
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrofluorochromism based on the valence change of europium complexes in electrochemical devices with Prussian blue as the counter electrode†
Ryoto
Yabuta
 ,
Norihisa
Kobayashi
and
Kazuki
Nakamura
*
,
Norihisa
Kobayashi
and
Kazuki
Nakamura
*
Graduate School of Engineering, Chiba University, 1-33, Yayoi-cho, Inage-ku, Chiba, 263-8522, Japan. E-mail: Nakamura.Kazuki@faculty.chiba-u.jp
First published on 1st October 2024
Abstract
The electrofluorochromism of Eu complexes based on the valence change between Eu3+ and Eu2+ is demonstrated in a two-electrode electrochemical device consisting of Prussian blue (PB) as the counter electrode. This study aims to improve the electrofluorochromic (EFC) performance of luminescence switching between Eu3+ and Eu2+ by enhancing the electrochemical reactivity of the EFC device. By introducing a PB film as a counter electrode in a two-electrode device, the redox reaction of Eu3+/2+ is promoted because of charge compensation by the counter PB film. The increase in the reaction charge enables faster changes in the photoluminescence from Eu3+ to Eu2+ and an increase in the blue luminescence intensity from the Eu2+ state. This approach achieves a lowered driving voltage, accelerates the electrochemical redox reaction of the Eu complex, and enhances the reversibility of the valence change of the Eu ion.
1 Introduction
Chromogenic materials, which can alter their optical properties such as luminescence and absorption in response to external stimuli such as light, heat, and electricity, have promising applications in various fields, including chemical sensors,1,2 biochemical labels,3 molecular memory,4 and display devices.5,6 Among these materials, electrofluorochromic (EFC) materials stand out for their innovative ability to control luminescence colors through electrochemical redox reactions, rapidly converting electrical inputs into visual signals.4,7–10 These novel materials include various molecules,11–15 conjugated polymers,16–20 inorganic compounds,21,22 and metal complexes.23–29Ln(III) complexes consist of luminescent Ln(III) ions encircled by antenna ligands with sufficient light-absorbing capacities. Their fluorescence is bolstered by efficient intramolecular energy transfer from the antenna ligands to the Ln(III) center, resulting in vivid luminescence.30–33 Additionally, Ln(III) complexes exhibit appealing photoluminescence characteristics, such as distinct and narrow emission bands in the visible-near infrared (vis-NIR) range owing to Ln(III), prolonged emission lifetimes, and high visibility in the visible spectrum due to a significant pseudo-Stokes shift.34 Compared to Ln(III) complexes, Eu complexes exhibit outstanding luminescence properties with higher stability in their trivalent (Eu3+) and divalent (Eu2+) states. Eu3+ is known for its intense and enduring red luminescence through f–f transitions and has been extensively applied in biosensors and luminescent materials.35 In contrast, Eu2+ is known to show broad blue luminescence induced by d–f transitions from the excited state of 4f65d1 to the ground level state of 8S7/2 (4f7),36 and the emission of Eu2+ in inorganic matrices is applied in various phosphor materials.37–40 However, Eu2+ tends to be unstable in air and common solutions, posing challenges to observing its luminescence. Therefore, in previous studies, Eu2+ was doped into inorganic materials such as glass,41,42 ceramics,43 and porous inorganic host materials (such as zeolite and gadolinium silicate)44,45 at high temperatures and reducing atmospheres to produce stable Eu2+.
Although the electrochemical dynamic control of both the trivalent and divalent states of the Eu ion in solution has been exceedingly challenging, the dynamic and reversible EFC phenomena caused by the valence changes of Eu ions have rarely been reported because of the instability of the Eu2+ state. The lanthanide ions maintain their oxidation valence in the trivalent state of Ln3+. The Eu3+ state is preferred as ions in solution and complexes for europium ions.
In our previous research, stable Eu2+ was successfully generated electrochemically in polyethylene glycol (PEG), enabling control over the red and blue luminescence derived from the Eu3+ and Eu2+ states of the β-diketonate Eu complex.46 However, to obtain luminescence from Eu2+ for voltage applications, a long time is required due to the lower redox activity of the Eu ions. Because the redox reaction of Eu3+/2+ requires electron transfer between the inner shell 4f orbital and the electrode surface, the redox activity (i.e., the electron transfer rate) of the reaction tends to be suppressed.47
In this study, we aimed to achieve faster and more obvious control of the EFC reaction of Eu3+/2+. We introduced a highly reactive counter electrode material to improve the electrochemical reactivity of the EFC device.
In our previous study,46 we did not introduce a suitable counter electrode reaction material into the electrolyte solution because the resulting undesired electron transfer, energy transfer, and absorption of excitation from its dissolution make it challenging to observe the luminescence from the Eu complex. Therefore, immobilizing redox-active materials on the counter electrode is essential for our EFC device. We employed a redox-active Prussian blue (PB) film as a suitable counter electrode reaction material.
PB has a structure with divalent and trivalent iron atoms and is expected to undergo reversible electrochemical reactions with cations such as Li+ and K+ by injecting electrons from the electrode.48
| FeIII4[FeII(CN)6]3 (cyan) + 4e− + 4M+ ⇆ MFeII4[FeII(CN)6]3 (transparent) (M = Li+, etc.) |
PB exhibits high stability during long-term device operation through this reversible redox reaction. Moreover, because of the porous structure of the PB film, an increase in the redox capacity of the EFC device is expected. Modifying the counter electrode with a PB film provides a counteractive material to compensate for the charge generated by the reactions at the working electrode.49 When Eu3+ converts to Eu2+ upon electrochemical reduction, the PB-modified counter electrode is oxidized to its cyan-colored form; the transformation of both Eu3+ and Eu2+ is thought to be improved through the electrochromism of the PB film on the counter electrode.
2 Experimental
2.1 Materials
Potassium chloride, tetra-n-butylammonium perchlorate (TBAP), and silver nitrate were purchased from Kanto Chemical Industry Co., Japan. Hexafluoroacetylacetone (hfa-H) was purchased from Tokyo Chemical Industry Co., Japan. Lithium trifluoromethanesulfonate, lithium nitrate, polyethylene glycol 400 (PEG400), potassium hexacyanoferrate(II) trihydrate, and iron(III) chloride hexahydrate were purchased from FUJIFILM Wako Pure Chemical Co., Japan. Carbon paste (G7710) was purchased from EM Japan Co., Ltd. The supporting electrolyte CF3SO3Li was used without further purification. Indium tin oxide (ITO) (10 Ω sq−1; Yasuda, Japan) was used as an electrode after washing and ozonation.2.2 Synthesis of the luminescent Eu(III) complex
Tris(hexafluoroacetylacetonate)europium (Eu(hfa)3(H2O)2) was prepared according to a literature method.50 Europium acetate n-hydrate was dissolved in deionized water at room temperature, and three equivalents of liquid hfa-H were added dropwise. After 3 h of stirring, a white precipitate was filtered and purified by recrystallization using methanol/water. Yield: 80%. Anal. calcd for C15H7O8F18Eu: C, 22.48; H, 0.88%. Found: C, 22.12; H, 1.05%.2.3 Preparation of electrolyte solutions
The electrochemical redox behavior of the β-diketonate Eu complex was investigated using a PEG400 electrolyte solution containing Eu(hfa)3(H2O)2 (10 mM) and LiCF3SO3 (500 mM). A PEG400 solution containing only LiCF3SO3 (500 mM) was prepared for comparison. The solutions for optical measurements were purged with nitrogen gas for 20 min before each measurement.2.4 Preparation of modified electrodes
The PB-modified and CM electrodes were prepared as counter electrodes for the EFC devices. The PB-modified electrode was obtained by electrodeposition of a PB layer on an ITO electrode using an aqueous solution containing K3[Fe(CN)6] (10 mM), FeCl3·6H2O (10 mM), and KCl (50 mM).48 The condition for constant current electrolysis of a cathodic current was 59 μA cm−2 for 200 s. After removing the electrodeposition solution, the obtained PB-modified electrode was rinsed with pure water and ethanol. Consequently, the electrode was dried at 70 °C for 1 h. Before assembling the EFC device, the PB-modified electrode was reduced to the bleached state. The crystal structure of the PB-modified electrode was characterized by X-ray diffraction (XRD) spectroscopy51 (Fig. S1, ESI†).The CM electrodes were prepared according to a previously reported method as follows:52 a carbon paste was coated directly onto the ITO substrate by blade coating, and the gap between the blade and the ITO substrate was 180 μm. The obtained porous CM electrode was heated to 250 °C for 1 h in an electron furnace under an ambient atmosphere. The film thicknesses were measured using a surface profiler (ET 4000; Kosaka Laboratory, Japan).
2.5 Fabrication of EFC devices
The EFC devices were fabricated by sandwiching an electrolyte solution containing the Eu complex between a flat ITO electrode as the working electrode and the prepared functional counter electrodes. A flat ITO electrode (ITO–ITO device), a PB electrode in the bleached state (ITO–PB device), and a CM electrode (ITO–CM device) were used as the counter electrodes. The distance between the electrodes was fixed at 300 μm with a silicon spacer (Mitsubishi Chemical, Japan). The active area of the device was 1 cm × 1 cm.2.6 Electrochemical measurements
For electrochemical measurements using a three-electrode cell, the PB electrode or ITO electrode was used as the working electrode, Pt wire (ϕ = 1 mm) was used as the counter electrode, and an Ag/Ag+ electrode was used as the reference electrode. The reference electrode was prepared by injecting an Ag+ solution (10 mM AgNO3 and 100 mM TBAP in acetonitrile) into a sample holder with ion-permeable glass (BAS, Japan) and sealed with a cap containing an Ag wire (ϕ = 1 mm).Cyclic voltammetry (CV) and chronoamperometry experiments were performed using a potentiostat/galvanostat (ALS440A; CH Instruments, Inc., Austin, TX, USA) controlled by a computer. The scan rate under conventional measurement conditions was 50 or 100 mV s−1. During potential or voltage sweeping, the absorption spectra of the three- and two-electrode devices were recorded using a fiber-optic spectrometer system (USB2000; Ocean Optics, Orlando, FL, USA). We monitored the change in the absorbance of the PB-modified electrode during potential sweeping to evaluate its electrochemical reactivity. The monitoring wavelength was selected as 700 nm, which corresponds to the peak absorbance wavelength of the PB film. The polarity of the voltage applied to the two-electrode device was considered positive when the working ITO electrodes were connected to the anode.
2.7 Photophysical measurements
The photoluminescence (PL) spectra of Eu(hfa)3(H2O)2 in the PEG400 electrolyte solution were measured using a spectrofluorometer (FP-6600, JASCO, Japan). In situ luminescence spectra were obtained during voltage sweeping using an epifluorescence unit (EFA-133, JASCO, Japan) with an excitation wavelength of 365 nm for the two-electrode EFC devices.2.8 Electrode potential measurements
Electrode potential measurements were performed to characterize the redox behavior of the Eu(hfa)3(H2O)2 solution using two electrochemical analyzers: HZ-7000 (Hokuto Denko Co., Japan) and ALS 440A, as described in our previous studies (Scheme 1).53 The ITO electrode was used as the working electrode, and the bleached state of the PB, ITO, or CM electrode was used as the counter electrode at a distance of approximately 1 cm. A reference Ag/Ag+ electrode was placed between the working and counter electrodes.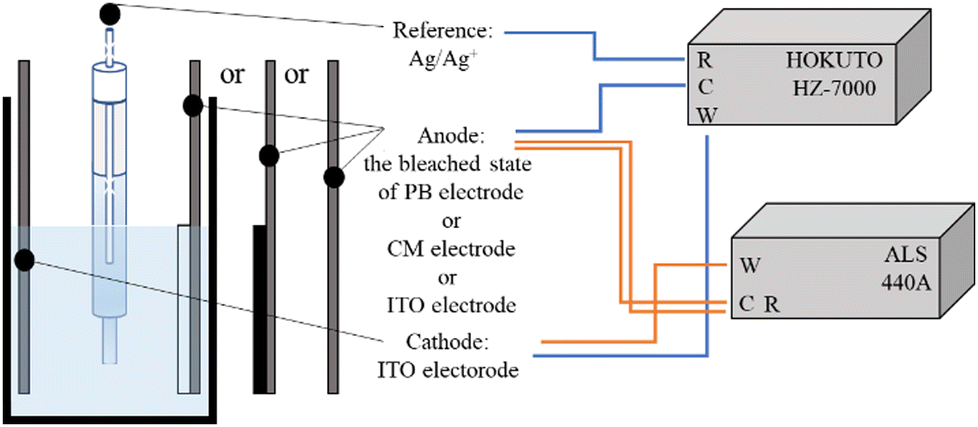 | ||
| Scheme 1 Schematic illustration of the electrochemical system for measuring electrode potential using two electrochemical analyzers. | ||
3 Results and discussion
3.1 Electrochemical analysis of the PB layer in PEG400
First, we evaluated the electrochemical reactivities of the prepared PB-modified electrodes in an EFC solution. The absorption spectra of the PB-modified electrodes are presented in Fig. S2 (ESI†). A broad absorption peak was observed at 700 nm. This broad absorption is attributed to FeIII4[FeII(CN)6]3. The electrochemical properties, including electrochromic behavior, of the PB-modified electrodes were investigated using a three-electrode electrochemical cell. A PEG400 solution was used as the electrolyte, containing LiCF3SO3 (500 mM) and Eu(hfa)3(H2O)2 (0 or 10 mM). PEG400 is the longest polyethylene glycol chain among the PEGs under liquid conditions at ambient temperature. Since a longer PEG chain is expected to contribute to stabilization of the Eu2+ state,36 we used PEG400 as the solvent in this research. LiCF3SO3 was used because the Li+ cation was necessary for the electrochromic change of Prussian blue. In addition, the Li+ ion is known to show higher solubility in the PEG matrix because ether oxygen atoms on the PEO chains well coordinate with the Li+ ions.54The Eu(hfa)3(H2O)2 complex exhibits relatively good luminescence characteristics and high solubility in various organic solvents including PEG compounds.
Fig. 1 shows the CV curves of the PB-modified electrode and the absorbance changes at 700 nm during the potential scan.
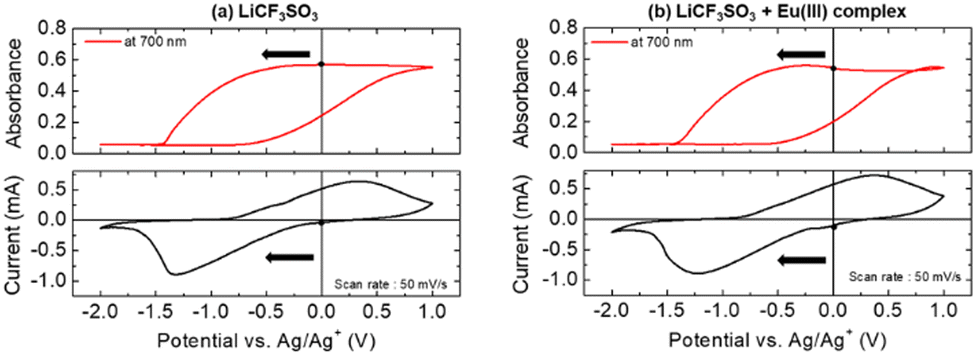 | ||
| Fig. 1 Changes in the absorption at 700 nm (top) and the cyclic voltammogram (bottom) of the PB-modified electrode in PEG400 electrolyte solution containing (a) LiCF3SO3 (500 mM) and (b) LiCF3SO3 (500 mM) and Eu(hfa)3(H2O)2 (10 mM). | ||
For the electrolyte solution without the Eu complex (Fig. 1(a)), when the potential was swept in the negative direction, a cathodic current was observed at 0 V. This electrochemical reduction corresponds to the transformation from the PB state to the bleached state, leading to a decrease in absorbance at 700 nm. Conversely, during the positive sweep, an oxidation current was observed from −0.75 V, accompanied by an increase in absorbance as the bleached state was oxidized to PB. Few tens of the reversible EC reaction of the PB film were obtained. Therefore, the PB film is expected to be a suitable counter redox material in this system. The absorption spectra of the bleached and PB states obtained during CV measurements are shown in Fig. S3 (ESI†). By changing PB to the bleached state, the absorption bands of the PB film around 350 nm and over 500 nm decreased, resulting in no absorption bands in the visible region. These electrochromic reactions of the PB film aligned with the results obtained from the solution containing the Eu complex (Fig. 1(b)). Furthermore, the CVs of PB in PEG solution containing only the Eu3+ ion (Eu(NO3)·6H2O) showed no redox current and absorbance change in this potential range (Fig. S4, ESI†). These results suggest that the electrochemical reactivity of the PB-modified electrode was not affected by the Eu(III) complex and we are assuming that intercalation and exchange of Eu3+/Eu2+ ions into the PB layer can be almost negligible in this study.
3.2 Evaluation of a two-electrode device with a PB electrode as a counter electrode
Fig. 2 shows the results of CV curves and absorption spectrum measurements using a two-electrode device prepared by sandwiching the PEG electrolyte solution containing the Eu complex between a flat ITO electrode as the working electrode and a bleached PB electrode as the counter electrode (ITO–PB device).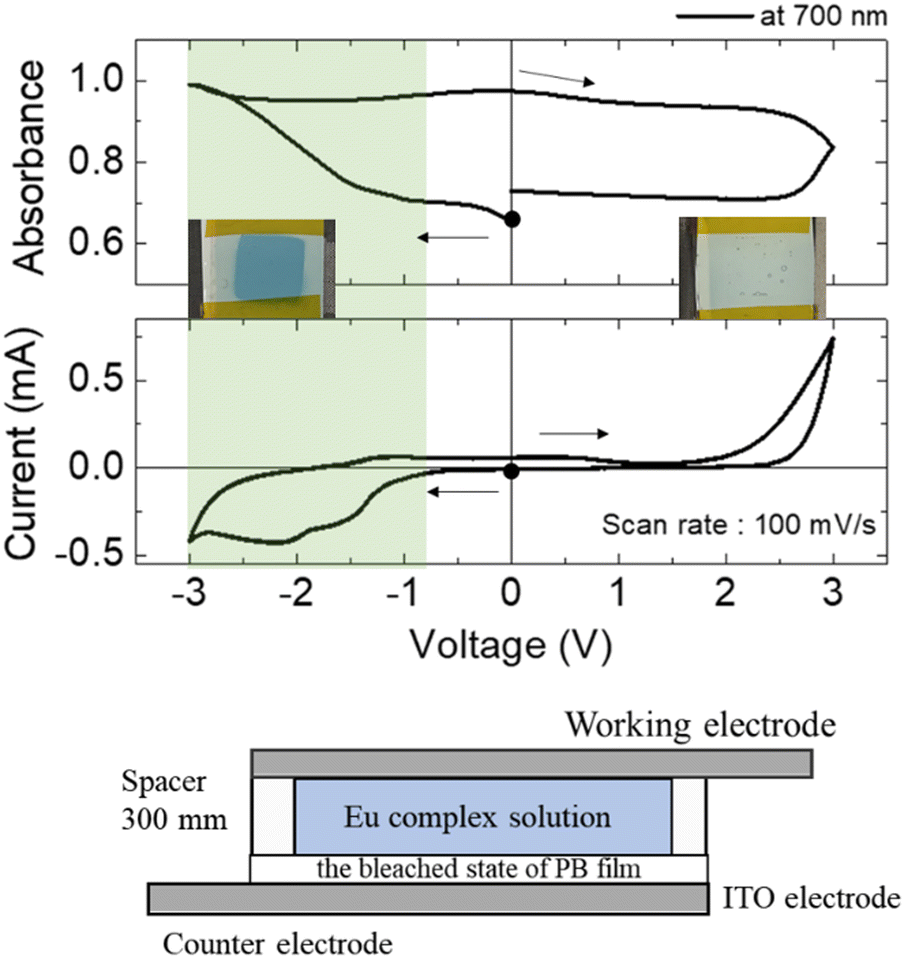 | ||
| Fig. 2 Changes in the absorption at 700 nm (top) and the cyclic voltammogram (middle) of the ITO–PB device. The bottom picture is the ITO–PB device image. | ||
When the voltage was scanned in the negative direction, a redox current was observed from −0.8 V, and the absorbance at 700 nm began to increase, leading to a cyan color. This redox reaction corresponds to the electrochemical oxidation of the bleached state of PB to the colored PB state on the counter electrode. Thus, reversible changes in the PB and bleached states of PB were observed in the ITO–PB devices. Because the redox reaction on the working electrode at this voltage was still unclear, the electrode potential of each electrode in the device during voltage sweeping was monitored using a combination of the two potentiostats. Fig. 3 shows the electrode potential measurements (vs. Ag/Ag+) for the ITO–PB device.
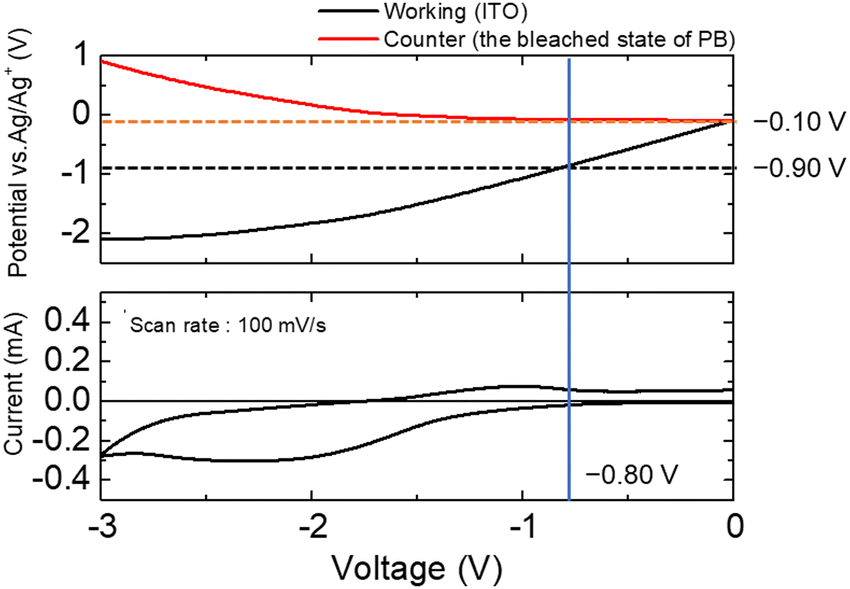 | ||
| Fig. 3 Electrode potential changes of the ITO electrode as the working electrode and the PB electrode as the counter electrode during voltage sweeping between the two electrodes. | ||
The potentials of the working and counter electrodes in the electrochemical cell (vs. Ag/Ag+) were monitored during voltage scanning between the working electrode (WE) and the counter electrode (CE). When the applied voltage reached −0.8 V, as observed in Fig. 2, the redox current began to flow. The electrode potential of the working electrode at that time was found to be −0.9 V (vs. Ag/Ag+), whereas the electrode potential of the counter electrode was −0.1 V (vs. Ag/Ag+). To determine the redox potential of the Eu ions, a three-electrode CV curve of the Eu(NO3)3 solution was obtained (Fig. S5, ESI†). As shown in the figure, the reduction potential of the Eu(III) ions was approximately −0.9 V (vs. Ag/Ag+), which was consistent with our previous research. When the voltage of −0.8 V was applied between the WE and the CE, the counter electrode potential was −0.1 V (vs. Ag/Ag+), which corresponded to the oxidation potential of the bleached state of PB, as shown in Fig. 1. Therefore, when a voltage of −0.8 V was applied to the ITO–PB device, the reduction of Eu3+ began on the working electrode, and the bleached state of PB was oxidized on the counter electrode simultaneously.
To confirm the improvement in the electrochemical reactivity of the EFC device by introducing the PB film onto the counter electrode, we compared the electrochemical properties of this device with those of an ITO or a carbon-modified electrode (CM). Fig. 4 shows the electrode potential (vs. Ag/Ag+) measurement results for the two electrode devices (ITO–ITO, ITO–CM, and ITO–PB devices).
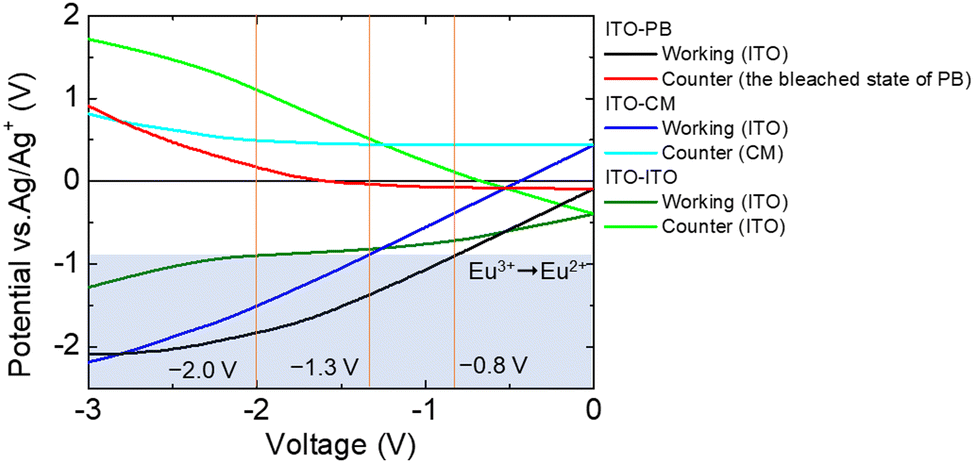 | ||
| Fig. 4 Electrode potential measurements using the two-electrode devices of the Eu(hfa)3(H2O)2 solution: (ITO–PB (ITO as the WE: the black line and the bleached state of PB as the CE: the red line), ITO–CM (ITO as the WE: the blue line and CM as the CE: the light blue line), ITO–ITO (ITO as the WE: the deep green line and ITO as the CE: the green line). | ||
When the electrochemical reduction of Eu3+ started (electrode potential: −0.9 V vs. Ag/Ag+), the driving voltage between the working and counter electrodes had reached −2.0 V for the ITO–ITO device and −1.3 V for the ITO–CM device. These driving voltages of the devices were higher than that of the ITO–PB device (−0.8 V). By introducing a suitable counter electrode reaction material, the potential of the working electrode can be efficiently controlled by changing the driving voltage between the working and counter electrodes. Thus, the ITO–PB device exhibits a lower redox voltage and better electrochemical properties than the ITO–ITO and ITO–CM devices. Next, the emission of the two-electrode device with the PB electrode as the counter electrode (ITO–PB device) was evaluated.
Fig. 5 shows the emission spectra of the Eu(hfa)3(H2O)2 solution in the ITO–PB device at various applied bias voltages.
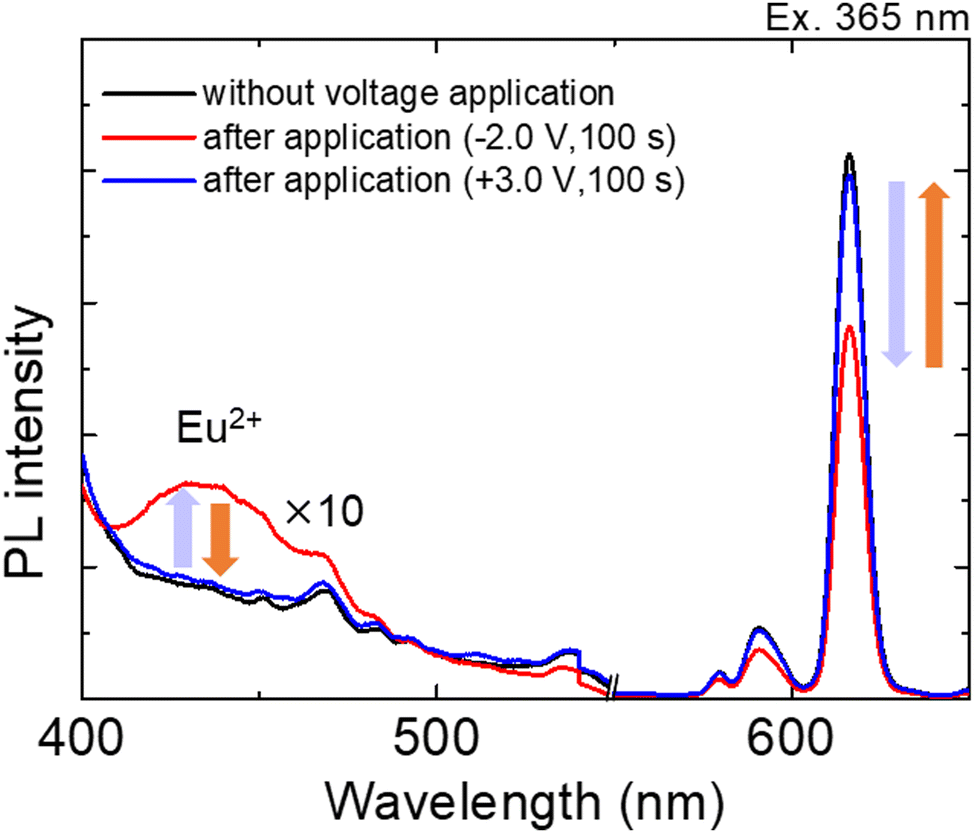 | ||
| Fig. 5 Luminescence spectra from the ITO–PB device before and after applying reduction and oxidation voltages (reduction voltage: −2.0 V for 100 s and oxidation voltage: +3.0 V for 100 s). The excitation wavelength was 365 nm. | ||
Before applying the reduction voltage, an obvious sharp red emission due to f–f transitions from the excited state of Eu3+ was observed upon photoexciting the hfa ligand moiety. When sufficient reduction voltage (−2.0 V) was applied for 100 s, the red emission band from the Eu3+ state (around 600 nm) decreased, and an emission band arising from the d–f transition in the Eu2+ state (430 nm) simultaneously increased. The subsequent oxidation of Eu2+ ions occurred at an oxidation voltage of +3.0 V, as the emission band from the Eu2+ state (430 nm) decreased and the emission band from the Eu3+ state (615 nm) increased. For clear visibility of the luminescence color change, we applied a reduction voltage to Eu(hfa)3(H2O)2 in the ITO–PB device for a longer time (Fig. 6).
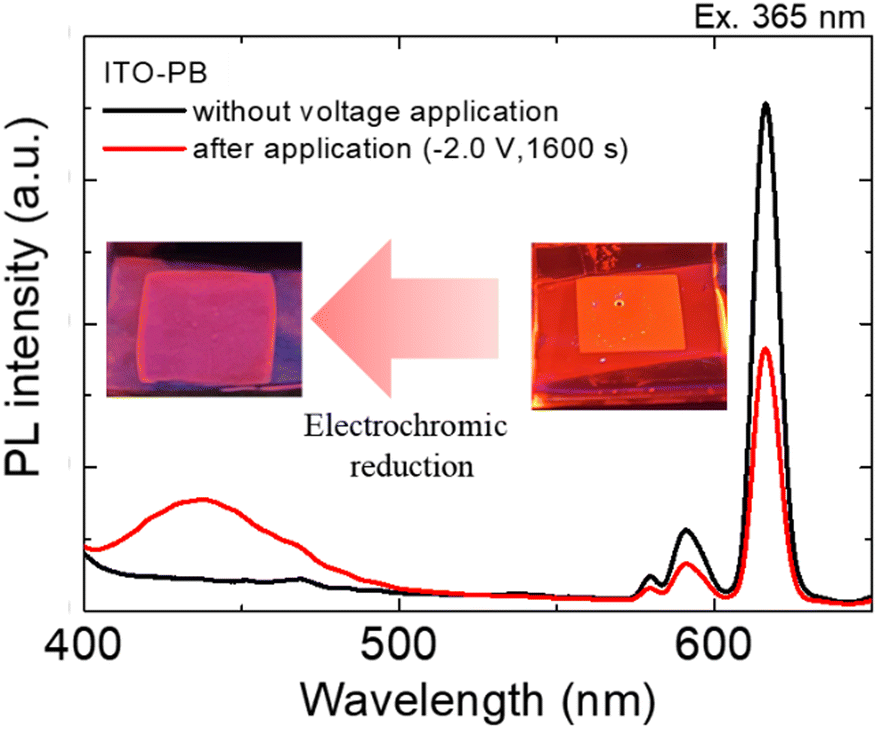 | ||
| Fig. 6 Emission spectra of the Eu(hfa)3(H2O)2 solution in PEG400 using the ITO–PB device before and after the application of reducing voltages. The excitation wavelength was 365 nm. | ||
When a reduction voltage of −2.0 V was applied for 1600 s, while the emission bands from the Eu2+ state (430 nm) significantly increased significantly, the red emission from the Eu3+ state (616 nm) simultaneously decreased. The intensity ratio of Eu2+ emission (430 nm) to Eu3+ emission (616 nm) reached 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.3 under application of the reduction voltage. The X–Y coordinates in the CIE chromaticity diagram changed from (0.49, 0.29) to (0.31, 0.18), and we could recognize the change in luminescence colors. (Fig. 6 photographs). From these results, electrochemical photoluminescence control based on the valence change of the Eu ion, that is, the EFC reaction of the Eu ion, was successfully observed when the PB electrode was introduced as the counter electrode. To investigate the EFC properties of the device in detail, the CVs and in situ luminescence intensities of both Eu3+ and Eu2+ were synchronously recorded (Fig. 7).
:2.3 under application of the reduction voltage. The X–Y coordinates in the CIE chromaticity diagram changed from (0.49, 0.29) to (0.31, 0.18), and we could recognize the change in luminescence colors. (Fig. 6 photographs). From these results, electrochemical photoluminescence control based on the valence change of the Eu ion, that is, the EFC reaction of the Eu ion, was successfully observed when the PB electrode was introduced as the counter electrode. To investigate the EFC properties of the device in detail, the CVs and in situ luminescence intensities of both Eu3+ and Eu2+ were synchronously recorded (Fig. 7).
 | ||
| Fig. 7 Changes in the luminescence of Eu2+ at 430 nm (top) and Eu3+ at 615 nm (middle) during the cyclic voltammogram measurement (bottom) using the ITO–PB device. The excitation wavelength was 365 nm. The scan rate was 100 mV s−1. | ||
When the voltage was scanned in the negative direction, the reduction current began to flow around −0.8 V, and the emission intensity of Eu3+ at 615 nm decreased while that of Eu2+ at 430 nm increased. By scanning back towards the positive direction, the oxidation current began to flow from around −1.0 V, the 615 nm emission intensity of Eu3+ increased, and the 430 nm emission intensity of Eu2+ decreased. As the oxidation reaction proceeded, the emission intensities of both Eu3+ and Eu2+ returned to their original values. This result indicates that the redox reaction of the two-electrode device was due to the Eu3+/2+ reaction in the device, leading to a corresponding luminescence change in each state.
The EFC performance of the ITO–PB device was then compared with those of other EFC devices with different counter electrode materials (ITO–ITO and ITO–CM devices). Fig. 8 shows the emission spectra of Eu(hfa)3(H2O)2 in the two electrode devices (ITO–ITO, ITO–CM, and ITO–PB).
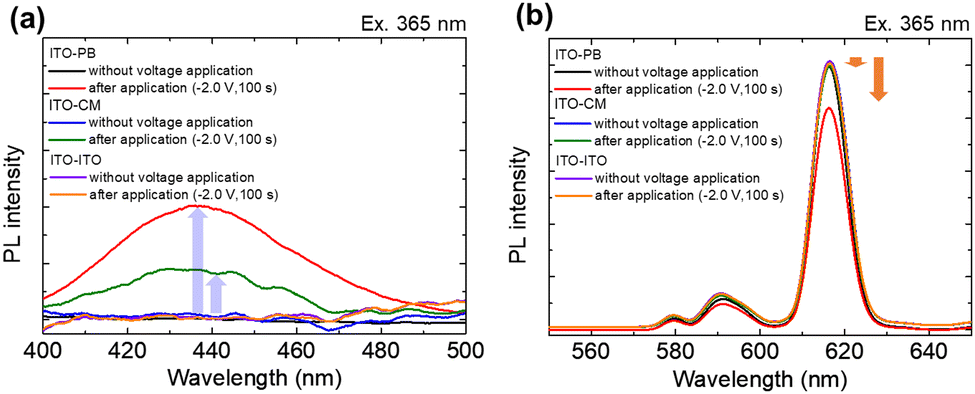 | ||
| Fig. 8 Emission spectra of the Eu complex in the two-electrode devices (ITO–PB, ITO–CM, and ITO–ITO device) before and after applying a reduction voltage of −2.0 V for 100 s at approximately (a) 430 nm and (b) 615 nm. The excitation wavelength was 365 nm. | ||
When a reduction voltage (−2.0 V) was applied for 100 s, a broad emission band appeared at approximately 430 nm for the ITO–PB and ITO–CM devices (Fig. 8(a)). Simultaneously, the red emission from the excited state of Eu3+ at approximately 615 nm decreased (Fig. 8(b)). For the ITO–PB device, the emission changes in both emission bands (615 nm for Eu3+ and 430 nm for Eu2+) were the largest compared with those of the other devices. These differences in the change in the luminescence intensity can be discussed in terms of the difference in the charge amount of the redox reaction in the EFC devices. Table 1 summarizes the charge amount of the redox reaction under the application voltage of −2.0 V for 100 s to each device.
| Construction of devices | Charge amount/mC |
|---|---|
| ITO–PB | 5 |
| ITO–CM | 2.9 |
| ITO–ITO | 0.5 |
The charge amounts of the redox reaction in the device were 0.5, 2.9, and 5.0 mC for the ITO–ITO, ITO–CM, and ITO–PB devices, respectively. These results show that the reaction charge of the ITO–PB device was approximately 10 times larger than that of the ITO–ITO device and approximately twice as large as that of the ITO–CM device. Fig. S6 (ESI†) shows the two-electrode CV curves of the EFC devices. As can be seen in the CVs, the cathodic current of the ITO–PB device at −2.0 V was approximately −0.26 mA, which is more significant than those of the other EFC devices. These tendencies would be attributed to the electrode potential of the working electrode under the application of a driving voltage of −2.0 V to the EFC device. As shown in Fig. 4, the electrode potential of the working electrodes was found to be −1.8 V for the ITO–PB device, −1.5 V for the ITO–CM device, and −0.8 V for the ITO–ITO device. The ITO–PB device experienced a greater modulation of the photoluminescence and a higher electrochemical reactivity as a result of the significant differences in the electrode potential. Nevertheless, luminescence modulation from the ITO–ITO device necessitates the application of a higher voltage (>−3.0 V). The electrolyte solution suffers substantial damage under such a high voltage, leading to decreased repetition stability. Comparing the fluorescence switching responses of the ITO–ITO and ITO–PB devices, the reversible performance of the ITO–PB device was higher than that of the ITO–ITO device. (Fig. S7, ESI†) Further improvement of EFC properties would require immobilization of Eu ions on the electrode. In conclusion, the introduction of a suitable counter electrode material against the redox reaction of Eu3+/2+ compensated for the reaction charge required for the valence change of the Eu complex on the working electrode, resulting in increased redox activities and photoluminescence modulation properties between Eu3+ and Eu2+.
4 Conclusions
In this study, we aimed to improve the EFC performance based on the valence change between Eu3+ and Eu2+ by enhancing the electrochemical reactivity of the EFC device. A two-electrode EFC device consisting of a PEG400 electrolyte solution and an Eu complex was fabricated by introducing a PB film as the counter electrode. The counter electrode modified with the PB film successfully compensated for the reaction charge required for the redox reaction of the Eu complex on the working electrode. The reaction charge of the ITO–PB device was approximately 10 times larger than that of the ITO–ITO device and approximately twice as large as that of the ITO–CM device. The increase in the reaction charge enabled faster changes in the photoluminescence from Eu3+ to Eu2+ and increased the blue luminescence intensity from the Eu2+ state. These findings will contribute to developing novel imaging-related applications, such as chemical or biological sensors, molecular logic gates, security materials, and display devices.Author contributions
Ryoto Yabuta: investigation and writing – original draft. Norihisa Kobayashi: writing – review and editing. Kazuki Nakamura: project administration and writing – review and editing.Data availability
All data will be made available by the authors upon request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partly supported by JSPS KAKENHI (17H06377, 20K05641, and 23K04871), New Energy and Industrial Technology Development Organization (JPNP20004), Izumi Science and Technology Foundation, and Iketani Science and Technology Foundation. This work was also supported by JST SPRING, Grant Number JPMJSP2109. We would like to thank Editage (https://www.editage.com) for English language editing.Notes and references
- D. Tyler McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537 CrossRef PubMed.
- R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419 CrossRef PubMed.
- M. A. Rizzo, G. H. Springer, B. Granada and D. W. Piston, Nat. Biotechnol., 2004, 22, 445 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, Nature, 2002, 420, 759 CrossRef CAS PubMed.
- A. A. Argun, P. H. Aubert, B. C. Thompson, I. Schwendeman, C. L. Gaupp, J. Hwang, N. J. Pinto, D. B. Tanner, A. G. MacDiarmid and J. R. Reynolds, Chem. Mater., 2004, 16, 4401 CrossRef CAS.
- C. Bechinger, S. Ferrere, A. Zaban and J. Sprague, Nature, 1996, 383, 608 CrossRef CAS.
- P. Audebert and F. Miomandre, Chem. Sci., 2013, 4, 575 RSC.
- H. J. Yen and G. S. Liou, Chem. Commun., 2013, 49, 9797 RSC.
- X. Yang, S. Seo, C. Park and E. Kim, Macromolecules, 2014, 47, 7043 CrossRef CAS.
- H. J. Nie, W. W. Yang, J. Y. Shao and Y. W. Zhong, Dalton Trans., 2016, 45, 10136 RSC.
- M. Chang, W. Chen, H. Xue, D. Liang, X. Lu and G. Zhou, J. Mater. Chem. C, 2020, 8, 16129 RSC.
- M. H. Chua, Q. Zhu, K. W. Shah and J. Xu, Polymers, 2019, 11, 98 CrossRef PubMed.
- H. Al-Kutubi, H. R. Zafarani, L. Rassaei and K. Mathwig, Eur. Polym. J., 2016, 83, 478 CrossRef CAS.
- F. Miomandre, Curr. Opin. Electrochem., 2020, 24, 56 CrossRef CAS.
- S. Kim and Y. You, Adv. Opt. Mater., 2019, 7, 1900201 CrossRef.
- K. Su, N. Sun, Z. Yan, S. Jin, X. Li, D. Wang, H. Zhou, J. Yao and C. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 22099 CrossRef CAS PubMed.
- C. P. Kuo, C. L. Chang, C. W. Hu, C. N. Chuang, K. C. Ho and M. K. Leung, ACS Appl. Mater. Interfaces, 2014, 6, 17402 CrossRef CAS PubMed.
- J. H. Wu and G. S. Liou, Adv. Funct. Mater., 2014, 24, 6422 CrossRef CAS.
- A. Beneduci, S. Cospito, M. La Deda and G. Chidichimo, Adv. Funct. Mater., 2015, 25, 1240 CrossRef CAS.
- C. P. Kuo and M. K. Leung, Phys. Chem. Chem. Phys., 2014, 16, 79 RSC.
- W. Gao, T. Yu, Y. Du, R. Wang, L. Wu and L. Bi, ACS Appl. Mater. Interfaces, 2016, 8, 11621 CrossRef CAS PubMed.
- B. Wang, L. H. Bi and L. X. Wu, J. Mater. Chem., 2011, 21, 69 RSC.
- Y. Kim, H. Ohmagari, A. Saso, N. Tamaoki and M. Hasegawa, ACS Appl. Mater. Interfaces, 2020, 12, 46390 CrossRef CAS PubMed.
- J. Lehr, M. Tropiano, P. D. Beer, S. Faulkner and J. J. Davis, Chem. Commun., 2015, 51, 6515 RSC.
- T. W. Ngan, C. C. Ko, N. Zhu and V. W. W. Yam, Inorg. Chem., 2007, 46, 1144 CrossRef CAS PubMed.
- F. Miomandre, R. B. Pansu, J. F. Audibert, A. Guerlin and C. R. Mayer, Electrochem. Commun., 2012, 20, 83 CrossRef CAS.
- M. Tropiano, N. L. Kilah, M. Morten, H. Rahman, J. J. Davis, P. D. Beer and S. Faulkner, J. Am. Chem. Soc., 2011, 133, 11847 Search PubMed.
- M. Yano, K. Matsuhira, M. Tatsumi, Y. Kashiwagi, M. Nakamoto, M. Oyama, K. Ohkubo, S. Fukuzumi, H. Misaki and H. Tsukube, Chem. Commun., 2012, 48, 4082 RSC.
- T. Sato and M. Higuchi, Tetrahedron Lett., 2019, 60, 940 CrossRef CAS.
- J. C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53 CrossRef PubMed.
- J. C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC.
- S. Faulkner, S. J. A. Pope and B. P. Burton-Pye, Appl. Spectrosc. Rev., 2005, 40, 1 CrossRef CAS.
- K. Miyata, Y. Konno, T. Nakanishi, A. Kobayashi, M. Kato, K. Fushimi and Y. Hasegawa, Angew. Chem., Int. Ed., 2013, 52, 6413 Search PubMed.
- A. Ishii and M. Hasegawa, Sci. Rep., 2015, 5, 11714 CrossRef PubMed.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- J. Jiang, N. Higashiyama, K.-I. Machida and G.-Y. Adachi, Coord. Chem. Rev., 1998, 170, 1 CrossRef CAS.
- W. Chen, Y. Ouyang, M. Mo, H. Zhang and Q. Su, J. Lumin., 2021, 229, 117672 CrossRef CAS.
- W. Liu, L. Liu, Y. Wang, L. Chen, J. A. McLeod, L. Yang, J. Zhao, Z. Liu, J. Diwu, Z. Chai, T. E. Albrecht-Schmitt, G. Liu and S. Wang, Chem. – Eur. J., 2016, 22, 11170 CrossRef CAS PubMed.
- A. Acharjya, B. A. Corbin, E. Prasad, M. J. Allen and S. Maity, J. Photochem. Photobiol., A, 2022, 429, 113892 CrossRef CAS.
- X. Yang, T. S. Tiam, X. Yu, H. V. Demir and X. W. Sun, ACS Appl. Mater. Interfaces, 2011, 3, 4431 CrossRef CAS PubMed.
- E. Malchukova and B. Boizot, Divalent Europium in β-irradiated aluminoborosilicate glass, J. Am. Ceram. Soc., 2010, 93, 4005–4007 CrossRef CAS; E. Malchukova and B. Boizot, J. Am. Ceram. Soc., 2010, 93, 4005 CrossRef.
- Y. Tratsiak, M. Buryi, V. Babin, M. Korjik and E. Trusova, Opt. Mater., 2019, 94, 356–362 CrossRef CAS.
- F. Meng, J. Zhang, Z. Zhang, H. J. Seo and X. Zhang, Ceram. Int., 2015, 41, 11726 CrossRef CAS.
- S. Thongchant, S. Katagiri, Y. Hasegawa, Y. Wada, S. Watase, M. Nakamoto, T. Sakata, H. Mori and S. Yanagida, Bull. Chem. Soc. Jpn., 2004, 77, 807 Search PubMed.
- F. A. Sigoli, M. R. Davolos and M. Jafelicci, J. Alloys Compd., 2002, 344, 308 Search PubMed.
- R. Yabuta, N. Kobayashi and K. Nakamura, Phys. Chem. Chem. Phys., 2023, 25, 25979 Search PubMed.
- V. Balzani, F. Scandola, G. Orlandi, N. Sabbatini and M. T. Indelli, J. Am. Chem. Soc., 1998, 103, 3370 CrossRef.
- S. Kimura, K. Nakamura and N. Kobayashi, Sol. Energy Mater. Sol. Cells, 2020, 205, 110247 CrossRef CAS.
- S. W. Tong, J. Chai, M. F. Ng, W. Fu, W. P. Goh and C. Jiang, ACS Appl. Opt. Mater., 2024, 2, 539 CrossRef CAS.
- K. Nakamura, K. Kanazawa and N. Kobayashi, J. Photochem. Photobiol., C, 2022, 50, 100486 CrossRef CAS.
- X. H. Yan, T. S. Zhao, L. An, G. Zhao and L. Shi, Int. J. Hydrog., 2016, 41, 5135 Search PubMed.
- Z. Liang, K. Nakamura and N. Kobayashi, Sol. Energy Mater. Sol. Cells, 2019, 200, 109914 CrossRef CAS.
- R. Ozawa, K. Nakamura, T. Tachikawa and N. Kobayashi, J. Phys. Soc. Japan, 2022, 61, 562 CAS.
- Y. Zhao, Y. Bai, W. Li, M. An, Y. Bai and G. Chen, Chem. Mater., 2020, 32, 6811 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03155e |
| This journal is © the Owner Societies 2024 |