
Unprecedented selectivity behavior in the direct dehydrogenation of n-butane to n-butenes with similar active Pt nanoparticle size: unveiling structural and electronic characteristics of supported monometallic catalysts
Gustavo Enrique
Ramos Montero
 *ab,
Adriana Daniela
Ballarini
a,
María Julia
Yañez
c,
Sergio Rubén
de Miguel
a,
Sonia Alejandra
Bocanegra
a and
Patricia Daniela
Zgolicz
ab
*ab,
Adriana Daniela
Ballarini
a,
María Julia
Yañez
c,
Sergio Rubén
de Miguel
a,
Sonia Alejandra
Bocanegra
a and
Patricia Daniela
Zgolicz
ab
aInstituto de Investigaciones en Catálisis y Petroquímica “Ingeniero José M. Parera” (INCAPE), Facultad de Ingeniería Química, Universidad Nacional del Litoral – CONICET, Centro Científico Tecnológico CONICET Santa Fe (CCT-SF), Santa Fe, Argentina. E-mail: geramos@fiq.unl.edu.ar
bPhysicochemistry Department, Facultad de Ingeniería, Universidad Nacional de Entre Ríos, Paraná, Entre Ríos, Argentina
cCentro Científico Tecnológico CONICET Bahía Blanca (CCT-BB), Camino La Carrindanga, Km 7, (8000) Bahía Blanca, Argentina
First published on 14th October 2024
Abstract
In this work, supported Pt monometallic catalysts were prepared using oxide and carbon supports by conventional impregnation methods. Similar Pt metallic nanoparticle sizes (mean sizes about 1.8–2 nm) have been obtained using different Pt precursor loadings (0.3 to 5 wt%). For comparison, catalysts with larger nanoparticle sizes were prepared using the liquid phase reduction method. Characterization results indicate different electronic and structural characteristics for the Pt nanoparticles, comparing nanoparticles with similar and different sizes, implying that both the Pt loading and the preparation method affect the formation of different metallic phases. We used the direct dehydrogenation of n-butane to n-butenes reaction as a test reaction to study the catalytic behavior of the Pt nanoparticles obtained at different Pt atomic concentrations. Surprisingly, Pt catalysts with the lowest metallic loading show the highest selectivities to olefins. Besides, Pt catalysts supported on carbon materials showed higher selectivity to butenes than those supported on oxide materials, this was attributed to a higher electron density in the Pt active sites. Likewise, at low Pt loadings, the CNP-supported Pt nanoparticles could be confined at the defect in the nanotube structure as crystalline agglomerates of atoms with few layers or monolayers with very few surface adatom or stepped adatom nanostructures or simply as a group of atoms, thus creating active Pt sites that favor the dehydrogenation reaction over secondary reactions.
Introduction
Heterogeneous catalytic systems based on supported noble metal nanoparticles are important industrial catalysts used in the petrochemical industry and in the production of intermediate products of the plastic-petrochemical value chains, such as olefins obtained from selective hydrocarbon dehydrogenation reactions.1–4 In recent years, the growing demand for olefins has driven the rapid development of dehydrogenation technologies as the direct catalytic dehydrogenation of light alkanes to alkenes.1,3,5–8 In this sense, butenes (n-butene, butadiene, and isobutylene) are one of the main building blocks for petrochemicals; it is an important raw material for the production of chemicals and polymers.2,5,9–13The metallic particle size is a key point in the research on designing novel catalysts to promote a highly efficient dehydrogenation process.14–19 Several reasons explain this asseveration: the particle size determines (i) the number of surface active atoms at a given metal loading, (ii) the electronic properties of the metallic phase, and (iii) the structural properties of the active surface. In relation to the active atoms surface, it is logical to think (according to numerical calculations) that smaller particle sizes and higher dispersions will be obtained with low metallic loadings, a positive factor in the design of a catalyst for economic reasons. On the other hand, it must be noted that the electronic properties refer to the amount of packed metallic atoms and metallic bonds in the crystalline particle, and the superficial metallic structure (or geometry of the active surface) refers to particle shape.17,18,20–23 Electronic properties and superficial metallic structure are also important for the adequate development of an active surface because these factors determine the properties of the active sites for a given reaction.17,18,24,25 However, the geometrical and electronic influences for a given particle cannot be separated as independent parameters since increasing the size of the metallic particles results in an electron bandwidth increase, as well as the nature of the exposed planes and the topology of the superficial sites changes.17 Likewise, the amount of surface atoms, seen as coordinatively unsaturated and saturated metallic sites, changes with the particle size, also evidencing different electronic properties for these site types.17,26,27
Thus, the research about the preparation of nanoparticles for catalytic applications has received intensive attention in the last decades due to the importance of the properties mentioned above; the effect of the particle size is the main parameter studied on the catalytic performance.19,26,28–31 Parallel, the influence of the surface structure of a metal particle on its catalytic properties has also been extensively reported.20,26,32–34 However, since the catalytic activity of metal nanoparticles is generally linked to the particle size as the fraction of metal sites on the surface, such as corners, edges, and planes, the effect of surface structure has been related to the so-called “size effect”.17,18,20 Thus, several studies have revealed that a large number of reactions have been shown to be strongly dependent on the particle size, whereas others are independent of the particle size. Besides, the general concept of structure-sensitivity, reported by Boudart in 1969, arises depending on whether the metallic structure affects the rate per active metal atom.35 Since the structure is linked to the particle size, the reactions, in consequence, were classified as structure sensitive or insensitive, or also structure demanding or facile.27,35
There are at least two reasons why these concepts have been historically maintained for decades. First, the structure, as the fraction of surface metal sites such as corner, edge, and plane, can be estimated using a nanocrystal model.26,36,37 Second, the effect of metal nanoparticle size on catalytic activity is better understood nowadays than the influence of the shape or structure surface because size control is easier to achieve than structure control.20 Thus, for example, considering the Pt atomic radius and using a Pt nanocrystal model as a cuboctahedron particle, the Pt particle size dependence on the fraction of Pt surface sites can be estimated.27,34,36,37 Murata et al. used data from these estimations to represent the fraction of site surface vs. nanocrystal size, thus showing that the fraction of Pt(111) and Pt(100) planar sites in the Pt nanoparticle increases with the increasing of the crystal size and that the fraction of edges and corners are higher as the size decrease.26 That is, considering a theoretical crystalline model particle, the structure of the metallic phase will be unique in relation to the fraction of metallic sites as corners, edges, and planes for a determined particle size and crystalline shape (i.e., cubic, octahedron, cuboctahedron). Besides, from this analysis, a size of 2 nm seems to be critical: for Pt nanoparticles of >2 nm in size, the planes were found to be dominant, while Pt nanoparticles of <2 nm in size feature an abundance of corners and edges. It must be noted that the 2 nm value seems to coincide with the value range of particle size, which is reported as the transition between clusters and nanoparticles.17,34,38,39 As the size of metal particles decreases, the continuous valence band of bulk metal starts to separate in a discrete state, thus losing the true metallic bonds.34
On the other hand, Somorjai in 1977 reported that the structure-sensitive character of a given reaction is related to the structure-sensitive character of the chemical bond formed between the reactants and the catalytic surface.27 Since the bond strength is affected by the local surface structure, the relative concentration of steps, corners, or planes is very important for catalytic activity. This point of view agrees with the ensemble effect proposed by Sachtler in 1981, through which an ensemble of surface atoms is essential for the adsorption of reactants and the subsequent catalytic transformations.40 So, preferential exposure of certain facets of a metallic nanoparticle could increase the reaction rate, change reaction paths, or also improve selectivity and yields to desired products.
It must be noted that according to the concepts initially introduced by Boudart, the structure sensitive–insensitive character of a reaction is linked to the size effect since, ideally, there would be a unique structure for a model nanoparticle with a determinate size.35 Somorjai used non-supported metallic single crystals in their studies.27 However, the surface of dispersed metallic catalytic nanoparticles on a determined support is imperfect, and also, as the particle size decreases, the nanoparticles lose their regular atomic arrangements and exhibit disordered atomic arrangements.26,27,33,41 These disordered atomic arrangements could include a higher or lower amount of steps, corners, and edges than those nanoparticles with the ideal arrangement in the crystalline structure. Besides, imperfect surfaces could include other arrangements such as kinks, adatoms, step-adatoms, and terrace vacancies.27 So, these disordered atomic arrangements would represent not only a complex system of facets and entwining of atoms but also a different amount of coordinatively unsaturated sites on the surface of the supported metallic particles.26,42 What is more, no basis indicates that these arrangements in supported imperfect catalytic nanoparticles must be equal for the same particle size. Nevertheless, for the same particle size, the relative abundance of coordinatively saturated and unsaturated atoms on the local surface structure could be achieved by different preparation methods or using different metallic loading, for example. In addition to this, the material used to support the nanoparticles could affect the geometric and electronic structure and change the properties through metal–support interactions.24,25 So, several works have studied the structure of the metallic phase at different particle sizes, but there are practically no works that specifically study the effect of the metallic phase structure considering only a specific particle size, and even fewer that study the effect of the catalytic support using materials with different properties. In addition, carbon supports have been scarcely studied for catalysts in dehydrogenation reactions.9,13,43
On the other hand, the growing interest in the dehydrogenation of light alkanes to cover the increasing demand for polymers, solvents, and gasoline additives relies on developing novel catalytic processes involving non-oxidative direct catalytic dehydrogenation.1–4 For economic reasons, this process is preferable to the oxidative one; however, as the direct process is endothermic, it usually requires high temperatures to obtain high olefin yields. Under the condition of high temperatures, catalytic systems mainly based on monometallic catalysts lead to undesirable reactions, such as the formations of light cracking and isomerization products and coking deposits, being the products from these reactions responsible both for the loss of selectivity and activity of the catalysts.3,6,7,44,45 At this point, it is essential to highlight that the desired dehydrogenation reaction is considered an insensitive reaction, while the hydrogenolysis (or cracking) and coking formation reactions are sensible to the structure. Subsequently, this creates an interesting situation of compromise since, in general, it has been seen that a low particle size is detrimental to reaction selectivity because of the higher amount of the unsaturated coordinatively metallic atoms in small particles, which has been demonstrated that promoting side reactions such hydrogenolysis.27
In this work, we prepared Pt monometallic catalysts supported on oxide and carbonaceous materials for the direct dehydrogenation of n-butane to n-butenes. We used different Pt loadings and obtained catalysts with similar Pt nanoparticle sizes (about 2 nm for catalysis supported on carbon materials and about 1.8 nm for catalysis supported on oxide materials). These results revealed unprecedented selectivity behavior for both monometallic catalysts supported on carbon and those supported on oxide materials, showing the highest selectivities the catalysts with the lowest Pt loading. Besides, the best yields towards n-butenes, with selectivities greater than 80%, were obtained with catalysts supported on carbon. So, here we report these interesting results and analyze the relationship between the characteristics of the metallic phase and the catalytic behavior. Thus, this work aims to clarify some aspects concerning the catalytic behavior of Pt nanoparticles on hydrocarbon transformations under dehydrogenation conditions, which involve structure-sensitive and structure-insensitive reactions. Likewise, it is also an aim of this study to contribute to the general understanding of both the structural and electronic properties of the Pt active metallic sites that lead to the production of olefins and of those properties that allow the formation of side products in monometallic catalysts supported both on oxide and carbons materials. In conclusion, the results of the characterization of the catalyst will allow these objectives to be achieved through the analysis and correlation between the characteristics of the metallic phase and the catalytic behavior. So, since the results obtained for monometallic Pt catalysts supported on oxide and carbon materials would allow to shed light on the aspects of the structural effect and the electronic properties of the supported Pt nanoparticles on the selectivity of the n-butane dehydrogenation reaction, this would be evidence of how these results could be extrapolated to other catalytic systems.
Materials and methods
The following commercially available materials were used as supports:(i) Multiwalled carbon nanotubes (MWCN from Sunnano, purity > 90%, diameter: 10–30 nm, length: 1–10 μm) purified according to the technique reported in previous works.46 This support was called CNP.
(ii) Carbon black (Cabot Corp. Vulcan carbon XC −72, purity > 99%); this support was called CV.
(iii) Commercial γ-Al2O3 (CK 300 from Cyanamid Ketjen) previously calcined at 650 °C.
(iv) MgAl2O4 with a spinel structure. It was prepared by co-precipitation method from solutions of Mg(NO3)2·6H2O (Merck, purity 99%), Al(NO3)3·9H2O (Merck, purity 98.5%), and ammonia solution (Merck, 28%, analytical grade) as described in a previous work.47
To prepare the monometallic Pt catalysts, the supports were conventionally impregnated with an aqueous solution of H2PtCl6 at room temperature. To obtain a uniform contact between the solid and the impregnating solution, an impregnation volume/support weight ratio of 30 4 mL g−1 and 4 mL g−1 was used for the carbon and oxide supports, respectively. The Pt concentration in the impregnation solution was that required to achieve a Pt loading of 0.3, 0.5, or 5 wt%. After impregnation, the samples were dried overnight at 110 °C and ground into powder.
Additionally, two impregnation methods with liquid phase reduction were used: (i) for the sodium borohydride reduction method,48 carbon nanotubes were dispersed into water and stirred for 30 min at 55 °C. Then, calculated amounts of H2PtCl6 solutions were added to the above mixture and stirred for 30 min. A freshly prepared 0.4 M NaBH4 solution (in 1 M NaOH) was added drop by drop into the above solution under vigorous stirring. After stirring for 1 h, the obtained mixture was cooled to room temperature, filtered, and washed repeatedly with deionized water. The formed powder was dried at 70 °C for 2 h in a vacuum oven. The catalysts are denoted as Pt/CNP-BH, (ii) for the formic acid reduction method, a suspension of the support and formic acid was prepared (1 g of support and 50 mL of formic acid 0.3 M) by sonication at room temperature for 15 min. The suspension was thermostatic under stirring (300 rpm) at 50–60 °C for 30 min. Then, the H2PtCl6 solution was added in three steps under stirring. The contact time for each addition of H2PtCl6 was at least 1 h. After 1 h of the first addition of the Pt precursor, 10 mL of formic acid was added to ensure the total reduction of the metallic compound. After each H2PtCl6 addition, a reducibility test was carried out using KI and starch (indicator).49
The specific surface areas (SBET) of the different supports were determined by adsorption of N2 at −196 °C in a Micromeritics ASAP 2020 instrument. Before the adsorption isotherms were determined, the samples were degassed at 250 °C.
The surface chemistry of the supports was determined by temperature-programmed desorption (TPD) performed in a differential flow reactor coupled to a thermal conductivity detector. Samples (0.300 g) were heated from 25 to 750 °C in an electric furnace at 6 °C min−1. During the TPD experiments, He was passed through the reactor at a 9 mL min−1 flow rate. Before the TPD experiments, the samples were stabilized with He at room temperature for 1 h.
The isoelectric points of the carbon supports were determined by titration or neutralization at constant pH in an aqueous KNO3 solution. Equilibration and titrations were performed under a nitrogen atmosphere to eliminate the influence of atmospheric CO2.50 First, the aqueous solution of KNO3 (0.1 N) was stirred while purified N2 bubbled through the system at room temperature. The pH of the solution was kept constant (at pH = 7) with nitric acid or potassium hydroxide. Then, the samples (500 mg) were suspended in the aqueous solution and equilibrated to obtain the corresponding value of the isoelectric point.
In order to characterize the acid properties of the oxide supports, 2-propanol dehydration experiments in a continuous flow reactor at atmospheric pressure were carried out. Prior to the reaction, samples were reduced “in situ” with H2 at 500 °C.
The alcohol was vaporized in an H2 stream (H2/2-propanol molar ratio = 19) and fed to the reactor with a space velocity of 0.52 mol alcohol per h per gcat. The sample weighed 100 mg, and the reaction temperature was 200 °C.
The Raman spectra were recorded using a LabRam spectrometer (Horiba–Jobin–Yvon) coupled to an Olympus confocal microscope (a 100× objective lens was used for simultaneous illumination and collection), equipped with a CCD detector cooled to about −70 °C using the Peltier effect. The excitation wavelength was, in all cases, 532 nm (diode-pumped solid-state laser). The laser power measured above the sample cell was 5 mW, with a collection time = 10 s and 10 accumulations.
The characterization of the metallic phase was carried out using different techniques. The following test reactions were used: cyclohexane dehydrogenation (CHD) at 250 °C and cyclopentane hydrogenolysis (CPH) at 500 °C, both performed in a differential reactor. The activation energy for the CHD reaction was calculated by linear regression using the reaction rates measured at three temperatures (250, 240, and 230 °C) and the Arrhenius equation. In both reactions, the catalysts were previously reduced “in situ” in H2 at 500 °C for 2 h. The reaction was carried out at a flow rate of 6 cm3 h−1 (molar ratios H2/CH = 29 and H2/CP = 26). The competitive hydrogenation of toluene + benzene mixtures was performed in a conventional flow reactor at 110 °C and atmospheric pressure. The partial pressure ratio of toluene and benzene was varied between 0 and 1. From the kinetic measurements and following the method proposed by Tri et al.51 the ratio of toluene to benzene adsorption coefficients (KT/B) was calculated. The reaction products were analyzed by CG.
H2 chemisorption was carried out at 25 °C in a static volumetric system. Adsorption isotherms were performed in the pressure range (50–250 torr) in samples previously reduced under flowing H2 and degassed (evacuated at 5.10–5 torr). The amount of chemisorbed hydrogen was calculated by extrapolating the isotherm at zero pressure.
Temperature-programmed reduction (TPR) experiments were performed. The samples were heated at 6 °C min−1 in a stream (9 cm3 min−1) of the gas mixture (H2 (5% v/v)/N2) from room temperature to 800 °C.
The previously reduced catalysts were characterized by XPS. For each sample, spectral regions corresponding to C 1s, O 1s, Al 2p, Pt 4f, and Pt 4d were registered. The areas of the peaks were calculated by fitting the experimental results to the Lorentz–Gauss curves using the software CASA XPS.
Transmission electron microscopy (TEM) measurements were performed on a JEOL 100 CX II microscope with a nominal resolution of 0.6 nm. The instrument was operated with an accelerating voltage of 100 kV and a magnification range of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× to 100000×. High-resolution transmission electron microscopy (HR-TEM) was performed on a JEOL JEM-2100 Plus microscope. The observations were carried out in HRTEM and HAADF mode, and images and X-ray spectra were also obtained using an accelerating voltage of 200 kV with magnification ranging from 600000 to 1500000×. In both cases, a drop of a suspension of the reduced samples in ethanol was placed on a carbon–copper grid, and the solvent was later evaporated. A significant number of Pt particles (over 150) were measured for each catalyst to obtain the size distributions. The mean metal particle diameter (D) was calculated as
000× to 100000×. High-resolution transmission electron microscopy (HR-TEM) was performed on a JEOL JEM-2100 Plus microscope. The observations were carried out in HRTEM and HAADF mode, and images and X-ray spectra were also obtained using an accelerating voltage of 200 kV with magnification ranging from 600000 to 1500000×. In both cases, a drop of a suspension of the reduced samples in ethanol was placed on a carbon–copper grid, and the solvent was later evaporated. A significant number of Pt particles (over 150) were measured for each catalyst to obtain the size distributions. The mean metal particle diameter (D) was calculated as 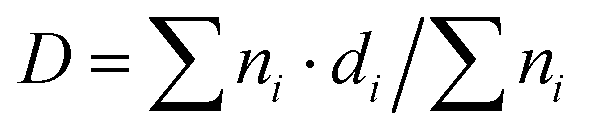 , where ni is the number of particles with diameter di.
, where ni is the number of particles with diameter di.
Catalytic performance was evaluated using n-butane dehydrogenation in a continuous flow reactor at 530 °C fed with a 1.25:1 mixture of H2/C4H10. Prior to the reaction, each catalyst was reduced in H2 stream for 2 h at 530 °C. The first sample of the reactor effluent was taken after 10 min from the start of the reaction. The reaction products (methane, ethane, ethylene, propane, propene, 1-butene, cis- and trans-2-butene, and butadiene) were analyzed in a gas chromatograph with a FID detector. The n-butane conversion was calculated as the sum of the percentages of the chromatographic areas of all reaction products corrected by the corresponding response factor. Selectivity to all butenes was calculated as the ratio of moles of total butenes (1-butene, cis- and trans-2-butene, and butadiene) to moles of all reaction products.
Catalysts were also tested in the n-butane dehydrogenation reaction in a pulse equipment to analyze the catalytic behavior at the beginning of the dehydrogenation. The pulse experiment was carried out at 530 °C by injecting successive pulses of pure n-butane into the catalytic bed, which was maintained under He flow between two successive pulses. The catalytic samples were previously reduced under an H2 flow for 2 h at 530 °C. The reaction products were analyzed by a GC system with an FID detector.
Results and discussion
Two carbon materials (CNP and CV) and two oxide materials (γ-Al2O3 and MgAl2O4) were used to prepare four series of catalysts by the conventional impregnation method with different Pt loadings. For comparison, two additional series of catalysts were prepared using the liquid phase reduction method using NaBH4 or formic acid as reducer agents and CNP support (see Table 1).| Catalyst | Preparation method | Support |
|---|---|---|
| X: 0.3, 0.5 or 5 wt%. | ||
| XPt/CNP | Purified carbon nanotubes | |
| XPt/CV | (i) Conventional impregnation | Vulcan carbon |
| XPt/Al2O3 | (ii) H2 reductive treatment at 530 °C for 2 h | γ-Alumina |
| XPt/MgAl2O4 | Mg-spinel | |
| XPt/CNP-BH | (i) Liquid phase reduction with NaBH4 | Purified carbon nanotubes |
| (ii) H2 reductive treatment at 530 °C for 2 h | ||
| XPt/CNP-FA | (i) Liquid phase reduction with formic acid | |
| (ii) H2 reductive treatment at 530 °C for 2 h | ||
Table 2 shows the textural characteristics of the supports. The results indicate that all the supports have reasonably high areas to disperse metallic nanoparticles in the studied loading range (0.3 to 5 wt%). The CNP and CV supports are graphitic-type nanostructured carbon materials. Both materials have mesoporous structure due to the exceptional properties of the graphitic concentric layers.46,50,52 CNP exhibits a structure of nested concentric tubes, which evidences a high length-to-diameter aspect ratio and provides a high external surface area to deposit metallic nanoparticles.53,54 Vulcan carbon is a granulated graphite-type carbon black but with a nanostructure of concentric onion-type layers, which also provide them with a high external surface area.46,55 It must be noted that both structures could be imperfect at the surface, thus leading to π-basic sites defects (or sites with unsaturated atoms) and sites with carbon atoms bonded to oxygen atoms (forming oxygenates surface groups), which could serve to anchor metallic nanoparticles.50,56,57
| Support | S BET (m2 g−1) | V p (cm3 g−1) | IP |
|---|---|---|---|
| n.m.: not measured. | |||
| Al2O3 | 180 | 0.5 | n.m. |
| MgAl2O4 | 108 | 0.33 | n.m. |
| CN | 211 | 0.46 | 4.6 |
| CNP | 179 | 0.39 | 4.7 |
| CV | 240 | 0.36 | 7.4 |
Table 2 also shows the isoelectric point values of the supports. The results reveal a more acidic character for the CNP than CV. However, despite the neutral characteristics of CV support, it shows an amphoteric behavior and displays good metal adsorption during the impregnation step.
Since the acidity depends on the amount and type of surface acid oxygenated groups, these results can also be evidenced by TPD experiments. Surface oxygenated groups decompose upon heating and can be desorbed as CO2 at low temperatures from stronger acid groups and as CO at high temperatures from weaker acid groups.46,50,58 In this sense, carboxylic and anhydride groups are considered stronger acid groups, while lactone, phenol, and carbonyl groups are considered weaker.58Fig. 1 shows the amount of desorbed CO and CO2 as a function of the temperature for the carbon supports. It can be seen that the original carbon nanotubes desorb a low amount of weak acid groups, and after purification, this support additionally desorbs a low amount of strong acid groups and a higher amount of weak acid ones. Besides, the CV sample does not desorb oxygenated surface groups. The proper structure of carbon nanotubes with a high length-to-diameter relation would evidence the possible formation of a higher number of defects at the surface and an easier formation of oxygenated groups (strong or weak) in unsaturated sites than in the Vulcan carbon. In addition, the proper secondary structure of Vulcan carbon as spherical aggregates is maintained by certain groups of heteroatoms placed in the perimeter of each basal layer, being these groups not very acidic since they allow the stabilization of the aggregates.46 Thus, the strong acid surface groups could contribute to the acidity of CNP, while the oxygenated groups at the edges of the basal plane structure of Vulcan carbon contribute to its neutral character.
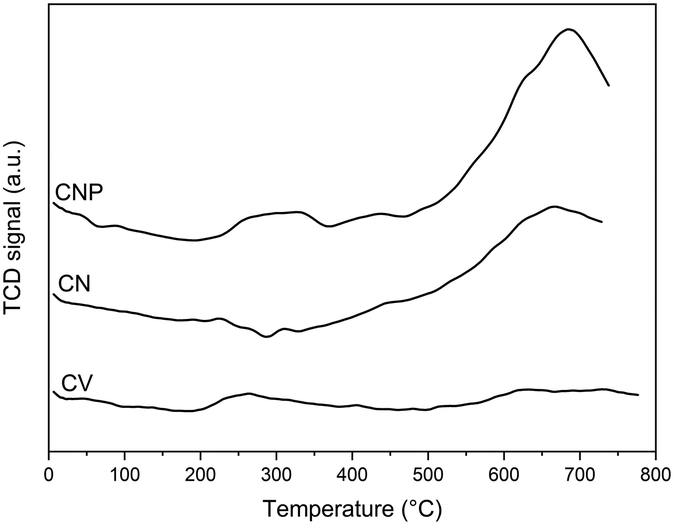 | ||
| Fig. 1 TPD profiles of the supports. | ||
Raman spectra of the catalysts supported on carbons were also obtained. Raman spectroscopy is an attractive method for the structural characterization of carbon-based materials, as it can detect the degree of carbon structural disorder.59 Besides, this technique is highly sensitive to the structural and chemical features of the catalysts, so the results were used to characterize carbon-based materials and the anchoring of Pt particles to the support.60,61Fig. 2 shows Raman spectra obtained for the supports and catalysts based on CNP and CV. It can be observed in these spectra the characteristic bands, D and G, obtained for type-graphitic carbon samples. The D band at ∼1345 cm−1 is due to the disordered carbon structure with defects and sp3 hybridization, while the G band observed at ∼1580 cm−1 is due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in sp2-hybridized carbon structure, which corresponds to an intrinsic lattice-type vibrational mode for the perfect graphite structure.
C bonds in sp2-hybridized carbon structure, which corresponds to an intrinsic lattice-type vibrational mode for the perfect graphite structure.
 | ||
| Fig. 2 Raman spectra (a) samples on CNP and (b) samples on CV. | ||
Moreover, in Fig. 2(a), in addition to the two peaks representing the D (so-called defect) and G (so-called graphite) bands, the D′ band was also observed as a shoulder of the G band around 1610 cm−1, leading to a broadened G band. As the D band, the D′ band is also associated with lattice domains and is related to the degree of disorder. However, the D band is associated with a vibration mode corresponding to the lattice-graphene layer edge, while the D′ band can be related to the lattice-surface graphene layer.59,60,62–64
Fig. 2(a) and (b) show the Raman spectra of 0.3Pt/CNP and 0.3Pt/CV, respectively, before and after the reduction step. It can be seen in Fig. 2(a) that the peaks of the D and G bands of the samples with Pt are shifted to higher wavenumbers. This could be associated with the existence of strong interactions and even bonding between the carbon nanotubes and Pt.59,64–66 Consequently, the upshift in the bands in the spectra of 0.3Pt/CNP could be related to strong bonding between carbon nanotubes defects and Pt atoms. Furthermore, Pt atoms could not only form bonds with surface oxygenated groups but also saturate the defect sites; i.e., the deposition of Pt atoms can saturate the vacancy. In this last case, the formation of pseudo hexagons by Pt atoms that saturate the dangling bonds of adjacent carbon atoms in the vacancy could alter the vibrational modes as new bonds are formed.65,66 Besides, the intensity of the D′ band slightly increased (the relative area increased by about 5%). It can also be observed that 0.3Pt/CNP presents an additional shoulder in the D band centered at about 1310 cm−1. This shoulder was reported as a band corresponding to a non-homogeneous defect associated with solids with impurities as ions.67 Thus, this could be related to the deposition of precursor ionic species on the nanotubes before reduction. In the spectra of the sample 0.3Pt/CNP-red, the shoulder at about 1310 cm−1 is absent, as was expected from the reduction of the Pt precursor complex. However, the shoulder at 1610 cm−1 remains. This, together with the upshift of the D and G bands, can be explained by the fact that the Pt atoms that saturated the defect sites remain anchored to the carbon nanotube surface and could serve as seeds that allow the growth of metallic nanoparticles or simply the grouping of a certain amount of Pt atoms.
Similarly, these results can be verified in CV-supported catalysts, nevertheless, some differences must be mentioned. The absence of the D′ band in all samples associated with the lattice-surface graphene layer would be related to the characteristic structure of Vulcan carbon, which, compared to nanotubes, does not present concentric graphene layers as nested concentric tubes but as onion-like concentric layers. So, these results agree with those mentioned above that the imperfections predominate at the graphene layer edges for CV support. Likewise, the upshift in the bands in the spectrum of 0.3Pt/CV, before and after the reduction step, would also be related to strong bonding between carbon Vulcan defects and Pt atoms, being mainly these defects at graphene layer edges. In addition to the CV support characterization, it must be noted that the band at approximately 1500 cm−1 would be associated with amorphous carbon.59,67,68
On the other hand, since the D and G band areas are known to vary significantly on each sample, the D band was integrated and further normalized with respect to the G band.65,69 So, the D-to-G integrated peak area ratios (ID/IG) can be used to measure the degree of structural perfection when comparing graphitic carbons. The ID/IG ratios were calculated for CNP and CV supports. From these data, it can be observed that the disordered sp3 state is higher in CV support (ID/IG = 2.04) than for CNP support (ID/IG = 1.66), thus indicating a greater amount of defects in CV support, which would be associated at graphene layer edges.
Table 2 also shows adequate areas for the oxide supports to deposit metallic nanoparticles in the range of Pt loadings studied.45,47 γ-Al2O3 and MgAl2O4 are well-known materials as catalytic supports for dehydrogenation reactions. They present regular arrangements and well-defined crystallographic phases.70–72 Besides, γ-Al2O3 is a material with acidic characteristics while MgAl2O4 is a support with a neutral character.73–75 The acid–base characteristics can be evidenced by the 2-propanol dehydration reaction test. Fig. 3 shows the 2-propanol dehydration conversion as a function of reaction time for γ-Al2O3 and MgAl2O4 supports, considering the conversion as a measurement of the total acid character of the support. It can be observed that γ-Al2O3 has a higher acid character than MgAl2O4, as expected.
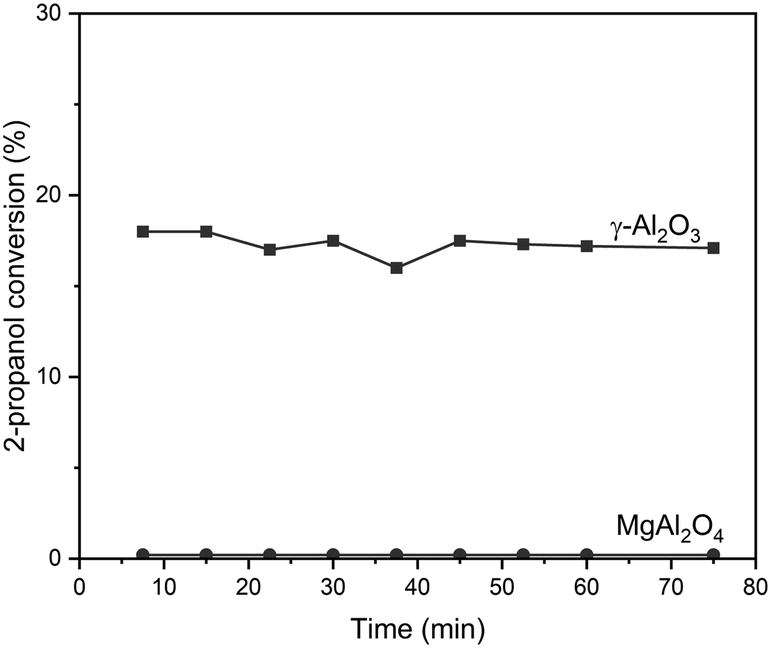 | ||
| Fig. 3 2-Propanol conversion versus time for Al2O3 and MgAl2O4. | ||
Regarding the binding of metals that can act as catalysts to nonreducible metal oxides, it is assumed to occur at the exposed cation of the oxide.76,77 In the case of γ-Al2O3, a model of binding where penta-coordinated Al3+ sites (acid Lewis sites) at the surface interact strongly with Pt or PtO species was proposed.76–78 However, these sites would not be present in the MgAl2O4. Nonetheless, since it seems that the cations of the oxide constitute the probable binding sites of catalytic metals, being these sites in abundance and regular form on the oxide structures. These sites have been postulated that could produce important metal–support interactions.24,26,76,77,79
Further insights about the nature of the support and metal–support interaction can be evidenced from TPR results. Fig. 4 shows the TPR profiles of the four catalytic series prepared by conventional impregnation. These results show that all the catalysts show a main reduction peak at low temperatures (between 100 and 350 °C). These peaks appear in a characteristic reduction zone for supported metallic platinum and would correspond to the decomposition and reduction of the metallic complexes adsorbed on the supports during the impregnation with Pt precursor (H2PtCl6) and also its drying.46,50,70,80,81 A comparative analysis between the TPR profiles of catalysts supported on oxide and carbon materials reveals that the maximum of the Pt reduction peaks for the catalysts supported on carbon appeared around 175–225 °C, while the maximum of these peaks for catalysts supported on oxide materials appear around 225–275 °C. It is well known that the higher the reduction temperature, the stronger the interaction between the metallic species and the support,43,82 in consequence there would be a higher interaction of Pt nanoparticles in catalysts supported on oxide materials than those supported on carbons. Besides, a more detailed analysis for each series shows that the lower the Pt loading, the higher the interaction of metal particles with the support would be. These features of the nanoparticles could produce noticeable effects on catalytic activity.82
 | ||
| Fig. 4 TPR profiles of the samples. | ||
Additional information can be obtained from TPR profiles. Both the TPR profiles of catalysts based on oxide and carbon supports display the existence of other H2 consumption zones at temperatures above 350 °C. For catalysts supported on carbons, these facts would indicate that the Pt could produce a catalytic effect on the decomposition of the surface oxygenated groups present in each support.46,50 This is obvious for weak acid groups for CNP and CV supports, but also, the small shoulder between 275 and 375 °C in catalysts supported on CNP would be evidence of a catalytic effect on the strong acid groups. These TPR results are in agreement with those related to the acidity properties of the carbon supports. Besides, it must be noted that at the end of the catalyst preparation, strong acid groups can be desorbed or could form PtO bonds since a catalyst reduction temperature of 530 °C was used. Unlike carbon supports, the other H2 consumption zones at temperatures above 350 °C in TPR profiles of catalysts based on oxide supports can be explained by the reduction of different oxychlorinated species of Pt with different adsorption strengths.47,80,83
In order to determine the degree of reducibility of Pt in the catalysts, XPS measurements were performed in samples supported on oxide and carbon supports and prepared by the impregnation method. The binding energy (BE) values of Pt 4f7/2 (for catalysts supported on carbons) and Pt 4d5/2 (for catalysts supported on oxides) levels and the percentages of reduced and oxidized species are shown in Table 3. It must be noted that the Pt 4f signal, the most intense for platinum, overlaps with two Al 2p3/2 signals coming from the oxide supports. So, to obtain more precision about the oxidation states of the Pt, the deconvolution of the Pt 4d signals for the catalysts supported on oxide materials was carried out. As seen in Table 3, from Pt 4d5/2 spectra of all the catalyst samples based on oxide supports, the presence of one peak (at about 313.6–314.8 eV) was found. This peak can be assigned to Pt0, thus indicating 100% reducibility in catalysts supported on oxide supports, except for catalysts with 5% Pt that achieve a reduction of approximately 95%.80,81,84 On the other hand, Pt 4f signals of all the catalysts supported on carbons show an asymmetry at about 75–80 eV (see Fig. 5 for the 0.3Pt/CNP catalyst, which was selected as an example). This fact would indicate that Pt(II) and Pt(IV) ionic species are masked with the signal corresponding to metallic Pt at about 71–72 eV.50 Therefore, from the deconvolution of the Pt 4f spectra of reduced catalysts, three doublets were obtained, one at low binding energy, which represents about 64–70% of the total area (zerovalent Pt), and two doublets at higher binding energies corresponding to the rest of the total area (ionic Pt). These results are in agreement with those reported in the literature, which indicates the presence of Pt oxide species in a Pt(0):Pt(ox) ratio of about 70:30 in catalysts supported on graphitic carbons (such as nanotubes and Vulcan carbon) regardless of the metallic loading of the catalysts.46,50,69,85,86 It must be noted that these Pt ionic species could be bonded to surface oxygenated groups of the carbon supports.
| Catalyst | 4f7/2 | Catalyst | 4d5/2 | ||
|---|---|---|---|---|---|
| BE | Pt0–Ptoxi (%) | BE | Pt0–Ptoxi (%) | ||
| 0.3Pt/CNP | 71.5 | 64.3–35.7 | 0.3Pt/Al2O3 | 314.2 | 100–0 |
| 0.5Pt/CNP | 71.8 | 65.5–34.5 | 0.5Pt/Al2O3 | 313.9 | 100–0 |
| 5Pt/CNP | 71.7 | 63.6–36.4 | 5Pt/Al2O3 | 313.6 | 95.3–4.7 |
| 0.3Pt/CV | 71.2 | 66.5–33.5 | 0.3Pt/MgAl2O4 | 314.8 | 100–0 |
| 0.5Pt/CV | 72.1 | 69.4–30.6 | 0.5Pt/MgAl2O4 | 313.5 | 100–0 |
| 5Pt/CV | 71.7 | 66.2–33.8 | 5Pt/MgAl2O4 | 313.9 | 94.6–5.4 |
 | ||
| Fig. 5 XPS spectra of 0.3Pt/CNP. | ||
TEM analysis was used to determine the particle sizes of the catalysts studied. Fig. 6 shows the TEM images and the corresponding histograms of the particle size distribution for the different catalysts supported on oxide and carbon materials and prepared by impregnation. The mean particle size was calculated from the histograms, and the corresponding values are summarized in Table 4. Surprisingly, these results show that the mean particle size does not significantly change with the Pt loading in each series. Also, the average diameters of Pt nanoparticles for catalytic series prepared by the impregnation are very similar, about 2 nm for catalysts supported on carbons and about 1.8 nm for catalysts supported on oxides. In addition, catalysts supported on oxide materials seem to exhibit a narrower and more homogeneous distribution than those supported on carbons. This fact would be related to the more homogeneous distribution of anchoring sites for nanoparticles and, as a consequence, to a more homogeneous dispersion of the active phase on oxide supports. Thus, these results agree with those mentioned above, which indicated that the surface oxygenated groups and defect randomly arranged on the surface on the carbons, or regular arrangement of the cations of the oxides can anchorage and disperse the Pt nanoparticles.
 | ||
| Fig. 6 TEM images of the catalysts prepared by conventional impregnation. | ||
| Catalyst | D (nm) | Catalyst | D (nm) |
|---|---|---|---|
| 0.3Pt/CNP | 2.1 | 0.3Pt/CV | 2.1 |
| 0.5Pt/CNP | 2.2 | 0.5Pt/CV | 2.0 |
| 5Pt/CNP | 2.3 | 5Pt/CV | 2.1 |
| 0.3Pt/Al2O3 | 1.7 | 0.3Pt/MgAl2O4 | 1.7 |
| 0.5Pt/Al2O3 | 1.7 | 0.5Pt/MgAl2O4 | 1.8 |
| 5Pt/Al2O3 | 1.8 | 5Pt/MgAl2O4 | 1.9 |
The different catalytic series were studied in the n-butane dehydrogenation reaction in a continuous flow reactor. Fig. 7 shows the total conversion of the n-butane and selectivities to all butenes as a function of the reaction time for the different catalytic series. Table 5 shows the results of the dehydrogenation reaction of n-butane for the different catalysts, being (X) the conversion values, (S) the selectivity values to the all butenes, and (ΔX) the deactivation parameter. The ΔX parameter considers the catalytic deactivation through the reaction time, and it was defined as ΔX = 100 × (X0 − Xf)/X0, being X0 and Xf the initial and final conversion values, respectively. From comparing the results of n-butane initial conversion for each Pt loading in all the series of catalysts prepared by impregnation, it can be observed that the catalysts supported on alumina are more active than those supported on carbons. However, although the catalyst with the highest loading of Pt (5 wt%) supported on the other oxide material, the Mg-spinel, is also more active than the corresponding catalysts supported on carbons, the 0.3Pt/MgAl2O4 catalyst showed the lowest initial activity. On the other hand, unlike the Pt catalysts supported on oxides, which exhibit a significant drop in the conversion along the time, the catalysts supported on carbons maintain better activity throughout the reaction time, especially this can be seen for catalysts with low Pt loadings. In effect, as it was observed in Table 5, independently of the Pt loading, the deactivation parameter values for the catalysts supported on oxide materials are higher than those corresponding to catalysts supported on carbons (except for the 5Pt/CNP catalyst). Moreover, for catalysts supported on oxide materials, the higher the Pt loading, the lower the deactivation parameter values, the opposite being true for catalysts supported on carbons.
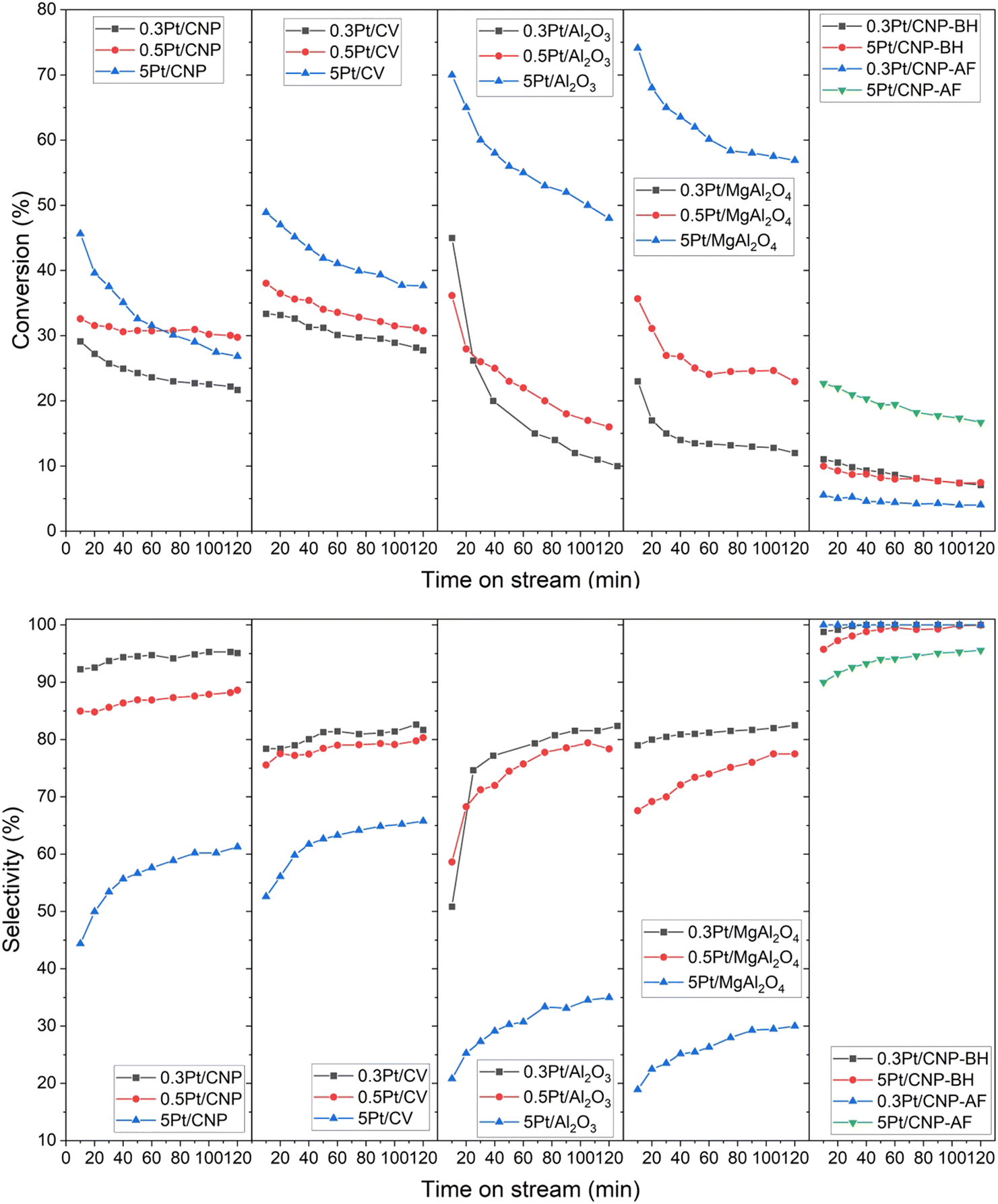 | ||
| Fig. 7 Conversion and selectivity for all catalysts for n-butane dehydrogenation. | ||
| Catalyst | X 0 | X f | S 0 | S f | ΔX (%) | Catalyst | X 0 | X f | S 0 | S f | ΔX (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.3Pt/CNP | 29.1 | 21.7 | 92.3 | 95.1 | 25.4 | 0.3Pt/CV | 33.4 | 27.7 | 78.4 | 81.7 | 17.1 |
| 0.5Pt/CNP | 32.6 | 29.7 | 84.9 | 88.6 | 8.9 | 0.5Pt/CV | 38.0 | 30.7 | 75.6 | 80.3 | 19.2 |
| 5Pt/CNP | 45.6 | 26.8 | 44.4 | 61.2 | 42.1 | 5Pt/CV | 48.9 | 37.7 | 52.6 | 65.8 | 22.9 |
| 0.3Pt/Al2O3 | 44.9 | 10.2 | 50.8 | 82.3 | 77.2 | 0.3Pt/MgAl2O4 | 23.2 | 11.8 | 79.1 | 82.5 | 49.1 |
| 0.5Pt/Al2O3 | 36.1 | 15.9 | 58.6 | 78.4 | 55.9 | 0.5Pt/MgAl2O4 | 35.7 | 22.9 | 67.6 | 77.5 | 35.8 |
| 5Pt/Al2O3 | 70.1 | 48.0 | 20.8 | 35.4 | 31.5 | 5Pt/MgAl2O4 | 74.1 | 56.9 | 18.9 | 29.9 | 36.7 |
In relation to the selectivities to all butenes, high values were found for the catalysts with Pt low loadings supported on CNP, which corresponds to a fact unanticipated for monometallic catalysts since typical monometallic catalysis for this type of reaction has been reported not to be very selective.1,3,13,20,44,45,47,80,81 It must be noted that the 0.3Pt/CNP displayed the highest selectivity values around 90%. Moreover, an unexpected selectivity behavior for all the catalysts prepared by impregnation was also found. Indeed, as observed, the selectivity values are greater at low Pt loading than at high Pt loading despite a similar Pt nanoparticle size found in each catalytic series. Furthermore, it can also be seen that the difference in selectivities between low and high Pt loadings is greater for catalysts supported on oxide materials.
The key role of a dehydrogenation catalyst is to accelerate the main reaction while controlling other side reactions. As mentioned above, it is known that supported monometallic catalysts are not very selective to dehydrogenation and have not been well studied on carbons.13,20,44 For example, it was widely reported that unmodified alumina-supported Pt catalysts and Pt supported on other oxide materials are active but not selective to dehydrogenation.1,3,80 Since the main reaction in catalytic dehydrogenation is the formation of mono-olefins, other reactions such as consecutive dehydrogenation, hydrogenolysis or cracking, isomerization and coke formation occur in detrimental of the selectivity. Then, as various by-products are formed, the catalyst is deactivated due to fouling by heavy carbonaceous materials. The consecutive reactions, the dehydrogenation of mono-olefins to diolefins, and polymers are catalyzed on the same active metallic sites as the dehydrogenation of paraffin to mono-olefins, these reactions promote coke formation. Since the olefin reaction on metallic nanoparticles is faster than that of paraffin (because olefins interact with metallic phases more strongly than paraffins), the role of the metallic phase is to weaken the metal–olefin interaction selectively without affecting the metal–paraffin interaction.3,27 However, even when this fact occurs, other side reactions, such as hydrogenolysis, decrease the selectivity. Also, catalysts supported on acidic supports such as alumina have acidic sites that accelerate isomerization, cracking, oligomerization, and polymerization of olefinic materials, enhancing coke formation and decreasing selectivity.1,3,44
The main problem regarding the simultaneous occurrence of secondary reactions, such as hydrogenolysis and coke formation, is that some are thermodynamically favored over the dehydrogenation reaction at the temperature at which the process is carried out. Indeed, the dehydrogenation reaction is endothermic and should be carried out at high temperatures, so a compromise situation occurs since the higher the reaction temperature, the higher the amount of side products.1,3,44
It must be noted that the X0 values correspond to the first sample taken from the reactor effluent 10 min from the start of the reaction, that is, once the steady state is reached. However, carbon deposits and coke formation are always formed more significantly at the beginning of the reaction.80,84,87 Likewise, the intrinsic selectivity can also be evaluated when starting the reaction, since at this time there would be no active sites blocked by carbon deposits.
So additionally, the catalysts were tested in n-butane dehydrogenation reaction in a pulse equipment to obtain information about the initial coke deposition and the catalytic behavior in the first steps of the reaction, which cannot be observed in continuous flow experiments. In this sense, it must be considered that the injection of the first pulse of n-butane is produced on a clean surface free of carbon, in contrast to the flow experiments, where the initial activity (taken to at 10 min of the reaction time) corresponds to the reaction of n-butane on a partially deactivate surface by carbon deposition Fig. 8(a)–(c) show the n-butane conversion, selectivity to n-butenes, and carbon retention in the catalysts with the lowest Pt loadings as a function of the number of pulses. The C retention was calculated by a carbon balance between the inlet and the exit of the reactor. From these results, it can be noted that the n-butane conversion is very high with the first pulse for 0.3Pt/Al2O3 and 0.3Pt/MgAl2O4 samples (Fig. 8(a)). These catalysts also display very low selectivity to butenes at the first pulse and, very high selectivity to hydrogenolytic products (Fig. 8(b)). As the number of pulses increases, the conversion decreases, and the selectivity to all butenes slightly increases (with the consequent decrease of the hydrogenolytic selectivity). However, the selectivity to n-butenes reached very low values in the last pulses. Hence, these facts would indicate that in the first step of the dehydrogenation reaction, the selectivity to the hydrogenolysis products is very high in these catalysts supported on oxide materials. These effects are accompanied by important carbon retention (as carbon deposition or coke formation) during the first pulses (Fig. 8(c)), with the cumulative amount of carbon after each pulse being higher in the catalyst supported on alumina than in the catalyst supported on Mg-spinel. Besides, for both samples, it is observed that the carbon deposition decreases with the number of pulses. Indeed, the percentage of carbon retained in each pulse decreases with respect to the previous pulse, which is more pronounced after various pulses of n-butane injected into the reactor with the catalysts supported on both oxide materials. So, it can be deduced that the monometallic catalysts supported on oxides have a certain amount of very active hydrogenolytic sites, which produce an important hydrogenolysis or C–C bond breaking during the first butane pulses. Since an extensive carbon deposition is simultaneously produced on the Pt surface, it can be concluded that the hydrogenolytic sites would also promote coke formation. Thus, the carbon formation would produce a selective poisoning of the hydrogenolytic sites decreasing the selectivity to hydrogenolysis products and increasing the selectivity to n-butenes in the first steps of the reaction. Likewise, the results indicate that at the beginning of the reaction (first pulses), the metallic phase supported on oxide materials shows very high activity, but it is quickly poisoned by carbon with the consequent catalyst deactivation (last pulses). Besides, it must be noted that the values of the deactivation parameters obtained in continuous flow experiments for these catalysts are very important, thus indicating that the retention of carbon continues along the reaction time. Since the selectivity to n-butenes in flow experiments also simultaneously increases as the conversion decreases, the results would also indicate that there would be many hydrogenolytic sites in these catalysts supported on oxide materials.
 | ||
| Fig. 8 n-Butane dehydrogenation reaction results as a function of the number of pulses. | ||
On the other hand, the catalysts supported on carbons, showed at the beginning of the n-butane dehydrogenation reaction in pulse experiments (Fig. 8(a)), conversion values slightly higher than the corresponding values found in continuous flow experiments. Likewise, these conversion values are lower than those found for catalysts supported on oxides. So, while the main reaction route in the first moments of the reaction is the hydrogenolysis for the catalysts supported on oxides, but the dehydrogenation competes with hydrogenolysis in catalysts supported on carbons. In these catalysts, the selectivity values to n-butenes for the first pulse are lower than those found in flow experiments but also significantly higher than those found for catalysts supported on oxide materials in the pulse experiments (Fig. 8(b)). As the number of pulses increases, the behavior of the catalysts supported on carbons also differs from those supported on oxide materials. It must be noted that the catalysts supported on carbon quickly reached a quasi-stationary state after 4–5 pulses, and then the conversion values slightly decrease in the following pulses. This behavior is accompanied by a significant increase in the selectivity values to n-butenes in the first pulses, which is less important in the last pulses, reaching values similar to those found after 10 minutes of reaction in flow experiments. As seen in Fig. 8(c), the carbon retention in the first pulses for the catalysts supported on carbon are similar to those of the catalyst supported on Mg-spinel but lower than the catalyst supported on alumina. Also, as for the catalysts supported on oxide materials, these facts would be indicating that the carbon deposited would produce poisoning of hydrogenolytic sites, with the subsequent catalyst deactivation and increase of the selectivity to n-butenes. However, the selectivity values only slightly increase when the last pulses are injected to the reactor. This behavior is also observed in flow experiments, in spite of the formation of coke and continuing deactivation along the reaction time, the selectivity to n-butenes only slightly increases. These facts could indicate that initially there would be a lower amount of hydrogenolytic sites in catalysts supported on carbons than in those supported on oxides and/or that these sites would have a different activity. Besides, these hydrogenolytic sites with different activity (highly reactive and small sites), in turn, would be less active in catalysts supported on carbons than in catalysts supported on oxides. Thus, the most active hydrogenolytic sites could predominate in catalysts supported on oxides, defining these catalysts as intrinsically hydrogenolytics.
Furthermore, the results of the flow and pulse experiment indicate that the secondary reactions of hydrogenolysis and coke formation are inevitable. In all catalysts, although hydrogenolysis decreases as dehydrogenation progresses, coke deposition appears to be constant. The catalysts were tested in the dehydrogenation reaction with a longer reaction time than those initially used. After 24 h of reaction, the activity of the 0.3Pt/Al2O3 catalyst was insignificant, practically indicating the total deactivation of the catalyst. For the 0.5Pt/CNP catalyst, the deactivation parameter increases from 8.2 (at 2 h) to 54.2 (at 24 h). However, the catalytic phases of these catalysts appear to remain unchanged after the reaction period. The XPS results after 2 h of reaction showed no change in either the Pt 4f BE values or the oxidation states, indicating that there would be no electronic density changes. TEM measurements also did not show any change in particle size. Thus, these results would indicate that the catalytic phases does not change due to the reaction conditions and are only blocked by coke deposits.
Raman measurements of the catalysts with low and high Pt loading supported on carbon nanotubes and alumina were also carried out after 2 h of reaction. As mentioned above, Raman spectroscopy can be used to detect the degree of structural disorder of carbons and is a sensitive technique for evaluating structural changes.59,60 Raman spectroscopic measurements can be carried out on supported Pt catalysts during durability tests, either to evaluate the corrosion reaction on the support and/or the amount and type of coke formed on the surface of the catalysts, performing coke characterizations in catalysts supported on both oxide and carbon materials.59,88–90
Fig. 9 shows the Raman spectra of the catalysts supported on carbon nanotubes and alumina materials. For catalysts supported on alumina, the results show the appearance of characteristic bands for sp2 and sp3 carbons, which would directly confirm the formation of carbon deposits on the catalysts. Furthermore, the D and G bands are shifted to higher wavenumbers and the values of the ID/IG ratios are greater than 1.5. These results could provide additional information about of the nature of carbon deposits.59,89 The G band corresponds to the stretching vibration of any pair of sp2 sites, either in CC chains or in aromatic rings.89,91 However, the D band corresponds to the vibration mode of sp3 sites only at the boundary of the rings, not in the chains.89,91 In addition, these bands can appear in the Raman spectra of carbons with some graphitic structure but even without a particular graphitic order. On the other hand, it is known that the formation of carbon deposits on the catalysts is produced by the polymerization of the olefins formed during dehydrogenation.3,44,80,84,92 These deposits would initially form polyenes and then evolve into oligomers or polymers and aromatics, thus becoming more graphitic.92,93 Thus, according to these results, after two hours on stream, the nature of the coke deposited on catalysts supported on alumina would correspond to a graphitic carbon but, since the ID/IG ratio is higher than that of ordered graphitic carbon such as carbon nanotubes, with a high degree of disorder. The results for catalysts supported on carbon are more difficult to interpret due to the overlap of the signals from the carbon deposits with those from the support. However, some conclusions can be drawn. Since the values of the ID/IG ratios decrease with respect to those of the support, these results could indicate that only the intensity of the G bands increases due to the formation of a type of disordered aliphatic carbon, or also that a part of the formed carbon could be deposited at the boundary of the Pt particles, contributing to reducing defects in the structure of the carbon nanotubes. In any case, these results would indicate that the nature of the coke deposited on catalysts supported on carbon is different from those supported on alumina. Since coke formation is common during hydrocarbon reactions, we will do more studies on the nature of coke and catalyst deactivation, as this knowledge is necessary when regeneration strategies are to be developed.
 | ||
| Fig. 9 Raman spectra of the catalysts after n-butane dehydrogenation reaction. | ||
It is worth mentioning that the dehydrogenation of light paraffins is considered a structure-insensitive reaction, i.e., all the atoms of the metallic phase are considered as active sites,17,18,27,35 however, the secondary reactions that take place on the metallic phase of a dehydrogenation catalyst are structure-sensitive reaction; it requires a certain ensemble or a certain type of atoms.17,27 It must be noted that for a certain nanoparticle, the surface amount of these ensembles or type of atoms are related to the geometry and shape of the nanoparticle. Thus, the surface structure as the fraction of surface metal sites such as corner, edge and plane, can be estimate using a nanocrystal model which represent the shape of the nanoparticle.26,36,42 So, as consequence of these fundaments, Somorjai carried out studies of dehydrogenation and hydrogenolysis reactions on Pt crystallographic phases with different relative concentrations of atoms in terraces, steps and kinks.27 These studies show that the hydrogenolysis rate increases with the kink density, while the dehydrogenation rate remains unaffected. The independence of the dehydrogenation rate from the step and kink density indicate that the reaction is indeed structure insensitive, while the hydrogenolysis reaction is structure sensitive. Besides, from these studies also was found that the chemisorption of hydrocarbons on Pt surface requires low activation energy, however, it was observed that the activation energy for surface reactions such as those that involve rupture of C–H and C–C bonds is greatly reduced at atomic steps on the Pt surface. These facts indicate that the dehydrogenation can occur rapidly at a step, but also that the steps are sites primarily responsible for C–H bond breaking and kinks for C–C bond breaking, since the hydrogenolysis is promoted at kink in the atomic steps. Additionally, it has also been reported that because the average bond energy of C–C bond (347 kJ mol−1) is weaker than that of C–H bond (414 kJ mol−1), the cleavage of C–C bonds occurs more easily.5,94 It must be noted that the difference in the bond-breaking ability between various surface sites can be distinguishable by the number of nearest neighbouring atoms. While corners, steps and kinks are sites with low coordination number, terraces or planes represent sites with high coordination numbers. This is important because each site has both a unique local structure and charge density. The charge density has been calculated at the corner for various metallic nanoparticles.27,95 For Pt the results indicate that at the corner there is a net positive charge density which could help further to polarize incoming molecules to the surface and promote the adsorption and breaking of C–H and C–C bonds. These results are in agreement with those that indicate a higher reactivity for bond breaking at sites as corner, step and kink than the atomic terraces sites.
In relation to the sensitive or insensitive character of the reaction, the same was historically linked to the size effect because of the supposition that a nanoparticle with a certain size would have a unique structure based on relative amount of atoms at corner, edges and terraces or planes. Besides, because the smaller particle sizes the higher the percentage of corners and edges in relation to those on the planes, it is believed low particles sizes is detrimental to reaction selectivity. However, we found a different activity and selectivity for catalytic each series with different Pt loading and similar particle size. Moreover, the dehydrogenation results also show that the support could have an important influence on the reactivity of the sites which catalyze side reactions. In consequence, these results indicate that a different structure of the catalytic phase is obtained for each catalyst with a Pt loading and a similar particle size.
Accordingly, we studied the catalysts by other characterization techniques with the aim of obtaining more information about the metallic phases and allowing to shed light on aspects of the structure effects and electronic properties of supported Pt nanoparticles. We carried out H2 chemisorption measurements to obtain dispersion data, test reactions of the metallic phase to obtain more information about the geometric and electronic characteristics of the metallic phases, and HRTEM to elucidate aspect about the geometry and shape of the nanoparticles. Additionally, for the sake of comparison, catalysts were prepared by liquid phase reduction method with the objective to obtain particles size greater that the size obtain by the conventional impregnation method.
Test reactions (cyclohexane dehydrogenation –CHD–, cyclopentane hydrogenolysis –CPH– and benzene–toluene competitive hydrogenation) of the metallics phase and H2 chemisorption measurements were carried out for the catalytic series prepared by conventional impregnation. Table 6 shows the results of some of these techniques. CHD reaction is considered a structure-insensitive reaction,27,35,96 which involves a metallic Pt exposed sites, while CPH is a structure-sensitive reaction which is carried out on ensembles of a certain number of active atoms.27,35,46,97 Both the reaction rate and the activation energy are important for CHD. The reaction rate can be taken as an indirect measurement of exposed surface Pt atoms, the activation energy value can be modified if the nature of the metallic site is changed by some effect such as the presence of a promoter or the different metal–support interactions. For CPH, a change in the reaction rate can also be observed due to a modification of the metallic phase due to some geometric or electronic effect.
| Catalyst | R 0CHD (mol h−1 gcat−1) | E aCHD (kcal mol−1) | R 0CPH (mol h−1 gcat−1) | % Dispersion |
|---|---|---|---|---|
| 0.3Pt/Al2O3 | 108.0 | 23 | 4.9 | 86 |
| 0.5Pt/Al2O3 | 99.2 | 23 | 3.5 | 81 |
| 5Pt/Al2O3 | 17.6 | 22 | 2.1 | 43 |
| 0.3Pt/MgAl2O4 | 52.1 | 16 | 7.6 | 31 |
| 0.5Pt/MgAl2O4 | 33.0 | 19 | 5.8 | 31 |
| 5Pt/MgAl2O4 | 9.9 | 21 | 2.2 | 32 |
| 0.3Pt/CNP | 12.0 | 41 | 10.7 | 92 |
| 0.5Pt/CNP | 9.6 | 43 | 6.1 | 87 |
| 5Pt/CNP | 1.8 | 46 | 8.9 | 30 |
| 0.3Pt/CV | 9.0 | 39 | 7.3 | 55 |
| 0.5Pt/CV | 7.9 | 43 | 6.7 | 41 |
| 5Pt/CV | 5.7 | 45 | 0.5 | 27 |
The CDH results show that the activation energy values are higher for catalysts supported on carbons than those corresponding to catalysts supported on oxides. This result would indicate that the dehydrogenation reaction is easier to carry out on catalysts supported on oxide than on catalysts supported on carbon. In consequence, this results also reveal that a different metal–support interaction occurs for these catalysts supported on oxide and carbons. It was widely reported that graphitic supports can donate electronic density to metallic nanoparticles, while in catalysts supported on acidic materials, such as alumina, a transfer of electrons from the metal to the support occurs.46,97–99 Hence, this effect could be responsible for the increase in polarity in coordinately unsaturated Pt sites in catalysts supported on oxide, while the atoms on the surface of the Pt particle in catalysts supported on carbon would not show much variation in the charge density with respect to the atoms in the distant surface sites of steps and corners. Consequently, coordinately unsaturated Pt sites in catalysts supported on oxide would turn more active to bonds breaking than those corresponding to catalysts supported on carbons. Besides, it must also be noted from Ea values that the activation energy of CDH for catalysts supported on carbons increase as Pt loading decreases. This fact could be indicating that Pt nanoparticles formed at low loading would have even greater interaction with the support and a different electronic density than those formed at high Pt loading. These results are in agreement with those found from TPR results. It must also be noted that the dehydrogenation of light paraffins is also a structure-insensitive reaction, so these observations for catalysts about CHD reaction can be extrapolated to the n-butane dehydrogenation. Thus, the results are in agreement with those found for n-butane dehydrogenation which evidence a high activity for catalysts supported on oxides, as well as, a great hydrogenolytic capacity.
In relation to initial rates for CDH and CPH, it can be observed that in each series the R0CHD and R0CPH decrease as the Pt loading increases. These facts are in agreement with those of H2 chemisorption and dispersion values. It is observed that for each series the dispersion decreases with the increase of the Pt loading. Hence, the higher the dispersion, the higher the amount of Pt sites and Pt ensembles for CHD and CPH reactions respectively. In addition, it is observed that catalysts supported on CNP and alumina supports show very high dispersion at low Pt loadings. The higher the dispersion, the larger the fraction of the metal atoms in contact with the support, and consequently, the stronger the metal–support interaction would be. Likewise, since the particle size is similar in each catalytic series but the dispersion changes with the Pt loading, the results would evidence that Pt particles with high loading would have more atomic layers in their structure and Pt particles with low Pt loading there would have a very few layers.
Toluene and benzene competitive hydrogenation was carried out to analyze the Pt nanoparticles electronic properties. Table 7 shows the kinetic parameters of benzene–toluene competitive hydrogenation for all the catalytic series prepared by conventional impregnation: adsorption coefficient (KT/B), and relative reactivities (RT/B). As the benzene hydrogenation reaction is the inverse of CHD, this reaction is considered a structure-insensitive reaction. Besides, because of structural similarity between benzene and toluene, the toluene hydrogenation reaction is also considered a structure-insensitive.27 As it was reported,51 assuming a competitive adsorption of benzene and toluene for the same sites on the metal surface, the rate of benzene and toluene hydrogenation can be given by the Langmuir rates laws as VB = (kBKBPB)/1 + KBPB + KTPT and VT = (kTKTPT)/1 + KBPB + KTPT respectively, where k, K and P are the benzene (B) and toluene (T) rate constant, adsorption coefficient and partial pressure, respectively. The ratio of rates VB/VT is a linear function of PB/PT, being the slope RB/T = kBKB/kTKT the relative reactivities of the hydrocarbons. Besides, the rates ratio V0B/VB(T) (where V0B and VB(T) are the rates of hydrogenation of benzene at P = 0 and P = variable respectively) is also a linear function of PT/PB, being the slope KT/B (relative adsorption coefficient). So, the plot of these linear functions allows to obtain the KT/B and RT/B values. The KT/B values are used to characterize the electronic structure of Pt nanoparticles induced by size or environmental effects. The different electronic structures of the toluene and benzene indicate that toluene is a better electron donor (it has a higher ionization potential and a stronger basicity than benzene).51,100–104 Subsequently, as the electron donation is easier for toluene, a lower KT/B value suggests a higher electron density of Pt nanoparticles, and vice versa. As shown in Table 7, in each series the lowest KT/B values correspond to the highest Pt loading, thus indicating the Pt metallic phase has a higher electronic density. Since, for each series similar particle size have been obtained, the results would be indicating a greater metallic character of the nanoparticles for catalysts with higher Pt loading. Considering that a same particle size is formed on each support but different amounts of Pt atoms are dispersed on this support, it is logical to think that the higher metallic loading the more amount of Pt atoms would be available to form the bulk of the metallic nanoparticles. It must be noted that the true band structure which form the bonds in metal particles involve a high grouping of atoms. On the contrary, catalysts with lowest Pt loading show the highest KT/B values in each series, thus indicating a lower electronic density and a lower metallic character in these catalysts. However, the change of electronic density on Pt nanoparticles also can be induce by environmental effects such as metal–support interaction. As it was mentioned above, graphitic materials can donate electronic density while acidic support can take it from of Pt nanoparticles.46,97–99 It is important to clarify that the CNP support does not have acidic groups that catalyze secondary reactions unlike alumina. Besides, the small amount of strong acid groups could serve as anchoring sites of Pt but also these groups are desorbed at the catalyst reduction temperature. KT/B values obtained for catalysts supported on carbons materials are in agreement with those reported in the literature which show that the KT/B values are influence by the donor effect from of support due to the metal–support interaction.105 On the other hand, the KT/B values found for catalysts supported on oxide materials are not comparable to those KT/B values corresponding to catalysts supported on carbons. However, KT/B values are comparable for a catalytic series prepared on the same support.51,101,103 It was reported that the KT/B values obtained for catalysts supported on oxides with certain acidy must be interpreted with caution because the acid sites on the support also catalyze side reactions. A kinetic study shows that the KT/B values would be masking additional adsorption from the support.106 Thus, the relative reactivities (RT/B) between catalysts supported on the different supports were compared. It was found RT/B values above one for all catalysts, thus indicating that toluene is more reactive than benzene on Pt catalysts as it was reported. However, the catalysts supported on oxides show higher RT/B values in relation to those supported on carbon, thus also indicating that the Pt nanoparticles supported on oxides would lower electronic density than those supported on carbons. This is important because the catalytic behavior in the studied dehydrogenation system can be related with the electronic density of the Pt atoms. Pt sites with positive or low electronic density are very active both in the dehydrogenation and hydrogenolysis, the sites with the lowest electron density being those coordinately unsaturated located at edges, steps and corners in the structure of the metal particle.27,95 So, the results from test reaction would explain the dehygrogenation results which indicate that the catalysts supported on oxides are more active and less selective to olefins that those supported on carbons.
| Catalyst | K T/B | R T/B | Catalyst | K T/B | R T/B |
|---|---|---|---|---|---|
| 0.3Pt/Al2O3 | 1.6 | 2.3 | 0.3Pt/CNP | 5.3 | 1.6 |
| 0.5Pt/Al2O3 | 0.9 | 2.0 | 0.5Pt/CNP | 4.9 | 1.4 |
| 5Pt/Al2O3 | 0.6 | 1.9 | 5Pt/CNP | 4.2 | 1.4 |
| 0.3Pt/MgAl2O4 | 2.1 | 1.9 | 0.3Pt/CV | 5.6 | 1.6 |
| 0.5Pt/MgAl2O4 | 1.8 | 1.9 | 0.5Pt/CV | 5.5 | 1.6 |
| 5Pt/MgAl2O4 | 1.2 | 1.7 | 5Pt/CV | 4.7 | 1.5 |
In summary, from the results of activation energy of test reactions, it can be inferred that catalysts supported on oxides dehydrogenate n-butane more easily than catalysts supported on carbons. The ability could be related to an effect of the support (metal–support interaction) which would imply that the support takes electronic density from metallic phase, leaving active sites both on coordinatively saturated and unsaturated Pt atoms with a lower charge density than on other supports. This positive charge density was confirmed by results from benzene–toluene competitive hydrogenation. These sites also would allow consecutive dehydrogenations and hydrogenolysis reactions, thus decreasing the selectivity and the stability of the catalyst. Besides, it is possible that consecutive dehydrogenations could lead to the formation of polymers and coke, resulting in catalyst deactivation. Indeed, the deactivation occur in a significative proportion in catalysts supported on oxides and the coke formation was confirmed by Raman results. On the other hand, it was also deduced from dispersion and test reaction results that the higher Pt loading, the higher the amount of Pt atoms grouped in nanocrystals and the metallic character of the nanoparticles. So, considering conventional particles models, these nanocrystals would have theoretically less amount of coordinatively unsaturated atoms. Since this type of sites have been found to be very reactive to side reactions, the catalysts with highest Pt loading should show the highest selectivities to the dehydrogenation to olefins. So, since this fact does not occur the behaviour of selectivity in relation to the different Pt loadings could not be explained.
In contrast, for catalysts supported on carbons the n-butane dehydrogenation is more difficult to be carried out as can be deduced from the activation energy results from CHD test reaction. The active sites have a higher electron density because of the donor effect of the graphitic materials. The negative charge density was confirmed by results from benzene–toluene competitive reaction. These more electronegative catalytic sites are turned less active due to the higher electronic density which affect the ability to adsorb and break bonds both for dehydrogenation and hydrogenolysis reactions. Besides, the higher the density on the metallic phase, lower the adsorption of olefins. So, once the olefins are formed, they are easily desorbed and do not remain adsorbed on the surface of the catalyst. Thus, the increased electron density of Pt nanoparticles attenuates the binding energy particularly that of CC bond, and in consequence, consecutive dehydrogenation reactions leading to coke formation can be partially inhibited. In this sense, Raman results show a different nature of the coke formed on catalysts supported on carbon. Since there would be less coke formation on the metallic phase, the catalysts prepared on carbon would not deactivate as those supported on oxides. Indeed, this fact is evidence of n-butane dehydrogenation results, showing the catalysts have high stability and also high selectivities. In relation to the selectivity behaviour with different Pt loading, the same conclusions that for catalysts supported on oxides can be reached. As the crystallinity increase, less amount of coordinatively unsaturated atoms and the selectivity behaviour could not be explained by simply crystal models.
Therefore, the local Pt nanoparticles structure on the catalysts prepared with different Pt loadings was investigated with help of HRTEM measurements. It must be noted that the results indicate that Pt nanoparticles with a high fraction of coordinatively unsaturated sites with positive density are highly active for side reactions in the n-butane dehydrogenation.17,27 Considering the conventional particle models, such as cuboctahedron and truncated octahedron, the size dependence on the surface sites fraction indicate that conventional particles show the maximum fraction of the unsaturated sites such as corner or edges at particle of about 2 nm.17,26,27,32,35,36 Also, it must be noted that, the edge sites may be regarded as the step sites and all the sites that are not part of the planar surface (except edges and corners) are considered as coordinatively unsaturated sites. As it was found, for the catalysts supported on carbons and oxides, the mean particle size was about 2 and 1.8 nm, respectively. So, the nanoparticles would have the same diameter but different grouping of Pt atoms according to the Pt loading. Therefore, as the Pt loading decreases, the nanoparticles lose the regular atomic arrangement but could exhibit an atomic arrangement which could include less coordinatively unsaturated sites.26,27,33,41 At lower Pt loading, less atoms would be available to form layers with crystallographic arrangements. In relation to this fact, it must be noted that catalysts with low Pt loading supported on alumina and CNP, show very high Pt dispersion. In these catalysts, a percentage of Pt atoms less than 10% of the total atoms that make up the particle would be blocking sites on the surface, but these atoms would also be forming other active sites randomly arranged on the surface that would more likely include kinks, adatoms, step-adatoms and terrace vacancies than corners and edges.27 Furthermore, the corner and edge sites could represent a higher percentage in particles providing the catalysts with a low Pt loading and lower dispersion (those supported on CV and Mg-spinel) a greater crystallographic disposition. So, these corresponding nanoparticles formed with low Pt loadings (especially those supported on CNP and alumina) must have lower crystallographic arrangements and less amount of coordinatively unsaturated sites.26,27,33,41,79 In all cases, taking into account the Pt atom size, the bond distance between two Pt atoms in the metallic phase,76 the relatively few amount of Pt atoms that would form the particle with a size about of 2 nm and low Pt loading,35 a particle could thus be described as a cluster of Pt atoms belonging to an incomplete crystalline structure with a structural base with a complete rows of Pt atoms and, a few layers with incomplete rows of Pt atoms which give rise to roughness surface.33,42,79,107 In addition, nanoparticles on CNP could be confined at defects in the surface graphene layers as a monolayer, monolayers with very few adatom or step-adatom nanostructures, or as simply cluster of atoms, as it was also suggested from Raman results.65 This fact could differentiate these nanoparticles from those supported on other supports, since the Pt atoms at the boundaries of the particles can interact with adjacent C atoms at the boundaries of the surface defects,65 thus decreasing the amount of coordinately unsaturated atoms in catalysts that are active for secondary reactions.27 It should be noted that the CV support predominantly has defects at the edges of its structure, so fewer atoms would be blocked from the interaction of the Pt atoms nanoparticles with the C atoms in the defect. On the other hand, the formation of Pt nanoparticles with a raft-type structure anchored to Al3+ atoms in alumina was reported.76,77 These structures (which could be formed on the catalysts supported on oxides) would have less coordinately unsaturated Pt atoms blocked due to the interaction of the nanoparticle with the oxide support.
Fig. 10 shows HRTEM images of the catalysts with different Pt loadings supported on CNP and alumina and the illustrations corresponding to the Pt nanoparticles structure model proposed. The catalysts with low Pt loading exhibited small nanoparticles, which predominate as structures with low crystallinity where the lattice fringes of the Pt nanoparticles cannot be resolved as well as in particles with good crystallographic arrangement.26,33,108 The 0.3Pt/CNP catalyst shows a typical example in which the Pt particles appear to be embedded in the structure of the surface layers of a nanotube. For the 0.3Pt/Al2O3 catalyst, the microphotograph of the amplified nanoparticle shows not only that the atomic columns cannot be resolved to evidence the lattice spacing of the particle, but also that different intensities provide the apparent position of the atoms that make up the atomic columns of the particle.107 These details in the microphotograph of particles formed at low Pt loading are in agreement with the features mentioned above regarding the type of Pt sites present on the particles (involving poor crystallography arrangements and surface roughness).24,108–111 Therefore, the HRTEM results allow us to confirm the structural models proposed for the nanoparticles. Furthermore, it was reported that a strong metal–support interaction could contribute to reducing the crystallinity of the particles and affecting the morphology of the nanoparticle.24,108–111 In this sense, the TPR results also show a stronger nanoparticles interaction with low loading than with those formed with high loading.
 | ||
| Fig. 10 HAADF STEM and HRTEM for the catalysts supported on CNP and Al2O3. Illustration of the structure of Pt nanoparticles: yellow and grey spheres represent the surface and bulk Pt atoms, respectively. | ||
On the other hand, the higher the Pt loading, the larger the row of Pt atoms that complete the crystal structure and form the bulk of the particle. In accordance with these features, the catalysts with high Pt loading showed lower dispersion than the catalysts with low Pt loading. Therefore, good crystallinity is to be expected. However, these particles would be anchored to the same sites on the support (which act as seeds for particle growth) and would have more atom layers with the same average size than those with low Pt loading. Consequently, these particles would also have a lower percentage of Pt sites in corners and edges considering a model particle, but in addition, as mentioned above, since the support influences the growth of the nanoparticles, they could grow imperfectly and therefore also have a high surface roughness. In agreement with this particle model, the HRTEM results shown that catalysts with high Pt loading exhibited small nanoparticles with round shape, which predominate as crystalline-like structures that show characteristic lattice fringes. However, most of these particles do not clearly exhibit a combination of lattice facets (they do not appear showing several facets as in particles with a well-defined shape). It was reported that small Pt nanoparticles are more likely to exist as a truncated octahedron than faceted nanocubes or cuboctahedral structures.19,21,22,33,112 Furthermore, it was reported that metal particles tightly adhered to the support with support-nanoparticle contact angles <90° tend to form flattened hemispherical particles.34 Additionally, the visualization of small nanoparticles by HRTEM as an amorphous structure or with lattice fringes is not well resolved, suggesting that metallic nanocrystallites are covered with amorphous layers or form nanoparticles with a rough surface.42,79,108,113
So, the illustration of the corresponding models for 5Pt/CNP and 5Pt/Al2O3 catalysts in Fig. 10 indicates the nanoparticles with round shape are considered to be the key to the generation of Pt step sites in a high fraction from the rough surface layer. Namely, these particles would show a rough or amorphous-like structure near the surface. This layer is formed from the imperfect growth of nanoparticles on the support, which allows the deposit of particles with a distorted shape. In addition, given that the average particle size in the catalysts supported on oxides is slightly smaller than that corresponding to the catalysts supported on carbons, it can be concluded that the catalysts supported on oxides would have even more coordinately unsaturated sites.
These results can then be correlated with the catalytic behavior. The larger the coordinately unsaturated sites, the higher the percentage of secondary products and, consequently, the lower the selectivity towards n-olefins. Therefore, catalysts with high Pt loading would be less selective and catalysts with low Pt loading would be more selective. Moreover, given that the type of support influences the growth and electronic properties of the nanoparticles, in turn, for the same Pt loading, catalysts on carbon materials would be more selective to n-olefins than those supported on oxides.
In addition, other studies were carried out with the aim of investigating the influence of smooth surfaces on the secondary reaction and coke formation. Another catalyst preparation method was used, the liquid phase reduction method. It was known that from this technique, well-faceted metallic particles can be obtained, as is characteristic of crystalline models, since the nanoparticles are formed in most of the reducing solution without any or little influence of the support.48,49 So, catalysts supported on CNP support with Pt loadings of 0.3 wt% and 5 wt% were prepared using two reduction agents (NaBH4 and formic acid). It was found from HRTEM results that because of poor interaction between Pt atoms and support during step reduction, the Pt nanoparticles are formed as crystals with well-faceted shapes (see Fig. 10). In this microphotograph it can be observed both the shapes (at low magnification) and the lattice fringes of the facets providing of nanoparticle (at high magnification). Besides, these nanoparticles show higher mean particle size, above 3.0 nm. It must be also noted that because of the preparation method, particles with few imperfections and smooth surfaces were obtained, where lattice fringes are well resolved. So, these particles can be represented by a crystal model without a surface roughness layer, as shown in Fig. 10.
These results can be correlated with those of n-butane dehydrogenation and CHD test reaction (Table 6). It must be noted that all the catalysts prepared by liquid phase reduction were submitted to a thermal treatment with H2 at the temperature at which the dehydrogenation reaction was carried out after their preparation. TEM measurements indicate that the average particle size increases after the thermal treatment. For example, 5Pt/CNP-BH increased from 3.3 to 4.4 nm. These results would indicate that these nanoparticles would not interact strongly with the support, and sintering would occur. CHD test reaction (Table 8) results reveal that the initial rates for catalysts prepared by liquid phase reduction are lower than those corresponding to the homologous catalysts prepared by conventional impregnation. As it is evident, these results would indicate a lower Pt dispersion on these catalysts. Besides, the activation energy values for catalysts prepared by liquid phase reduction are also lower. This would indicate that the dehydrogenation reaction is carried out easily on these catalysts but also that because of a minor interaction with the support, the metallic phase is not as electronegative as the metallic phase formed by the conventional impregnation.
Nevertheless, these characteristics of the nanoparticles are similar to those corresponding to catalysts supported on oxides and prepared by conventional impregnation, which are very active both in secondary reactions and in dehydrogenation, and therefore not very selective. However, the opposite is observed in the dehydrogenation results with catalysts prepared by the liquid phase reduction method (see Fig. 7), which show a low conversion and a very high selectivity in relation to the corresponding homologous catalysts prepared by the conventional impregnation method. Therefore, we focus the studies on the correlation between the type of active sites of the nanoparticles with the catalytic behavior. It must be considered that well-faceted particles present typical characteristics of metallic phases.17,18,27,35,113 In Fig. 8 it can be seen the shape of the nanoparticles that are assigned to an octahedron model, since they do not have rounded or cubic shapes.20,21,23,33,34 It is known that octahedron particles mainly consist of a mixture of Pt(100) and Pt(111) facets where Pt(100) and Pt(111) family of planes are confined in the particle in thermodynamic equilibrium.19,33 First, according to conventional particle models, in-plane Pt atoms are dominant sites for particles with particle size >2 nm.26,34 Besides as the particle size increases, the fraction of coordinatively unsaturated Pt sites such as edges and corners sites significantly decreases. So, as Pt nanoparticles prepared by liquid phase reduction would have a small amount of coordinatively unsaturated atoms, the side reactions would inhibit, increasing the selectivity in the n-butane dehydrogenation. In relation to these facts, it should be added that estimates of the fraction of surface sites in an ideal Pt crystal as an octahedron or truncated octahedron also show that the number of surface atoms in Pt(111) facets in a Pt nanoparticle is much larger than that corresponding to Pt(100) facets (more than an order of magnitude), this fact being independent of the particle size.19,26,36,37 Furthermore, it should be noted that the Pt atoms in Pt(111) facets have a higher coordination number than in Pt(100) facets and consequently the dense Pt(111) surfaces would also have a higher electron density. Also, it was previously reported that these facets of Pt may be related to the typical dehydrogenation mechanism.5,17,27,35,114–116 These studies show that Pt nanoparticles show strong structurally sensitive behavior both in the activation of the C–H bond to produce olefins and in the activation of the C–C bond to produce side products. In this sense, kinetic studies combined with density functional theory calculations show that the dehydrogenation activation energies for the first and second activation of the C–H bond are lower in the corners, edges and in the Pt(100) facets than in the Pt(111) surfaces, thus indicating that Pt catalysts with a high fraction of edges, corners and Pt(100) facets would lead to a lower dehydrogenation activation energy and, therefore, a higher activity.114,115 Besides, in Pt nanoparticles of around 3 nm in size, the first activation of the C–H bond is probably the rate-determining step at Pt(100) edges, corners, and surfaces, while in large Pt nanoparticles of around 9 nm size is more reasonable to expect that activation of the second C–H bond is the rate-determining step.114,115 Thus, these results would help to explain the catalytic behavior in relation to the low activity found with the catalysts prepared by the liquid phase reduction method, since in the nanoparticles obtained by this method a low number of edges, corners and Pt(100) facets without a rough surface were found.
Furthermore, the greater participation of dense Pt(111) surfaces in the dehydrogenation as active sites would also help to explain the low deactivation and the very high selectivity to n-butenes. It was also reported that on Pt(100) facets and low coordinated atoms such as corners and steps, the adsorption of CC double bonds (i.e., adsorption of the dehydrogenation product) is easier than on Pt(111) surfaces, which in turn it is not favorable to the CC coordination due to the grouping of atoms and the high electronic density that would produce electronic repulsion.5,20–22,115,116 It should be noted that the resorption of olefins could lead to further dehydrogenations and C–C bond breaking and, consequently, a greater amount of coke precursors or hydrogenolysis products and coke.5,114,115 Moreover, the ease of desorption of the olefins formed also matters.5,20,114 If the interaction between the C atom and the active site is very strong, it will be difficult for the adsorbed olefin to desorb when the second C–H is cleaved and two carbon atoms bind to the surface. Therefore, it was reported that the activation energy difference between olefin dehydrogenation and olefin desorption can be a selectivity descriptor.5,114–117 For example, the desorption barrier of propylene on low coordinated Pt atoms is higher than the activation energy for propylene dehydrogenation, leading to a strong preference for the formation of deeply dehydrogenated intermediates. However, on Pt(111) surfaces, the difference in activation energy between dehydrogenation and desorption of propylene is smaller, so as a result, the selectivity toward propylene is predicted to be higher on this surface.114 Therefore, these conclusions reached in the aforementioned investigations are consistent with our experimental results. These results clearly indicate that the number of Pt(111) facets composing the metallic Pt particle, prepared by liquid phase reduction methods, plays a crucial role in increasing the selectivity towards n-butenes. The Pt(111) facets in the nanoparticles would also be responsible for a low activity in dehydrogenation when using catalysts prepared by the liquid phase reduction method. Furthermore, these results are also in agreement with the results obtained from catalysts prepared by conventional impregnation, since a lower amount of Pt(111) surfaces would imply that other facets with lower coordination of Pt atoms and other coordinately unsaturated sites would be exposed as very active sites in dehydrogenation, but also in hydrogenolysis and coke formation to the detriment of selectivity towards olefins.
Therefore, the morphological effects revealed in this study would resolve the contradictions between previous experimental results on the size effect and may therefore contribute to obtaining a unified theory of the size effect and morphology of Pt nanoparticles on the dehydrogenation of light alkanes.
In conclusion, the results obtained with catalysts by liquid phase reduction not only confirm the results found for catalysts prepared by conventional impregnation but also show that the selectivity of the bond-breaking processes for dehydrogenation and side reactions at different atomic surface sites on Pt nanoparticles is significant in that the atomic surface structure could be appropriately tailored according to the support and the preparation method used to provide desire selectivity.
Conclusions
The catalytic activity of monometallic Pt catalysts supported on carbon materials showed higher selectivity, this was attributed to a higher electron density in the Pt active sites, which would attenuate the binding energy of the CC bond of monoolefins and, therefore, inhibits consecutive dehydrogenation reactions. Furthermore, the average particle size of the catalysts supported on carbons and oxides was approximately 2 and 1.8 nm, regardless of the metal loading. Therefore, depending on the Pt loading, the nanoparticles would have the same diameter but a different grouping of Pt atoms. At low Pt loadings, the CNP-supported Pt nanoparticles could be confined to the defect as a monolayer with very few adatom or stepped adatom nanostructures or simply as a group of atoms. Thus, the nanoparticles supported on carbons, specially those with lower Pt loading, could have a lower number of coordinately unsaturated sites, explaining the higher intrinsic selectivity to dehydrogenation and higher stability.
Author contributions
Gustavo Ramos Montero: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing. María Julia Yañez: investigation, validation. Adriana Ballarini: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, validation, visualization, writing – review & editing. Sergio de Miguel: conceptualization, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, validation. Sonia Bocanegra: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, validation, visualization, writing – review & editing. Patricia Zgolicz: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, validation, visualization, writing – original draft, writing – review & editing.Data availability
The authors declare that the data supporting the findings of this study are available within the paper, raw data is available at Zenodo at https://doi.org/10.5281/zenodo.13144746.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors are especially grateful for the collaboration of Maria Fernanda Mori from CENACA and Yohana Martinez from SECEGRIN for their professional and scientific contribution to the development of this work. This work was done with the financial support of Universidad Nacional del Litoral (Project CAI + D) and CONICET – Argentina.References
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Catalytic dehydrogenation of light alkanes on metals and metal oxides, Chem. Rev., 2014, 114, 10613–10653 CrossRef PubMed.
- Z. Nawaz, F. Baksh, J. Zhu and F. Wei, Dehydrogenation of C3-C4 paraffin's to corresponding olefins over slit-SAPO-34 supported Pt-Sn-based novel catalyst, J. Ind. Eng. Chem., 2013, 19, 540–546 CrossRef CAS.
- M. M. Bhasin, J. H. Mccain, B. V. Vora, T. Imai and P. R. Pujadó, Dehydrogenation and oxydehydrogenation of paraffins to olefins, 2001, vol. 221 Search PubMed.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Propane dehydrogenation: Catalyst development, new chemistry, and emerging technologies, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- C. Li and G. Wang, Dehydrogenation of light alkanes to mono-olefins, Chem. Soc. Rev., 2021, 50, 4359–4381 RSC.
- W. Tolek, K. Suriye, P. Praserthdam and J. Panpranot, Effect of preparation method on the Pt-In modified Mg(Al)O catalysts over dehydrogenation of propane, Catal. Today, 2020, 358, 100–108 CrossRef CAS.
- E. C. Wegener, Z. Wu, H.-T. Tseng, J. R. Gallagher, Y. Ren, R. E. Diaz, F. H. Ribeiro and J. T. Miller, Structure and reactivity of Pt–In intermetallic alloy nanoparticles: Highly selective catalysts for ethane dehydrogenation, Catal. Today, 2018, 299, 146–153 CrossRef CAS.
- M.-L. Yang, J. Zhu, Y.-A. Zhu, Z.-J. Sui, Y.-D. Yu, X.-G. Zhou and D. Chen, Tuning selectivity and stability in propane dehydrogenation by shaping Pt particles: A combined experimental and DFT study, J. Mol. Catal. A: Chem., 2014, 395, 329–336 CrossRef CAS.
- J. Zhang, X. Cai, K. H. Wu, Y. Zhang, J. Wang, J. Diao, N. Wang, H. Liu and D. Su, Nanodiamond-Core-Reinforced, Graphene-Shell-Immobilized Platinum Nanoparticles as a Highly Active Catalyst for the Low-Temperature Dehydrogenation of n-Butane, ChemCatChem, 2018, 10, 520–524 CrossRef CAS.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degrève, P. A. Jacobs and B. F. Sels, Review of old chemistry and new catalytic advances in the on-purpose synthesis of butadiene, Chem. Soc. Rev., 2014, 43, 7917–7953 RSC.
- Q. Zhu, G. Wang, H. Zhang, X. Zhu and C. Li, n-Butane dehydrogenation over Ni-Sn/SiO2: Adsorption modes and reaction paths of n-butane and 1-butene, Appl. Catal., A, 2018, 566, 113–120 CrossRef CAS.
- F. T. Zangeneh, A. Taeb, K. Gholivand and S. Sahebdelfar, The effect of mixed HCl–KCl competitive adsorbate on Pt adsorption and catalytic properties of Pt–Sn/Al2O3 catalysts in propane dehydrogenation, Appl. Surf. Sci., 2015, 357, 172–178 CrossRef CAS.
- J. C. Serrano-Ruiz, A. Sepúlveda-Escribano and F. Rodríguez-Reinoso, Bimetallic PtSn/C catalysts promoted by ceria: Application in the nonoxidative dehydrogenation of isobutane, J. Catal., 2007, 246, 158–165 CrossRef CAS.
- J. Zhu, K. Kailasam, A. Fischer and A. Thomas, Supported Cobalt Oxide Nanoparticles As Catalyst for Aerobic Oxidation of Alcohols in Liquid Phase, ACS Catal., 2011, 1, 342–347 CrossRef CAS.
- J. P. Den Breejen, P. B. Radstake, G. L. Bezemer, J. H. Bitter, V. Frøseth, A. Holmen and K. P. De Jong, On the Origin of the Cobalt Particle Size Effects in Fischer–Tropsch Catalysis, J. Am. Chem. Soc., 2009, 131, 7197–7203 CrossRef CAS PubMed.
- M. Che and C. O. Bennett, Advances in Catalysis, Elsevier, 1989, vol. 36, pp. 55–172 Search PubMed.
- B. Coq and F. Figueras, Structure–activity relationships in catalysis by metals: some aspects of particle size, bimetallic and supports effects, Coord. Chem. Rev., 1998, 178–180, 1753–1783 CrossRef CAS.
- M. Boudart, Heterogeneous catalysis by metals, J. Mol. Catal., 1985, 30, 27–38 CrossRef CAS.
- W. Chen, J. Ji, X. Feng, X. Duan, G. Qian, P. Li, X. Zhou, D. Chen and W. Yuan, Mechanistic Insight into Size-Dependent Activity and Durability in Pt/CNT Catalyzed Hydrolytic Dehydrogenation of Ammonia Borane, J. Am. Chem. Soc., 2014, 136, 16736–16739 CrossRef CAS PubMed.
- J. Serranoruiz, A. Lopezcudero, J. Sollagullon, A. Sepulvedaescribano, A. Aldaz and F. Rodriguezreinoso, Hydrogenation of α, β unsaturated aldehydes over polycrystalline, (111) and (100) preferentially oriented Pt nanoparticles supported on carbon, J. Catal., 2008, 253, 159–166 CrossRef CAS.
- S. Kato, J. Ohyama, M. Machida and A. Satsuma, Gas-phase synthesis of morphology-controlled Pt nanoparticles and their impact on cinnamaldehyde hydrogenation, Catal. Sci. Technol., 2019, 9, 2097–2102 RSC.
- J. Ohyama, S. Kato, M. Machida and A. Satsuma, Shape Control Preparation of Supported Platinum Nano-octahedra by Ethylene Treatment for Enhancement of Selective Hydrogenation of Cinnamaldehyde, Chem. Lett., 2019, 48, 1203–1205 CrossRef CAS.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Platinum Nanoparticle Shape Effects on Benzene Hydrogenation Selectivity, Nano Lett., 2007, 7, 3097–3101 CrossRef CAS PubMed.
- T. W. Van Deelen, C. Hernández Mejía and K. P. De Jong, Control of metal–support interactions in heterogeneous catalysts to enhance activity and selectivity, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- G. Kumar, L. Tibbitts, J. Newell, B. Panthi, A. Mukhopadhyay, R. M. Rioux, C. J. Pursell, M. Janik and B. D. Chandler, Evaluating differences in the active-site electronics of supported Au nanoparticle catalysts using Hammett and DFT studies, Nat. Chem., 2018, 10, 268–274 CrossRef CAS PubMed.
- K. Murata, J. Onoda, Y. Yamamoto, A. Oda, J. Ohyama and A. Satsuma, Enhancement of toluene hydrogenation activity of supported Pt nanoparticles with increasing the crystallinity of Pt, Appl. Catal., A, 2022, 629, 118425 CrossRef CAS.
- G. A. Somorjai, Advances in Catalysis, Elsevier, 1977, vol. 26, pp. 1–68 Search PubMed.
- J. Ohyama, A. Esaki, T. Koketsu, Y. Yamamoto, S. Arai and A. Satsuma, Atomic-scale insight into the structural effect of a supported Au catalyst based on a size-distribution analysis using Cs-STEM and morphological image-processing, J. Catal., 2016, 335, 24–35 CrossRef CAS.
- F. Auer, A. Hupfer, A. Bösmann, N. Szesni and P. Wasserscheidpeter, Influence of the nanoparticle size on hydrogen release and side product formation in liquid organic hydrogen carrier systems with supported platinum catalysts, Catal. Sci. Technol., 2020, 10, 6669–6678 RSC.
- M. Haruta, Catalysis of Gold Nanoparticles Deposited on Metal Oxides, ChemInform, 2002, 33, 260 CrossRef.
- J. Teddy, A. Falqui, A. Corrias, D. Carta, P. Lecante, I. Gerber and P. Serp, Influence of particles alloying on the performances of Pt–Ru/CNT catalysts for selective hydrogenation, J. Catal., 2011, 278, 59–70 CrossRef CAS.
- M. J. Kale and P. Christopher, Utilizing Quantitative in Situ FTIR Spectroscopy To Identify Well-Coordinated Pt Atoms as the Active Site for CO Oxidation on Al2O3-Supported Pt Catalysts, ACS Catal., 2016, 6, 5599–5609 CrossRef CAS.
- S. I. Sanchez, M. W. Small, E. S. Bozin, J.-G. Wen, J.-M. Zuo and R. G. Nuzzo, Metastability and Structural Polymorphism in Noble Metals: The Role of Composition and Metal Atom Coordination in Mono- and Bimetallic Nanoclusters, ACS Nano, 2013, 7, 1542–1557 CrossRef CAS PubMed.
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes, Chem. Rev., 2020, 120, 464–525 CrossRef CAS PubMed.
- M. Boudart, Catalysis by Supported Metals, Adv. Catal., 1969, 20, 153–166 CAS.
- R. Van Hardeveld and F. Hartog, The statistics of surface atoms and surface sites on metal crystals, Surf. Sci., 1969, 15, 189–230 CrossRef CAS.
- J. Ohyama, T. Nishiyama and A. Satsuma, Formation of Rhodium Metal Ensembles that Facilitate Nitric Oxide Reduction over Rhodium/Ceria in a Stoichiometric Nitric Oxide–Carbon Monoxide–Propene–Oxygen Reaction, ChemCatChem, 2018, 10, 1651–1656 CrossRef CAS.
- A. W. Castleman and P. Jena, Clusters: A bridge between disciplines, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10552–10553 CrossRef CAS PubMed.
- J. F. Hamilton and R. C. Baetzold, Science, 1979, 205, 1213–1220 CrossRef CAS PubMed.
- W. M. H. Sachtler, The Second Rideal Lecture. What makes a catalyst selective?, Faraday Discuss. Chem. Soc., 1981, 72, 7 RSC.
- L. Li, L.-L. Wang, D. D. Johnson, Z. Zhang, S. I. Sanchez, J. H. Kang, R. G. Nuzzo, Q. Wang, A. I. Frenkel, J. Li, J. Ciston, E. A. Stach and J. C. Yang, Noncrystalline-to-Crystalline Transformations in Pt Nanoparticles, J. Am. Chem. Soc., 2013, 135, 13062–13072 CrossRef CAS PubMed.
- J. Ohyama, T. Sato, Y. Yamamoto, S. Arai and A. Satsuma, Size Specifically High Activity of Ru Nanoparticles for Hydrogen Oxidation Reaction in Alkaline Electrolyte, J. Am. Chem. Soc., 2013, 135, 8016–8021 CrossRef CAS PubMed.
- J. Liu, Y. Yue, H. Liu, Z. Da, C. Liu, A. Ma, J. Rong, D. Su, X. Bao and H. Zheng, Origin of the Robust Catalytic Performance of Nanodiamond–Graphene-Supported Pt Nanoparticles Used in the Propane Dehydrogenation Reaction, ACS Catal., 2017, 7, 3349–3355 CrossRef CAS.
- G. J. Siri, J. M. Ramallo-López, M. L. Casella, J. L. G. Fierro, F. G. Requejo and O. A. Ferretti, XPS and EXAFS study of supported PtSn catalysts obtained by surface organometallic chemistry on metals: Application to the isobutane dehydrogenation, Appl. Catal., A, 2005, 278, 239–249 CrossRef CAS.
- S. Bocanegra, A. Ballarini, P. Zgolicz, O. Scelza and S. de Miguel, Highly selective and stable bimetallic catalysts supported on different materials for n-butane dehydrogenation, Catal. Today, 2009, 143, 334–340 CrossRef CAS.
- J. P. Stassi, P. D. Zgolicz, S. R. De Miguel and O. A. Scelza, Formation of different promoted metallic phases in PtFe and PtSn catalysts supported on carbonaceous materials used for selective hydrogenation, J. Catal., 2013, 306, 11–29 CrossRef CAS.
- S. A. Bocanegra, A. D. Ballarini, O. A. Scelza and S. R. de Miguel, The influence of the synthesis routes of MgAl2O4 on its properties and behavior as support of dehydrogenation catalysts, Mater. Chem. Phys., 2008, 111, 534–541 CrossRef CAS.
- V. I. Rodríguez, N. S. Veizaga and S. R. de Miguel, Effect of the Preparation Method on the Electrocatalytic Activity of Pt-Sn/Nanotubes Catalysts Used in DMFC, J. Electrochem. Soc., 2017, 164, F1524 CrossRef.
- N. Veizaga, J. Fernandez, M. Bruno, O. Scelza and S. De Miguel, Deposition of Pt nanoparticles on different carbonaceous materials by using different preparation methods for PEMFC electrocatalysts, Int. J. Hydrogen Energy, 2012, 37, 17910–17920 CrossRef CAS.
- P. D. Zgolicz, J. P. Stassi, M. J. Yañez, O. A. Scelza and S. R. De Miguel, Influence of the support and the preparation methods on the performance in citral hydrogenation of Pt-based catalysts supported on carbon nanotubes, J. Catal., 2012, 290, 37–54 CrossRef CAS.
- T. Tri, J. Massardier, P. Gallezot and B. Imelik, Studies in Surface Science and Catalysis, Elsevier, 1982, vol. 11, pp. 141–148 Search PubMed.
- P. Serp, M. Corrias and P. Kalck, Carbon nanotubes and nanofibers in catalysis, Appl. Catal., A, 2003, 253, 337–358 CrossRef CAS.
- J.-M. Nhut, L. Pesant, J.-P. Tessonnier, G. Winé, J. Guille, C. Pham-Huu and M.-J. Ledoux, Mesoporous carbon nanotubes for use as support in catalysis and as nanosized reactors for one-dimensional inorganic material synthesis, Appl. Catal., A, 2003, 254, 345–363 CrossRef CAS.
- Q.-H. Yang, P.-X. Hou, S. Bai, M.-Z. Wang and H.-M. Cheng, Adsorption and capillarity of nitrogen in aggregated multi-walled carbon nanotubes, Chem. Phys. Lett., 2001, 345, 18–24 CrossRef CAS.
- J. L. G. de la Fuente, M. V. Martínez-Huerta, S. Rojas, P. Terreros, J. L. G. Fierro and M. A. Peña, Methanol electrooxidation on PtRu nanoparticles supported on functionalised carbon black, Catal. Today, 2006, 116, 422–432 CrossRef.
- H. E. Van Dam and H. Van Bekkum, Preparation of platinum on activated carbon, J. Catal., 1991, 131, 335–349 CrossRef CAS.
- O. V. Kharissova and B. I. Kharisov, RSC Adv., 2014, 4, 30807–30815 RSC.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Modification of the surface chemistry of activated carbons, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- M. Hara, M. Lee, C. H. Liu, B. H. Chen, Y. Yamashita, M. Uchida, H. Uchida and M. Watanabe, Electrochemical and Raman spectroscopic evaluation of Pt/graphitized carbon black catalyst durability for the start/stop operating condition of polymer electrolyte fuel cells, Electrochim. Acta, 2012, 70, 171–181 CrossRef CAS.
- Y. Wang, D. C. Alsmeyer and R. L. McCreery, Raman Spectroscopy of Carbon Materials: Structural Basis of Observed Spectra, Chem. Mater., 1990, 2, 557–563 CrossRef CAS.
- S. J. P. Gamage, K. Yang, R. Braveenth, K. Raagulan, H. S. Kim, Y. S. Lee, C. M. Yang, J. J. Moon and K. Y. Chai, MWCNT coated free-standing carbon fiber fabric for enhanced performance in EMI shielding with a higher absolute EMI SE, Materials, 2017, 10, 1350 CrossRef.
- G. Katagiri, H. Ishida and A. Ishitani, Raman spectra of graphite edge planes, Carbon, 1988, 26, 565–571 CrossRef CAS.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- M. Marcinek, L. J. Hardwick, G. Z. Zukowska and R. Kostecki, Microwave plasma chemical vapor deposition of graphitic carbon thin films, Carbon, 2010, 48, 1552–1557 CrossRef CAS.
- S. J. Kim, Y. J. Park, E. J. Ra, K. K. Kim, K. H. An, Y. H. Lee, J. Y. Choi, C. H. Park, S. K. Doo, M. H. Park and C. W. Yang, Defect-induced loading of Pt nanoparticles on carbon nanotubes, Appl. Phys. Lett., 2007, 90, 94–97 Search PubMed.
- A. M. Rao, P. C. Eklund, S. Bandow, A. Thess and R. E. Smalley, Evidence for charge transfer in doped carbon nanotube bundles from Raman scattering, Nature, 1997, 191, 257–259 CrossRef.
- A. Cuesta, P. Dhamelincourt, J. Laureyns, A. Martínez-Alonso and J. M. D. Tascón, Raman microprobe studies on carbon materials, Carbon, 1994, 32, 1523–1532 CrossRef CAS.
- U. J. Kim, C. A. Furtado, X. Liu, G. Chen and P. C. Eklund, Raman and IR spectroscopy of chemically processed single-walled carbon nanotubes, J. Am. Chem. Soc., 2005, 127, 15437–15445 CrossRef CAS PubMed.
- R. V. Hull, L. Li, Y. Xing and C. C. Chusuei, Pt Nanoparticle Binding on Functionalized Multiwalled Carbon Nanotubes, Chem. Mater., 2006, 18, 1780–1788 CrossRef CAS.
- M. Acikgoz, J. Harrell and M. Pavanello, Seeking a Structure–Function Relationship for γ-Al2O3 Surfaces, J. Phys. Chem. C, 2018, 122, 25314–25330 CrossRef CAS.
- M.-H. Lee, C.-F. Cheng, V. Heine and J. Klinowski, Distribution of tetrahedral and octahedral A1 sites in gamma alumina, Chem. Phys. Lett., 1997, 265, 673–676 CrossRef CAS.
- S. M. Hosseini, Structural, electronic and optical properties of spinel MgAl2O4 oxide, Phys. Status Solidi B, 2008, 245, 2800–2807 CrossRef CAS.
- P. D. Zgolicz, V. I. Rodríguez, I. M. J. Vilella, S. R. De Miguel and O. A. Scelza, Catalytic performance in selective hydrogenation of citral of bimetallic Pt-Sn catalysts supported on MgAl2O4 and γ-Al2O3, Appl. Catal., A, 2011, 392, 208–217 CrossRef CAS.
- H. Pines and W. O. Haag, Alumina: Catalyst and Support. I. Alumina, its Intrinsic Acidity and Catalytic Activity, J. Am. Chem. Soc., 1960, 82, 2471–2483 CrossRef CAS.
- P. J. Kropp, G. W. Breton, S. L. Craig, S. D. Crawford, W. F. Durland, J. E. Jones and J. S. Raleigh, Surface-Mediated Reactions. 6. Effects of Silica Gel and Alumina on Acid-Catalyzed Reactions, J. Org. Chem., 1995, 60, 4146–4152 CrossRef CAS.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Coordinatively Unsaturated Al3+ Centers as Binding Sites for Active Catalyst Phases of Platinum on γ-Al2O3, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- K. Murata, T. Shiotani, J. Ohyama, R. Wakabayashi, H. Maruoka, T. Kimura and A. Satsuma, Relationship between penta-coordinated Al3+ sites in the Al2O3 supports and CH 4 combustion activity of Pd/Al2O3 catalysts, Catal. Sci. Technol., 2021, 11, 2374–2378 RSC.
- D. Mei, J. H. Kwak, J. Hu, S. J. Cho, J. Szanyi, L. F. Allard and C. H. F. Peden, Unique Role of Anchoring Penta-Coordinated Al3+ Sites in the Sintering of γ-Al2O3-Supported Pt Catalysts, J. Phys. Chem. Lett., 2010, 1, 2688–2691 CrossRef CAS.
- K. Murata, Y. Mahara, J. Ohyama, Y. Yamamoto, S. Arai and A. Satsuma, The Metal–Support Interaction Concerning the Particle Size Effect of Pd/Al 2 O 3 on Methane Combustion, Angew. Chem., Int. Ed., 2017, 56, 15993–15997 CrossRef CAS PubMed.
- S. R. de Miguel, S. A. Bocanegra, I. M. J. Vilella, A. Guerrero-Ruiz and O. A. Scelza, Characterization and Catalytic Performance of PtSn Catalysts Supported on Al2O3 and Na-doped Al2O3 in n-butane Dehydrogenation, Catal. Lett., 2007, 119, 5–15 CrossRef CAS.
- A. D. Ballarini, P. Zgolicz, I. M. J. Vilella, S. R. de Miguel, A. A. Castro and O. A. Scelza, n-Butane dehydrogenation on Pt, PtSn and PtGe supported on γ-Al2O3 deposited on spheres of α-Al2O3 by washcoating, Appl. Catal., A, 2010, 381, 83–91 CrossRef CAS.
- M. C. Román-Martínez, D. Cazorla-Amorós, H. Yamashita, S. De Miguel and O. A. Scelza, XAFS study of dried and reduced PtSn/C catalysts: nature and structure of the catalytically active phase, Langmuir, 2000, 16, 1123–1131 CrossRef.
- L. Huang, B. Xu, L. Yang and Y. Fan, Propane dehydrogenation over the PtSn catalyst supported on alumina-modified SBA-15, Catal. Commun., 2008, 9, 2593–2597 CrossRef CAS.
- S. A. Bocanegra, A. Guerrero-Ruiz, S. R. de Miguel and O. A. Scelza, Performance of PtSn catalysts supported on MAl2O4 (M: Mg or Zn) in n-butane dehydrogenation: characterization of the metallic phase, Appl. Catal., A, 2004, 277, 11–22 CrossRef CAS.
- A. K. Shukla, M. Neergat, P. Bera, V. Jayaram and M. S. Hegde, An XPS study on binary and ternary alloys of transition metals with platinized carbon and its bearing upon oxygen electroreduction in direct methanol fuel cells, J. Electroanal. Chem., 2001, 504, 111–119 CrossRef CAS.
- N. H. Tran, M. A. Wilson, A. S. Milev, J. R. Bartlett, R. N. Lamb, D. Martin and G. S. K. Kannangara, Photoemission and absorption spectroscopy of carbon nanotube interfacial interaction, Adv. Colloid Interface Sci., 2009, 145, 23–41 CrossRef CAS PubMed.
- S. A. Bocanegra, A. Guerrero-Ruiz, O. A. Scelza and S. R. De Miguel, MgAl2O4 spinel prepared by mechanochemical synthesis used as a support of multimetallic catalysts for paraffin dehydrogenation, Catal. Ind., 2013, 5, 61–73 CrossRef.
- Y. Hiramitsu, H. Sato, H. Hosomi, Y. Aoki, T. Harada, Y. Sakiyama, Y. Nakagawa, K. Kobayashi and M. Hori, Influence of humidification on deterioration of gas diffusivity in catalyst layer on polymer electrolyte fuel cell, J. Power Sources, 2010, 195, 435–444 CrossRef CAS.
- A. Iglesias-Juez, A. M. Beale, K. Maaijen, T. C. Weng, P. Glatzel and B. M. Weckhuysen, A combined in situ time-resolved UV-Vis, Raman and high-energy resolution X-ray absorption spectroscopy study on the deactivation behavior of Pt and PtSn propane dehydrogenation catalysts under industrial reaction conditions, J. Catal., 2010, 276, 268–279 CrossRef CAS.
- T. Yoda, H. Uchida and M. Watanabe, Effects of operating potential and temperature on degradation of electrocatalyst layer for PEFCs, Electrochim. Acta, 2007, 52, 5997–6005 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Raman Spectrum of Graphene and Graphene Layers, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- S. A. Bocanegra, A. A. Castro, A. Guerrero-Ruíz, O. A. Scelza and S. R. De Miguel, Characteristics of the metallic phase of Pt/Al2O3 and Na-doped Pt/Al2O3 catalysts for light paraffins dehydrogenation, Chem. Eng. J., 2006, 118, 161–166 CrossRef CAS.
- R. A. Comelli, S. A. Canavese, C. A. Querini and N. S. Fígoli, Coke deposition on platinum promoted WOx–ZrO2 during n-hexane isomerization, Appl. Catal., A, 1999, 182, 275–283 CrossRef CAS.
- R. Georgiadis, E. R. Fisher and P. B. Armentrout, Neutral and Ionic Metal–Hydrogen and Metal–Carbon Bond Energies: Reactions of Co+, Ni+, and Cu+ with Ethane, Propane, Methylpropane, and Dimethylpropane, J. Am. Chem. Soc., 1989, 111, 4251–4262 CrossRef CAS.
- L. L. Kesmodel and L. M. Falicov, The electronic potential in a metal close to a surface edge, Solid State Commun., 1975, 16, 1201–1204 CrossRef.
- D. W. Blakely and G. A. Somorjai, The dehydrogenation and hydrogenolysis of cyclohexane and cyclohexene on stepped (high miller index) platinum surfaces, J. Catal., 1976, 42, 181–196 CrossRef CAS.
- G. E. Ramos Montero, J. P. Stassi, S. R. De Miguel and P. D. Zgolicz, Hydrogenation of citral and carvone on Pt and PtSn supported metallic catalysts. A comparative study on the regioselectivity and chemoselectivity, React. Chem. Eng., 2023, 8, 3133–3149 RSC.
- H. Vu, F. Gonçalves, R. Philippe, E. Lamouroux, M. Corrias, Y. Kihn, D. Plee, P. Kalck and P. Serp, Bimetallic catalysis on carbon nanotubes for the selective hydrogenation of cinnamaldehyde, J. Catal., 2006, 240, 18–22 CrossRef CAS.
- S. Santiago-Pedro, V. Tamayo-Galván and T. Viveros-García, Effect of the acid–base properties of the support on the performance of Pt catalysts in the partial hydrogenation of citral, Catal. Today, 2013, 213, 101–108 CrossRef CAS.
- I. S. Mashkovsky, G. N. Baeva, A. Yu Stakheev, T. V. Voskoboynikov and P. T. Barger, Pd/Al2O3 catalyst for selective hydrogenation of benzene in benzene–toluene mixture, Mendeleev Commun., 2009, 19, 108–109 CrossRef CAS.
- J. M. Grau, L. Daza, X. L. Seoane and A. Arcoya, Effect of Ba and rare earths cations on the properties and dehydrocyclization activity of Pt/K-LTL catalysts, Catal. Lett., 1998, 53, 161–166 CrossRef CAS.
- G. Larsen and G. L. Haller, Metal–support effects in Pt/L-zeolite catalysts, Catal. Lett., 1989, 3, 103–110 CrossRef CAS.
- T. Phuong, Competitive hydrogenation of benzene and toluene on group VIII metals: Correlation with the electronic structure, J. Catal., 1986, 102, 456–459 CrossRef.
- C. Minot and P. Gallezot, Competitive hydrogenation of benzene and toluene: Theoretical study of their adsorption on ruthenium, rhodium, and palladium, J. Catal., 1990, 123, 341–348 CrossRef CAS.
- Z. Guo, Y. Chen, L. Li, X. Wang, G. L. Haller and Y. Yang, Carbon nanotube-supported Pt-based bimetallic catalysts prepared by a microwave-assisted polyol reduction method and their catalytic applications in the selective hydrogenation, J. Catal., 2010, 276, 314–326 CrossRef CAS.
- D. Poondi and M. Albert Vannice, Competitive Hydrogenation of Benzene and Toluene on Palladium and Platinum Catalysts, J. Catal., 1996, 161, 742–751 CrossRef CAS.
- M. J. Yacamán, R. Herrera, S. Tehuacanero, A. Gómez and L. B. Del Río, Surface roughness studies in small particles using HREM, Ultramicroscopy, 1990, 33, 133–141 CrossRef.
- L. Zhang, H. Liu, X. Huang, X. Sun, Z. Jiang, R. Schlögl and D. Su, Stabilization of Palladium Nanoparticles on Nanodiamond–Graphene Core–Shell Supports for CO Oxidation, Angew. Chem., Int. Ed., 2015, 54, 15823–15826 CrossRef CAS PubMed.
- C.-T. Kuo, Y. Lu, L. Kovarik, M. Engelhard and A. M. Karim, Structure Sensitivity of Acetylene Semi-Hydrogenation on Pt Single Atoms and Subnanometer Clusters, ACS Catal., 2019, 9, 11030–11041 CrossRef CAS.
- K. Murata, K. Ogura, J. Ohyama, K. Sawabe, Y. Yamamoto, S. Arai and A. Satsuma, Selective Hydrogenation of Cinnamaldehyde over the Stepped and Plane Surface of Pd Nanoparticles with Controlled Morphologies by CO Chemisorption, ACS Appl. Mater. Interfaces, 2020, 12, 26002–26012 CrossRef CAS PubMed.
- S. J. Tauster, S. C. Fung, R. T. K. Baker and J. A. Horsley, Strong Interactions in Supported-Metal Catalysts, Science, 1981, 211, 1121–1125 CrossRef CAS PubMed.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Pd-Pt Bimetallic Nanodendrites with High Activity for Oxygen Reduction, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- L. Bai, X. Wang, Q. Chen, Y. Ye, H. Zheng, J. Guo, Y. Yin and C. Gao, Explaining the Size Dependence in Platinum-Nanoparticle-Catalyzed Hydrogenation Reactions, Angew. Chem., Int. Ed., 2016, 55, 15656–15661 CrossRef CAS PubMed.
- J. Zhu, M. L. Yang, Y. Yu, Y. A. Zhu, Z. J. Sui, X. G. Zhou, A. Holmen and D. Chen, Size-Dependent Reaction Mechanism and Kinetics for Propane Dehydrogenation over Pt Catalysts, ACS Catal., 2015, 5, 6310–6319 CrossRef CAS.
- Z. Lian, S. Ali, T. Liu, C. Si, B. Li and D. S. Su, Revealing the Janus Character of the Coke Precursor in the Propane Direct Dehydrogenation on Pt Catalysts from a kMC Simulation, ACS Catal., 2018, 8, 4694–4704 CrossRef CAS.
- A. W. Hauser, P. R. Horn, M. Head-Gordon and A. T. Bell, A systematic study on Pt based, subnanometer-sized alloy cluster catalysts for alkane dehydrogenation: Effects of intermetallic interaction, Phys. Chem. Chem. Phys., 2016, 18, 10906–10917 RSC.
- M.-L. Yang, Y.-A. Zhu, C. Fan, Z.-J. Sui, D. Chen and X.-G. Zhou, DFT study of propane dehydrogenation on Pt catalyst: effects of step sites, Phys. Chem. Chem. Phys., 2011, 13, 3257 RSC.
| This journal is © the Owner Societies 2024 |