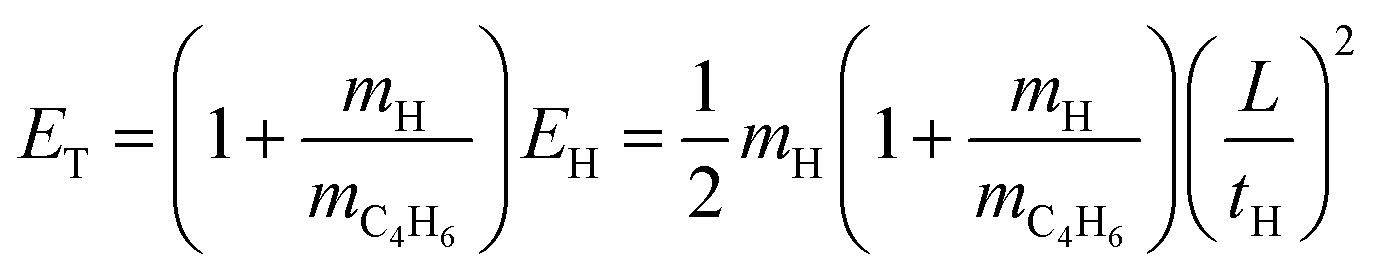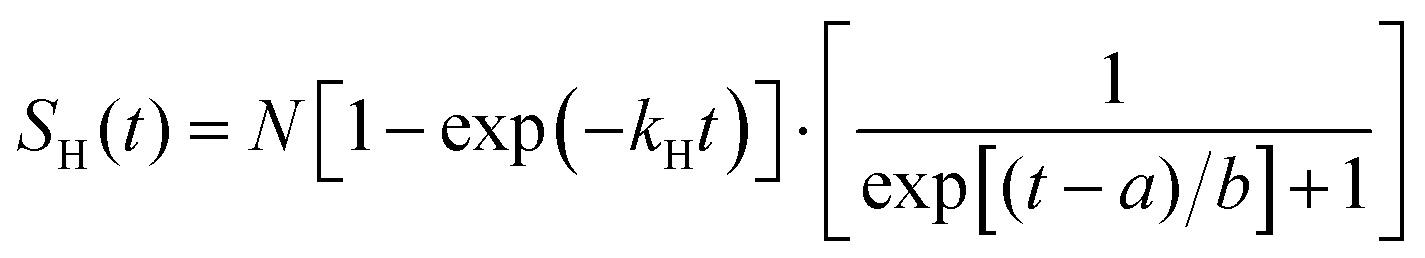Ultraviolet photochemistry of the 2-buten-2-yl radical†
Received
3rd August 2024
, Accepted 12th October 2024
First published on 15th October 2024
Abstract
The ultraviolet (UV) photodissociation dynamics of the 2-buten-2-yl (C4H7) radical were studied using the high-n Rydberg atom time-of-flight (HRTOF) technique in the photolysis region of 226–246 nm. 2-Buten-2-yl radicals were generated by 193 nm photodissociation of the precursor 2-chloro-2-butene. The H-atom photofragment yield (PFY) spectrum of 2-buten-2-yl is broad, peaking at 234 nm. Quantum chemistry calculations show that the UV absorption is due to the 3py and 3px Rydberg states (parallel to the plane of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond). The translational energy distributions of the H-atom loss product channel, P(ET)'s, of 2-buten-2-yl show a bimodal distribution indicating two dissociation pathways. The major pathway peaks at ET ∼ 7 kcal mol−1 with a nearly constant fraction of average ET in the total excess energy, 〈fT〉, at ∼0.11–0.12. This main pathway has an isotropic product angular distribution with β ∼ 0, consistent with the unimolecular dissociation of a hot 2-buten-2-yl radical following internal conversion from the electronically excited state, resulting in the formation of 2-butyne + H (∼84%) and 1,2-butadiene + H (∼16%). Additionally, there is a minor non-statistical pathway with an isotropic angular distribution. The minor pathway peaks at ET ∼ 35 kcal mol−1 in the P(ET) distributions and exhibits a large 〈fT〉 of ∼0.40–0.46. This fast pathway suggests a direct dissociation of the methyl H-atom on a repulsive excited state surface or on the repulsive part of the ground state surface, forming 1,2-butadiene + H. The fast/slow pathway branching ratio is in the range of 0.03–0.08.
C double bond). The translational energy distributions of the H-atom loss product channel, P(ET)'s, of 2-buten-2-yl show a bimodal distribution indicating two dissociation pathways. The major pathway peaks at ET ∼ 7 kcal mol−1 with a nearly constant fraction of average ET in the total excess energy, 〈fT〉, at ∼0.11–0.12. This main pathway has an isotropic product angular distribution with β ∼ 0, consistent with the unimolecular dissociation of a hot 2-buten-2-yl radical following internal conversion from the electronically excited state, resulting in the formation of 2-butyne + H (∼84%) and 1,2-butadiene + H (∼16%). Additionally, there is a minor non-statistical pathway with an isotropic angular distribution. The minor pathway peaks at ET ∼ 35 kcal mol−1 in the P(ET) distributions and exhibits a large 〈fT〉 of ∼0.40–0.46. This fast pathway suggests a direct dissociation of the methyl H-atom on a repulsive excited state surface or on the repulsive part of the ground state surface, forming 1,2-butadiene + H. The fast/slow pathway branching ratio is in the range of 0.03–0.08.
Introduction
The C4H7 radicals are prevalent intermediates in combustion environments, including combustion of isobutene and tert-butyl ethers. In the perspective of astrochemistry, C4H7 radicals also play a critical role in the reaction between CH and propylene, where the production of 1,3-butadiene is pivotal for the formation of mono- and bicyclic aromatic molecules in various extraterrestrial environments.
Among the C4H7 isomers, 1-methylallyl (CH2CH–˙CH–CH3, 1-MA) and 2-methylallyl (CH2C(CH3)–˙CH2, 2-MA) radicals are the most researched due to their resonance-stabilized nature. Various spectroscopic studies have extensively examined both radicals.1–19 The first ultraviolet (UV) absorption spectra of 2-MA were measured in 220–260 nm,1 with a primary feature peaking at 238 nm.4,10 The 1 + 1 resonance-enhanced multiphoton ionization (REMPI) spectra of 2-MA revealed a prominent band at 260 nm, assigned to the ![[B with combining tilde]](https://www.rsc.org/images/entities/char_0042_0303.gif) 3s Rydberg excited state (2A1),3,8,11,14,20 and a broad absorption starting at ∼240 nm attributed to transitions to the 3pz and/or 3px Rydberg states.21 For 1-MA, a broad absorption feature was observed in 226–238 nm, peaking at 237.8 nm.1 The 2 + 1 REMPI spectra of cis and trans-1-MA radicals centered at 472.5 nm for cis and ∼470 nm for trans, indicating resonance to the 2B1 (3p) Ryberg state.5 The 1 + 1′ REMPI and H-atom photofragmentation yield spectra of 1-MA at the à ←
3s Rydberg excited state (2A1),3,8,11,14,20 and a broad absorption starting at ∼240 nm attributed to transitions to the 3pz and/or 3px Rydberg states.21 For 1-MA, a broad absorption feature was observed in 226–238 nm, peaking at 237.8 nm.1 The 2 + 1 REMPI spectra of cis and trans-1-MA radicals centered at 472.5 nm for cis and ∼470 nm for trans, indicating resonance to the 2B1 (3p) Ryberg state.5 The 1 + 1′ REMPI and H-atom photofragmentation yield spectra of 1-MA at the à ← ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) transition were investigated between 23
transition were investigated between 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 cm−1 and 24600 cm−1.12
900 cm−1 and 24600 cm−1.12
The photodissociation dynamics of the two methylallyl radicals have been investigated at different wavelengths.11,12,14,22,23 For the 3s state of 2-MA radical at 258 nm, H-loss dissociation mainly occurred on the ground state following internal conversion, producing methylenecyclopropane + H, with possible isomerization to 1-MA forming 1,3-butadiene + H.11 In the 226–244 nm UV region, 2-MA dissociation via the 3p Rydberg state showed bimodal H-atom product translational energy and angular distributions.22 The slow H-atom product with isotropic angular distribution (anisotropy parameter β ≈ 0) resulted from internal conversion followed by dissociation of vibrationally excited hot 2-MA radical on the electronic ground state to H + methylenecyclopropane, while the faster H-atom product showing a slightly anisotropic angular distribution (β ≈ −0.2) likely arose from direct H-atom loss from the 3p Rydberg state or a repulsive part of the ground state to the 1,3-butadiene + H products. For 1-MA, at the à ← transition (23900–24600 cm−1),12 the main H-loss product channel was 1,3-butadiene + H. UV photodissociation of 1-MA at 226–244 nm revealed two H-loss channels: a slow, isotropic channel indicating statistical dissociation to 1,3-butadiene + H on the ground state after internal conversion, and high-energy, anisotropic products (β ≈ −0.23) from non-statistical dissociation, occurring either on the excited state or within a repulsive ground state region, possibly involving a conical intersection, leading to 1,3-butadiene + H formation.23
Compared to 1-MA and 2-MA, the studies on other C4H7 isomers are limited. McCunn et al. and Miller et al. investigated the secondary dissociation of 2-buten-2-yl (CH3–˙CCH–CH3) and 1-buten-2-yl (CH2˙C–CH2–CH3) from the 193 nm photodissociation of 2-chloro-2-butene and 2-bromo-1-butene, respectively.24,25 Following the loss of Cl from 2-chloro-2-butene, 2-buten-2-yl exhibits three competing dissociation channels: the loss of CH3 resulting in propyne + CH3, H-atom loss leading to 1,2-butadiene + H, or 2-butyne + H formation. The CH3 loss channel exhibited a significant preference in production compared to the H-atom loss channels. However, according to the Rice–Ramsperger–Kassel–Marcus (RRKM) calculations, the significance of H-atom loss channels increases with higher internal energy.24 Similar to the findings for 2-buten-2-yl, the secondary photodissociation of 1-buten-2-yl was primarily characterized by the prevalence of the CH3 loss channel, resulting in the formation of CH3 + allene. Additionally, 1-buten-2-yl also exhibited the potential to undergo H-atom loss, leading to the formation of 1-butyne + H or 1,2-butadiene + H.25 Experiments conducted with synchrotron radiation vacuum ultraviolet (VUV) photoionization in the high-energy range of 9.6–10.6 eV for 1-buten-2-yl revealed a notable propensity for a direct H-atom loss from the excited state. This process was found to be considerably more favorable, leading to the formation of 1,2-butadiene + H.25
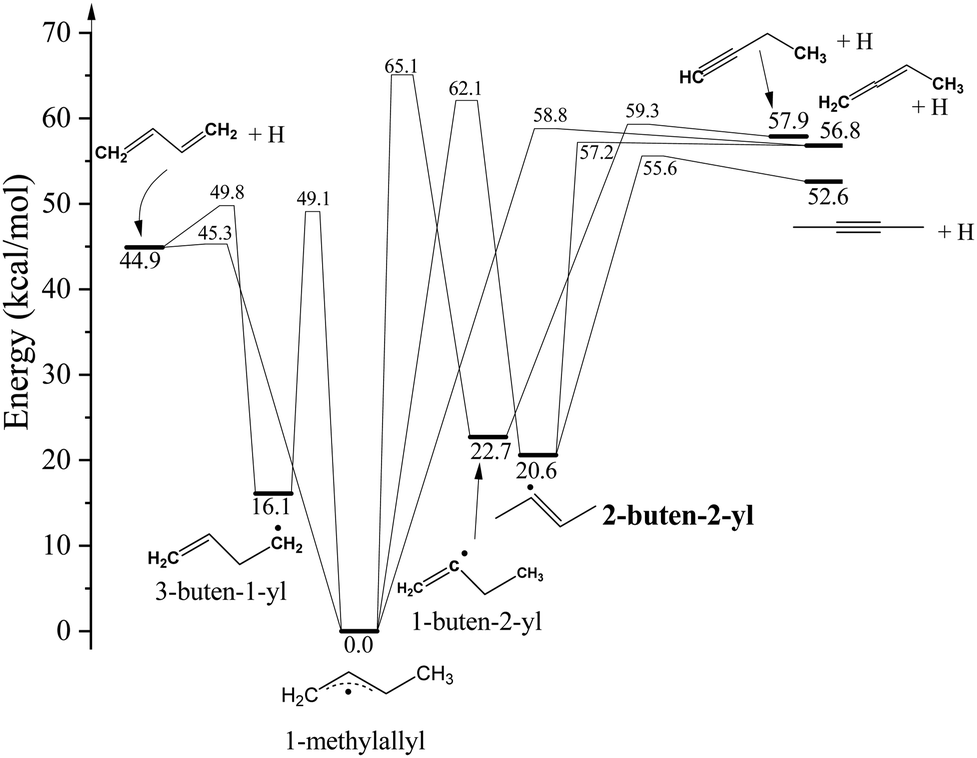
As 2-buten-2-yl acts as the intermediate in the unimolecular dissociation pathways of 1-MA to the 2-butyne + H and 1,2-butadiene + H channels, it shares the same H-loss dissociation channels as 1-MA (Fig. 1).11,26–31 The energy data in this paper were taken from Miller's work,26 which includes all the involved species and are close to the Active Thermochemical Tables (ATcT) database (difference less than 0.8 kcal mol−1).32 The lower energetic channel of 2-buten-2-yl is the loss of a β H-atom to form 2-butyne + H with a barrier of 35.0 kcal mol−1. The other H-atom product channel is the formation of 1,2-butadiene + H from the C–H bond fission of the terminal β H-atom (with a barrier of 36.6 kcal mol−1). After isomerization to 1-MA over a relatively high barrier (41.5 kcal mol−1), it can undergo the direct C–H bond fission of the methyl H to form 1,3-butadiene + H, or the direct loss of the central allylic H-atom producing 1,2-butadiene + H. In addition, 1-MA can further isomerize to 3-buten-1-yl (CH2CH–CH2–˙CH2) through a 1,2-hydrogen shift from the methyl group, or undergo a 1,2-hydrogen shift from the allyl backbone and isomerize to the 1-buten-2-yl radical. A second pathway to the 1,3-butadiene + H products is via dissociation of the 3-buten-1-yl radical. The isomer 1-buten-2-yl can undergo the H-atom loss to form 1,2-butadiene + H, or the loss of the terminal β H-atom resulting in the formation of 1-butyne + H.
| 2-buten-2-yl + hν → 2-butyne + H ΔHrxn,0 = 32.0 kcal mol−1 |
| → 1,2-butadiene + H ΔHrxn,0 = 36.2 kcal mol−1 |
| → 1,3-butadiene + H ΔHrxn,0 = 24.3 kcal mol−1 |
| → 1-butyne + H ΔHrxn,0 = 37.3 kcal mol−1 |
In addition to the H-atom product channels mentioned above, there are also some methyl loss channels that can compete with the H-atom loss. For instance, 2-buten-2-yl can undergo CH
3 elimination, leading to the formation of propyne + CH
3. This product channel is 8.5 kcal mol
−1 lower than the formation of 2-butyne + H, while both pathways have approximately equal transition state barriers.
26 For the 1-buten-2-yl isomer, the formation of allene + CH
3 can occur through a C–C bond fission, with a transition state barrier that is ∼15 kcal mol
−1 lower than the transition state barrier for the production of 1,2-butadiene + H and 1-butyne + H.
26
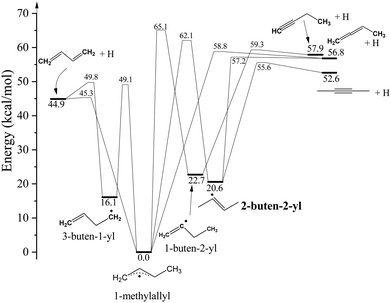 |
| | Fig. 1 Potential energy diagram of C4H7 dissociation pathways. The energetics and pathways are based on the theoretical calculations in ref. 26. | |
Based on the earlier report on the decomposition of 2-buten-2-yl, which focused on the secondary decomposition of 2-chloro-2-butene with 193 nm radiation to produce hot 2-buten-2-yl radical in the ground electronic state,24 the current study explores the UV photodissociation dynamics of jet-cooled 2-buten-2-yl radicals in the region of 226–246 nm using the high-n Rydberg time-of-flight (HRTOF) technique. In this work, the H-atom photofragment yield (PFY) spectra of 2-buten-2-yl radical were obtained. The center-of-mass (CM) translational energy distributions and the angular distributions of the H + C4H6 products were obtained from the H-atom TOF spectra. Additionally, this work reports the non-statistical H-atom photodissociation dynamics of the 2-buten-2-yl radical for the first time.
Experimental methods
The details of the HRTOF technique and experimental setup have been described in previous studies.33–36 To generate the 2-buten-2-yl radical, the precursor 2-chloro-2-butene (cis isomer, >97%, TCI) was employed.24 A pulsed beam of 2-buten-2-yl radicals was generated by photolyzing a ∼2% mixture of the precursor seeded in He (at a total pressure of ∼120 kPa) with 193 nm radiation from an ArF excimer laser. The production of the 2-buten-2-yl radical beam was characterized by the 121.6 nm vacuum ultraviolet (VUV) photoionization TOF mass spectrometry (TOFMS). Following the generation from the precursor, 2-buten-2-yl radicals underwent photodissociation under a slightly focused UV photolysis laser radiation (at 226–246 nm, 0.25–1.5 mJ per pulse, linewidth 0.3 cm−1). For the measurements of the angular distribution of the H-atom products, a Fresnel-Rhomb achromatic λ/2 plate was utilized to rotate the polarization of the photolysis radiation. To tag the H atoms produced from the photodissociation of 2-buten-2-yl, a two-color resonant excitation process was employed. This involved transitioning the H atoms from the 12S to 22P state through the H-atom Lyman-α transition at 121.6 nm, followed by further excitation to a high-n Rydberg state using UV radiation at 366.3 nm. A minor portion of the metastable Rydberg H atoms drifted with their nascent velocities toward a microchannel plate (MCP) detector, which was oriented perpendicular to the molecular beam. Subsequently, these atoms underwent field ionization in front of the detector and were then detected. The nominal flight length was calibrated to be 37.2 cm. The ion signals were amplified by a fast preamplifier, and the H-atom TOF spectra were recorded and averaged using a multichannel scaler. The recorded TOF spectra were based on a variable number of laser shots, ranging from 100 to 500k.
Theoretical methods
The equation of motion coupled cluster with single and double excitations (EOM-CCSD) calculations by Koziol et al.37 indicated that the vertical excitation energies of several electronic transitions of 2-butene-2-yl lie near the UV region covered in this study: π* ← n (5.10 eV), π* ← π (5.62 eV), 3s ← n (5.64 eV), 3py ← n (5.64 eV), and 3px ← n (5.64 eV). To complement previous theoretical findings,37 we investigated the electronic transitions from the ground state to excited states of trans-2-buten-2-yl utilizing a range of quantum chemistry methodologies. Initially, ground state structures of the trans-2-buten-2-yl were optimized employing CCSD/aug-cc-pVDZ. Subsequently, vertical excitation energies from the ground state to the eight lowest excited states were computed with the benchmark method EOM-CCSD38 and time-dependent density functional theory (TDDFT, CAM-B3LYP39 for comparison only), with various basis sets (including d-aug-cc-pVXZ, X = D, T, Q).40 To offer a qualitative understanding of the electronic excitations, the natural transition orbitals (NTOs)41 were analyzed with CAM-B3LYP/6-31++G. All the ab initio calculations were conducted using Gaussian 16, with optimized geometries, excitation energies, transition dipole moments and NTO analysis results presented in ESI† (Tables S1–S5 and Fig. S2).
Results
Fig. 2 displays the net VUV photoionization mass spectrum of the 2-chloro-2-butene precursor. The TOF mass spectrum is the difference between the spectrum obtained during radical production photolysis with 193-nm radiation on and off. The production of 2-buten-2-yl is illustrated by the peak at 55 m/z, and the depletion of the 2-chloro-2-butene precursor is shown at 90 and 92 m/z. The peak at 54 m/z is attributed to a close–shell C4H6 byproduct. However, it is anticipated that this byproduct will not interfere with the H-atom photodissociation signal of the 2-buten-2-yl radical in the experiment, due to the much higher C–H bond strength in the close–shell C4H6 and thus their low H-atom product translational energy (<20–30 kcal mol−1) and, more importantly, the much lower absorption cross sections of the C4H6 species compared with the 2-butene-2yl radical (estimated to be 102–103 times smaller at the UV wavelength of interest).
 |
| | Fig. 2 121 nm VUV photoionization mass spectrum of the 2-buten-2-yl radical molecular beam using 2-chloro-2-butene precursor in the He carrier gas. The net spectrum (b) was obtained by subtracting the data with 193 nm photolysis laser off from the spectrum with 193 nm on in (a). The main product is 2-buten-2-yl at m/z = 55 amu, while the 2-chloro-2-butene parent peak at m/z = 90 and 92 amu (the positive intensity was probably due to imperfect measurements and subtraction of the large parent peaks). | |
The TOF spectra of the H-atom products from the photodissociation of 2-buten-2-yl were recorded in the photolysis wavelength range of 226–246 nm, with the polarization of the photolysis laser set both parallel and perpendicular to the flight path. It was determined that the primary background comprised H-atom products originating from the photodissociation of the 2-chloro-2-butene precursor (see details in Fig. S1 in the ESI†), and this background has been properly removed in the H-atom TOF spectra.33,36,42,43 The H-atom TOF spectrum for 2-buten-2-yl at 236 nm is shown in Fig. 3. The spectrum displays a broad feature with a peak around 37 μs and a preceding shoulder at ∼20 μs. The TOF spectra at the other wavelengths in this region exhibit a similar pattern, characterized by a broad feature accompanied by a shoulder on the earlier time side of the broad feature. The sharp peaks at ∼25 μs in Fig. 3(b) at the perpendicular polarization are caused by photodissociation of HCl, which is a byproduct of photodissociation of the 2-chloro-2-butene precursor. Fig. 4 shows the H-atom TOF spectra and the power dependence at 236 nm, normalized by the number of laser shots and plotted on the same scale. The TOF signals exhibit a linear dependence on the photolysis laser power, in agreement with a one-photon photodissociation process.
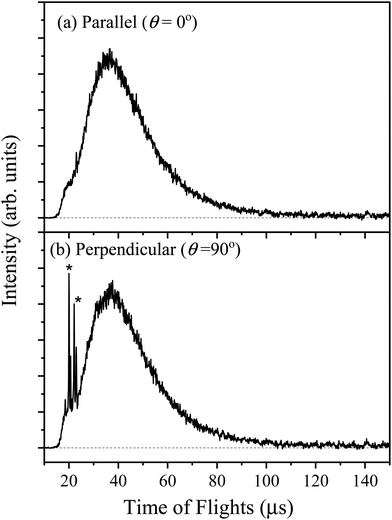 |
| | Fig. 3 H-atom TOF spectrum in the photodissociation of jet-cooled 2-buten-2-yl radical at 236 nm, produced from 193 nm photolysis of 2-chloro-2-butene. This is the net H-atom TOF with the 193 nm photolysis radiation on minus off. The polarization E vector of the 236 nm radiation is parallel (θ = 0°) (a) and perpendicular (θ = 90°) (b) to the TOF axis. The sharp peaks (*) are caused by the photodissociation of HCl, the byproduct from photodissociation of the 2-chloro-2-butene precursor. | |
 |
| | Fig. 4 Photolysis laser power dependence: the H-atom product TOF spectra of the 2-buten-2-yl at 236 nm with photolysis energy 0.4 and 1.2 mJ per pulse. The TOF spectra are normalized by the number of laser shots and are plotted on the same scale. The H-atom product signals have a linear dependence on the photolysis laser power. | |
In Fig. 5, the H-atom PFY spectrum (action spectrum) is presented for the photolysis wavelength range starting from 226 nm to 246 nm. Based on the conformational structure and previous investigations on cis/trans isomerization of 2-chloro-2-butene,44 the primary product from photolysis of cis-2-chloro-2-butene should be trans-2-buten-2-yl. The 2-buten-2-yl radicals were generated with low internal energy from the 193-nm photolysis of the precursor24 and underwent further cooling in the molecular beam. The spectrum was derived by integrating the net H-atom TOF spectra across the photolysis wavelength in this range. The H-atom signals at 236 nm were employed as a reference to monitor experimental drift and for normalization. The resulting action spectrum reveals a broad feature in this region, with a peak occurring around 234 nm. To interpret the observed action spectrum, the excited electronic states of 2-buten-2-yl were calculated using two quantum chemical methods: TDDFT and EOM-CCSD. Detailed calculation results, including the structures of 2-buten-2-yl, excited state energies, and transition dipole moments, can be found in the ESI† (Tables S1–S5 and Fig. S2). The computed excited energies for the eight excited states (D1–D8) of 2-buten-2-yl indicate that the UV excitation observed in the experiments in the region of 226–246 nm can be attributed to the fifth and sixth excited states (D5 and D6). This is mainly due to their much larger oscillator strengths, along with their electronic excitation energies being reasonably close to the observed UV absorption feature (Fig. 5). Several methods and basis were utilized in the above predictions, such as CAM-B3LYP/d-aug-cc-pVDZ, CAM-B3LYP/aug-cc-pVDZ, and EOM-CCSD/d-aug-cc-pVDZ. Specifically, the EOM-CCSD/d-aug-cc-pVTZ method calculates the vertical and adiabatic energy to be 196.7 nm and 237.8 nm for the excited D5 state, and 193.2 nm and 233.4 nm for the D6 state, respectively. The d-aug-cc-pVXZ augments, incorporating 2 shells of each angular momentum, are more accurate for Rydberg excited states compared to the aug-cc-pVXZ augments, which only include 1 shell of each angular momentum. The notable difference between the vertical excitation energy and the adiabatic energy arises from the significant shift in the minimum energy geometry between the ground state and the excited state. The optimization of the electronic state structures in Fig. S2 (ESI†) reveals that the angle of CH3–C–CH is 137.2° in the ground state but shifts to 173.8° at the D5 state. The comparison between the calculated vertical excitation energy and the peak position of the observed absorption feature suggests that the CAM-B3LYP and EOM-CCSD methods overpredict the electronic excitation energies of D5 and D6. Examination of NTOs indicates that both the D5 and D6 states exhibit a predominant 3p Rydberg character, arising from the n → 3py (D5) and n → 3px (D6) transitions (Table S5, ESI†). The associated Rydberg orbitals are located in the plane of the CC double bond, consistent with previous studies on the excited states of trans-2-buten-2-yl conducted by Koziol et al.37 The transition dipole moments from the ground state to the D5 state and to the D6 state are both predicted to lie within the CC plane (Table S5 and Fig. S2, ESI†).
 |
| | Fig. 5 H-atom product yield (PFY) spectrum as a function of photolysis excitation energy in the region of 226–246 nm. The solid squares (■) represents the integrated HRTOF signals using the 2-chloro-2-butene precursor. | |
The net H-atom TOF spectra from the photodissociation of jet-cooled 2-buten-2-yl are converted to the product center-of-mass (CM) translational energy distributions, P(ET)'s. The CM translational energy of the products, ET, is calculated from the H-atom flight time, tH, using the following equation:
| | 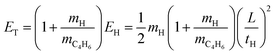 | (1) |
Here,
EH represents the laboratory translational energy of the H-atom photofragment, and
L denotes the length of the TOF path. In
Fig. 6, the distributions of product CM translational energy,
P(
ET), resulting from the photodissociation of 2-buten-2-yl at 236 nm (with polarization parallel (a) and perpendicular (b) to the TOF axis) are presented. The parallel distribution
P‖(
ET) in
Fig. 6(a) is derived from the H-atom TOF spectrum shown in
Fig. 3 utilizing the
eqn (1). The
P(
ET) reveals a moderate release of translational energy, peaking at ∼7 kcal mol
−1 and extending to ∼90 kcal mol
−1. Due to the small intensity, the shoulder in the TOF spectrum is not obvious in the
P(
ET). The product translational energy distributions agree with the maximum available energy for the 2-butyne + H (89.2 kcal mol
−1) product channel. The maximum available energy for other possible H-atom product channels at 236 nm are: 1-butyne + H (83.9 kcal mol
−1), 1,2-butadiene + H (85.0 kcal mol
−1), and 1,3-butadiene + H (96.9 kcal mol
−1). The maximum available energy is determined by subtracting the dissociation energy of the channel from the photon energy used in photolysis, under the assumption that the internal energies of the radicals and the photodissociation fragments are ∼0.
 |
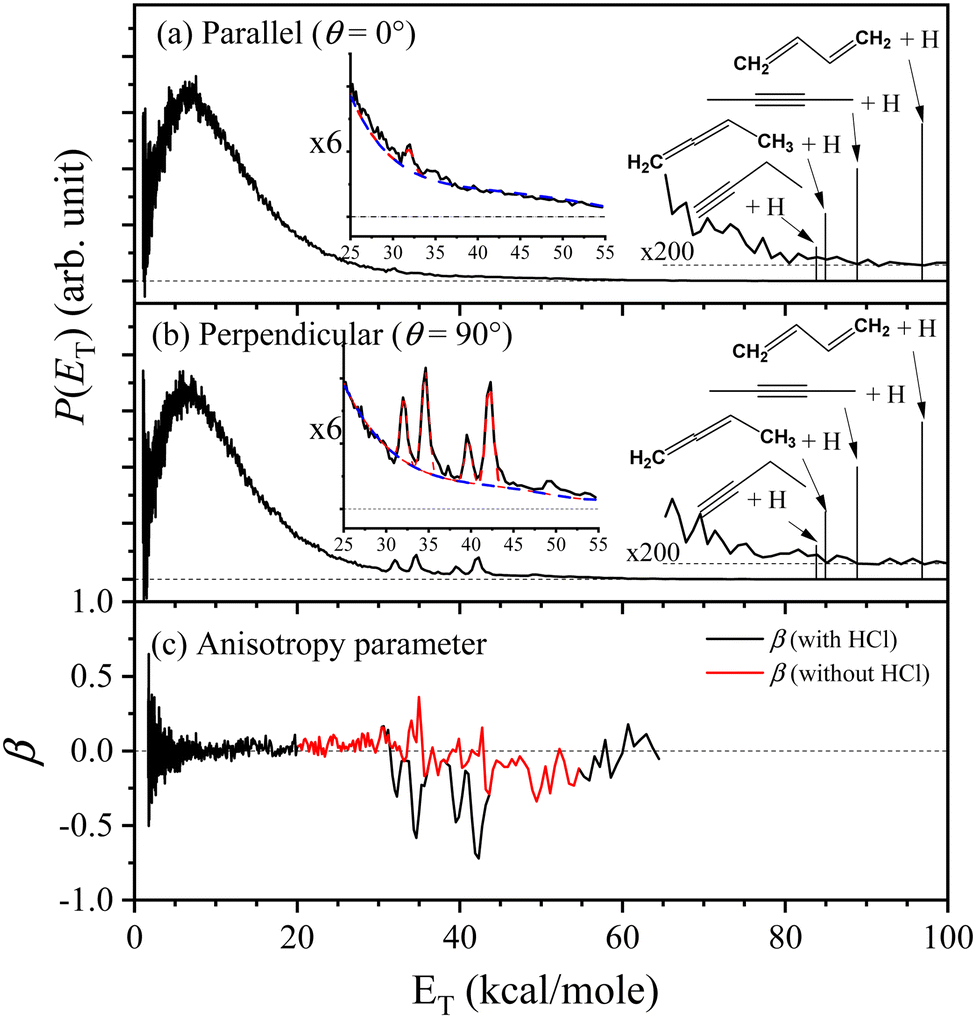
| | Fig. 6 Center-of-mass product translational energy distribution, P(ET), of 2-buten-2-yl radical at 236 nm. The 236 nm radiation is polarized (a) parallel and (b) perpendicular to the TOF axis. The sharp peaks between 35–45 kcal mol−1 are caused by the photodissociation of HCl, which occurs due to H-atom abstraction from the parent 2-chloro-2-butene. The vertical lines in (a) and (b) denote the maximum translational energies associated with the H-atom product channels. Additionally, the anisotropy parameter in (c) is calculated based on data from (a) and (b). See the text for details. | |
Linearly polarized light tends to selectively excite radicals whose electronic transition dipole moment aligns parallel to the electric vector of the polarized radiation. The H-atom product angular distributions in the UV photodissociation of 2-buten-2-yl were studied with the photolysis radiation polarized parallel and perpendicular to the TOF axis. The translational energy distributions, P(ET)'s, depicted in Fig. 6(a) and (b) for both polarizations exhibit a similar pattern, suggesting an isotropic angular distribution. In Fig. 6 (b), under perpendicular polarization, the distinct peaks of P⊥(ET) observed between 30–45 kcal mol−1, situated at the short-time shoulder in the perpendicular TOF spectrum, arise from HCl byproduct in the 193-nm photolysis of the 2-chloro-2-butene precursor. Subsequently, the photodissociation of HCl (v′ = 2–3) contributes to these peaks. The angular distribution of photofragments is described by.
| | | I(θ) = (1/4π)[1 + βP2(cosθ)] | (2) |
where the anisotropy parameter
β is defined within the range of −1 to 2,
θ is the angel between the electric vector of the polarized laser radiation
E and the recoiling velocity vector of the H-atom product (the direction of the TOF axis), and
P2(cos
θ) represents the second Legendre polynomial. By applying this equation to the H-atom
P(
ET) spectra presented in
Fig. 6(a) and (b), the anisotropy parameter
β was determined as
β(
ET) = 2[
P‖(
ET) −
P⊥(
ET)]/[
P‖(
ET) + 2
P⊥(
ET)], displayed in
Fig. 6(c). The derived anisotropy parameter
β reveals two different
β values: the main peak (below 30 kcal mol
−1) exhibits an isotropic distribution with
β ∼ 0, while the fast component (above 30 kcal mol
−1) has a negative
β parameter indicating an anisotropic distribution. For the perpendicular distribution
P⊥(
ET), both dissociation of 2-buten-2-yl and direct dissociation of HCl make contributions to the fast component. Direct dissociation of HCl should give a negative
β value,
45,46 consistent with the sharp peak regions (30–36 kcal mol
−1 and 38–44 kcal mol
−1) of the
β including the HCl signals in
Fig. 6(c). To remove the contributions from direct dissociation of HCl, one and four Gaussian functions were used to fit the peaks in this energy region at the parallel and perpendicular polarization (displayed in the insects in
Fig. 6(a) and (b)), respectively. After removing the contributions from HCl (subtracting the fitted peaks with the baseline removed), the resultant
β (the red line in
Fig. 6(c)) remains approximately 0 (although noisy due to the subtraction procedure) in the region of 30–45 kcal mol
−1. Thus, it can be concluded that dissociation of 2-buten-2-yl has a
β value of ∼0 for both the slow and fast component regions.
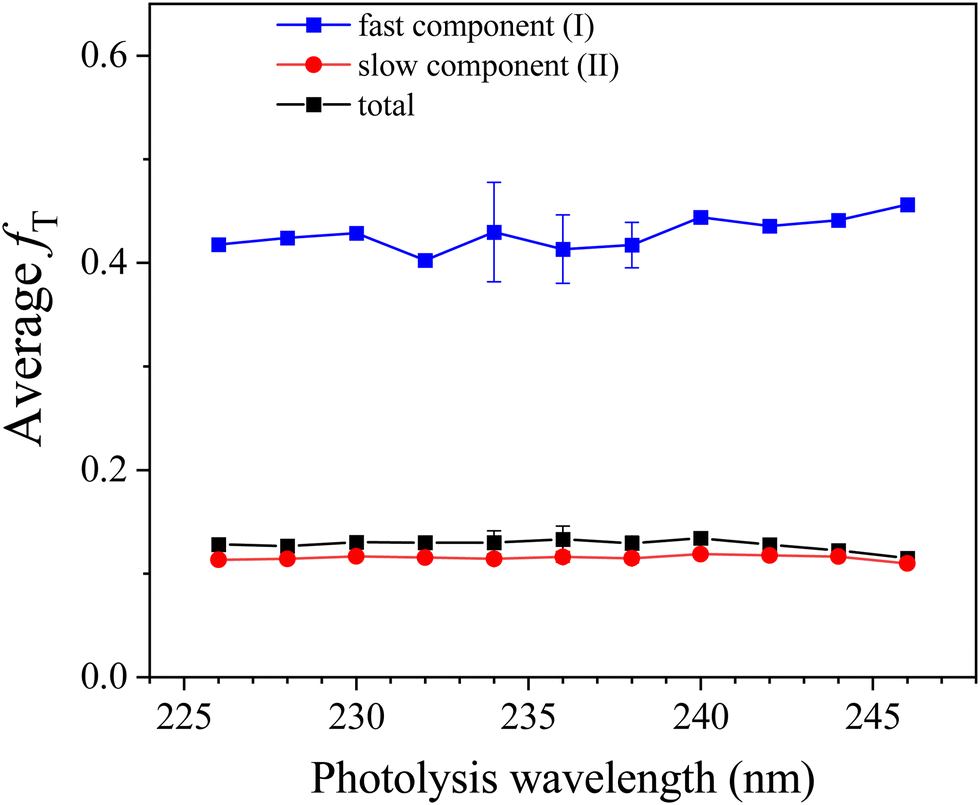
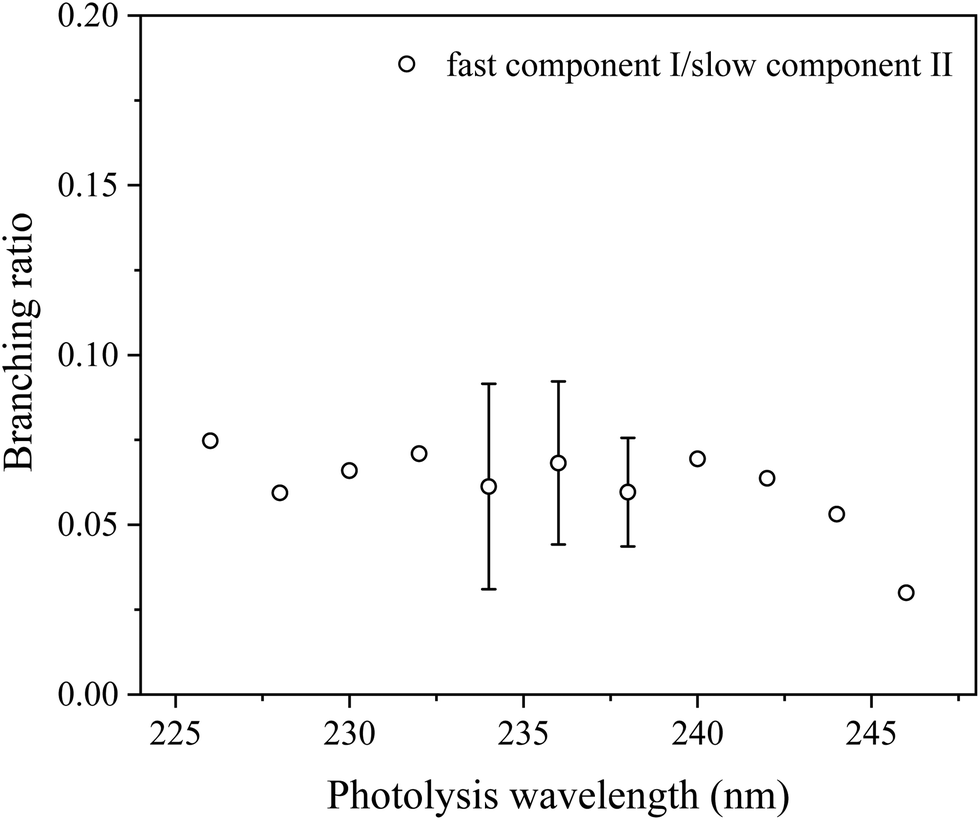
As the dissociation of HCl has little contributions to the parallel distribution P‖(ET) and the parallel H-atom TOF spectrum exhibits a fast shoulder (Fig. 3), it suggests two different photodissociation processes during dissociation of 2-buten-2-yl. To deconvolute the fast and slow components, two Rice–Ramsperger–Kassel (RRK)-type functions, P(ET) = A(ET)p(E0 − ET)q are utilized.47 Compared to the parallel distribution P‖(ET), the signal of the fast component is more prominent in the TOF spectrum. Thus, two reformed RRK functions are employed to separate the parallel TOF spectrum into a fast and slow components part: I(t) = C·P(ET)(ET)3/2. The parameters A, p, q and C are variable, while E0 represents the maximum available energy, which is fixed for the fast component (e.g. 89.2 kcal mol−1 at 236 nm) and can be adjusted for the slow component. The two fitted components (I for the fast one and II for the slow one) are illustrated in Fig. 7. From the fitted components, the average translational energy release, 〈ET〉, and the fraction of the average translational energy release in the total available energy, 〈fT〉, can be calculated for each component. At 236 nm, the fast channel (I) exhibits 〈ET〉 = 35.1 kcal mol−1 and 〈fT〉 = 0.41 (assume the H + 1,2-butadiene channel, discussed later), while the slow channel (II) shows 〈ET〉 = 10.3 kcal mol−1 and 〈fT〉 = 0.12 (assume the H + 2-butyne channel, discussed later). The overall 〈fT〉 value of the H-loss product channel is ∼0.13 at 236 nm, indicating a moderate release of overall translational energy. Fig. 8 displays the 〈fT〉 values of the H-loss product channels across photolysis wavelengths of 226–246 nm. Throughout the studied photolysis wavelength range, the overall 〈fT〉 varies between 0.12 and 0.14. The modest 〈fT〉 values indicate a high level of internal excitation within the C4H6 product, supporting a mechanism of hot radical unimolecular dissociation. The 〈fT〉 of the fast component (I) increases slightly from 0.40 to 0.46 in the wavelength range from 226 nm to 246 nm, with an average of 0.43. The 〈fT〉 of the slow component (II) undergoes slight changes from 0.11 to 0.12 in the 226–246 nm range, with an average of 0.12. The integrated signals of components I and II from the P‖(ET) distribution in Fig. 7 provide an estimated branching ratio of the fast to slow component of ∼0.07 at 236 nm. Similarly, the branching ratios of the fast to slow components at other photolysis wavelengths from 226 nm to 246 nm are also estimated and are summarized in Fig. 9. These branching ratio values vary between 0.03 and 0.08, with an average of 0.06.
 |
| | Fig. 7 The 236-nm photodissociation of 2-buten-2-yl with the polarization of the linearly polarized photolysis radiation parallel (‖, θ = 0°) to the TOF axis in the top panel, deconvolution fitting of the TOF spectrum with a fast component (I, red line) and a slow component (II, blue line). The deconvolution fitting results in the CM translational energy distribution is displayed in the bottom panel. The vertical lines in the bottom panel denote the maximum translational energies associated with the H-atom product channels. See text for more details. | |
 |
| | Fig. 8 Photolysis wavelength and fraction of average translational energy release in the total available energy, 〈fT〉, in the UV photodissociation of the 2-buten-2-yl radical. The average translational energies are calculated from the experimental P(ET) distributions. The total available energy at each photolysis wavelength is derived from the corresponding photon energy and the dissociation energy of 2-buten-2-yl to 2-butyne + H. The error bars (where available) represent the statistical uncertainty (1σ) from multiple measurements. The error scale for the remaining data could be similar to those indicated by the available error bars. | |
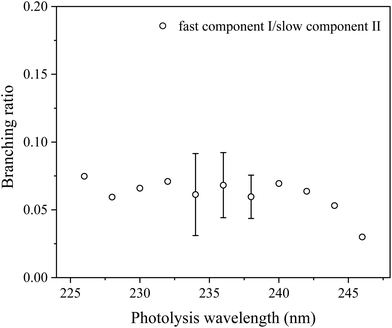 |
| | Fig. 9 Photolysis wavelength and branching ratio of the fast and slow components in the UV photodissociation of the 2-buten-2-yl radical. The intensities of the fast and slow components are obtained from integrating the deconvoluted fast and slow components in the fitted P(ET) distributions (for example, in Fig. 7 for 236 nm). The error bars (where available) represent the statistical uncertainty (1σ) from multiple measurements. The error scale for the remaining data could be similar to the available error bars. | |
The H-atom yield time profile in the UV photolysis of 2-butne-2-yl was investigated by changing the time delay between the photolysis laser and probe laser. Fig. 10 illustrates the H-atom yield time profile at 236 nm photolysis radiation, derived by integrating the HRTOF spectra concerning the photolysis-probe delay time. This profile of delay time offers insights into the microcanonical rate of the unimolecular dissociation of 2-buten-2-yl. The initial rise of the signal represents the rate of the H-atom generation from 2-buten-2-yl, while the signal's decline is attributed to the flight of H-atoms out of the interaction region between the two laser beams. The time profile of the H-atom signals, SH(t), is fitted using an expression derived from prior research conducted by Chen's group48 to estimate the unimolecular dissociation rate of the 2-buten-2-yl radical:
| | 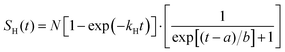 | (3) |
Here,
kH is the unimolecular dissociation rate constant for the H-atom formation from the 2-buten-2-yl radical, and
a and
b are constants that describe the width of the plateau region and the decay of the signal. The fitting (solid line) depicted in
Fig. 10 yields a dissociation rate constant
kH of ∼1.3 × 10
7 s
−1 at 236 nm. As the time resolution of the pump–probe experiment was ∼10 ns (limited by the laser pulse widths), the measurement could not detect rate constant faster than ∼10
8 s
−1. This value of
kH would represent approximately a lower limit of the actual dissociation rate constant.
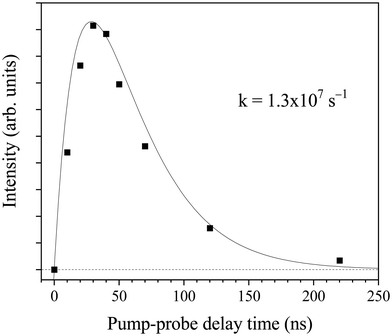 |
| | Fig. 10 H-atom product signal as a function of photolysis and probe delay time at 236 nm. The signals are obtained by integrating the HRTOF spectra at the various photolysis-probe delay times. | |
Discussion
The UV photodissociation of the jet-cooled 2-buten-2-yl radical was studied in the range of 226–246 nm for the first time. The C4H6 + H dissociation channels were directly observed in the H-atom TOF spectra. The TOF spectrum from the 2-chloro-2-butene precursor, as shown in Fig. 3, recorded at 236 nm for 2-buten-2-yl, displays a broad peak at ∼37 μs, along with a minor peak near ∼20 μs. The action spectrum (Fig. 5) displays a broad feature within the UV photolysis range of 226–246 nm, with a peak at ∼234 nm. No UV absorption spectrum for the 2-buten-2-yl radical has been reported prior to this study. This research presents, for the first time, the UV absorption feature of the 2-buten-2-yl radical. If the main dissociation channel involved the H-atom product, the H-atom PFY spectrum would reflect the UV absorption spectra. However, this may not be the case due to the energetically favored CH3 product channels predicted by Miller26 and experimentally reported by McCunn et al.24 Our electronic state calculations show that the D5 and D6 states are likely the excited states involved in the photolysis range of 226–246 nm, due to their much larger oscillator strengths than other excited states and their excitation energies being reasonably close to the observed absorption feature.
The TOF spectra in the region of 226–246 nm exhibits a similar pattern, characterized by a broad peak with a shoulder appearing at shorter flight time. As shown in Fig. 7, after conversion from the TOF spectrum at 236 nm, the CM translational energy distribution of the H-atom product channel, P(ET), exhibits a bimodal distribution. This observation implies the presence of two distinct dissociation pathways in the photodissociation of 2-buten-2-yl. However, the angular distribution is nearly isotropic for both the fast and slow component in Fig. 6. The larger (slow) feature peaks at ∼7 kcal mol−1 with a small translational energy release (〈fT〉 ∼ 0.12 in Fig. 8). This low ET energy, broad distribution is characteristic of a statistical dissociation mechanism. The isotropic angular distribution of the slow component (II) suggests that this H-loss channel of 2-buten-2-yl has a dissociation time scale exceeding one rotational period of the 2-buten-2-yl radical (∼10 ps using the predicted rotational constants by Miller26). This time scale is again consistent with unimolecular dissociation of the hot radical, which involves the dissociation of 2-buten-2-yl from a highly vibrational ground electronic state following internal conversion from the excited D5 and D6 (the 3p Rydberg orbitals in the carbon skeleton plane) states. The preferred product of this mechanism is the pathway with the lowest energy transition state barrier. In the case of 2-buten-2-yl, the lowest energy H-loss route involves the direct release of the β H-atom, forming the 2-butyne + H product (with a barrier of 35 kcal mol−1). Slightly higher in energy barrier (∼36 kcal mol−1), the direct loss of a β H-atom from the methyl group, leading to the production of the 1,2-butadiene + H, would compete at the studied photolysis energies (116.2–126.5 kcal mol−1). Additionally, 2-buten-2-yl can undergo a 1,2-hydrogen shift to isomerize to 1-MA, requiring 42 kcal mol−1 to overcome the barrier and most likely resulting in the generation of 1,3-butadiene + H. McCunn et al.'s prior RRKM rate calculations for the ground state 2-buten-2-yl radical,24 with internal energy ranging from 32 to 54 kcal mol−1, indicated that direct dissociation to CH3 + propyne prevails (>90%) over other dissociation reactions. However, as internal energy increases (>45 kcal mol−1), the contributions of H-loss channels become significantly more pronounced. Given similar energy barriers, all three direct dissociation pathways (CH3 + propyne, 2-butyne + H, and 1,2-butadiene + H) are expected to have considerable involvements in the dissociation of 2-buten-2-yl within the studied energy range of this work. To evaluate the branching ratios between 2-butyne + H and 1,2-butadiene + H in the dissociation of ground state 2-buten-2-yl, RRKM rate calculations are applied in the wavelength region of 226–246 nm. Assuming the excitation energy is applied as the internal energy to the ground state 2-buten-2-yl, the unimolecular decomposition rate constants to the 2-butyne + H and 1,2-butadiene + H products are calculated (see more details in the ESI†). Based on the calculated relative population over time (Fig. S3 and S4, ESI†), between the two lowest energy H-loss pathways, ∼84% of the 2-buten-2-yl radicals dissociate into 2-butyne + H with a rate constant k = 4.5 × 1011 s−1 at 246 nm, and the branching ratio for 1,2-butadiene + H channel is predicted to be ∼16% (k = 8.5 × 1011 s−1 at 246 nm). These RRKM rate constants are higher than the measured lower limit of the dissociation rate constant of kH ∼ 1.3 × 107 s−1 at 236 nm.
As the 2-buten-2-yl radical can be viewed as a 1-propenyl radical with an α H atom replaced by a methyl group, it is worthwhile to compare the dissociation dynamics of these two radicals in the similar energy region. This comparison can help explore the effects of methyl substitution on the photochemistry of the 1-propenyl radical. The statistical distribution for the slow component in the P(ET) of 2-buten-2-yl is similar to previous findings on photodissociation of 1-propenyl in 224–248 nm,49 in which a 〈fT〉 value of ∼0.12 was reported by Lucas et al.49 The similarity between the low-energy P(ET) feature in 2-buten-2-yl and earlier investigations of the photodissociation of 1-propenyl suggests that this reaction pathway is governed by a statistical mechanism. This process involves H-atom dissociation subsequent to internal conversion from the electronically excited state to a highly vibrationally excited ground electronic state. The slow component in this work mostly corresponds to the production of 2-butyne + H and 1,2-butadiene + H, with 2-butyne + H being the dominant channel according to the RRKM calculations. The peak (∼7 kcal mol−1) of the P(ET) of the slow component suggests a modest exit channel barrier of several kcal mol−1. This also supports that the 2-butyne + H product channel (with an exit channel barrier of ∼3 kcal mol−1) is more important than the 1,2-butadiene + H channel (with an exit channel barrier of ∼0.4 kcal mol−1).
The fast component in the CM translational energy distribution peaks at ∼35 kcal mol−1 and extends to ∼90 kcal mol−1, the maximum available energy for the H + 2-butyne product channel (Fig. 7). The 〈fT〉 for the fast component has a large value in the range of 0.40–0.46 in the photolysis region of 226–246 nm (Fig. 8). The presence of a high-energy peak in the P(ET) distributions and the large 〈fT〉 values of the fast component suggest a nonstatistical, repulsive product energy distribution in the photodissociation of 2-buten-2-yl. In Fig. 6, after removing the contributions from photodissociation of HCl, the β value is ∼0 between 10–70 kcal mol−1 (the energy region for the fast components). The absorption of 2-buten-2-yl at 226–246 nm likely involves with two Rydberg states, D5 (3py) and D6 (3px). This high-energy feature of the P(ET) suggests a nonstatistical distribution, resulting from direct dissociation on the electronically excited state or a repulsive segment of the ground state (D0). This process is possibly facilitated by a conical intersection connecting the 3p Rydberg states, leading to the formation of 2-butyne + H and 1,2-butadiene + H. The structure of 2-buten-2-yl and 1-propenyl differs at the terminal CH3 group of 2-buten-2-yl. As the previous study on the photodissociation of 1-propenyl did not show a discernible high-energy feature, this suggests that the fast component is likely due to the removal of an H-atom from the methyl group in 2-buten-2-yl, rather than from the allylic backbone. This would lead to the direct formation of the 1,2-butadiene + H product channel. A similar observation was made when comparing the dissociation of 1-MA and allyl. The UV photodissociation of allyl did not display a discernible high-energy feature,43 while dissociation of 1-MA showed a fast component in 226–244 nm.23 It is believed that the β H elimination from the CH3 group accounts for the fast channel in 1-MA, which aligns with the behavior of 2-buten-2-yl forming 1,2-butadiene + H. Although the fast component in the P(ET) distributions reached the maximum available energy for the 2-butyne + H product channel, it is likely that the elimination of the β H atom on the CC bond forming 2-butyne + H would not significantly contribute to the fast component. The observed nearly isotropic angular distribution might still be attributed to the prompt β H dissociation, arising from a combination of the two D0 → D5 or D0 → D6 transitions and various β H eliminations on the CH3 group, which reduces the anisotropy to a nearly isotropic angular distribution.
Overall, the UV photodissociation of 2-buten-2-yl has similarities with those of its isomers, 1-MA and 2-MA, which also exhibit two distinct features within the similar photodissociation wavelength region. All three C4H7 systems show a broad slow component in the H-elimination product P(ET) distributions with an isotropic angular distribution. The 〈fT〉 for the isotropic, slow component is 0.16–0.19 for 2-MA and 0.13–0.17 for 1-MA. For both methylallyl radicals, the small fast component located at ∼50 kcal mol−1 with 〈fT〉 = 0.58–0.68 and β = −0.2 for 2-MA22 and 〈fT〉 = 0.62–0.72 and β = −0.23 for 1-MA.23 For 2-buten-2-yl, the fast component peaked at ∼ 35 kcal mol−1 with 〈fT〉 = 0.40–0.46 and an isotropic angular distribution. 1-MA has a slightly higher fast/slow component ratio (∼0.03–0.08) than 2-MA (∼0.02–0.03). This difference is attributed to the more straightforward direct H-loss pathways in 1-MA, whereas 2-MA must undergo isomerization before releasing an H atom. 2-Buten-2-yl is supposed to undergo direct β H loss on the CH3 group and produce 1,2-butadiene + H, leading a comparable fast/slow ratio of ∼0.03–0.08, although the different deconvolution methods may affect the determination of the branching ratio values. It can be concluded that the overall photodissociation outcomes are primarily determined by the energetics and isomerization pathways involved. Based on our theoretical calculations, the UV excitation of 2-buten-2-yl in this study is attributed to the fifth (D5) and sixth (D6) excited states, which predominantly exhibit the 3p Rydberg character. The involved Rydberg orbital is predicted to lie in the CC plane, which is different to those for 1-MA and 2-MA, where the 3p orbital is calculated to be perpendicular to the plane. For 1-MA, we proposed that the prompt H-atom dissociation originates from the direct removal of the out-of-plane hydrogen on the CH3 group, resulting in the formation of 1,3-butadiene + H. Likewise, the fast component of 2-buten-2-yl, with an isotropic angular distribution, is likely due to the production of 1,2-butadiene + H via β H elimination on the CH3 group, although the exact geometry during dissociation remains unclear.
Conclusion
The H-atom product channels in the UV photodissociation of jet-cooled 2-buten-2-yl, were studied in the photolysis wavelength region of 226–246 nm using the HRTOF technique. Similar to the photodissociation of 1-MA and 2-MA, the H-atom product photodissociation of 2-buten-2-yl also shows a bimodal translational energy distribution indicating two photodissociation pathways. The slow component (II) is a broad peak at ET ∼ 7 kcal mol−1 while the fast component (I) centers at ET ∼ 35 kcal mol−1, and both components display an isotropic angular distribution (β ∼ 0). The fraction of the average translational energy release in the total available energy, 〈fT〉, is ∼0.11–0.12 for the slow pathway (II) and 0.40–0.46 for the fast pathway (I). The slow component is consistent with the nonradiative decay of the excited electronic state to the ground electronic state followed by H-atom dissociation on the highly vibrationally excited ground state, most likely producing 2-butyne + H (∼84%) and 1,2-butadiene + H (∼16%). The fast component suggests the formation of 1,2-butadiene + H directly on the excited electronic state or a repulsive part of the ground state surface, possible through a conical intersection. The statistical unimolecular dissociation channels of 2-buten-2-yl are consistent with the photodissociation dynamics of previously reported results of the 1-MA and 2-MA radicals.11,12,14,23,24 The non-statistical photodissociation dynamics of the 2-buten-2-yl radical are reported for the first time.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
Authors declare no competing financial interest.
Acknowledgements
This work was financially supported by the US National Science Foundation (no. CHE-2155232). Yuan Qin acknowledges support from a UC Riverside Dissertation-Year Fellowship.
References
- A. B. Callear and H. K. Lee, Trans. Faraday Soc., 1968, 64, 308–316 RSC.
- J. C. Schultz, F. A. Houle and J. L. Beauchamp, J. Am. Chem. Soc., 1984, 106, 7336–7347 CrossRef CAS.
- J. W. Hudgens and C. S. Dulcey, J. Phys. Chem. A, 1985, 89, 1505–1509 CrossRef CAS.
- N. Nakashima and K. Yoshihara, Laser Chem., 1987, 7, 177–196 CrossRef CAS.
-
B. P. Tsai, R. D. Johnson III and J. W. Hudgens, Resonance Ionization Spectroscopy and Its Applications, Institute of Physics, Bristol, UK, 1989, p. 129 Search PubMed.
- D. H. Tarrant, J. D. Getty, X. Liu and P. B. Kelly, J. Phys. Chem., 1996, 100, 7772–7777 CrossRef CAS.
- J. D. Getty, X. Liu and P. B. Kelly, J. Chem. Phys., 1996, 104, 3176–3180 CrossRef CAS.
- C.-C. Chen, H.-C. Wu, C.-M. Tseng, Y.-H. Yang and Y.-T. Chen, J. Chem. Phys., 2003, 119, 241–250 CrossRef CAS.
- K.-C. Lau, Y. Liu and L. J. Butler, J. Chem. Phys., 2006, 125, 144312 CrossRef PubMed.
- F. Bayrakçeken, Z. Telatar, F. Arı and A. B. Bayrak, Spectrochim. Acta, Part A, 2007, 67, 1276–1280 CrossRef.
- M. Gasser, A. Bach and P. Chen, Phys. Chem. Chem. Phys., 2008, 10, 1133–1138 RSC.
- M. Gasser, J. A. Frey, J. M. Hostettler and A. Bach, Chem. Commun., 2011, 47, 301–303 RSC.
- M. Bahou, J.-Y. Wu, K. Tanaka and Y.-P. Lee, J. Chem. Phys., 2012, 137, 084310 CrossRef PubMed.
- J. Herterich, T. Gerbich and I. Fischer, ChemPhysChem, 2013, 14, 3906–3908 CrossRef CAS PubMed.
- M. Lang, F. Holzmeier, P. Hemberger and I. Fischer, J. Phys. Chem. A, 2015, 119, 3995–4000 CrossRef CAS PubMed.
- A. R. Brown, P. R. Franke and G. E. Douberly, J. Phys. Chem. A, 2017, 121, 7576–7587 CrossRef CAS PubMed.
- A. Röder, J. Petersen, K. Issler, I. Fischer, R. Mitric and L. Poisson, J. Phys. Chem. A, 2019, 123, 10643–10662 CrossRef.
- S. Hartweg, J.-C. Loison, S. Boye-Peronne, B. Gans, D. M. P. Holland, G. A. Garcia, L. Nahon and S. T. Pratt, J. Phys. Chem. A, 2020, 124, 6050–6060 CrossRef CAS.
- T. Preitschopf, F. Hirsch, A. K. Lemmens, A. M. Rijs and I. Fischer, Phys. Chem. Chem. Phys., 2022, 24, 7682–7690 RSC.
- A. Röder, K. Issler, L. Poisson, A. Humeniuk, M. Wohlgemuth, M. Comte, F. Lepetit, I. Fischer, R. Mitric and J. Petersen, J. Chem. Phys., 2017, 147, 013902 CrossRef PubMed.
- M. Gasser, J. A. Frey, J. M. Hostettler and A. Bach, J. Mol. Spectrosc., 2010, 263, 93–100 CrossRef CAS.
- M. Lucas, Y. Qin, M. Chen, G. Sun and J. Zhang, Chin. J. Chem. Phys., 2024, 37, 255–263 CrossRef CAS.
- M. Lucas, Y. Qin, L. Yang, G. Sun and J. Zhang, J. Phys.
Chem. A, 2024, 128, 5556–5566 CrossRef CAS.
- L. R. McCunn, M. J. Krisch, Y. Liu, L. J. Butler and J. Shu, J. Phys. Chem. A, 2005, 109, 6430–6439 CrossRef CAS.
- J. L. Miller, M. J. Krisch, L. J. Butler and J. Shu, J. Phys. Chem. A, 2005, 109, 4038–4048 CrossRef CAS PubMed.
- J. L. Miller, J. Phys. Chem. A, 2004, 108, 2268–2277 CrossRef CAS.
- Y. Li, H. Liu, Z. Zhou, X. Huang and C. Sun, J. Phys. Chem. A, 2010, 114, 9496–9506 CrossRef CAS PubMed.
- J. M. Ribeiro and A. M. Mebel, J. Phys. Chem. A, 2016, 120, 1800–1812 CrossRef CAS.
- Y. Li, S. J. Klippenstein, C.-W. Zhou and H. J. Curran, J. Phys. Chem. A, 2017, 121, 7433–7445 CrossRef CAS.
- C. Huang, B. Yang and F. Zhang, Combust. Flame, 2017, 181, 100–109 CrossRef CAS.
- J. Cho, A. W. Jasper, Y. Georgievskii, S. J. Klippenstein and R. Sivaramakrishnan, Combust. Flame, 2022, 257, 112502 CrossRef.
-
B. Ruscic and D. H. Bross, Active Thermochemical Tables (ATcT) Thermochemical Values ver. 1.130, Argonne National Lab.(ANL), Argonne, IL (United States), 2023 Search PubMed.
- Y. Song, M. Lucas, M. Alcaraz, J. Zhang and C. Brazier, J. Chem. Phys., 2012, 136, 044308 CrossRef.
- G. Amaral, K. Xu and J. Zhang, J. Chem. Phys., 2001, 114, 5164–5169 CrossRef CAS.
- K. Xu, G. Amaral and J. Zhang, J. Chem. Phys., 1999, 111, 6271–6282 CrossRef CAS.
- Y. Song, X. Zheng, M. Lucas and J. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 8296–8305 RSC.
- L. Koziol, S. V. Levchenko and A. I. Krylov, J. Phys. Chem. A, 2006, 110, 2746–2758 CrossRef CAS.
- V. Riffet, D. Jacquemin, E. Cauët and G. Frison, J. Chem. Theory Comput., 2014, 10, 3308–3318 CrossRef CAS.
- A. D. Laurent and D. Jacquemin, Int. J. Quantum Chem., 2013, 113, 2019–2039 CrossRef CAS.
- D. E. Woon and T. H. Dunning Jr., J. Chem. Phys., 1994, 100, 2975–2988 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- M. Lucas, J. Minor, J. Zhang and C. Brazier, J. Phys. Chem. A, 2013, 117, 12138–12145 CrossRef CAS.
- Y. Song, M. Lucas, M. Alcaraz, J. Zhang and C. Brazier, J. Phys. Chem. A, 2015, 119, 12318–12328 CrossRef CAS.
- H. Noller, H. Hantsche and P. Andreu, J. Catal., 1965, 4, 354–362 CrossRef CAS.
- P. M. Regan, S. R. Langford, D. Ascenzi, P. A. Cook, A. J. Orr-Ewing and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 1999, 1, 3247–3251 RSC.
- P. M. Regan, D. Ascenzi, A. Brown, G. G. Balint-Kurti and A. J. Orr-Ewing, J. Chem. Phys., 2000, 112, 10259–10268 CrossRef CAS.
- Y. Song, X. Zheng, W. Zhou, M. Lucas and J. Zhang, J. Chem. Phys., 2015, 142, 224306 CrossRef PubMed.
- H.-J. Deyerl, I. Fischer and P. Chen, J. Chem. Phys., 1999, 110, 1450–1462 CrossRef CAS.
- M. Lucas, Y. Song, J. Zhang, C. Brazier, P. L. Houston and J. M. Bowman, J. Phys. Chem. A, 2016, 120, 5248–5256 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on background removal in the H-atom TOF spectrum, the structure, energies of excited states, transition dipole moments and the NTO analysis of 2-buten-2-yl calculated at different levels of theories, and the rate constant calculations of 2-buten-2-yl to 2-butyne + H and 1,2-butadiene + H using the RRKM theory. See DOI: https://doi.org/10.1039/d4cp03076a |
| ‡ These two authors contributed equally. |
| § Present address: Department of Chemistry, University of Hawaii at Manoa, Honolulu, HI 96822, USA. |
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Ge
Sun
and
Jingsong
Zhang
,
Ge
Sun
and
Jingsong
Zhang