
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn active machine learning discovery platform for membrane-disrupting and pore-forming peptides†
Alexander
van Teijlingen
 a,
Daniel C.
Edwards
b,
Liao
Hu
b,
Annamaria
Lilienkampf
b,
Scott L.
Cockroft
b and
Tell
Tuttle
*a
a,
Daniel C.
Edwards
b,
Liao
Hu
b,
Annamaria
Lilienkampf
b,
Scott L.
Cockroft
b and
Tell
Tuttle
*a
a1Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: tell.tuttle@strath.ac.uk
bEaStCHEM School of Chemistry, Joseph Black Building, University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK
First published on 30th May 2024
Abstract
Membrane-disrupting and pore-forming peptides (PFPs) play a substantial role in bionanotechnology and can determine the life and death of cells. The control of chemical and ion transport through cell membranes is essential to maintaining concentration gradients. Likewise, the delivery of drugs and intracellular proteins aided by pore-forming agents is of interest in treating malfunctioning cells. Known PFPs tend to be up to 50 residues in length, which is commensurate with the thickness of a lipid bilayer. Accordingly, few short PFPs are known. Here we show that the discovery of PFPs can be accelerated via an active machine learning approach. The approach identified 71 potential PFPs from the 25.6 billion octapeptide sequence space; 13 sequences were tested experimentally, and all were found to have the predicted membrane-disrupting ability, with 1 forming highly stable pores. Experimental verification of the predicted pore-forming ability demonstrated that a range of short peptides can form pores in membranes, while the positioning and characteristics of residues that favour pore-forming behaviour were identified. This approach identified more ultrashort (8-residues, unmodified, non-cyclic) PFPs than previously known. We anticipate our findings and methodology will be useful in discovering new pore-forming and membrane-disrupting peptides for a range of applications from nanoreactors to therapeutics.
Introduction
Membrane active peptides have a wide range of applications from investigating chemical and physical phenomena,1 and single-molecule sensing2,3 to being leading candidates against the increasing prevalence of antibiotic-resistant bacteria that pose a major threat to global health.4–8 The utility of lipid-peptide interactions in nature arises from the ability to provide channels through ordinarily impermeable membranes to allow the flow of ions, water, and biologically relevant small molecules.An archetypical example of a membrane protein nanopore is provided by alpha-hemolysin (α-HL), which is produced by the bacterium Staphylococcus aureus. α-HL is produced as a monomer (33.2 kDa) that self-assembles into a heptameric pore when inserted into a membrane.1 Insertion of α-HL into many cell membranes is profoundly damaging due to the swift passage of water, K+ ions, ATP, and from small molecules to those as large as 4 kDa through the newly formed channel.9 This transmembrane leakage results in osmotic swelling and cell death through rupture of the membrane. The broad natural scope of pore-forming biomolecules includes functions as cell receptors, molecular transporters, and ion channels for cell regulation.
Structurally, transmembrane channels possess certain characteristic features, which allow for their high stability as oligomers formed in membranes. Membranes are usually constructed of a lipid bilayer formed from phospholipids, sphingolipids, or glycolipids. Hence, nanopores require a variation of hydrophobic and hydrophilic surface functionality for interaction with the amphiphilic membrane constituents and insertion into the lipid layer.10
De novo or consensus design of membrane-spanning peptides has been employed, but these approaches often rely on knowledge of known PFPs.7,10–17 Our approach differs from those used previously as it does not use pre-existing databases of known PFPs. Rather, the active machine learning algorithm predicts the pore-forming ability of all octapeptides and then benchmarks the quality of these predictions using molecular dynamics simulations on the top ten predicted sequences. The complete sequence space of 25.6 billion octapeptides is explored in our approach via iterative feedback cycles of simulation and prediction.
PFPs are a diverse group of peptides that can create pores in the cell membrane of microorganisms, leading to their destruction. Naturally occurring pore-forming peptides (PFPs) are typically short (10 to 50 residues),18 cationic (+2 to +9),14,18 and amphiphilic.13 PFPs usually take one of four 3D structures: α-helical such as melittin19 (Apis mellifera) and temporin-SHf20 (a C-terminus modified octapeptide); β-stranded such as human α-defensin;15 αβ which contain both α and β regions and “other” such as the 13-residue peptide indolicidin, which is active against pathogenic bacteria, fungi and HIV.21 Typically, PFPs disrupt the membrane of a cell or viral envelope via an initial surface aggregation step that induces structural changes and/or assembly of the peptides. With increasing surface concentration, the peptides aggregate further and shift from parallel conformations to transmembrane conformations (barrel stave mechanism).22 Alternatively, peptides may assemble hydrophobically but leave hydrophilic regions exposed to solution such that they can sink through the bilayer (sinking raft mechanism).23 Toroidal pores differ from these two mechanisms as they are formed by the re-arrangement of hydrophilic phospholipid head groups into the centre of the pore. This re-arrangement is facilitated by attraction to polar or cationic peptide residues in the centre of the pore, this provides a mechanism by which less hydrophobic peptides such as magainin II24 and melittin22 can act as PFPs. Xu et al.25 showed that pores are stabilised by the reduced diffusion coefficient of phospholipid molecules associated with the pore complex. The fourth model of membrane-disruption is the carpet model. In this model, the peptides act as a detergent and destroy the bilayer by breaking it down into micelles and other phospholipid–peptide complexes, which typically requires a much higher concentration of peptides than the other models.26
Access to a greater array of robust channel-forming peptides has long been desired for both therapeutic and analytical purposes.4 Previous studies looking at designing PFPs have tended to focus on larger peptides that form pores via the assembly of a small number of monomers. For instance Vorobieva et al. used Rossetta protein structure predictions to design an octamer β-barrel that aggregates on the surface of the bilayer in an unfolded form that span the bilayer and folds into transmembrane β-barrel.27 Other existing approaches that utilize genetic algorithms and machine learning also rely on known PFPs as a starting point for their optimization approach.7,10–16,28–32 Recently, Woolfson and co-workers33 reported the de novo design of a water-soluble 30 amino acid peptide that formed membrane-spanning α-helical peptide barrels. Whilst transmembrane peptide channels have been designed, the successful designs tend to be at the longer end of the sequence length range14,33–37 (i.e., commensurate with the thickness of the bilayer) and accordingly require comprehensive synthetic and purification procedures.
Herein we provide a method for searching the octapeptide chemical space for peptides that change the bilayer morphology such as inducing curvature or perpendicular pressure. The extent of morphological change is assessed using the area per lipid (APL), these scores are then fed into an active learning cycle. The model is only trained on data that it itself selects. This cycle is then iterated until high-scoring peptides are identified by the active machine learning model (Fig. 1). This is achieved by using the extreme gradient boosting tree-based learning algorithm (XGB)38 to score each of the sequences. The area per lipid (APL) of the ten top-scoring peptides was then calculated using coarse grain molecular dynamics simulations (details of the selection procedure are provided in the ESI†). An active learning approach was employed in which the sequences selected for simulations were self-directed via an iterative feedback cycle (Fig. 1). Such an approach was chosen due to the computational expense of the simulations and the size of the chemical space (25.6 billion possible octapeptide sequences). Hence, the active learning approach helps to generate a dataset with more informative training points than the alternative of randomly or uniformly sampling of the vast sequence space, which likely contains a very large majority of non-pore forming peptides. In this work we specifically focus on discovering shorter peptides capable of assembling into membrane-spanning pores. To allow an unbiased search of this sequence space we did not place any constraints on the peptide sequence other than the length. We targeted octapeptides for this study since they would be easy to synthesize, but large enough to facilitate self-assembly into membrane-spanning superstructures. Meanwhile, the 25.6 billion octapeptide sequence space provides an excellent opportunity for demonstrating the ability of machine learning to accelerate sequence design.
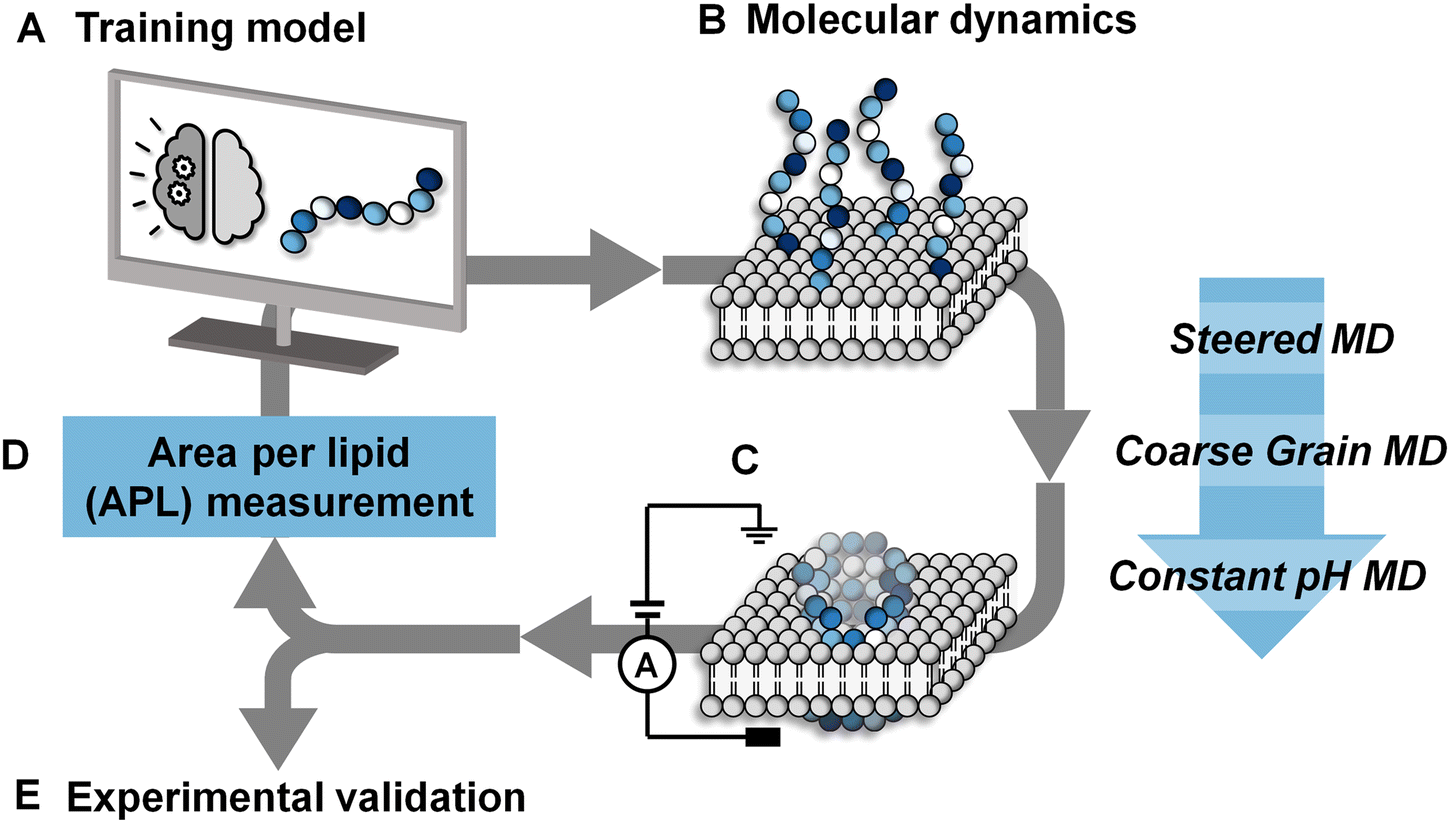 | ||
| Fig. 1 Active machine learning for discovery of pore-forming octapeptides (A). A total sequence space of 208 octapeptides was generated. Full details of the active machine learning method are available in the ESI.† (B) 10 octapeptide sequences selected by the training model were subjected to three-stage molecular dynamics simulations to determine their ability to penetrate a phosphatidylcholine/1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPC/POPS) membrane contained within a water box. Details of the system setup and simulation details are available in the Methods section. (C) The pore-forming ability of each peptide was scored based on the average area per lipid in the final simulation frame across duplicate runs. (D) Area per lipid scores were fed back into the training model for the selected sequences. (E) The pore-forming ability of a selection of high scoring peptides after 7 iterative cycles were tested experimentally in planar lipid bilayers. | ||
Methods
Molecular dynamics and constant pH
The molecular dynamics simulations for pore formation are inherently challenging as this requires the disruption of a stable bilayer and transition of the peptide molecules from a hydrophilic to a hydrophobic environment. To achieve this, we first used a steered molecular dynamics simulation to bring 80 copies of each peptide sequence into contact with the surface of the bilayer. A POPC/POPS 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 bilayer was used to further accelerate the binding of peptides to the surface of the bilayer by electrostatic interactions as has been used in previous computational works.22,39,40 Following this, the steering bias on the peptides was released and the system allowed to equilibrate so that any peptides that did not interact favourably with the surface can escape back into the surrounding aqueous medium. The equilibration was then followed by a constant pH molecular dynamics (CpHMD) simulation,41,42 which was necessary to account for the significant change in the dielectric environment that the peptide experiences when moving from the solvent to the interior of the bilayer. We also tested neutralized peptides with MD rather as opposed to CpHMD, however we found that method to be overly permissive in allowing peptides to enter the bilayer (peptide 3 formed a pore using that method while CpHMD and experimental studies confirmed it should not). At the completion of this simulation the area per lipid (APL) score for the lipid bilayer was calculated, compared and fed back into the active machine learning algorithm, which is retrained using the new data. The use of APL is based on the proposal that membrane thinning precedes pore formation.43 Hence, membrane thinning and the associated increase in APL should predict the pore-forming ability of a peptide. Other measurements such as bilayer surface area were tested and were equally indicative of pore-formation (ESI,† Fig. S17–S20). The train-predict-test loop was then repeated until the APL score converged. CGMD simulations were performed using the GROMACS44 software package and CpHMD simulations were performed using NAMD.45 The phospholipid bilayers were built using INSANE.46 All simulations were performed using the MARTINI (v2.1) forcefield47,48 with helical secondary structure for the peptides. Full details of the workflow, comparison of machine learning models and the simulations are available in the ESI.† APL values were calculated for each system as the multiple for the X and Y dimensions divided by the number of lipids per leaflet (η/2).
:20 bilayer was used to further accelerate the binding of peptides to the surface of the bilayer by electrostatic interactions as has been used in previous computational works.22,39,40 Following this, the steering bias on the peptides was released and the system allowed to equilibrate so that any peptides that did not interact favourably with the surface can escape back into the surrounding aqueous medium. The equilibration was then followed by a constant pH molecular dynamics (CpHMD) simulation,41,42 which was necessary to account for the significant change in the dielectric environment that the peptide experiences when moving from the solvent to the interior of the bilayer. We also tested neutralized peptides with MD rather as opposed to CpHMD, however we found that method to be overly permissive in allowing peptides to enter the bilayer (peptide 3 formed a pore using that method while CpHMD and experimental studies confirmed it should not). At the completion of this simulation the area per lipid (APL) score for the lipid bilayer was calculated, compared and fed back into the active machine learning algorithm, which is retrained using the new data. The use of APL is based on the proposal that membrane thinning precedes pore formation.43 Hence, membrane thinning and the associated increase in APL should predict the pore-forming ability of a peptide. Other measurements such as bilayer surface area were tested and were equally indicative of pore-formation (ESI,† Fig. S17–S20). The train-predict-test loop was then repeated until the APL score converged. CGMD simulations were performed using the GROMACS44 software package and CpHMD simulations were performed using NAMD.45 The phospholipid bilayers were built using INSANE.46 All simulations were performed using the MARTINI (v2.1) forcefield47,48 with helical secondary structure for the peptides. Full details of the workflow, comparison of machine learning models and the simulations are available in the ESI.† APL values were calculated for each system as the multiple for the X and Y dimensions divided by the number of lipids per leaflet (η/2).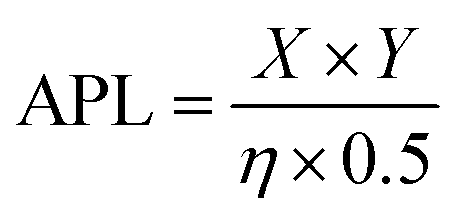
Solid-phase peptide synthesis
Peptides were synthesized on a Wang-linker functionalized polystyrene resin (200 mg, 0.9 mmol g−1, 35–100 mesh) using standard Fmoc chemistry. N-Fmoc-amino acid (3 equiv.) and Oxyma (3 equiv.) were dissolved in N,N-dimethyl formamide (DMF) (0.1 M) and the solution was stirred for 10 min. N,N′-Diisopropylcarbodiimide (DIC)(3 equiv.) was added and the solution was stirred for 3 min. This solution was added to the resin (pre-swollen in dichloromethane, DCM) and the mixture was stirred for 40 min at 50 °C. The solution was drained, and the resin was washed with DCM (3 × 10 mL), DMF (3 × 10 mL), and MeOH (3 × 10 mL). For the attachment of the first amino acid to the Wang-linker, 4-dimethylaminopyridine (DMAP) (0. 1 equiv.) was added to the coupling reaction. 20% of piperidine in DMF (2.5 mL) was added to the resin and the mixture was shaken (2 × 15 min) at room temperature. The solution was drained, and the resin was washed with DCM (3 × 10 mL), DMF (3 × 10 mL), and MeOH (3 × 10 mL). Trifluoroacetic acid/H2O/phenol/thioanisole/1,2-ethanedithiol (89.5:3:3:3:1.5, 1 mL per 100 mg resin) were added to the resin (pre-swollen in DCM) and the mixture was shaken for 3 h at rt. The solution was filtered into ice-cold Et2O (50 mL) and the precipitate collected by centrifugation (15 min at ∼7200 rpm). The crude peptides were purified by semi-preparative RP-HPLC using an Agilent 1100 system (detection at 220 nm) with a Zorbax Eclipse XDB-C18 RP column (250 × 9.4 mm, 5 μm), with a flow rate of 2 mL min−1, eluting with a gradient of H2O and acetonitrile (from 5/95 to 95/5) over 20 min, followed by a 5-min isocratic elution. The lyophilized peptides were characterized by analytical RP-HPLC (Agilent 1100 modular HPLC system (detection at 220 nm) with a Phenomenex Kinetex® 5 μm XB-C18 100 Å column (5 cm × 4.6 mm) with a flow rate of 1 mL min−1, eluting with a gradient of H2O and acetonitrile (from 5/95 to 95/5) over 6 min, followed by a 3 min isocratic elution). The peptides 1 and 3–13 were characterized by matrix-assisted laser desorption/ionisation–time of flight mass spectrometry (Bruker UltrafleXtreme MALDI TOF/TOF mass spectrometer) using α-cyano-4-hydroxycinnamic acid as a matrix. The instrument was calibrated by the ‘nearest neighbour’ method, using Bruker peptide calibration standard II as reference masses. Peptide 2 was characterized using electrospray ionization mass spectrometry (ESI-MS) on an Agilent Technologies LC/MSD quadrupole 1100 series mass spectrometer.
Planar lipid bilayer recordings
Planar lipid bilayer recordings were performed in a custom Teflon cell equipped with two 1 mL compartments separated by a 20 μm thick Teflon film (Goodfellow) with an ∼100 μm diameter aperture (ESI,† Fig. S1). A hanging drop of hexadecane in n-pentane (5 μL, 10%, v/v) was touched on each side of the Teflon sheet containing an aperture and allowed to dry for 1 min. KCl/MOPS buffer (600 μL) was added to the well on each side of the aperture. POPC lipid (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) (approximately 8 μL, 5 μg μL−1 in n-pentane) was added to each side of the well and left for ∼5 min to allow the pentane to evaporate. The cell was subsequently placed into a Faraday cage, and Ag/AgCl electrodes (Warner) connected to a patch clamp amplifier (Axopatch 200B, molecular devices) were suspended either side of the Teflon sheet. The buffer solution on both sides of the Teflon sheet was aspirated and dispensed using a Hamilton syringe to paint a phospholipid bilayer across the aperture. A ±1 mV pulse was applied at 1333 Hz to determine when a bilayer was obtained (capacitance >40 pF). The membrane was characterized with successive 2 s sweeps under an applied potential ranging from +100 to −100 mV. The membrane seal was deemed acceptable if the range of current flow across the membrane measured <1.5 pA. Under an applied voltage (+10 mV), a solution of peptide (10 μL, 50 μM, final concentration ∼0.8 μM) was added to the trans well of a membrane-containing system, subsequent aliquots were added in the same fashion. Signals were digitized using a molecular devices digidata 1322A digitizer and recorded using the pCLAMP 10.4 software. Data were analysed and plotted using Clampfit 10.6. POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and DPhPC (1,2-diphytanoyl-sn-glycero-3-phosphocholine) lipids were obtained as powders from Avanti polar lipids and used without further purification. All other reagents and buffer components were purchased from Sigma Aldrich, UK.Machine learning
Implementations of the machine learning algorithms were accessed via the scikit-learn (version 1.0.1)49 and extreme gradient boosting (XGBoost, version 1.5.1)50 Python modules. XGBoost was used, after hyperparameter optimization, with a learning rate of 0.3, root mean squared error was used as the evaluation metric. A maximum tree depth of 2 was used with a maximum of 10 epochs and a squared error training loss. When training on the on-the-fly more expensive dataset, after hyperparameter optimization, the extra trees regressor (ETR) model was trained to fit 100 trees with a maximum depth of 5 and the minimum number of samples at a leaf node of 1 and a maximum of 10 leaf nodes per tree. Squared error was used as the training criterion. When training on the Judred dataset, after hyperparameter optimization, the maximum tree depth was set to 20, with a minimum of 2 samples per leaf, a maximum depth of 20 and the training criterion was the absolute error.Hyperparameter optimization
The sample dataset of 200 octapeptide simulations was run in triplicate and used to fit each model's hyperparameters. Randomized search with 5-fold cross-validation was used to scan a wide range of hyperparameter combinations of each model. The results of each model's predictions made for a separate validation set can be found in the ESI,† Table S3.Machine learning model selection
The models were chosen based on the score of each machine learning regression model (RMSE & r2), trained on the random set run in triplicate, against a previously unseen high APL validation set of 200 systems run in duplicate and reduced to only those systems with above average APL, see ESI,† Table S3. This was done to prioritize the accurate prediction of the best performers (highest APL) at the expense of accurately predicting how bad an octapeptide performs (low APL).Results and discussion
Experimental validation of machine learning selected peptide sequences
After 7 iterations of the active machine learning protocol, the APL score of the top-scoring peptides began to stabilize. We therefore selected a range of PFPs based on their predicted APL and sequence diversity to test experimentally, along with a series of positive and negative control sequences (Fig. 2). The Phe-rich Temporin-SHf peptide 1 (FFFLSRIF) was chosen as a positive control in experiments due to its previously documented activity against dimyristoyl-based lipids.20 Temporin-SHf was predicted by the ML algorithms to have a high APL score, but not selected out of the millions of high APL predictions by the active learning model in any iteration. Peptide 2 (GSGTGSGT) was chosen as a negative control peptide, and peptide 3 (CFTYFFRV) was also tested as a negative control as it was predicted to be inactive by the algorithm despite the characteristic cationic, amphiphilic nature of the sequence with one small polar residue. Peptide sequences 4–11 were all selected by the active machine learning algorithm and visually confirmed to cause membrane-disruption in the molecular dynamics simulations. These peptides were selected due to their varying chemistry to maximize the probability of discovering a hit in the planar lipid bilayer experiments.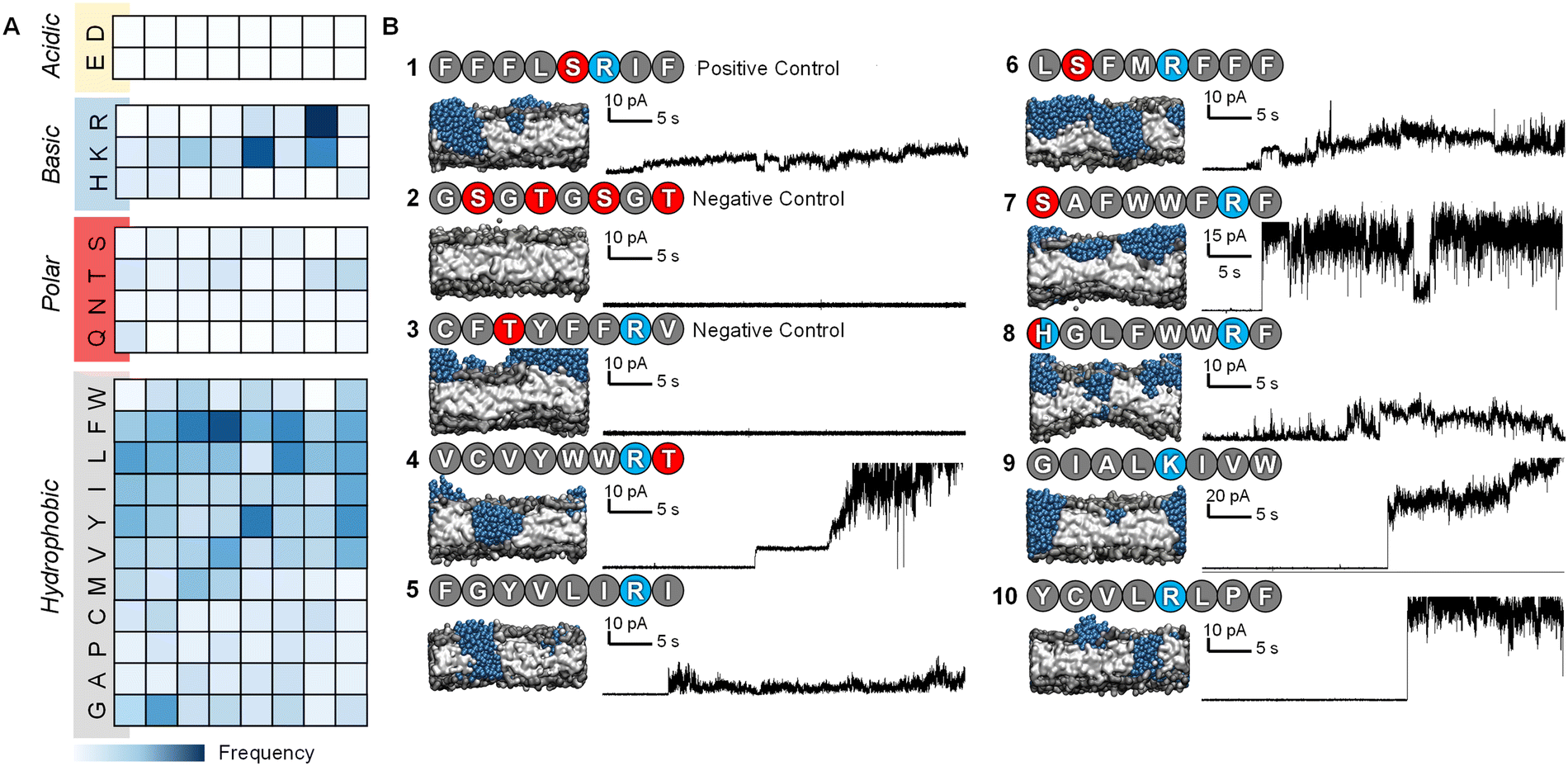 | ||
| Fig. 2 Characteristics and experimental validation of hit peptides. (A) Probability heatmap of amino acid residues, categorized by residue characteristic, and position in octapeptide of hit peptides selected by the active machine learning cycle (ESI,† Table S4). (B) Planar lipid bilayer ion current recordings of octapeptides 1–3 (positive and negative controls) and sequences selected by the active machine learning algorithm 4–10. Each peptide was added (10 μL of 50 μM, final concentration ∼0.8 μM) into a buffer-containing well (1 M KCl, 10 mM MOPS, pH 7.4) on the side of the POPC planar lipid bilayer containing the positive electrode. A potential difference +10 mV was applied, and the resulting ion currents shown were recorded (2 kHz lowpass Bessel filter). Snapshots of the final frame from the molecular dynamics’ simulations of peptide-membrane interactions; peptides are represented as blue beads with the bilayer in grey, and water beads are omitted for clarity. | ||
The selected peptides were obtained as lyophilized solids with >90% purity following solid-phase peptide synthesis and HPLC purification (ESI†). The ability of the peptides to form pores in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers was assessed using ion currents recorded using a patch-clamp amplifier (ESI†).33,51,52
POPC lipid membranes exhibited disruption and leakage upon addition of 1 as determined by the observation of current flow through the membrane (0.8 μM peptide conc. in well, Fig. 2B). Subsequent addition of further aliquots of the positive control led to complete destruction of the membrane (peptide conc. in well ∼3 μM, ESI,† Fig. S4). Reassuringly, the lipid membrane remained experimentally unperturbed for over an hour at +10 mV following the addition of the negative control peptide 2, even at 10-times the concentration used for the positive control peptide (to a final conc. of 8 μM in the well added over 5 minutes). The second negative control peptide 3 that was predicted not to form channels by the MD simulations was also confirmed experimentally (Fig. 2B).
Peptides 4–10 were all experimentally observed to cause membrane-disruption as indicated by significant current flow upon the addition of peptide-containing solutions. In some cases, such as that of peptide 4 (VCVYWWRT), stable discrete current levels were observed, which supports the hypothesis of stable channel formation. However, the appearance of apparent discrete channels was generally observed prior to major membrane-disruption and eventual bursting of the bilayer even under relatively low applied potential difference (10–50 mV, see ESI,† Fig. S5). Whilst the ion traces indicate that the channels are transient and stochastic, it is remarkable that all selected sequences demonstrated disruptive potential against stable POPC bilayers.
Efficacy of active machine learning derived peptide design principles
The characteristics of the 71 hit sequences output by the active machine learning model (ESI,† Table S4) were analysed to see whether this provided insight into the design principles favouring pore-formation.Model A (see ESI,† Table S2 for models tested), repeatedly selected octapeptides with a YYYY motif, which despite producing relatively high APL scores, did not form pores in the simulations, except in one case. Models B and C yielded a far more varied selection of amino acids and motifs. Most of the short peptide sequences identified through our active machine learning protocol have a net charge of +1, which contradicts previous rules defined for longer peptides. However, since the aim is to identify short sequences, the overall positive charge per amino acid is still relatively high. For example, an octapeptide with a +1 charge has a charge per amino acid of +0.125. In comparison, melittin has five positively charged residues with a charge per amino acid of +0.19, LL-37 has a charge per amino acid of +0.16, and magainin-II has a charge per amino acid of +0.13. Based on these observations, a PFP should have a charge per amino acid of approximately +0.1 to +0.2.
Previous work has investigated specific sequences and demonstrated the different role that specific amino acids can have on the ability of peptides to form pores. For example, Cutrona, et al.53 demonstrated that arginine improves membrane translocation relative to lysine and MacCallum, et al.54 have provided residue-specific membrane interaction scales that report the relative affinity of peptide side chain mimicking small molecules for different portions of the bilayer. The analysis of the sequences obtained through the active machine learning protocol show that the four amino acids with the highest relative affinity for the bilayer core (I, V, L, & F) also contribute the most to area per lipid (Fig. 2A).55 Additionally, we observed that anionic residues (D/E), followed by polar uncharged residues (N, Q, S, T & H), have the least effect on APL due to their lower affinity for the bilayer.
The values for the amino acid positional probabilities within the octapeptide were calculated (Fig. 2A) over all systems studied, including both the random selection and active machine learning selections. These probability distributions suggested that the positioning of specific residues within the hit sequences may also be important. It is worth noting that a residue at a terminal position does not necessarily indicate its presence within the head group region of the bilayer, as multiple octapeptides are required to bridge the bilayer due to their short length. Our analysis suggests that cationic residues contribute more to pore-forming ability when positioned in the latter half of the octapeptide, closer to the C-terminus. However, it must be noted that these design rules are very generic and peptides such as peptide 3, which seem like perfect candidates based on these rules, may not be active. The specific order of the amino acids is also a very important characteristic, as shown in Fig. 3, two isomers of peptide 11 were tested and found not to produce stable pores.
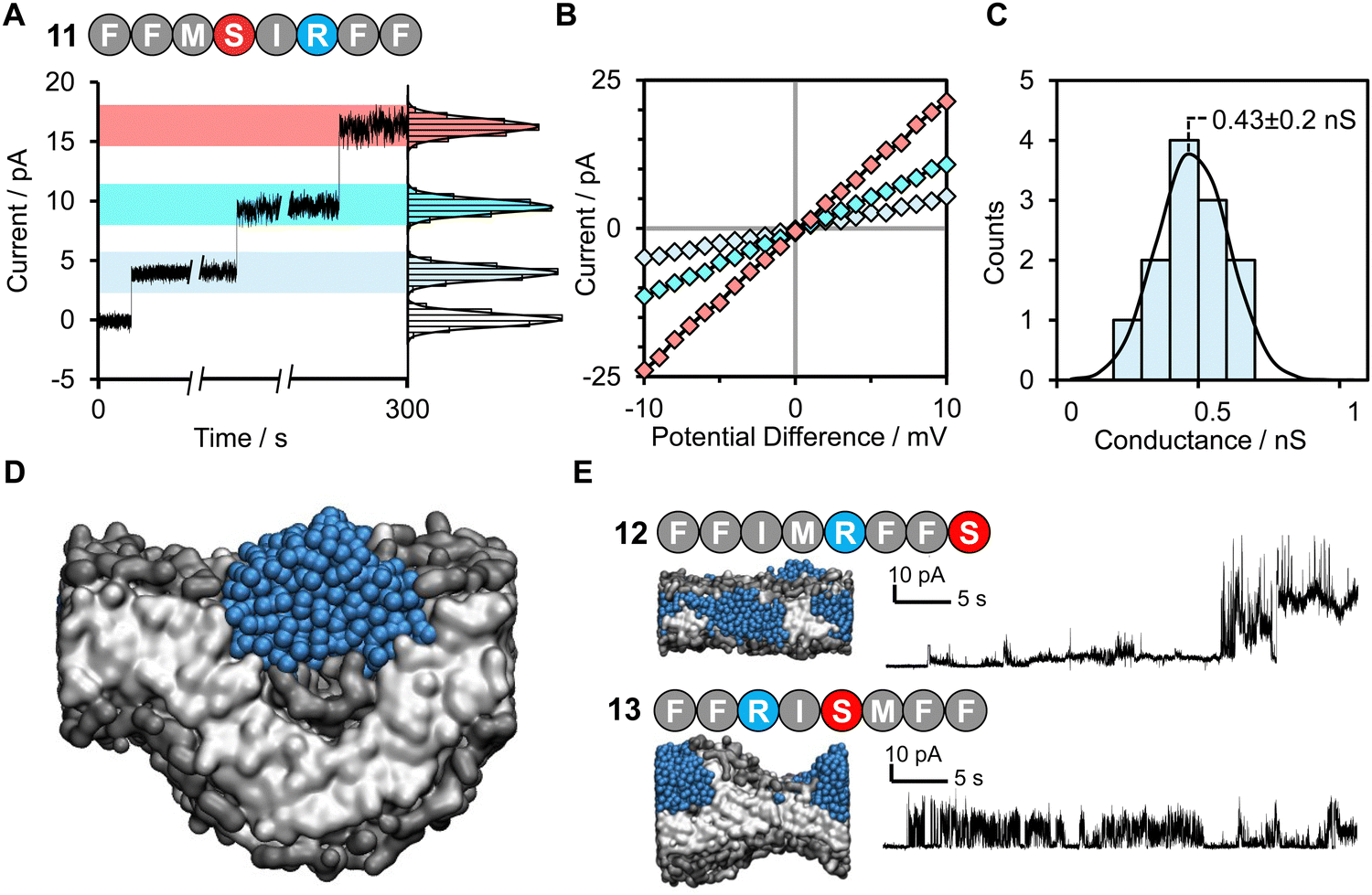 | ||
| Fig. 3 Discrete channel formation of active machine learning hit peptide 11 FFMSRIFF. (A) Discrete channel formation following addition of peptide 11 FFMSRIFF (10 μL, 50 μM, final concentration ∼0.8 μM) to the well using 1 M KCl 30 mM MOPS pH 7.4 buffer under +10 mV applied potential difference. Entries of multiple channels or enlargement of a single channel observed as indicated by instantaneous increases in current. Histograms show distribution of residual current at each level. (B) I/V sweep characterization of different channels from –10 mV to +10 mV. (C) Histogram analysis of open-channel conductance observed following the addition of peptide 11 at +10 mV in 1 M KCl, 10 mM MOPS, pH 7.4, determined from 12 individual insertions from the zero-current, unperturbed membrane, Fig. S13. This histogram has a normal distribution with a mean channel conductance of 0.43 nS. (D) Coarse grained molecular dynamics simulation visualization of 11 causing bulbous membrane-disruption. (E) Octapeptides 12 and 13 are scrambled/reversed sequences of peptide 11. | ||
Remarkably, peptide 11 was found to form discrete transmembrane channels, as indicated by the stepwise ∼4 pA increases in the current shown in Fig. 3A. Histogram analysis revealed the conductance of the three steps corresponded to 0.4, 1, and 1.7 nS (by comparison, α-hemolysin has a conductance of 1 nS). This suggests that either three different sizes of pores were inserted or that a single pore was changing its size. However, histogram analysis of the current change upon initial channel formation from an intact membrane had a normal distribution across a relatively narrow conductance range (Fig. 3C). This may point towards subsequent current increases being attributed to a single pore increasing in size during a sinking raft mechanism, as suggested by the MD simulation (Fig. 3D). The current/voltage response of these channels (Fig. 3B) was linear and symmetrical in the positive and negative regions (+10 to –10 mV), which indicates a lack of ion selectivity and a randomized orientation of the peptides in the bilayer. Since the octapeptides are too short (∼15 Å) to span the bilayer (∼25–30 Å) and were only added on one side of the bilayer, this indicates that the peptides must be able to diffuse between both leaves of the membrane. This diffusion requirement may also explain why largely apolar sequences with a charge per residue of +0.1 to +0.2 are favoured for channel formation. Continued exposure of the lipid bilayer to 1.6 μM peptide 11 led to rupture of the membrane within 5 minutes, which could not be reformed. To our knowledge this is the first observation of discrete single-channels formed from an 8-residue peptide. Similar behaviour was observed in 1 M KCl, NaCl and CsCl albeit with decreased channel stability (ESI,† Fig. S6). Channel formation was also observed in 1,2-diphytanoyl-sn-glycero-3-phosphocholine bilayers (DPhPC, ESI,† Fig. S7), which is frequently used in single-channel detection methods.51
Both peptide 11 and the positive control peptide 1 contain R in position 6, which is not particularly favoured in the probability heat map shown in Fig. 2A, yet channel-forming ability was confirmed in the experiments and MD simulations (Fig. 2B and 3). Hence, we synthesized two sequence-scrabbled variants of peptide 11 and performed retrospective MD modelling on them. The MD simulations correctly predicted that both the scrambled (12) and reversed (13) peptides possessed membrane-disrupting capacity but lacked the remarkable pore-forming ability of the parent peptide 11 to form discrete stepwise channels (Fig. 3E).
Conclusions
Here we present the discovery of nine (4–10, 12,13) membrane-disrupting octapeptides and the first ultra short unmodified pore-forming peptide known to be capable of producing discrete membrane channels. These peptides were designed by an active machine learning algorithm that targeted membrane perturbation. Experimental validation confirmed that our approach successfully predicted active and inactive sequences, even where the sequences shared similar physiochemical features. This approach differs from those previously developed to identify pore-forming peptides, and the results show that these peptides can be identified through an active machine learning approach. The approach does not rely on prior knowledge of pore-forming peptides and can explore the complete sequence space. Our work challenges the thinking that it is not worth exploring the pore-forming abilities of peptides that are too short to span the bilayer or possess significant secondary/tertiary structures. Based on an analysis of the sequences identified, we propose that the charge per residue rather than overall charge is a crucial component in membrane-disrupting behaviour;14,33–37 active PFPs possess a charge per amino acid of between +0.1 and +0.2, and this charge tends to be located towards the C-terminus, but not at the C-terminal amino acid. Acidic residues were entirely absent from those peptides predicted to be pore-forming, while they were present in peptides predicted to be non-pore forming (Table S5, ESI†). Moreover, only ∼1 polar amino acid was tolerated in the channel-forming peptides identified. This work provides a starting point for the discovery of novel PFPs with improved antimicrobial activity and selectivity, and the approach may have broad implications for the discovery of other bioactive peptides and pore-forming agents with a range of applications from nanoreactors to therapeutics. While the methods employed here reliably predicted pore-forming ability, predicting pore quality and stability will provide a challenge that will drive the development of future approaches.Author contributions
A. V. T designed the machine learning and simulation methodology and performed the analyses of computational data. L. H. synthesized and characterized the peptides. D. E. performed the PLB experiments and characterized the pores formed. T. T. conceived this research and S. L. C., A. L. and T. T. supervised the work. A. V. T. and T. T. wrote the manuscript to which all authors contributed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Results were obtained using the EPSRC-funded ARCHIE-WeSt High-Performance Computer (https://www.archie-west.ac.uk; EPSRC grant no. EP/K000586/1). We are also grateful of funding from the Leverhulme Trust (Philip Leverhulme Prize) and BBSRC grant no. BB/M010996/1.References
- M. M. Haugland, S. Borsley, D. F. Cairns-Gibson, A. Elmi and S. L. Cockroft, ACS Nano, 2019, 13, 4101–4110 CrossRef CAS PubMed.
- S. Fujita, I. Kawamura and R. Kawano, ACS Nano, 2023, 17, 3358–3367 CrossRef CAS PubMed.
- Y.-L. Ying, Z.-L. Hu, S. Zhang, Y. Qing, A. Fragasso, G. Maglia, A. Meller, H. Bayley, C. Dekker and Y.-T. Long, Nat. Nanotechnol., 2022, 17, 1136–1146 CrossRef CAS PubMed.
- B. P. Lazzaro, M. Zasloff and J. Rolff, Science, 2020, 368, eaau5480 CrossRef CAS PubMed.
- G. Liu, D. B. Catacutan, K. Rathod, K. Swanson, W. Jin, J. C. Mohammed, A. Chiappino-Pepe, S. A. Syed, M. Fragis, K. Rachwalski, J. Magolan, M. G. Surette, B. K. Coombes, T. Jaakkola, R. Barzilay, J. J. Collins and J. M. Stokes, Nat. Chem. Biol., 2023, 19, 1342–1350 CrossRef CAS PubMed.
- H. W. Huang, Biochemistry, 2000, 39, 8347–8352 CrossRef CAS PubMed.
- M. Yoshida, T. Hinkley, S. Tsuda, Y. M. Abul-Haija, R. T. McBurney, V. Kulikov, J. S. Mathieson, S. Galiñanes Reyes, M. D. Castro and L. Cronin, Chem, 2018, 4, 533–543 CAS.
- A. Luther, M. Urfer, M. Zahn, M. Muller, S. Y. Wang, M. Mondal, A. Vitale, J. B. Hartmann, T. Sharpe, F. L. Monte, H. Kocherla, E. Cline, G. Pessi, P. Rath, S. M. Modaresi, P. Chiquet, S. Stiegeler, C. Verbree, T. Remus, M. Schmitt, C. Kolopp, M. A. Westwood, N. Desjonqueres, E. Brabet, S. Hell, K. LePoupon, A. Vermeulen, R. Jaisson, V. Rithie, G. Upert, A. Lederer, P. Zbinden, A. Wach, K. Moehle, K. Zerbe, H. H. Locher, F. Bernardini, G. E. Dale, L. Eberl, B. Wollscheid, S. Hiller, J. A. Robinson and D. Obrecht, Nature, 2019, 576, 452–458 CrossRef CAS PubMed.
- H. Ostolaza, D. González-Bullón, K. B. Uribe, C. Martín, J. Amuategi and X. Fernandez-Martínez, Toxins, 2019, 11, 354 CrossRef CAS PubMed.
- S. Howorka, Nat. Nanotechnol., 2017, 12, 619–630 CrossRef CAS PubMed.
- K. Shimizu, B. Mijiddorj, M. Usami, I. Mizoguchi, S. Yoshida, S. Akayama, Y. Hamada, A. Ohyama, K. Usui, I. Kawamura and R. Kawano, Nat. Nanotechnol., 2022, 17, 67–75 CrossRef CAS PubMed.
- E. Zakharova, M. Orsi, A. Capecchi and J. L. Reymond, ChemMedChem, 2022, 17, e202200291 CrossRef CAS PubMed.
- E. Y. Lee, B. M. Fulan, G. C. Wong and A. L. Ferguson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13588–13593 CrossRef CAS PubMed.
- C. D. Fjell, J. A. Hiss, R. E. W. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2012, 11, 37–51 CrossRef CAS PubMed.
- Y. Fan, X. D. Li, P. P. He, X. X. Hu, K. Zhang, J. Q. Fan, P. P. Yang, H. Y. Zheng, W. Tian, Z. M. Chen, L. Ji, H. Wang and L. Wang, Sci. Adv., 2020, 6, eaaz4767 CrossRef CAS PubMed.
- S. André, S. K. Washington, E. Darby, M. M. Vega, A. D. Filip, N. S. Ash, K. A. Muzikar, C. Piesse, T. Foulon, D. J. O’Leary and A. Ladram, ACS Chem. Biol., 2015, 10, 2257–2266 CrossRef PubMed.
- G. Bhardwaj, J. O’Connor, S. Rettie, Y. H. Huang, T. A. Ramelot, V. K. Mulligan, G. G. Alpkilic, J. Palmer, A. K. Bera, M. J. Bick, M. Di Piazza, X. Li, P. Hosseinzadeh, T. W. Craven, R. Tejero, A. Lauko, R. Choi, C. Glynn, L. Dong, R. Griffin, W. C. van Voorhis, J. Rodriguez, L. Stewart, G. T. Montelione, D. Craik and D. Baker, Cell, 2022, 185, 3520–3532 CrossRef CAS PubMed.
- N. Mookherjee, M. A. Anderson, H. P. Haagsman and D. J. Davidson, Nat. Rev. Drug Discovery, 2020, 19, 311–332 CrossRef CAS PubMed.
- H. Gong, M. Liao, X. Hu, K. Fa, S. Phanphak, D. Ciumac, P. Hollowell, K. Shen, L. A. Clifton, M. Campana, J. R. P. Webster, G. Fragneto, T. A. Waigh, A. J. McBain and J. R. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 44420–44432 CrossRef CAS PubMed.
- F. Abbassi, O. Lequin, C. Piesse, N. Goasdoue, T. Foulon, P. Nicolas and A. Ladram, J. Biol. Chem., 2010, 285, 16880–16892 CrossRef CAS PubMed.
- R. E. Hancock and M. G. Scott, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8856–8861 CrossRef CAS PubMed.
- A. Tuerkova, I. Kabelka, T. Kralova, L. Sukenik, S. Pokorna, M. Hof and R. Vacha, eLife, 2020, 9, e47946 CrossRef CAS PubMed.
- P. G. Dougherty, A. Sahni and D. Pei, Chem. Rev., 2019, 119, 10241–10287 CrossRef CAS PubMed.
- S. J. Ludtke, K. He, W. T. Heller, T. A. Harroun, L. Yang and H. W. Huang, Biochemistry, 1996, 35, 13723–13728 CrossRef CAS PubMed.
- C. Xu, K. Yang and B. Yuan, J. Phys. Chem. Lett., 2023, 14, 854–862 CrossRef CAS PubMed.
- Y. Huan, Q. Kong, H. Mou and H. Yi, Front. Microbiol., 2020, 11, 582779 CrossRef PubMed.
- A. A. Vorobieva, P. White, B. Liang, J. E. Horne, A. K. Bera, C. M. Chow, S. Gerben, S. Marx, A. Kang, A. Q. Stiving, S. R. Harvey, D. C. Marx, G. N. Khan, K. G. Fleming, V. H. Wysocki, D. J. Brockwell, L. K. Tamm, S. E. Radford and D. Baker, Science, 2021, 371, eabc8182 CrossRef PubMed.
- J. R. Randall, C. D. DuPai, T. J. Cole, G. Davidson, K. E. Groover, S. L. Slater, D. A. I. Mavridou, C. O. Wilke and B. W. Davies, Sci. Adv., 2023, 9, eade0008 CrossRef PubMed.
- E. Y. Lee, G. C. L. Wong and A. L. Ferguson, Bioorg. Med. Chem., 2018, 26, 2708–2718 CrossRef CAS PubMed.
- H. Fu, Z. Cao, M. Li and S. Wang, BMC Genom., 2020, 21, 597 CrossRef CAS PubMed.
- B. Manavalan, S. Subramaniyam, T. H. Shin, M. O. Kim and G. Lee, J. Proteome Res., 2018, 17, 2715–2726 CrossRef CAS PubMed.
- H. Li and C. Nantasenamat, PeerJ, 2019, 7, e8265 CrossRef PubMed.
- A. J. Scott, A. Niitsu, H. T. Kratochvil, E. J. M. Lang, J. T. Sengel, W. M. Dawson, K. R. Mahendran, M. Mravic, A. R. Thomson, R. L. Brady, L. Liu, A. J. Mulholland, H. Bayley, W. F. Degrado, M. I. Wallace and D. N. Woolfson, Nat. Chem., 2021, 13, 643–650 CrossRef CAS PubMed.
- G. Wang, J. Biol. Chem., 2008, 283, 32637–32643 CrossRef CAS PubMed.
- M. Pirtskhalava, A. A. Amstrong, M. Grigolava, M. Chubinidze, E. Alimbarashvili, B. Vishnepolsky, A. Gabrielian, A. Rosenthal, D. E. Hurt and M. Tartakovsky, Nucleic Acids Res., 2021, 49, D288–D297 CrossRef CAS PubMed.
- C. Aisenbrey, M. Amaro, P. Pospil, M. Hof and B. Bechinger, Sci. Rep., 2020, 10, 1–13 CrossRef PubMed.
- C. H. Chen, C. G. Starr, E. Troendle, G. Wiedman, W. C. Wimley, J. P. Ulmschneider and M. B. Ulmschneider, J. Am. Chem. Soc., 2019, 141, 4839–4848 CrossRef CAS PubMed.
- T. Chen and C. Guestrin, presented in part at the Proc. 22nd ACM SIGKDD Int. Conf. Knowl. Discov. Data Min., aug, 2016.
- S. Ludtke, K. He and H. Huang, Biochemistry, 1995, 34, 16764–16769 CrossRef CAS PubMed.
- J. R. Brender, A. J. McHenry and A. Ramamoorthy, Front. Immunol., 2012, 3, 195 Search PubMed.
- B. K. Radak, C. Chipot, D. Suh, S. Jo, W. Jiang, J. C. Phillips, K. Schulten and B. Roux, J. Chem. Theory Comput., 2017, 13, 5933–5944 CrossRef CAS PubMed.
- A. van Teijlingen, H. W. A. Swanson, K. H. A. Lau and T. Tuttle, J. Phys. Chem. Lett., 2022, 13, 4046–4051 CrossRef CAS PubMed.
- H. W. Huang, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1292–1302 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hnin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kalé, K. Schulten, C. Chipot and E. Tajkhorshid, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- T. A. Wassenaar, H. I. Inglfsson, R. A. Bckmann, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2015, 11, 2144–2155 CrossRef CAS PubMed.
- S. J. Marrink, H. J. Risselada, S. Yefimov and D. P. A. Tieleman, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- L. Monticelli, S. K. Kandasamy, X. Periole, R. G. Larson, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2008, 4, 819–834 CrossRef CAS PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- T. Chen and C. Guestrin, presented in part at the Proc. ACM SIGKDD Int. Conf. Knowl. Discov. Data Min., San Francisco, California, USA, 2016.
- K. R. Mahendran, Building Synthetic Transmembrane Peptide Pores, in Nanopore Technology. Methods in Molecular Biology, ed. M. A. Fahie, Humana, New York, NY, 2021, vol. 2186 Search PubMed.
- D. P. August, S. Borsley, S. L. Cockroft, F. Della Sala, D. A. Leigh and S. J. Webb, J. Am. Chem. Soc., 2020, 142, 18859–18865 CrossRef CAS PubMed.
- K. J. Cutrona, B. A. Kaufman, D. M. Figueroa and D. E. Elmore, FEBS Lett., 2015, 589, 3915–3920 CrossRef CAS PubMed.
- J. L. MacCallum, W. F. Bennett and D. P. Tieleman, Biophys. J., 2008, 94, 3393–3404 CrossRef CAS PubMed.
- V. Kubyshkin, Org. Biomol. Chem., 2021, 19, 7031–7040 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Peptide synthesis with RP-HPLC and MS characterisation, machine learning procedures, experimental details and additional computational and planar lipid bilayer experiment results. See DOI: https://doi.org/10.1039/d4cp01404a |
| This journal is © the Owner Societies 2024 |