decades (though not always in an isolated molecular form). This includes C72,66 C74,67 C76,68 C80,69 C82,68 C84,63 C86 and C88,70 as well as C90 to C106.71 Synthesis of lower fullerenes (those made of fewer than 60 carbon atoms) is more complex, probably due to their higher curvature (which is thought to make them more interesting72). For instance, syntheses of C20,73 C32,65,74 C3675 and C5076,77 have been attained (again, not necessarily in molecular form). Notwithstanding their non-availability, many authors have published theoretical research based on not-yet-synthesized fullerenes. Note that even the first discovered fullerene was theoretically proposed before its first synthesis.78 Examples of simulations of non-synthesized-and-isolated fullerenes are given in ref. 72 and 79–85, among many others. For example, ref. 84 displays computed properties of 271 isomers of the C50 fullerene, ref. 86 simulates 2385 isomers of C62 and ref. 85 performs calculations of a not-yet-synthesized solid which involves C32. Finding their properties in silico is considered a good way to estimate which ones have more promising features, and hence in which syntheses special efforts should be made. For example, fullerenes with strong electron–phonon interactions may be good candidates for high temperature superconductors.72In the present paper, we use coordinates of fullerenes mainly generated by geometrical means to generate a family of molecules, which are expected to have similar features that can be extracted using statistics and machine learning. We present our procedure as a proof-of-concept, i.e. an example to be applied to other sets of systems which share a similar structure among them.
Our calculations consist of two clearly different stages: (i) quantum mechanical; (ii) ML-based. The first one consists of ab initio computations of electronic eigenvalues and the subsequent evaluation of renormalizations due to the interactions between electrons and vibrating nuclei87 using the method presented in ref. 26. The second stage uses the information outputted in the first stage as its input.
The input data for the first stage (ab initio) is merely the set of atomic coordinates of the analysed molecules, together with the chosen set of input parameters for the calculation (e.g. pseudopotentials, plane wave cutoff, etc.).
For the second stage (ML-based) we have considered numerous variables as possible inputs for the employed regression and classification methods. We classify them into eleven sets of features: electronic structure features (selected, few, all), geometric features, phonon features, bond length features and bond order features (Mayer, GJ, NM1, NM2, NM3). Comprehensive explanations about them can be found in the ESI.‡ The electronic structure features are obtained from the output of the DFT ground state calculation. These are easy to obtain from a human viewpoint; moreover, they are computationally cheap, because just one PBE-based relaxation, one PBE-based ground state calculation and one B3LYP-based calculation are necessary (see Section SII of the ESI‡). Many of the electronic structure features are differences between electronic eigenvalues (e.g. HOMO–LUMO gap using PBE and B3LYP functionals, εHOMO − εHOMO−1, …, εHOMO − εHOMO−6, εLUMO+1 − εLUMO, …, εLUMO+6 − εLUMO) as well as their inverses (e.g. (εHOMO − εHOMO−1)−1). The reason for considering the inverses of differences between eigenvalues is that such terms appear in the renormalizations given by many-body perturbation theory according to the Allen–Heine–Cardona formalism.26,88,89 We also consider the mean, variance, skewness and kurtosis coefficients of the electronic eigenvalues, as well as the inverses of the average occupied and unoccupied eigenvalues. We analyse three sets of regressors containing electronic structure features. One of them (all, consisting of 38 variables) includes all them; another one (few) includes 10 of the variables; finally, the feature set called electronic structure selected consists of a lower number of regressors, which were chosen based on their predictive power (further remarks on these variables can be found in Section IV.B and in the ESI‡).
Another set of variables which were tried as input (regressors) of the machine learning methods corresponds to phonon features. It includes the minimum and maximum phonon frequencies, as well as the mean, variance skewness and kurtosis coefficients of the set of phonon frequencies of the fullerene. Note that the evaluation of these frequencies is numerically more complex (i.e. it requires more computing time) than a single ground state calculation, and hence one of the goals of ML-based forecasting of renormalizations would be to avoid performing the phonon calculations (i.e. to avoid solving the dynamical equation90).
The geometric features include the number of atoms and the number of hexagons occurring in the fullerene (the number of pentagons is constant, 12, for all them91), as well as the surface and volume of the molecule and the quotient between both.
The bond length features are a collection of regressors calculated from the lengths of the bonds of each molecule. Finally, there are five bond order features (Mayer, GJ, NM1, NM2 and NM3), which were calculated using the information on the bond orders of the molecules. These five features differ in the way the bond orders are calculated (see the ESI‡ for further details).
So far we have listed the regressors (input columns) of our dataset. The regressands (i.e. the quantities to forecast) are the renormalizations of the HOMO, LUMO and HOMO–LUMO gap. The rows of our dataset are those displayed in Table 2, excluding the C62-C2v (which is singular because it contains a ring of just 4 carbons) as well as the fullerene derivatives ([6,6]-phenyl-C61-butyric acid methyl ester [60]PCBM, [70]PCBM and indene IC60BA) which due to the attached atoms are expected to behave slightly differently than pristine fullerenes. We have also discarded three of the fullerenes (C28-D2, C30-C2v-b, C58-C3), considering them outliers, because the values of the LUMO renormalization that they provide strongly differ from the rest. Their names appear in italics in Table 2.
III. Methods
A. Renormalization of electronic eigenvalues
Our calculations are based on the frozen-phonon method26,92,93 to calculate the variation of electronic eigenvalues (renormalization) due to the interaction among electrons and phonons. This method consists of calculating the ground state of the system with relaxed nuclear positions and with positions displaced in the directions given by the nuclear vibrations (normal modes). This provides the renormalizations for both zero (zero-point renormalization) and nonzero temperatures. The calculation of eigenvalue renormalizations using the frozen-phonon method (as presented in ref. 26 and 92) is more efficient and less complex than the explicit calculation of electron–phonon couplings24,88 because it does not require summations in unoccupied eigenstates, which converge very slowly. We performed our calculations following an accurate version of the frozen-phonon method recently developed by us;26 this method avoids distortions arising from mixing of states (anticrossing). We used the Quantum Espresso (v.5.3.0) DFT code94 with HGH pseudopotentials.95 For efficiency's sake we have performed all the calculations necessary to evaluate renormalizations using the GGA-PBE96 exchange correlation functional and a basis set with a plane-wave cutoff of 30 Ry (see Section SI of the ESI‡ for remarks about convergence). In addition, we have run ground state calculations using the B3LYP functional,97,98 so that the HOMO–LUMO gap calculated with B3LYP can be used as a regressor in our subsequent machine learning-based analysis. Despite our choice of GGA due to its low computational cost, the conclusions presented in this article are expected to hold for renormalizations calculated using more accurate theory levels, like GW. Our conclusions indicate that the renormalizations can be forecast using just ground state properties, and we find no reason to expect that this does not hold if the regressors and regressands are calculated with a higher accuracy (this should indeed reduce noise and hence improve the predictability).We relaxed the geometry of the systems with PBE until individual forces on the atoms of the order of 10−6 Ry bohr−1 were reached. Phonon frequencies and normal modes were calculated using density-functional perturbation theory.99 These quantities, as well as relaxed geometries, if calculated with PBE, are thought to be nearly as accurate as if calculated with methods like DFT-B3LYP or GW (ref. 30, ESI‡).
The atomic geometries of some analysed systems were taken from experimental references.18,19,31,69,71 Other initial geometries were obtained from further works.86,100 The rest were generated by geometrical means, and taken from the database of ref. 101; among them, an important part had been post-processed using force fields,102 and extracted from ref. 103. For the sake of a broader scope we included the renormalizations of a singular fullerene which contains a 4-carbon ring.104 The geometries of all the simulated systems can be downloaded from ref. 105. The input and output files of the ab initio (DFT) calculation can be found in ref. 106.
B. Forecasting through machine learning
The relatively high numerical complexity of the calculations of the electron–vibrational renormalization of electronic bands, together with their need to master the phonon theory, may discourage many research groups from evaluating such renormalization. Nevertheless, as stated in Section I, this renormalization can have a large size and a potentially strong impact on technological applications like photovoltaics. In this section we present methods to make estimations of the electron–vibrational renormalization of electronic bands without the need for performing complicated and costly calculations. In our analysis we have used different sets of regressors and applied both regression (Section III.B.1) and classification (Section III.B.2) methods.(i) Using the (bare) ML method to forecast the renormalizations (as found in our frozen-phonon calculations, i.e. as they appear in Table 2);
(ii) Using an ordinary least squares linear regression (LR);
(iii) Using the ML method to forecast the residuals of linear regressions performed (ii), this is applying ML on top of linear regression.
In the latter case (iii), we first apply LR to the training dataset; we then calculate the difference between the result of this linear regression and the outputs, i.e. the residuals; then, the ML method (e.g. RF) learns these residuals, rather than the renormalizations themselves. Finally, the forecasting of the test dataset is performed by adding the outputs from both LR and ML methods (whose parameters were obtained in the training stage).
As already mentioned, we performed ML calculations using different sets of input variables. Among them, the set of regressors which performed best for the linear regression (LR) and random forests (RF) calculations corresponds to the electronic structure selected dataset, whose constituents are presented in Table 1. In it GapPBE and GapB3LYP stand for the HOMO–LUMO gaps calculated using PBE and B3LYP, respectively, ε indicates electronic eigenvalues, and AvgOcc and AvgEmpty indicate the average value of occupied and unoccupied electronic eigenvalues (see their definitions in Section SII of the ESI‡).
| Predicted property | Model | Input regressors |
|---|---|---|
| HOMO renorm. | LR, RF | GapPBE, AvgOcc, (εHOMO − εHOMO−1), …, (εHOMO − εHOMO−5), (εHOMO − εHOMO−1)−1, …, (εHOMO − εHOMO−5)−1, (εLUMO − εHOMO−1)−1, (εLUMO − εHOMO−2)−1, (εLUMO+1 − εHOMO)−1, (εLUMO+2 − εHOMO)−1 |
| LUMO renorm. | LR | GapPBE, AvgEmpty, (εHOMO − εHOMO−1), (εLUMO+2 − εLUMO)−1, (εLUMO+1 − εLUMO)−1 |
| RF | GapPBE, GapB3LYP, AvgEmpty, (εLUMO+4 − εLUMO)−1, …, (εLUMO+1 − εLUMO)−1 | |
| GAP renorm. | LR | GapPBE, AvgOcc, AvgEmpty, (εHOMO − εHOMO−1)−1, (εLUMO+1 − εLUMO)−1 |
| RF | GapPBE, GapB3LYP, AvgEmpty, AvgOcc, (εHOMO − εHOMO−1)−1, …, (εHOMO − εHOMO−4)−1, (εLUMO+4 − εLUMO)−1, …, (εLUMO+1 − εLUMO)−1 | |
In order to evaluate the predicting power of each regression method we executed 1000 trials. In each of them a training dataset and a test dataset were randomly generated (shuffle). In order to perform comparisons on equal footing, these datasets are equal for the three mentioned kinds of calculations and for the different analysed ML methods. We finally evaluated the predictive power of a regression method as the mean for the 1000 trials of the average absolute error out-of-sample (this is in the test dataset).
We have run the RF algorithm with 250 estimators, making the maximum number of features equal to 3, and minimum number of samples required to split an internal node equal to 4. We have executed the NN algorithm with two layers of neurons. For the forecast without using linear regression (i.e. using bare NNs) they consisted of 400 and 200 neurons, respectively. For the forecast performed on top of linear regression (i.e. forecasting of residuals) the first layer consisted of 100 neurons, while the second one consisted of just one neuron. Using different hyperparameters for the NNs in these two cases leads to more accurate results. For both cases the activation function was the logistic function, and the chosen solver was ADAM109 using up to 10k iterations; the α was set to 0.01, the learning rate was set to 0.0015 and the momentum was set to 0.6. Concerning KNN we have taken into account 22 neighbors and we have set the Minkowski parameter to 1. We were unable to attain reasonable predicting power using other popular regression methods, like kernel ridge or support vector machines. Details on the way our calculations were performed can be viewed by analysing our code.107 We have also written and made publicly available108 a code which performs forecasts of the electron–phonon renormalizations of any given fullerene. The user must simply specify a few input variables which can be promptly calculated through a ground state DFT calculation (example input files as well as pseudopotentials are also provided). These codes were written in Python; their ML calculations are based on the functions provided in the scikit-learn library.109
Given a dataset with objects (rows) having known values in the dependent variable v, the first step is, for each object of the dataset, to use a regression model to predict a value for v for each object. The second step is, for each object, to calculate the prediction error (distance d, defined as the absolute value of the difference between the forecasted and the observed – i.e. ab initio calculated – renormalization) and to label the object as either acceptable (if d ≤ 10 meV) or unacceptable (if d > 10 meV). Finally, the third step consists of growing a regression tree with the labeled objects. In order to avoid overfitting, we decided to prune the regression tree so that it has a depth equal to one. This means that, in fact, only one independent variable (the most relevant one) is taken into account to determine the validity of the prediction. The explanation of how to grow a regression tree is beyond the scope of this article, but the reader can find the procedure in the description of the CART system.110
The reliability of our approach has been measured using 5-fold cross-validation, being the results of an average of five trials.
In the calculations involving classification we have used the following sets of regressors for the linear regression on top of which the random tree-based classification is performed:
• For the HOMO renormalization: (εHOMO − εHOMO−1), (εHOMO − εHOMO−2), (εHOMO − εHOMO−1)−1, (εHOMO − εHOMO−2)−1 and AvgOcc.
• For the LUMO renormalization: an ensemble of two regression models whose regressors are, respectively:
– {GapPBE, AvgEmpty, (εHOMO − εHOMO−1), (εHOMO − εHOMO−2)}
– {GapPBE, (εLUMO+1 − εLUMO)−1, (εLUMO+2 − εLUMO)−1}
• For the gap renormalization: an ensemble of two regression models whose regressors are, respectively:
– {GapPBE, AvgOcc, (εLUMO+1 − εLUMO)−1}
– {GapB3LYP, AvgOcc, (εLUMO+1 − εLUMO)−1}
Concerning the tree construction, it is important to select a subset of appropriate variables. We tried with (1) the complete set of variables describing the fullerenes (see Section II); (2) several subsets of variables; (3) principal component analysis (PCA) with several subsets of variables. After several preliminary calculations we decided to use the following variables to grow the regression trees:
• HOMO renormalization: PCA with 〈ε〉, GapB3LYP.
• LUMO renormalization: PCA with AvgOcc and AvgEmpty.
• Gap renormalization: PCA with AvgOcc and (εLUMO+1 − εLUMO)−1.
IV. Results and discussion
A. Renormalization of electronic eigenvalues
We present the results of our calculations of electronic eigenvalue renormalizations due to electron–vibrational interaction in Table 2. This table displays the HOMO–LUMO gap (calculated using B3LYP) and the renormalizations of HOMO, LUMO and gap (calculated using GGA-PBE) at zero-temperature of 163 fullerenes and fullerene derivatives. These data are also presented in Fig. 1, which displays the renormalizations as a function of the HOMO–LUMO gap calculated from B3LYP (see the ESI‡ for the corresponding plots as a function of the gap calculated with GGA-PBE). For the sake of completion and in order to avoid overcomplicating this table, we report the band gap obtained with PBE in Table SVI in the ESI.‡| Fullerene - symmetry | HOMO–LUMO gap [meV] | HOMO renorm [meV] | LUMO renorm [meV] | Gap renorm [meV] | Fullerene - symmetry | HOMO–LUMO gap [meV] | HOMO renorm [meV] | LUMO renorm [meV] | Gap renorm [meV] | Fullerene - symmetry | HOMO–LUMO gap [meV] | HOMO renorm [meV] | LUMO renorm [meV] | Gap renorm [meV] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C 28-D2 | 1495.2 | 25.7 | 11.2 | −14.5 | C56-C2v | 1264.8 | 10.0 | −22.0 | −32.0 | C84-D2 | 1241.8 | 28.7 | −27.3 | −56.0 |
| C30-C2v-a | 2200.9 | 65.7 | −74.6 | −140.3 | C56-Cs | 1727.6 | 20.6 | −45.4 | −66.0 | C84-D2d | 1936.4 | 37.5 | −29.6 | −67.1 |
| C 30-C2v-b | 1270.4 | 26.3 | 43.0 | 16.7 | C56-D2 | 1521.9 | 12.6 | −32.2 | −44.8 | C84-D3d | 1232.8 | 6.8 | −12.9 | −19.7 |
| C32-C2 | 1959.4 | 12.4 | −51.5 | −63.9 | C56-Td | 1707.2 | 11.0 | −13.3 | −24.3 | C84-D6h | 2198.9 | 73.7 | −36.7 | −110.4 |
| C32-D3 | 2535.8 | 48.3 | −56.3 | −104.6 | C58-C1 | 996.4 | 17.3 | −6.8 | −24.1 | C84-Td | 2493.3 | 75.2 | −39.1 | −114.3 |
| C32-D3h | 2541.5 | 16.6 | −59.3 | −75.9 | C 58-C3 | 909.1 | −84.0 | 33.9 | 117.9 | C86-C1 | 1091.4 | 5.3 | −31.8 | −37.1 |
| C34-C2 | 1420.6 | −14.6 | −29.3 | −14.7 | C58-Cs | 911.2 | 7.1 | −0.9 | −8.0 | C86-C2 | 1408.6 | 14.4 | −24.9 | −39.3 |
| C36-C2 | 1341.7 | 2.2 | −20.4 | −22.5 | C60-C1 | 1611.5 | 25.6 | −42.6 | −68.2 | C86-C2v | 1017.1 | −2.0 | −16.4 | −14.4 |
| C36-D2 | 1544.5 | 4.6 | −59.3 | −63.9 | C60-C2 | 1389.4 | 21.5 | −18.4 | −39.9 | C86-D3 | 964.3 | 9.1 | −37.3 | −46.4 |
| C36-D2d | 1290.1 | −25.2 | −47.9 | −22.7 | C60-C2v | 1795.6 | 35.2 | −31.5 | −66.7 | C88-C1 | 927.6 | 31.5 | −7.6 | −39.1 |
| C36-D6h | 1159.1 | 30.4 | −33.8 | −64.2 | C60-C3v | 1951.6 | 50.2 | −29.2 | −79.4 | C88-C2 | 632.4 | 10.6 | 0.3 | −10.3 |
| C38-C2-a | 1675.0 | 22.4 | −68.5 | −90.9 | C60-Cs | 1852.0 | 75.0 | −35.3 | −110.3 | C88-T | 725.9 | −25.9 | −14.3 | 11.6 |
| C38-C2-b | 1662.7 | 37.2 | −56.0 | −93.2 | C60-D2h | 1446.7 | 38.9 | −17.2 | −56.1 | C90-C1-a | 1156.2 | 61.2 | −9.1 | −70.3 |
| C38-C2v | 1012.2 | 26.7 | −15.7 | −42.4 | C60-D5 | 887.8 | −13.9 | −28.4 | −14.5 | C 90 -C 1 -b | 987.7 | 84.0 | −0.6 | −84.6 |
| C40-C1 | 1441.0 | 42.1 | −19.3 | −61.4 | C 60 -I h | 2634.6 | 3.0 | −33.4 | −36.4 | C90-C2 | 1535.6 | 39.5 | −20.4 | −60.0 |
| C40-D2 | 1859.2 | 55.7 | −59.9 | −115.6 | C60-S4 | 1134.8 | 10.6 | −18.9 | −29.5 | C90-C2v | 1645.1 | 46.6 | −20.7 | −67.3 |
| C40-D5d | 1983.4 | 24.1 | −7.2 | −31.3 | C62-C1 | 1206.2 | 62.6 | −14.7 | −77.3 | C90-Cs | 1635.8 | 15.3 | −35.6 | −50.9 |
| C40-Td | 1352.0 | 23.9 | −9.2 | −33.1 | C62-C2 | 931.7 | 24.6 | 0.1 | −24.5 | C92-C1 | 859.7 | 3.1 | −10.6 | −13.7 |
| C42-C1 | 1197.5 | 18.8 | −2.8 | −21.6 | C 62-C2v | 1704.2 | 5.3 | −37.0 | −42.4 | C92-C2 | 1099.5 | 2.7 | −18.3 | −21.0 |
| C42-C2 | 1012.1 | 23.5 | 0.8 | −22.7 | C64-C2 | 1729.1 | 37.0 | −25.5 | −62.5 | C92-Cs | 1428.4 | 0.6 | −68.0 | −68.6 |
| C42-Cs | 1074.1 | −3.7 | 7.1 | 10.8 | C64-C3v | 1043.2 | −8.9 | 2.1 | 11.0 | C92-D2 | 1420.0 | 25.6 | −32.6 | −58.2 |
| C42-D3 | 1854.3 | 33.7 | −36.9 | −70.6 | C64-D2 | 2098.3 | 21.5 | −53.1 | −74.6 | C92-D2h | 1029.5 | 3.4 | −16.3 | −19.7 |
| C44-C1 | 1317.4 | 28.0 | −15.2 | −43.2 | C66-C2v | 1020.2 | 16.1 | −5.0 | −21.1 | C92-S4 | 995.2 | 11.6 | −11.5 | −23.1 |
| C44-C2 | 1379.4 | 27.6 | −14.4 | −42.0 | C66-Cs | 1811.1 | 60.9 | −24.8 | −85.7 | C92-T | 1111.2 | −3.9 | −6.4 | −2.5 |
| C44-C2v | 1955.0 | 115.3 | −36.7 | −152.0 | C68-C1 | 1207.0 | 27.2 | −11.4 | −38.6 | C92-Td | 1158.5 | 12.3 | −5.8 | −18.1 |
| C44-D2 | 1741.4 | 116.8 | −22.7 | −139.5 | C68-C2 | 1744.3 | 44.8 | −25.5 | −70.3 | C94-C2 | 1063.9 | 9.7 | −28.0 | −37.7 |
| C44-D3 | 1011.9 | 8.4 | −4.3 | −12.7 | C68-D2 | 1271.9 | 10.7 | −62.9 | −73.6 | C94-C2v | 1022.7 | 37.6 | −23.0 | −60.6 |
| C44-D3d | 1704.4 | 21.2 | −15.9 | −37.1 | C68-S4 | 1411.6 | 8.7 | −88.2 | −96.9 | C96-C2 | 1239.3 | 24.3 | −29.0 | −53.3 |
| C44-T | 1990.3 | 105.6 | −13.9 | −119.5 | C68-S6 | 1581.2 | 102.8 | −18.8 | −121.6 | C96-C2v | 972.1 | 7.1 | −9.9 | −17.0 |
| C46-C1 | 1338.1 | 18.9 | −11.3 | −30.2 | C68-T | 1158.5 | −14.8 | −11.5 | 3.3 | C96-Cs | 1168.5 | 2.6 | −29.6 | −32.2 |
| C46-C2 | 1244.3 | 28.9 | −13.4 | −42.3 | C70-C1 | 968 | −1.3 | −15.8 | −14.5 | C96-D2h | 1927.7 | 18.6 | −110.4 | −129.0 |
| C46-C2v | 1546.4 | 25.6 | −36.9 | −62.5 | C70-C2 | 1538 | 36.4 | −31.2 | −67.6 | C96-D3h | 2259.9 | 44.6 | −72.3 | −116.9 |
| C46-Cs | 1522.8 | 27.3 | −20.9 | −48.2 | C 70 -D 5h | 2622.6 | 59.4 | −76.5 | −135.9 | C96-D6h | 1409.3 | 21.1 | −26.1 | −47.2 |
| C48-C1 | 1451.9 | 11.6 | −47.6 | −59.2 | C72-C2v | 1459.2 | −11.1 | −32.0 | −20.9 | C98-C1-a | 923.4 | 35.6 | −5.6 | −41.2 |
| C48-C2-a | 1475.8 | 45.7 | −19.3 | −65.0 | C72-D6d | 2326.8 | 22.2 | −56.4 | −78.6 | C98-C1-b | 1199.2 | 23.0 | −27.8 | −50.9 |
| C48-C2-b | 1923.9 | 78.2 | −29.6 | −107.9 | C74-C1 | 887.8 | 22.3 | −1.5 | −23.8 | C98-C2 | 1286.1 | 21.6 | −25.5 | −47.1 |
| C48-Cs | 1490.0 | 6.3 | −58.4 | −64.7 | C74-C2 | 1364.1 | 7.7 | −25.5 | −33.2 | C98-C2v | 989.3 | 21.9 | −8.9 | −30.8 |
| C48-D2 | 1634.1 | −6.7 | −92.1 | −85.4 | C74-Cs | 1133.1 | −11.4 | −16.2 | −4.8 | C98-C3 | 1385.7 | 90.2 | −22.5 | −112.7 |
| C48-D2h | 1244.3 | 23.8 | −28.5 | −52.3 | C76-C1 | 1100.9 | 5.0 | −19.4 | −24.4 | C98-D3 | 867.8 | 4.3 | −20.8 | −25.1 |
| C48-D6d | 1732.9 | 8.8 | −23.6 | −32.4 | C76-C2 | 944.4 | 14.0 | −13.4 | −27.4 | C100-C2 | 995.7 | 4.5 | −26.3 | −30.9 |
| C50-C1-a | 1215.7 | −0.7 | −23.7 | −22.9 | C76-C3v | 813.1 | 31.3 | 6.6 | −24.7 | C100-C2v | 1747.7 | 51.6 | −30.3 | −81.9 |
| C50-C1-b | 1551.2 | 13.0 | −28.9 | −41.9 | C76-D2 | 1855.2 | 17.0 | −67.3 | −84.3 | C100-Cs | 834.0 | 71.0 | 4.7 | −66.3 |
| C50-C2 | 1543.5 | 14.0 | −38.3 | −52.3 | C76-S4 | 1288.9 | 16.3 | −16.1 | −32.4 | C100-D2 | 1077.0 | −3.5 | −34.4 | −30.9 |
| C50-Cs | 1494.1 | 8.9 | −18.6 | −27.5 | C78-C2v | 1545.1 | 17.1 | −55.3 | −72.4 | C100-S4 | 1069.8 | −6.6 | −23.9 | −17.3 |
| C50-D3 | 2230.3 | 42.7 | −48.4 | −91.1 | C78-D3 | 1505.2 | 3.7 | −37.8 | −41.5 | C100-T | 721.9 | −34.2 | −27.2 | 7.0 |
| C50-D5h | 1193.9 | 5.8 | −12.6 | −18.4 | C78-D3h | 1406.9 | 19.0 | −30.7 | −49.7 | C100-Td | 957.2 | 51.4 | 12.0 | −39.4 |
| C52-C1 | 1242.0 | −11.8 | −24.1 | −12.3 | C80-C2v | 848.8 | 22.4 | −4.0 | −26.4 | C104-C1 | 967.0 | 5.5 | −20.8 | −26.3 |
| C52-C2 | 1062.3 | 39.4 | −5.4 | −44.8 | C80-D2 | 1217.6 | −3.8 | −27.4 | −23.6 | C104-S4 | 1099.5 | 15.1 | −8.2 | −23.3 |
| C52-Cs | 1302.2 | 36.0 | −5.0 | −41.0 | C80-Ih | 781.6 | −17.0 | −29.3 | −12.3 | C104-T | 1357.7 | 79.4 | −11.4 | -90.8 |
| C52-D2 | 1115.8 | −7.8 | −47.9 | −40.1 | C80-S4 | 1148.7 | 39.6 | −12.9 | −52.5 | C180-Ih | 2244.0 | 38.0 | −38.3 | −76.4 |
| C52-D2d | 1080.2 | −16.3 | −29.3 | −13.0 | C82-C2 | 1179.9 | 60.5 | −15.5 | −76.0 | |||||
| C54-C1 | 1144.0 | −6.1 | −24.6 | −18.5 | C82-C3v | 770.0 | 20.7 | 4.1 | −16.5 | |||||
| C54-C2 | 990.8 | 7.0 | −14.7 | −21.8 | C82-Cs | 1451.5 | 40.2 | −30.8 | −71.0 | [60]PCBM | 2475.5 | 45.8 | −74.4 | −120.2 |
| C56-C1 | 1051.2 | −1.2 | −10.8 | −9.6 | C84-C2v | 1172.3 | 5.9 | −25.6 | −31.5 | [70]PCBM | 2444.0 | 68.8 | −103.0 | −171.8 |
| C56-C2 | 1480.2 | 25.4 | −27.0 | −52.4 | C84-Cs | 1426.5 | 17.3 | −30.9 | −48.2 | IC60BA | 2489.4 | 37.7 | −79.6 | −117.3 |
 | ||
| Fig. 1 Frozen-phonon renormalizations (calculated using the GGA-PBE functional) in 163 fullerenes and fullerene derivatives as a function of the HOMO–LUMO gap (calculated using B3LYP). Top: HOMO renormalization; center: LUMO renormalization; bottom: gap renormalization. | ||
In the literature there exist many research papers, both theoretical and experimental, which analyse the electron–phonon interaction in buckminsterfullerene. However, they usually focus on given electron–phonon couplings,30,112–116 which correspond to specific electronic levels and vibrational modes, due to their interest for analysing superconductivity. It is therefore hard to find an analysis of the gap renormalization as the one that we display in Table 2.
From the results shown in Fig. 1 we notice that most of the renormalizations of the HOMO are positive, and most of the renormalizations of the LUMO are negative, as expected from the Allen–Heine–Cardona theory for systems with a gap. This theory states that the renormalization of a given eigenvalue εn is given by:
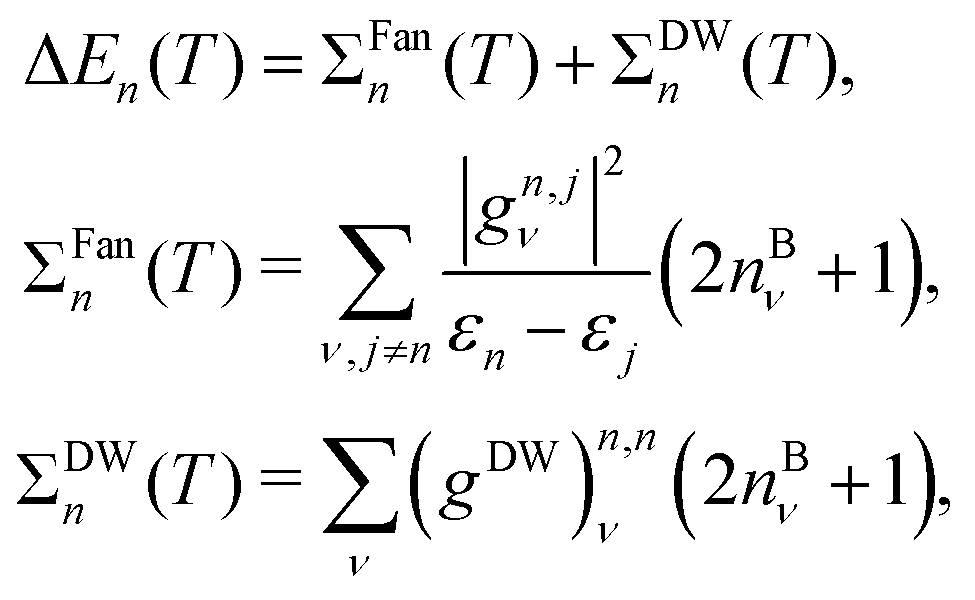 | (1) |
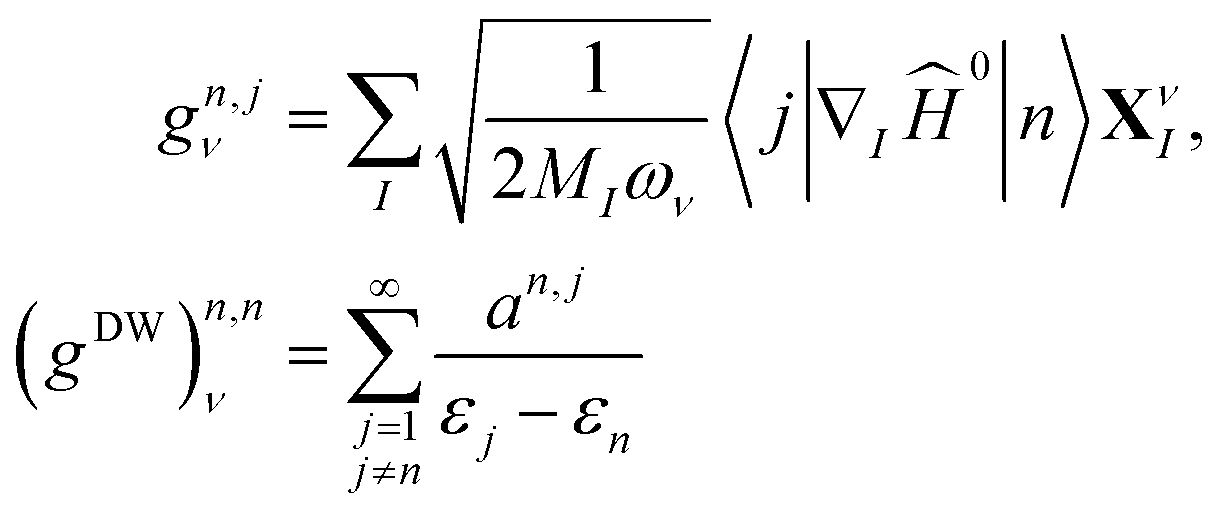 | (2) |
Since the renormalization is proportional to the inverse of the eigenvalue difference (εn − εj)−1, considering only the linear term in nuclear displacements (ΣFan) and assuming that all the matrix elements g have an equal value if i, j are both occupied (also another equal value if both are unoccupied), and a lower value if i is occupied and j is unoccupied (which is a coarse-grain approximation), the gap makes the contribution of empty states low for the HOMO renormalization. Analogously, the contribution of occupied states is expected to be low for the LUMO renormalization. Under these assumptions, eqn (1) implies that the HOMO renormalization is expected to be positive, while the LUMO renormalization is expected to be negative. The fact that this holds for most of the dots displayed in Fig. 1 indicates the appropriateness of this approximation. Eqn (1) also indicates that the lower the gap, the stronger the negative (positive) contribution of the unoccupied (occupied) states to the HOMO (LUMO) renormalization, and hence the more likely a negative HOMO (positive LUMO) renormalization is. This is also confirmed by our results; as can be viewed in Fig. 1, the negative HOMO and positive LUMO renormalizations concentrate in the region of low and middle-sized gap.
The fact that a higher gap correlates with a higher gap renormalization can be partly due to eqn (1). Higher gaps tend to damp negative (positive) renormalization of HOMO (LUMO) due to unoccupied (occupied) states, which increases the size of the gap renormalization. Despite mentioning eqn (1), note that our calculations were not performed using many-body perturbation theory (that is, they are not using that equation). Our calculations used the frozen-phonon approach instead (that is, they were based on the calculation of electronic eigenvalues, not on the calculation of electron–phonon matrix elements g, gDW).
Other authors have also found a linear relationship between the gap renormalization and the gap itself,61 which is attributed to the electric permittivity. The Penn model117 establishes an inverse relationship between the relative permittivity and the (average, in a solid) gap between valence and conduction bands. Avoiding approximations made by Penn, the equations also display an inverse relationship between the permittivity and differences between electronic energies.118 A small gap will thus tend to imply a high permittivity, which implies that the electric interaction will propagate more weakly (that is, the electric fields will be weaker, and the interaction will be screened). The screened interaction will mitigate the effect of the wavefunctions of positively charged nuclei on the electrons, thus leading to a weaker variation in the electronic eigenvalues. This will lower the concavities of ε vs. h (eigenvalue vs. displacement size, see eqn (3) of ref. 26), which in turn will lead to a lower renormalization. At nonzero temperatures, the amplitude of the nuclear vibrations (term proportional to the Bose–Einstein distribution in eqn (3) of ref. 26 and eqn (1) of this paper) will also be affected by the screening, also lowering the renormalization for low gaps.
Fig. 1 indicates that, despite the fact that some data points correspond to either a negative HOMO renormalization or a positive LUMO renormalization, just a few (7) of them present a positive gap renormalization. This indicates that renormalizations with an unexpected sign for the HOMO or LUMO are largely canceled out by each other. Moreover, if we discard the molecules with T symmetry, just three have a gap renormalization with an unexpected (positive) sign.
The linear relationship of renormalizations with gap displayed in Fig. 1 indicates that it is possible to make a coarse-grain estimate of the gap renormalization due to electron–vibrational interaction in fullerenes very efficiently, without performing any actual phonon calculation. Fittings are presented in the ESI.‡ The gap renormalizations of 96% of the fullerenes with C, D or S symmetries lay between −2% and −18% of the gap calculated with the PBE-GGA exchange correlation functional.
The results from Fig. 1 also show that the zero-point renormalization of the HOMO–LUMO gap of fullerenes is smaller than that of other carbon compounds, like diamond and diamondoids. Ref. 119 determined experimental values for the zero-point renormalization of bulk diamond between −320 and −450 meV. Ref. 26 found theoretical gap renormalizations up to −370 meV in diamondoids. However, due to the fact that the gap itself is smaller in fullerenes than in bulk diamond and diamondoids, the average quotient between the absolute value of the renormalization and the gap have comparable sizes: 8.1% in diamond25 (which is considered very high24), about 4.2% in diamondoids26 and about 3.5% in fullerenes on average.120 Nonetheless, nearly all of the most prominent of the analysed systems (in bold in Table 2), with wide technological applications, present non-negligible renormalizations. The module of LUMO renormalizations of the analysed fullerene derivatives is above 70 meV, which can have a strong effect on their capabilities as an acceptor in photovoltaics.31
Note that the renormalizations of IC60BA and [60]PCBM have similar sizes, and the renormalizations of C70 and [70]PCBM have similar sizes as well. This hints that the addition of atoms to form derivatives has a limited impact on the phonon-based renormalization. Also note that the results of pristine C60 with Ih symmetry clearly differ from those of IC60BA and [60]PCBM. We deem it to be an exception due to geometrical properties: pristine C60 has 5-fold degeneracy in its HOMO and 3-fold degeneracy in its LUMO; hence it is wise to calculate the renormalization as an average of states,26 which lowers the renormalization. In the analysed derivatives of C60, the addition of further atoms broke the symmetry, thus leading to a different behaviour. If we calculate the renormalizations of IC60BA as an average of 5 and 3 states for the HOMO and the LUMO respectively, we obtain renormalizations similar to those of pristine C60 (+8 and −52 meV).
The relationship of the gap renormalization with the temperature for some representative systems can be viewed in Fig. 2. From it we note that the variation of the renormalization between 0 and 300 K is relatively low, about one order of magnitude lower than the zero-point renormalization (e.g. 10 meV for [60]PCBM and 12 meV for [70]PCBM). This low variability agrees with previous calculations and observations of carbon-based materials.26,35,119
 | ||
| Fig. 2 Renormalization of the band gap of selected fullerenes and derivatives as a function of the temperature. | ||
B. Forecasting through machine learning
| Renorm. of | LR | KNN | KNN@LR | NN | NN@LR | RF | RF@LR |
|---|---|---|---|---|---|---|---|
| HOMO | 5.80 | 11.58 | 5.46 | 8.04 | 5.68 | 7.62 | 5.56 |
| LUMO | 6.47 | 11.69 | 6.11 | 7.17 | 6.25 | 6.95 | 5.58 |
| Gap | 8.55 | 17.40 | 8.24 | 14.02 | 8.44 | 10.48 | 7.75 |
These results indicate that the renormalizations of electronic eigenvalues due to electron–phonon interactions can be forecast with a low error (whose absolute value is below 8 meV on average) from data obtained in a single ground state calculation. In addition, our results indicate that an ordinary least squares linear regression provides a fair approximation to renormalizations, which can be improved by applying random forests on top of it.
Fig. 3 and 4 present further information on the accuracy for prediction of renormalizations of the random forests on top of linear regression. Analogous plots for neural networks and k-nearest neighbours can be viewed in the ESI.‡ The calculations represented in Fig. 3 are based on the electronic structure selected set of features (see Table 1). In Fig. 3 the scatter plots (around the diagonal line) present the values of the renormalizations; the x axis corresponds to the values obtained in complete frozen-phonon ab initio calculations, while the y axis corresponds to the forecasts, which were made using machine learning on top of linear regression using the selected regressors. The differences between both quantities are presented in the histograms of Fig. 3. The data correspond to 1000 random choices of the training and test datasets, which consist of 126 and 31 molecules (80% and 20%, respectively). In order to make the graphs clearer, we only display 300 (randomly chosen) dots for the training data and 150 dots for the test data in the scatter plots. As it can be viewed in the subplots, the ML algorithm can predict the renormalization of the HOMO and LUMO with an error lower than 15 meV for the majority of the cases, which is lower than the typical errors introduced by DFT as compared with higher accuracy methods like GW25 (indeed for the HOMO and LUMO renormalizations most of the test errors lie in the ±7.5 meV range). Hence the data that we report here do provide a reasonable estimate of the impact of the electron–vibrational interaction on electronic bands in generic fullerenes. The forecasts are expected to be more accurate for molecules not tackled in this article. This is because, when performing the training of the machine learning methods in order to perform the corresponding forecast, the whole dataset of Table 2 is available, not just part (80%) of it.
 | ||
| Fig. 3 Scatter plots: predictions of the renormalizations using random forests on top of linear regression vs. ab initio results. Histograms: training and test errors. Top: HOMO; center: LUMO; bottom: gap. | ||
 | ||
| Fig. 4 Test errors of predictions of renormalizations using different feature sets (predictions made using random forests on top of linear regression). Boxes: first and third quantiles; lines: mean and median; whiskers: 95% confidence interval. Top: HOMO; center: LUMO; bottom: gap. | ||
We have also analysed the predictive power of our forecasts if using different sets of regressors (the complete lists, together with an analysis of feature importance, are presented in the ESI‡). We display box plots of them in Fig. 4. The box itself indicates the limits of the first and third quantiles. The mean and the median are represented by dotted and solid lines, respectively. The most external levels (whiskers) indicate the limits of the 95% confidence interval. Fig. 4 indicates that fair forecasts are obtained using linear regression and random forests with the electronic structure variables. Neural networks and k-nearest neighbours do still provide reasonable forecasts if using the electronic structure variables as regressors. However, other regressors lack predictive power. Among them we list geometric variables and features of phonons, bond lengths and bond orders.
The very different sizes of boxes in Fig. 4 (some being less than 10 meV, others being much larger) for different sets of features indicate that properties which are important in other contexts, like bond lengths, bond orders, number of atoms, area and volume, do not have a close relationship with the renormalization of electronic eigenvalues due to electron–vibrational interaction. Conversely, renormalization seems to be rather a function of the electronic eigenvalues themselves. The similar sizes of the three boxes which consist of electronic structure variables indicate that introducing further regressors neither improves nor worsens the predictive power. That is, using random forests the further regressors do not contain much useful information, but the efficacy of the regression does not drop due to the noise that they introduce.
The fact that average, maximum, minimum, and other regressors based on phonon frequencies lack any predicting power for the electron–phonon renormalization might be seen as counterintuitive, especially noting that ων appears in the denominator of the right-hand side of eqn (2). However, this is supported by recent research: ref. 61 also found negligible predictive power of minimum phonon frequencies, average of phonon frequencies and number of atoms, while finding significant predicting power for the band gap. Our hypothesis is that, since the range of phonon frequencies is nearly the same for all fullerenes, it is not a quantity that can give an account for the differences in their renormalizations.
 121) of the bare LR and of the data subset selected as acceptable by the decision tree.
121) of the bare LR and of the data subset selected as acceptable by the decision tree.  has been computed as the percentage of objects that the method has considered as having an acceptable value (i.e., d ≤ 10 meV) but it is not (i.e., d ≥ 10 meV). Both
has been computed as the percentage of objects that the method has considered as having an acceptable value (i.e., d ≤ 10 meV) but it is not (i.e., d ≥ 10 meV). Both  and
and  are thus percentages of false positives. Their values, as well as those of the other columns, correspond to the average of the test data of a 5-fold cross validation calculation. In addition to the false positives, one must take the false negatives into consideration. These are predictions which the tree has wrongly marked as being unacceptable. The fifth column of Table 4 (Marked) displays the percentage of results that the tree has considered as (suspicious to be) unacceptable. Column
are thus percentages of false positives. Their values, as well as those of the other columns, correspond to the average of the test data of a 5-fold cross validation calculation. In addition to the false positives, one must take the false negatives into consideration. These are predictions which the tree has wrongly marked as being unacceptable. The fifth column of Table 4 (Marked) displays the percentage of results that the tree has considered as (suspicious to be) unacceptable. Column  presents the percentage of marked results that correspond to false negatives (i.e. they are marked as possibly unacceptable, but are indeed acceptable). This column informs that many of the data which are selected by the tree as unacceptable are indeed unacceptable, and hence it is advisable to discard them from any further analysis. Finally, column
presents the percentage of marked results that correspond to false negatives (i.e. they are marked as possibly unacceptable, but are indeed acceptable). This column informs that many of the data which are selected by the tree as unacceptable are indeed unacceptable, and hence it is advisable to discard them from any further analysis. Finally, column  represents the percentage of false negatives with respect to the whole test set.
represents the percentage of false negatives with respect to the whole test set.
| Quantity | R 2 (LR) |
|
|
Marked (%) |
|
|
|---|---|---|---|---|---|---|
| HOMO ren. | 0.891 | 17.13 | 15.51 | 6.23 | 39.46 | 4.78 |
| LUMO ren. | 0.74, 0.79 | 17.59 | 13.62 | 6.37 | 32.07 | 2.41 |
| Gap ren. | 0.86, 0.87 | 31.36 | 26.99 | 14.78 | 48.63 | 5.87 |


To sum up, if we have a fullerene whose electron–phonon renormalizations have not been calculated ab initio then we can decide to forecast them using linear regression, which will be fairly accurate. We can also decide to discard this forecast if the decision trees indicate that it is unreliable; in this manner the forecast will be more accurate, as indicated by the third and fourth columns of Table 4.
V. Conclusion
In this article we have presented ab initio calculations of the renormalizations of HOMO, LUMO and gap due to electron–vibrational interaction in a remarkable class of molecules – fullerenes – showing that the typical size of such renormalization is large enough to have an impact on photovoltaics. This is expected to provide the scientific community with a useful view on properties of these carbon-based systems. Our subsequent machine learning-based analysis indicates which features are closely related with renormalizations and which ones are not, which we expect will help readers to acquire deeper insights about this issue. In addition to the presented calculations and conclusions, we have provided a computing code which forecasts renormalizations – to a reasonable degree of accuracy – using the outputs of simple ground state calculations (with relaxed nuclear positions) as its input. Calculations that determine electron–vibrational renormalization of electronic bands are complex and require a deep understanding of phonon theory, which may discourage many research groups from pursuing such investigations. The approach presented here is expected to enable researchers to swiftly and easily obtain estimates of the renormalizations. Finally, our analysis highlights the possibility that features arising from complex quantum phenomena can be forecast from ground state calculations, even with training sets of relatively low size. Our results are expected to provide a proof-of-concept for this fact.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Peter Schwerdtfeger (Massey University, New Zealand), Miguel Pruneda (ICN2, Barcelona) and Ghanshyam Pilania and Michael Hartman (Los Alamos National Laboratory, USA) for interesting discussion. We are also grateful to the University of Zaragoza for providing computing facilities.References
- A. Castro, M. A. L. Marques, J. A. Alonso, G. F. Bertsch, K. Yabana and A. Rubio, Can optical spectroscopy directly elucidate the ground state of C20?, J. Chem. Phys., 2002, 116(5), 1930–1933 CrossRef CAS.
- H. Schwertfeger and P. R. Schreinter, Chemie mit Nano-Juwelen: Diamantoide, Chem. Unserer Zeit, 2010, 44, 248 CrossRef CAS.
- S. Latil, L. Henrard, C. Goze Bac, P. Bernier and A. Rubio, 13C NMR Chemical Shift of Single-Wall Carbon Nanotubes, Phys. Rev. Lett., 2001, 86, 3160–3163 CrossRef CAS PubMed.
- F. Langa and J. F. Nierengarten, Fullerenes: principles and applications, RSC Publishing, 2007 Search PubMed.
- T. Ishiguro, K. Yamaji and G. Saito, Organic Superconductors, Springer, 2nd edn, 2012 Search PubMed.
- N. Tagmatarchis and H. Shinohara, Fullerenes in medicinal chemistry and their biological applications, Mini-Rev. Med. Chem., 2001, 1(4), 339–348 CAS.
- A. Astefanei, O. Nuñez and M. T. Galceran, Characterisation and determination of fullerenes: A critical review, Anal. Chim. Acta, 2015, 882, 1–21, DOI:10.1016/j.aca.2015.03.025.
- R. Gordon, I. Podolski, E. Makarova, A. Deev, E. Mugantseva and S. Khutsyan, et al., Intrahippocampal Pathways Involved in Learning/Memory Mechanisms are Affected by Intracerebral Infusions of Amyloid-β25–35 Peptide and Hydrated Fullerene C60 in Rats, J. Alzheimer's Dis., 2017, 58, 711 CAS.
- X. Yang, A. Ebrahimi, J. Li and Q. Cui, Fullerene-biomolecule conjugates and their biomedicinal applications, Int. J. Nanomed., 2014, 9, 77–92 CrossRef PubMed.
- H. M. Kuznietsova, O. V. Lynchak, N. V. Dziubenko, V. L. Osetskyi, O. V. Ogloblya and Y. I. Prylutsky, et al., Water-soluble C60 fullerenes reduce manifestations of acute cholangitis in rats, Appl. Nanosci., 2019, 9, 601–608 CrossRef CAS.
- Y. He and Y. Li, Fullerene derivative acceptors for high performance polymer solar cells, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Origin and elimination of photocurrent hysteresis by fullerene passivation in CH3NH3PbI3 planar heterojunction solar cells, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- P. W. Liang, C. C. Chueh, S. T. Williams and A. K. Y. Jen, Roles of Fullerene-Based Interlayers in Enhancing the Performance of Organometal Perovskite Thin Film Solar Cells, Adv. Energy Mater., 2015, 5, 10 Search PubMed.
- M. C. Scharber, On the Efficiency Limit of Conjugated Polymer:Fullerene-Based Bulk Heterojunction Solar Cells, Adv. Mater., 2016, 28(10), 1994–2001 CrossRef CAS PubMed.
- Y. Bai, Q. Dong, Y. Shao, Y. Deng, Q. Wang and L. Shen, et al., Enhancing stability and efficiency of perovskite solar cells with crosslinkable silane-functionalized and doped fullerene, Nat. Commun., 2016, 7, 12806 CrossRef CAS PubMed.
- K. R. Graham, C. Cabanetos, J. P. Jahnke, M. N. Idso, A. El Labban and G. O. Ngongang Ndjawa, et al., Importance of the Donor:Fullerene Intermolecular Arrangement for High-Efficiency Organic Photovoltaics, J. Am. Chem. Soc., 2014, 136(27), 9608–9618 CrossRef CAS PubMed.
- T. Mikie, A. Saeki, Y. Yamazaki, N. Ikuma, K. Kokubo and S. Seki, Stereochemistry of Spiro-Acetalized [60]Fullerenes: How the Exo and Endo Stereoisomers Influence Organic Solar Cell Performance, ACS Appl. Mater. Interfaces, 2015, 7(16), 8915–8922 CrossRef CAS PubMed.
- M. T. Rispens, A. Meetsma, R. Rittberger, C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Influence of the solvent on the crystal structure of PCBM and the efficiency of MDMO-PPV:PCBM ‘plastic’ solar cells, Chem. Commun., 2003, 2116–2118 RSC.
- T. Umeyama, T. Miyata, A. C. Jakowetz, S. Shibata, K. Kurotobi and T. Higashino, et al., Regioisomer effects of [70]fullerene mono-adduct acceptors in bulk heterojunction polymer solar cells, Chem. Sci., 2017, 8, 181–188, 10.1039/C6SC02950G.
- P. Han and G. Bester, First-principles calculation of the electron-phonon interaction in semiconductor nanoclusters, Phys. Rev. B, 2012, 85, 235422, DOI:10.1103/PhysRevB.85.235422.
- E. Mostaani, B. Monserrat, N. D. Drummond and C. J. Lambert, Quasiparticle and excitonic gaps of one-dimensional carbon chains, Phys. Chem. Chem. Phys., 2016, 18, 14810–14821 RSC.
- The open circuit voltage of photovoltaic devices depends on the difference between the eigenvalues of the HOMO of the donor and LUMO of the acceptor. If the fullerene acts as acceptor, a reduction of its LUMO eigenvalue leads to a lower open circuit voltage31.
- Z. Wang, M. Rafipoor, P. García-Risueño, J. P. Merkl, P. Han and H. Lange, et al., Phonon-assisted Auger process enables ultrafast charge transfer in CdSe quantum dot/organic molecule, J. Phys. Chem. C, 2019, 123(28), 17127–17135 CrossRef CAS.
- F. Giustino, S. G. Louie and M. L. Cohen, Electron-Phonon Renormalization of the Direct Band Gap of Diamond, Phys. Rev. Lett., 2010, 105, 265501 CrossRef PubMed.
- G. Antonius, S. Poncé, P. Boulanger, M. Côté and X. Gonze, Many-Body Effects on the Zero-Point Renormalization of the Band Structure, Phys. Rev. Lett., 2014, 112, 215501 CrossRef.
- P. García-Risueño, P. Han, S. Kumar and G. Bester, Frozen-phonon method for state anticrossing situations and its application to zero-point motion effects in diamondoids, Phys. Rev. B, 2023, 108, 125403, DOI:10.1103/PhysRevB.108.125403.
- A. Galli, T. Demján, M. Vörös, G. Thiering, E. Cannuccia and A. Marini, Electron-vibration coupling induced renormalization in the photoemission spectrum of diamondoids, Nat. Commun., 2016, 7, 11327 CrossRef PubMed.
- E. H. Hwang, R. Sensarma and S. Das Sarma, Plasmon-phonon coupling in graphene, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 195406 CrossRef.
- B. Chakraborty, A. Bera, D. V. S. Muthu, S. Bhowmick, U. V. Waghmare and A. K. Sood, Symmetry-dependent phonon renormalization in monolayer MoS2 transistor, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 161403 CrossRef.
- C. Faber, J. L. Janssen, M. Côté, E. Runge and X. Blase, Electron-phonon coupling in the C60 fullerene within the many-body GW approach, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 155104 CrossRef.
- R. Tao, T. Umeyama, T. Higashino, T. Koganezawa and H. Imahori, A single cis-2 regioisomer of ethylene-tethered indene dimer-fullerene adduct as an electron-acceptor in polymer solar cells, Chem. Commun., 2015, 51, 8233–8236, 10.1039/C5CC01712B.
- A Primer in Density Functional Theory, ed. C. Fiolhais, F. Nogueira and M. A. L. Marques, Springer, Series: Lecture Notes in Physics, 1st edn, 2003, vol. 620 Search PubMed.
- C. Friedrich and A. Schindlmayr, Many-Body Perturbation Theory: The GW Approximation, in Computational Nanoscience: Do It Yourself!, ed. J. Grotendorst, S. Blügel and D. Marx, NIC Series, John von Neumann Institute for Computing, Jülich, 2006, vol. 31, pp. 335–355 Search PubMed.
- T. Kotani, M. van Schilfgaarde and S. V. Faleev, Quasiparticle self-consistent GW method: A basis for the independent-particle approximation, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 165106, DOI:10.1103/PhysRevB.76.165106.
- C. E. Patrick and F. Giustino, Quantum nuclear dynamics in the photophysics of diamondoids, Nat. Commun., 2013, 4(1), 2006 CrossRef PubMed.
- T. Hastie, R. Tibshirani and J. Friedman, The elements of statistical learning: data mining, inference, and prediction, Springer series in statistics, Springer, New York, 2009 Search PubMed.
- R. Ramprasad, R. Batra, G. Pilania, A. Mannodi-Kanakkithodi and C. Kim, Machine learning in materials informatics: recent applications and prospects, npj Comput. Mater., 2017, 3(1), 54 CrossRef.
- G. R. Schleder, A. C. Padilha, C. M. Acosta, M. Costa and A. Fazzio, From DFT to machine learning: recent approaches to materials science-a review, J. Phys. Mater., 2019, 2(3), 032001 CrossRef CAS.
- C. Draxl and M. Scheffler, NOMAD: The FAIR concept for big data-driven materials science, MRS Bull., 2018, 43(9), 676–682 CrossRef.
- L. M. Ghiringhelli, J. Vybiral, S. V. Levchenko, C. Draxl and M. Scheffler, Big data of materials science: critical role of the descriptor, Phys. Rev. Lett., 2015, 114(10), 105503 CrossRef PubMed.
- J. Jumper, R. Evans, A. Pritzel and T. Green, et al., Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596(7873), 583–589 CrossRef CAS PubMed.
- C. Kim, R. Batra, L. Chen, H. Tran and R. Ramprasad, Polymer design using genetic algorithm and machine learning, Comput. Mater. Sci., 2021, 186, 110067 CrossRef CAS.
- J. Schmidt, M. Fadel and C. L. Benavides-Riveros, Machine learning universal bosonic functionals, Phys. Rev. Res., 2021, 3(3), L032063 CrossRef CAS.
- G. Hegde and R. C. Bowen, Machine-learned approximations to density functional theory Hamiltonians, Sci. Rep., 2017, 7(1), 42669 CrossRef CAS PubMed.
- D. Di Sante, M. Medvidović, A. Toschi, G. Sangiovanni, C. Franchini and A. M. Sengupta, et al., Deep Learning the Functional Renormalization Group, Phys. Rev. Lett., 2022, 129, 136402 CrossRef CAS PubMed.
- J. Kirkpatrick, B. McMorrow, D. H. Turban, A. L. Gaunt, J. S. Spencer and A. G. Matthews, et al., Pushing the frontiers of density functionals by solving the fractional electron problem, Science, 2021, 374(6573), 1385–1389 CrossRef CAS PubMed.
- J. C. Snyder, M. Rupp, K. Hansen, K. R. Müller and K. Burke, Finding density functionals with machine learning, Phys. Rev. Lett., 2012, 108(25), 253002 CrossRef PubMed.
- F. Brockherde, L. Vogt, L. Li, M. E. Tuckerman, K. Burke and K. R. Müller, Bypassing the Kohn-Sham equations with machine learning, Nat. Commun., 2017, 8(1), 872 CrossRef PubMed.
- J. R. Moreno, G. Carleo and A. Georges, Deep learning the Hohenberg-Kohn maps of density functional theory, Phys. Rev. Lett., 2020, 125(7), 076402 CrossRef CAS PubMed.
- L. E. Ratcliff, T. Oshima, F. Nippert, B. M. Janzen, E. Kluth and R. Goldhahn, et al., Tackling Disorder in γ-Ga2O3, Adv. Mater., 2022, 34(37), 2204217 CrossRef CAS PubMed.
- S. A. Tawfik, O. Isayev, C. Stampfl, J. Shapter, D. A. Winkler and M. J. Ford, Efficient prediction of structural and electronic properties of hybrid 2D materials using complementary DFT and machine learning approaches, Adv. Theory Simul., 2019, 2(1), 1800128 CrossRef.
- W. Pronobis, K. T. Schütt, A. Tkatchenko and K. R. Müller, Capturing intensive and extensive DFT/TDDFT molecular properties with machine learning, Eur. Phys. J. B, 2018, 91(8), 178 CrossRef.
- K. Hansen, G. Montavon, F. Biegler, S. Fazli, M. Rupp and M. Scheffler, et al., Assessment and validation of machine learning methods for predicting molecular atomization energies, J. Chem. Theory Comput., 2013, 9(8), 3404–3419 CrossRef CAS PubMed.
- R. Jinnouchi and R. Asahi, Predicting catalytic activity of nanoparticles by a DFT-aided machine-learning algorithm, J. Phys. Chem. Lett., 2017, 8(17), 4279–4283 CrossRef CAS PubMed.
- G. Pilania, C. Wang, X. Jiang, S. Rajasekaran and R. Ramprasad, Accelerating materials property predictions using machine learning, Sci. Rep., 2013, 3(1), 2810 CrossRef PubMed.
- A. M. Alvertis and E. A. Engel, Importance of vibrational anharmonicity for electron-phonon coupling in molecular crystals, Phys. Rev. B, 2022, 105(18), L180301 CrossRef CAS.
- Y. Sun, F. Zhang, C. Z. Wang, K. M. Ho, I. I. Mazin and V. Antropov, Electron-phonon coupling strength from ab initio frozen-phonon approach, Phys. Rev. Mater., 2022, 6(7), 074801 CrossRef CAS.
- J. Park, W. A. Saidi, B. Chorpening and Y. Duan, Applicability of Allen–Heine–Cardona Theory on MOx Metal Oxides and ABO3 Perovskites: Toward High-Temperature Optoelectronic Applications, Chem. Mater., 2022, 34(13), 6108–6115 CrossRef CAS.
- Y. Nomura, Machine learning quantum states—extensions to fermion–boson coupled systems and excited-state calculations, J. Phys. Soc. Jpn., 2020, 89(5), 054706 CrossRef.
- Z. Shi, M. Dao, E. Tsymbalov, A. Shapeev, J. Li and S. Suresh, Metallization of diamond, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(40), 24634–24639 CrossRef CAS PubMed.
- A. Haldar, Q. Clark, M. Zacharias, F. Giustino and S. Sharifzadeh, Machine learning electron–phonon interactions in 2D materials, 2023 DOI:10.21203/rs.3.rs-3253133/v2.
- A. Kundu and G. Galli, Quantum Vibronic Effects on the Excitation Energies of the Nitrogen-Vacancy Center in Diamond, J. Phys. Chem. Lett., 2024, 15(3), 802–810 CrossRef CAS PubMed.
- S. Margadonna, C. M. Brown, T. J. S. Dennis, A. Lappas, P. Pattison and K. Prassides, et al., Crystal structure of the higher fullerene C84, Chem. Mater., 1998, 10(7), 1742–1744 CrossRef CAS.
- Prof. Peter Schwerdtfeger (private communication).
- H. Kietzmann, R. Rochow, G. Ganteför, W. Eberhardt, K. Vietze and G. Seifert, et al., Electronic structure of small fullerenes: evidence for the high stability of C32, Phys. Rev. Lett., 1998, 81(24), 5378 CrossRef CAS.
- C. Zhao, M. Nie, H. Meng, C. Wang and T. Wang, Synthesis and Structural Studies of Two Paramagnetic Metallofullerenes with Isomeric C72 Cage, Inorg. Chem., 2019, 58(12), 8162–8168 CrossRef CAS PubMed.
- H. Nikawa, T. Kikuchi, T. Wakahara, T. Nakahodo, T. Tsuchiya and G. A. Rahman, et al., Missing metallofullerene La@C74, J. Am. Chem. Soc., 2005, 127(27), 9684–9685 CrossRef CAS PubMed.
- H. Kawada, Y. Fujii, H. Nakao, Y. Murakami, T. Watanuki and H. Suematsu, et al., Structural aspects of C82 and C76 crystals studied by X-ray diffraction, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51(14), 8723 CrossRef CAS PubMed.
- T. Zuo, M. M. Olmstead, C. M. Beavers, A. L. Balch, G. Wang and G. T. Yee, et al., Preparation and Structural Characterization of the Ih and the D5h Isomers of the Endohedral Fullerenes Tm3N@C80: Icosahedral C80 Cage Encapsulation of a Trimetallic Nitride Magnetic Cluster with Three Uncoupled Tm3+ Ions, Inorg. Chem., 2008, 47(12), 5234–5244 CrossRef CAS PubMed.
- Y. Miyake, T. Minami, K. Kikuchi, M. Kainosho and Y. Achiba, Trends in structure and growth of higher fullerenes isomer structure of C86 and C88, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 2000, 340(1), 553–558 CrossRef CAS.
- S. Zhao, P. Zhao, W. Cai, L. Bao, M. Chen and Y. Xie, et al., Stabilization of Giant Fullerenes C2(41)-C90, D3(85)-C92, C1(132)-C94, C2(157)-C96, and C1(175)-C98 by Encapsulation of a Large La2C2 Cluster: The Importance of Cluster-Cage Matching, J. Am. Chem. Soc., 2017, 139(13), 4724–4728 CrossRef CAS PubMed.
- I. Spagnolatti, M. Bernasconi and G. Benedek, Electron-phonon interaction in the solid form of the smallest fullerene C20, Europhys. Lett., 2002, 59(4), 572 CrossRef CAS.
- Z. Wang, X. Ke, Z. Zhu, F. Zhu, M. Ruan and H. Chen, et al., A new carbon solid made of the world's smallest caged fullerene C20, Phys. Lett. A, 2001, 280(5–6), 351–356 CrossRef CAS.
- P. W. Dunk, N. K. Kaiser, M. Mulet-Gas, A. Rodrguez-Fortea, J. M. Poblet and H. Shinohara, et al., The smallest stable fullerene, M@C28 (M = Ti, Zr, U): stabilization and growth from carbon vapor, J. Am. Chem. Soc., 2012, 134(22), 9380–9389 CrossRef CAS PubMed.
- C. Piskoti, J. Yarger and A. Zettl, C36, a new carbon solid, Nature, 1998, 393(6687), 771–774 CrossRef CAS.
- S. Y. Xie, F. Gao, X. Lu, R. B. Huang, C. R. Wang and X. Zhang, et al., Capturing the labile fullerene[50] as C50Cl10, Science, 2004, 304(5671), 699 CrossRef CAS PubMed.
- J. H. Chen, Z. Y. Gao, Q. H. Weng, W. S. Jiang, Q. He and H. Liang, et al., Combustion synthesis and electrochemical properties of the small hydrofullerene C50H10, Chem. – Eur. J., 2012, 18(11), 3408–3415 CrossRef CAS PubMed.
- E. Ōsawa, The evolution of the football structure for the C60 molecule: a retrospective, Philos. Trans. R. Soc. London, Ser. A, 1993, 343(1667), 1–8 CrossRef.
- N. Shao, Y. Gao and X. C. Zeng, Search for lowest-energy fullerenes 2: C38 to C80 and C112 to C120, J. Phys. Chem. C, 2007, 111(48), 17671–17677 CrossRef CAS.
- N. Shao, Y. Gao, S. Yoo, W. An and X. C. Zeng, Search for lowest-energy fullerenes: C98 to C110, J. Phys. Chem. A, 2006, 110(24), 7672–7676 CrossRef CAS PubMed.
- G. Sun, M. C. Nicklaus and R.-h. Xie, Structure, Stability, and NMR Properties of Lower Fullerenes C38–C50 and Azafullerene C44N6, J. Phys. Chem. A, 2005, 109(20), 4617–4622 CrossRef CAS PubMed.
- E. Małolepsza, H. A. Witek and S. Irle, Comparison of geometric, electronic, and vibrational properties for isomers of small fullerenes C20-C36, J. Phys. Chem. A, 2007, 111(29), 6649–6657 CrossRef PubMed.
- D. L. Wang, H. L. Xu, Z. M. Su and D. Y. Hou, Ab initio and density functional study on fullerene C44 and its derivatives, Comput. Theor. Chem., 2011, 978(1–3), 166–171 CrossRef CAS.
- X. Zhao, On the structure and relative stability of C50 fullerenes, J. Phys. Chem. B, 2005, 109(11), 5267–5272 CrossRef CAS PubMed.
- Y. Chang, A. F. Jalbout, J. Zhang, Z. Su and R. Wang, Theoretical study on C32 fullerenes and derivatives, Chem. Phys. Lett., 2006, 428(1–3), 148–151 CrossRef CAS.
- Y. H. Cui, W. Q. Tian, J. K. Feng and D. L. Chen, Structures, stabilities, electronic, and optical properties of C64 fullerene isomers, anions (C642− and C644−), metallofullerene Sc2@C64, and Sc2C2@C64, J. Comput. Chem., 2008, 29(16), 2623–2630 CrossRef CAS PubMed.
- A remark about nomenclature: for some authors, the word phonon does, strictly speaking, refer to quanta of vibration in periodic lattices. Therefore such a term would not be appropriate for vibrations in molecules such as the ones analysed in the present article (which may be called vibrons instead). Nevertheless, since this strict convention is frequently overridden, in this article we refer to vibration quanta as phonons, that its contents can be more easily understandable for a wide number of readers.
- P. B. Allen, Solids with thermal or static disorder. I. One-electron properties, Phys. Rev. B: Solid State, 1978, 18, 5217–5224 CrossRef CAS.
- P. B. Allen and V. Heine, Theory of the temperature dependence of electronic band structures, J. Phys. C: Solid State Phys., 1976, 9(12), 2305 CrossRef CAS.
- P. Han and G. Bester, Band gap renormalization of diamondoids: vibrational coupling and excitonic effects, New J. Phys., 2016, 18(11), 113052 CrossRef.
- P. Schwerdtfeger, L. N. Wirz and J. Avery, The topology of fullerenes, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5(1), 96–145 CAS.
- R. B. Capaz, C. D. Spataru, P. Tangney, M. L. Cohen and S. G. Louie, Temperature Dependence of the Band Gap of Semiconducting Carbon Nanotubes, Phys. Rev. Lett., 2005, 94, 036801 CrossRef PubMed.
- S. Poncé, G. Antonius, Y. Gillet, P. Boulanger, J. Laflamme Janssen and A. Marini, et al., Temperature dependence of electronic eigenenergies in the adiabatic harmonic approximation, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 214304, DOI:10.1103/PhysRevB.90.214304.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car and C. Cavazzoni, et al., QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21(39), 395502, DOI:10.1088/0953-8984/21/39/395502.
- C. Hartwigsen, S. Goedecker and J. Hutter, Relativistic separable dual-space Gaussian pseudopotentials from H to Rn, Phys. Rev. B: Condens.Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789, DOI:10.1103/PhysRevB.37.785.
- S. Baroni, S. de Gironcoli, A. Dal Corso and P. Giannozzi, Phonons and related crystal properties from density-functional perturbation theory, Rev. Mod. Phys., 2001, 73, 515–562 CrossRef CAS.
- D. Vukicevic, F. Cataldo, O. Ori and A. Graovac, Topological efficiency of C66 fullerene, Chem. Phys. Lett., 2011, 501(4), 442–445 CrossRef CAS.
- P. Schwerdtfeger, L. Wirz and J. Avery, Fullerene – A Software Package for Constructing and Analyzing Structures of Regular Fullerenes, J. Comput. Chem., 2013, 34, 1508–1526 CrossRef CAS PubMed.
- D. Tománek, Guide Through the Nanocarbon Jungle, IOP Publishing, Bristol, UK, 2014, pp. 2053–2571 Search PubMed.
- Database from the University of Michigan, https://nanotube.msu.edu/fullerene/.
- W. Qian, M. D. Bartberger, S. J. Pastor, K. N. Houk, C. L. Wilkins and Y. Rubin, C62, a Non-Classical Fullerene Incorporating a Four-Membered Ring, J. Am. Chem. Soc., 2000, 122(34), 8333–8334 CrossRef CAS.
- https://github.com/pablogr/ML_fullerenes/tree/main/FULLERENE_XYZ_FILES .
- https://doi.org/10.5281/zenodo.10059442 .
- https://github.com/pablogr/ML_fullerenes/tree/d7f658381c3e69695aa8e021c2880ecc51afc81e/CODE_FOR_CALCULATIONS_OF_THE_PAPER .
- https://github.com/pablogr/ML_fullerenes/tree/d7f658381c3e69695aa8e021c2880ecc51afc81e/CODE_FOR_FORECASTING .
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion and O. Grisel, et al., Scikit-learn: Machine Learning in Python, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- L. Breiman, J. Friedman, C. J. Stone and R. A. Olshen. Classification and Regression Trees, Chapman and Hall/CRC, 1984 Search PubMed.
- E. Armengol, Estimation of Prediction Error with Regression Trees, in Modeling Decisions for Artificial Intelligence – 19th International Conference, MDAI 2022, Sant Cugat, Spain, August 30 – September 2, 2022, Proceedings, ed. V. Torra and Y. Narukawa, Lecture Notes in Computer Science, Springer, 2022, vol. 13408, pp. 193–202 DOI:10.1007/978-3-031-13448-7_16.
- J. L. Janssen, M. Côté, S. G. Louie and M. L. Cohen, Electron-phonon coupling in C60 using hybrid functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81(7), 073106 CrossRef.
- M. Saito, Electron-phonon coupling of electron- or hole-injected C60, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65(22), 220508 CrossRef.
- R. S. Wang, D. Peng, J. W. Hu, L. N. Zong and X. J. Chen, Orientational ordering and electron-phonon interaction in K3C60 superconductor, Carbon, 2022, 195, 1–8 CrossRef CAS.
- O. Gunnarsson, H. Handschuh, P. S. Bechthold, B. Kessler, G. Ganteför and W. Eberhardt, Photoemission Spectra of C60−: Electron-Phonon Coupling, Jahn-Teller Effect, and Superconductivity in the Fullerides, Phys. Rev. Lett., 1995, 74(10), 1875 CrossRef CAS PubMed.
- I. D. Hands, J. L. Dunn, C. A. Bates, M. J. Hope, S. R. Meech and D. L. Andrews, Vibronic interactions in the visible and near-infrared spectra of C60− anions, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77(11), 115445 CrossRef.
- D. R. Penn, Wave-number-dependent dielectric function of semiconductors, Phys. Rev., 1962, 128(5), 2093 CrossRef CAS.
- P. Yu and M. Cardona, Fundamentals of semiconductors: physics and materials properties, Springer Science & Business Media, 2010 Search PubMed.
- S. Logothetidis, J. Petalas, H. M. Polatoglou and D. Fuchs, Origin and temperature dependence of the first direct gap of diamond, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 4483–4494 CrossRef CAS PubMed.
- For diamondoids and fullerenes, these percentages were calculated using the gap from B3LYP and the gap renormalization from GGA-PBE.
- P. García-Risueño, A. Syropoulos and N. Vergés, New ideograms for physics and chemistry, Found. Phys., 2016, 46, 1713 CrossRef.
Footnotes |
| † PACS numbers: 63.22.Kn, 71.38.-k, 81.05.uj, 65.80.-g. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00632a |
| This journal is © the Owner Societies 2024 |