
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploration of carvacrol aggregation by laser spectroscopy†
Paúl
Pinillos‡
 ,
Fernando
Torres-Hernández‡
,
Imanol
Usabiaga
,
Pablo
Pinacho
* and
José A.
Fernández
*
,
Fernando
Torres-Hernández‡
,
Imanol
Usabiaga
,
Pablo
Pinacho
* and
José A.
Fernández
*
Department of Physical Chemistry, Faculty of Science and Technology, University of the Basque Country (UPV/EHU), B° Sarriena, S/N, Leioa, 48940, Spain. E-mail: josea.fernandez@ehu.eus; pablo.pinacho@ehu.eus
First published on 9th September 2024
Abstract
Carvacrol is an aromatic monoterpenoid found in thyme oil. Due to its implications for human health, it is important to elucidate its structure and its intramolecular interactions. We have characterised the carvacrol monomer, its complex with water, its dimer, and even its trimer in a supersonic expansion using mass-resolved laser spectroscopy techniques complemented by quantum-chemical computations. The resonance-enhanced multiphoton ionisation spectrum of the monomer features several transitions, which were assigned to the same conformer, confirmed by ion-dip infrared spectroscopy. However, a conclusive assignment of the infrared bands to one of the four conformations of carvacrol remains elusive. The experimental spectra for the monohydrated, the homodimer, and the homotrimer point to the detection of the lowest energy isomer in each case. Their structures are governed by a balance of intramolecular interactions, specifically hydrogen bonding and dispersion forces. Comparison with other similar systems demonstrates that dispersion interactions are key to the stabilisation of the aggregates, being present in all the structures. However, the hydrogen bonding is the dominant force as observed in the lowest-energy conformations.
Introduction
Monoterpenes are a family of naturally occurring compounds with a shared chemical structure, abundant in essential oils of many plants, fruits, vegetables, and herbs.1 While the main application of essential oils is for food seasoning and the perfume industry,2 they have been used in traditional medicine to treat several diseases and alleviate pain.1,2 Notably, monoterpenoids, a subset of monoterpenes, are potential precursors of novel drug compounds with promising properties.1,3 Their versatility and plant-based origin position them as a cheap and sustainable source for new potential medicines.Carvacrol (5-isopropyl-2-methylphenol, C10H14O) is a phenolic monoterpenoid known for its biological activity. As an isomer of thymol (2-isopropyl-5-methylphenol), carvacrol can be found in the essential oil of thyme, oregano, or wild bergamot.4–6 Carvacrol is widely used in the medical and pharmaceutical fields because of its analgesic, antibacterial, and antifungal properties.1,4,7,8 In the last years essential oils rich in carvacrol have shown to exhibit a potent antimicrobial activity, even against bacterial strains resistant to erythromycin.9 Of special relevance is the anticancer activity of carvacrol inhibiting the proliferation of metastatic cell lines, and inducing the apoptosis of cancer cells.10–12
Therefore, carvacrol can become a cheap and renewable source of potential new drugs to control pathogens and diseases.1,9 The effectiveness of carvacrol lies in the establishment of non-covalent interactions (NCI) with the molecular receptors, making it crucial to have a precise description of its inherent structure and how it associates with other molecules. Carvacrol, along with other phenolic monoterpenoids (thymol, eugenol, guaiacol, Scheme S1, ESI†) have hydrophobic and hydrophilic moieties: the aromatic ring itself and a hydroxyl residue (Scheme 1). Each part gives rise preferentially to a type of interaction with the surrounding molecules to form the NCI13–15 that bind the monoterpenoid to the cell receptors. However, the asymmetry of carvacrol and the flexibility of the isopropyl chain complicate the prediction of its behaviour. Also, the conformation of the isopropyl group and the methyl group in the benzene ring can affect the disposition of other molecules to form interactions. For that reason, we conducted a comprehensive analysis of the interplay between functional groups in carvacrol. Such studies nicely complement past work on the aggregation of aromatic alcohols.16–20
 | ||
| Scheme 1 Carvacrol (5-isopropyl-2-methylphenol). | ||
Spectroscopic techniques such as nuclear magnetic resonance, X-ray diffraction, Fourier-transform infrared spectroscopy, microwave spectroscopy, or resonance-enhanced multiphoton ionisation (REMPI), stand out as powerful methods to determine the structure and NCI of biologically relevant molecules in any of the main states of matter (solid, liquid, and gas). Among those, the gas-phase techniques prove to be the most adequate to characterise the inherent structure of the molecules and the intermolecular interactions in their clusters, laying the foundations for a deep understanding of their biological activity.
A previous gas-phase study on carvacrol using microwave spectroscopy reported the detection of four conformers in the conditions of a supersonic expansion.21 The four conformers arise from the different orientations of the hydroxyl and isopropyl groups and are connected by the motion of those groups. Since the microwave spectroscopy study described in detail the structure of carvacrol, our work focuses on the aggregation processes. For doing so, we generated complexes of carvacrol with water and itself and used mass-resolved spectroscopic techniques to obtain physical observables. Comparison between the experimental data and the quantum mechanical predictions obtained at M06-2X/6-311++G(d,p) and B3LYP-D3BJ/def2-TZVP levels enabled us to propose a family of structures for each experimentally detected species. Such structures, served us to explore the balance between the competition and cooperation of hydrophobic and hydrophilic groups in carvacrol. The dimer and the trimer are especially suitable to analyse the intricate interplay between hydrogen bonding and dispersion forces. The relative position of the substituents seems to be crucial for cluster formation, influencing the three-dimensional shape of the aggregates.
Experimental and theoretical procedures
Experimental section
The experiments were performed in a supersonic expansion chamber coupled with an in-house designed time-of-flight mass spectrometer (TOF-MS, R. M. Jordan). The sample was held in a reservoir upstream of the gas line, and heated to 70 °C to transfer it into the gas phase. It was diluted in a mixture of He with 10% of Ar at a backing pressure of around 2.5 bar. The gas was introduced into the TOF vacuum chamber by a pulsed valve (R. M. Jordan), generating a supersonic expansion, which cools the rotational and vibrational degrees of freedom of the molecules and complexes to a few K. The molecular beam was probed by UV dye nanosecond lasers (Q-Scan Dye Laser, Quantel and LiopStar, LIOP-TECH) to collect the REMPI spectra using the equipment described elsewhere.22 One- and two-colour REMPI spectra were collected for carvacrol and its aggregates. However, only the two-colour spectra are reproduced in this work because of their better signal-to-noise ratio (S/N) and resolution. For the ion-dip infrared spectroscopy (IDIRS)23 an IR laser (LaserVision OPO/A, ∼5 mJ per pulse, 6 cm−1 bandwidth) was used. A complete description of the experimental setup can be found elsewhere.22,24Computational section
The spectroscopic information was compared with quantum-chemical computations. The procedure to scan for the possible conformations consisted of several steps as described in previous works.25–27 Initially, a rapid exploration of the potential energy surface (PES) was performed using molecular mechanics.28 Three different force fields (OPLS3e, MMFFs, and AMBER)29–32 integrated into Macromodel from the Schrödinger suite were employed.33 Next, the outcome from the conformational search was evaluated and re-optimised by quantum-chemical computations in the harmonic approximation using Gaussian 16.34 In this work, we optimised the geometries of the carvacrol monomer (carOH), the carvacrol–water complex (carOH⋯w), the carvacrol dimer ((carOH)2), and the carvacrol trimer ((carOH)3) using two levels of theory: M06-2X/6-311++G(d,p)35,36 and B3LYP-D3BJ/def2-TZVP.37–40 The levels of theory chosen, enhanced by the corrections, have demonstrated to provide good results in predicting the geometries and spectra for this type of molecular systems.25,26,41 The results from both levels of theory were compared between them and with the experimental spectra to obtain the best agreement, leading to the determination of the conformations responsible for the spectral signatures. In the final step, the IR spectra were simulated for each candidate geometry. The assignment of the carOH spectrum was straightforward and served as a reference for scaling the frequencies of C–H and O–H stretches for the aggregates. For the B3LYP-D3BJ/def2-TZVP level of theory a scale factor of 0.957 was applied for both CH and OH stretches to account for anharmonicity, while for M06-2X/6-311++G(d,p), factors of 0.946 and 0.932 were used for C–H and O–H, respectively. The simulation of the harmonic spectra was performed representing the predicted frequencies with a Lorentzian function, in which the line width depends on the type and strength of the interaction. The spectra were convoluted with a Gaussian function of a full-width half maximum (FWHM) of 6 cm−1 to account for the broadening induced by the laser's bandwidth.Binding energy (BE) and Gibbs energy were computed at 0 and 298 K. The values reported include zero-point energy (ZPE) and basis set superposition error (BSSE) corrections using the Boys and Bernardi method.42 The NCI surfaces were generated using the Multiwfn software,43 and they were visualised using Chimera.44
Results and discussion
Carvacrol monomer (carOH)
We explored first the results from the conformational search for carOH. Initially, 12 conformations were predicted by the molecular mechanics search. Some redundant geometries were identified and only four different structures remained after the optimisation step with each theory level (Fig. S1, ESI†). The conformational flexibility of carvacrol arises from the orientation of the hydroxyl and isopropyl groups, resulting in the four predicted geometries. In the most stable forms the –OH group is in trans orientation relative to the methyl group, being the only difference in the orientation of the isopropyl group (labelled as A or B, Fig. S1, ESI†). The other two isomers also differ in the orientation of the isopropyl group but with the –OH in a cis configuration. Both levels of theory (B3LYP-D3BJ/def2-TZVP and M06-2X/6-311++G(d,p)) predicted the same energy ordering for the four structures of carOH, with differences within computation accuracy. All the conformers were predicted to be very close in energy (within 1.5 kJ mol−1) at both 0 and 298 K.CarOH exhibited a clean and well-resolved vibronic spectrum, indicating the absence of excited state dynamics that could interfere with the observation (Fig. 1). The red-most band observed at 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 916 cm−1 was tentatively assigned to the
916 cm−1 was tentatively assigned to the  transition (Fig. 1a). The spectrum extended for more than 1000 cm−1, despite that it is well-known that carvacrol reacts after absorption of a UV photon.45 In a recent study, carvacrol was trapped in a cryogenic argon matrix and irradiated with UV light of wavelengths lower than 200 nm. Under such conditions, carvacrol underwent a dissociation of the OH group, followed by recombination, resulting in alkyl-substituted cyclohexadienones.45 In our experimental setup, the observation of transitions for carvacrol for more than 1000 cm−1, between 35800 and 37000 cm−1, seemed to indicate that carvacrol does not experience any reaction or fragmentation process.
transition (Fig. 1a). The spectrum extended for more than 1000 cm−1, despite that it is well-known that carvacrol reacts after absorption of a UV photon.45 In a recent study, carvacrol was trapped in a cryogenic argon matrix and irradiated with UV light of wavelengths lower than 200 nm. Under such conditions, carvacrol underwent a dissociation of the OH group, followed by recombination, resulting in alkyl-substituted cyclohexadienones.45 In our experimental setup, the observation of transitions for carvacrol for more than 1000 cm−1, between 35800 and 37000 cm−1, seemed to indicate that carvacrol does not experience any reaction or fragmentation process.
 | ||
| Fig. 1 (a) One- and two-colour REMPI spectra of carvacrol monomer recorded under four different conditions; (b) zoom into the 36165 cm−1 band showing the two nearby transitions. | ||
The same IDIR spectrum was recorded probing the bands at 36165, 36265, 36883, and 36980 cm−1, suggesting that they either belonged to the same conformation of carvacrol or to very similar conformational isomers. Given the close similarity of the four structures for carOH, the predicted IR spectra were almost identical (Fig. 2 and Fig. S2, ESI†). To check for the presence of higher energy conformers we performed additional experiments, varying the carrier gas. This resulted in a change in the observed intensities, confirming the presence of more than one peak in the spectrum recorded with Ne (Fig. 1b). We performed additional UV/UV and IR/UV hole burning experiments to discern the origin of the observed bands (Fig. S3, ESI†), showing that either all the bands belonged to the same isomer of carvacrol, or that if more than one isomer was present, their spectra would overlap within the spectral resolution of our experimental setup. Based on the predicted Gibbs energies, it seems possible that the four conformers were present in the supersonic expansion and contribute to the experimental spectrum. The observed change in the intensities with different carrier gases points out to the presence of more than one isomer, in line with the microwave spectroscopy study.21 The microwave study also reports the barriers for the conversion between the four conformers by the scan of the isopropyl and the hydroxyl groups. The barrier for the conversion between A and B conformers imply the motion of the isopropyl chain and passes by a potential energy barrier of around 10 kJ mol−1, while the barrier between cis and trans forms is of 15 kJ mol−1. The values of the barrier for both motions are in line with the observation of the four conformers in the supersonic expansion.21
 | ||
| Fig. 2 (a) Experimental (black trace) and simulated (coloured traces) IDIR spectra for (b) the four carOH isomers. The simulations were done using the M06-2X structures using scaling factors of 0.946 and 0.932 for C–H and O–H stretching modes, respectively, to account for anharmonicity. The relative Gibbs free energies (kJ mol−1) are given at the M06-2X/6-311++G(d,p) level of theory at 0 and 298 K. | ||
Carvacrol⋯H2O aggregate (carOH⋯w)
Introduction of one molecule of water substantially complicated the conformational landscape. Both carvacrol and water can serve as either proton-acceptor or donor, and the molecule of water can establish NCI with any part of carvacrol, giving rise to numerous possible structures. The exploration of this conformational panorama allowed us to discern the most favourable interactions. The search procedure yielded 60 potential geometries, which were reduced to 17 distinct conformers in an energy window of 20 kJ mol−1 after the optimisation step. Both levels of theory predict the same structures with different relative energy ordering (Fig. S4, ESI†).The aggregates of carvacrol with water (carOH⋯w) were grouped into families based on the interactions between both molecules (Fig. S4, ESI†). In the most stable complexes, the interaction involves an O–H⋯Ow hydrogen bond with water acting as the proton-acceptor and carvacrol as the donor. A weak C–H⋯Ow dispersion interaction contributes to the stability of those isomers. The presence of the methyl group in the ortho position to the –OH prevents the water molecule from reaching the optimum position for the formation of an O−H⋯OH2 hydrogen bond, and thus, the preferred position for water is opposite to the CH3 residue. The orientation of the substituents in each of the isomers of carvacrol gives rise to a slightly different environment for the water molecules. This results in distinct relative stability of the aggregates (Fig. S4, ESI†). As in the monomer the most stable structures correspond to trans-carOH followed by the cis forms.
The water molecule can also interact with the most hydrophobic side of carvacrol. Those structures feature OH⋯π interactions, or hydrogen bonding with water acting as the proton-donor and carvacrol as the acceptor. As could be expected, those structures are higher in energy than the ones stabilised by hydrogen bonding with water acting as the proton acceptor. The orientation of the ring substituents does not seem to influence substantially the solvation process, and thus, the energy difference between the remaining isomers is mostly due to the relative stability of the carOH isomers (Fig. S4, ESI†). The BE for the four clusters were obtained considering the BSSE corrections at the M06-2X/6-311++G(d,p) level of theory (Fig. 3). The values obtained reflected the stability of the aggregates and explained their energetic order. The most stable carOH⋯w structure, stabilised by a strong hydrogen bond, had the highest BE, with decreasing values for the other conformations.
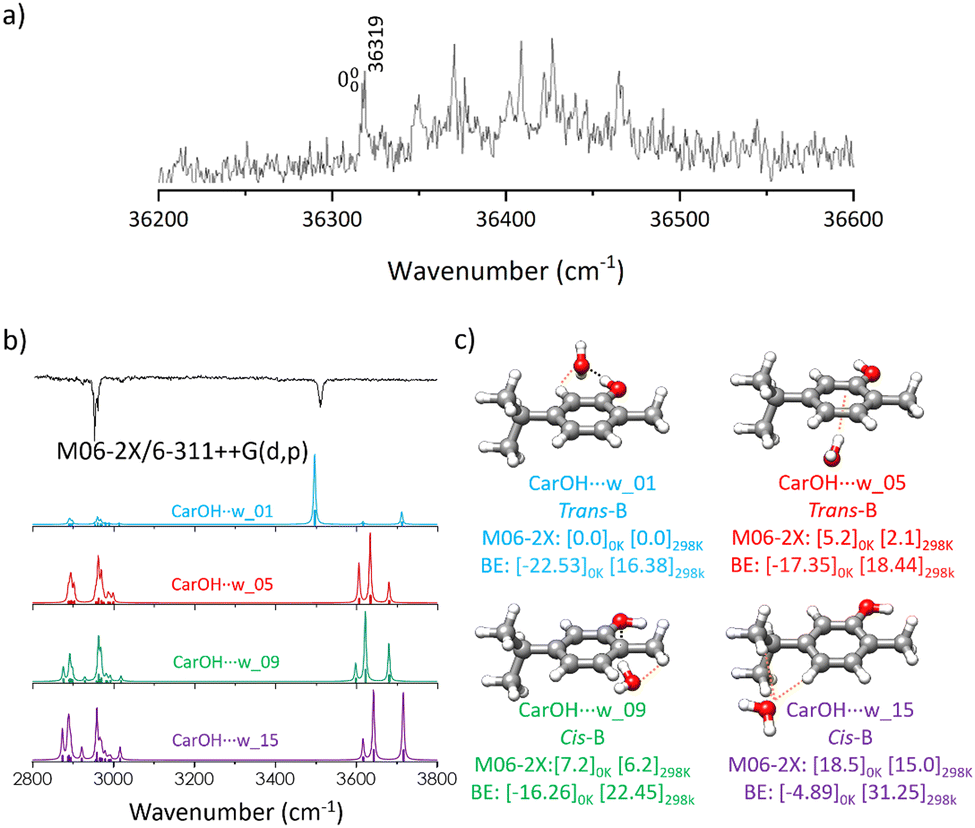 | ||
| Fig. 3 (a) Two-colour REMPI spectrum of the carvacrol–water complex (carOH⋯w) (b) experimental (black trace) and simulated (coloured traces) IDIR spectra for (c) four representative carOH⋯w isomers. The simulations were done based on the M06-2X structures using scaling factors of 0.946 and 0.932 for C–H and O–H stretching modes, respectively, to account for anharmonicity. The relative Gibbs free energy and BE (kJ mol−1) are given at the M06-2X/6-311++G(d,p) level of theory at 0 and 298 K. | ||
The 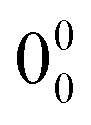 transition of carOH⋯w appeared at 36319 cm−1 in the two-colour REMPI spectrum (Fig. 3), which means a blue shift of ca. 403 cm−1 with respect to the
transition of carOH⋯w appeared at 36319 cm−1 in the two-colour REMPI spectrum (Fig. 3), which means a blue shift of ca. 403 cm−1 with respect to the  transition of the monomer, indicating either a decrease in binding energy in the electronic excited state or that the true origin band was not located due to its low intensity. Comparison with other similar systems such as phenol/phenol–w,16–18 eugenol/eugenol–w,19 or propofol/propofol–w,20 showed always a red-shift (Table 1). As in carOH⋯w, the experimental complexes with propofol and phenol correspond to O–H⋯Ow interactions with water as proton-acceptor; while in the aggregate with eugenol, water acts as the proton donor. Phenol, propofol and carvacrol are photoacids, i.e.: their proton-donation ability increases upon electronic excitation, justifying a red shift.
transition of the monomer, indicating either a decrease in binding energy in the electronic excited state or that the true origin band was not located due to its low intensity. Comparison with other similar systems such as phenol/phenol–w,16–18 eugenol/eugenol–w,19 or propofol/propofol–w,20 showed always a red-shift (Table 1). As in carOH⋯w, the experimental complexes with propofol and phenol correspond to O–H⋯Ow interactions with water as proton-acceptor; while in the aggregate with eugenol, water acts as the proton donor. Phenol, propofol and carvacrol are photoacids, i.e.: their proton-donation ability increases upon electronic excitation, justifying a red shift.
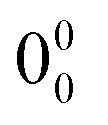 transition for carvacrol and similar molecules from REMPI experiments, and position of the OH vibration band in the IR spectra for the species reported in this work. All values are given in cm−1
transition for carvacrol and similar molecules from REMPI experiments, and position of the OH vibration band in the IR spectra for the species reported in this work. All values are given in cm−1
|
|
||||
|---|---|---|---|---|
| Monomer | Water complex | Dimer | Trimer | |
| a This work. b Ref. 19 and 46. c Ref. 46. d Ref. 20 and 47. e Ref. 16–18. f Ref. 48. g Ref. 49. h Ref. 25. | ||||
| Carvacrola | 35916 |
36319 |
35705 |
35909 |
| Eugenolb | 35202 |
34908 |
34698 |
— |
| Guaiacolc | 35929 |
— | 35549 |
— |
| Propofold | 36222 |
36029 |
36169 |
36267 |
| Phenole | 36348 |
35994 |
— | — |
| Benzyl alcoholf | 37528 |
— | 37482 |
37457 |
| 1-naphtholg | 31457 |
— | 31207 |
31380 |
| 2-phEtOHh | 37627 |
— | 37460 |
37330 |
| Δ mono-w | Δ mono-di | Δ mono-tri | ||
|---|---|---|---|---|
| Carvacrol | 403 | −211 | −7 | |
| Eugenol | −294 | −504 | — | |
| Guaiacol | — | −308 | — | |
| Propofol | −193 | −53 | 45 | |
| Phenol | −354 | — | — | |
| Benzyl alcohol | — | −46 | −71 | |
| 1-naphthol | — | −250 | −77 | |
| 2-phEtOH | — | −167 | −297 |
| ν(OH) carvacrol | ||||
|---|---|---|---|---|
| Monomer | Water complex | Dimer | Trimer | |
| 3640 | 3510 | 3513 | 3401 | |
| Δ mono-w | Δ mono-di | Δ mono-tri | ||
|---|---|---|---|---|
| −130 | −127 | −239 |
The IDIR technique unequivocally confirmed that all the transitions observed in the two-colour REMPI spectrum resulted from the same carOH⋯w conformation. One could expect three bands in the IR spectrum corresponding to the vibration of the OH in carvacrol and water. However, in the experimental IR spectrum (Fig. 3) only a band at 3510 cm−1, attributed to the stretching vibration of the O–H in carvacrol, was observed. Higher energy carOH⋯w structures were predicted to exhibit more peaks in the 3600–3800 cm−1 region and were rejected from consideration (Fig. 3). The IR spectrum of carOH⋯w was better simulated from the prediction at M06-2X/6-311++G(d,p) than at B3LYP-GD3BJ/def2-TZVP (Fig. S5, ESI†). Although it is challenging to precise which one of the four structures is responsible for the observed bands in the IR spectra, it was unambiguously determined that it was formed by a hydrogen bond interaction between water acting as a proton-acceptor and carvacrol as a donor. In carOH⋯w the OH vibration was red-shifted around 130 cm−1 with respect to the monomer (Table 1). A similar shift of 130 cm−1 was also observed for the phenol/phenol–w system,16–18 attributed to the formation of a strong hydrogen bond. On the other hand, the shift from monomer to monohydrated complex in propofol20 was of only 100 cm−1 and correlated with a weaker hydrogen bond due to the steric hindrance of the two isopropyl groups. Contrary to the propofol case, the location of water in carOH⋯w was not hindered by any group, allowing for the formation of a strong hydrogen bond.
Carvacrol dimer – (carOH)2
The substantially larger size of the dimer compared to the monohydrate complicated the exploration of the conformational landscape. The isomers for the homodimer were built from the four available conformations of the monomer, offering a wide range of interaction possibilities, highlighted by 152 structures obtained in the conformational search step. The isopropyl group influenced the potential energy surface (PES) of the dimer with its steric hindrance preventing the formation of certain intermolecular interactions.Geometry optimisation reduced the number of species to 50 distinct isomers for the dimer (carOH)2, 18 of them within the 0 to 5 kJ mol−1 range. We started the exploration of the dimer focusing on those low-energy isomers. They were labelled based on their order in energy and further identified by the carvacrol monomer conformation (Fig. S6, ESI†). While both levels of theory (B3LYP-GD3BJ/def2-TZVP and M06-2X/6-311++G(d,p)) yielded similar isomers for the dimer, they exhibited different energy orders (Fig. S6, ESI†), due to the parametrisation potentials of each functional. The two levels agreed on the global minimum, which was predicted as the lowest energy structure and with similar interactions for both. Predictions with M06-2X seemed to be more accurate for the dimer (see below), and therefore we used those predictions as a guide for the discussion.
The global minimum presents an O–H⋯O hydrogen bond that guides the aggregation process. This structure is further stabilised through C–H⋯π contacts between the alkyl groups of one carvacrol unit and the aromatic ring of the other. The contribution of the interaction due to dispersion forces is at least of similar importance to the hydrogen bond, as highlighted by the second most stable isomer, which is stabilised mainly by dispersion interactions. In the remaining structures until 5 kJ mol−1, dispersion forces seem to play a dominant role. The main difference between the structures stabilised by dispersion is the relative position of the two carvacrol molecules (see NCI section). This behaviour was confirmed by the values of the BE for the four homodimers at M06-2X/6-311++G(d,p) (Fig. 4c).
 | ||
| Fig. 4 (a) Two-colour REMPI spectrum of the carvacrol dimer complex (carOH)2 (b) experimental (black trace) and simulated (coloured traces) IDIR spectra for (c) four representative (carOH)2 isomers. The simulations were done based on the M06-2X structures using scaling factors of 0.946 and 0.932 for C–H and O–H stretching modes, respectively, to account for anharmonicity. The relative Gibbs free energies and BE (kJ mol−1) are given at the M06-2X/6-311++G(d,p) level of theory at 0 and 298 K. | ||
The presence of a second chromophore resulted in more complicated dynamics of the electronic excited state. In the two-colour REMPI spectra of (carOH)2 several transitions were evident at 35705, 35765, 35801, and 35873 cm−1 (Fig. 4a), all attributed to the same conformation of the dimer. The red-most band, at 35705 cm−1 was assigned to the  transition, which was red-shifted from the monomer by around 211 cm−1. In contrast to the water complex, carvacrol homodimer followed the same tendency as other similar systems, since a red-shift was also observed in eugenol,46 guaiacol,46 or propofol.47,50
transition, which was red-shifted from the monomer by around 211 cm−1. In contrast to the water complex, carvacrol homodimer followed the same tendency as other similar systems, since a red-shift was also observed in eugenol,46 guaiacol,46 or propofol.47,50
The band at 35801 cm−1 was selected to record the IR spectrum of (carOH)2, yielding valuable structural information about the dimer. Once more, the agreement with the experimental IR spectrum seemed to be better for the predictions at the M06-2X/6-311++G(d,p) level, although both methods reproduced nicely the experimental spectrum using the structure of the global minimum. Predictions for other isomers did not agree with the experimental bands. Representative isomers with different types of interactions were selected to simulate their IR spectra (Fig. 4 and Fig. S7, ESI†). Interestingly, the assignment using M06-2X results was more conclusive, as B3LYP predicted that some other isomers higher in energy could also reproduce the experimental trace. More precisely, the spectrum predicted for structure 06 was shockingly similar to that predicted for the global minimum. That structure is held by dispersion forces between the two aromatic rings (Fig. S7, ESI†), and therefore, one would not expect a similar shift to that observed for the global minimum, which presents a clear hydrogen bond.
If one accepts the assignment to the global minimum, the band at 3630 cm−1 (Fig. 4b) should be assigned to the vibration of the proton-acceptor hydroxyl group, while the band at 3513 cm−1 (Fig. 4b) would correspond to the vibration of the proton-donor –OH group in the dimer. This band was almost in the same position as in the spectrum of carOH⋯w (3510 cm−1), in which carvacrol also acts as proton-donor. Apparently, despite the large volume of the interacting molecules and the steric hindrance, the hydroxyl groups were able to reach a position in which they can establish a hydrogen bond of similar strength to that in the monohydrate.
Carvacrol trimer – (carOH)3
The conformational landscape of the carvacrol trimer is complicated due to the multiple potential arrangements for the three carvacrol units. The steric hindrance of the isopropyl groups influenced the stability of the aggregates, favouring cyclic geometries. The conformational search identified 234 possible isomers for the trimer, composed by the four conformations of the carvacrol monomer. We focused on the lowest energy structures, grouping them into four families and attending to their intermolecular interactions, either hydrogen bonding, dispersion forces, or a combination of both. The most stable member of each family was included in our analysis and compared with the experimental spectrum.The trimer global minimum exhibited a cyclic structure with three sequential O–H⋯O–H⋯O–H⋯O hydrogen bonds, reinforced by C–H⋯π interactions between the isopropyl groups and the aromatic rings (Fig. 5). This aggregate consisted of three identical molecules of carvacrol in the trans-A conformation resulting in a symmetric geometry. The orientation of the OH groups was optimal for the interaction with the next molecule forming the sequential cycle, while the orientation of the isopropyl groups maximised the dispersion interactions. Additional structures with three O–H⋯O hydrogen bonds were identified, however, the slightly different alkyl chain arrangements resulted in higher energies. For the remaining families of (carOH)3, the carvacrol molecules are held together by either one or two O–H⋯O hydrogen bonds in linear arrangements, with additional O–H⋯π contacts (see NCI section). The most stable aggregates from each family are depicted in Fig. 5c and Fig. S8 (ESI†). In the homotrimer case, the binding energy (BE) confirmed the energy ordering for each family of interactions. The most stable structure, stabilised by three cooperative hydrogen bonds, exhibited the highest BE. In the following aggregates, the relative energy increased and the BE decreased with the decrease in the number of O–H⋯O hydrogen bond interactions (Fig. 5c).
 | ||
| Fig. 5 (a) Two-colour REMPI spectrum of the carvacrol trimer (carOH)3 (b) experimental (black trace) and simulated (coloured traces) IDIR spectra for (c) four representative (carOH)3 isomers. The simulations were done based on the B3LYP-D3BJ structures using a scaling factor of 0.957 for C–H and O–H stretching modes, to account for anharmonicity. The relative Gibbs free energies and BE (kJ mol−1) are given at the B3LYP-D3BJ/def2-TZVP level of theory at 0 and 298 K. | ||
Although the REMPI spectrum of the carvacrol trimer is more noisy than that of the monomer and dimer, bands at 35909, 35959, 35991, and 36018 cm−1 were identified (Fig. 5a). The 35909 cm−1 band was assigned to the  transition, with all bands attributed to a single isomer of the trimer. The structure of (carOH)3 was compared to other similar trimers (Fig. S9, ESI†), such as those of 1-naphthol,49 phenol,51 propofol,47 cyclohexanol,52 benzyl alcohol,48 or 2-phenylethanol (2-phEtOH).25 This comparison allowed us to investigate the effect of the substituent groups on the final geometry of the aggregate and its spectrum. Similarly to (carOH)3, (1-naphthol)3,49 (phenol)3,51 (propofol)3,47 and (cyclohexanol)3,52 displayed cyclic structures with three cooperative hydrogen bonds, along with dispersion interactions (Fig. S9, ESI†). On the other side, the observed structures for (benzyl alcohol)348 or (2-phEtOH)325 were not cyclic, with two O–H⋯O hydrogen bonds and an O–H⋯π interaction (Fig. S9, ESI†). The trimers formed by molecules in which the OH group is directly attached to the aromatic ring seemed to prefer the cyclic arrangements. On the other hand, in both (benzyl alcohol)3 and (2-phEtOH)3 there was an alkyl substituent between the benzyl ring and the OH, granting some flexibility. Those molecules tend to form linear trimers rather than cooperative cyclic aggregates.
transition, with all bands attributed to a single isomer of the trimer. The structure of (carOH)3 was compared to other similar trimers (Fig. S9, ESI†), such as those of 1-naphthol,49 phenol,51 propofol,47 cyclohexanol,52 benzyl alcohol,48 or 2-phenylethanol (2-phEtOH).25 This comparison allowed us to investigate the effect of the substituent groups on the final geometry of the aggregate and its spectrum. Similarly to (carOH)3, (1-naphthol)3,49 (phenol)3,51 (propofol)3,47 and (cyclohexanol)3,52 displayed cyclic structures with three cooperative hydrogen bonds, along with dispersion interactions (Fig. S9, ESI†). On the other side, the observed structures for (benzyl alcohol)348 or (2-phEtOH)325 were not cyclic, with two O–H⋯O hydrogen bonds and an O–H⋯π interaction (Fig. S9, ESI†). The trimers formed by molecules in which the OH group is directly attached to the aromatic ring seemed to prefer the cyclic arrangements. On the other hand, in both (benzyl alcohol)3 and (2-phEtOH)3 there was an alkyl substituent between the benzyl ring and the OH, granting some flexibility. Those molecules tend to form linear trimers rather than cooperative cyclic aggregates.
The IDIR spectrum of (carOH)3 recorded from the 35959 cm−1 band was compared with the simulations at both levels of theory (Fig. 5b and Fig. S8, ESI†). The simulated IDIR spectra for the lowest-energy conformations for each family always produced the same spectrum (Fig. 5b and Fig. S8, ESI†). Surprisingly, contrary to other species reported in this work, B3LYP-D3BJ/def2-TZVP performed better than M06-2X/6-311++G(d,p) in the prediction of the IR spectrum (Fig. S8, ESI†). The spectrum for the global minimum, with cooperative hydrogen bonding, presented a strong band for the OH vibration, in good agreement with the experimental findings. Thus, the experimental band at 3401 cm−1 was assigned to the symmetric OH stretching in the cyclic (carOH)3 structure. Simulations for higher-energy conformations with other interactions presented three bands for the OH vibration, reflecting the different environments for the hydroxyl groups. A similar behaviour with one peak has only been observed for (1-naphthol)3,49 whereas the other trimers; (propofol)3,47 (cyclohexanol)3,52 (benzyl alcohol)3,48 and (2-phEtOH)325 presented multiple stretching bands, illustrating the difference in their chemical environment. This could be rationalised based on the conformers within the trimer. In the observed (carOH)3 the three carvacrol molecules are in trans-A conformation, building a symmetric trimer, as the one observed for 1-naphthol. For the other systems, the three molecules forming the trimer adopted slightly different conformations, resulting in the observed spectra.
Non-covalent interactions analysis
NCI analysis is a convenient way to visualise the strength and distribution of intra- and inter-molecular interactions in molecular aggregates. Fig. 6 and Fig. S10–S13 (ESI†) present the NCI plots for the most relevant isomers for carOH, carOH⋯w, (carOH)2, and (carOH)3. The second eigenvalue of the electron density of the Hessian matrix, λ2, indicates the nature of the interaction. Negative values of λ2 are associated with strong attractive non-covalent interactions, typically hydrogen bonds (blue surfaces in Fig. 6). Values around 0 are interpreted as weakly attractive forces, such as London dispersion interactions (green surfaces in Fig. 6). Finally, positive values of λ2 imply repulsive forces (red surfaces in Fig. 6).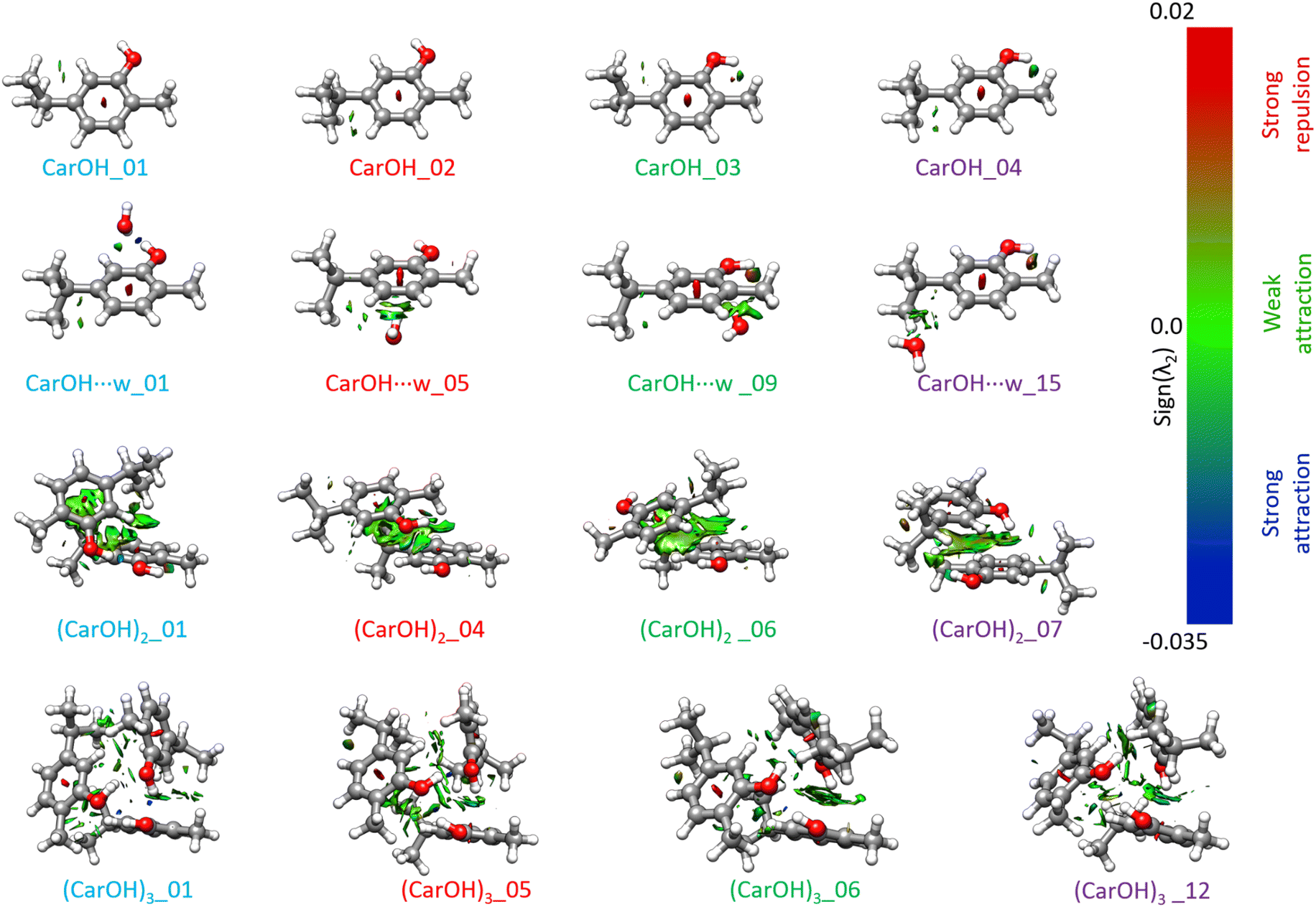 | ||
| Fig. 6 NCI plots for selected isomers for carOH, carOH⋯w, (carOH)2, and (carOH)3. λ2 is the second eigenvalue of the electron density Hessian matrix. Positive values (red areas) are associated with strong repulsions; negative values (blue areas) are associated with strong attractions; while values close to 0 (green surfaces) indicate the presence of weak interactions. Please note that the colour bar scale is not linear. | ||
As expected, in the trans monomer, the only relevant interaction is a weak C–H⋯H dispersion force between the isopropyl group and the aromatic ring. The cis isomers presents additional interactions between the hydroxyl group and the methyl group in ortho.
The NCI plot of the carOH⋯w complex supported the conclusions from the electronic and IR spectra. Water and carvacrol formed a hydrogen bond, with water as a proton-acceptor, as observed in the shift of the OH vibration. The second interaction, a C–H⋯O contact, contributed to the stability of this structure. In the dimer, the panorama of intermolecular interactions become complicated. For the lowest energy structure, the main interaction is a hydrogen bond between the two hydroxyl groups, but it seems to be weaker than the hydrogen bond formed with water. On the other hand, there is an almost-continuous surface of interaction between the two aromatic rings and the alkyl groups in both carvacrol molecules. The higher energy structures of the dimer seemed to be stabilised by dispersion forces, or even one dipole–dipole interaction between the two units (Fig. 6).
The NCI plot of the carvacrol trimer confirmed the assignment to the lowest energy structure, characterised by three cooperative hydrogen bonds in a cyclic, symmetric arrangement (Fig. 6), in agreement with the observation of a single band in the IR spectrum. The NCI plots for the remaining homotrimer structures revealed additional O–H⋯π interactions, resulting in higher relative energy and lower BE (Fig. 5). The main difference with the structures of the homodimer is the lack of dispersion interactions, evidenced by smaller green surfaces between the three carvacrol units (Fig. 6). The analysed structures for the trimer are all formed by carvacrol monomers in the trans configuration, while the dimers presented also cis forms. Such difference could be the origin of the smaller contribution of dispersion interactions, highlighting the importance of the monomer configuration in the aggregates.
Conclusions
We present a detailed spectroscopic investigation of carvacrol, its homodimer, homotrimer, and monohydrated complex combined with state-of-art quantum-chemical computations. In each case, the predicted most stable structure agrees with the experimental spectra, although higher-energy forms may also be present due to the similarity of the predicted spectra. The spectrum of the monomer is compatible with the presence of the four conformations reported previously by a microwave study. Those four conformations arise from the different orientation of the hydroxyl and isopropyl groups and are present in the monomer and the different aggregates. Our findings seem to suggest that the structure of carvacrol remains almost unaffected by interactions with nearby molecules during complexation. In the monohydrate complex, the red-shift observed in the OH stretching vibration, similar to that reported for phenol–water, is associated with a strong hydrogen bond. This interaction is compared with propofol–water, a similar but more sterically crowded system, resulting in a weaker hydrogen bond. The outcome of this work indicates that hydrogen bonding is the main stabilising force defining the structures of the low-energy conformers. However, dispersion interactions between the aromatic rings and the isopropyl groups are of great importance in determining the dimer structure. As highlighted by the NCI plots, dispersion interactions are important for the stabilisation of the aggregates. Interestingly, mixing cis and trans units in the dimer results in an extensive interaction domain between the carvacrol units. As the aggregates grow in size and with only trans monomers, the dispersion interactions seem to be less critical for stabilisation. In the homotrimer of carvacrol, the observed structure corresponds to a cyclic, symmetric geometry with three cooperative hydrogen bonds. Our comparative study with other trimers shows that the substituents on the aromatic ring are not as determinant for forming cyclic or linear trimers. The key parameter for the formation of cyclic structures seems to be the connectivity of the OH group to the aromatic ring. If the OH is directly attached to the benzyl ring cyclic structures are favoured, while if there are alkyl chains that allow for some flexibility, then linear structures with O–H⋯π interactions are preferred.Author contributions
Paúl Pinillos: investigation, formal analysis, writing – original draft, writing – review & editing. Fernando Torres-Hernández: investigation, formal analysis, writing – review & editing. Imanol Usabiaga: conceptualisation, formal analysis, writing – review & editing. Pablo Pinacho: formal analysis, writing – original draft, writing – review & editing. José A. Fernández: conceptualisation, funding acquisition, project administration, formal analysis, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.† The data can be requested from the corresponding authors.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
This work was supported by grant PID2021-127918NB-I00 funded by MCIN/AEI/10.13039/501100011033 and, by “ERDF A way of making Europe” and Grant IT1491-22 funded by the Basque Government. F. T. H. acknowledges financial support from Spanish Ministry of Science and Innovation under the FPI predoctoral program. P. P. acknowledges a Maria Zambrano grant (UPV/EHU, Ministry of Universities, Recovery, Transformation, and Resilience Plan—Funded by the European Union—Next Generation EU, MAZAM22/16). We also thank the SGIKER (UPV/EHU, MICIU-FEDER) for the computational and laser resources.References
- N. F. Salakhutdinov, K. P. Volcho and O. I. Yarovaya, Pure Appl. Chem., 2017, 89, 1105–1117 CrossRef CAS.
- A. K. Pandey, P. Kumar, P. Singh, N. N. Tripathi and V. K. Bajpai, Front. Microbiol., 2017, 7, 2161 Search PubMed.
- A. Mathur, A. Meena and S. Luqman, Phytother. Res., 2023, 38, 939–969 CrossRef PubMed.
- M. Sharifi-Rad, E. M. Varoni, M. Iriti, M. Martorell, W. N. Setzer, M. M. Contreras, B. Salehi, A. Soltani-Nejad, S. Rajabi, M. Tajbakhsh and J. Sharifi-Rad, Phytother. Res., 2018, 32, 1675–1687 CrossRef CAS.
- M. Bertuola, N. Fagai and M. Fernández Lorenzo de Mele, Heliyon, 2020, 6, e03714 CrossRef CAS.
- E. L. Malankina, A. N. Kuzmenko, B. T. Zaitchik, A. O. Ruzhitskiy, A. A. Evgrafov and L. N. Kozlovskaya, Mosc. Univ. Chem. Bull., 2020, 75, 391–394 CrossRef.
- A. Ben Arfa, S. Combes, L. Preziosi-Belloy, N. Gontard and P. Chalier, Lett. Appl. Microbiol., 2006, 43, 149–154 CrossRef CAS PubMed.
- W.-X. Du, C. W. Olsen, R. J. Avena-Bustillos, T. H. McHugh, C. E. Levin and M. Friedman, J. Agric. Food Chem., 2008, 56, 3082–3088 CrossRef CAS PubMed.
- G. Magi, E. Marini and B. Facinelli, Front. Microbiol., 2015, 6, 165 Search PubMed.
- K. M. Arunasree, Phytomedicine, 2010, 17, 581–588 CrossRef CAS.
- S. J. Mehdi, A. Ahmad, M. Irshad, N. Manzoor and M. M. A. Rizvi, Biol. Med., 2011, 3, 307–312 CAS.
- G. Magi, E. Marini and B. Facinelli, Front. Microbiol., 2015, 6, 165 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
- E. Garand, M. Z. Kamrath, P. A. Jordan, A. B. Wolk, C. M. Leavitt, A. B. McCoy, S. J. Miller and M. A. Johnson, Science, 2012, 335, 694–698 CrossRef CAS.
- C. Puzzarini, L. Spada, S. Alessandrini and V. Barone, J. Phys.: Condens. Matter, 2020, 32, 343002 CrossRef CAS.
- A. Oikawa, H. Abe, N. Mikami and M. Ito, J. Phys. Chem., 1983, 87, 5083–5090 CrossRef CAS.
- R. M. Helm and H. J. Neusser, Chem. Phys., 1998, 239, 33–47 CrossRef CAS.
- T. Watanabe, T. Ebata, S. Tanabe and N. Mikami, J. Chem. Phys., 1996, 105, 408–419 CrossRef.
- A. Longarte, I. Unamuno, J. A. Fernández, F. Castaño and C. Redondo, J. Chem. Phys., 2004, 121, 209–219 CrossRef CAS PubMed.
- I. Leon, E. J. Cocinero, J. Millán, S. Jaeqx, A. M. Rijs, A. Lesarri, F. Castaño and J. A. Fernández, Phys. Chem. Chem. Phys., 2012, 14, 4398–4409 RSC.
- D. Schmitz, V. A. Shubert, B. M. Giuliano and M. Schnell, J. Chem. Phys., 2014, 141, 034304 CrossRef CAS.
- I. Usabiaga, J. González, P. F. Arnáiz, I. León, E. J. Cocinero and J. A. Fernández, Phys. Chem. Chem. Phys., 2016, 18, 12457–12465 RSC.
- C. J. Gruenloh, F. C. Hagemeister, J. R. Carney and T. S. Zwier, J. Phys. Chem. A, 1999, 103, 503–513 CrossRef CAS.
- A. Camiruaga, I. Usabiaga, A. Insausti, I. León and J. A. Fernández, Phys. Chem. Chem. Phys., 2017, 19, 12013–12021 RSC.
- A. Camiruaga, R. T. Saragi, F. Torres-Hernández, M. Juanes, I. Usabiaga, A. Lesarri and J. A. Fernández, Phys. Chem. Chem. Phys., 2022, 24, 24800–24809 RSC.
- P. Pinillos, A. Camiruaga, F. Torres-Hernández, F. J. Basterrechea, I. Usabiaga and J. A. Fernández, Phys. Chem. Chem. Phys., 2023, 25, 7205–7212 RSC.
- F. Torres-Hernández, P. Pinillos, W. Li, R. T. Saragi, A. Camiruaga, M. Juanes, I. Usabiaga, A. Lesarri and J. A. Fernández, J. Phys. Chem. Lett., 2024, 15, 5674–5680 CrossRef.
- P. G. Mezey, Potential Energy Hypersurfaces, Elsevier, Amsterdam, 1987 Search PubMed.
- K. Roos, C. Wu, W. Damm, M. Reboul, J. M. Stevenson, C. Lu, M. K. Dahlgren, S. Mondal, W. Chen, L. Wang, R. Abel, R. A. Friesner and E. D. Harder, J. Chem. Theory Comput., 2019, 15, 1863–1874 CrossRef CAS PubMed.
- T. A. Halgren, J. Comput. Chem., 1999, 20, 720–729 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS.
- Schrödinger, Schrödinger Release 2019-4, Glide. LLC, New York, 2019 Search PubMed.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. J. Krieg, Chem. Phys., 2010, 132, 154101 Search PubMed.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 122, 154101 CrossRef.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- P. Pinacho, J. C. López and S. Blanco, J. Mol. Spectrosc., 2017, 337, 145–152 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
- A. J. Lopes Jesus, R. Fausto and I. Reva, J. Phys. Chem. A, 2021, 125, 8215–8229 CrossRef CAS PubMed.
- A. Longarte, C. Redondo, J. A. Fernández and F. Castaño, J. Chem. Phys., 2005, 122, 164304 CrossRef.
- I. León, J. Millán, E. J. Cocinero, A. Lesarri and J. A. Fernández, Angew. Chem., Int. Ed., 2013, 25, 7772–7775 CrossRef.
- R. Medel, A. Camiruaga, R. T. Saragi, P. Pinacho, C. Pérez, M. Schnell, A. Lesarri, M. A. Suhm and J. A. Fernández, Phys. Chem. Chem. Phys., 2021, 23, 23610–23624 RSC.
- M. Saeki, S.-I. Ishiuchi, M. Sakai, K. Hashimoto and M. Fujii, J. Phys. Chem. A, 2010, 114, 11210–11215 CrossRef CAS PubMed.
- I. León, J. Millán, F. Castaño and J. A. Fernández, ChemPhysChem, 2012, 13, 3819–3826 CrossRef PubMed.
- N. A. Seifert, A. L. Steber, J. L. Neill, C. Pérez, D. P. Zaleski, B. H. Pate and A. Lesarri, Phys. Chem. Chem. Phys., 2013, 15, 11468–11477 RSC.
- I. León, R. Montero, A. Longarte and J. A. Fernández, Phys. Chem. Chem. Phys., 2015, 17, 2241–2245 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures of conformers, and predicted IR spectra. See DOI: https://doi.org/10.1039/d4cp02945c |
| ‡ Shared first authors. |
| This journal is © the Owner Societies 2024 |