
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputationally aided design of defect-appended aliphatic amines for CO2 activation within UiO-66†
Gerard
Pareras
 ab,
Albert
Rimola
b,
Marco
Taddei
*c and
Davide
Tiana
*a
ab,
Albert
Rimola
b,
Marco
Taddei
*c and
Davide
Tiana
*a
aSchool of Chemistry, University College Cork, College Road, Cork, Ireland. E-mail: davide.tiana@ucc.ie
bDepartament de Química, Universitat Autònoma de Barcelona, 08193 Bellaterra, Catalonia, Spain
cDipartimento di Chimica e Chimica Industriale, Università di Pisa, Via Giuseppe Moruzzi 13, 56124 Pisa, Italy. E-mail: marco.taddei@unipi.it
First published on 11th October 2024
Abstract
The introduction of aliphatic amine groups in metal–organic frameworks (MOFs) can improve their ability to capture CO2 at low pressures, driven by chemisorptive formation of C–N bonds. Understanding the chemistry of amine-CO2 interaction within the confined porous space in MOFs is key to design and develop effective CO2 adsorbents. Here, we report a computational study of CO2 adsorption and subsequent formation of carbamic acid within defective UiO-66 functionalised with a series of four amino acids of varying aliphatic chain length (glycine, beta-alanine, gamma-aminobutyric acid and 5-aminovaleric acid). Periodic density functional theory (DFT) calculations suggest that CO2 can be activated by the aliphatic amines only when they are sufficiently close to each other to form hydrogen bonds and stabilise the adduct with CO2, a condition met only by UiO-66 functionalised with gamma-aminobutyric acid and 5-aminovaleric acid. The proposed mechanism involves the formation of a carbamate zwitterionic intermediate, which evolves via a simultaneous double hydrogen transfer with a proximal amine group to a carbamic acid. For the 5-aminovaleric acid case, it is suggested that even the functionalisation of just 16% of the available defective sites can be sufficient to form the CO2-amine adduct. Finally, we also investigate the effect of a possible protonation of the amine groups by the hydroxyl groups in the clusters, finding that this could lead to even more favourable interaction with CO2.
Introduction
The use of aliphatic amines to capture and/or activate CO2 is one of the oldest and most successful methods, via either chemical absorption (amine scrubbing)1 or chemical adsorption (solid-supported amines).2 In the case of primary and secondary amines in anhydrous conditions, a plausible mechanism involves the formation of a C–N bond between CO2 and one amine group, yielding a zwitterionic Lewis adduct with carbamate-like structure (Fig. 1, Step 1).3 From this carbamate-like structure, it is possible to obtain the carbamic acid from a double hydrogen transfer. This double transfer is happening when one hydrogen is transferred from the ammonium carbamate to a neighbour amine which transfers one of its hydrogens to the activated CO2, thus compensating the previously created zwitterionic structure (Fig. 1, Step 2).4 The intra-hydrogen-transfer within the same ammonium carbamate is usually not favoured, meaning that two amine groups are required to capture one CO2 molecule and promote the inter-hydrogen-transfer between them. The proximity of these groups is key to reach the most stable state. Note that here we represented step 2 with two structures, but there is the possibility that it occurs in a one single step (involving only one transition state).5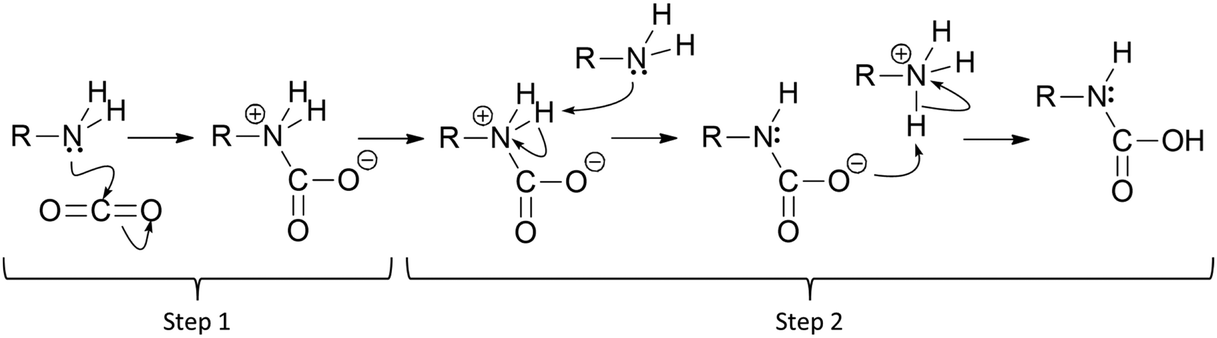 | ||
| Fig. 1 Schematic representation of the CO2 activation and subsequent carbamic acid formation with primary aliphatic amines. | ||
In this work, we computationally explored the possibility of forming carbamate from CO2 and aliphatic amines grafted and the posterior hydrogenation leading the carbamic acid, onto the pore surface of metal–organic frameworks (MOFs). Grafting of aliphatic amines within MOFs has been achieved in various fashions, involving either functionalisation of the organic linker or grafting on the inorganic part of the framework. A successful approach involves the introduction of methyleneamine groups on the aromatic ring of the linker in a pore-expanded MOF-74 analogue.6,7 A few strategies have been proposed that exploit modifications of the inorganic unit, such as grafting of diamines on the open metal sites found in MOF-74 analogues8 or in MIL-101,9 and installation of ethanolamine in place of μ3-OH groups on the metal clusters of UiO-66.10 A remarkable example form literature is the work of Yaghi and co-workers in which MOF-808 functionalised with glycine by grafting on the Zr-oxyhydroxide clusters was found to be able to capture and release CO2 under humid coal flue gas conditions via formation of ammonium bicarbonate.11 A subsequent work by the same group exploited a two-step approach that involved the grafting of 3-chloropropionic acid to the clusters, followed by nucleophilic substitution with aliphatic polyamines, to produce a sorbent able to capture CO2 directly from humid air.12
Herein, we propose to include aliphatic amines in the internal surface of Zr-based UiO-66 using a defect-engineering approach. In fact, after having created missing cluster defects in UiO-66, functionalisation with a range of monocarboxylic acids, including amino acids, is relatively simple to achieve via post-synthetic defect exchange (PSDE).13–15 Four aliphatic amino acids were chosen here to functionalise the internal surface of UiO-66: glycine (gly), beta-alanine (ala), gamma-aminobutyric acid (gaba) and 5-aminovaleric acid (ava). The rationale behind this choice is to evaluate the effect of increasing degrees of freedom in the amino acid chain and to see if, by increasing its length, it is possible to promote cooperative effects between amine groups that lead to formation and stabilisation of carbamates in anhydrous conditions.
Computational details
The CO2 adsorption and activation were studied using periodic density functional theory (DFT) simulations using the CP2K software.16 The initial UiO-66 unit cell parameters were obtained from our previous publication.17 An inorganic node was then removed from the centre of the unit cell to simulate the missing cluster defect (see Fig. 2).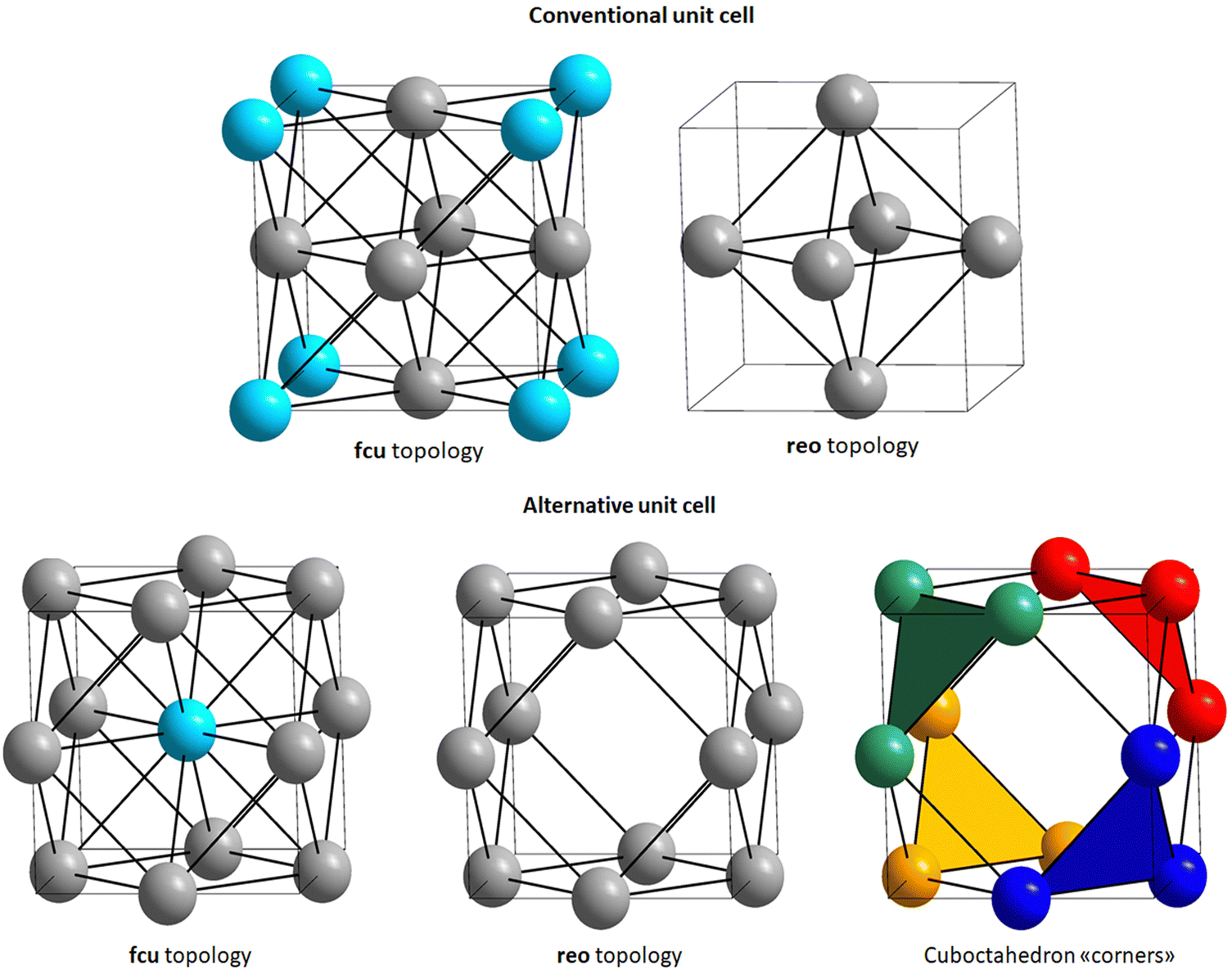 | ||
| Fig. 2 Top: Simplified representation of a conventional UiO-66 unit cell where each sphere represents an hexanuclear [(Zr6O4(OH)4)]12+ cluster (left). This connectivity gives rise to a fcu topology that becomes reo after removing one cluster per unit cell (right). The removed cluster, originally sitting on the cell corner, is depicted in light blue in the left figure. Bottom: Alternative unit cell with shifted origin (+1/2; +1/2; +1/2) for fcu topology (left) and reo topology (centre). The removed cluster, originally sitting on the cell centre, is depicted in light blue in the left figure. The remaining clusters in the reo topology are grouped in triplets of adjacent clusters, defining four cuboctahedron “corners” (right). | ||
As an initial step, a full cell optimization was performed, relaxing both the internal atomic positions and the lattice parameters. From this point on, subsequent modifications (i.e., functionalisation of the defects and CO2 reaction) were performed relaxing only the atomic positions (i.e., keeping the cell parameters fixed). Geometry optimizations were carried out using the semi-local Perdew–Burke–Ernzerhof (PBEsol) functional, combined with a double-ζ basis set (DZVP-MOLOPT-SR-GTH Gaussian basis set) for all the atom types, and a cutoff set at 500 Ry for the plane wave auxiliary basis set. Core electrons were described with the Goedecker–Teter–Hutter pseudopotentials,18 and valence ones with a mixed Gaussian and plane-wave (GPW) approach.19 Solvation effects were considered by performing single point energy calculations on the optimized systems adopting the self-consistent continuum solvation (SCCS) model as implemented in CP2K, the solvent of choice was water.20
Transition states search has been performed by means of the Climbing image nudged elastic band approach.21 To determine the nature of the stationary points of the potential energy surfaces (i.e., local minima and saddle points) the corresponding vibrational harmonic frequencies were calculated at the PBE/DZVP level using the finite differences method. A partial Hessian approach was used to reduce the computational cost of the calculations and the vibrational frequencies were calculated on only on a fragment of the system that represents the most chemically relevant part (i.e., the reactive atoms and the amine chains).
Finally, the interaction energy was calculated defining two fragments, A and B. One fragment being the functionalized MOF (A) and a second fragment being the CO2 molecule (B)
| Eint = EAB − (EA + EB) | (1) |
A counterpoise approach was employed in the calculations of the interaction energies to avoid basis set superposition errors.
Results and discussion
MOF functionalization benchmark
We started by simulating the ideal situation in which all the defective sites available in the unit cell of reo UiO-66 are functionalised. To relieve the computational cost of our simulations, we approximated this situation by functionalising three sites belonging to one “corner” of the cell, out of a total of twelve available sites associated with one defect. Statistically, this would correspond to the ideal situation in which all the twelve defective sites created by a missing cluster are functionalised with amine groups. As explained later in the paper, we also simulated situation with a lower degrees of defect functionalisation.Standard MOFs scaffolds are quite stiff and, normally, their structures can be reasonably approximated as rigid. The situation changes when aliphatic amino acid groups are inserted into the framework, because the aliphatic chains are flexible and fluctuate due to thermal motions. To partially account for this effect, and to check that our starting configuration was a reasonably snapshot of the vibration the occur at thermal energy, we analysed several configurations for each defect-engineered MOF with random orientation of the aliphatic chains. Once the geometry of every configuration was optimised, we found that many configurations were stabilised by the presence of hydrogen bonds, and they were within a difference in energy of a range from 5 to 15 kcal mol−1, which is less than the thermal energy that the system would have in real conditions. This guarantees that our starting configuration was representative of a snapshot of the system (few configurations where no hydrogen bonds were present were found to be higher in energy). We picked the geometry with the lowest energy for each of the four defect-engineered UiO-66 (energies reported in Table S1, ESI†) using it as starting structure. To be noted, these were only guess geometries to produce reasonable starting configurations for the reactions. Further geometry optimisations were performed for all the studied systems in the following steps.
CO2 activation
Once the most stable structure from the preliminary optimisation was selected, we continued our study with the binding of CO2 to the amine group to produce the carbamate (Fig. 1, Step 1). Our calculations show that the binding of CO2 depends on the length of the amine group (all energetic values have been gathered in Table S2, ESI†). On one hand, we found that UiO-66_gly and UiO-66_ala do not show strong binding sites (average adsorption energies for UiO-66_gly and UiO-66_ala are −2.66 and −4.43 kcal mol−1, respectively) and the CO2 is never activated (see Fig. 3A and B). On the other hand, we found that CO2 can be chemically adsorbed in both UiO-66_gaba and UiO-66_ava. A geometrical analysis of the chemically adsorbed configurations, i.e., configurations in which N⋯C distance is within the order of a chemical bond and there is an elongation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distance in CO2 (Table S2, ESI†), indicates that this is promoted by a cooperative effect between the amine groups that create a network of hydrogen bonds (Fig. 3C and D). Thus, proximity of the amine groups plays a central role, enabling the establishment of intermolecular interactions that contribute to stabilise the zwitterionic product.
O bond distance in CO2 (Table S2, ESI†), indicates that this is promoted by a cooperative effect between the amine groups that create a network of hydrogen bonds (Fig. 3C and D). Thus, proximity of the amine groups plays a central role, enabling the establishment of intermolecular interactions that contribute to stabilise the zwitterionic product.
 | ||
| Fig. 3 Detail of the periodic unit cells for the optimized geometries of: UiO-66_gly_6 (A), UiO-66_ala_5 (B), UiO-66_gaba_5 (C) and UiO-66_ava_2 (D). Relevant distances are depicted in angstroms (Å). Hydrogen bonds are represented as dotted lines. Colour scheme: C (grey), H (white), O (red), N (blue) and Zr (cyan). | ||
Carbamic acid formation through hydrogen transfer mechanism
From all the different structures where CO2 was successfully activated (i.e., the carbamate zwitterionic intermediate was obtained), we selected the UiO-66_ava_2 to study the hydrogen transfer mechanism that leads to the carbamic acid. The reaction was found to be energetically favourable, with the product being 3.82 kcal mol−1 more stable than the reactants. Our calculations indicate that the hydrogen transfer is happening in one single step. Interestingly, the transition state shows a double hydrogen transfer: one from the amino carbamate to the neighbour amine and another transfer from that amine to one of the oxygens in the carbamate (see Fig. 4A). We noted that the third aliphatic amine does not intervene in the transition state, and the previously formed hydrogen bond with the other amine is no longer existent (Fig. 4B). This suggests that only two amine groups (i.e., only two H bonds) are required for the reaction to proceed. The calculated activation energy was found to be ca. 8 kcal mol−1. The transition state structure was confirmed by the analysis of the frequencies that had only one imaginary frequency showing the double hydrogen transfer mentioned before. For completeness, we also computed the activation energy for the system UiO-66_gaba_7, as it showed a strong activation of the CO2 molecule, finding that the reaction evolves in the same way as the ava system.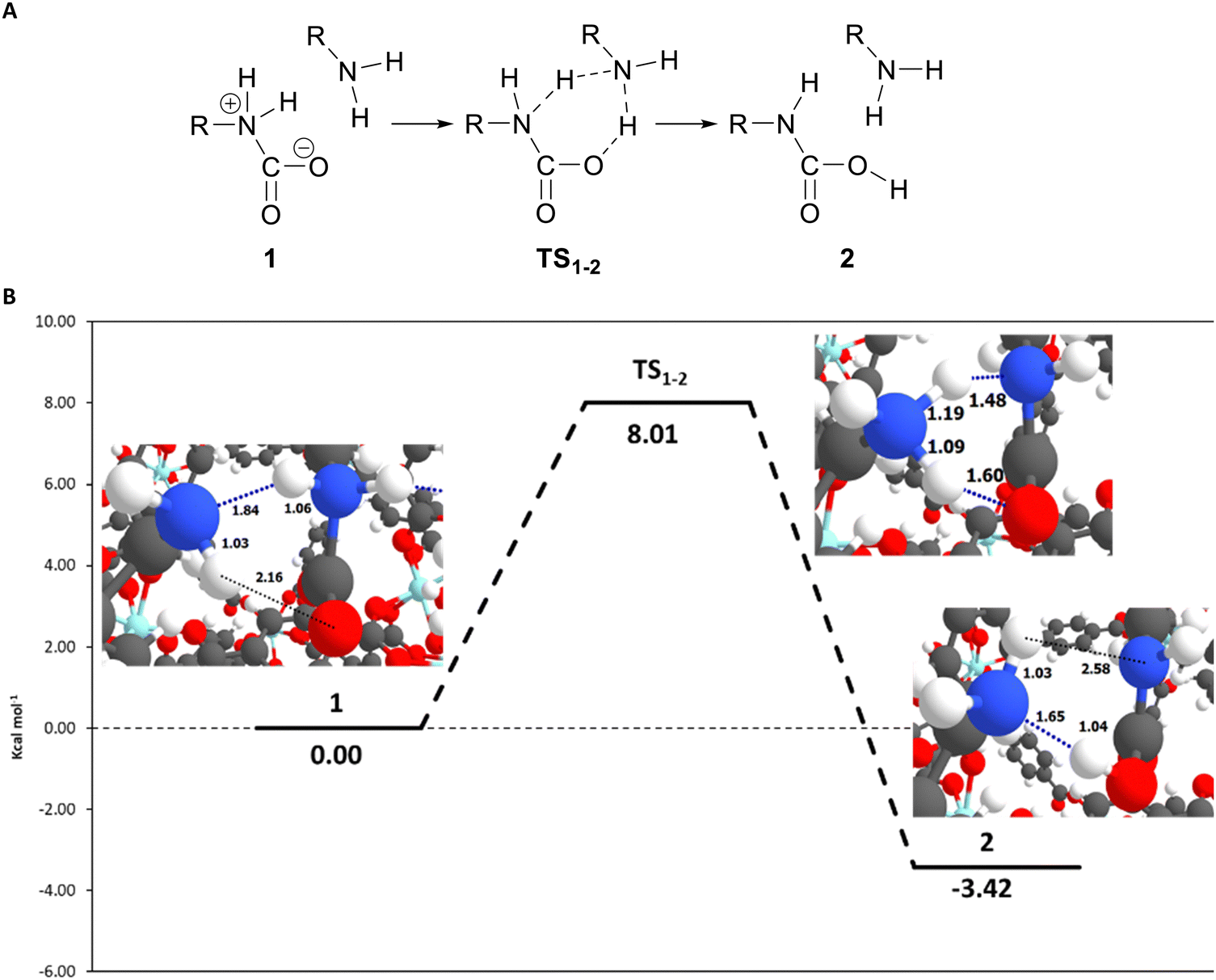 | ||
| Fig. 4 Panel (A): proposed reaction mechanism that involves only one transition state for the formation of the carbamate acid. Panel (B): potential energy surface (PES) for the proton transfer mechanism leading to the formation of the carbamic acid for the system UiO-66_ava_2. Relative energies are in kcal mol−1. Relevant distances are in angstroms (Å). Hydrogen bonds are represented as dotted lines. Colour scheme: C (grey), H (white), O (red), N (blue) and Zn (cyan). | ||
Beyond the ideal situation
At this point, we considered the possibility that not all of the defective sites are functionalised with amino acids. This situation might be realistic for the pendant amino acids with the longest chains, i.e., gaba and ava, due to steric constraints within the confined pore space that might prevent from achieving full functionalisation.First of all, we simulated the adsorption of CO2 for UiO-66_gaba and UiO-66_ava where 5 out of 12 sites are functionalised, corresponding to a degree of functionalisation of ca. 42%. As for the previous case, we approximated the system functionalising two groups in the same corner (this would correspond to the situation in which one of the four corners has two sites functionalised with amine groups while the other three corners have only one amine. Situations with lower degree of functionalisation will be also discussed later). In this case, we found that CO2 can still be activated forming the zwitterionic carbamate intermediate. Binding energies in Table S3 (ESI†) demonstrate that selected structures with a degree of functionalisation of 42% are still in the range of chemisorption. Hydrogen transfer to form the carbamic acid is confirmed to be energetically favourable for all these structures (Table S4, ESI†).
Secondly, we analysed the situation in which the pore can still be functionalised with two amino acid chains per pore, corresponding to a degree of functionalisation of ca. 17%. Here, we have different scenarios depending on where these chains are located (Fig. 5). Configuration A is the ideal situation where functionalization has happened in the same corner which would give the same results discussed earlier for a degree of functionalisation of 42%, but is the least probable to occur in the real case. Other possibilities, displayed in Fig. 5, include finding the amino acid in opposite corners of the reo unit cell (configuration B, with distance between the carboxylic groups of amino acids of ca. 20 Å), or in contiguous corners (configuration C, with distance between the carboxylic groups of amino acids of ca. 14 Å; configuration D, with distance between the carboxylic groups of amino acids of ca. 17 Å).
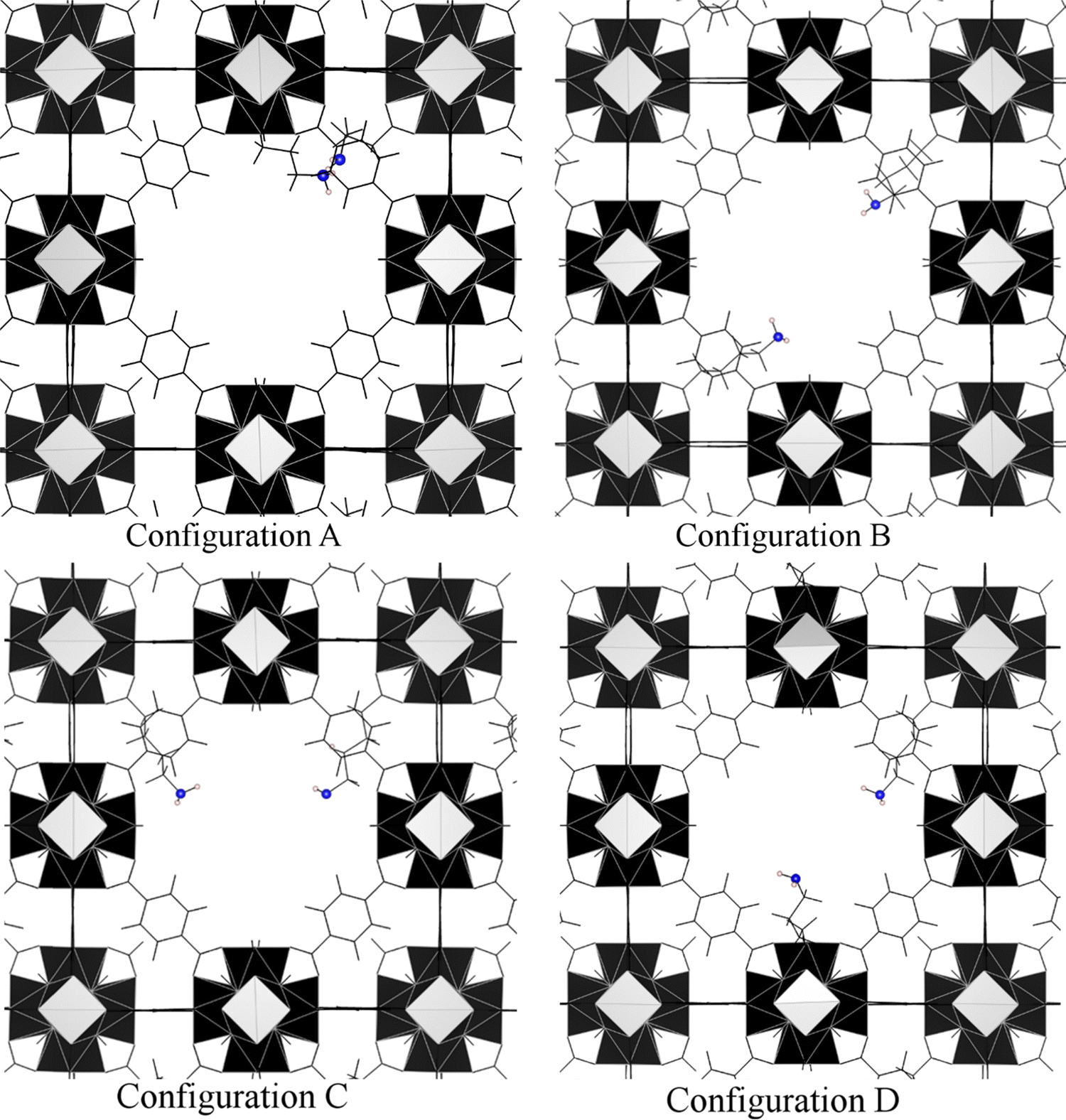 | ||
| Fig. 5 Missing cluster UiO-66 functionalised with two ava groups. Atom colours: N = blue, H = beige. The black blocks are the ZrO inorganic clusters. | ||
Data collected in Table S5 (ESI†) demonstrate that only when the amine group are in adjacent corners (i.e., configuration C), CO2 can be activated with a binding energy of 21.50 kcal mol−1 (see Fig. S1, ESI†). In all the other configurations, it is not possible to activate CO2 due to the long distance between the amine chains that prevents the formation of H bonds.
Finally, we analysed the situation in which the pore is only functionalised with an 8% degree (only one amino acid per pore), finding that only one amino acid is not able to activate CO2. Reported binding energies in Table S3 (ESI†) demonstrate that the interactions are not in the range of chemical bonding.
From all these non-ideal situations we have studied here, we can conclude that, although the degree of functionalization of the MOF can be very low and/or not ideally allocated, activation of CO2 could still happen as soon as at least two amine chains are close enough to form hydrogen bonds. It is clear, then, that the cooperation between chains is essential for the activation of CO2. A functionalised MOF structure able to activate CO2 by means of the cooperation between amine chains will then promote the following hydrogen transfer forming the carbamic acid.
CO2 activation on the metal node
Until now, we have only considered the possibility of CO2 being activated by the amine groups, however defective metal nodes contain μ3-OH groups that could also interact with CO2, competing with the amine chains. In view of that, we analysed the interaction of CO2 with the μ3-OH groups in the inorganic node. We found the binding energies are small (average of −2.03 kcal mol−1) and CO2 is not activated (Table S6, ESI†). This is in line with what previously found in literature,22,23 and corroborates the experimental observation of Shearer et al. that the isosteric heat of adsorption is only marginally increased when serine (an α amino acid) is grafted at missing cluster defects in UiO-66.13MOF intra-protonation
Although the μ3-OH moieties are not able to activate the CO2, they have a pKa (about 3.5)24,25 that is much lower than the one of the primary amine groups (9.78 for gly, 10.24 for ala, 10.56 for gaba and 10.77 for ava). For this reason, we considered the possibility of having an intra-proton transfer from μ3-OH groups on the metal node to the –NH2 groups residing in the pore forming a –NH3+ group. The clusters and the amine groups are separated by a distance across which it is highly unlikely for a proton to hop. However, the presence of solvents could act as proton carriers realising the proposed scenario. Solvents such as water and alcohols, are often used as media for post-synthetic exchange reactions or for workup procedures. Here, the solvent is present during the PSDE process and before the MOF is activated prior to its use as a sorbent. Such scenario could be possible in practice if solvent removal during activation occurs rapidly, quenching the system in a state where the proton stays on the amine group (e.g., kinetic crystallisation).To simulate the proposed scenario, we calculated the stability of the –NH3+versus the initial state using the continuum solvent approximation (see Computational details, ESI†). Table S7 (ESI†) gathers all the energetic values for the –NH3+ protonation. We considered that the proton can come from a hydroxyl group on the same metal node on which the amino acid is grafted (labelled HIN in Table S7, ESI†), or from a hydroxyl group of another node (labelled HOUT in Table S7, ESI†). Our calculations suggest that the protonation of an amine group is only favourable for the solvated UiO-66_ava (averaged value Table S7, ESI†), where the formed NH3+ systems are thermodynamically more stable than the unprotonated system. We also found that the origin (HIN or HOUT) of the proton does not imply any significant difference. On top of the effect of the solvent, which is present and partially stabilises also the other functionalised MOFs (UiO-66_gly, UiO-66_ala, UiO-66_gaba), the reason why the amine is protonated in the solvated UiO-66_ava appears to be the formation of hydrogen bonding between the protonated –NH3+ and the two neighbouring, neutral –NH2 groups (see Fig. S2, ESI†). We can then conclude that the proximity of amine groups is crucial to allow the stabilisation of the system upon proton transfer. For UiO-66_ava, we also checked if further amine groups could be protonated, finding that further proton transfers are not favourable (see Table S8, ESI†).
CO2 activation on the protonated systems
Following the previous consideration on protonation, we simulated the possibility that the amino group remains quenched in the protonated form after crystallisation and the possibility that this form could activate CO2 (Fig. 1, Step 1). Results showed that, if one of the amine groups is protonated, the adsorption of CO2 becomes stronger (average of ca. −56 kcal mol−1 and interactions up to ca. −100 kcal mol−1 for the protonated UiO-66_ava compared to an average of ca. −20 kcal mol−1, max −50 kcal mol−1 for the non-protonated form, see Tables S2 and S9, ESI†). A geometrical analysis of the configurations reveals that a hydrogen bond between activated CO2 and NH3+ is formed, resulting in an enhanced stabilisation of the system (see Fig. 6 on the right and Fig. S3, ESI†).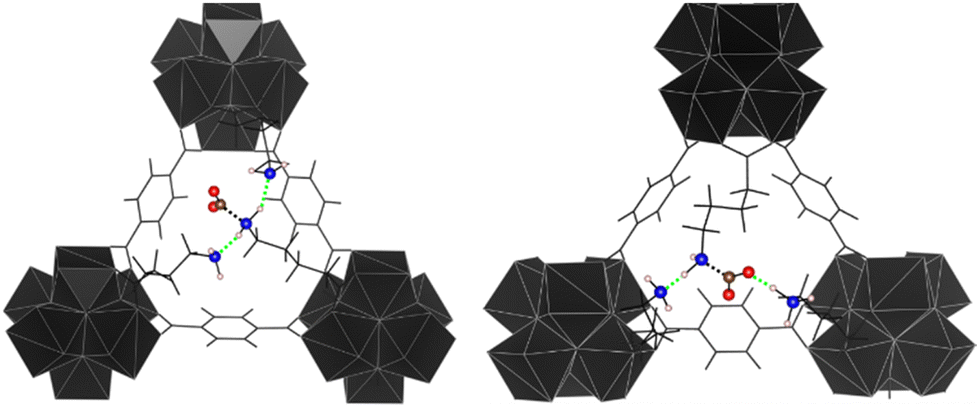 | ||
| Fig. 6 Example of configuration in which CO2 has been activated. On the left CO2 adsorbed in UiO-66_ava. On the right CO2 adsorbed in the amine protonated UiO-66_ava_HOUT_5. Atom colours: N = blue, C = brown, O = red, H = beige. Hydrogen bonds are highlighted with green dashed lines, N C interaction black dashed line. | ||
To be noted that, as for the non-protonated case, a degree of functionalisation of at least 5/12 (42%) of the defective sites in the pore is necessary for having at least one corner with two pendant amine groups. The results further corroborate the idea that a cooperative effect between amine groups is necessary to activate CO2. The adsorption energies are smaller for all the configurations with only two amines and no chemisorption can occur when there is only one amino acid (corresponding to a functionalisation of ca. 8% of the pore, Table S10, ESI†).
Carbamic acid formation in protonated systems
Following the same methodology as proposed before, we simulated the formation of the carbamic acid towards the double hydrogen transfer from the nitrogen atom in the carbamate to a neighbour aliphatic amine and from the NH3+ moiety to one of the O atoms of CO2 (Fig. 1, Step 2). Given the known lability of the zwitterionic Lewis adduct,26 we expected the system to become more stable upon proton transfer. As described earlier, CO2 activation leading to the carbamate structure is over-stabilized here due to a hydrogen bonding with the CO2. The mechanism is similar to the one explained above, although here the proton transfer involves the transfer from the NH3+ to the CO2 (see Fig. 7A).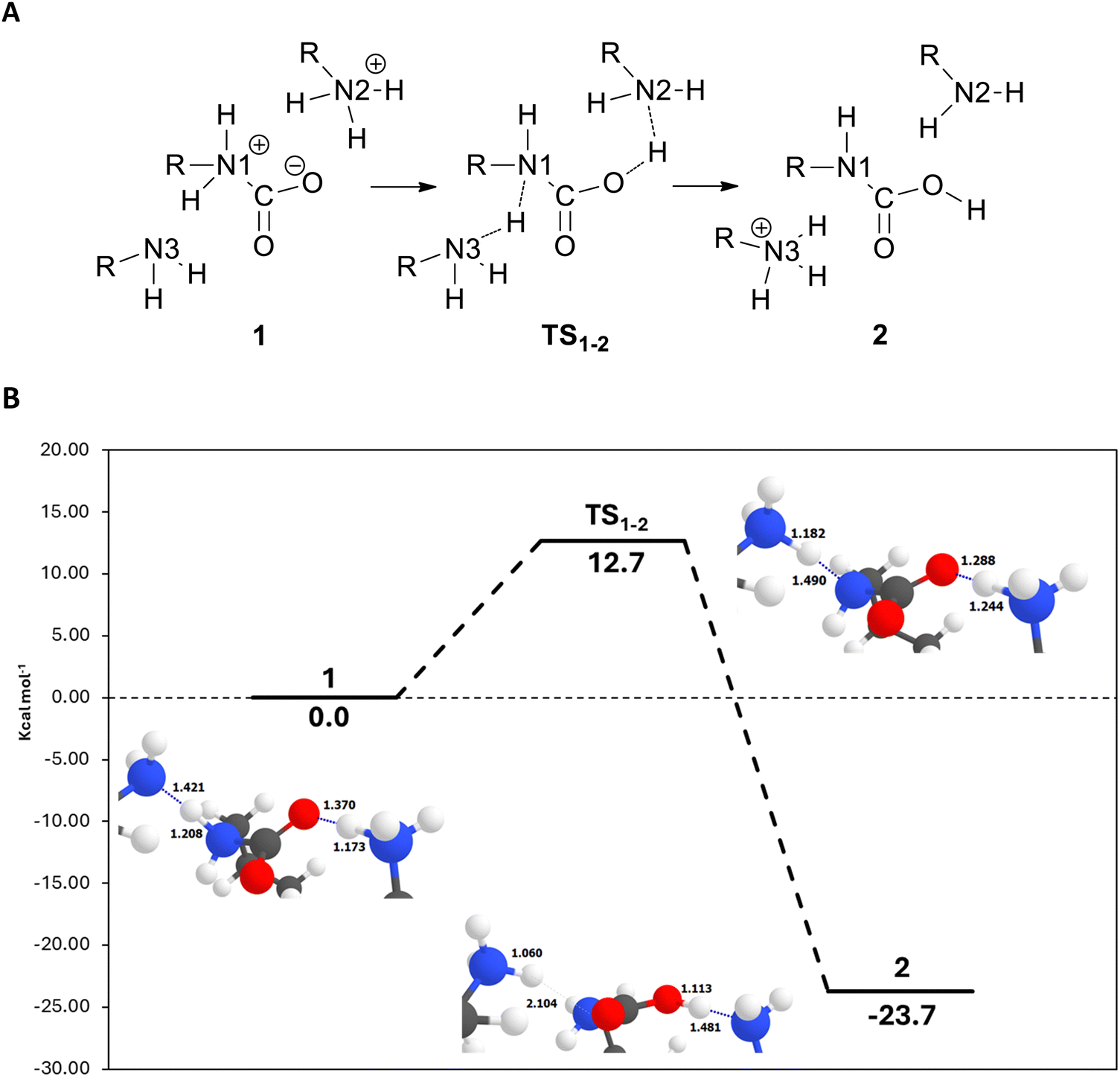 | ||
| Fig. 7 Panel (A): proposed reaction mechanism that involves only one transition state for the formation of the carbamate acid. Panel (B): potential energy surface (PES) for the proton transfer mechanism leading to the formation of the carbamate acid for the system UiO-66_ava_HOUT_6. Relative energies are in kcal mol−1. Relevant distances are in angstroms (Å). Hydrogen bonds are represented as dotted lines. Colour scheme: C (grey), H (white), O (red), N (blue) and Zr (cyan). | ||
Interestingly, protonated systems lead to more stable products (a similar observation was reported for the amine-functionalised expanded MOF-74 analogue when working in dry conditions),8 however the energetic barrier is substantially higher than the previously studied mechanism (to be noted that, to save computational resources, we only calculated one activation energy per proposed system as done in the sections before). Here, we could consider as a rule of thumb that the activation of CO2 would lead to the carbamic acid formation.
Finally, there is one last question to be answered yet: would it be possible to get that proton from the NH3+ back to the metal node? As expected, vacuum calculations show that the restoration of the OH moiety in the metal node is energetically favourable, as the neutral products are more stable than the zwitterion ones (NH3+ + O−) (see Tables S11 and S12, ESI†). Nevertheless, we acknowledge that, in the real case of a MOF exposed to a dry stream containing CO2, the proton back transfer to the inorganic cluster might as well not be realised due to the lack of a suitable proton carrier, such as the solvent during the initial proton transfer from the cluster to the amine group occurring during synthesis.
Conclusions
To conclude, a computationally aided design procedure was used to engineer the internal surface of UiO-66 to promote CO2 activation. The internal surface was decorated functionalising missing cluster defects with aliphatic amino acids with increasing chain length: glycine (gly), beta-alanine (ala), gamma-aminobutyric acid (gaba) and 5-aminovaleric acid (ava). It was found that, to promote CO2 activation, hydrogen bonds are required between the different amine groups (cooperative effect). This is possible only for the longer-chain residues gaba and ava. For the former, at least 2/3 of the same corner of the pore must be functionalised (i.e., at least 5 out of 12 defective sites within a unit cell), which corresponds to a degree of functionalisation higher than 42%. For the latter, being longer, only two sites out of 12, corresponding to a degree of functionalisation of 16%, may be sufficient within the pore to chemically adsorb and activate CO2. With these concepts in mind, the synthesis of UiO-66_gaba and UiO-66_ava, and the experimental verification of the reaction, are under progress.Data availability
The data supporting this article have been included as part of the ESI.† The code used for the calculations can be found at https://www.cp2k.org/ with DOI: https://doi.org/10.1063/5.0007045. The version of the code employed for this study is version 9.1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication has emanated from research conducted with the financial support of Science Foundation Ireland under Grant No. 12/RC/2275_P2. G. P. thanks the Irish Research Council that funded his PhD. D. T. thanks the University of Pisa for a Visiting Fellow grant that allowed his stay at the University of Pisa. Science Foundation Ireland (SFI) for financial support under grant number 12/RC/2275_P2 (SSPC). The computing resources was provided by the SFI/Higher Education Authority Irish Centre for High-End Computing (ICHEC).References
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef PubMed.
- L. B. Hamdy, C. Goel, J. A. Rudd, A. R. Barron and E. Andreoli, Mater. Adv., 2021, 2, 5843–5880 RSC.
- R. Ben Said, J. M. Kolle, K. Essalah, B. Tangour and A. Sayari, ACS Omega, 2020, 5, 26125–26133 CrossRef PubMed.
- P. V. Kortunov, M. Siskin, L. S. Baugh and D. C. Calabro, Energy Fuels, 2015, 29, 5940–5966 CrossRef.
- E. F. Da Silva and H. F. Svendsen, Ind. Eng. Chem. Res., 2004, 43, 3413–3418 CrossRef.
- R. W. Flaig, T. M. Osborn Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef PubMed.
- A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gándara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef CAS.
- J. S. Yeon, W. R. Lee, N. W. Kim, H. Jo, H. Lee, J. H. Song, K. S. Lim, D. W. Kang, J. G. Seo, D. Moon, B. Wiers and C. S. Hong, J. Mater. Chem. A, 2015, 3, 19177–19185 RSC.
- A. Lan, K. Li, H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. Hong and J. Li, Angew. Chem., 2009, 121, 2370–2374 CrossRef.
- L. J. Li, P. Q. Liao, C. T. He, Y. S. Wei, H. L. Zhou, J. M. Lin, X. Y. Li and J. P. Zhang, J. Mater. Chem. A, 2015, 3, 21849–21855 RSC.
- H. Lyu, O. I. F. Chen, N. Hanikel, M. I. Hossain, R. W. Flaig, X. Pei, A. Amin, M. D. Doherty, R. K. Impastato, T. G. Glover, D. R. Moore and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 2387–2396 CrossRef CAS PubMed.
- O. I. F. Chen, C. H. Liu, K. Wang, E. Borrego-Marin, H. Li, A. H. Alawadhi, J. A. R. Navarro and O. M. Yaghi, J. Am. Chem. Soc., 2024, 146, 2835–2844 CrossRef CAS.
- G. C. Shearer, J. G. Vitillo, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 7190–7193 CrossRef CAS.
- A. Koutsianos, E. Kazimierska, A. R. Barron, M. Taddei and E. Andreoli, Dalton Trans., 2019, 48, 3349–3359 RSC.
- M. Taddei, G. M. Schukraft, M. E. A. Warwick, D. Tiana, M. J. McPherson, D. R. Jones and C. Petit, J. Mater. Chem. A, 2019, 7, 23781–23786 RSC.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. Vandevondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- M. Taddei, D. Tiana, N. Casati, J. A. Van Bokhoven, B. Smit and M. Ranocchiari, Phys. Chem. Chem. Phys., 2017, 19, 1551–1559 RSC.
- S. Goedecker and M. Teter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef PubMed.
- G. Lippert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–488 CrossRef.
- O. Andreussi, I. Dabo and N. Marzari, J. Chem. Phys., 2012, 136, 064102 CrossRef PubMed.
- V. Ásgeirsson, B. O. Birgisson, R. Bjornsson, U. Becker, F. Neese, C. Riplinger and H. Jónsson, J. Chem. Theory Comput., 2021, 17, 4929–4945 CrossRef.
- X. Wu, Z. Bao, B. Yuan, J. Wang, Y. Sun, H. Luo and S. Deng, Microporous Mesoporous Mater., 2013, 180, 114–122 CrossRef.
- T. G. Grissom, D. M. Driscoll, D. Troya, N. S. Sapienza, P. M. Usov, A. J. Morris and J. R. Morris, J. Phys. Chem. C, 2019, 123, 13731–13738 CrossRef.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC.
- F. G. Cirujano and F. X. Llabrés I Xamena, J. Phys. Chem. Lett., 2020, 11, 4879–4890 CrossRef.
- K. Li, J. D. Kress and D. S. Mebane, J. Phys. Chem. C, 2016, 120, 23683–23691 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03223c |
| This journal is © the Owner Societies 2024 |