
Dry desulphurisation of gas streams using KCC-1 mesoporous silica functionalised with deep eutectic solvents
Mohd Saiful Adli
Azizman
 a,
Muhammad Adli
Hanif
a,
Naimah
Ibrahim
*ab,
Ayu Wazira
Azhari
ab,
Wan Khairunnisa
Wan Ramli
c,
Aishah
Abdul Jalil
de,
Nurul Sahida
Hassan
de,
Fazilah Farhana
Abdul Aziz
de and
Raja Nazrul Hakim
Raja Nazri
f
a,
Muhammad Adli
Hanif
a,
Naimah
Ibrahim
*ab,
Ayu Wazira
Azhari
ab,
Wan Khairunnisa
Wan Ramli
c,
Aishah
Abdul Jalil
de,
Nurul Sahida
Hassan
de,
Fazilah Farhana
Abdul Aziz
de and
Raja Nazrul Hakim
Raja Nazri
f
aFaculty of Civil Engineering & Technology, Universiti Malaysia Perlis, Arau, Perlis 02600, Malaysia. E-mail: naimah@unimap.edu.my
bCentre of Excellence for Water Research and Environmental Sustainability Growth (WAREG), Universiti Malaysia Perlis, Arau, Perlis 02600, Malaysia
cFaculty of Chemical Engineering & Technology, Universiti Malaysia Perlis, Arau, Perlis 02600, Malaysia
dFaculty of Chemical and Energy Engineering, Universiti Teknologi Malaysia, Johor Bahru, Johor 81310, Malaysia
eCentre of Hydrogen Energy, Institute of Future Energy, 81310, UTM, Johor Bahru, Johor, Malaysia
fUniversiti Kuala Lumpur, Branch Campus Malaysian Institute of Chemical and Bioengineering Technology, Lot 1988, Kawasan Perindustrian Bandar Vendor, Alor Gajah, Melaka 78000, Malaysia
First published on 23rd September 2024
Abstract
Sulphur dioxide, a toxic gas pollutant, is mainly generated by the combustion of fossil fuels and the smelting of sulphur-bearing mineral ores. Removal of SO2 gas or desulphurisation can be accomplished in industries using a variety of processes; the most efficient is wet flue gas desulphurisation (FGD). However, wet FGD has challenges, such as the requirement for wastewater treatment, excessive water usage, and the necessity for chloride protective coating. Despite having a lesser adsorption capacity than wet FGD, dry FGD can efficiently remove SO2 from the effluent gas stream and avoid the issues associated with wet FGD, provided that the sorbents are modified and regenerable. An alternative dry desulphurisation strategy by using fibrous mesoporous silica (KCC-1) modified with deep eutectic solvents (DES), choline chloride–glycerol (DES1) and choline chloride–ethylene glycol (DES2) is studied in this paper. KCC-1 modified with DES1 is found to increase SO2 adsorption capacity to 4.83 mg g−1, which is 1.73 times greater than unmodified KCC-1 and twice higher than KCC-1 modified with DES2 attributed to the sorbent's high porosity. Increasing reaction temperature and SO2 concentration reduce the adsorption capacity to 1.73 mg g−1 and 2.73 mg g−1, respectively. The Avrami kinetic model and the Toth isotherm model best reflect SO2 adsorption on the modified KCC-1, indicating that SO2 molecules are adsorbed exothermically in multilayer adsorption on a heterogeneous surface through a combination of physical and chemical processes. The higher SO2 adsorption capacity of the modified KCC-1 suggests that choline chloride–glycerol can provide additional sites for SO2 adsorption in dry FGD technology.
1. Introduction
Clean air is vital for all living things in order to sustain human health, protect ecosystems, and help preserve building structures. Unfortunately, fossil fuel utilisation in road transportation, power generation and industrial activities produces reactive air pollutants, including nitrogen oxides (NOx), carbon monoxide (CO), particulate matter (PM) and sulphur dioxide (SO2) that pose a significant threat to human health and the environment.1 While CO has the most immediate impact on health due to its capability to bind in red blood cells and rapidly reduce oxygen delivery2 and PM exposure has the most significant long-term health risks for cardiovascular disease and cancer,3 SO2 primarily affects the respiratory systems, causing irritation, coughing and asthma. In addition, SO2 also contribute to the formation of acid rain, harming forests, reducing soil fertility and damaging aquatic ecosystems.4Since the late 1960s, flue gas desulphurisation (FGD) technology has reduced emissions from coal-fired power plants. FGD systems typically comprise a vertical, cylindrical tower or column in which the solvent is brought into contact with the pollutants to be removed from the exhaust gas. These systems are frequently employed in industry for gas stream purification and separation, product recovery, and pollution management. Wet limestone, seawater-based, ammonia-based, and dry FGD are common methods used in power stations to reduce SO2 emissions. These methods are proven technologies and commercially available. The capacity to remove sulphur, dependability, spatial requirements, and reagent availability are the primary technical, economic, and commercial variables influencing the choice of a suitable FGD technology.5 Wet FGD systems generally achieve higher SO2 removal efficiency (exceeding 90%) than dry FGD systems (approximately 80%). However, the large amount of wastewater produced by wet FGD systems must be treated before disposal, requiring additional cost and time.6,7
Among the variety of sorbents in dry FGD, activated carbon is a commonly studied material which can be derived from various carbonaceous sources like agricultural wastes. Unfortunately, the application of activated carbon in up-scaled SO2 treatment is frequently limited by high flue gas temperatures and acidic gas compositions.8 Mesoporous silica (MS) based sorbents offer more extensive applications, including in medicine delivery, wastewater treatment, indoor air purification, catalysis, and SO2 removal from flue gas.9 MS are superior because of their unique features, which include organised pore architectures, high specific surface areas, and the ability to be synthesised in various morphologies, including spheres, rods, discs, powders, etc.
One of the newest members of the MS materials group, KAUST Catalyst Centre-1 (KCC-1), was initially synthesised in 2010 by Polshettiwar et al.10 In contrast to the conventional MS materials such as Fudan University-12 (FDU-12), Korea Institute of Science and Technology-5 and 6 (KIT-5, KIT-6), Santa Barbara Amorphous-15 and 16 (SBA-15, SBA-16), Mobil Composition of Matter-41 and 48 (MCM-41, MCM-48), KCC-1 has concentric and regular fibres that developed radially from the centre of the silica spheres to the outside of the spheres, which accounts for its high surface area.11,12 As a result, KCC-1 could be an excellent alternative for catalysis applications and adsorption that require easily accessible active sites.12–15 Silica-based sorbents require modification to enhance removal efficiencies and improve catalytic activities, as limited active sites will reduce their accessibility when significant mass transport is vital.16 Modification of silica-based materials has been shown to improve the adsorption activity of the sorbents in a recent work by Lai et al.17
On the other hand, the use of deep eutectic solvent (DES) for CO2 capture and SO2 sequestration has also grown in interest.18 DES has a high distribution coefficient of solutes, which means it is particularly efficient in storing solutes with properties and behaviours similar to ionic liquids (ILs). DES has been widely employed in organic synthesis, metal electrodepositions and extractions, and CO2 absorptions due to its low cost, low volatility, non-toxicity and ease of synthesis. Yang et al.19 reported that DES achieves high SO2 sorption capacity through significant charge transfer interactions between chloride anion and SO2 molecules. Furthermore, the interaction may be tuned to control SO2 desorption, so that the sorbent can be cycled multiple times. In addition, DES is advantageous in FGD applications due to its good thermal stability and the ability to maintain stability.13 Motivated by the excellent properties of KCC-1 and DES, the suitability of KCC-1 modified with DES for SO2 removal was investigated in this work.
2. Materials and methods
2.1 Materials
2.2 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (HBA:HBD) by stirring for 60 min at 60 °C (with glycerol) and 70 °C (with ethylene glycol). This specific ratio was selected based on preliminary findings indicating optimal performance with minimised chemical usage. A ratio of 1:1 is inapplicable as the produced DES will self-crystallise during storage. The mixtures were agitated until clear and homogeneous. Afterwards, the mixtures were allowed to cool to room temperature and then stored in sealed vials, kept in a desiccator.
000 h−1, characteristic of an exhaust gas condition.
:2 (HBA:HBD) by stirring for 60 min at 60 °C (with glycerol) and 70 °C (with ethylene glycol). This specific ratio was selected based on preliminary findings indicating optimal performance with minimised chemical usage. A ratio of 1:1 is inapplicable as the produced DES will self-crystallise during storage. The mixtures were agitated until clear and homogeneous. Afterwards, the mixtures were allowed to cool to room temperature and then stored in sealed vials, kept in a desiccator.
000 h−1, characteristic of an exhaust gas condition.
In this study, the breakthrough point was considered achieved once the gas analyser measured 5% of the initial SO2 concentration in the exit gas stream. To ease comparison with samples that took too long to reach complete saturation, the capacity of SO2 removal was computed at a fractional concentration (C/C0) equal to 0.95 using eqn (1), assuming all samples have reached bed saturation. C and C0 are the SO2 concentrations at time t and initial SO2 concentration (mg L−1), respectively; yt is the gas molar fraction, Qf is the gas flow rate (L min−1), and mc is the mass of the sorbent bed (g).
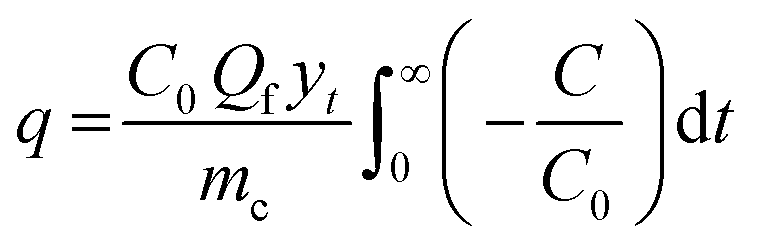 | (1) |
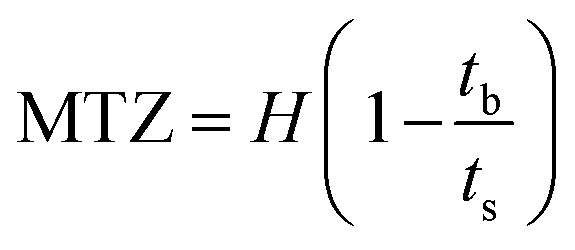 | (2) |
000. Meanwhile, Fourier Transform Infrared Spectroscopy (FTIR) via attenuated total reflection (ATR) was used to examine the surface chemistry of the samples using a Frontier PerkinElmer instrument, with polystyrene film NIST as the verification material. The surface area and porosity of fresh and modified KCC-1 were determined through N2 adsorption–desorption using Micromeritics 3Flex at −196 °C. Before measurement, the samples were degassed at 150 °C for 1 hour. The surface area, average pore size and pore volume of all samples were determined by Brunauer–Emmett–Teller (BET) analysis and the Medek model as well as the adsorbed N2 volume calculation at standard temperature and pressure (STP), respectively.
| qt = qe[1 − exp(−K1t)] | (3) |
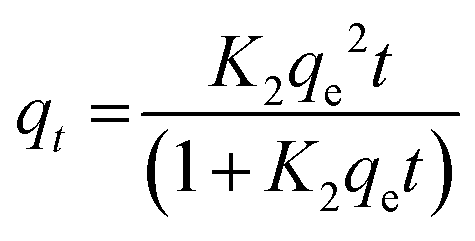 | (4) |
| qt = qe[1 − exp(−K3tNA)] | (5) |
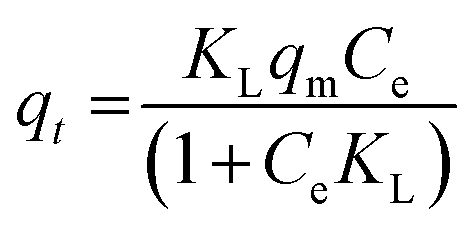 | (6) |
| qt = KFCe1/n | (7) |
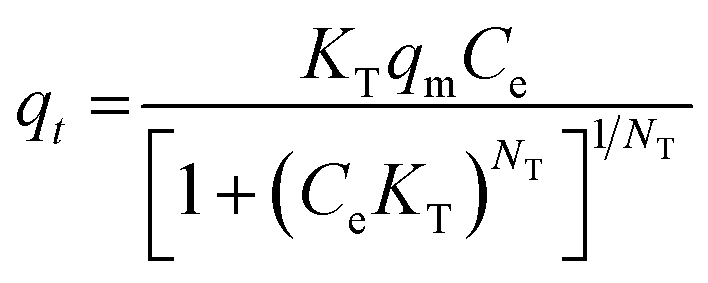 | (8) |
Freundlich's reaction rate constant is denoted as KF (L mg−1), while the adsorption affinity is represented by 1/n (dimensionless). On the other hand, KT (mg g−1) and NT (mg g−1) are Toth rate and isotherm constants, respectively. The adsorption process is evaluated based on the value of 1/n. A 1/n < 1 indicates chemisorption, or alignment with the conventional Langmuir isotherm, whereas a 1/n > 1 indicates cooperative adsorption. The Toth equation describes a heterogeneous, multilayer adsorption system and serve as a specialised form of Langmuir isotherm with minimal validity. The system heterogeneity is represented by the parameter NT. The further its deviation from unity, the greater the heterogeneity within the system.
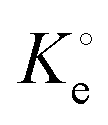 (dimensionless) is the adsorption thermodynamic equilibrium constant.27
(dimensionless) is the adsorption thermodynamic equilibrium constant.27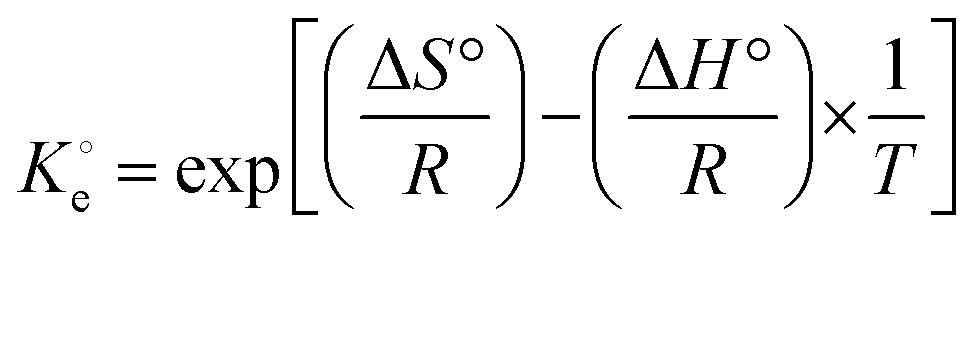 | (9) |
3. Results and discussion
3.1 DES synthesis
After heating, the initial attempt to prepare the DES solution using a 1:1 molar ratio of HBA to HBD resulted in a colourless, homogeneous liquid. However, a white precipitate formed in both DES1 and DES2 mixtures after cooling to room temperature. As noted by Manurung et al., the formation of precipitates in a DES mixture can be attributed to excess halide anions, which can occur when the concentration of the HBA is insufficient to form hydrogen bonds with all the halide anions.28 Furthermore, excess salts may also lead to an increase in the freezing point of the mixture, resulting in precipitation, as mentioned by Makos et al. and Qin et al., highlighting that the molar ratio of the HBA to HBD significantly influences the freezing point of the DES.29,30 By adjusting the molar ratio to 1:2, a clear and homogeneous DES solution was achieved at room temperature, indicating a more balanced interaction between the HBA and HBD components, as shown in Fig. 1(a).
 | ||
| Fig. 1 Physical appearance: (a) DES solution at room temperature, (b) post-mixing of modified catalyst and (c) post-calcination of modified KCC-1. | ||
3.2 KCC-1 optimization and characterization
The SO2 breakthrough study produced a breakthrough-time curve (C/C0versus t), where C0 is the initial concentration (ppm) of SO2 passed through the adsorbent bed and C is the outlet concentration (ppm) recorded by the gas analyser. Both fresh and DES-modified KCC-1 samples (KCC-1/DES1 and KCC-1/DES2) were screened, and the sample exhibiting the highest adsorption capacity was selected for further optimisation by varying reaction temperatures and inlet concentrations. Fig. 2 shows the breakthrough curves of all three samples under 200 mL min−1 flow of 1500 ppm SO2, while Table 1 summarises the adsorption capacity and breakthrough data for these samples.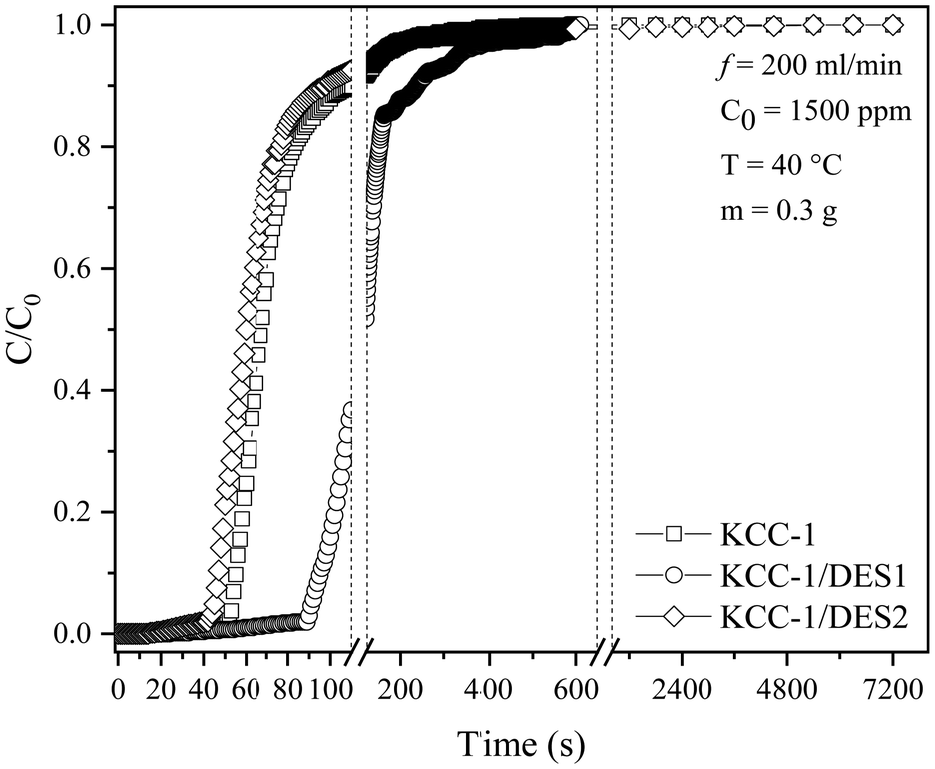 | ||
| Fig. 2 SO2 breakthrough curves of fresh KCC-1 and DES-modified KCC-1. | ||
| Sample | Breakthrough time at C/C0 = 0.05 (s) | Time at C/C0 = 0.95 (s) | MTZ (%) | Adsorption capacity at C/C0 = 0.95 (mg SO2 per g sample) |
|---|---|---|---|---|
| KCC-1 | 54 | 159 | 87.21 | 2.79 |
| KCC-1/DES1 | 91 | 332 | 92.17 | 4.84 |
| KCC-1/DES2 | 46 | 144 | 87.95 | 2.41 |
Table 1 and Fig. 2 show that KCC-1/DES1 has the longest adsorption breakthrough time of 91 seconds compared to the other samples under the same experimental conditions. On the other hand, KCC-1/DES2 has a faster breakthrough time than fresh KCC-1 and KCC-1/DES1, respectively, which is less preferred, as the slower the breakthrough time, the longer the adsorption bed can serve.
The breakthrough investigation of fresh and modified KCC-1 samples reveals SO2 adsorption capacities (at C/C0 = 0.95) ranging between 2.41 and 4.84 mg g−1. KCC-1/DES1 exhibits the highest SO2 adsorption capacity, which is 1.73 times higher than that of fresh KCC-1 and 2 times higher than KCC-1/DES2. The results also show that fresh KCC-1 has the lowest MTZ of 87.21% compared to KCC-1/DES1 and KCC-1/DES2, with MTZ values of 92.17 and 87.95%, respectively. A low MTZ is theoretically caused by a high flow rate, which shortens the time it takes for the adsorbate to diffuse into the pores of the sorbent materials as the adsorbate solution (or gas mixture in this case) leaves the column before reaching equilibrium, causing breakthrough time to appear relatively faster.31
According to recent studies in gas adsorption, choline chloride-based DES is deemed more popular than the other types for CO2 adsorption. For instance, the modification of mesoporous silica gel with choline chloride improved its CO2 adsorption capacity at 25 °C to 51 mg g−1.32 In addition, a nano-composite adsorbent functionalised with DES mixture (ChCl–urea)33 also produced a maximum capacity of 23.0 mg g−1 in a comparable work. Zulkurnai et al.21 also investigated and analysed the CO2 adsorption capacity of choline chloride-based DES. Compared to raw activated carbon, the DES-based activated carbon had a higher CO2 adsorption removal at 9.851 mg g−1.21 Chloride–glycerol DES showed a higher CO2 adsorption capacity (20.1 mg g−1) than choline chloride–ethylene glycol at a capacity of 18.8 mg g−1.34 In the case of SO2 removal here, KCC-1/DES1 (utilising choline chloride–glycerol) also shows better performance than fresh KCC-1 and KCC-1/DES2 (with choline chloride–ethylene glycol).
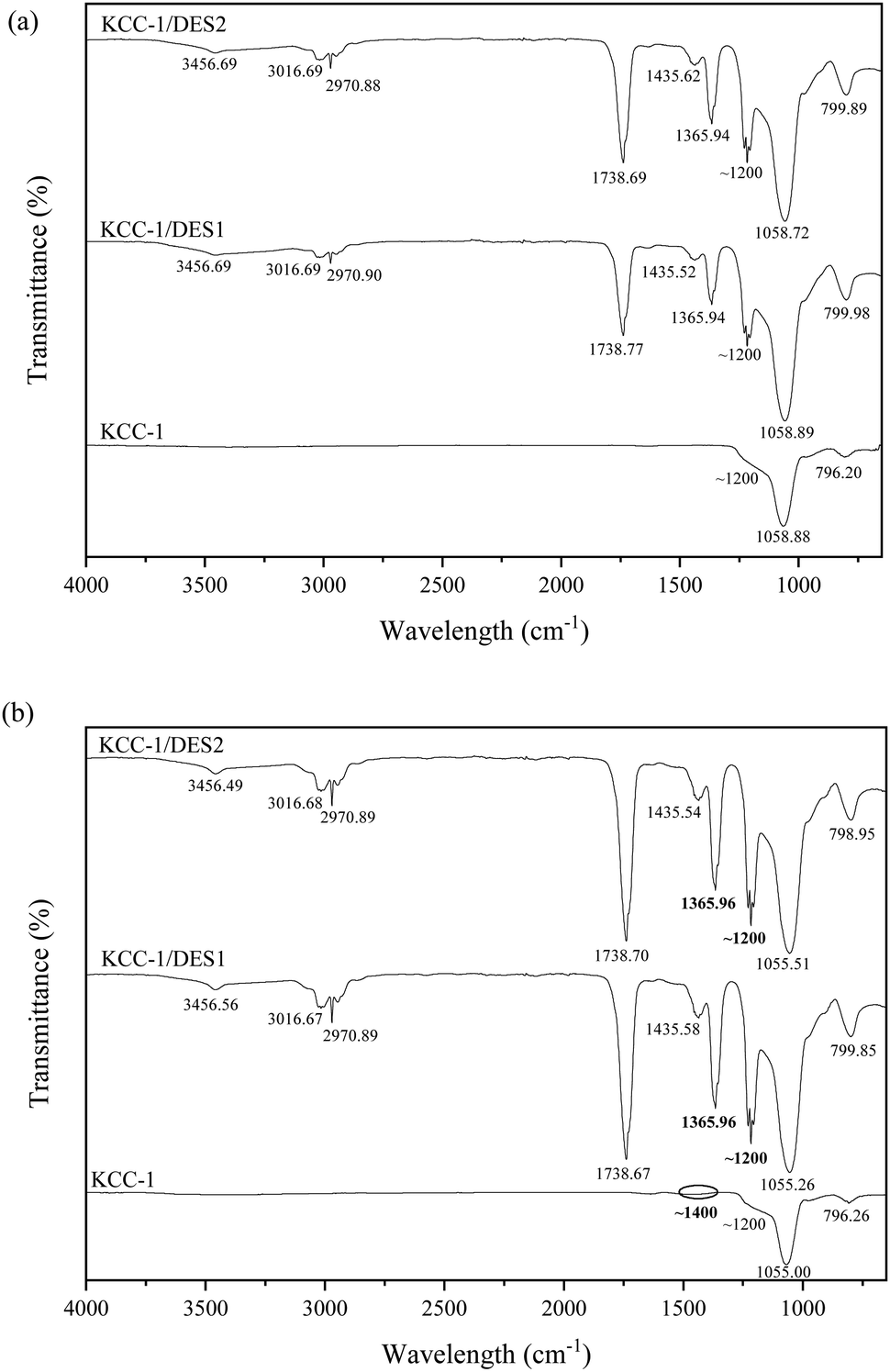 | ||
| Fig. 3 FTIR spectra of all samples (a) before and (b) after SO2 adsorption. | ||
| Possible functional group | Peak wavelength (cm−1) | |||||
|---|---|---|---|---|---|---|
| KCC-1 | KCC-1/DES1 | KCC-1/DES2 | ||||
| Before | After | Before | After | Before | After | |
| Si–O–H, O–H group, N–H group | — | — | 3455.69 | 3456.56 | 3456.37 | 3456.49 |
| O–H group | — | — | 3016.69 | 3016.67 | 3016.69 | 3016.68 |
| C–H symmetric and asymmetric stretching vibration | — | — | 2970.90 | 2970.89 | 2970.88 | 2970.89 |
| –COOH group | — | — | 1738.77 | 1738.67 | 1738.69 | 1738.70 |
| C–H bending vibration | — | — | 1435.62 | 1435.58 | 1435.52 | 1435.54 |
| SO2 | — | ∼1200, ∼1400 | — | ∼1200, ∼1360 | — | ∼1200, ∼1360 |
| O–H group | — | — | 1365.94 | 1365.96 | 1365.94 | 1365.96 |
| Si–O stretching vibration, C–O stretching vibration | 1228.86, 1217.09, 1206.16 | 1228.86, 1217.11, 1206.16 | 1228.41, 1217.05, 1205.98 | 1228.77, 1217.10, 1206.04 | 1228.64, 1217.07, 1205.79 | 1228.76, 1217.10, 1206.01 |
| Si–O asymmetric vibration, O–H group | 1058.85 | 1055.00 | 1058.89 | 1055.26 | 1058.72 | 1055.51 |
| Si–O symmetric vibration | 796.20 | 796.26 | 799.98 | 799.85 | 799.89 | 798.95 |
Additionally, six new peaks appear on the FTIR spectra of the DES-modified KCC-1 samples, corresponding to the presence of various functional groups. A stretching bond can be seen around 3455 cm−1, corresponding to SiO–H associated with silanol, O–H group indicating the presence of water and N–H stretching pertaining to amides and amine groups.36 The peak around 3016 cm−1 can also be attributed to water adsorbed on the surface of the modified KCC-1.37 The presence of water on the surface of the adsorbent may result in a slower adsorption rate and limit the mass transfer capacity.38 Water molecules can preferentially compete with SO2 for adsorption sites if the adsorbent has a higher affinity for water, leaving fewer sites available for the SO2 molecules.39 In addition, a slower diffusion rate can limit the overall adsorption capability as the water molecules create a barrier of diffusion, hindering the movement of SO2 molecules into the pores of the adsorbent.40 Nonetheless, based on the spectra, the transmittance intensity related to water is significantly lower than the other compounds; therefore, it can be concluded that the effect of water on SO2 adsorption is minimal.
On the other hand, the observed N–H stretching proves the presence of choline chloride on the surface of DES-modified KCC-1 samples. The N-element is expected to aid in SO2 capture and successful bonding to the surface of KCC-1 samples.21 A sharp, small peak corresponding to C–H symmetric and asymmetric stretching vibrations associated with alkanes and alkenes is detected around 2970 cm−1 across all samples.41
A significant peak is observed in the FTIR spectra of all DES-modified KCC-1 samples at around 1738 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, suggesting the presence of saturated aliphatic compounds from alkane groups probably originating from both HBA and HBD elements of the DES.22 Meanwhile, due to the chemical nature of HBD component, all samples show C–H bonding in the 1435 cm−1 range, possibly ascribed to an alkane group.42
O stretching, suggesting the presence of saturated aliphatic compounds from alkane groups probably originating from both HBA and HBD elements of the DES.22 Meanwhile, due to the chemical nature of HBD component, all samples show C–H bonding in the 1435 cm−1 range, possibly ascribed to an alkane group.42
The chemical heterogeneity of the sorbent's surface also determines the extent and feasibility of an adsorption mechanism. The presence of heterocyclic compounds containing sulphur (S), nitrogen (N) and oxygen (O) influences the surface chemical heterogeneity. The specific quantity and form of heteroatoms depend on the modification methods and materials from which the atoms are derived. The presence of basic groups with strong affinity improves interaction; however, this is not the point at which adsorption becomes entirely chemisorption.43 On the other hand, an acidic character is caused by oxygen-containing functionalities that form during oxidation synthesis, giving the sorbents electron-acceptor properties. In contrast, N-containing functionalities are typically basic in nature. These functionalities also have electron-donor properties, making them attractive sites for the electron-deficient S-atom in SO2.44 At low temperatures, imines, amines, amides, imides, and nitriles will predominate in specific N-functionalities produced during modification. Although both O- and N-surface functional groups can improve SO2 adsorption, the presence of O prior to N-doping can further enhance the effectiveness of N-functionalisation significantly.45
Oxygen-containing groups influence two key properties of sorbents: their hydrophilicity or hydrophobicity, and their basicity or acidity. The number of polar O-groups influences the degree of hydrophilicity. Carbon oxidation introduces various O-functionalities and endows the surface with electron-acceptor properties. For instance, lactone and phenol groups contribute to surface acidity, whereas surface basicity is caused by pyrone, carbonyl, benzopyran and alcohol groups.46,47 Nitrogen surface functional groups are not the only species with basic properties; other O-containing groups with basic properties include alcohols, carbonyls, and ethers, which contain electron-donating O-atoms capable of having electrostatic interactions with SO2, thereby enhancing adsorption.48,49 The framework whereby SO2 is adsorbed due to these functional groups can be evaluated by classifying them according to their intrinsic heteroatoms. These functional groups are as complicated as the techniques utilised to introduce them. It is difficult to assume that these groups are entirely independent of one another, and further research is still required to clarify any potential antagonistic effects among these groups in the context of SO2 adsorption.45
Based on the literature, peaks related to SO2 can be observed in typical FTIR spectra around 1150–1210 cm−1 and 1340–1400 cm−1 corresponding to the symmetric and asymmetric vibrations of SO2 molecules, respectively.50–52 In this work, a tiny peak can be observed in Fig. 3(b) around 1400 cm−1 on the unmodified KCC-1 spectra, while a significantly enhanced peak can be seen around 1365 cm−1, corresponding to the asymmetric vibration of adsorbed SO2 molecules in both DES-modified samples. The presence of these new vibrational peaks may result from the formation of a charge-transfer complex due to the interactions between SO2 molecules and lone pair of electrons on N (N:), leading to the formation of an antibonding SO2 orbital (N → SO2).52 On the other hand, the presence of symmetric SO2 molecules is more difficult to determine, as the peak could have merged with the adjacent peaks around 1200 cm−1, corresponding to Si–O and C–O stretching vibrations. Nonetheless, the sharp increment of the transmittance intensity at this frequency strongly suggests the presence of symmetric SO2 on the adsorbent. This is an indication that SO2 molecules are adsorbed on active sites comprising silica-based radicals (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O˙ and Si˙) on KCC-1, similar to that suggested by literature.53 The minimal change in FTIR spectra of the unmodified KCC-1 in comparison to the DES-modified samples after SO2 adsorption demonstrates the importance of DES in the process as the interaction between SO2 molecules and unmodified KCC-1 is very weak52
Si–O˙ and Si˙) on KCC-1, similar to that suggested by literature.53 The minimal change in FTIR spectra of the unmodified KCC-1 in comparison to the DES-modified samples after SO2 adsorption demonstrates the importance of DES in the process as the interaction between SO2 molecules and unmodified KCC-1 is very weak52
 | ||
| Fig. 4 FESEM micrographs for (a) KCC-1, (b) KCC-1/DES1 and (c) KCC-1/DES2. | ||
In each figure, the colloidal nanospheres reveal well-defined fibrous structures made of three-dimensional arrangements of dendrimeric fibres. Because of the existence of these dendrimeric fibres, the available high surface area of fresh and modified KCC-1 samples may become easily accessible.10 As analysed in Section 3.1, KCC-1/DES1 (with choline chloride) has the highest adsorption capacity of 191.88 mg g−1, surpassing the other samples. On the other hand, KCC-1/DES2 modified with ethylene glycol has lower adsorption capacity despite having the largest size (average diameter of 301.70 nm), as shown in Fig. 4(b), indicating that the larger the diameter of the nanospheres, the lower the saturation surface concentration. This is because larger particle sizes have less curvature, lower deviation from a flat surface, fewer number of active site densities, and thus less surface adsorbates.54 In addition, unmodified KCC-1 has the smallest sphere diameter with an average of 281.55 nm; however, the adsorption capacity is also lower by 3.92 times than the modified KCC-1/DES1. This indicates that the nature of interaction between unmodified KCC-1 and SO2 is weak, and there is a shortage of active sites.55
The surface properties of nanospheres are size-dependent, with smaller particles having a higher affinity to adsorb SO2 and decontaminating more slowly, as evidenced by a longer saturation time. A possible explanation of the increase in KCC-1 particle size upon DES modification, is that the high concentration of the modified sample allows a faster rate of precursor hydrolysis. This rapid hydrolysis contributes more to particle size growth than nucleation rate, causing mesoporous silica particle to increase in size with higher concentration; one of the major variables influencing particle size.56 Furthermore, the catalyst molar ratio also contributed to the increase in the particle size of the mesosphere.57
N2 adsorption–desorption isotherm of KCC-1 and modified KCC-1 samples are shown in Fig. 5, while the properties of the samples (surface area, total pore volume and average pore size) are summarized in Table 3.
| Sample | Surface area (m2 g−1) | Total pore volume (cm3 g−1) | Average pore size (nm) |
|---|---|---|---|
| KCC-1 | 553.07 | 0.1170 | 2.186 |
| KCC-1/DES1 | 528.78 | 0.1138 | 2.208 |
| KCC-1/DES2 | 524.43 | 0.1123 | 2.201 |
The isotherm plots are offset along the y-axis for better comparison among the samples. The N2 adsorption–desorption isotherm of KCC-1 and DES-modified KCC-1 shown in Fig. 5 can be classified as type IV(a) isotherm with H3 hysteresis loop in P/P0 range of 0.4–1.0. This implies that the pores of all samples demonstrate mesoporous properties with characteristics of capillary condensation and non-uniform slit-shaped pores.58 The N2 uptake by the samples follows the order of KCC-1 > KCC-1/DES1 > KCC-1/DES2, where slight reduction demonstrated by the DES-modified KCC-1 can be attributed to blockage of interparticle pores due to the introduction of DES onto the KCC-1 surface.59 As shown in Table 3, the surface area and total pore volume of KCC-1 samples slightly decrease upon modification with DES, possibly due to the dispersion of DES, which partially blocks the available pores, as mentioned earlier. On the other hand, the average pore size of KCC-1 samples shows a minor increment with the presence of DES, possibly due to partial breakage of the silica framework, which may also contribute to the reduced surface area.60
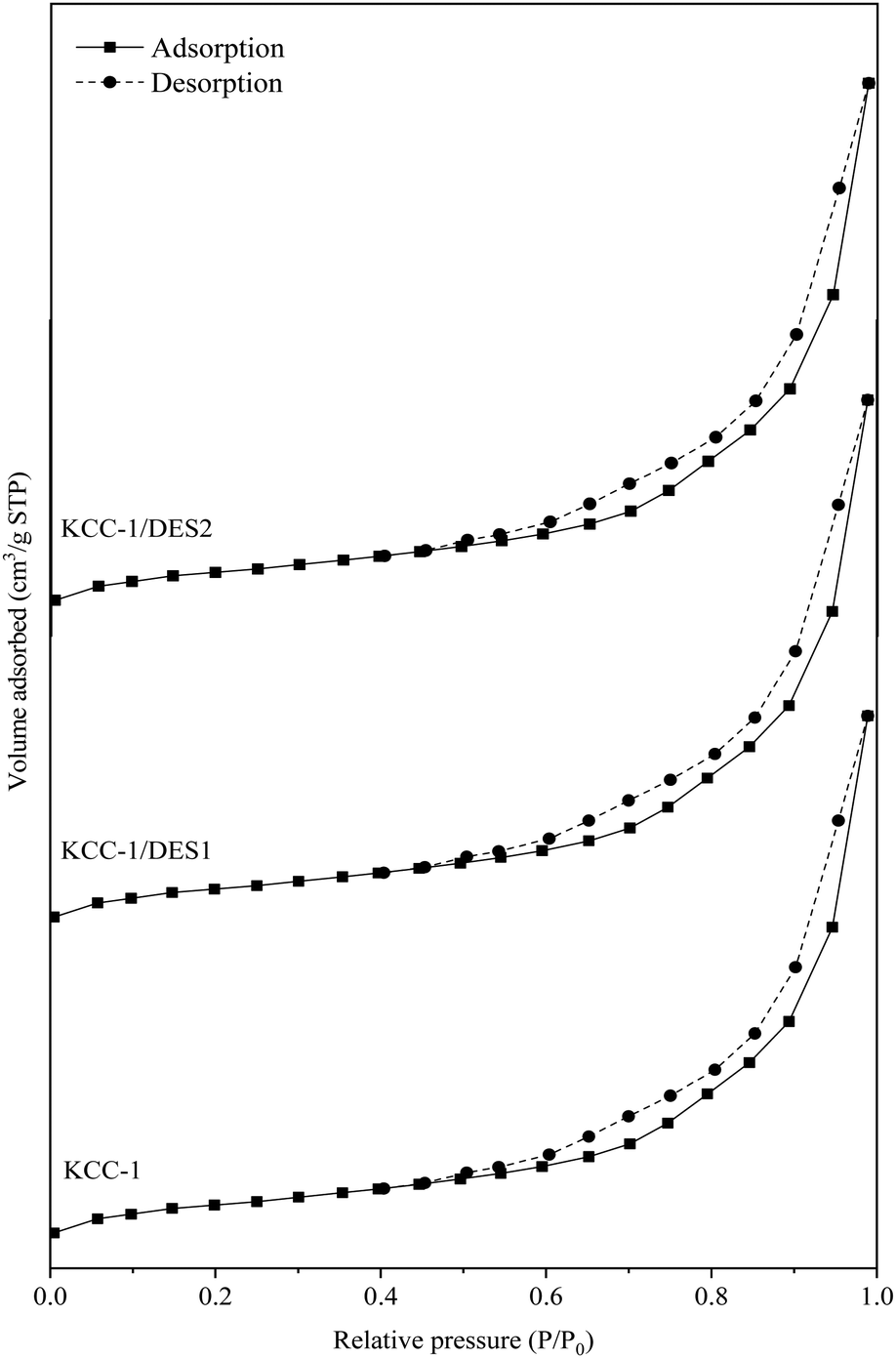 | ||
| Fig. 5 N2 adsorption–desorption isotherms for KCC-1 and DES-modified KCC-1. | ||
3.3 Effects of different inlet concentrations
The effect of inlet SO2 concentration was investigated using KCC-1/DES1 (chosen based on the breakthrough study in Section 3.1) at various concentrations ranging from 1500 to 2000 ppm, reflecting typical SO2 concentrations released by medium to high sulphur-content fossil fuels.61Fig. 6 shows the breakthrough curves of SO2 adsorption at varying SO2 concentrations, while Table 4 summarizes breakthrough time, MTZ and adsorption capacity at different inlet SO2 concentrations. As the SO2 concentration is already relatively high at 1500 ppm, further increase to 2000 ppm with the same amount of adsorbent decreases the adsorption capacity of KCC-1/DES1 from 4.84 to 2.73 mg g−1. The decrease in SO2 adsorption capacity with an increase in SO2 concentration or the uptake of SO2 molecules supplied per g of adsorbent could be attributed to the higher number of SO2 molecules available, which may remain unabsorbed at a constant adsorbent mass. This results in quick occupancy of the available active sites, which subsequently causes rapid breakthrough and saturation of the adsorbent bed.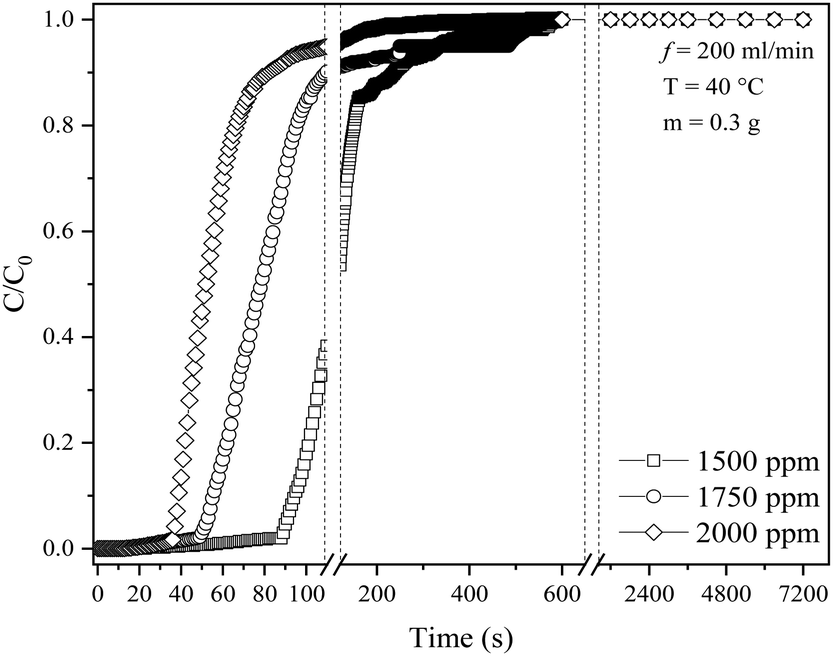 | ||
| Fig. 6 SO2 breakthrough curves of KCC-1/DES1 at different SO2 concentrations. | ||
| Inlet concentration (ppm) | Breakthrough time at C/C0 = 0.05 (s) | Time at C/C0 = 0.95 (s) | MTZ (%) | Adsorption capacity at C/C0 = 0.95 (mg SO2 per g sample) |
|---|---|---|---|---|
| 1500 | 91 | 332 | 92.17 | 4.84 |
| 1750 | 53 | 250 | 90.80 | 3.49 |
| 2000 | 38 | 105 | 83.81 | 2.73 |
A higher SO2 concentration also results in a greater driving force due to the steeper concentration gradient between SO2 in the gas phase and on the adsorbent's surface. This gradient acts as a driving force for diffusion, causing SO2 molecules to move from the gas phase towards the adsorbent,62 inducing an effect similar to the influence of a high SO2 flow rate on MTZ, as mentioned in Section 3.2. The limitation in the capacity of the adsorbent bed as the SO2 concentrations increase has also been reported by others.24,63 However, a fixed-bed desulphurisation efficiency is also influenced by numerous other factors such as the preparation method, activity, the type of desulphurising agent, SO2 concentration, pressure and bed temperature. Based on the results, it can be concluded that an optimum capacity for SO2 removal could be attained under lower concentration conditions, emphasising the crucial role that concentration plays for effective adsorption.64
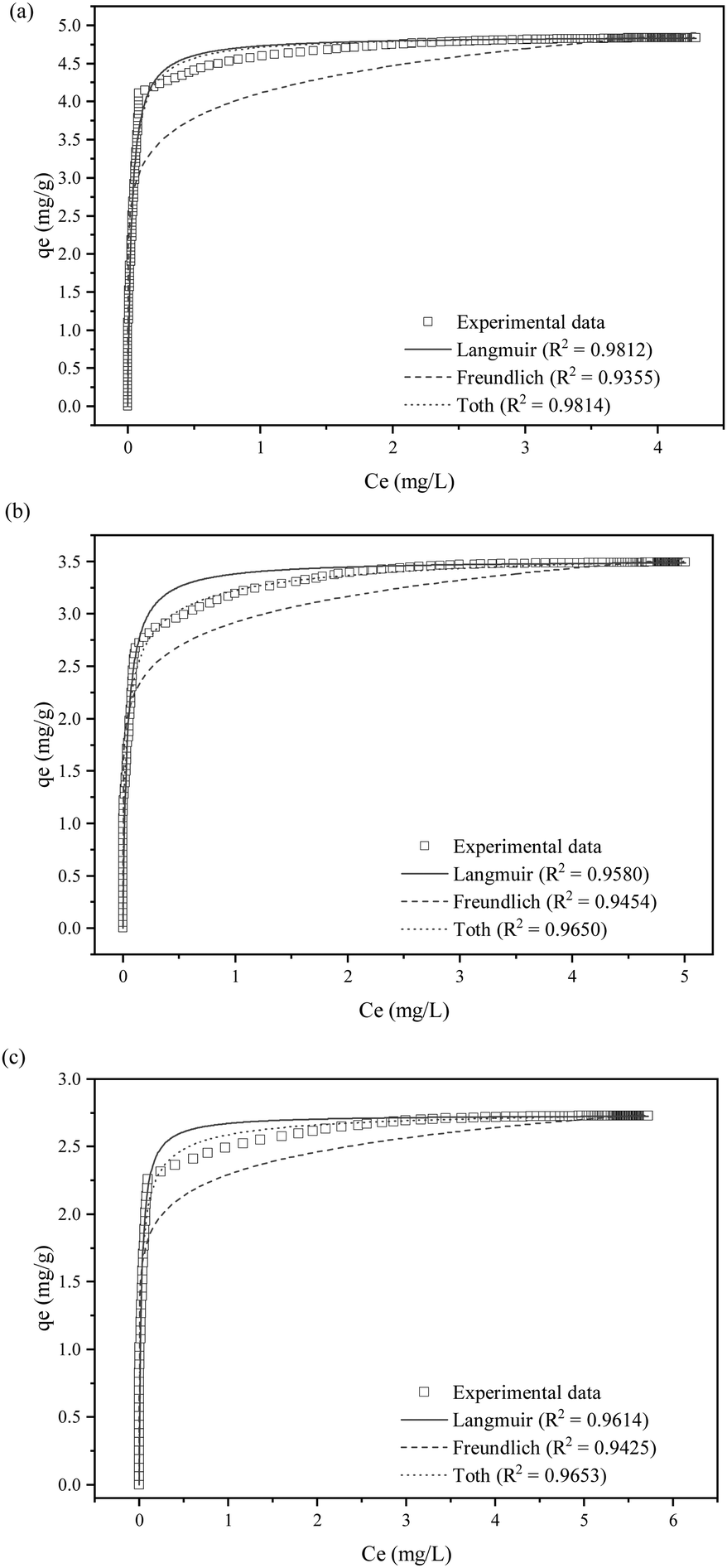 | ||
| Fig. 7 Nonlinear fittings of SO2 removal at (a) 1500 ppm, (b) 1750 ppm and (c) 2000 ppm SO2 initial concentrations with Langmuir, Freundlich and Toth isotherm model. | ||
| Isotherm model | SO2 concentration (ppm) | ||
|---|---|---|---|
| 1500 | 1750 | 2000 | |
| Langmuir | |||
| R 2 | 0.9812 | 0.9580 | 0.9614 |
| K L | 35.424 | 25.320 | 41.089 |
| q m | 4.8635 | 3.5148 | 2.7359 |
| Freundlich | |||
| R 2 | 0.9355 | 0.9454 | 0.9650 |
| K F | 4.1103 | 2.9195 | 2.2928 |
| Toth | |||
| R 2 | 0.9814 | 0.9650 | 0.9653 |
| K T | 40.409 | 115.84 | 117.64 |
| N T | 0.9208 | 0.5301 | 0.6217 |
| q m | 4.8801 | 3.7305 | 2.8049 |
All samples fit well with the Toth isotherm, with NT values less than 1, which indicates localized multilayer adsorption of interacting particles onto heterogeneous surface systems.65 The NT parameter quantifies the adsorption system's heterogeneity, and a deviation from unity suggests that the system is heterogeneous.66 On the other hand, the Toth equation simplifies to the Langmuir isotherm when NT = 1. A study by Avijegon et al.67 also concluded that the adsorption of natural gas may be well characterized by a Toth isotherm model, with parameters derived from data regression for CO2, CH4, and N2 mixtures within the same pressure and temperature ranges, along with the experimental binary and ternary adsorption equilibrium data.67
Based on Table 5, a fascinating trend can be observed concerning the role of SO2 concentration in the adsorption process, i.e. NT decreases with increasing SO2 concentration (0.92 at 1500 ppm, 0.53 at 1750 ppm, and 0.62 at 2000 ppm). As mentioned earlier, an NT value closer to 1 at a lower SO2 concentration signifies a relatively uniform surface, consistent with the Langmuir model. However, the subsequent decrease in NT values with increasing SO2 concentrations suggests a shift towards a more heterogeneous surface. This trend indicates that the higher SO2 concentrations may be altering the surface properties of the adsorbent as the SO2 molecules preferentially occupy the most favourable sites at lower concentrations, leaving a wider distribution of lower-energy sites that contribute to adsorption at higher concentrations.
Furthermore, the interaction of KCC-1/DES1 with SO2 may modify its surface chemistry, creating new adsorption sites with varied energies. The observed decreasing trend in NT values of Toth isotherm provides compelling evidence that high SO2 concentrations lead to a more heterogeneous surface, resulting in non-uniform adsorption behaviour. Similar interaction patterns between adsorption behaviour and SO2 concentration are also observed in other studies.68–71
3.4 Effects of different reaction temperatures
SO2 adsorption was studied at various temperatures for kinetic investigation, and the resulting breakthrough curves are presented in Fig. 8. The MTZ and adsorption capacity at all studied temperatures are summarized in Table 6. The SO2 adsorption capacity is observed to decrease by 2.51 and 2.80 times with an increase in the reaction temperature from 40 °C to 80 and 120 °C, respectively. This is expected as increased temperatures generally inhibit gas adsorption; therefore, the greater the temperature, the lower the gas adsorption capacity, whereas elevated pressures promote adsorption.72 As the temperature increases from 40 to 120 °C, the percentage of MTZ also decreases after a contact period of 91, 36 and 29 s under the three different temperatures, respectively, indicating that the adsorption process is optimum at lower temperatures. Similar findings was reported by Delgado et al.,63 who observed that the rise in temperature from 298 K to 308 K decreased the breakthrough time and adsorption capacity by 21.6% and 23.6%, respectively.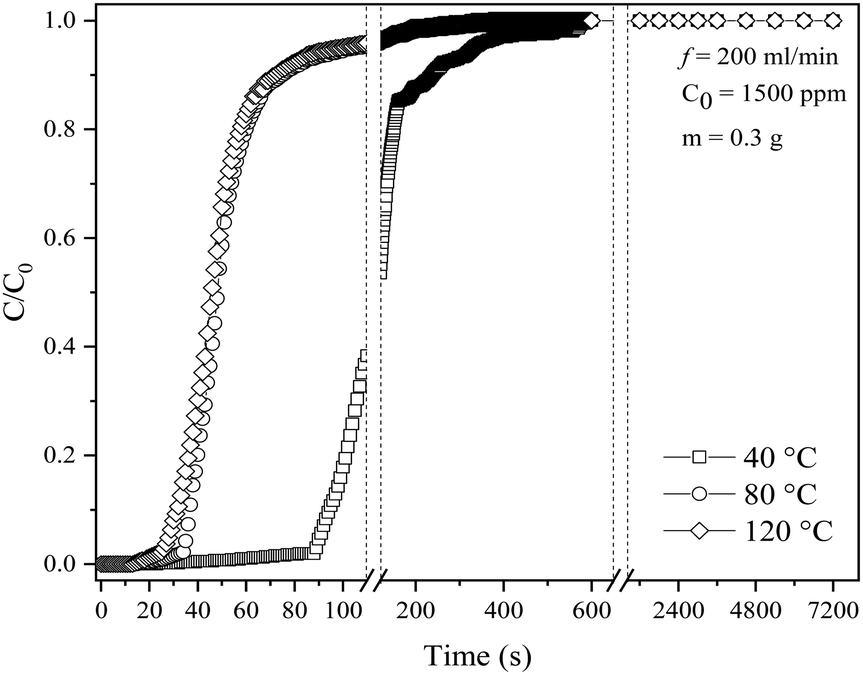 | ||
| Fig. 8 SO2 breakthrough curves of KCC-1/DES1 at different temperatures. | ||
| Temperature (°C) | Breakthrough time at C/C0 = 0.05 (s) | Time at C/C0 = 0.95 (s) | MTZ (%) | Adsorption capacity at C/C0 = 0.95 (mg SO2 per g sample) |
|---|---|---|---|---|
| 40 | 91 | 332 | 92.17 | 4.84 |
| 80 | 36 | 106 | 83.02 | 1.93 |
| 120 | 29 | 87 | 76.00 | 1.73 |
At higher temperatures, the breakthrough time and total adsorption rate decrease faster than the growth rate. As a result, the adsorption capacity and breakthrough time decrease by 34.3% and 27.8%, respectively, as the temperature rises from 40 °C to 120 °C. It can be inferred that the temperature increase has a negative impact on the adsorption performance,63 influenced by two main factors: the exothermic nature of the adsorption process and kinetic effects. As the temperature increases, the system becomes less favourable for the adsorption reaction. The adsorbate molecules (SO2) have additional thermal energy, making them less likely to adhere to the adsorbent surface and increasing their tendency to desorb into the gas phase. Moreover, the adsorbate molecules have higher kinetic energy at high temperatures, leading to more frequent collisions with the surface, making it harder to achieve successful adsorption as they may collide and bounce off the surface instead of forming stable interactions.73,74
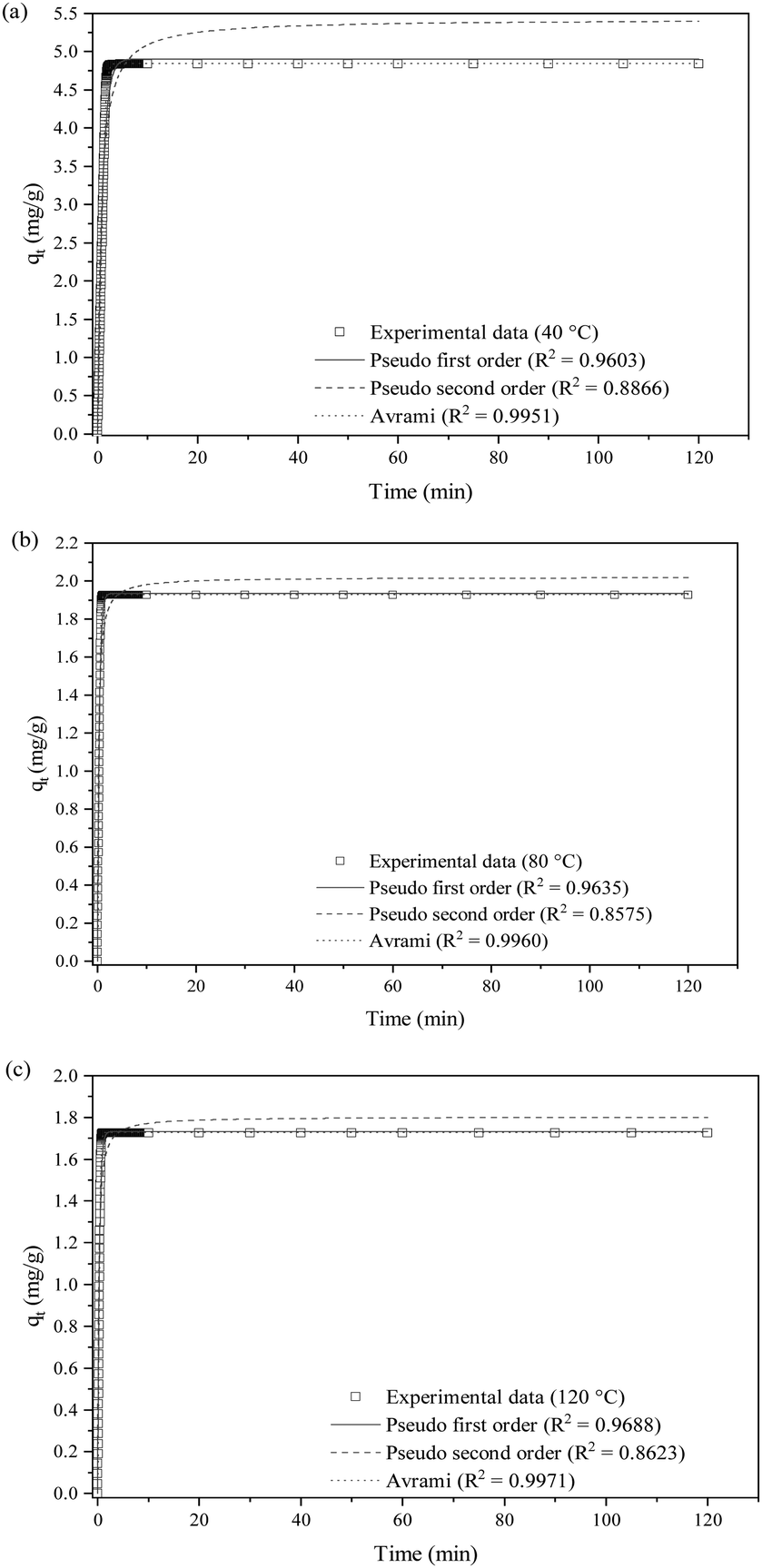 | ||
| Fig. 9 Non-linear fittings of SO2 removal at (a) 40 °C, (b) 80 °C and (c) 120 °C with P1st, P2nd and Avrami kinetic models. | ||
| Temperature (°C) | Kinetic model | ||||||
|---|---|---|---|---|---|---|---|
| Pseudo-first-order (P1st) | Pseudo-second-order (P2nd) | Avrami | |||||
| R 2 | K 1 | R 2 | K 2 | R 2 | K 3 | N A | |
| 40 | 0.961 | 1.040 | 0.887 | 0.278 | 0.995 | 0.986 | 1.642 |
| 80 | 0.963 | 2.681 | 0.857 | 2.484 | 0.996 | 2.486 | 1.640 |
| 120 | 0.969 | 2.930 | 0.862 | 3.105 | 0.997 | 2.728 | 1.576 |
The adsorption rate for P1st, K1, increases from 1.040 to 2.930 with the temperature increase. A similar rise in adsorption rate is also recorded for P2nd's K2 and Avrami's K3. The nonlinear Avrami kinetic plots show R2 values above 0.99 across all tested temperatures, higher than P1st (above 0.96) and P2nd (between 0.85 and 0.88), indicating that the experimental data fit better to the Avrami model. The Avrami model assumes that adsorption happens through both chemisorption and physisorption pathways. Avrami exponents (NA) that range from 1.576 to 1.642 indicate a one-dimensional growth of adsorbed SO2 molecules on the accessible active sites of the KCC-1/DES1 surface. In SO2 adsorption mechanisms, NA > 1 also implies the coexistence of physisorption and chemisorption.69
Previous studies on gas adsorption, including CO2, NO and H2S have demonstrated the suitability of explaining the adsorption kinetics using Avrami kinetic model.75–79 However, the validity of this model in SO2 adsorption study is limited. In addition, SO2 adsorption is mutually reported by various studies as exothermic, implying that the process is negatively governed by the reaction temperature.20
The P1st kinetic model is generally suitable to describe purely physical adsorption processes characterised by low surface coverage and fairly describe the reversible interactions between the adsorbate and adsorbent surfaces, without accounting for chemical bonding or interactions80 between SO2 molecules and the surface of KCC-1/DES1. The model's fit improves as the adsorption temperature increases and the surface coverage is reduced due to the thermodynamic limitations associated with the exothermicity of the adsorption process. On the other hand, Fig. 9(a)–(c) and Table 7 reveal that the P2nd model does not fit well with the experimental data compared to the P1st model. As the P2nd kinetic model is more suitable to describe chemisorption processes induced by strong chemical bonds between molecules and the sorbent surface rather than a process with simultaneous occurrence of physisorption and chemisorption,81 the model is not examined further for SO2 adsorption on KCC-1/DES1.
Meanwhile, the Avrami model fits well with the experimental data across all studied temperatures. The excellent fit is believed to be due to its capacity to account for both physical and chemical adsorption mechanisms.82 Since the SO2 adsorption mechanism on KCC-1/DES1 is neither purely chemisorption nor physisorption, the SO2 uptake may originate from two different pathways.80 Initially, SO2 molecules may adsorb onto the surface through weak van der Waals interactions (physisorption). As the surface coverage increases, some of these molecules may undergo a transition to chemisorption, forming stronger chemical bonds with the surface.80,83 This hybrid adsorption mechanism is further supported by the comparable fit of the adsorption data to both P1st (R2 ≈ 0.96) and Avrami (R2 ≈ 0.99) kinetic models, suggesting contributions from both physical and chemical interactions. In addition, the kinetic constants (k1, k2 and k3) reported in Table 7 show that the kinetic parameters increase with temperatures, indicating that the adsorption rate is favoured at higher temperatures. However, the adsorption rate becomes faster (lower amount of SO2 is adsorbed on KCC-1/DES1; lower adsorption capacity) at high temperatures, as tabulated in Table 6. Therefore, the adsorption of SO2 onto KCC-1/DES1 is characterized by a kinetic-thermodynamic trade-off. At higher temperatures, the adsorption rate increases due to kinetic factors, but the thermodynamically unfavourable exothermic process limits the overall adsorption capacity.
Moreover, the NA values shown in Table 7 are always larger than 1, suggesting that SO2 adsorption is not homogeneous (not adsorbed with the same probability over different regions of the sorbent surface).84,85 These results are consistent with the findings reported in previous thermodynamic study,20 confirming that SO2 adsorption on KCC-1 is heterogeneous. Most likely, the initial occupation of adsorption sites is uniform; however, as the adsorption proceeds, additional adsorption preferentially occurs near existing adsorption sites, resulting in deviations from the uniformity of adsorption sites and a value of NA greater than 1.86,87 More specifically, NA is always around 1.6, indicating that the adsorption rate decreases gradually with the one-dimensional growth of the adsorbed nuclei, and the little variations of NA with increasing temperatures suggest that the underlying adsorption mechanism does not change.
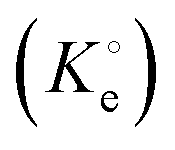 versus temperature (T), while Table 8 lists the adsorption thermodynamics parameters. The adsorption process can be assumed spontaneous based on the negative Gibbs free energy (ΔG°), indicating a favourable interaction between SO2 and the KCC-1/DES1 adsorbent.88 The ΔH° value for KCC-1/DES1 adsorbent is −18.685 kJ mol−1, which aligns with values reported in other SO2 adsorption studies using siliceous-based materials (−4.59 to −20.2 kJ mol−1).89,90 The ΔH° value suggests that physisorption is the primary adsorption mechanism as physical adsorption has been defined in literature as having an activation energy of less than 40 kJ mol−1.91,92 However, since findings from the kinetic and isotherm studies have indicated that the SO2 removal process may occur through both physical and chemical adsorptions, it may be safe to assume that SO2 removal occurs through both pathways, but the physical route predominates.
versus temperature (T), while Table 8 lists the adsorption thermodynamics parameters. The adsorption process can be assumed spontaneous based on the negative Gibbs free energy (ΔG°), indicating a favourable interaction between SO2 and the KCC-1/DES1 adsorbent.88 The ΔH° value for KCC-1/DES1 adsorbent is −18.685 kJ mol−1, which aligns with values reported in other SO2 adsorption studies using siliceous-based materials (−4.59 to −20.2 kJ mol−1).89,90 The ΔH° value suggests that physisorption is the primary adsorption mechanism as physical adsorption has been defined in literature as having an activation energy of less than 40 kJ mol−1.91,92 However, since findings from the kinetic and isotherm studies have indicated that the SO2 removal process may occur through both physical and chemical adsorptions, it may be safe to assume that SO2 removal occurs through both pathways, but the physical route predominates.
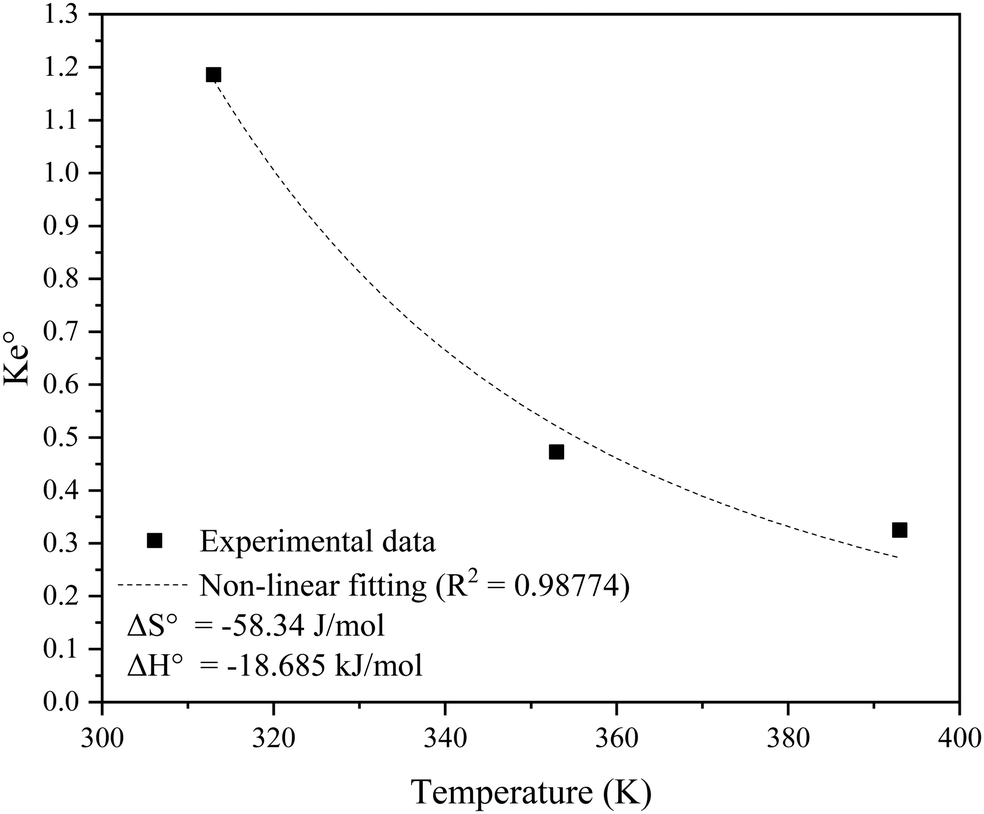 | ||
| Fig. 10 Non-linear Van’t Hoff plot of SO2 adsorption onto KCC-1/DES1. | ||
| Temperature (°C) | Temperature (K) | ΔS° (J mol−1 K−1) | ΔH° (kJ mol−1) | ΔG° (kJ) |
|---|---|---|---|---|
| 40 | 313 | −58.347 | −18.685 | −0.4224 |
| 80 | 353 | 1.9115 | ||
| 120 | 393 | 4.2454 |
In general, adsorption of molecules on an adsorbent surface minimises the degree of irregularity. The stronger contact between adsorbed SO2 molecules and the adsorbent surface compared to interactions with free SO2 molecules may be responsible for the observed negative ΔH° value. The adsorption process is exothermic, as evidenced by the negative ΔH° value, while the low ΔH° absolute value indicates a weak connection between the adsorbent and adsorbate, characteristic of physisorption. The negative entropy change (ΔS°) suggests that the adsorption process is becoming more ordered with temperature.93
The calculated Gibbs free energy (ΔG°) for SO2 adsorption on KCC-1 at different temperatures range between −0.4224 and 4.2454 kJ. The adsorption process at 40 °C can be assumed spontaneous based on the negative ΔG° value.88 However, the process becomes non-spontaneous with an increase in temperature to 80 °C and 120 °C as evidenced by the shift to positive ΔG° values. The sorption process is driven by both entropy and enthalpy, according to the values of ΔG°, ΔH°, and ΔS°.94 The term enthalpy-driven process refers to a process in which the system achieves a more stable state through releasing heat energy. Entropy-driven processes, in addition, indicate that the system evolves in such a way that the arrangement of molecules becomes more random or chaotic. The comparison of the absolute values of ΔH° and ΔS° serves as the selection indicator. It should be noted that the value of ΔG° increases when the temperature rises, implying that the adsorption process is more feasible at lower temperatures.95
4. Conclusions
The possibility of using modified fibrous mesoporous silica, KCC-1, as an alternative sorbent for dry removal of SO2 is evidenced in this study. The modified sorbent is shown to outperform the unmodified KCC-1 due to a shortage of accessible active sites and the weak interaction between KCC-1 and SO2. KCC-1 was impregnated with deep eutectic solvents (DES) to compensate for this weakness. The presence of DES1 (choline chloride and glycerol) produces a higher number of accessible active sites, resulting in 3.91 times higher SO2 removal capacity over unmodified KCC-1 and 4.97 times over KCC-1 modified with DES2 (choline chloride and ethylene glycol). Choline chloride and glycerol based DES impregnation results in more significant mesoporosity growth and higher SO2 adsorption capability. The Avrami kinetic model, which assumes numerous physisorption and chemisorption pathways, best represent SO2 adsorption on optimised KCC-1/DES1 across all studied reaction temperatures. The adsorption process progresses from a random stage on the surface of KCC-1 to a more ordered stage with higher degrees of freedom as the adsorbed SO2 becomes more dispersed. Based on the Gibbs free energy, the process is spontaneous and highly favourable at lower reaction temperatures. The experimental data at varying inlet SO2 concentrations are consistent with the Toth isotherm model, indicating the presence of localised multilayer adsorption onto a heterogeneous surface. Overall, these findings suggest that SO2 adsorption is favoured at lower reaction temperatures and incoming SO2 concentrations, due to the exothermic nature of the process and the slower sorbent saturation.Data availability
All data generated or analysed during this study are included in this article. Raw data are available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support from the Faculty of Civil Engineering & Technology, Universiti Malaysia Perlis and Universiti Teknologi Malaysia for providing the mesoporous silica KCC-1.References
- World Health Organization, Ambient (outdoor) air pollution, 2022.
- U.S. Centers For Disease Control and Prevention, Carbon Monoxide Poisoning, 2024.
- United States Environmental Protection Agency, Particulate Matter (PM) Pollution, 2024.
- United States Environmental Protection Agency, Sulfur Dioxide (SO2) Pollution, 2024.
- K. Naderi, M. S. Kalami Yazdi, H. Jafarabadi, F. Bahmanzadegan, A. Ghaemi and M. R. Mosavi, Sci. Rep., 2024, 14, 954 Search PubMed.
- United States Environmental Protection Agency, Air pollution control technology fact sheet, 2022.
- A. Upadhyay, Electrical India, 2020 Search PubMed.
- M. A. Hanif, N. Ibrahim and A. Abdul Jalil, Environ. Sci. Pollut. Res., 2020, 27, 27515–27540 Search PubMed.
- X. Li, L. Zhang, Y. Zheng and C. Zheng, Energy Fuels, 2015, 29, 942–953 Search PubMed.
- V. Polshettiwar, D. Cha, X. Zhang and J. M. Basset, Angew. Chem., Int. Ed., 2010, 49, 9652–9656 Search PubMed.
- N. Bayal, B. Singh, R. Singh and V. Polshettiwar, Sci. Rep., 2016, 6, 24888 Search PubMed.
- R. Soltani, A. Marjani, M. Hosseini and S. Shirazian, Chemosphere, 2020, 239, 124735 Search PubMed.
- S. Sun, Y. Niu, Q. Xu, Z. Sun and X. Wei, Ind. Eng. Chem. Res., 2015, 54, 8019–8024 Search PubMed.
- F. Zarei, A. Marjani and R. Soltani, Eur. Polym. J., 2019, 119, 400–409 Search PubMed.
- K. Yu, X. Zhang, H. Tong, X. Yan and S. Liu, Mater. Lett., 2013, 106, 151–154 Search PubMed.
- H. S. Oboudatian and J. Safaei-Ghomi, Sci. Rep., 2022, 12, 2381 Search PubMed.
- L. W. Lai, L. P. Teh, S. N. Timmiati, N. H. N. Kamarudin and H. D. Setiabudi, J. Environ. Chem. Eng., 2023, 11, 111295 Search PubMed.
- M. B. Singh, V. S. Kumar, M. Chaudhary and P. Singh, J. Indian Chem. Soc., 2021, 98, 100210 Search PubMed.
- D. Yang, S. Zhang, D. Jiang and S. Dai, Phys. Chem. Chem. Phys., 2018, 20, 15168–15173 Search PubMed.
- M. A. Hanif, N. Ibrahim, K. Md Isa, F. Muhammad Ridwan, T. A. Tuan Abdullah and A. A. Jalil, Microporous Mesoporous Mater., 2022, 330, 111610 Search PubMed.
- N. Z. Zulkurnai, U. F. Md Ali, N. Ibrahim and N. S. Abdul Manan, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 206, 012001 Search PubMed.
- C. Y. Lim, M. F. Majid, S. Rajasuriyan, H. F. Mohd Zaid, K. Jumbri and F. K. Chong, Environments, 2020, 7, 97 Search PubMed.
- S. H. Ammar and S. A. Jaffar, Eng. Technol. J., 2017, 35, 856–863 Search PubMed.
- M. A. Hanif, N. Ibrahim, K. Md. Isa, U. F. Md. Ali, T. A. Tuan Abdullah and A. A. Jalil, J. Porous Mater., 2022, 29, 501–514 Search PubMed.
- R. Guo, S. Li, L. Zhao, Y. Zhao, W. Lu, P. Yuan, P. Deng and F. Liao, J. Zhejiang Univ., Sci., B, 2007, 8, 867–874 Search PubMed.
- T. R. Sahoo and B. Prelot, Nanomaterials for the Detection and Removal of Wastewater Pollutants, Elsevier, 2020, pp. 161–222 Search PubMed.
- R. Chang and J. W. J. Thoman, Physical Chemistry for the Chemical Sciences, Universiti Science Books, 2014 Search PubMed.
- R. Manurung, G. C. Simanjuntak, R. N. Perez, A. Syahputra, M. A. Alhamdi, H. Siregar and R. R. Syahputri Zuhri, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 505, 012134 Search PubMed.
- H. Qin, X. Hu, J. Wang, H. Cheng, L. Chen and Z. Qi, Green Energy Environ., 2020, 5, 8–21 Search PubMed.
- P. Makoś, E. Słupek and J. Gębicki, Microchem. J., 2020, 152, 104384 Search PubMed.
- D. T. Mekonnen, E. Alemayehu and B. Lennartz, Materials, 2021, 14, 5466 Search PubMed.
- Z. Ghazali, N. Suhaili, M. N. A. Tahari, M. A. Yarmo, N. H. Hassan and R. Othaman, J. Mater. Res. Technol., 2020, 9, 3249–3260 Search PubMed.
- Z. Ghazali, M. A. Yarmo, N. H. Hassan, L. P. Teh and R. Othaman, Arab. J. Sci. Eng., 2020, 45, 4621–4634 Search PubMed.
- Z. Ghazali, M. A. Yarmo and R. Othaman, AIP Conf. Proc., 2019, 2111, 050014 Search PubMed.
- R. Soltani, R. Pelalak, M. Pishnamazi, A. Marjani, A. B. Albadarin, S. M. Sarkar and S. Shirazian, Sci. Rep., 2021, 11, 2716 Search PubMed.
- J. Luo, O. Conrad and I. F. J. Vankelecom, J. Mater. Chem., 2012, 22, 20574 Search PubMed.
- G. Kaur, N. Singh, A. Rajor and J. P. Kushwaha, J. Contam. Hydrol., 2021, 242, 103847 Search PubMed.
- C. K. Rojas-Mayorga, I. A. Aguayo-Villarreal, J. Moreno-Pérez, R. Muñiz-Valencia, M. Á. Montes-Morán and R. Ocampo-Pérez, J. Environ. Chem. Eng., 2021, 9, 104810 Search PubMed.
- X. Yang, Q. Wang, J. Chen, H. Liu, L. Xu and M. Rao, Processes, 2024, 12, 1547 Search PubMed.
- Y. Sabahi, M. Razmkhah and F. Moosavi, Results Chem., 2022, 4, 100283 Search PubMed.
- R. K. Ibrahim, A. El-Shafie, L. S. Hin, N. S. B. Mohd, M. M. Aljumaily, S. Ibraim and M. A. AlSaadi, J. Environ. Manage., 2019, 235, 521–534 Search PubMed.
- B. H. Stuart, Infrared Spectroscopy: Fundamentals and Applications, John Wiley & Sons Ltd, 2004 Search PubMed.
- Y.-C. Chiang and R.-S. Juang, J. Taiwan Inst. Chem. Eng., 2017, 71, 214–234 Search PubMed.
- A. A. Adelodun, K.-H. Kim, J. C. Ngila and J. Szulejko, Appl. Energy, 2015, 158, 631–642 Search PubMed.
- B. Petrovic, M. Gorbounov and S. Masoudi Soltani, Carbon Capture Sci. Technol., 2022, 3, 100045 Search PubMed.
- C. Goel, H. Bhunia and P. K. Bajpai, RSC Adv., 2015, 5, 93563–93578 Search PubMed.
- D. Saha and M. J. Kienbaum, Microporous Mesoporous Mater., 2019, 287, 29–55 Search PubMed.
- M. G. Plaza, K. J. Thurecht, C. Pevida, F. Rubiera, J. J. Pis, C. E. Snape and T. C. Drage, Fuel Process. Technol., 2013, 110, 53–60 Search PubMed.
- Y. Liu and J. Wilcox, Environ. Sci. Technol., 2013, 47, 95–101 Search PubMed.
- M. G. Rabbani, R. K. Sasse, S. Behera, P. Jena, J. Liu, P. K. Thallapally, T. Islamoglu, M. K. Shehab, M. M. Kaid, O. K. Farha and H. M. El-Kaderi, Langmuir, 2024, 40, 8024–8034 Search PubMed.
- M. McAnally, J. Bocková, A. Herath, A. M. Turner, C. Meinert and R. I. Kaiser, Nat. Commun., 2024, 15, 4409 Search PubMed.
- E. Martínez-Ahumada, D. He, V. Berryman, A. López-Olvera, M. Hernandez, V. Jancik, V. Martis, M. A. Vera, E. Lima, D. J. Parker, A. I. Cooper, I. A. Ibarra and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 17556–17563 Search PubMed.
- M. Y. S. Hamid, M. L. Firmansyah, S. Triwahyono, A. A. Jalil, R. R. Mukti, E. Febriyanti, V. Suendo, H. D. Setiabudi, M. Mohamed and W. Nabgan, Appl. Catal., A, 2017, 532, 86–94 Search PubMed.
- H. Wang and F. Shadman, AIChE J., 2013, 59, 1502–1510 Search PubMed.
- L. Gang, H. Shaoguang and K. Qian, Silicon, 2022, 14, 2225–2233 Search PubMed.
- Á. A. Beltrán-Osuna, J. L. Gómez Ribelles and J. E. Perilla, J. Nanopart. Res., 2017, 19, 381 Search PubMed.
- Y. Chen, X. Shi, B. Han, H. Qin, Z. Li, Y. Lu, J. Wang and Y. Kong, J. Nanosci. Nanotechnol., 2012, 12, 7239–7249 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 Search PubMed.
- F. Ariffin, S. M. Izan, A. A. Jalil, C. R. Mamat, N. Basar, J. Jaafar, H. Hamdan and S. Triwahyono, Malaysian J. Catal., 2017, 2, 86–90 Search PubMed.
- M. Y. Shahul Hamid, S. Triwahyono, A. A. Jalil, N. W. Che Jusoh, S. M. Izan and T. A. Tuan Abdullah, Inorg. Chem., 2018, 57, 5859–5869 Search PubMed.
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. Wang, A. I. Cooper, R. Dawson, X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815–2823 Search PubMed.
- L. H. Macfarlan, M. T. Phan and R. Bruce Eldridge, Chem. Eng. Sci., 2023, 275, 118720 Search PubMed.
- J. A. Delgado, M. A. Uguina, J. L. Sotelo and B. Ruíz, Sep. Purif. Technol., 2006, 49, 91–100 Search PubMed.
- H. Ning, R. Tang, C. Li, X. Gu, Z. Gong, C. Zhu, J. Li, K. Wang and J. Yu, Sep. Purif. Technol., 2025, 353, 128425 Search PubMed.
- A. N. Módenes, F. B. Scheufele, F. R. Espinoza-Quiñones, P. S. Carraro de Souza, C. R. B. Cripa, J. Dos Santos, V. Steffen and A. D. Kroumov, Int. J. Bioautomotion, 2015, 19, 187–206 Search PubMed.
- S. Tazikeh, A. Shafiei, T. Yerkenov, A. Abenov, N. Seitmaganbetov and T. Sh. Atabaev, Fuel, 2022, 329, 125379 Search PubMed.
- G. Avijegon, G. Xiao, G. Li and E. F. May, Adsorption, 2018, 24, 381–392 Search PubMed.
- J. H. Carter, X. Han, F. Y. Moreau, I. da Silva, A. Nevin, H. G. W. Godfrey, C. C. Tang, S. Yang and M. Schröder, J. Am. Chem. Soc., 2018, 140, 15564–15567 Search PubMed.
- T. Hou, W. Zeng and Q. Zhou, Chemosensors, 2022, 10, 279 Search PubMed.
- Y. Song, J. Cheng, Y. Miao, W. Guo and J. Zhou, ACS Sustainable Chem. Eng., 2021, 9, 5580–5589 Search PubMed.
- H. Wang, J. Li and Z. Zhang, Nanomaterials for Carbon Dioxide Capture and Conversion Technologies, Elsevier, 2023, pp. 261–275 Search PubMed.
- J. Zhou, J. Ma and Z. Wang, Fuel Process. Technol., 2022, 227, 107133 Search PubMed.
- G. Cui, S. Lyu, F. Zhang, H. Wang, Z. Li, Y. Li and J. Wang, Ind. Eng. Chem. Res., 2020, 59, 21522–21529 Search PubMed.
- H. Mashhadimoslem, M. Safarzadeh Khosrowshahi, M. Jafari, A. Ghaemi and A. Maleki, ACS Omega, 2022, 7, 18409–18426 Search PubMed.
- M. K. A. Asman, N. A. Lutpi, Y. Wong, M. A. Hanif, S. Ong, F. A. Dahalan, N. I. Iberahim and M. Hamdzah, Environ. Quality Manage., 2024, 33, 81–89 Search PubMed.
- N. Ahmad, N. Ibrahim, P. Yun Fu and R. Ahmad, J. Teknol., 2020, 82(5), 127–133 Search PubMed.
- X. Zhang, H. Xu, W. Xiang, X. You, H. Dai and B. Gao, Biochar, 2024, 6, 22 Search PubMed.
- B.-C. Alcántar-Vázquez and R.-M. Ramírez-Zamora, Adsorption, 2020, 26, 687–699 Search PubMed.
- B. Guo, Y. Wang, X. Shen, X. Qiao, L. Jia, J. Xiang and Y. Jin, Materials, 2020, 13, 877 Search PubMed.
- F. Raganati, M. Alfe, V. Gargiulo, R. Chirone and P. Ammendola, Chem. Eng. J., 2019, 372, 526–535 Search PubMed.
- J. Wang and X. Guo, J. Hazard. Mater., 2020, 390, 122156 Search PubMed.
- D. Tiwari, C. Goel, H. Bhunia and P. K. Bajpai, Sep. Purif. Technol., 2017, 181, 107–122 Search PubMed.
- B. Li and C. Ma, Energy Proc., 2018, 153, 471–477 Search PubMed.
- R. Serna-Guerrero and A. Sayari, Chem. Eng. J., 2010, 161, 182–190 Search PubMed.
- Y. Liu and X. Yu, Appl. Energy, 2018, 211, 1080–1088 Search PubMed.
- Y. Chen, J. Wu, X. Wang, M. Liu and Y. Liu, Molecules, 2022, 27, 3429 Search PubMed.
- J. Rojas, D. Suarez, A. Moreno, J. Silva-Agredo and R. A. Torres-Palma, Appl. Sci., 2019, 9, 5337 Search PubMed.
- Y. Liu and X. Yu, Appl. Energy, 2018, 211, 1080–1088 Search PubMed.
- Q. Zhang, J. Wei, C. Gao, Y. Zheng, Y. Xiao, F. Liu and L. Jiang, J. Cleaner Prod., 2024, 436, 140633 Search PubMed.
- H. Deng, H. Yi, X. Tang, Q. Yu, P. Ning and L. Yang, Chem. Eng. J., 2012, 188, 77–85 Search PubMed.
- M. Ghaedi, A. G. Nasab, S. Khodadoust, M. Rajabi and S. Azizian, J. Ind. Eng. Chem., 2014, 20, 2317–2324 Search PubMed.
- A. A. A. Darwish, M. Rashad and H. A. AL-Aoh, Dyes Pigm., 2019, 160, 563–571 Search PubMed.
- A. A. El-Bindary, A. Z. El-Sonbati, A. A. Al-Sarawy, K. S. Mohamed and M. A. Farid, Spectrochim. Acta, Part A, 2015, 136, 1842–1849 Search PubMed.
- S. Yang, L. Li, Z. Pei, C. Li, J. Lv, J. Xie, B. Wen and S. Zhang, Colloids Surf., A, 2014, 457, 100–106 Search PubMed.
- S. Banerjee, G. C. Sharma, M. C. Chattopadhyaya and Y. C. Sharma, J. Environ. Chem. Eng., 2014, 2, 1870–1880 Search PubMed.
| This journal is © the Owner Societies 2024 |