
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental and theoretical investigation of the Auger electron spectra of isothiocyanic acid, HNCS†
Dorothee
Schaffner
 a,
Marius
Gerlach
a,
Emil
Karaev
a,
John
Bozek
b,
Ingo
Fischer
*a and
Reinhold F.
Fink
*c
a,
Marius
Gerlach
a,
Emil
Karaev
a,
John
Bozek
b,
Ingo
Fischer
*a and
Reinhold F.
Fink
*c
aInstitute of Physical and Theoretical Chemistry, University of Würzburg, 97074 Würzburg, Germany. E-mail: ingo.fischer@uni-wuerzburg.de; Tel: +49 931 31 86360
bSynchrotron SOLEIL, L'Orme des Merisiers, 91192 Gif-sur-Yvette, France
cInstitut für Physikalische und Theoretische Chemie, Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: reinhold.fink@uni-tuebingen.de; Tel: +49 7071 29 76593
First published on 17th October 2024
Abstract
We investigate isothiocyanic acid, HNCS, by resonant and nonresonant Auger electron spectroscopy at the K-edge of carbon and nitrogen, and the L2,3-edge of sulfur, employing soft X-ray synchrotron radiation. The C1s and N1s ionization energies as well as the S2s and S2p ionization energies are determined and X-ray absorption spectra reveal the transitions from the core to the virtual orbitals. Final states for all normal Auger electron spectra and the resonant ones recorded at the carbon and nitrogen edge are assigned and rationalized with theoretical spectra obtained with a wave-function based protocol. The latter is based on ab initio configuration–interaction representations of the bound part of the electronic wave functions, the one-center approximation for Auger intensities, and a moment theory for band shapes. The computed spectra are in very good agreement with the experimental data and most of the relevant signals are assigned. The double ionization energy is derived from the S2p1/2 spectrum and in good agreement with a recently determined value. The Auger electron spectra are compared with those of the congener HNCO. A similar shape of the normal Auger electron spectra was found for the low binding energy final states, while intensities differed. Similarities are less pronounced in the resonant Auger electron spectra.
1 Introduction
Isothiocyanic acid, HNCS, is the simplest isothiocyanate and the sulfur analog of isocyanic acid, HNCO. Although formally a closed-shell molecule with 1A′ electronic ground state, the molecule is unstable at room temperature and thus challenging to study. Originally due to its unknown molecular structure and its analogy to isocyanic acid, later due to its role in astrochemistry,1 isothiocyanic acid was subjected to several spectroscopic studies. The structure was investigated by experimentalists2,3 but only the first microwave spectrum of the reaction product of potassium thiocyanate with phosphorous acid showed that the vapor was mainly composed of isothiocyanic acid, HNCS, rather than its isomer thiocyanic acid, HSCN.4HNCS was found to be a quasi-linear molecule5,6 with a trans bent conformation and an HNC angle of 132°.7 The photoionization of isothiocyanic acid was studied by He I photoelectron spectroscopy8–10 and photoionization mass spectrometry.11 From the latter, approximate fragment appearance energies were determined. Isothiocyanic acid and its isomers were also the subject of several theoretical studies concerning their structure, relative stability and isomerization reactions.12–14 Isothiocyanic acid was found to be the most stable isomer of the [C, H, N, S] tetrade followed by thiocyanic acid, HSCN. This order of stability is analogous to the respective oxygen congeners isocyanic acid and cyanic acid.15
Interest in HNCS arises from its observation in the interstellar medium. In 1979 it was detected for the first time towards the molecular cloud Sagittarius (Sgr) B2(OH) by its rotational transitions.16 The detection of HNCS in space is intriguing, because it consists of four elements which are essential for organic life. Its oxygen congener HNCO is a well-known astrochemical molecule17–19 and could be related to the formation of biomolecules.20,21 As a sulfur-containing molecule, HNCS is relevant to astrochemistry, because sulfur chemistry in interstellar environments and planetary science is not well understood.22 A proper understanding of the chemistry of interstellar molecules requires to investigate their interaction with high-energy radiation.23,24 Several UV absorption spectra of HNCS have been reported,25,26 before contaminations were identified and band assignment finally settled.27 The dissociation from electronically excited states was investigated by flash photolysis at ≥190 nm.28 Furthermore, Northrup and Sears studied the photodissociation at 248 and 193 nm, investigated several fragmentation channels by laser induced fluorescence and compared them to the corresponding alkyl(iso)thiocyanates.29,30 Very recently, Wallner et al. investigated the double ionization of HNCS at 40.8 eV with a He discharge lamp and at 90 eV by synchrotron radiation.31 The double ionization energy and fragmentation patterns as well as a computational study of low-lying doubly ionized states were reported. Despite this work, which was carried out in parallel to our efforts, spectroscopy of HNCS with soft X-ray radiation is not well studied. The overall lack of X-ray data recently motivated theoretical work on X-ray absorption spectra (XAS) of sulfur-containing molecules, with the goal to provide reference data for observations with X-ray telescopes.32 Only few reactive molecules have been explored in the X-ray regime. Previously, we investigated HNCO by Auger electron spectroscopy and compared its spectra as well as its fragmentation dynamics with the isomer HCNO, fulminic acid.33–36 Only a few further spectroscopic studies on reactive molecules have been reported, including XAS of the radicals OH,37 methyl,38 allyl39 and tert-butyl.40
Here, we will explore the changes in the normal (AES) and resonant (RAES) Auger electron spectra upon replacing the oxygen atom in HNCO by a sulfur atom. An important pillar of our program is the cooperation between experiment and theory with the aim to compare experimental spectra with high level computations. We demonstrate that this helps to rationalize the observed transitions and to gain insight into the underlying processes in the core hole and the final states. Recent computations on HCNO, using the same approach as in the present work showed excellent agreement between computed and measured Auger electron spectra and a large number of transitions was assigned to the final states of the ion.35 We will show that the method employed for second-row compounds like HNCO33 and HCNO35 can be extended to a molecule with a third-row heavy atom with similar accuracy.
2 Methods
2.1 Experimental
The experiments were carried out at the high resolution soft X-ray beamline PLÉIADES41 at the French national synchrotron facility SOLEIL. Isothiocyanic acid samples were prepared by slowly treating a saturated solution of potassium thiocyanate with an excess of phosphoric acid (85%). The solution was kept at 0 °C and stirred while the evolving gas was condensed into a liquid nitrogen cooling trap using the vacuum of a forepump. Carbonyl sulfide (OCS), which was identified as side product of this reaction was removed at −50 °C (ethanol/dry ice) by vacuum due to its higher vapour pressure. As HNCS rapidly decomposes at temperatures above −20 °C the samples were kept at −35 °C during the experiments. Soft X-ray radiation was generated by a permanent magnet undulator that allows for different beam polarizations. Linearly polarized light was diffracted off a 600 lines per mm grating and passed through a differentially pumped gas cell containing the sample aligned in front of the acceptance of the electron analyzer. To measure the kinetic energy of the electrons a high resolution (VG-SCIENTA R4000) hemispherical electron analyzer was used that was mounted perpendicular to the propagation direction of the light. Normal Auger electron spectra were recorded with linear vertically polarized light (0° between the light polarization and electron detection axis). The pass energy of the electron analzyer was set to 20 eV and the entrance slit width to 0.8 mm leading to a kinetic energy resolution of 40 meV. Resonant Auger electron spectra were recorded with linearly polarized light oriented at the magic angle (54.7°) with respect to the electron detection axis in order to avoid anisotropy effects of the electron emission. For resonant measurements a pass energy of 100 eV and an entrance slit width of 0.3 mm (0.5 mm) on the N1s and S2p (C1s) edge were applied corresponding to an electron kinetic energy resolution of 75 meV (125 meV). The photon bandwidth was 140 meV around the carbon 1s edge and 100 meV and 60 meV around the nitrogen 1s and sulfur 2p edge. To measure near-edge X-ray absorption fine structure (NEXAFS) spectra the total ion yield was recorded with a negatively biased channeltron detector at the downstream exit tube of the gas cell while stepping the photon energy. The electron kinetic energy and the photon energy were calibrated using reference spectra of N2,42,43 CO242,44 and Argon.452.2 Theoretical details
Theoretical Auger electron spectra were determined in a similar fashion as described in ref. 33, 35 and 46–51. As shown in the ESI,† the geometrical structure, vibrational frequencies and coordinates, which were obtained by density functional theory (DFT) using the B3LYP52–55 functional and the def2-TZVP (triple-zeta valence polarization) basis set56 are in reasonable agreement with experimental data.The intermediate and final state wave functions were obtained with the configuration interaction (CI) method using Hartree–Fock- and virtual valence50-orbitals as obtained with the correlation consistent polarized valence triple zeta (cc-pVTZ)57 basis. The shape of the valence and the lowest virtual valence orbitals are shown in Fig. 1. Further information on the MOs are collected in the ESI.† The intermediate states were represented with the CI method using all configurations with up to three electrons in the virtual valence orbitals and one hole in the 1s or any of the three 2p core orbitals. The respective final state configuration space included all configurations with up to two electrons in the two lowest excited orbitals 13a′ and 4a′′. Please note, that the electronic states resulting from these CI calculations are principally mixtures of all configurations with the same symmetry and the contribution of the leading configuration is in many cases less than 50%. Nevertheless, the states are designated with their leading configurations in the following as this frequently (but not always) allows to rationalize their properties such as the Auger transition rates. As before,33,35 the final state energies range was empirically squeezed by multiplying the energies with a factor of 0.85.
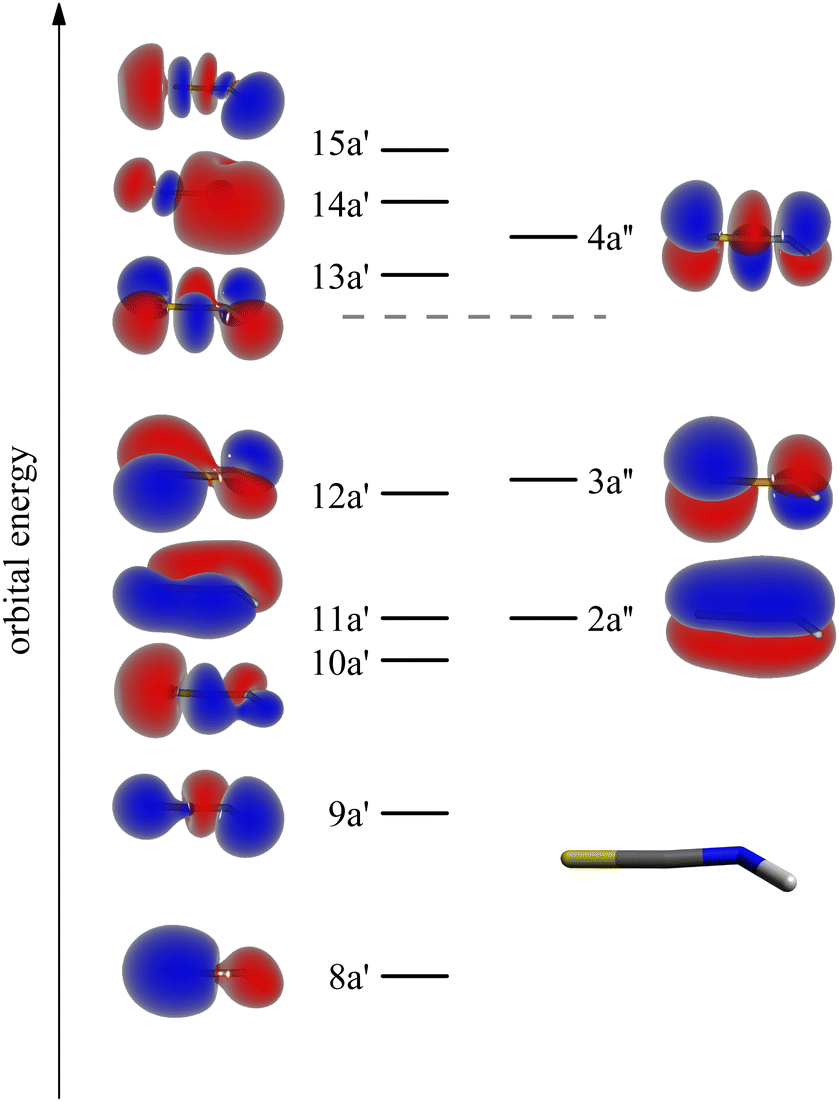 | ||
| Fig. 1 Shape and energetic order of the molecular orbitals (MOs) of the isothiocyanic acid molecule which were used for calculating the Auger electron spectra. In this scheme the S-atom points to the left, the H-atom to the right. See text for details of the determination of the orbitals. The orbital energies are given in Table S3 (ESI†). | ||
Accurate estimates of the vertical energy differences of the ground state, the intermediate states and the lowest final states have been obtained with the averaged coupled-pair functional (ACPF)58 and multi-configuration coupled electron pair approximation (MCCEPA)59 approaches which have been proven to allow for realistic estimates of such energies with typical errors in the order of a few tenths of an electron volt.33,35,47,48,50,60 In the present work we determined these states with the correlation consistent polarized weighted core-valence quadruple zeta (cc-pwCVQZ)61 basis set and restricted open-shell Hartree–Fock (ROHF) reference wave functions that were optimized for the respective state (see ESI,† where further details on a new iterative procedure for core hole states are provided). Excitations from all orbitals were included from these single configuration reference wave functions. For the highly excited core hole states, excitations which increase the number of electrons in the core hole shell were excluded as in previous works.48,62 The calculated HF, ACPF and MCCEPA excitation energies are collected in Table 1. For the normal Auger electron spectra, the vertical double ionization energy of the lowest singlet state [ã1A′(3a′′−2)] was set to the respective MCCEPA values in Table 1. The same was done for the resonant Auger electron spectra with the lowest doublet state [![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 2A′′(3a′′−1)].
2A′′(3a′′−1)].
| State | HF | ACPF | MCCEPA | Expt. |
|---|---|---|---|---|
| a From S2p1/2 AES. | ||||
| 3A′′(12a′−13a′′−1) |
26.38 | 27.87 | 27.75 | 27.6a |
| ã1A′(3a′′−2) | 27.40 | 28.63 | 28.75 | |
| N1s−1 | 405.55 | 405.64 | 405.65 | 405.7 |
| C1s−1 | 295.12 | 293.91 | 294.18 | 293.8 |
| S2p3/2−1 | 169.30 | 169.78 | 169.83 | 169.7 |
| S2p1/2−1 | 170.50 | 170.98 | 171.03 | 170.9 |
| 2A′′(3a′′−1) |
9.14 | 9.81 | 9.76 | 10.0 |
| N1s−113a′ | 399.78 | 399.53 | 399.49 | 399.4 |
| N1s−14a′′ | 400.43 | 401.29 | 400.24 | 400.1 |
| C1s−113a′ | 287.89 | 287.31 | 287.30 | 286.7 |
| C1s−14a′′ | 288.88 | 288.24 | 288.24 | 287.8 |
| S2p3/2−113a′ | 163.56 | 163.68 | 163.66 | 163.4 |
| S2p1/2−113a′ | 164.76 | 164.88 | 164.86 | 164.6 |
| S2p3/2−14a′′ | 164.71 | 164.77 | 164.78 | 164.6 |
| S2p1/2−14a′′ | 165.91 | 165.97 | 165.98 | 165.7 |
Transition rates between the intermediate and final states are obtained with the one-center approximation33,35,47,48,63–66 as implemented for KVV,46,47,50 L2,3VV,48 and M4,5VV67 Auger electron spectra in the local program package (wavels)46,68–71 of one of the authors. Here we followed the approach described before for the 1s core hole states.33,34,46,47 The S2p core hole states are split by about 1.2 eV due to spin–orbit coupling.48,49,62,72 into 2p1/2 and 2p3/2 components. Since the molecular field splitting of the 2p3/2 component is small (≈0.14 eV) compared to the vibrational broadening, it is neglected in the following. Thus, the theoretical S2p Auger electron spectra are obtained as the average of the calculated spectra of the three nonrelativistic core hole states which are multiplied by two (one) and shifted by 0.4 eV (−0.8 eV) in binding energy for the S2p3/2 (S2p1/2) component. The same concept was used before in ref. 73.
Apart from the energetic positions of the initial and final states and the transition rates between them, the appearance of Auger electron spectra is furthermore affected by the line shapes of the electronic transitions. Here, the moment method of Cederbaum et al.74–76 was used where the bands are represented by Gaussian distributions whose centers and widths are determined50,76 from the lifetime energy width of the intermediate state, the gradients of the intermediate and final states with respect to the normal coordinates of the ground state, and the ground state vibrational frequencies. An important result of the moment method is that narrow bands are obtained if the intermediate state gradients are small or similar to those of the final state. Furthermore, bands are shifted to lower binding energies if the gradient of the intermediate state is more negative or more positive than that of the final state and to higher binding energies in the opposite cases.
3 Results
3.1 Normal Auger spectroscopy
Fig. 2 shows the normal Auger electron spectra of HNCS at the sulfur 2p, carbon 1s and nitrogen 1s edge recorded at photon energies of 240, 375 and 485 eV, respectively. Experimentally, the Auger electron yield is measured as a function of the kinetic energy of the Auger electrons. To compare Auger–Meitner processes that lead to the same dicationic final states after the decay of core holes on different atomic sites, the kinetic energy of the Auger electrons can be converted into the binding energy EB. EB is given by the difference of the respective core orbital ionization energy IE(core) and the kinetic energy of the Auger electron Ekin| EB = IE(core) − Ekin. | (1) |
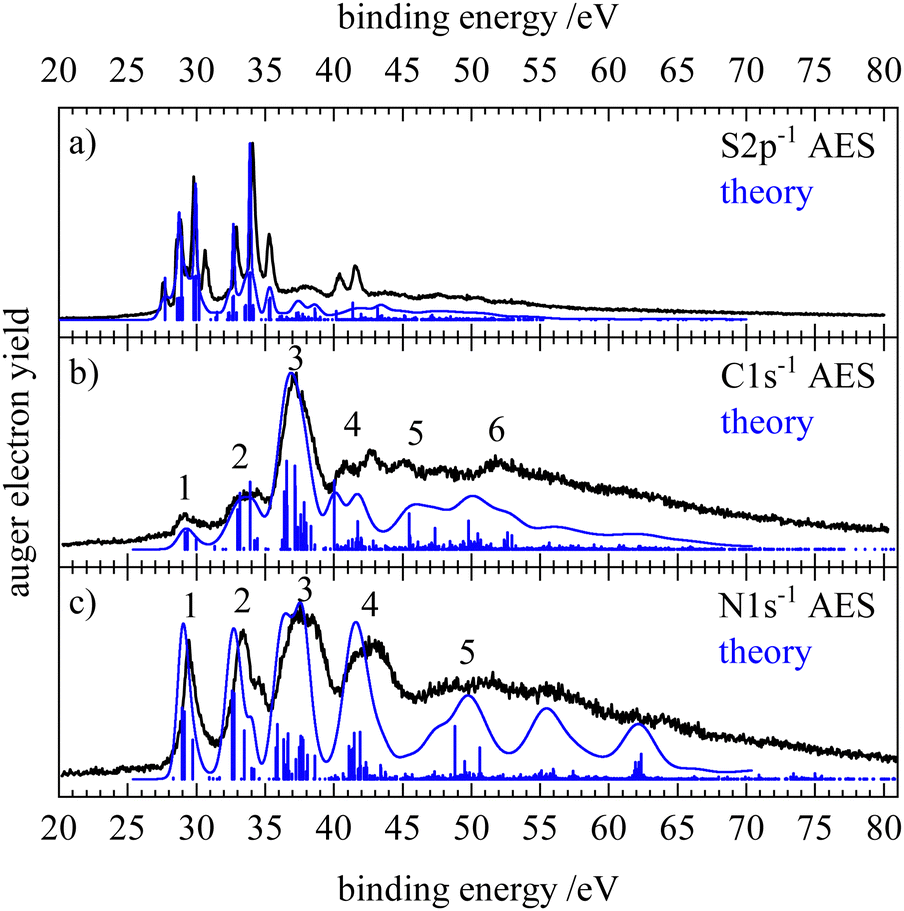 | ||
| Fig. 2 Normal Auger electron spectra of HNCS at the (a) sulfur 2p, (b) carbon 1s and (c) nitrogen 1s edge, shown in black. The blue stick spectra show the calculated energies and intensities of the individual transitions while the blue lines provide the resulting calculated spectra. | ||
The Auger electron spectra at the different edges (Fig. 2) are remarkably different: while the spectra after carbon and nitrogen 1s ionization show comparably broad features, the spectrum after sulfur 2p ionizing shows a number of narrow bands. Due to the ionization from the S2p1/2 and S2p3/2 orbital with different IEs, the S2p AES is a superposition of processes with different core holes. For isothiocyanic acid we observe processes involving two weakly bound outer valence shell electrons as distinct bands up to a binding energy of 45 eV. At higher binding energies where the spectra become featureless and less intense, processes with one weakly and one strongly bound electron appear. Final states with two inner-valence shell vacancies are shifted to even higher binding energies and are difficult to identify.42 The theoretical Auger electron spectra of HNCS are shown in Fig. 2 as blue curves and stick spectra, while the energy position, intensity, line width and orbital occupation of the final states are summarized in Table 2. The simulated spectrum of the S2p edge is separately shown in Fig. 3 for the S2p1/2 and S2p3/2 component. As the calculated final states show multiconfigurational character the orbital occupations given in Table 2 refer to the most dominant occupation found for the respective final state. More than two thousand final states were calculated, so only final states with significant contribution to the AES are listed in Table 2. For comparison, the computed energies for the lowest dicationic final states that were calculated by Wallner et al. are also given.31 A very good agreement is evident. The ground state configuration of HNCS is: (core) 7a′28a′29a′210a′211a′22a′′212a′23a′′2, where core includes S1s, N1s, C1s, S2s and three S2p orbitals. 7a′ and 8a′ are inner-valence shell orbitals constructed mainly of s-orbitals (compare MO coefficients in Table S3, ESI†).
| E f(0) | N1s−1 | C1s−1 | S2p1/2−1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. 31 | This work | I | 〈Ef〉 | W | f | I | 〈Ef〉 | W | f | I | 〈Ef〉 | W | f | |
| 3A′′(12a′−13a′′−1) | 27.77 | 28.04 | 4 | 28.32 | 1.19 | 0 | 28.53 | 1.62 | 313 | 27.72 | 0.85 | 1 | ||
| 1A′(3a′′−2) | 28.57 | 28.75 | 187 | 28.96 | 0.84 | 1 | 26 | 29.14 | 1.19 | 1 | 166 | 28.74 | 0.20 | 2 |
| 1A′′(12a′−13a′′−1) | 28.70 | 28.87 | 178 | 29.12 | 1.10 | 1 | 28 | 29.34 | 1.54 | 1 | 161 | 28.60 | 0.77 | 2 |
| 1A′(12a′−2) | 29.57 | 29.44 | 103 | 29.72 | 1.28 | 1 | 15 | 29.97 | 1.80 | 1 | 169 | 29.00 | 1.23 | 2 |
| 3A′′(2a′′−112a′−1) | 31.63 | 31.48 | 4 | 31.67 | 1.03 | 0 | 32.14 | 1.88 | 58 | 31.51 | 0.78 | |||
| 1A′(2a′′−13a′′−1) | 32.47 | 32.31 | 232 | 32.59 | 1.21 | 2 | 60 | 32.99 | 1.92 | 2 | 55 | 32.37 | 0.60 | |
| 1A′′(2a′′−112a′−1) | 32.63 | 32.42 | 226 | 32.72 | 1.30 | 2 | 85 | 33.16 | 2.11 | 2 | 49 | 32.31 | 0.77 | |
| 3A′′(10a′−13a′′−1) | 33.14 | 32.74 | 0 | 32.97 | 0.89 | 0 | 33.10 | 1.13 | 178 | 32.69 | 0.15 | 3 | ||
| 3A′(10a′−112a′−1) | 33.44 | 33.00 | 0 | 33.27 | 1.20 | 2 | 33.46 | 1.58 | 168 | 32.63 | 0.96 | 3 | ||
| 1A′(11a′−112a′−1) | 33.70 | 33.12 | 127 | 33.48 | 1.50 | 2 | 102 | 33.89 | 2.28 | 2 | 54 | 32.91 | 0.86 | 3 |
| 1A′′(10a′−13a′′−1) | 34.06 | 33.89 | 29 | 34.01 | 0.54 | 13 | 34.22 | 0.93 | 83 | 34.16 | 0.65 | 4 | ||
| 1A′(10a′−112a′−1) | 34.37 | 34.06 | 26 | 34.19 | 0.68 | 16 | 34.44 | 1.18 | 75 | 34.09 | 0.55 | 4 | ||
| 1A′(2a′′−112a′−13a′′−113a′) | 36.17 | 35.70 | 82 | 35.79 | 1.20 | 51 | 36.37 | 1.97 | 1 | 36.35 | 1.65 | |||
| 3A′(10a′−111a′−1) | 36.63 | 35.77 | 1 | 35.90 | 0.71 | 15 | 36.24 | 1.33 | 26 | 36.08 | 0.79 | |||
| 1A′′(11a′−13a′′−24a′′1) | 36.31 | 35.81 | 144 | 35.88 | 1.16 | 3 | 87 | 36.38 | 1.80 | 3 | 4 | 36.43 | 1.61 | |
| 3A′′(10a′−12a′′−1) | 35.87 | 1 | 36.00 | 0.78 | 13 | 36.39 | 1.47 | 27 | 36.20 | 0.88 | 5 | |||
| 1A′(11a′−2) | 36.82 | 36.11 | 104 | 36.33 | 1.14 | 3 | 134 | 36.55 | 1.49 | 3 | 22 | 36.55 | 1.26 | 5 |
| 1A′′(9a′−13a′′−1) | 36.92 | 36.15 | 37 | 36.43 | 1.30 | 66 | 36.59 | 1.54 | 3 | 10 | 36.66 | 1.51 | ||
| 1A′(2a′′−112a′−13a′′−113a′1) | 37.17 | 36.47 | 90 | 36.61 | 1.18 | 3 | 127 | 37.14 | 1.97 | 3 | 10 | 36.95 | 1.35 | |
| 1A′(11a′−112a′−13a′′−14a′′1) | 36.54 | 21 | 36.73 | 1.29 | 34 | 37.35 | 2.28 | 3 | 36.82 | 1.16 | ||||
| 1A′′(2a′′−13a′′−213a′1) | 37.26 | 36.56 | 120 | 36.66 | 1.21 | 3 | 96 | 37.19 | 1.92 | 3 | 3 | 37.20 | 1.64 | |
| 1A′′(2a′′−13a′′−213a′1) | 36.99 | 33 | 37.22 | 1.27 | 23 | 37.68 | 1.99 | 7 | 37.47 | 1.32 | ||||
| 1A′′(9a′−13a′′−1) | 37.17 | 84 | 37.48 | 1.24 | 3 | 53 | 37.56 | 1.41 | 3 | 42 | 37.40 | 0.93 | 5 | |
| 1A′(10a′−111a′−1) | 37.34 | 113 | 37.60 | 1.20 | 3 | 71 | 37.83 | 1.64 | 3 | 27 | 37.47 | 0.79 | ||
| 1A′(9a′−112a′−1) | 37.44 | 109 | 37.68 | 1.23 | 3 | 40 | 37.97 | 1.72 | 10 | 37.81 | 1.16 | |||
| 1A′(12a′−13a′′−213a′1) | 37.65 | 104 | 37.76 | 1.17 | 3 | 35 | 38.32 | 1.98 | 1 | 38.17 | 1.43 | |||
| 1A′′(12a′−13a′′−24a′′1) | 37.87 | 63 | 38.08 | 1.19 | 1 | 38.61 | 2.09 | 1 | 38.08 | 0.94 | ||||
| 1A′(12a′−23a′′−14a′′1) | 38.42 | 61 | 38.62 | 1.29 | 2 | 39.25 | 2.38 | 0 | 38.38 | 1.26 | ||||
| 1A′(10a′−2) | 39.79 | 10 | 40.09 | 1.31 | 104 | 40.03 | 1.20 | 4 | 64 | 40.16 | 1.35 | 6 | ||
| 1A′′(9a′−112a′−23a′′−113a′1) | 40.95 | 87 | 41.09 | 1.66 | 4 | 16 | 41.34 | 1.88 | 2 | 42.06 | 2.84 | |||
| 1A′(12a′−23a′′−213a′2) | 41.12 | 76 | 41.18 | 1.24 | 4 | 17 | 41.65 | 1.78 | 2 | 41.90 | 1.92 | |||
| 1A′′(2a′′−23a′′−113a′1) | 41.26 | 80 | 41.36 | 1.81 | 4 | 11 | 41.80 | 2.22 | 0 | 42.35 | 2.80 | |||
| 1A′(9a′−112a′−213a′1) | 41.31 | 121 | 41.46 | 1.56 | 4 | 14 | 41.64 | 1.72 | 4 | 42.24 | 2.47 | |||
| 1A′(9a′−110a′−1) | 41.50 | 16 | 41.79 | 1.32 | 42 | 41.73 | 1.21 | 4 | 43 | 41.98 | 1.59 | 6 | ||
| 1A′(10a′−12a′′−13a′′−113a′1) | 41.56 | 26 | 41.76 | 1.28 | 15 | 42.01 | 1.61 | 4 | 14 | 42.24 | 1.82 | |||
| 1A′(9a′−112a′−13a′′−14a′′1) | 41.65 | 30 | 41.81 | 1.57 | 4 | 42.29 | 2.19 | 2 | 42.32 | 1.92 | ||||
| 1A′′(9a′−112a′−13a′′−113a′1) | 41.69 | 124 | 41.89 | 1.81 | 4 | 18 | 41.99 | 1.89 | 4 | 0 | 42.76 | 2.94 | ||
| 1A′(9a′−112a′−13a′′−14a′′1) | 42.16 | 44 | 42.34 | 1.49 | 5 | 42.86 | 2.23 | 1 | 42.72 | 1.67 | ||||
| 1A′′(8a′−12a′′−1) | 44.77 | 2 | 45.04 | 1.46 | 54 | 45.47 | 2.14 | 5 | 2 | 45.28 | 1.50 | |||
| 1A′(10a′−12a′′−112a′−14a′′1) | 44.84 | 2 | 44.98 | 1.38 | 21 | 45.51 | 2.09 | 5 | 1 | 45.49 | 1.75 | |||
| 1A′(9a′−110a′−112a′−113a′1) | 46.99 | 20 | 47.30 | 1.86 | 32 | 47.36 | 1.95 | 5 | 1 | 47.87 | 2.67 | |||
| 1A′(9a′−2) | 48.49 | 139 | 48.81 | 2.81 | 5 | 12 | 48.45 | 2.62 | 0 | 49.92 | 4.52 | |||
| 1A′(8a′−110a′−1) | 49.21 | 47 | 49.50 | 1.59 | 5 | 43 | 49.80 | 2.06 | 6 | 9 | 49.78 | 1.76 | ||
| 1A′(9a′−110a′−13a′′−14a′′1) | 50.36 | 82 | 50.60 | 2.06 | 5 | 10 | 50.62 | 2.09 | 2 | 51.39 | 3.06 | |||
| 1A′(9a′−110a′−112a′′−113a′14a′′1) | 52.04 | 12 | 52.29 | 1.92 | 25 | 52.64 | 2.37 | 0 | 52.86 | 2.42 | ||||
| 1A′(7a′−110a′−1) | 52.35 | 2 | 52.59 | 1.47 | 21 | 52.93 | 1.96 | 9 | 52.97 | 1.77 | ||||
| 1A′′(8a′−111a′−112a′−14a′′1) | 55.73 | 26 | 55.96 | 2.16 | 6 | 56.41 | 2.67 | 0 | 56.78 | 2.93 | ||||
| 1A′(9a′−110a′−111a′−12a′′−113a′14a′′1) | 61.64 | 44 | 61.94 | 2.49 | 4 | 62.22 | 2.84 | 0 | 62.75 | 3.32 | ||||
| 1A′(7a′−19a′−1) | 61.93 | 44 | 62.17 | 2.55 | 2 | 62.55 | 2.97 | 0 | 63.09 | 3.41 | ||||
| 1A′(9a′−110a′−111a′−12a′′−113a′14a′′1) | 62.09 | 66 | 62.36 | 2.57 | 3 | 62.64 | 2.91 | 0 | 63.16 | 3.31 | ||||
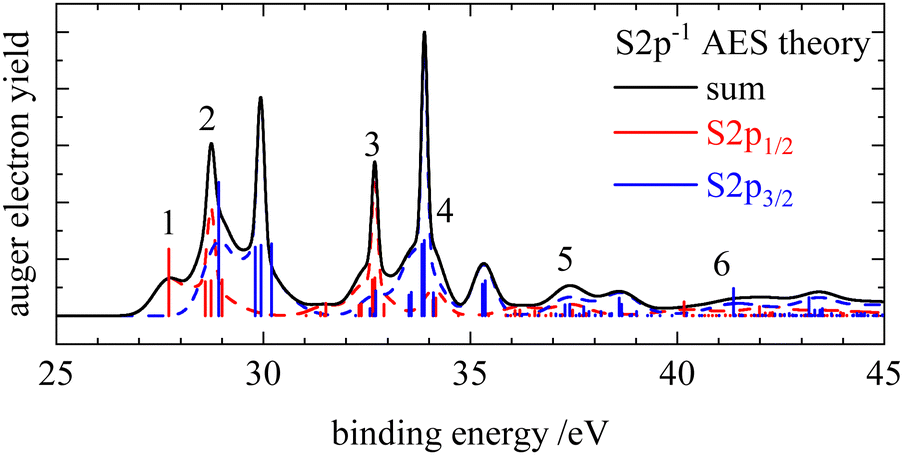 | ||
| Fig. 3 Calculated S2p Auger electron spectra of isothiocyanic acid in black. The contributions of the individual components, S2p1/2 and S2p3/2, are shown in red and blue. Only transitions from the S2p1/2 are labelled, transitions from the S2p3/2 component appear shifted by +1.2 eV to higher binding energies. | ||
In the Auger electron spectra of isothiocyanic acid the feature at the lowest binding energy (up to 31 eV) can be assigned to a decay into triplet and singlet final states with two holes in the highest occupied molecular orbitals, 12a′ and 3a′′, corresponding to 3A′′(12a′−13a′′−1), 1A′(3a′′−2 and 12a′−2) and 1A′′(12a′−13a′′−1) final states (see Table 2). On the sulfur edge this feature is split into several bands, see top trace of Fig. 2. The first one is experimentally found at 27.6 eV and corresponds to the S2p1/2−1 → 3A′′(12a′−13a′′−1) decay that is calculated to occur at an average binding energy of 27.7 eV (feature 1 in Fig. 3). These values of the double ionization energy (DIE) of HNCS warrant comparison to the recent DIE values of Wallner et al.31 From the onset of the HNCS2+ signal in their experiments they derived an adiabatic DIE of 27.1 ± 0.1 eV, which agrees well with their computed value of 27.2 eV. Their computed vertical DIE of 27.77 eV is on the other hand in excellent agreement with our value. This confirms the agreement between the two computational and experimental approaches.
While the lowest 3A′′(12a′−13a′′−1) final state gives rise to an intense signal in the S2p AES the corresponding transition is basically invisible in the N1s and C1s spectra. This can be explained with symmetry arguments. For the neon atom, the Auger–Meitner decay of the 1s hole state to the 3P(2p−2) final state is parity forbidden in LS coupling.77,78 Thus, for low Z elements KLL Auger transitions of atoms are extremely weak if they generate two triplet coupled holes in orbitals with 2p character. The one-center approximation assumes that molecular Auger transition rates are essentially determined by contributions of atomic orbitals (AOs) of the involved MOs.34,46,63,66 Accordingly, KLL transition rates are predicted to be small if a triplet final state is reached which has two holes in MOs with large 2p character at the core hole atom. This applies to the atomic orbital coefficients of the 12a′ and 3a′′ MOs of HNCS (see Table S3, ESI†) as for MOs with higher energy in general which avoid contributions of the low energy 2s AOs. The weak intensity of the triplet ground state in the experimental KLL AES of HNCS and of many low binding energy triplet states in molecules33,46,63,79 is well established and fully in line with the predictions of the one-center approximation. On the contrary, low lying triplet states give rise to strong signals in the S2p AES of HNCS and other L2,3MM transitions of atoms and molecules48,80,81 as they are here symmetry allowed.
The S2p1/2 transitions into the singlet states (feature 2 in Fig. 3) are shifted to higher binding energies compared to the triplet ground state and overlap with the 3A′′(12a′−13a′′−1) transition of the S2p3/2 component that shows the same transitions shifted by +1.2 eV. The simulation describes the narrow shape of the bands on the sulfur edge but does not reproduce the fourth band of the first multiplet that is found at 30.6 eV. A comparison of our vertical binding energies for the states contributing to feature 2 and the signal at about 30 eV in the theoretical S2p AES (Fig. 3) with the values of Wallner et al.31 indicates that we underestimate the relative energy of the 1A′(12a′−2) final state by about 0.3 eV as compared to the other states involved. This provides confidence to assign the signal in the experimental AES at 30.6 eV to the S2p3/2-1A′(12a′−2) decay. On the carbon and nitrogen edge only a single feature (labelled 1) is found in the region below 31 eV.
On the carbon edge we observe a much lower intensity for low binding energy features than on the sulfur and nitrogen edge. As the holes of the final states occur in the HOMO (3a′′) and/or HOMO−1 (12a′) which both have a nodal plane near the carbon atom (compare Fig. 1), the absolute value of the MO coefficient of the C2p atomic orbital is much smaller than for the S2p and N2p atomic orbitals (compare Table S3, ESI†). The decay rate depends on the MO coefficients of the core ionized atom to the involved molecular orbitals,35 so the decay rate for the Auger–Meitner process involving HOMO and HOMO−1 is much lower on the carbon edge compared to the sulfur and nitrogen edge. The second band on the nitrogen and carbon edge between 32 and 35 eV (feature 2) corresponds to transitions into 1A′(2a′′−13a′′−1), 1A′′(2a′′−112a′−1) and 1A′(11a′−112a′−1) final states. Again, one observes a lower intensity on the carbon edge due to final states with one hole in the HOMO or HOMO−1. On the nitrogen edge a shoulder appears at 34.7 eV as the 1A′(11a′−112a′−1) final state is shifted to higher energies. As a result of the narrower peak width on the nitrogen edge compared to the carbon edge, this feature can only be resolved on the nitrogen edge. On the sulfur edge these final states have low intensity due to the small absolute values of the 2a′′ and 11a′ MO coefficients of sulfur. Instead the two triplet states 3A′′(10a′−13a′′−1) and 3A′(10a′−112a′−1) dominate in this energy region. The experimental feature at 32.9 eV originates from the decay of the S2p1/2 hole (feature 3 in Fig. 3) while the most intense signal at 34.1 eV is caused by the decay of the S2p3/2 hole. For the latter signal we also find a weaker contribution of S2p1/2 transitions to the corresponding singlet states (feature 4 in Fig. 3). In the region 35–40 eV we observe the highest intensity in the AES on the carbon and nitrogen edge which is in accordance with the prediction from the calculation. Transitions in this region show dominant final state occupations with holes in the nearly degenerate 2a′′ and 11a′ orbitals including excited (shake-up) configurations where electrons are excited from the highest occupied to the lowest unoccupied MOs. On the carbon edge we find a particularly high intensity for a 1A′(11a′−2) final state. As both 2a′′ and 11a′ are mainly located on the carbon and nitrogen atom we observe a much lower intensity in the S2p AES in this region (feature 5 in Fig. 3). In the higher binding energy region from 40–45 eV a doublet is observed on the sulfur edge. Our calculations assign these features at 40.4 and 41.6 eV mainly to the S2p1/2 and S2p3/2 decay to the 1A′(10a′−2) and 1A′(9a′−110a′−1) final states. The same final states contribute to feature 4 in the carbon AES that also exhibits a double peak structure.
At binding energies >45 eV the Auger electron spectra show less pronounced features. Transitions to final states that exhibit one hole in inner-valence orbitals (8a′, 7a′) become more important in this energy region. For all edges an increase in the width of the final state signals at high binding energies is predicted (compare Table 2). Together with an increased density of final states at higher binding energies, this results in broad, overlapping features and explains the loss of structure in the AES. In this binding energy region final states with a hole in the 8a′ orbital, 1A′′(8a′−12a′′−1) and 1A′(8a′−110a′−1), contribute to feature 5 and 6 on the carbon edge. On the nitrogen edge feature 5 corresponds mainly to a 1A′(9a′−2) final state. Its high intensity is due to high MO coefficients of the 9a′ orbital at the nitrogen atom.
The assignments of the normal AES of isothiocyanic acid are confirmed by a comparison to the isosteric molecule carbonyl sulfide, OCS, whose Auger electron spectra are well studied.73,81,82 While the linear OCS molecule has two sets of doubly degenerate π-orbitals, their degeneracy is lifted when going from the linear geometry in OCS to the bent geometry in HNCS. A full comparison of the Auger electron spectra of the two compounds can be found in Section S4 of the ESI.†
It should be noted, that the measurement of the S2s Auger electron spectrum was not possible in this experiment. This is due to the short decay time of the sulfur 2s core hole that results from a fast L1L2,3V Coster–Kronig transition, where an electron from a 2p shell fills the 2s hole. From the sulfur 2s X-ray photoelectron spectrum (see Fig. S4, ESI†) an IE(core) of 234.3 eV was determined. Furthermore it is possible to extract the lifetime of the S2s ionized state from the Lorentzian part of the experimental photoelectron line. To do so the curve was fitted to a Voigt function with a Gaussian width of 0.13 eV which corresponds to the superposition of the photon bandwidth (120 meV), the Doppler broadening83 (25 meV) and the resolution of the electron spectrometer (40 meV).84 A Lorentzian width of 1.72 eV was determined from which a 2s core hole lifetime of 0.4 fs is extracted using the uncertainty principle. For sulfur 2s core holes in other molecules such as CS2 or H2S lifetime widths of 1.85 or 1.8 eV were extracted, respectively.85,86
3.2 NEXAFS/XAS spectra
In order to investigate transitions from core orbitals into unoccupied orbitals we measured X-ray absorption (XAS or NEXAFS) spectra of HNCS which are shown in Fig. 4. On the nitrogen edge the signals indicated by an asterisk result from a N2 contamination. In order to measure a spectrum free of N2 the partial electron yield was recorded and is shown as grey dashed curve in Fig. 4(c). Contaminations from residual N2 in the experimental chamber are only visible in the total ion yield spectrum as the channeltron that was used to monitor the total ion yield is located further away from the molecular beam compared to the electron analyzer. The partial electron yield only represents the low photon energy part of the NEXAFS spectrum well, because only low binding energy electrons (9–18 eV) were recorded. Vertical transition energies into different core excited states were also evaluated at different levels of theory and are given in Table 1.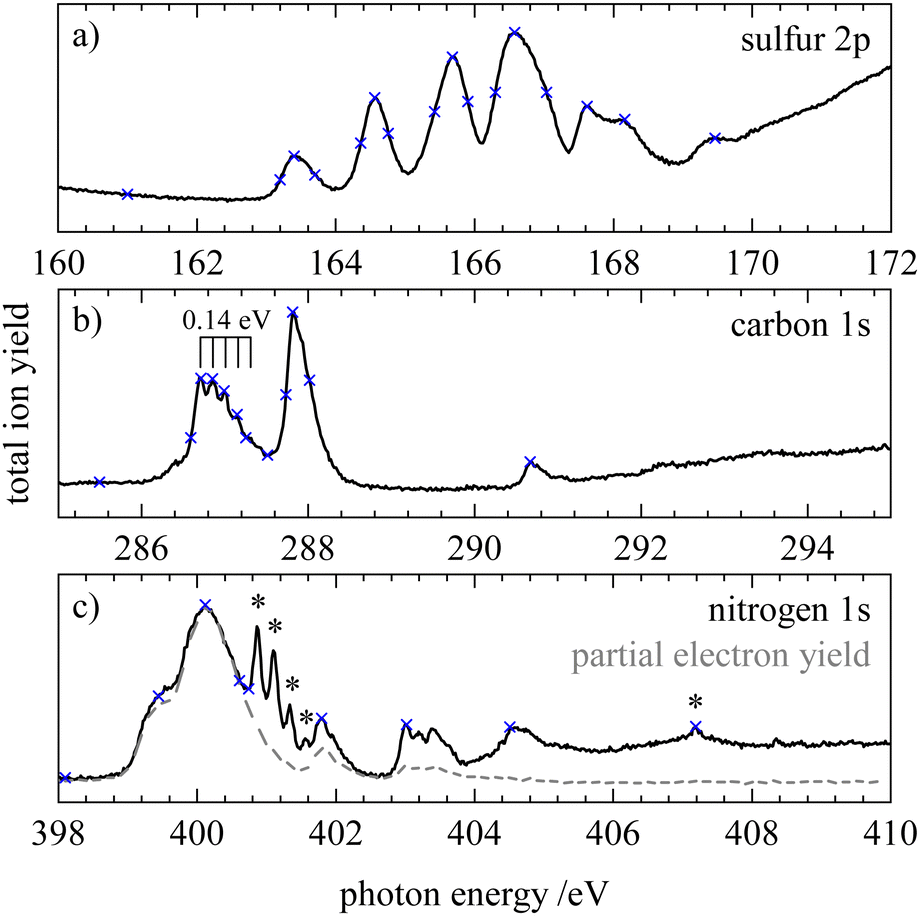 | ||
| Fig. 4 NEXAFS spectra of HNCS at the (a) sulfur 2p, (b) carbon 1s and (c) nitrogen 1s edge in black and partial electron yield spectrum at the (c) nitrogen 1s edge in grey. Blue crosses indicate photon energies where (off-) resonant Auger electron spectra were measured. Asterisks mark signals from a N2 contamination. | ||
On the sulfur 2p edge (Fig. 4a) we observe several bands between 163 and 170 eV. They result from transitions of the S2p3/2 and S2p1/2 level, whereby the latter appear shifted by 1.2 eV to higher photon energies. The first band at 163.4 eV belongs to the S2p3/2 → 13a′(LUMO) transition and is in good agreement with the calculated value of 163.66 eV for this transition on the MCCEPA level of theory. The second band at 164.6 eV is a superposition of the corresponding transition of the S2p1/2 component (S2p1/2 → 13a′) and the S2p3/2 transition into 4a′′(LUMO+1) which is calculated to lie 1.1 eV higher than the transition into the LUMO. The next bands indicate transitions into higher core excited states. By comparison with the photoabsorption spectrum of OCS87,88 we tentatively assign these bands to the transition into the 14a′ and into s- and d-Rydberg series. While the energy separation of the low-lying core excited states is close to the value of the spin–orbit splitting, broader bands at higher photon energies result from a different energy separation of high-energy core-excited states. In the NEXAFS spectrum below the sulfur 2s edge (not shown) we observed only a single broad signal at 227.8 eV.
At the carbon edge (Fig. 4b) the two intense bands at 286.7 and 287.8 eV are assigned to the C1s → 13a′ and C1s → 4a′′ transition, respectively. The first band shows a progression with a spacing of 0.14 eV. In contrast to the sulfur 2p spectrum the energies for these transitions are less well reproduced by our calculations. A possible explanation for this disagreement is that the potential energy surfaces for these core-excited states have a maximum at the ground state structure with respect to the N–C–S bending motion. In the OCS congener it is well established that the corresponding C1s−14π states show a Renner–Teller effect with this behavior.89 At higher photon energies a third small signal is observed at 290.7 eV, coinciding with the position of the broad 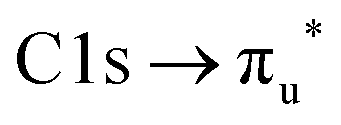 transition of CO2.90 However, as we see no signs of CO2 when we compare our resonant Auger electron spectrum at this photon energy with the one of pure CO2, we tentatively assign this band to a C1s transition in HNCS.
transition of CO2.90 However, as we see no signs of CO2 when we compare our resonant Auger electron spectrum at this photon energy with the one of pure CO2, we tentatively assign this band to a C1s transition in HNCS.
At the nitrogen edge (Fig. 4c) the most intense signal is observed at 400.1 eV and features a shoulder at lower photon energies (399.4 eV). Again, we attribute these features to the transitions into the 4a′′(13a′) orbital, which is calculated to have a vertical excitation energy of 400.24 eV (399.49 eV) at the MCCEPA level. Transitions to higher core excited states are observed at 401.8, 403.0 and 404.5 eV which are presumably Rydberg states. The band at 403.0 eV exhibits a progression of approximately 190 meV.
We investigated the resonant Auger electron spectra after excitation at the photon energies indicated with blue crosses in the NEXAFS spectra in Fig. 4. These spectra will be discussed in the following section.
3.3 Resonant Auger spectroscopy
Fig. 5–7 show the resonant Auger electron spectra of HNCS for the excitation into the two lowest unoccupied orbitals, 13a′ and 4a′′, at the C1s, N1s and S2p edge. For resonant processes binding energies are determined as difference of the excitation energy and the measured kinetic energy of the Auger electrons. In order to investigate which processes are resonantly enhanced, off-resonant spectra were measured below the first resonance at each edge and are shown in the top trace of the respective figure. A complete discussion of all RAES indicated in Fig. 4 can be found in Section S5 of the ESI.† Calculations of the RAES were carried out for the carbon and nitrogen edge and computed spectra are shown in the respective figures in blue. Corresponding calculated data is given in Table 3.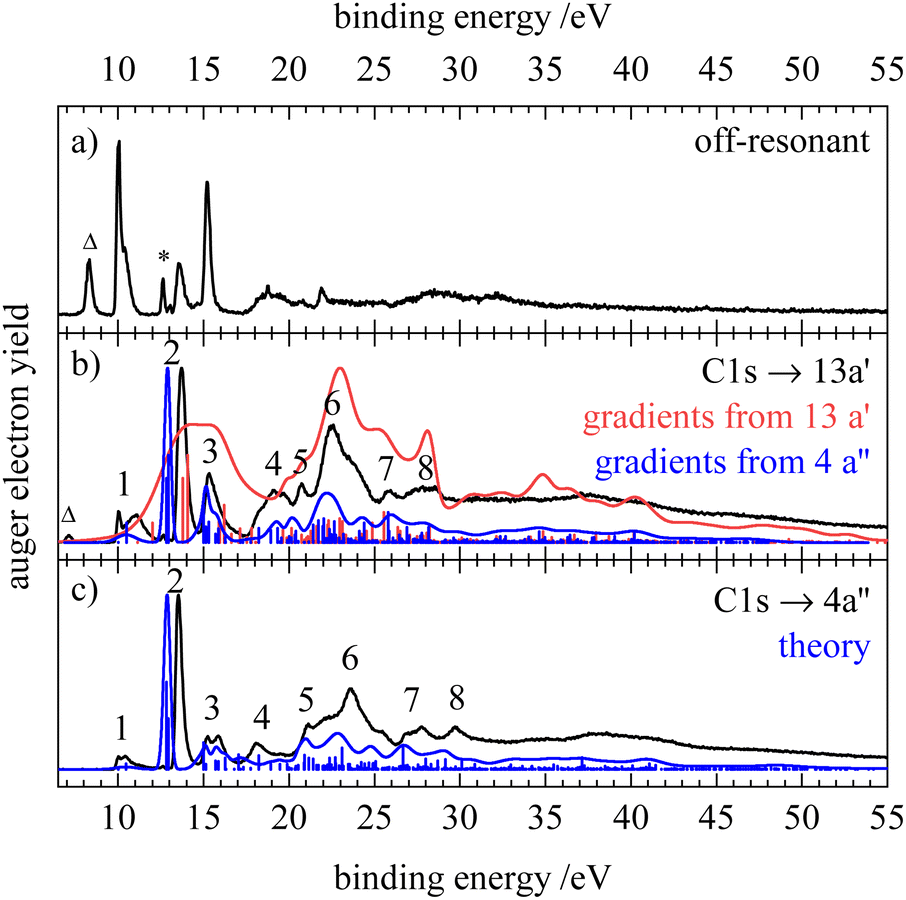 | ||
| Fig. 5 Off-resonant spectrum at 285.49 eV (a) and resonant Auger electron spectra of HNCS at 286.71 eV (b) and at 287.81 eV (c) at the carbon edge in black. The asterisk at 12.62 eV indicates a signal from water. The band marked by a triangle is due to carbon 1s ionization by second harmonic light. The computed spectra and individual final states are shown in blue and red. | ||
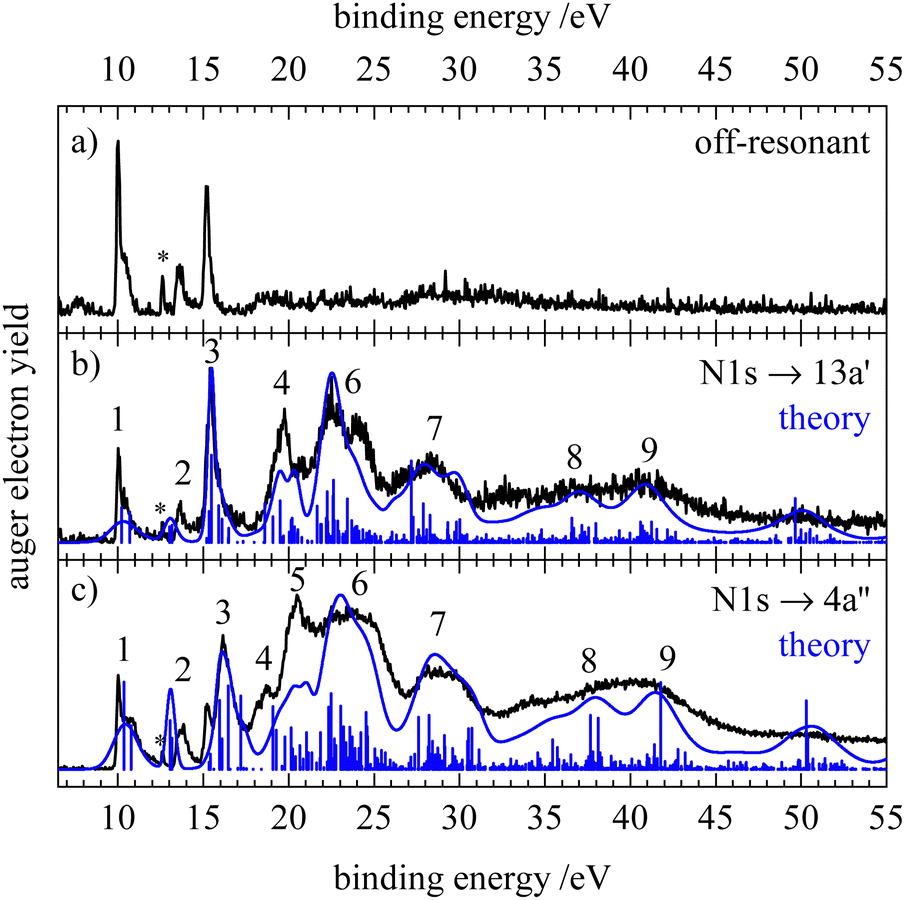 | ||
| Fig. 6 Off-resonant spectrum at 398.12 eV (a) and resonant Auger electron spectra of HNCS at 399.42 eV (b) and 400.12 eV (c) at the nitrogen edge. The asterisk at 12.62 eV indicates a signal from water. The computed spectra and individual final states are shown in blue. | ||
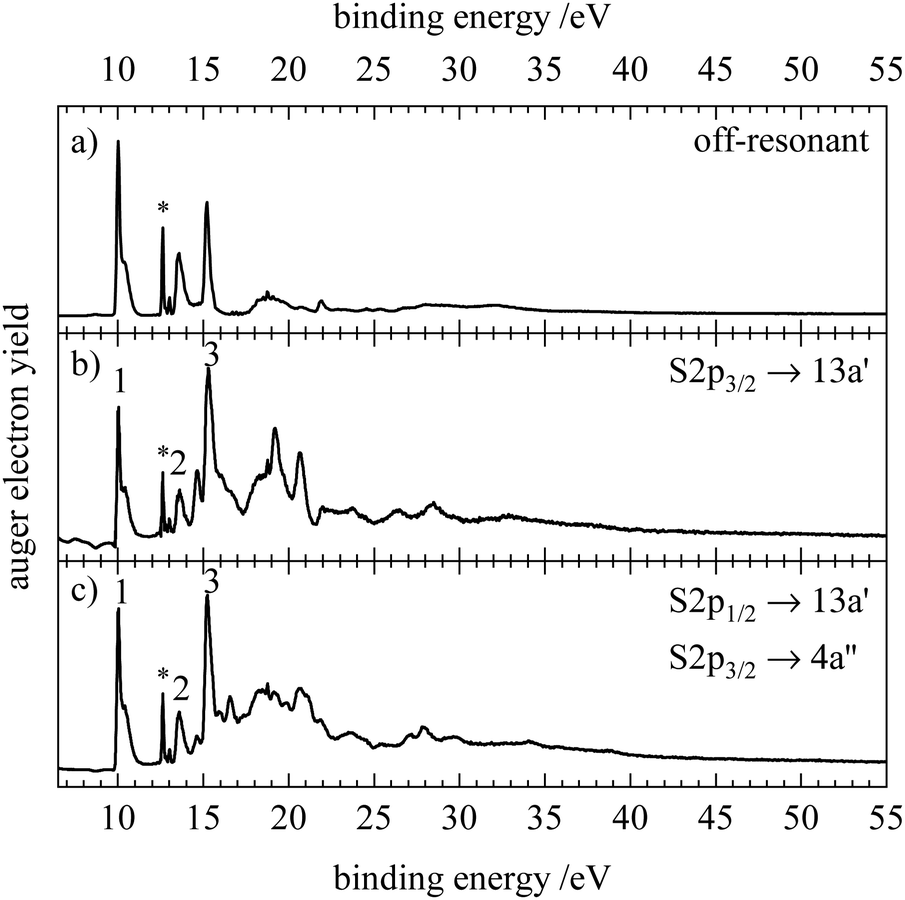 | ||
| Fig. 7 Off-resonant spectrum at 161.00 eV (a) and resonant Auger electron spectra of HNCS at 163.40 eV (b) and 164.56 eV (c) at the sulfur edge. Spectra b and c were corrected for the contribution from direct photoemission. The asterisk at 12.62 eV indicates a signal from water. | ||
| E f(0) | N1s−113a′1 | N1s−14a′′1 | C1s−113a′1 | C1s−14a′′1 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | 〈Ef〉 | W | f | I | 〈Ef〉 | W | f | I | 〈Ef〉 | W | f | I | 〈Ef〉 | W | f | ||
| 2A′′(3a′′−1) | 9.76 | 82 | 10.22 | 1.58 | 1 | 136 | 10.36 | 1.52 | 1 | 1 | 10.03 | 0.78 | 1 | 1 | 10.03 | 0.78 | 1 |
| 2A′(12a′−1) | 10.06 | 71 | 10.69 | 2.28 | 1 | 68 | 10.78 | 1.93 | 1 | 63 | 10.49 | 1.29 | 1 | 23 | 10.06 | 1.28 | 1 |
| 2A′′(12a′′−1) | 12.95 | 36 | 13.03 | 0.71 | 2 | 76 | 13.05 | 0.53 | 2 | 208 | 12.82 | 0.44 | 2 | 369 | 12.82 | 0.44 | 2 |
| 2A′(11a′−1) | 12.98 | 40 | 13.16 | 0.97 | 2 | 48 | 13.16 | 0.60 | 2 | 281 | 12.93 | 0.39 | 2 | 214 | 12.93 | 0.38 | 2 |
| 2A′(10a′−1) | 14.67 | 10 | 15.19 | 1.80 | 3 | 11 | 15.33 | 1.72 | 3 | 120 | 15.00 | 0.96 | 3 | 113 | 15.00 | 0.96 | 3 |
| 2A′′(12a′−13a′−113a′1) | 15.21 | 73 | 15.32 | 0.56 | 3 | 24 | 15.42 | 0.67 | 3 | 51 | 15.13 | 0.33 | 3 | 22 | 15.13 | 0.33 | 3 |
| 2A′′(3a′′−213a′1) | 15.47 | 204 | 15.48 | 0.41 | 3 | 1 | 15.61 | 0.69 | 66 | 15.30 | 0.57 | 3 | 0 | 15.30 | 0.57 | ||
| 2A′′(12a′−13a′′−113a′1) | 15.82 | 86 | 15.90 | 0.65 | 3 | 108 | 15.95 | 0.59 | 3 | 27 | 15.70 | 0.44 | 3 | 36 | 15.70 | 0.44 | 3 |
| 2A′(12a′−13a′′−14a′′1) | 15.90 | 54 | 16.10 | 1.10 | 3 | 48 | 16.08 | 0.67 | 3 | 44 | 15.87 | 0.51 | 3 | 35 | 15.87 | 0.51 | 3 |
| 2A′(12a′−13a′′−14a′′1) | 16.31 | 34 | 16.50 | 1.10 | 131 | 16.47 | 0.67 | 3 | 17 | 16.27 | 0.55 | 48 | 16.27 | 0.55 | 3 | ||
| 2A′′(12a′−24a′′1) | 16.97 | 3 | 17.32 | 1.86 | 114 | 17.19 | 1.02 | 3 | 0 | 17.06 | 1.02 | 62 | 17.05 | 1.02 | |||
| 2A′(9a′−1) | 18.54 | 91 | 18.60 | 2.07 | 4 | 98 | 19.07 | 2.43 | 4 | 47 | 18.23 | 1.97 | 4 | 57 | 18.24 | 1.96 | 4 |
| 2A′′(12a′′−112a′−113a′1) | 18.92 | 60 | 19.07 | 0.73 | 4 | 61 | 19.26 | 1.10 | 4 | 40 | 18.94 | 0.71 | 4 | 22 | 18.94 | 0.71 | |
| 2A′(11a′−112a′−113a′1) | 19.37 | 98 | 19.49 | 0.66 | 4 | 5 | 19.62 | 0.89 | 46 | 19.34 | 0.60 | 4 | 0 | 19.35 | 0.60 | ||
| 2A′(12a′−13a′′−14a′′1) | 19.68 | 24 | 20.04 | 1.39 | 65 | 20.15 | 1.32 | 5 | 7 | 19.88 | 0.88 | 23 | 19.88 | 0.88 | |||
| 2A′(2a′′−13a′′−113a′1) | 19.97 | 52 | 20.14 | 0.75 | 4 | 13 | 20.25 | 0.88 | 8 | 19.98 | 0.53 | 1 | 19.98 | 0.53 | |||
| 2A′′(2a′′−13a′′−14a′′1) | 20.00 | 57 | 20.19 | 0.93 | 4 | 32 | 20.23 | 0.76 | 20 | 19.99 | 0.50 | 5 | 7 | 19.99 | 0.50 | ||
| 2A′(10a′−112a′−113a′1) | 20.41 | 31 | 20.54 | 0.70 | 38 | 20.66 | 0.86 | 37 | 20.39 | 0.57 | 5 | 13 | 20.39 | 0.57 | |||
| 2A′′(10a′−13a′′−113a′1) | 20.57 | 22 | 20.51 | 0.61 | 4 | 20.69 | 0.89 | 24 | 20.34 | 0.82 | 5 | 3 | 20.34 | 0.81 | |||
| 2A′′(11a′−112a′−14a′′1) | 20.89 | 4 | 21.10 | 1.26 | 58 | 21.05 | 0.74 | 5 | 1 | 20.88 | 0.70 | 29 | 20.87 | 0.69 | |||
| 2A′(10a′−13a′′−14a′′1) | 21.09 | 1 | 21.12 | 0.69 | 26 | 21.15 | 0.67 | 2 | 20.90 | 0.62 | 60 | 20.90 | 0.62 | 5 | |||
| 2A′′(10a′−112a′−14a′′1) | 21.09 | 3 | 21.35 | 1.30 | 26 | 21.34 | 0.90 | 3 | 21.15 | 0.76 | 48 | 21.15 | 0.76 | 5 | |||
| 2A′′(11a′−12a′′−113a′1) | 21.81 | 85 | 21.54 | 1.26 | 6 | 10 | 21.52 | 1.42 | 57 | 21.30 | 1.37 | 44 | 21.30 | 1.37 | 5 | ||
| 2A′(11a′−213a′1) | 21.95 | 42 | 21.92 | 0.82 | 3 | 22.05 | 1.08 | 70 | 21.72 | 0.92 | 6 | 17 | 21.72 | 0.92 | |||
| 2A′′(2a′′−112a′−13a′′−113a′14a′′1) | 21.98 | 43 | 21.85 | 0.95 | 58 | 21.87 | 1.08 | 0 | 21.65 | 1.07 | 1 | 21.65 | 1.07 | ||||
| 2A′(10a′−111a′−113a′1) | 22.33 | 119 | 22.26 | 0.92 | 6 | 8 | 22.25 | 1.02 | 77 | 22.04 | 1.00 | 6 | 15 | 22.04 | 1.00 | ||
| 2A′′(2a′′−112a′−13a′′−113a′14a′′1) | 22.56 | 20 | 22.33 | 1.21 | 97 | 22.34 | 1.36 | 6 | 13 | 22.12 | 1.34 | 14 | 22.12 | 1.34 | |||
| 2A′(11a′−12a′′−14a′′1) | 22.56 | 15 | 22.56 | 1.02 | 118 | 22.47 | 1.17 | 6 | 13 | 22.23 | 1.11 | 20 | 22.24 | 1.11 | |||
| 2A′(2a′′−213a′1) | 22.70 | 108 | 22.59 | 1.04 | 6 | 48 | 22.67 | 1.24 | 43 | 22.37 | 1.14 | 6 | 49 | 22.37 | 1.14 | 6 | |
| 2A′′(2a′′−112a′−113a′1) | 22.71 | 145 | 22.63 | 0.92 | 6 | 16 | 22.65 | 1.05 | 10 | 22.41 | 1.01 | 14 | 22.41 | 1.01 | |||
| 2A′(9a′−112a′−113a′1) | 22.75 | 50 | 22.86 | 0.96 | 3 | 23.09 | 1.39 | 67 | 22.64 | 0.93 | 6 | 7 | 22.65 | 0.93 | |||
| 2A′(9a′−13a′′−14a′′1) | 22.97 | 26 | 22.93 | 1.03 | 56 | 23.11 | 1.32 | 16 | 22.70 | 1.09 | 49 | 22.71 | 1.09 | 6 | |||
| 2A′′(10a′−12a′′−113a′1) | 23.05 | 20 | 22.95 | 1.01 | 98 | 23.04 | 1.21 | 6 | 25 | 22.73 | 1.11 | 26 | 22.73 | 1.11 | |||
| 2A′′(9a′−112a′−14a′′1) | 23.27 | 3 | 23.36 | 1.12 | 66 | 23.58 | 1.51 | 1 | 23.11 | 1.07 | 91 | 23.12 | 1.07 | 6 | |||
| 2A′(11a′−112a′−13a′′−113a′14a′′1) | 23.58 | 102 | 23.43 | 1.25 | 6 | 31 | 23.35 | 1.39 | 27 | 23.17 | 1.34 | 13 | 23.17 | 1.34 | |||
| 2A′′(11a′−112a′−213a′14a′′1) | 24.73 | 2 | 24.70 | 1.38 | 89 | 24.54 | 1.28 | 6 | 2 | 24.43 | 1.30 | 23 | 24.43 | 1.29 | |||
| 2A′(10a′−213a′1) | 26.02 | 12 | 25.97 | 1.64 | 1 | 26.41 | 1.89 | 98 | 25.79 | 1.58 | 7 | 8 | 25.80 | 1.57 | |||
| 2A′′(10a′−24a′′1) | 26.90 | 1 | 26.93 | 1.11 | 16 | 27.15 | 1.47 | 0 | 26.68 | 1.10 | 104 | 26.68 | 1.10 | 7 | |||
| 2A′′(9a′−12a′′−113a′1) | 27.64 | 191 | 27.18 | 2.82 | 7 | 0 | 27.51 | 2.65 | 49 | 26.90 | 2.57 | 8 | 0 | 26.91 | 2.56 | ||
| 2A′(9a′−12a′′−14a′′1) | 27.76 | 13 | 27.51 | 1.59 | 82 | 27.60 | 1.78 | 7 | 10 | 27.25 | 1.65 | 7 | 27.26 | 1.64 | |||
| 2A′(9a′−111a′−113a′1) | 28.13 | 91 | 27.89 | 1.60 | 7 | 13 | 28.08 | 1.72 | 21 | 27.67 | 1.61 | 9 | 27.67 | 1.60 | |||
| 2A′(9a′−110a′−113a′1) | 28.37 | 17 | 28.40 | 1.29 | 33 | 28.72 | 1.68 | 49 | 28.16 | 1.28 | 8 | 1 | 28.16 | 1.28 | |||
| 2A′′(9a′−111a′−14a′′1) | 28.44 | 3 | 28.24 | 1.41 | 85 | 28.23 | 1.62 | 7 | 2 | 27.98 | 1.50 | 35 | 27.99 | 1.50 | 8 | ||
| 2A′(9a′−110a′−113a′1) | 28.46 | 62 | 28.28 | 2.09 | 7 | 20 | 28.63 | 2.21 | 3 | 27.99 | 1.97 | 10 | 28.00 | 1.96 | |||
| 2A′′(9a′−110a′−14a′′1) | 29.24 | 5 | 29.42 | 1.08 | 33 | 29.60 | 1.41 | 1 | 29.15 | 0.88 | 42 | 29.15 | 0.88 | 8 | |||
| 2A′′(8a′−13a′′−113a′1) | 29.98 | 49 | 29.83 | 1.32 | 7 | 6 | 29.98 | 1.53 | 15 | 29.62 | 1.37 | 1 | 29.62 | 1.36 | |||
| 2A′(10a′−111a′−113a′1) | 30.11 | 53 | 30.01 | 1.18 | 7 | 9 | 30.07 | 1.41 | 6 | 29.74 | 1.25 | 10 | 29.75 | 1.25 | |||
| 2A′(9a′−12a′′−14a′′1) | 30.54 | 3 | 30.40 | 1.47 | 63 | 30.52 | 1.71 | 7 | 4 | 30.10 | 1.52 | 33 | 30.11 | 1.52 | |||
| 2A′′(10a′−111a′−13a′′−14a′′2) | 30.72 | 1 | 30.71 | 1.19 | 64 | 30.73 | 1.36 | 7 | 0 | 30.45 | 1.17 | 4 | 30.45 | 1.17 | |||
| 2A′(9a′−213a′1) | 36.88 | 56 | 36.57 | 2.18 | 8 | 2 | 36.79 | 2.27 | 17 | 36.30 | 2.08 | 1 | 36.32 | 2.07 | |||
| 2A′(9a′−110a′−13a′′−113a′′4a′′1) | 37.47 | 40 | 37.16 | 2.60 | 8 | 11 | 37.53 | 2.58 | 11 | 36.88 | 2.37 | 0 | 36.90 | 2.36 | |||
| 2A′′(7a′−110a′−14a′′1) | 37.66 | 0 | 37.48 | 2.01 | 78 | 37.68 | 2.24 | 8 | 0 | 37.13 | 1.98 | 51 | 37.14 | 1.97 | |||
| 2A′′(9a′−110a′−112a′−113a′14a′′1) | 38.10 | 0 | 37.79 | 2.68 | 80 | 38.12 | 2.73 | 8 | 0 | 37.47 | 2.46 | 5 | 37.48 | 2.46 | |||
| 2A′(8a′−111a′−13a′′−113a′14a′′1) | 38.18 | 45 | 37.98 | 1.78 | 8 | 11 | 38.19 | 1.96 | 15 | 37.74 | 1.74 | 2 | 37.75 | 1.73 | |||
| 2A′′(7a′−13a′′−213a′14a′′1) | 40.91 | 48 | 40.60 | 1.84 | 9 | 1 | 40.65 | 2.02 | 8 | 40.38 | 1.85 | 1 | 40.39 | 1.85 | |||
| 2A′(7a′−112a′−13a′′−113a′14a′′1) | 41.49 | 48 | 41.25 | 1.89 | 9 | 20 | 41.38 | 2.10 | 5 | 40.97 | 1.87 | 3 | 40.98 | 1.87 | |||
| 2A′′(7a′−111a′−14a′′1) | 41.93 | 1 | 41.78 | 1.86 | 135 | 41.79 | 2.17 | 9 | 0 | 41.43 | 1.89 | 17 | 41.43 | 1.89 | |||
In the resonant Auger electron spectra, participator states occur at the position of the valence photoelectron lines in the off-resonant spectrum. The off-resonant spectrum of HNCS (Fig. 5a) shows intense signals at 10.0, 13.5 and 15.2 eV which agrees with the transitions found at 10.05 ± 0.1, 13.33 ± 0.03 and 15.2 eV in the He I photoelectron spectrum.8 For the C1s → 13a′(LUMO) excitation of HNCS a resonant enhancement of the bands at binding energies of 13.5 and 15.2 eV (features 2 and 3 in Fig. 5b) is found. Furthermore, the bands at binding energies >18 eV are enhanced by resonant Auger–Meitner decay. Together with the experimental RAES the corresponding calculated spectrum is shown in red. The agreement between the two spectra is not satisfactory, because the calculation overestimates the widths of the bands significantly. The same effect was observed before for the RAES of the corresponding state in the HNCO molecule.33 As some of the virtual valence orbitals have very strongly antibonding (σ*-type) character, the gradients of the C1s−113a′ intermediate state along the vibrational modes are overestimated by the CI approach. A much better agreement was found when employing the gradients from the C1s−14a′′ final state calculations for the RAES of the C1s−113a′ core excited state (blue spectrum in Fig. 5b). Actually the gradients of the two core excited states can be expected to be rather similar but for the C1s−14a′′ state they are better predicted by our CI approach as the excited electron resides here in a 4a′′ orbital which is antisymmetric with respect to the molecular plane and thus cannot admix symmetric σ*-type character.
The spectrum depicted in blue in Fig. 5(b) allows for an assignment based on the calculation. The feature at lowest binding energies, 1, is assigned to single hole (1h) final states with a vacancy in one of the nearly degenerate orbitals 3a′′(HOMO) or 12a′(HOMO−1). The latter one is slightly shifted to higher energies and appears as a shoulder. The assignment of the states agrees again with the photoelectron spectrum.8 These bands show only small resonant enhancement, similar to the low intensity of the first band in the C1s AES, as the involved molecular orbitals have a nodal plane near the carbon atom and therefore only small contributions of carbon AOs. Compared to the off-resonant spectrum the high-energy shoulder of feature 1 is displaced to higher binding energy and broadened, probably due to a vibrational sequence with a shifted vertical ionization potential. Feature 2 at 13.5 eV corresponds to 1h states with 2a′′−1 and 11a′−1 occupation, which is again in line with the previous assignment.8 The participator decay to these final states is strongly enhanced due to the significant contribution of carbon AOs to the nearly degenerate 2a′′ and 11a′ orbitals and the simulation is able to reproduce the high intensity and sharpness of this peak. Feature 3 at 15.2 eV is dominated by the participator decay to a 2A′ final state with 10a′−1 occupation and experiences a lower enhancement compared to feature 2 as the electron density at the carbon atom is lower for the 10a′ orbital. In addition, the lowest spectator final states which are described by an occupation with two holes in the HOMO and HOMO−1 and a spectating electron in the 13a′ orbital are calculated to add intensity to feature 3 in the RAES. Their lower contribution to feature 3 compared to the 2A′(10a′−1) participator state is once more connected to the nodal plane of the HOMO and HOMO−1 at the carbon atom. Below 20 eV feature 4 with a slightly lower intensity is also attributed to a participator decay (9a′−1 occupation) and several spectator transitions to states with an occupation with holes in the 12a′ and 2a′′/11a′ orbitals and a spectating electron in the LUMO. At binding energies above 20 eV mainly spectator states contribute to the RAES as well as states with shake-up configurations. Feature 5 includes final states described by an orbital occupation with one vacancy in the lower-lying 10a′ orbital and one vacancy in the 12a′/3a′′ orbital. A strong enhancement in the region of spectator final states is found for feature 6 and the corresponding final state occupations exhibit at least one hole in one of the nearly degenerate orbitals 11a′, 2a′′ or in the 9a′ orbital, which all show significant density at the carbon atom. Features 7 and 8 are described by final state occupations that mainly have holes in 9a′ and 10a′ orbitals and a spectating electron in the LUMO.
Measurements via the different members of the vibrational progression of the C1s → 13a′(LUMO) excitation show changes in the participator region of the RAES (compare Fig. S6, ESI†). Features 1 and 3 split into multiple bands, while a broadening is observed for the most intense band (feature 2). This implies transitions between different vibrational levels of the core excited and final state.91
The RAES of the C1s → 4a′′(LUMO+1) excitation is shown in Fig. 5(c) together with the calculated spectrum. As mentioned above, the simulation describes the band shapes for the decay of the C1s−14a′′1 state much better than for the 13a′ excitation. The experimental spectrum is largely similar to the RAES of the C1s → 13a′(LUMO) excitation as expected from the small energy difference of the 13a′ and 4a′′ virtual orbitals. However, an even stronger enhancement of feature 2 is observed and a smaller linewidth indicates smaller changes in the geometry between the C1s−14a′′1 core excited and the ionic final state compared to the 13a′ excitation. Most prominently we observe a splitting of feature 3 that is also predicted by the simulation. For the excitation into the 4a′′ orbital, participator states are observed at the same binding energy as for 13a′ excitation (although different gradients can lead to small variations in their binding energy and shape) whereas spectator states shift to higher binding energies since the spectating electron occupies a higher virtual orbital. This decay to spectator states with higher binding energy leads to the observed splitting of feature 3 as the energy difference between participator and spectator states increases. Similarly, we observe changes in the shape and energetic position of features 4–8 resulting from the spectator decay to final states with higher binding energy. Measurements of resonant Auger electron spectra at the high and low-energy side of this feature as indicated in the NEXAFS spectrum (Fig. 4b) reveal only minor changes of the spectral features and are shown in Fig. S6 (ESI†). The strong enhancement of feature 2 that is found upon resonant C1s excitation into the 13a′ and 4a′′ orbitals is in accordance with a stronger enhancement of the Ã2Π(2π−1) state in OCS upon C1s → 4π(LUMO) excitation compared to other participator transitions.92
The resonant Auger electron spectra of HNCS for the N1s excitation to the 13a′ and 4a′′ orbitals are shown in Fig. 6. The shape of the spectra is similar to the C1s edge and shows an enhancement of feature 3 and of the bands at binding energies >18 eV. In contrast to the C1s excitation we observe no enhancement of the second feature as predicted by the calculation which also anticipates a larger bandwidth. Feature 3 appears as a single band for the N1s → 13a′ excitation. While this band is dominated by the 2A′(10a′−1) final state on the carbon edge, the lower contribution of nitrogen AOs to the 10a′ orbital leads to a reduced intensity of this participator decay. Instead spectator decays with two holes in the HOMO and HOMO−1 have a larger contribution to this band since these orbitals show a higher density at the nitrogen than on the carbon atom. For the N1s → 4a′′ excitation the lower band of feature 3 at 15.2 eV likely belongs to the 2A′(10a′−1) participator final state, which is not reproduced in the calculation probably as the line shape is not correctly represented. The band at 15.6 eV is attributed to a decay to higher energy spectator final states with the same vacancies as for 13a′ excitation but the spectating electron occupying the 4a′′ instead of the 13a′ virtual orbital, leading to the observed splitting of the feature. For feature 4 we observe a larger partial decay rate to the 2A′(9a′−1) participator final state than on the carbon edge. This is readily explained by the localisation of the 9a′ orbital at the nitrogen atom. For 13a′ excitation several spectator states characterized by occupations with vacancies in each set of the nearly degenerate orbitals 11a′/2a′′ and 12a′/3a′′ and a spectating electron in the 13a′ orbital contribute to feature 4. For the 4a′′ excitation, spectator decays to final states characterised by a spectating electron in the 4a′′ orbital become more important (feature 5).
Scanning the photon energy over the high energy part of the N1s → 4a′′ resonance leads to a stronger splitting of feature 3 in the RAES and significant changes on the high energy side of feature 1 indicating the excitation of different vibrational levels in the core excited state. Interestingly, excitation of the core excited state observed at 401.8 eV leads not only to an enhancement of feature 2 but also shows very sharp, intense features in the region of spectator final states (compare Fig. S7, ESI†).
Fig. 7 shows the RAES obtained upon resonant excitation of HNCS at the sulfur 2p edge. Here the spectra are strongly affected by the contribution from direct valence ionization. This contribution decreases with photon energy and is subsequently negligible on the carbon and nitrogen 1s edges. Therefore, only the resonant spectra at the sulfur edge were corrected for the contribution from direct photoemission by subtraction of a scaled off-resonant spectrum. The scaling factor was determined from the direct photoemission background in the sulfur 2p NEXAFS spectrum (Fig. 4a). The RAES recorded at 163.40 eV (Fig. 7b) corresponds to the S2p3/2 → 13a′(LUMO) excitation whereas the one at 164.56 eV (Fig. 7c) is the superposition of the S2p3/2 → 4a′′(LUMO+1) and S2p1/2 → 13a′(LUMO) excitation. From the comparison to the off-resonant spectrum an enhancement of feature 3 at 15.2 eV and of features at higher binding energies is observed after excitation into 13a′ and 4a′′. As no calculations of the RAES on the sulfur edge were performed, we assign feature 1–3 by the comparison to the carbon and nitrogen RAES and the off-resonant spectrum to the following participator decays: 2A′′(3a′′−1) and 2A′(12a′−1) (feature 1), 2A′′(2a′′−1) and 2A′(11a′−1) (feature 2) and 2A′(10a′−1) (feature 3) final states. As the 10a′ orbital is mainly located at the sulfur atom, a strong enhancement of the corresponding participator decay is expected and the sharp feature at 15.2 eV which is only slightly broadened compared to the valence photo line is likely to be the signal of this decay. Its small vibrational broadening suggests a similar geometry of the core excited states and this participator final state. The broad underlying feature 3 in the S2p3/2 → 13a′ RAES likely belongs to the low binding energy spectator states that were also observed at the carbon and nitrogen edge. Interestingly, we find an additional signal at 14.6 eV. The lowest spectator state observed at the other two edges appears at higher binding energies. In the S2p3/2 → 4π RAES of OCS a transition to a low binding energy, spin forbidden quartet state, 4Π(3π−24π1), is found as a result of spin–orbit interaction in the S2p core excited states.88 A similar transition to a low-lying quartet state might explain the observed feature at 14.6 eV. In contrast to the spectra on the carbon and nitrogen edge, distinguishable bands also appear in the binding energy region >18 eV, the most intense ones are found at 19.2 and 20.7 eV in the RAES recorded at 163.40 eV. These bands cannot be assigned without calculations and appear as broader features in the RAES at 164.56 eV due to the superposition of two different resonant excitations. For the S2p excitation into higher virtual orbitals (compare Fig. S8, ESI†) mainly changes in the region of spectator final states occur and distinct sharp signals can be observed. Additionally, scanning the photon energy over the most intense band in the S2p NEXAFS reveals a small signal that is assigned to the Auger–Meitner decay of a core-excited atomic sulfur fragment which is likely to be produced by ultrafast dissociation of HNCS (see Section S5, ESI†).
4 Discussion
A very good agreement with the simulation is found for both the normal and resonant Auger electron spectra allowing various features to be assigned. Although core ionization and core excitation result in different final states after the Auger–Meitner decay, the 2h final states populated by normal Auger–Meitner decay are expected to have a counterpart spectator (2h1p) final state in the resonant spectrum. From the kinetic energy difference of the 2h and 2h1p final states with similar double valence holes we can estimate the screening effect of the spectating electron. From the AES and RAES on the nitrogen edge of HNCS (see Fig. S9, ESI†), we deduce a screening energy of 7.7 eV for the two lowest core excited states (excitation into 13a′ and 4a′′). The screening effect is reduced dramatically for the excitation into higher unoccupied orbitals as the spectating electron is located in orbitals further away from the nucleus (compare Section S6, ESI†). On the binding energy scale, the shift between the normal AES and the RAES of the LUMO+1 (4a′′) excitation is determined to be 13.3 eV. Fig. 8 shows the corresponding comparison, where the RAES are shifted by this value. Especially on the nitrogen edge a high similarity between the normal AES and the spectator states in the RAES is observed, implying a relatively low influence of the spectating electron. Additional RAES-features at 28.5 and 31.5–32.0 eV are shown to belong to participator decays, namely to the final states with 10a′−1 and 9a′−1 occupation. This confirms our assignment of the splitting of feature 3 and 4 in the RAES (Fig. 5 and 6) to a separation into participator and spectator states when going from 13a′ excitation to 4a′′ excitation. While spectator transitions dominate the N1s−14a′′ RAES, their contribution is lower upon C1s excitation. In the carbon RAES we find sharper signals compared to the normal AES for the two lowest bands of the AES. This agrees with the smaller calculated bandwidths for the carbon RAES (compare Tables 2 and 3). The shift in the spectral position of the RAES feature at 34 eV is attributed to a decay to spectator final states with one hole in the 10a′ orbital that show nearly no intensity in the normal AES. For the C1s excitation into the 13a′ orbital no such shift is observed as similar valence holes are generated. The separation of the parent Auger lines that is observed at a binding energy of 35 eV in the C- and N-AES becomes blurred in the corresponding region of the RAES spectra. This could be a result of the spectating electron coupling to the double valence hole which leads to a splitting into several states and results in broader spectral features. Generally, the similarity between RAES and AES found on the carbon and nitrogen edge is less obvious at the sulfur edge. Nevertheless, we observe similar features in the spectator region compared to the normal AES and the spectrum is also dominated by participator decays. The RAES feature that was tentatively attributed to a transition to a quartet state (at 27.9 eV on this scale) coincides with the position of the triplet ground state of the AES.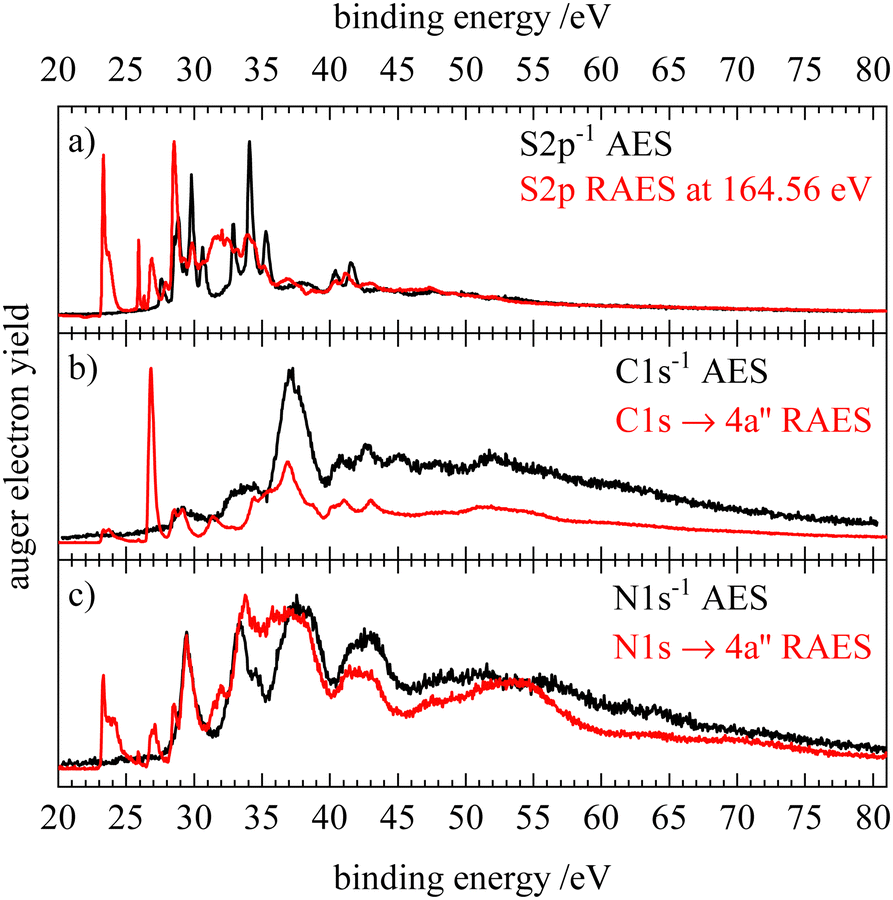 | ||
| Fig. 8 Comparison of the normal (black) and resonant (red) Auger electron spectra of isothiocyanic acid for S2p (a), C1s (b) and N1s (c) decay. The resonant spectra belong to the resonant 4a′′(LUMO+1) excitation and were each shifted by 13.3 eV to higher binding energy. | ||
Next, we compare isothiocyanic acid to its oxygen congener isocyanic acid, HNCO. A comparison of the XPS data of the two molecules shows a chemical shift of 2.1 eV for the C1s IE when going from HNCO (295.9 eV)33 to HNCS (293.8 eV). This is in line with the lower electronegativity of sulfur relative to oxygen. The latter thus draws electron density away from the carbon atom, which leads to a higher C1s IE, while in HNCS a higher electron density remains at the carbon atom. Consequently, the IE is lower. In contrast to the C1s values, a similar N1s IE (405.7 eV) is found for HNCO33 and HNCS. This confirms the stronger influence of the oxygen/sulfur substitution on the neighbouring carbon atom compared to the nitrogen atom as is expected from chemical intuition.
We compare the normal Auger electron spectra of isothiocyanic acid and isocyanic acid,33 in Fig. 9. Note that the S2p AES of HNCS is compared to the O1s AES of HNCO. The AES of HNCO were shifted to lower binding energies by 5.5 eV to account for the higher electronegativity of oxygen compared to sulfur. Like HNCS, HNCO is a bent molecule (CS symmetry) without degenerate orbitals. The most prominent difference between the spectra of the two molecules is the broader features on the oxygen edge compared to the sulfur edge. This implies stronger changes in the geometry leading to different gradients in the core excited and final state after O1s ionization. The lowest binding energy signals in the Auger electron spectra are shifted by up to 2 eV and we do not observe a separate signal of a low-lying triplet state on the oxygen edge that is found for the sulfur edge. As for isothiocyanic acid, the first band in the AES of HNCO corresponds to final states described by orbital occupations with holes in the HOMO (2a′′) and HOMO−1 (9a′) and has only a very low intensity on the carbon edge due to a nodal plane of the involved MOs at the carbon atom.33 This confirms a high similarity of the electronic structure of the two molecules. However, in HNCO this first band shows a high-energy shoulder at all three edges that is assigned to the final state occupation 9a′−2. This implies a larger energy separation of the HOMO and HOMO−1 in HNCO compared to HNCS. Also for the second band at 32–35 eV similar final state occupations are found for HNCS and HNCO. On the carbon edge these are mainly states with one hole in the HOMO/HOMO−1 and the other hole in the HOMO−2/HOMO−3. The higher intensity found for this band on the carbon edge in HNCO therefore suggests a higher density of the HOMO−2/HOMO−3 orbitals on the carbon atom than in HNCS. At higher binding energies the differences in energy positions and intensities increase. Especially in the AES on the oxygen edge we find a much higher intensity compared to the sulfur edge. This can be explained as follows: in this energy region final states with occupations with at least one hole in HOMO−2 and/or HOMO−3 occur. While these nearly degenerate orbitals have only very small MO coefficients at the sulfur atom in HNCS, the MO coefficients at the oxygen atom in HNCO are much higher. This is a result of the oxygen 2p valence orbitals lying lower in energy than the sulfur 3p valence orbitals which leads to a larger contribution of O2p orbitals to low-lying molecular orbitals. This shows that intensity differences between Auger electron spectra of second-row and third-row elements in molecules are a consequence of different MO coefficients arising from the elements′ different orbital energies.
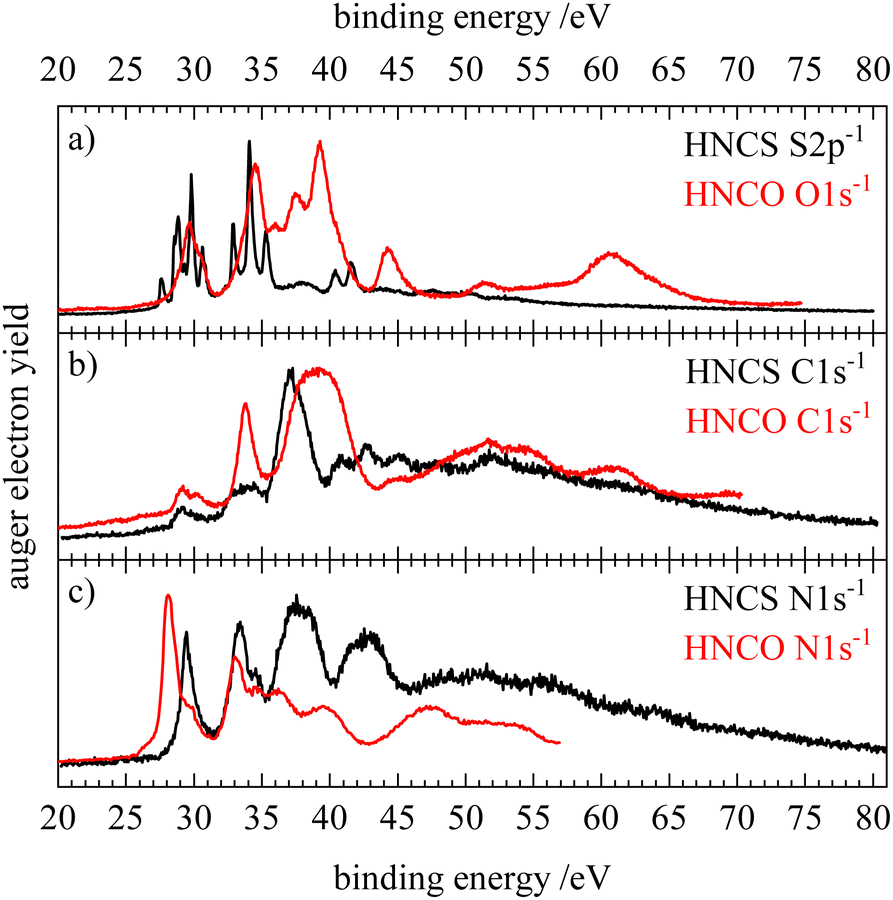 | ||
| Fig. 9 Comparison of the normal Auger electron spectra of isothiocyanic acid (black) and isocyianic acid (red) for S2p/O1s (a), C1s (b) and N1s (c) decay. The spectra of isocyanic acid were each shifted by 5.5 eV to lower binding energy. HNCO data reproduced from ref. 33 with permission of AIP. | ||
In addition, the resonant Auger electron spectra of the core excitation into the LUMO (13a′ or 10a′) of HNCS and HNCO are shown in Fig. 10. Here, the HNCO spectra33 were shifted by the difference in IE (1.6 eV) to lower binding energies. The comparison shows a similar participator region of the two isovalence electronic molecules: the first feature at 10 eV is assigned to the 1h final state with an occupation with the hole in the HOMO or HOMO−1 in both molecules. Below 15 eV the participator line for final states described by orbital occupations with a hole in one of the nearly degenerate orbitals 8a′ and 1a′′(HNCO) or 11a′ and 2a′′(HNCS) are found. At higher binding energies, a similar band in both spectra indicates the participator decay to a 2A′(10a′−1) final state for HNCS which corresponds to 2A′(7a′−1) in HNCO. The shift of two of the participator bands to higher binding energies compared to the HNCS RAES shows a comparatively larger energy separation between the 2a′′(HOMO) and the 8a′(HOMO−2), 1a′′(HOMO−3), 7a′(HOMO−4) orbitals for isocyanic acid. On the carbon edge of HNCO we observe a similar resonant enhancement of the second and third participator feature as for HNCS. A stronger enhancement of the band at 14.4 eV compared to the spectator region is found for HNCO which might support the assumption that the nearly degenerate HOMO−2 and HOMO−3 orbitals in HNCO have a higher density on the carbon atom. While we observe enhancements of the same features on the carbon edge, the O1s and S2p RAES of HNCO and HNCS differ to a higher extend. In the S2p RAES of HNCS we assigned the most intense feature to a resonantly enhanced participator decay to a state with 10a′−1 occupation, while in the oxygen RAES of HNCO no such enhancement of the corresponding 2A′(7a′−1) final state is observed.33 Furthermore, the spectator region shows a much higher intensity than in the S2p RAES of HNCS. This is similar to the differences in the normal AES of the two molecules on the sulfur and oxygen edges (Fig. 9a) and can also be explained by a preferred decay to final states with holes in the HOMO−2 and HOMO−3 orbitals at the oxygen edge of HNCO whereas these final states show only low intensity at the sulfur edge of HNCS (vide supra). On the nitrogen edge both molecules show a dominant decay to spectator final states. In the low EB region, the N1s RAES are very similar in shape and intensity, because the nitrogen atom is least influenced by the O/S substitution. However, in the region 15–21 eV differences are observed. Neither in HNCO nor in HNCS enhancement of the participator line at around 15 eV is found, which is in line with the low contribution of nitrogen atomic orbitals to the 7a′ orbital of HNCO and the corresponding 10a′ orbital of HNCS. For HNCS, the enhancement of the band is instead attributed to the population of low binding energy spectator final states in this energy region. In HNCO no such enhancement of the band is observed as the lowest spectator final states are shifted by around 2 eV to higher binding energies. These spectator decays are enhanced by the high density of the HOMO and HOMO−1 at the nitrogen atom in both molecules and in HNCO their shift to higher binding energies leads to an increased intensity at binding energies >17 eV in the N-RAES of HNCO. A similar shift of the lowest spectator final states to higher binding energies is also observed on the carbon and oxygen edge of HNCO.33 The smaller energy difference between the onset of participator and spectator states in HNCS indicates a smaller HOMO–LUMO gap compared to HNCO as the energy difference between the lowest participator and the lowest spectator final state can be seen as a rough approximation of the HOMO–LUMO gap.
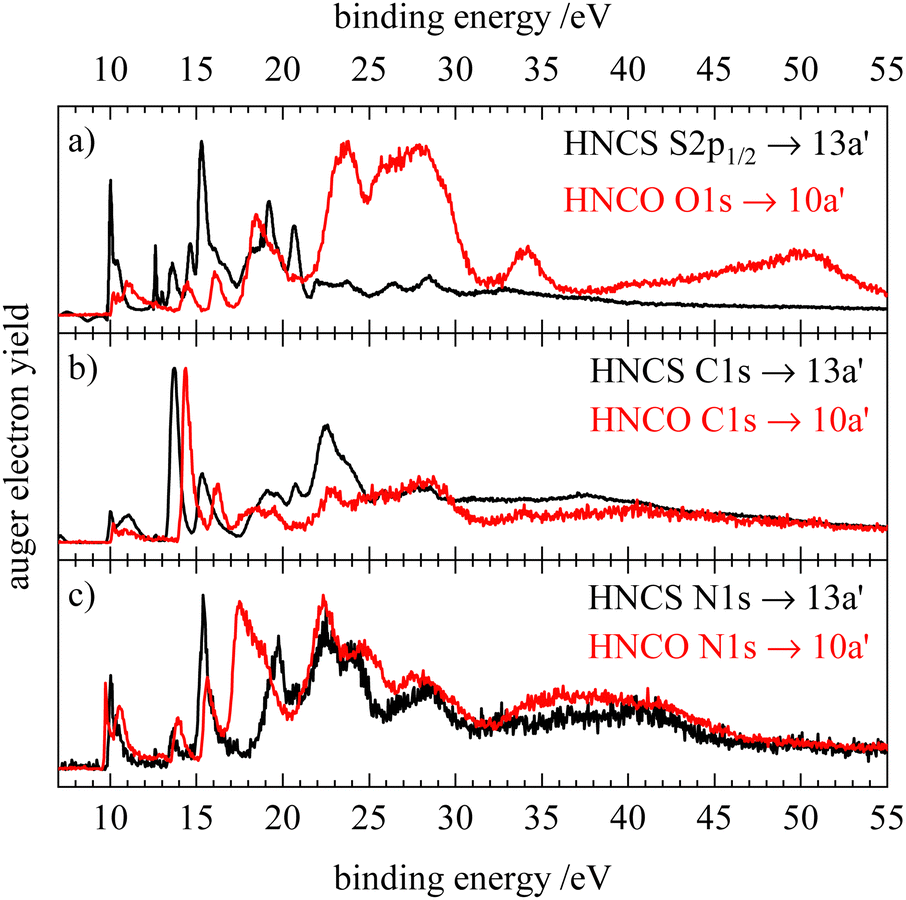 | ||
| Fig. 10 Comparison of the resonant Auger electron spectra of isothiocyanic acid (black) and isocyianic acid (red) for the excitation into the LUMO (13a′ in HNCS, 10a′ in HNCO) for S2p/O1s (a), C1s (b) and N1s (c) decay. The spectra of isocyanic acid were shifted by the difference in ionization energy (1.6 eV) to lower binding energy. HNCO data reproduced from ref. 33 with permission of AIP. | ||
5 Conclusions
Isothiocyanic acid was investigated in the soft X-ray regime by XPS, NEXAFS, normal and resonant Auger electron spectroscopy at the carbon and nitrogen 1s edge and the sulfur 2p edge. Core ionization energies of 405.7 eV (N1s), 293.8 eV (C1s), 234.3 eV (S2s), 170.9 eV (S2p1/2) and 169.7 eV (S2p3/2) were obtained. From the data, a spin–orbit splitting of 1.2 eV is deduced for the S2p levels, similar to related systems. In the normal Auger electron spectra (AES) the carbon and nitrogen KLL and sulfur L2,3MM Auger electron spectra show profound differences. In particular, narrower bands are found in the S2p AES as observed before for the isosteric molecule OCS. All bands are assigned by a comparison with ab initio calculations that show a very good agreement with the experiment and are able to reproduce the narrow features on the sulfur 2p edge. These bands result from final states with two holes in the non-bonding or weakly bonding 10–12a′ and 2–3a′′ orbitals. Thus, the resulting states have small gradients at the ground state structure causing small bandwidths.From the signal at the lowest binding energy in the S2p1/2 AES which corresponds to the dicationic ground state of HNCS, 3A′′, the double ionization energy of HNCS is determined to be 27.6 eV. On the carbon and nitrogen edge, no transition to the triplet ground state is observed. This is explained to be due to the almost atomic symmetry of the Auger–Meitner decay of a 1s core hole state which is parity forbidden if it leads to a final state with two triplet coupled p-orbitals. The transition to the lowest dicationic singlet final states shows very low intensity in the carbon AES, which is a result of a nodal plane of the HOMO and HOMO−1 at the carbon atom. In the electronically isovalent molecule isocyanic acid a similar effect was observed.
In the X-ray absorption spectra, the lowest resonant excitations of the core electrons were assigned to the transition into the 13a′(LUMO) and 4a′′(LUMO+1) orbitals at all three edges. The resonant enhancement of the Auger–Meitner decay to different final states at the different edges was rationalized in terms of the localisation of the involved molecular orbitals at the respective atomic site. As expected, the spectator decays show a close resemblance to the normal AES. Differences between the RAES of the 13a′ and 4a′′ excitation are mainly attributed to the occupation of higher energy spectator final states after 4a′′ excitation. The comparison to isocyanic acid shows differences in the relative energies of the molecular orbitals and a lower energy separation between participator and spectator final states in HNCS. For both resonant and normal Auger electron spectra intensity differences in the O1s and S2p spectra of the two molecules result from different energies of the atomic orbitals that consequently show different contributions to the molecular orbitals involved in the Auger–Meitner decay.
Author contributions
Dorothee Schaffner: investigation (experiment, theory, synthesis), formal analysis, writing – original draft; Marius Gerlach, Emil Karaev: investigation (experiment, synthesis); John Bozek: investigation (experiment), resources; Ingo Fischer: investigation (experiment), conceptualisation, writing – review and editing, supervision, project administration, funding acquisition; Reinhold Fink: investigation (theory), resources, conceptualisation, writing – original draft, supervision.Data availability
The data supporting this article have been included as part of the ESI.† Further data is available from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Experiments were carried out at the PLEIADES beamline of the SOLEIL storage ring, proposal ID 20221142. The work was supported by the Deutsche Forschungsgemeinschaft, contracts FI575/13-2 and 19-1. DS acknowledges a fellowship by the Fonds der Chemischen Industrie (FCI). We thank E. Robert for technical assistance and the SOLEIL staff for stable operation of the equipment and storage ring during the experiments. We thank T. X. Carroll for providing the Auger electron spectra of OCS.Notes and references
- B. A. McGuire, M.-A. Martin-Drumel, S. Thorwirth, S. Brünken, V. Lattanzi, J. L. Neill, S. Spezzano, Z. Yu, D. P. Zaleski, A. J. Remijan, B. H. Pate and M. C. McCarthy, Phys. Chem. Chem. Phys., 2016, 18, 22693–22705 RSC.
- M. Battegay and E. Hégazi, Helv. Chim. Acta, 1933, 16, 999–1008 CrossRef CAS.
- J. Goubeau and O. Gott, Ber. Dtsch. Chem. Ges., 1940, 73, 127–133 CrossRef.
- C. I. Beard and B. P. Dailey, J. Chem. Phys., 1947, 15, 762 CrossRef CAS.
- K. Yamada and M. Winnewisser, Z. Naturforsch., A: Phys. Sci., 1976, 31, 139–144 Search PubMed.
- K. Yamada, M. Winnewisser, G. Winnewisser, L. B. Szalanski and M. C. L. Gerry, J. Mol. Spectrosc., 1977, 64, 401–414 CrossRef CAS.
- K. Yamada, M. Winnewisser, G. Winnewisser, L. B. Szalanski and M. C. L. Gerry, J. Mol. Spectrosc., 1980, 79, 295–313 CrossRef.
- J. H. D. Eland, W. C. Price and D. W. Turner, Philos. Trans. R. Soc., A, 1970, 268, 87–96 CrossRef CAS.
- S. Cradock, E. A. V. Ebsworth and J. D. Murdoch, J. Chem. Soc., Faraday Trans. 2, 1972, 68, 86–100 RSC.
- T. Pasinszki, N. Kishimoto and K. Ohno, J. Phys. Chem. A, 1999, 103, 9195–9203 CrossRef CAS.
- B. Ruscic and J. Berkowitz, J. Chem. Phys., 1994, 101, 7975–7989 CrossRef CAS.
- B. Bak, J. J. Christiansen, O. J. Nielsen and H. Svanholt, Acta Chem. Scand., 1977, 31a, 666–668 CrossRef.
- M. Wierzejewska and J. Moc, J. Phys. Chem. A, 2003, 107, 11209–11216 CrossRef CAS.
- J. R. Durig, C. Zheng and H. Deeb, J. Mol. Struct., 2006, 784, 78–92 CrossRef CAS.
- D. Poppinger, L. Radom and J. A. Pople, J. Am. Chem. Soc., 1977, 99, 7806–7816 CrossRef CAS.
- M. A. Frerking, R. A. Linke and P. Thaddeus, Astrophys. J., 1979, 234, L143–L145 CrossRef CAS.
- L. E. Snyder and D. Buhl, Astrophys. J., 1972, 177, 619–623 CrossRef CAS.
- N.-Q. Rieu, C. Henkel, J. M. Jackson and R. Mauersberger, Astron. Astrophys., 1991, 241, L33–L36 Search PubMed.
- D. C. Lis, J. Keene, K. Young, T. G. Phillips, D. Bockelée-Morvan, J. Crovisier, P. Schilke, P. F. Goldsmith and E. A. Bergin, Icarus, 1997, 130, 355–372 CrossRef CAS.
- E. Mendoza, B. Lefloch, A. López-Sepulcre, C. Ceccarelli, C. Codella, H. M. Boechat-Roberty and R. Bachiller, Mon. Not. R. Astron. Soc., 2014, 445, 151–161 CrossRef CAS.
- M. Ferus, V. Laitl, A. Knizek, P. Kubelík, J. Sponer, J. Kára, J. E. Sponer, B. Lefloch, G. Cassone and S. Civiš, Astron. Astrophys., 2018, 616, A150 CrossRef.
- R. Le Gal, K. I. Öberg, R. A. Loomis, J. Pegues and J. B. Bergner, Astrophys. J., 2019, 876, 72 CrossRef CAS.
- E. F. van Dishoeck, B. Jonkheid and M. C. van Hemert, Faraday Discuss., 2006, 133, 231–243 RSC.
- H. Gao, Mol. Phys., 2021, 119, e1861354 CrossRef.
- J. R. McDonald, V. M. Scherr and S. P. McGlynn, J. Chem. Phys., 1969, 51, 1723–1731 CrossRef CAS.
- G. Chaturvedi, K. Subbaram, D. Ramachandra Rao and C. Rao, J. Mol. Spectrosc., 1971, 39, 242–246 CrossRef.
- W. Balfour, V. Bhujle, R. Whitlock, C. Rao and D. Rao, J. Mol. Spectrosc., 1972, 42, 577 CrossRef CAS.
- C. R. Boxall and J. P. Simons, J. Photochem., 1972, 1, 363–369 CrossRef.
- F. J. Northrup and T. J. Sears, J. Chem. Phys., 1990, 93, 2337–2345 CrossRef CAS.
- F. J. Northrup and T. J. Sears, J. Chem. Phys., 1990, 93, 2346–2356 CrossRef CAS.
- M. Wallner, E. Olsson, V. Ideböhn, M. Parriani, R. J. Squibb, S. Lundberg, D. Cole, S. Falcinelli, S. Stranges, B. Brunetti, J. M. Dyke, G. Nyman, J. H. D. Eland, M. Hochlaf and R. Feifel, J. Chem. Phys., 2024, 161, 044313 CrossRef CAS PubMed.
- G. Bilalbegović, A. Maksimović, L. A. Valencic and S. Lehtola, ACS Earth Space Chem., 2021, 5, 436–448 CrossRef PubMed.
- F. Holzmeier, T. J. A. Wolf, C. Gienger, I. Wagner, J. Bozek, S. Nandi, C. Nicolas, I. Fischer, M. Gühr and R. F. Fink, J. Chem. Phys., 2018, 149, 034308 CrossRef CAS PubMed.
- M. Gerlach, F. Fantuzzi, L. Wohlfart, K. Kopp, B. Engels, J. Bozek, C. Nicolas, D. Mayer, M. Gühr, F. Holzmeier and I. Fischer, J. Chem. Phys., 2021, 154, 114302 CrossRef CAS PubMed.
- M. Gerlach, T. Preitschopf, E. Karaev, H. M. Quitián-Lara, D. Mayer, J. Bozek, I. Fischer and R. F. Fink, Phys. Chem. Chem. Phys., 2022, 24, 15217–15229 RSC.
- M. Gerlach, D. Schaffner, T. Preitschopf, E. Karaev, J. Bozek, F. Holzmeier and I. Fischer, J. Chem. Phys., 2023, 159, 114306 CrossRef CAS PubMed.
- S. Stranges, R. Richter and M. Alagia, J. Chem. Phys., 2002, 116, 3676–3680 CrossRef CAS.
- U. Ekstrom, V. Carravetta, M. Alagia, M. Lavollee, R. Richter, C. Bolcato and S. Stranges, J. Chem. Phys., 2008, 128, 044302 CrossRef CAS PubMed.
- M. Alagia, E. Bodo, P. Decleva, S. Falcinelli, A. Ponzi, R. Richter and S. Stranges, Phys. Chem. Chem. Phys., 2013, 15, 1310–1318 RSC.
- D. Schaffner, T. Juncker von Buchwald, E. Karaev, M. Alagia, R. Richter, S. Stranges, S. Coriani and I. Fischer, J. Chem. Phys., 2024, 161, 034309 CrossRef CAS PubMed.
- PLÉIADES beamline at synchrotron SOLEIL, https://www.synchrotron-soleil.fr/en/beamlines/pleiades, (accessed October 2024).
- W. E. Moddeman, T. A. Carlson, M. O. Krause, B. P. Pullen, W. E. Bull and G. K. Schweitzer, J. Chem. Phys., 2003, 55, 2317–2336 CrossRef.
- M. Kato, Y. Morishita, M. Oura, H. Yamaoka, Y. Tamenori, K. Okada, T. Matsudo, T. Gejo, I. H. Suzuki and N. Saito, J. Electron Spectrosc. Relat. Phenom., 2007, 160, 39–48 CrossRef CAS.
- Y. Ma, C. T. Chen, G. Meigs, K. Randall and F. Sette, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 44, 1848–1858 CrossRef CAS PubMed.
- G. C. King, M. Tronc, F. H. Read and R. C. Bradford, J. Phys. B: At. Mol. Phys., 1977, 10, 2479 CrossRef CAS.
- R. Fink, J. Electron Spectrosc. Relat. Phenom., 1995, 76, 295–300 CrossRef CAS.
- R. Fink, J. Chem. Phys., 1997, 106, 4038–4052 CrossRef CAS.
- R. F. Fink, M. Kivilompolo, H. Aksela and S. Aksela, Phys. Rev. A: At., Mol., Opt. Phys., 1998, 58, 1988 CrossRef CAS.
- A. Machado Bueno, A. Naves de Brito, R. F. Fink, M. Bassler, O. Bjorneholm, F. Burmeister, R. Feifel, C. Miron, S. L. Sorensen, H. Wang and S. Svensson, Phys. Rev. A: At., Mol., Opt. Phys., 2003, 67, 022714 CrossRef.
- R. F. Fink, S. L. Sorensen, A. Naves de Brito, A. Ausmees and S. Svensson, J. Chem. Phys., 2000, 112, 6666–6677 CrossRef CAS.
- S. L. Sorensen, R. Fink, R. Feifel, M. N. Piancastelli, M. Bässler, C. Miron, H. Wang, I. Hjelte, O. Björneholm and S. Svensson, Phys. Rev. A: At., Mol., Opt. Phys., 2001, 64, 012719 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- R. J. Gdanitz and R. Ahlrichs, Chem. Phys. Lett., 1988, 143, 413–420 CrossRef CAS.
- R. Fink and V. Staemmler, Theor. Chim. Acta, 1993, 87, 129–145 CrossRef CAS.
- R. Püttner, I. Dominguez, T. J. Morgan, C. Cisneros, R. F. Fink, E. Rotenberg, T. Warwick, M. Domke, G. Kaindl and A. S. Schlachter, Phys. Rev. A: At., Mol., Opt. Phys., 1999, 59, 3415–3423 CrossRef.
- K. A. Peterson and T. H. Dunning, J. Chem. Phys., 2002, 117, 10548–10560 CrossRef CAS.
- K. J. Børve, Chem. Phys. Lett., 1996, 262, 801–806 CrossRef.
- H. Siegbahn, L. Asplund and P. Kelfve, Chem. Phys. Lett., 1975, 35, 330–335 CrossRef CAS.
- H. Ågren, S. Svensson and U. Wahlgren, Chem. Phys. Lett., 1975, 35, 336–344 CrossRef.
- E. Z. Chelkowska and F. P. Larkins, At. Data Nucl. Data Tables, 1991, 49, 121–206 CrossRef CAS.
- B. N. C. Tenorio, T. A. Voß, S. I. Bokarev, P. Decleva and S. Coriani, J. Chem. Theory Comput., 2022, 18, 4387–4407 CrossRef CAS PubMed.
- J. Palaudoux, T. Kaneyasu, L. Andric, S. Carniato, G. Gamblin, F. Penent, Y. Hikosaka, E. Shigemasa, K. Ito, S. Fritzsche, E. Kukk, S. Sheinerman, R. F. Fink, P. Lablanquie and R. Püttner, Phys. Rev. A, 2018, 98, 043406 CrossRef CAS.
- S. Behnle and R. F. Fink, J. Chem. Phys., 2022, 156, 124103 CrossRef CAS PubMed.
- U. Meier and V. Staemmler, Theor. Chim. Acta, 1989, 76, 95–111 CrossRef CAS.
- M. Jungen, Theor. Chim. Acta, 1981, 60, 369–377 CrossRef CAS.
- J. Wasilewski, Int. J. Quantum Chem., 1991, 39, 649–656 CrossRef CAS.
- F. Gel’mukhanov, H. Ågren, S. Svensson, H. Aksela and S. Aksela, Phys. Rev. A: At., Mol., Opt. Phys., 1996, 53, 1379–1387 CrossRef PubMed.
- D. Minelli, F. Tarantelli, A. Sgamellotti and L. S. Cederbaum, J. Chem. Phys., 1997, 107, 6070–6079 CrossRef CAS.
- L. S. Cederbaum, P. Campos, F. Tarantelli and A. Sgamellotti, J. Chem. Phys., 1991, 95, 6634–6644 CrossRef CAS.
- L. S. Cederbaum and F. Tarantelli, J. Chem. Phys., 1993, 98, 9691–9706 CrossRef CAS.
- L. S. Cederbaum and F. Tarantelli, J. Chem. Phys., 1993, 99, 5871–5884 CrossRef CAS.
- W. Asaad, Nucl. Phys., 1965, 66, 494–512 CrossRef CAS.
- H. Körber and W. Mehlhorn, Z. Phys., 1966, 191, 217–230 CrossRef.
- L. Inhester, C. F. Burmeister, G. Groenhof and H. Grubmüller, J. Chem. Phys., 2012, 136, 144304 CrossRef CAS PubMed.
- H. Pulkkinen, S. Aksela, O.-P. Sairanen, A. Hiltunen and H. Aksela, J. Phys. B, 1996, 29, 3033–3050 CrossRef CAS.
- P. Bolognesi, P. O'Keeffe and L. Avaldi, J. Phys. Chem. A, 2009, 113, 15136–15141 CrossRef CAS PubMed.
- T. X. Carroll, D. Ji and T. D. Thomas, J. Electron Spectrosc. Relat. Phenom., 1990, 51, 471–486 CrossRef CAS.
- P. Baltzer, L. Karlsson, M. Lundqvist and B. Wannberg, Rev. Sci. Instrum., 1993, 64, 2179–2189 CrossRef CAS.
- C. Nicolas and C. Miron, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 267–272 CrossRef CAS.
- H. Wang, M. Bässler, I. Hjelte, F. Burmeister and L. Karlsson, J. Phys. B: At., Mol. Opt. Phys., 2001, 34, 1745 CrossRef CAS.
- Y. Hikosaka, P. Lablanquie, F. Penent, J. G. Lambourne, R. I. Hall, T. Aoto and K. Ito, J. Electron Spectrosc. Relat. Phenom., 2004, 137–140, 287–291 CrossRef CAS.
- G. R. Wight and C. E. Brion, J. Electron Spectrosc. Relat. Phenom., 1974, 4, 335–345 CrossRef CAS.
- S. Masuda, T. Hatsui and N. Kosugi, J. Electron Spectrosc. Relat. Phenom., 2004, 137–140, 351–355 CrossRef CAS.
- J. Adachi, N. Kosugi, E. Shigemasa and A. Yagishita, J. Chem. Phys., 1997, 107, 4919–4926 CrossRef CAS.
- G. R. Wight and C. E. Brion, J. Electron Spectrosc. Relat. Phenom., 1974, 3, 191–205 CrossRef CAS.
- M. Neeb, J.-E. Rubensson, M. Biermann and W. Eberhardt, J. Electron Spectrosc. Relat. Phenom., 1994, 67, 261–274 CrossRef CAS.
- O. Travnikova, C. Miron, M. Bässler, R. Feifel, M. N. Piancastelli, S. L. Sorensen and S. Svensson, J. Electron Spectrosc. Relat. Phenom., 2009, 174, 100–106 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03897e |
| This journal is © the Owner Societies 2024 |