
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the effect of CaCl2 on amyloid β aggregation via supercritical angle Raman and fluorescence spectroscopy and microscopy†
Nathalia Simea
Münch
 ,
Subir
Das
and
Stefan
Seeger
*
,
Subir
Das
and
Stefan
Seeger
*
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. E-mail: sseeger@chem.uzh.ch
First published on 1st October 2024
Abstract
Amyloid β aggregation is an important factor in Alzheimer's disease. Since calcium homeostasis plays an important role in amyloid β aggregation, it is crucial to study the interaction between calcium ions and amyloid β directly at the surface of the lipid membrane. With supercritical angle techniques, the signal of interest at the surface is easily separated from the bulk solution, making them a powerful tool for aggregation study. In this work, the influence of calcium ions on amyloid β aggregation over different aggregation time periods is investigated with supercritical angle Raman and fluorescence spectroscopy and microscopy. Note that calcium ions have a larger influence on amyloid β1–42 than on the 40 amino acid variant. We found that a small layer of calcium ions significantly protects the lipid membrane against the protein insertion process.
Introduction
One of the main neurodegenerative diseases related to aging is Alzheimer's disease (AD), which affects approximately 50 million people worldwide. Two major pathological hallmarks are commonly associated with AD: (1) the aggregation of amyloid β (Aβ) to neurotoxic oligomers and fibrils with a subsequent accumulation of amyloid plaques outside the neurons.1–3 (2) The aggregation of tau protein that forms neurofibrillary tangles in the grey matter of the brain.4 Due to this protein deposition, the lipid membrane is disrupted, which leads to neural degeneration. The most important consequence is the loss of crucial brain functions such as motion, learning ability, memory or language skills.5 As a state-of-the-art diagnosis of AD, the detection of such Aβ aggregation is necessary for early diagnosis.6–8Calcium ions are important for modulating the functions of neuron cells, e.g. neurotransmitter release or forming new synapses.9 Therefore, Ca2+-binding proteins ensure calcium homeostasis inside neurons by regulating the amount of Ca2+ ions inside the cells. Otherwise, reactive oxygen species (ROS) accumulate on the neurons, which leads to cell death.2 Furthermore, calcium ions strongly interact with the membrane's lipid phosphate groups. Stronger interactions occur with negatively charged phosphatidylserine (PS), such as POPS or DOPS. These interactions decrease the negative charge of the lipid membrane.10,11 Due to the decrease of negative charges, the electrostatic attractions between lysine and the phosphate head groups are hindered, aggravating the approach of the peptide to the membrane. However, other positive amino acids such as histidine or arginine can also form attracting forces with negative lipid head groups, blocked by calcium ions. Y. Yang et al.11 showed a combination of hydrophobic and electrostatic interactions between membrane bound peptides and lipids via all-atom molecular dynamics simulations. The first step is the electrostatic approach of the peptide towards the lipid bilayer. Close to the membrane, the hydrophobic residues, especially phenylalanine, are anchored into the hydrophobic part of the lipid bilayer. The calculations further show a strong affinity of Ca2+ to the phosphate groups enhancing the positive charge of the membrane.11 Similar observations were also introduced by Sciacca et al.12 using NMR spectroscopy to test the insertion of amyloid peptides into lipid membranes. In the presence of calcium ions, the fibril insertion into the membrane is harder, leading to less membrane rupture than without Ca2+. On contrary, if the peptides already aggregate at the membrane before adding calcium ions, the aggregation and therefore the membrane disruption is increased.12 Other metal cations such as Cu2+, Fe2+ or Zn2+ are also interacting with Aβ and researchers investigated them more widely than Ca2+.13,14 However, one property of these ions is to form complexes with Aβ.15,16 On contrary, Ca2+ significantly block the interaction between the proteins and the lipid membrane. Because there are less studies investigating Ca2+ than Cu2+, we only focus on calcium ions in the current paper. The main advantage of Supercritical Angle microscopy is the simultaneous measurement of the bulk solution and molecules that are located close to the surface in two different channels due to separation of the signal coming from the surface above the critical angle. It is simpler to analyze surface bound molecules with this optical method. Furthermore, it is non-invasive to biological samples in aqueous solutions, which makes it applicable in different research areas such as biology, physics, surface-chemistry or material sciences.17,18 However, only Supercritical Angle Fluorescence (SAF) has a high enough intensity to measure under real biological conditions. The Raman scattering cross-sections is typically ∼10−29 to 10−31 cm2,19 which is lower than the fluorescence cross-section. Specifically when we deal with biological samples, the low concentration is a critical factor because of weak sensitivity of Raman spectroscopy. The advantage of conventional Raman spectroscopy over fluorescence is the label-free method, which is suitable to measure the secondary structure of proteins. The Raman scattering suffers low signal-to-noise (SNR) ratio. Therefore, classical assays such as ThioflavinT (ThT), circular dichroism (CD) and transmission electron microscopy (TEM) can be useful for Aβ protein characterization.20,21 Note that the characterization of Aβ proteins interacting with cations and nanoparticles using TEM have been reported.22,23 However, these methods allow measurements only in one environment, either with lipids or without lipids. Therefore, the direct comparison of proteins far and near of the membrane is not possible. Note that the supercritical angle Raman and supercritical angle fluorescence offers simultaneous measurement of the bulk solution and the surface. With this method, the structural changes near the lipid membrane, which are close to the microscope glass slide, can be observed with reduced photobleaching, improved SNR and axial resolution. One drawback of CD spectroscopy is the lack of high-resolution in complex systems such as proteins interacting with a lipid membrane.24 In addition, it is not possible with ThT assays to detect oligomers and protofibrils, since the dye only interacts with fibrils. ThT may also influence the aggregation of Aβ, which falsifies the results of the experiments.25
V. Dubois et al.26 used SAF microscopy to investigate the interaction between Aβ peptides and a solid supported lipid bilayer (SLB). The results showed a higher aggregation rate of Aβ1–42 compared to Aβ1–40. Furthermore, Aβ1–42 is more likely to bind to the SLB as Aβ1–40 and have a higher oligomerization. V. Dubois et al.27 also studied the interaction between Aβ and the SLB with supercritical angle Raman spectroscopy (SAR28), a label-free technique, which resolves the secondary structures in proteins and peptides. Their results showed a major number of β-sheets in Aβ1–40 whereas Aβ1–42 exploits more β-sheets in the bulk solution, while α-helices bind stronger to the surface of the lipid membrane.
In this current work, the interaction between the two Aβ peptides and a SLB is investigated with supercritical angle Raman (SAR) and fluorescence (SAF) spectroscopy and microscopy to study the aggregation at the surface compared with the bulk solution. The SLB is an easy membrane model, which is reproducible and therefore widely used to study the mechanism of the Aβ aggregation on membranes.26,29,30 In addition, the influence of calcium ions on the Aβ aggregation is studied. The aggregation of both peptides is compared, since some differences are already known, also with regard to AD: Aβ1–40 has a larger abundance, Aβ1–42 is more hydrophobic and more neurotoxic. In addition, Aβ1–42 undergoes more fibrillation than Aβ1–40.2 The studies were performed in vitro to investigate only the direct influence of calcium ions on the Aβ aggregation and omitting all other interactions within the cells.
Experimental
Materials
Aβ1–40 (APExBIO, Houston, Texas, United States), Aβ1–42 (Sigma-Aldrich, Pennsylvania, United States), ATTO633-COOH (ATTO-TEC, Siegen, Germany), 1,2-dioleoyl-sn-glycero-3-phospho-choline (DOPC, Avanti Polar Lipids, Alabaster, Alabama, United States) in chloroform, 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS, Avanti Polar Lipids, Alabaster, Alabama, United States) in chloroform, CaCl2·2H2O (Sigma-Aldrich CHEMIE, Steinheim Germany), Dulbeccos PBS (Sigma-Aldrich, Buchs, Switzerland), NaCl (Sigma-Aldrich, Buchs, Switzerland), tris(hydroxymethyl)aminomethane (tris, Sigma-Aldrich CHEMIE, Steinheim Germany), Nuclepore Track-Etch membrane (0.1 μm pore size, Whatman, Merck, Darmstadt, Germany), cover glass (MENZEL-Gläser, Braunschweig, Germany), Loctide 3105 (Henkel, Erlinsback, Switzerland), Deconex cip7 (Avan-tor, Dietikon, Switzerland), EtOH (REUSS-CHEMIE AG, Tägerig, Switzerland), Milli-Q-H2O (MQ H2O, deionisied H2O, filtered in Milli-Q system, Merck, Darmstadt, Germany), custom made metal plate (university intern workshop).Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35 (DOPC:DOPS). In earlier SLB-peptide studies, this ratio was optimized.31 The lipids (solution in chloroform) were vortexed, stirred under nitrogen and completely dried under vacuum (20 mbar) over night. Then, the lipids were mixed with 1 mL membrane buffer (100 mM NaCl, 5 mM CaCl2·2H2O, 10 mM tris, pH 7.4, filtered, degassed) and vortexed. To get homogenously distributed unilamellar vesicles of the same size, the suspension was extruded 25 times through a millipore membrane with 0.1 μm pore size and subsequent diluted to 0.1 mg mL−1 with membrane buffer. To build the measurement plate, a glass coverslip was washed in an ultrasonic bath with Deconex cip7, then EtOH and at the end with Milli-Q H2O and glued afterwards to the metal plate, which was washed in the same way. The liposomes were added to the measurement plate, which is connected to a peristaltic pump, for 15 min (200 μL min−1) to reach the critical concentration for fusion to the SLB on the 0.16 mm thick cover glass. The remaining vesicles were removed by washing with the membrane buffer at the same flow rate for 45 min and the SLB was stabilized for at least one hour in Dubelcos PBS before the measurements.
:35 (DOPC:DOPS). In earlier SLB-peptide studies, this ratio was optimized.31 The lipids (solution in chloroform) were vortexed, stirred under nitrogen and completely dried under vacuum (20 mbar) over night. Then, the lipids were mixed with 1 mL membrane buffer (100 mM NaCl, 5 mM CaCl2·2H2O, 10 mM tris, pH 7.4, filtered, degassed) and vortexed. To get homogenously distributed unilamellar vesicles of the same size, the suspension was extruded 25 times through a millipore membrane with 0.1 μm pore size and subsequent diluted to 0.1 mg mL−1 with membrane buffer. To build the measurement plate, a glass coverslip was washed in an ultrasonic bath with Deconex cip7, then EtOH and at the end with Milli-Q H2O and glued afterwards to the metal plate, which was washed in the same way. The liposomes were added to the measurement plate, which is connected to a peristaltic pump, for 15 min (200 μL min−1) to reach the critical concentration for fusion to the SLB on the 0.16 mm thick cover glass. The remaining vesicles were removed by washing with the membrane buffer at the same flow rate for 45 min and the SLB was stabilized for at least one hour in Dubelcos PBS before the measurements.
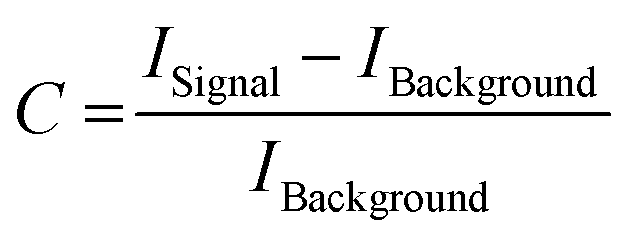 | (1) |
Results and discussion
The aggregation time was set to 72 h, since A. Itkin et al.35 observed the first major difference after this time, and it is important to discover the aggregation after a long time. Fig. 1 shows the UAR (black) and SAR (red) spectra of Aβ1–40 incubated for 72 h at 20 ± 1 °C in PBS (2.4 mg mL−1) and measured on the SLB. The two spectra differ from each other due to the interaction with the lipid membrane in the SAR channel. Most peaks in the SAR channel have a lower intensity, since the SAR method is less sensitive to the vibrational changes in the molecules than the UAR. The Raman shifts of each single peak are also slightly different in the UAR and SAR. The peak assignment to chemical bonds or amino acids/structures are as following: 762/763 and 837/846: Tyr, 939/940: ν(N–Cα–C) and Val/Ile, 1010/1005 and 1061/1052: Phe ring, 1140/1136: C–N, 1299/1311: amide III, 1368/—: δ(Cα–H), 1467/1460: δ(C–H), s1610/1615: Phe, 1674/1647: amide I, 1740/1757: Glu/Asp, 2874/2890 and 2952/2957: ν(C–H). All Raman shift values are represented in cm−1. The spectral variability is higher in the SAR than in the UAR spectrum due to the lower sensitivity of the SAR channel. Within the spectra, the largest variability is between 1550 and 1700 cm−1, in the amide I region (marked as a dark blue rectangle, Fig. 1). This region also differs the most between UAR and SAR. Due to the interaction with the lipid membrane, some peaks are shifted, some are not visible anymore and some new peaks arise. Mostly the peaks of hydrophobic amino acids are changed in the SAR due to interaction with the membrane. The hydrophobic Phe peak is almost not visible in the SAR, but it is visible in the UAR. Since both forms of the Aβ are important on the pathway of AD, Fig. 2 gives a comparison of the Raman spectra of both peptides with and without CaCl2. Fig. 2(A) shows the UAR spectra of Aβ1–40 (black), Aβ1–40 with CaCl2 (red), Aβ1–42 (blue) and Aβ1–42 with CaCl2 (green). All samples were measured on a SLB and incubated for 72 h without the SLB, before adding to the SLB for the measurements. Only the most relevant part of the spectra (465–1917 cm−1) is shown, since the spectra are normalized to the vibrational stretch of the C–H bond and therefore this band is the same for all spectra. The peak assignment is the same as above with some slight shifts in the wavenumbers. In Fig. 2(B), the spectra from Fig. 2(A) are magnified and overlaid for better comparison of the amide I peaks (1530–1800 cm−1).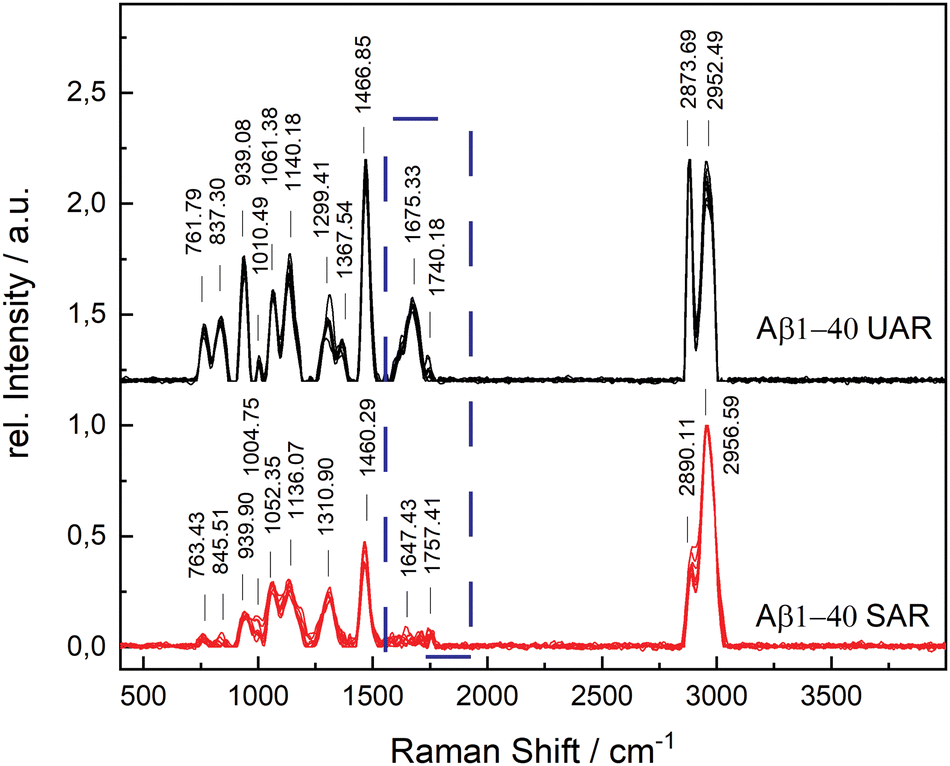 | ||
| Fig. 1 Difference of SAR (red) and UAR (black) spectra of Aβ1–40 incubated for 72 h at 20 ± 1 °C in MQ H2O and measured on the SLB. All the measured spectra are shown to display the spectral variability and were normalized to the highest peak intensity. Peak assignments in cm−1: 762/763 and 837/846: Tyr, 939/940: ν(N–Cα–C) and Val/Ile, 1010/1005 and 1061/1052: Phe ring, 1140/1136: C–N, 1299/1311: amide III, 1368/—: δ(Cα–H), 1467/1460: δ(C–H), 1610/1615: Phe 1675/1647: amide I, 1740/1757: Glu/Asp, 2874/2890 and 2952/2957: ν(C–H). The largest difference between UAR and SAR is in the range between 1530 and 1800 cm−1 (dark blue rectangle), which is also the most sensitive region in the protein Raman spectrum. | ||
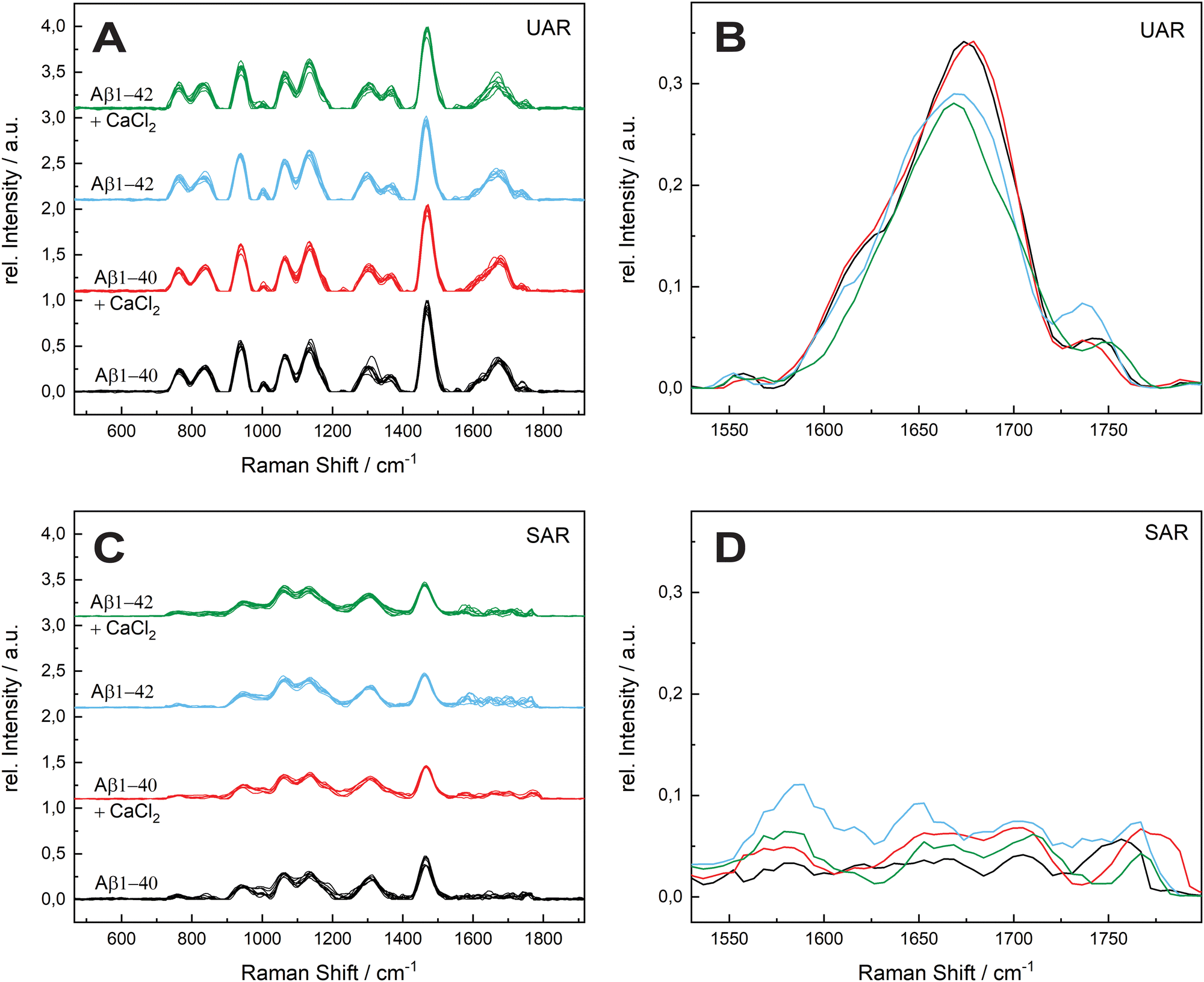 | ||
| Fig. 2 Comparison of the effect of calcium ions on the two forms of Aβ after 72 h. (A) Undercritical Angle Raman (UAR) spectra of 115 μM Aβ1–40 incubated in MQ H2O for 72 h at 20 ± 1 °C (black), 115 μM Aβ1–40 incubated with 25 mM CaCl2 for 72 h at 20 ± 1 °C (red), 111 μM Aβ1–42 incubated in MQ H2O for 72 h at 20 ± 1 °C (blue) and 111 μM Aβ1–42 incubated with 25 mM CaCl2 for 72 h at 20 ± 1 °C (green). (B) Magnification of the mean value of the spectra from A on range of the amide I band (1530–1800 cm−1). The spectra are overlaid for better comparison and the color assignment is the same as in A. (C) Supercritical angle Raman (SAR) spectra of the same samples as in A with the magnification of the amide I band (mean values) (D). All samples were measured on a SLB and normalized to the maximum peak intensity. The spectral variability is higher in the SAR spectra and within the spectra the highest variability is in the amide I region. | ||
The UAR spectra of Aβ1–40 are similar, no matter if CaCl2 is added or not. The Raman shifts of the amide I band are similar in both spectra, meaning the secondary structure remains the same, when it aggregates in the presence of calcium ions for 72 h. The main peak is at 1669.36 ± 2.51 cm−1 for Aβ1–40 without calcium and 1667.84 ± 3.65 cm−1 for Aβ1–40 with 25 mM CaCl2, meaning the peptides possess mainly β-sheet structures with a small shoulder in the α-helix region at 1621.55 cm−1. The intensity of the amide I peak is a bit increased, when aggregating in the presence of calcium ions. This result shows clearly that β-sheets are formed after 72 h of aggregation, even when calcium ions are present. Therefore, calcium does not prevent Aβ1–40 from aggregating in the bulk solution, since the aggregation after 72 h is almost the same as without calcium ions. The Raman measurements were also carried out with different concentrations of CaCl2 and Aβ. The results are presented in the ESI.†
The amide I band (UAR) of Aβ1–42 differs more when aggregated 72 h in the presence of CaCl2 compared to Aβ1–40. It also has the highest spectral variability of all samples. This band is flatter, but has on average more or less the same central wavenumber of the β-sheet peak (1667.19 ± 4.31 cm−1). Some of the single spectra are shifted slightly to lower wavenumbers. A lower wavenumber and a higher relative intensity mean a stronger bond in the beta sheet structure of Aβ1–42 in the bulk solution and therefore a stronger aggregation.
Fig. 2(C) shows the SAR spectra of the same samples with the same color code and same range in Raman shift as in Fig. 2(A). The magnification of the amide I region is presented in Fig. 2(D). The spectrum of Aβ1–40 (black) has a peak at 1680.42 ± 16.60 cm−1, indicating a β-sheet as in the UAR spectrum with a slightly higher shift, meaning a slightly weaker β-sheet at the surface. In addition, there is a peak in the α-helix range at 1652.16 ± 9.90 cm−1. When CaCl2 is added (red) for incubation, most of the peaks of the whole spectrum have a lower relative intensity and there is a shift of some individual peaks to higher wavenumbers. This result shows less aggregation with weaker bonds of Aβ1–40 at the surface of the lipid membrane, when incubated for 72 h in CaCl2 containing conditions. The Glu/Asp peak has a stronger shift to higher wavenumbers, compared to the rest of the spectrum, resulting in an ester bond at 1780 cm−1. This ester may result through binding to other amino acids or to lipids. One possible reason for this ester bond could be the reaction between the laser-activated lipid head groups and the calcium binding glutamate or aspartate. The amide I band of Aβ1–42 (blue) has a higher intensity as of Aβ1–40 in both, the β-sheet (1682.96 ± 15.65 cm−1) and the α-helix region (1649.70 ± 6.32 cm−1). However, the increase in the α-helix is much more intense than the β-sheet increase. This means, Aβ1–42 forms mostly stronger α-helices at the surface to get anchored into the lipid membrane, but also some β-sheets. In the bulk, Aβ1–42 forms only β-sheets. In contrast, Aβ1–40 forms more β-sheets over the whole space. This fact is consistent with the fact, that Aβ1–42 is more cytotoxic than Aβ1–40.1,36 When incubated for 72 h together with CaCl2 (green), there is not a large shift difference, but a decrease of intensity in the amide I region (1530–1800 cm−1), therefore there is less aggregation at the surface.
In Fig. 3, the mean values of the integrals of the deconvoluted amide I peaks for the spectra in Fig. 2 are shown to compare the number of α-helix and β-sheet bonds that are present in each sample. They are normalized to the integral of the β-sheet of Aβ1–40 without calcium ions for comparison for both UAR and SAR. Fig. 3(A) shows the integrals from the UAR spectra, the inset is a zoom-in for a better comparison of each bar, and similarly, Fig. 3(B) shows the integral from the SAR spectra. When the two conditions of Aβ1–40 after 72 h are compared, the peak area with CaCl2 increase in the UAR and in the SAR compared to the calcium free aggregation. There is a stronger aggregation, which is also described in the literature.37 Together with the higher shift in Fig. 2, Aβ1–40 incubated in CaCl2 forms more but weaker β-sheets than without CaCl2. Because calcium ions block some of the interactions between Glu/Asp and Lys, the β-sheets get weaker, since they are not bond tightly. However, due to more charges, they can be accumulated more easily, reaching higher integrals. As shown by Y. Yang et al.,11 most of the calcium ions stay near the surface close to the negatively charged lipids, hindering lysine to approach the membrane and therefore hindering aggregation at the surface of the membrane. The integral of Aβ1–42 reveals a much stronger aggregation with a lot more of α-helices at the surface than Aβ1–40.
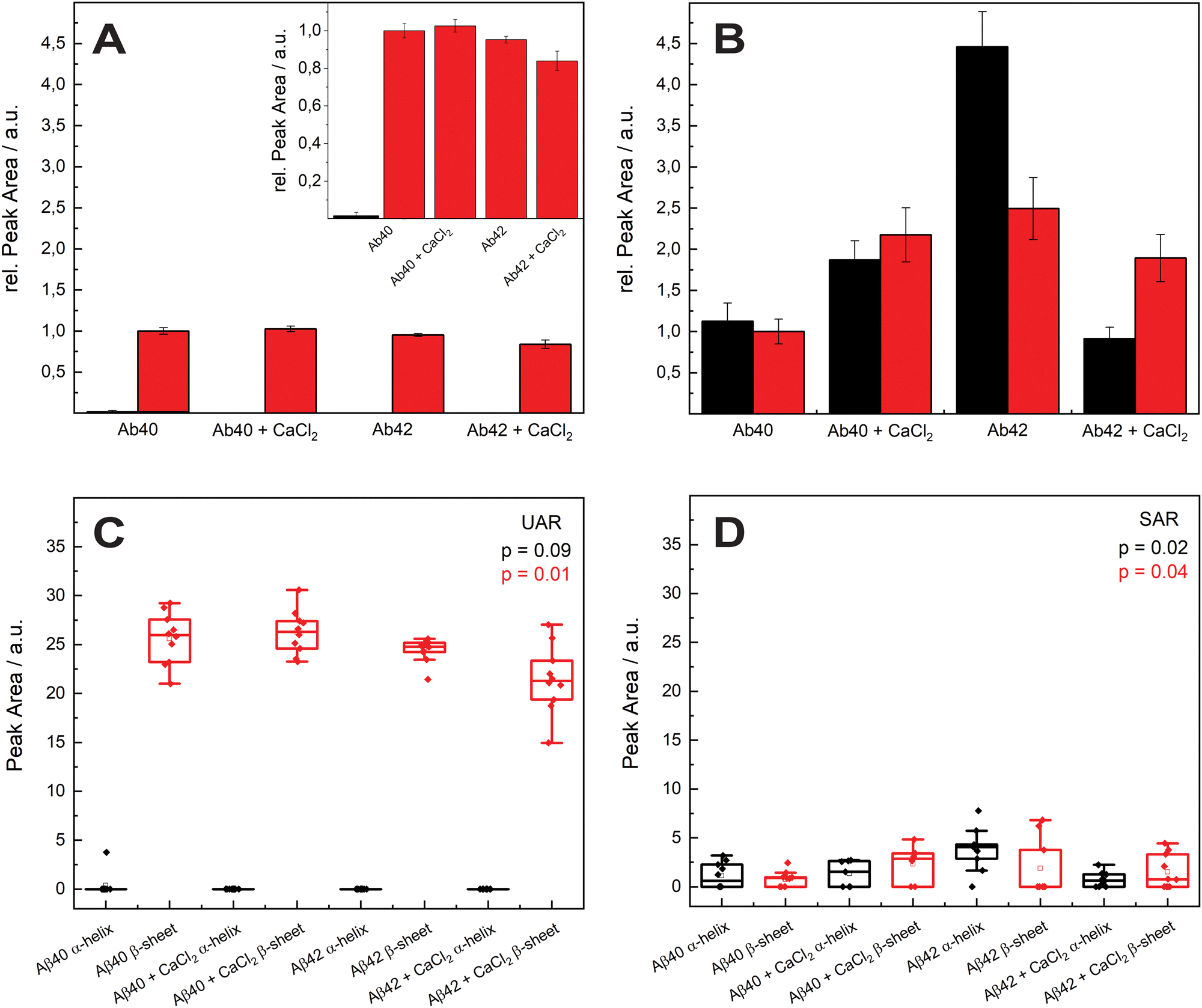 | ||
| Fig. 3 Mean values of the Integrals of the amide I peak (1640–1700 cm−1) of all samples. (A) The relative peak area of the amide I region (1640–1700 cm−1) in the UAR for the samples of 72 h incubation time. The black and red bars represent the α-helix and β-sheet peaks. The inset is a zoom-in for a better comparison of each bar. (B) The relative peak area of the amide I region (1640–1700 cm−1) in the SAR. The error bars are the standard deviations of each sample. (C) and (D) Represents the box charts of the absolute integrals of the amide peaks for the UAR and the SAR respectively. The statistically significance given by the p-value, which is lower than 0.05 (exception: UAR α-helix, due to an outlier). The UAR is slightly more significant than SAR, since SAR is more sensitive to the surface and little fluctuations may influence the values more than in the UAR, however the significance is still given by p < 0.05. The black and red boxes represents the α-helices β-sheets. | ||
These findings are consistent with the Raman shifts of the spectra, since Aβ1–42 forms mostly α-helices at the surface which leads to higher aggregation at the SLB due to anchoring of α-helices into the membrane.38,39 When Aβ1–42 aggregated in the presence of calcium ions, the relative peak intensity of the α-helix drastically decreased. Therefore, Aβ1–42 forms more β-sheets at the surface than α-helices when incubated with CaCl2. The integral of β-sheets is also a bit lower, when incubated in calcium ions containing conditions. This means that the barrier function of calcium ions works when Aβ1–42 approaches the membrane. With Aβ1–40 the barrier effect is not working well, probably since the peptide is more hydrophilic than the 42 amino acid variant.
Since Ca2+ is blocking the electrostatic attraction, which is the first important step in aggregation at the lipid membrane, the hydrophobic amino acids bury less into the membrane. Therefore, this effect is larger in the more hydrophobic Aβ1–42.
The aggregation process was even prolonged to 96 h incubation time (see Fig. 4). The spectral variability is in general higher after 96 h. A possible reason is a different aggregation time of each different protein in the samples. After 72 h most of the proteins are still in the aggregation process, therefore all the spectra are similar in the UAR. In the SAR channel, the case is a bit different, since the aggregation process takes place without SLB. The aggregated samples were added after the aggregation to the lipid membrane. Each protein can have different interactions with the lipids, no matter, if the aggregation time is 72 or 96 h.
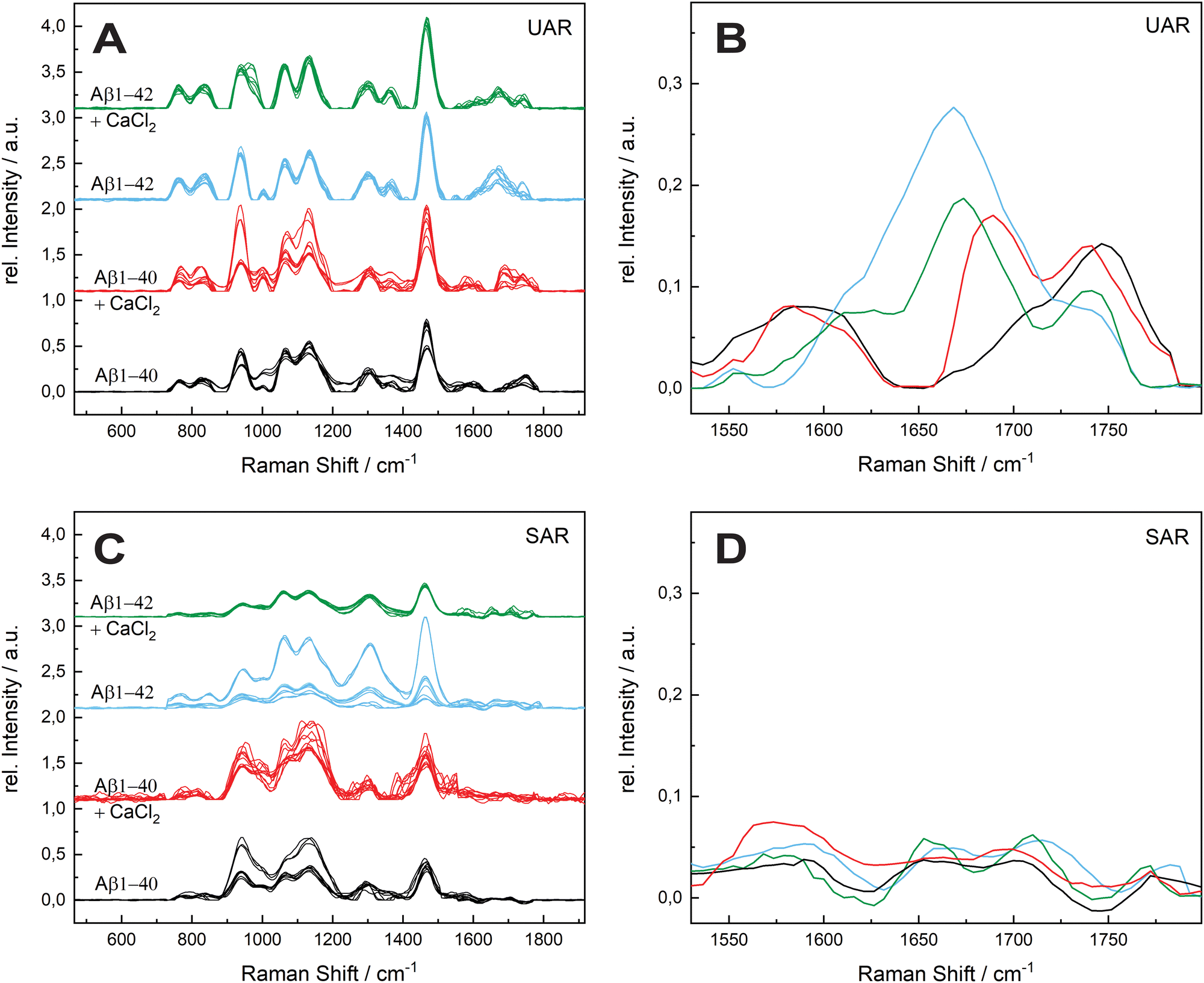 | ||
| Fig. 4 Comparison of the effect of calcium ions on the two forms of Aβ after 96 h. (A) Undercritical Angle Raman (UAR) spectra of 115 μM Aβ1–40 incubated in MQ H2O for 96 h at 20 ± 1 °C (black), 115 μM Aβ1–40 incubated with 25 Mm CaCl2 for 72 h at 20 ± 1°C (red), 111 μM Aβ1–42 incubated in MQ H2O for 96 h at 20 ± 1 °C (blue) and 111 μM Aβ1–42 incubated with 25 Mm CaCl2 for 96 h at 20 ± 1 °C (green). (B) Magnification of the mean value of the spectra from A on range of the amide I band (1530–1800 cm−1). The spectra are overlaid for better comparison and the color assignment is the same as in A. (C) Supercritical angle Raman (SAR) spectra of the same samples as in A with the magnification of the amide I band (mean values) (D). All samples were measured on a SLB and normalized to the maximum. The spectral variability is higher as in the aggregation after 72 h. | ||
The amide I region is the most important, therefore we concentrate on this region between 1530 and 1800 cm−1. The UAR spectra of the different samples after 96 h differs more than after 72 h. Furthermore, the relative intensity of the samples decreases after 96 h. For Aβ1–40, there is only a shoulder of the Glu/Asp peak around 1699 cm−1, which is still in the β-sheet region. One reason for loosing this peak is the accumulation of peptides. When incubated with CaCl2 for 96 h, the Aβ1–40 have a β-sheet peak that is shifted to higher wavelengths (1690.33 cm−1). This means a weaker bond due to the calcium ions, which hinders an accumulation and strong β-sheet formation. The spectra of Aβ1–42 incubated for 96 h with or without CaCl2 are similar to Aβ1–42 incubated for 72 h. The center of the main peak for both Aβ1–42 samples is at 1671 cm−1. Since Aβ1–42 forms fibrils in an early stage, the aggregation is already done after 72 h and there is not a large change after 96 h. In all spectra, the peak of the Glu/Asp interaction has a higher intensity after 96 h. The negatively charged amino acids get shielded with the positive calcium ions and therefore they interact less with other groups. Therefore, they are more distinct in the Raman spectra.
In the SAR spectra, the relative intensities of the amide I peak are similar after 96 h than after 72 h, even the relative intensities of the other peaks differs in some spectra. The center of the amide I peak for the Aβ1–40 without CaCl2 is at 1661 cm−1, a lower wavelength than after 72 h. This makes sense, since the aggregation after a longer time would be stronger. Incubated with CaCl2, the peak is shifted into the other direction towards weaker bonds. The interaction to the lipid membrane gets weaker due to the calcium ions on the lipid membrane. The spectra of Aβ1–42 with and without calcium ions are similar as Aβ1–40 without CaCl2, however the peak intensities are higher with the 42 amino acid variant.
The molecules that are already aggregated formed stronger bonds. The conclusion which is drawn from these results is a slower aggregation of Aβ1–42 in presence of calcium ions compared to Aβ1–40. This finding is consistent with the results from A. Itkin et al.35 and R. Lal et al.40 that Aβ1–40 forms first small oligomers, which aggregates further, while Aβ1–42 forms long fibrils, which need more time to form. In general, there is less aggregation at the surface when the Aβ was incubated for long time in the presence of calcium ions. This fact supports the theory of Y. Yang et al.11 that the calcium ions form a protecting layer over the membrane to prevent the peptide approaching towards the membrane, since the positively charged lysine amino acids are no longer attracted by the lipid molecules.
In addition to the Raman spectra, fluorescence images were recorded to further investigate the aggregation process and to give insights into the morphology of the aggregates. Fig. 5(A) shows the SAF image of 115 μM Aβ1–40 incubated for 72 h in Milli-Q H2O at 20 ± 1 °C and stained with ATTO633-COOH. The average length of all measured aggregates of this sample (on all recorded images) is 6.94 ± 6.53 μm. The size in the images is around 10-fold increased due to the diffraction limit. The 10-fold increase is determined through the 100 nm extruded liposomes, which have a size of around 1 μm in the UAF/SAF. The standard deviation is quite large, since the aggregates differ a lot in size and in shape. However, most of the aggregates have a length between 4 and 7 μm, which is congruent with the mean value of 6.94 μm. The number of aggregates in all images of this sample is n = 60. The image contrast was calculated using eqn (1), C = 12.6, using the aggregate with the highest intensity for Isignal.
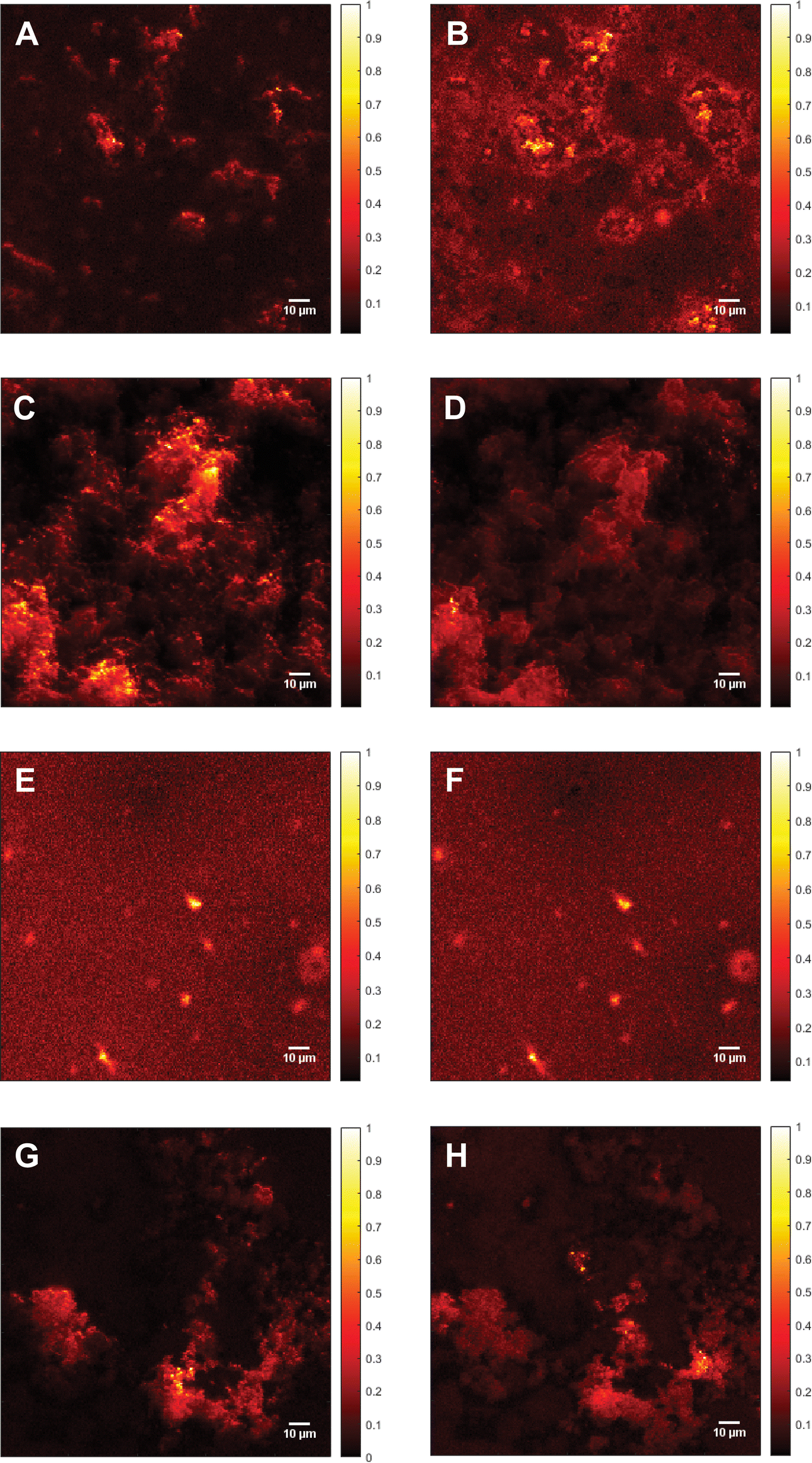 | ||
| Fig. 5 Fluorescence images of Aβ. (A) Supercritical angle fluorescence (SAF) image of 115 μM Aβ1–40 incubated for 72 h at 20 ± 1 °C and stained with ATTO633-COOH. Image contrast (calculated according to eqn (1)), C = 12.6. (B) Undercritical angle fluorescence (UAF) image of 115 μM Aβ1–40 incubated for 72 h at 20 ± 1 °C and stained with ATTO633-COOH, C = 7.87. (C) SAF image of 115 μM Aβ1–40 incubated with 25 mM CaCl2 for 72 h at 20 ± 1 °C and stained with ATTO633-COOH, C = 19.3. (D) UAF image of 115 μM Aβ1–40 incubated with 25 mM CaCl2 for 72 h at 20 ± 1 °C and stained with ATTO633-COOH, C = 12.0. (E) SAF image of 111 μM Aβ1–42 incubated for 72 h at 20 ± 1 °C and stained with ATTO633-COOH, C = 7.27. (F) UAF image of 111 μM Aβ1–42 incubated for 72 h at 20 ± 1 °C and stained with ATTO633-COOH, C = 11.5. (G) SAF image of 111 μM Aβ1–42 incubated with 25 mM CaCl2 for 72 h at 20 ± 1°C and stained with ATTO633-COOH, C = 17.7. (H) UAF image of 111 μM Aβ1–42 incubated with 25 mM CaCl2 for 72 h at 20 ± 1 °C and stained with ATTO633-COOH, C = 7.90. In the images, the oligomers seem to be around 10 times larger than the actual size due to the diffraction limit. All Samples were measured on a SLB and normalized to the highest fluorescent intensity. The color bars on the right of each image shows the relative fluorescence intensity. | ||
The UAF image (Fig. 5(B)) of 115 μM Aβ1–40 shows similar aggregates as the SAF (Fig. 5(A)), but a higher fluorescent background. The average length is 6.55 ± 2.45 μm (n = 145), resulting in similar aggregates in the SAR and the UAR. However, the UAR consists of around 2.5-fold the aggregates of the SAR. The UAR image have a lower contrast of C = 7.87, which means less aggregation in the bulk. The background fluorescence is higher in the bulk, since the UAF signal is stronger and more fluorescence in general can be detected in the UAF channel than in the SAF channel. Therefore, it is important to do the background correction and use the imaging contrast rather than the intensity itself. The aggregation at the surface is supported by negatively charged lipids, therefore the measured aggregation in the SAF channel is higher.
The fluorescence images of 115 μM Aβ1–40 incubated for 72 h in 25 mM CaCl2 and stained with ATTO633-COOH differ from them without CaCl2 (see Fig. 5(C) and (D)). In the SAF image (Fig. 5(C)), there are more abundant (n = 340) and smaller oligomers, which are close together with a size of 3.33 ± 1.36 μm and an intensity of 60.0 Cnts. The image contrast was calculated to C = 19.3. This image contrast is a bit higher as without calcium ions, consisting with a larger integral in Fig. 3(B).
As shown in the UAF image (Fig. 5(D)) the aggregates have an average length of 3.36 ± 1.72 μm and an intensity of up to 244 Cnts with an image contrast C = 12.0. The number of aggregates is lower as in the SAF channel (n = 190). Comparing the aggregates of Aβ1–40 incubated in 25 mM CaCl2 after 72 h of aggregation, the Aβ dimers and small oligomers accumulates to larger clusters (see Fig. 5(C) and (D)). That seems to be loosely bound and spread over almost the whole surface with probably weak binding to the surface.
The calculated contrast values of the four different images of Aβ1–40 shows the highest fluorescence intensity for the aggregates of Aβ1–40 with CaCl2 at the surface, followed by only Aβ1–40 at the surface. The lowest contrast value was calculated for Aβ1–40 incubated in H2O in the bulk solution. This means the aggregation with calcium ions is in general higher than without, but the aggregates cannot really stick to the surface, since the bonds are weakened due to the charges of the calcium ions. To have a comparison between the SAF and the UAF signal, the ratio r = SAF/UAF was calculated. rAβ1–40 = 1.6 and rAβ1–40+CaCl2 = 1.6, the aggregation is similar with or without CaCl2.
Fig. 5(E) shows the SAF image of Aβ1–42 incubated for 72 h in MQ H2O. The imaging contrast is 7.27 and the mean value of the length is 6.02 ± 3.21 μm, n = 75. In the UAF channel (Fig. 5(F)); the same aggregates are visible as in the SAF image, since the signal from the surface is also detected in the UAF up to a small proportion. The aggregates are similar in the UAR as in the SAR (C = 11.5, l = 6.93 ± 3.58 μm, n = 75). When aggregated in the presence of CaCl2 for 72 h (Fig. 5(G) and (H)), the aggregates are slightly larger (7.25 ± 4.22 μm, SAF and 9.61 ± 4.96 μm for UAF) with a much higher amount of these aggregates (n = 125 for SAF and n = 255 for UAF), but with a lower intensity in general (CSAF = 17.7 and CUAF = 7.90 for UAF). This result shows that the peptides cannot be anchored strongly into the membrane, when CaCl2 is present, due to this positively charged barrier above the membrane, therefore the peptides are spread more widely over the whole lipid bilayer, resulting in more lager aggregates with lower intensity.
A possible model for the aggregation on a molecular level is proposed. These models are based on the results of this work and the mechanism suggested by L. Yu et al.,41 R. Lal et al.40 and Y. Yang et al.11 The first step is the attraction of the lysine group, following by a subsequent insert of hydrophobic residues such as phenylalanine inside the inner part of the membrane. Due to the hydrophobic interactions, the peptide is anchored into the membrane. The calcium ions prevent this insertion into the membrane.
Conclusions
In summary, this paper demonstrates the optical spectroscopy and microscopy technique for a better understanding of the aggregation mechanism of Aβ with calcium ions. Even when the spectral variability is higher in the SAR, the method is still reliable and a good tool to investigate proteins at the lipid surface. However, the aggregation process with calcium is much more complicated, than described here, e.g. ion channels are formed through the membrane due to Aβ aggregation. The purpose of this paper is to simply present a comparison of the effect of calcium ions between the bulk solution and the surface. Our results show the time dependent Aβ ggregation in presence and absence of calcium ions. Calcium ions enhance the aggregation of Aβ1–42; however, it hinders the insertion into the membrane. The reason is the prevention of α-helix formation due to electrostatic hindrance. In addition, there is no strong aggregation at the surface, the peptides are spread over the whole membrane, forming large clusters. The effect of calcium ions is thus larger for Aβ1–42, since Aβ1–40 forms mostly β-sheets at the surface. A schematic representation of the effect of CaCl2 on amyloid β in different aggregation times is depicted in Fig. 6. Hence, the regulation of Ca2+ homeostasis is crucial in the beginning of the AD progress.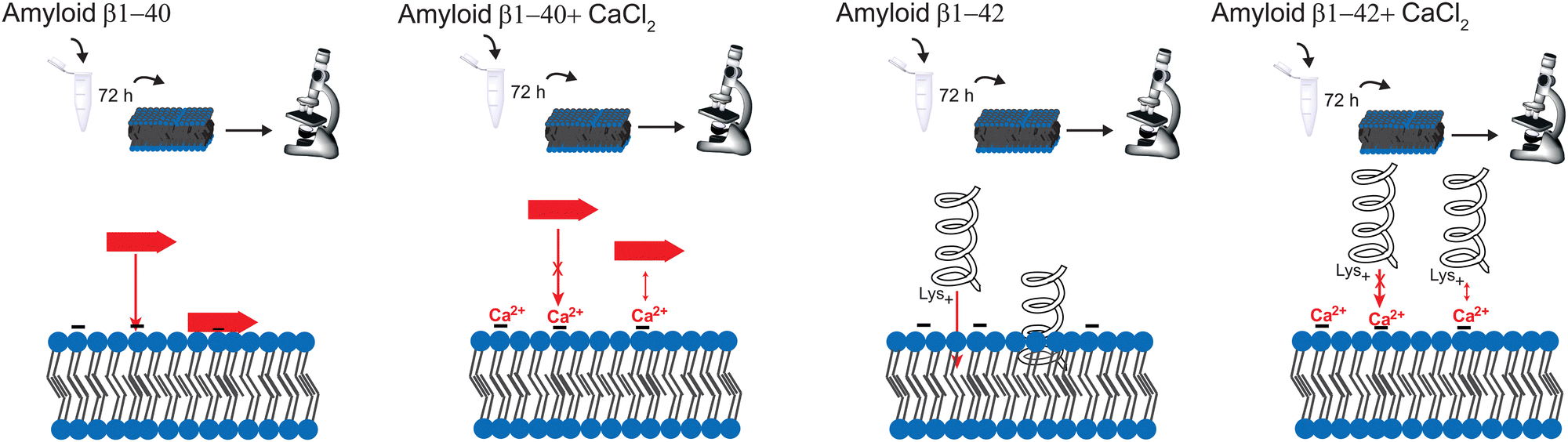 | ||
| Fig. 6 Schematic representation of the aggregation of Aβ. From left to right: Aβ1–40 forms mostly β-sheets on the surface. However, the calcium ions block the protein insertion into the membrane. Aβ1–42 forms more α-helices on the membrane, which inserts deeper into the membrane than β-sheets. This leads to a stronger effect of calcium ions blocking the insertion of Aβ1–42 into the membrane. | ||
Of course, in vitro aggregation cannot be directly translated in in vivo situation, since for the interaction with e.g. organelles cells are needed. Admittedly, the proposed models can be used to study the aggregation mechanism. Further studies will provide more information about the in vivo investigations with different Ca2+ concentrations. A detection system to monitor peptide aggregation inside AD patient's brains would also be helpful. For future work, the positive effect on Aβ aggregation, namely the barrier function could be enhanced by finding a way to prevent aggregates inserting into the membrane before they interact with calcium ions.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
All the raw data (Raman spectra and Fluorescence images) used in this article can be accessed from OSF repository at osf.io/3ku9n.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Swiss National Science Foundation (SNSF) and the University of Zurich (UZH) for financial support.Notes and references
- M. Zaman, A. N. Khan, S. M. Zakariya and R. H. Khan, Int. J. Biol. Macromol., 2019, 134, 1022–1037 CrossRef PubMed
.
- M. Pannuzzo, Alzheimer's & Dementia, 2022, 18(1), 191–196 Search PubMed
- H. Ashrafian, E. H. Zadeh and R. H. Khan, Int. J. Biol. Macromol., 2021, 167, 382–394 CrossRef PubMed
- K. Leroy, A. Bretteville, K. Schindowski, E. Gilissen, M. Authelet, R. De Decker, Z. Yilmaz, L. Buée and J.-P. Brion, Am. J. Pathol., 2007, 171(3), 976–992 CrossRef PubMed
- A. Serrano-Pozo, M. P. Frosch, E. Masliah and B. T. Hyman, Cold Spring Harbor Perspect. Med., 2011, 1(1), a006189 Search PubMed
- G. Lv, A. Sun, P. Wei, N. Zhang, H. Lan and T. Yi, Chem. Commun., 2016, 52, 8865 RSC
- R. Tao, N. Wang, T. Shen, Y. Tan, Y. Ren, W. Wei, M. Liao, D. Tan, C. Tang, N. Xu, H. Wang, X. Liu and X. Li, Theranostics, 2022, 12(6), 2549–2559 CrossRef PubMed
- H. Li, J. Wang, Y. Li, X. Chen, W. Zhang, Y. Zhao, G. Liu and J. Pan, Sens. Actuators, B, 2022, 358, 131481 CrossRef CAS
- S. J. Smith and G. J. Augustine, Trends Neurosci., 1988, 11(10), 458–464 CrossRef CAS
- J. M. Boettcher, R. L. Davis-Harrison, M. C. Clay, A. J. Nieuwkoop, Y. Z. Ohkubo, E. Tajkhorshid, J. H. Morrissey and C. M. Rienstra, Biochemistry, 2011, 50(12), 2264–2273 CrossRef CAS
- Y. Yang, S. Jalali, B. L. Nilsson and C. L. Dias, ACS Chem. Neurosci., 2021, 12(11), 2027–2035 CrossRef CAS
- M. F. M. Sciacca, D. Milardi, G. M. L. Messina, G. Marletta, J. R. Brender, A. Ramamoorthy and C. La Rosa, Biophys. J., 2013, 104(1), 173–184 CrossRef CAS
- A. I. Bush and R. E. Tanzi, Neurotherapeutics, 2008, 5(3), 421–432 CrossRef CAS PubMed
- A. Abelein, Acc. Chem. Res., 2023, 56(19), 2653–2663 CrossRef CAS PubMed
- C. D. Syme, R. C. Nadal, S. E. J. Rigby and J. H. Viles, J. Biol. Chem., 2004, 279(18), 18169–18177 CrossRef CAS PubMed
- J. Danielsson, R. Pierattelli, L. Banci and A. Gräslund, FEBS J., 2007, 274, 46–59 CrossRef CAS
- T. Ruckstuhl and D. Verdes, Opt. Express, 2004, 12(18), 4246–4254 CrossRef
- D. Verdes, T. Ruckstuhl and S. Seeger, J. Biomed. Opt., 2007, 12(3), 34012 CrossRef PubMed
- C. M. Penney, L. M. Goldman and M. Lapp, Nat. Phys. Sci., 1972, 235, 110–112 CrossRef CAS
- H. R. Kalhor, F. Shahin, M. H. Fouani and H. Hosseinkhani, Langmuir, 2011, 27(17), 10776–10784 CrossRef CAS
- S. Mittal and L. Rajendrakumar Singh, PLoS One, 2014, 9(12), e114029 CrossRef PubMed
- C. J. Matheou, N. D. Younan and J. H. Viles, J. Mol. Biol., 2016, 428(14), 2832–2846 CrossRef CAS
- Y. Kim, J. H. Park, H. Lee and J. M. Nam, Sci. Rep., 2016, 6(1), 19548 CrossRef CAS PubMed
- S. Munusamy, R. Conde, B. Bertrand and C. Munoz-Garay, Biochimie, 2020, 170, 173–202 CrossRef
- S. Freire, M. H. de Araujo, W. Al-Soufi and M. Novo, Dyes Pigm., 2014, 110, 97–105 CrossRef CAS
- V. Dubois, D. Serrano and S. Seeger, ACS Chem. Neurosci., 2019, 10(12), 4776–4786 CrossRef CAS
- V. Dubois, D. Serrano, X. Zhang and S. Seeger, Anal. Chem., 2020, 92(7), 4963–4970 CrossRef CAS
- D. Serrano and S. Seeger, Light: Sci. Appl., 2017, 6(10), e17066 CrossRef CAS
- S. Meker, H. Chin, T. N. Sut and N.-J. Cho, Langmuir, 2018, 34(32), 9548–9560 CrossRef CAS PubMed
- J. Robinson, N. Sarangi and T. E. Keyes, Phys. Chem. Chem. Phys., 2023, 25(11), 7648–7661 RSC
- N. P. Reynolds, A. Soragni, M. Rabe, D. Verdes, E. Liverani, S. Handschin, R. Riek and S. Seeger, J. Am. Chem. Soc., 2011, 133(48), 19366–19375 CrossRef CAS PubMed
- C. M. Winterflood, T. Ruckstuhl, D. Verdes and S. Seeger, Phys. Rev. Lett., 2010, 105(10), 108103 CrossRef PubMed
- G. H. Schulze, R. B. Foist, K. Okuda, A. Ivanov and R. F. B. Turner, Appl. Spectrosc., 2012, 66(7), 757–764 CrossRef PubMed
- S. Das, I.-C. Chen, K. U. Rehman, J.-L. Hsu, G.-Y. Zhuo and F.-J. Kao, J. Opt., 2019, 21(12), 125301 CrossRef
- A. Itkin, V. Dupres, Y. E. Dufrêne, B. Bechinger, J.-M. Ruysschaert and V. Raussens, PLoS One, 2011, 6(3), e18250 CrossRef
- K. Murakami, K. Irie, A. Morimoto, H. Ohigashi, M. Shindo, M. Nagao, T. Shimizu and T. Shirasawa, Biochem. Biophys. Res. Commun., 2002, 294(1), 5–10 CrossRef
- L. Canevari, A. Y. Abramov and M. R. Duchen, Neurochem. Res., 2004, 29, 637–650 CrossRef
- M. Vestergaard, T. Hamada and M. Takagi, Biotechnol. Bioeng., 2008, 99(4), 753–763 CrossRef
- N. S. Hatzakis, V. K. Bhatia, J. Larsen, K. L. Madsen, P.-Y. Bolinger, A. H. Kunding, J. Castillo, U. Gether, P. Hedegård and D. Stamou, Nat. Chem. Biol., 2009, 5(11), 835–841 CrossRef
- R. Lal, H. Lin and A. P. Quist, Biochim. Biophys. Acta, Biomembr., 2007, 1768(8), 1966–1975 CrossRef
- L. Yu, R. Edalji, J. E. Harlan, T. F. Holzman, A. Lopez, B. Labkovsky, H. Hillen, S. Barghorn, U. Ebert and P. L. Richardson, Biochemistry, 2009, 48(9), 1870–1877 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00996g |
| This journal is © the Owner Societies 2024 |