
The decisive role of Au in CO diffusion on Pt surfaces: a DFT study†
Ana Lucía
Reviglio
 *ab,
Paula S.
Cappellari
*ac,
German J.
Soldano
*de,
Marcelo M.
Mariscal
de and
Gabriel A.
Planes
ac
*ab,
Paula S.
Cappellari
*ac,
German J.
Soldano
*de,
Marcelo M.
Mariscal
de and
Gabriel A.
Planes
ac
aIITEMA, CONICET, Universidad Nacional de Río Cuarto, Ruta Nac. 36, Km 601, Río Cuarto, Córdoba 5800, Argentina. E-mail: areviglio@exa.unrc.edu.ar; pcappellari@exa.unrc.edu.ar
bDepartamento de Física, Facultad de Ciencias Exactas. Físico-Químicas y Naturales, Universidad Nacional de Río Cuarto, Ruta Nac. 36, Km 601, Río Cuarto, Córdoba 5800, Argentina
cDepartamento de Química, Facultad de Ciencias Exactas. Físico-Químicas y Naturales, Universidad Nacional de Río Cuarto, Ruta Nac. 36, Km 601, Río Cuarto, Córdoba 5800, Argentina
dINFIQC, CONICET, Universidad Nacional de Córdoba, XUA5000 Córdoba, Argentina
eDepartamento de Química Teórica y Computacional, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, XUA5000 Córdoba, Argentina
First published on 8th November 2024
Abstract
The modification of metallic surfaces with adsorbed atoms of a second metal is presented as an ideal method for producing electrocatalysts. In this work, we examined the role of Au atoms in the reactivity of Pt surfaces and their effect on the adsorption and diffusion of CO using first-principles calculations. Our comprehensive study utilized density functional theory (DFT) to analyze a variety of adsorption sites on single-crystal Pt structures, encompassing open and staggered configurations. The combined methodologies of the climbing image nudged elastic band (CI-NEB) and potential energy surfaces (PES) were employed to identify significant trends in the diffusional behavior of CO. These methods allowed for a thorough analysis of the movement and interaction of CO molecules, providing valuable insights into their diffusion properties. The findings of this study provide compelling evidence that the presence of Au inhibits the movement of CO towards highly reactive Pt sites. This contributes to a more thorough comprehension of polycrystalline metal surfaces with secondary metal deposits and their effectiveness in various electrocatalytic reactions.
1 Introduction
The advancement of renewable energy conversion and storage represents an emerging field of research that may offer an alternative to the use of fossil fuels in the transition towards the replacement of unsustainable energy sources by sustainable and clean technologies.1,2 In this context, fuel cells (FCs) are devices that convert chemical energy into electrical energy through electrochemical reactions, similar to batteries.3 The performance of fuel cells is largely dependent on the efficiency of the electrocatalyst, which is affected by a number of factors, including surface morphology, facet structure, ionomer/catalyst interaction, and chemical composition.4–6 The electrocatalytic properties of platinum (Pt) electrodes have been extensively studied with regard to their suitability for use in low-temperature FCs.7 One significant disadvantage of low-temperature FCs, such as direct formic acid or direct alcohol FCs (DFAFC or DAFC, respectively), is the formation of CO during the oxidation reactions taking place at the FCs device. The high adsorption affinity of CO on Pt surfaces is a well-established phenomenon.8–11 This results in the poisoning of the electrocatalyst material, which in turn affects the overall efficiency of the process and results in a decrease in its activity in low-potential fuel oxidation.9,12–14 Consequently, the study of the interaction between CO and the catalytic surface, as well as its removal (oxidation), is of crucial importance for the development of energy-efficient systems.A substantial literature review on CO oxidation on single-crystalline and staggered Pt surfaces has been conducted.15–17 It is widely agreed that low coordination number sites, such as steps, corners and defects, are the most active in the CO oxidation mechanism.17–21 Consequently, surface diffusion is a key factor in the CO electro-oxidation process, particularly in situations where the mobility of the reactive species is suspected to be limited by a potential barrier, as has recently been demonstrated.22–24 From chrono-amperometric measurements Koper and Garcia25 suggest that, in acidic media, the surface diffusion of CO should be very fast while in alkaline media this diffusion is slowed down. The rate at which CO diffuses is a crucial and determining factor in its subsequent oxidation or surface elimination.22,26
As a result, there is a renewed interest in the identification and development of advanced electrocatalysts with enhanced CO tolerance. In this regard, the combination of platinum with other metals can mitigate the effects of CO poisoning and improve the performance of anode materials.27,28 A particularly intriguing catalytic surface is that formed by a combination of Pt (active) and Au (inactive).29–32 It has been demonstrated that by introducing an ad-atom patch of Au onto a Pt surface, a synergistic effect can be achieved. The resulting catalytic activity is demonstrably superior to that which would be expected a priory, considering the catalytic properties of each individual surface.23,30,32 The enhanced performance observed in Pt/Au catalysts can be attributed to a change in the reaction mechanism. In particular, experimental results from our research group indicate that Pt/Au surfaces and Au ad-atoms act as a barrier, impeding the surface diffusion of CO from/to the Pt active sites.23,30,33
The objective of this study is to characterise and analyse the role of Au atoms in the adsorption and diffusion process of CO on stepped Pt surfaces. The primary aim is to identify potential explanations for the processes that occur between various oxidation reactions on Pt/Au surfaces, where CO plays an active intermediate. A series of computational studies based on DFT methodology were carried out in order to gain insight into the CO adsorption processes on monocrystalline and stepped Pt surfaces. In addition, the climbing image nudged elastic band (CI-NEB) and potential energy surfaces (PES) calculations were performed to explore the diffusion of CO on the surface. The findings indicate that CO diffusion on stepwise Pt is an anisotropic process that requires activation. In contrast, on open surfaces such as terraces or monocrystalline environments, diffusion is observed to occur freely and can be considered spontaneous. These data align with experimental findings where ‘slow’ diffusion is evident on Pt/Au surfaces compared to bare Pt surfaces. Furthermore, it was observed that the presence of Au atoms results in high activation barriers for the CO diffusive process.
2 Computational methods
Two main surfaces structures were considered: Pt(111) to account for densely packed terraces and stepped Pt(442) to account for surface defects. The slab approach was used with four layers for Pt(111) and five for Pt(442), both separated by at least 10 Å of vacuum and repeated in a super-cell geometry. The two uppermost layers were relaxed while the remaining layers were held at their truncated bulk positions. The unfixed layers, Au atoms, and CO adsorbed molecules were relaxed until the total force on all atoms was less than 0.1 eV Å−1. Different super-cells were employed according to Au coverage on the surfaces, ranging from a 2 × 2 × 4 Pt(111) super-cell [four Pt surface atoms, one Au atom, four Pt layers] and 4 × 1 × 4 Pt(442) super-cell [four surface atoms, one Au atom, five layers,] up to 3 × 3 × 4 and 1 × 3 × 4 super-cells, respectively.Density functional theory (DFT) calculations were performed through the Quantum Espresso code.34 The Kohn–Sham one-electron equations were solved self-consistently, the ionic cores being represented by Vanderbilt ultrasoft35 pseudopotentials with the Perdew–Burke–Ernzerhof (PBE)36 approximation. The GBRV pseudopotential library optimized for precision and efficiency was used.37 The energy cutoff was fixed at 50 Ry for wavefunctions and 500 Ry for charge density, with an Marzari/Vanderbilt smearing of 0.01 Ry. A Monkhorst/Pack scheme was used to sample the Brillouin zone with 12 × 12 × 1 k-points for the smallest Pt(111) surface cell, 2 × 12 × 1 k-points for the smallest stepped Pt(442) surface cell, and corresponding reciprocal values for super-cells multiple of these. Relaxations were converged when forces acting on atoms were weaker than 0.02 eV Å−1. Spin polarization calculations were performed for terrace and step surfaces but nor energetic difference with the unpolarized calculations neither magnetic moments were obtain so they were disregarded.
Adsorption energy (Ead) is a measure of how strongly a molecule binds to a surface, and it can affect catalysis processes. For our system, it can be calculated as:
| Ead = Esys − Esurf − Eadsor | (1) |
The climbing image nudged elastic band (CI-NEB)38 method was employed to calculate the reaction pathways and energy barriers associated with the CO diffusion process. This entailed interpolating between five, seven, or nine images contingent on the distance traversed through different surface sites. Five images were used for mobility between adjacent sites, and between seven and nine images for more distant sites. Convergence was achieved when the residual forces acting on each atom were weaker than 0.01 eV Å−1. The CI-NEB method was employed to identify the minimum energy paths, the transition states, and the energy barriers.38,39
Finally, potential energy surfaces (PES) were obtained by single point energy calculations at fixed xy positions of CO on the surfaces spaced in a square grid of around 0.5 Å per side. Both the C–O distance and its angle with respect to the metal surface were kept fixed. For each xy point, the equilibrium height was calculated with a 0.1 Å precision. The corresponding Ead was then assigned to each surface point, taking the lowest Ead of all the calculations as the reference.
3 Results and discussion
3.1 Pristine surfaces
The bibliography contains a large number of papers characterising ideal surfaces such as Pt(111);15,27,40 nevertheless, it has been demonstrated that the majority of electrodes employed in FC applications are constructed from polycrystalline Pt substrates. Consequently, the actual surfaces comprise a multitude of crystalline faces and numerous edges at their junctions. As an approach to model these non-ideal systems, we have consider the (442) stepped Pt surface, where the terrace and the step have a (111) and (100) local surface, respectively. Fig. 1 shows the Pt(111) and Pt(442) surfaces with and without Au considered in this work.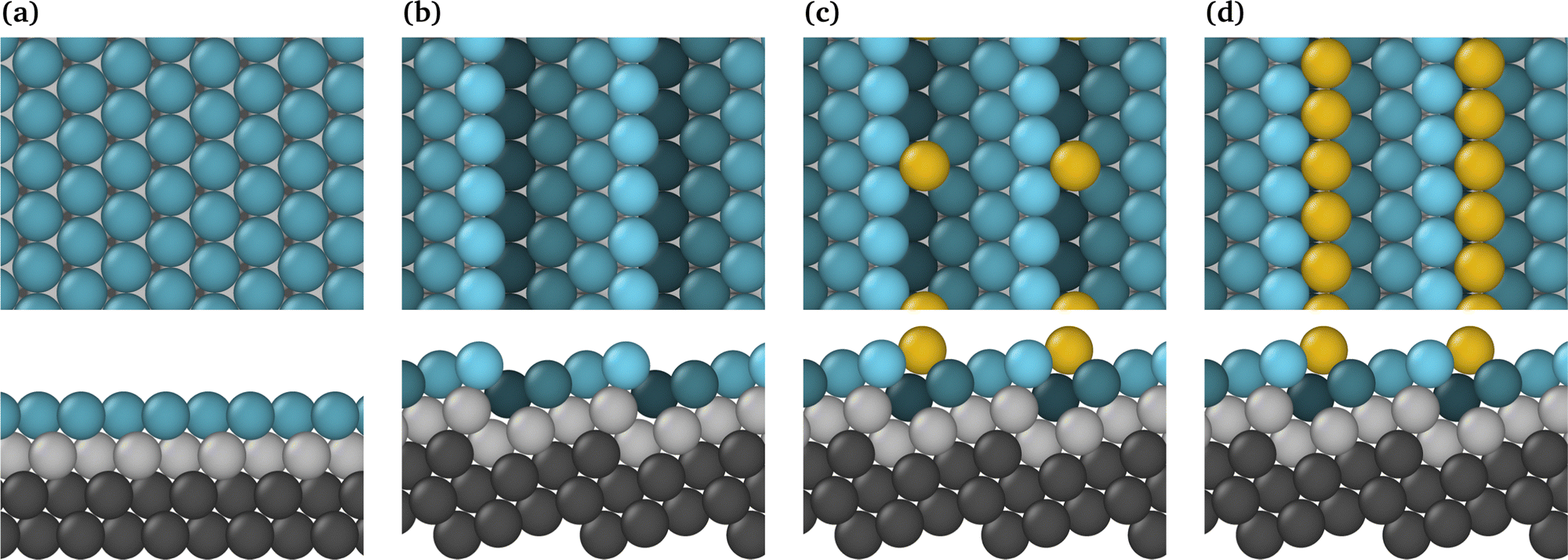 | ||
| Fig. 1 Top and side views of (a) Pt(111); (b) Pt(442); (c) Pt(442)/Au@at; (d) Pt(442)/Au@nw. The slab layers are distinguished with color coding: the bottom (fixed) layer is colored black, the second layer atoms are gray, and varying shades of light blue are used for the top layer to aid in highlighting the step. Au atoms are depicted in yellow. | ||
Presence of steps modifies the properties of the surface, such as the work function or the zero charge potential, which has been found to be directly proportional to the step density.41,42 Nevertheless, the linear relationship can be disrupted if the terrace surface is narrow enough to facilitate step–step interactions. Consequently, it is essential to compare the variables of interest between the terrace and the analogous planar counterpart, taking into account the presence of steps.43
For the purpose to facilitate a comparative and analytical assessment of the aforementioned, it was decided that an Ead analysis of the Au atom, as presented in Table 1, would be conducted for a planar surface of Pt(111), in addition to two stepped surfaces with terraces of different dimensions. The Pt(332) and Pt(442) surfaces. The final surface displays terraces of a narrower width and comprises two fewer atomic rows than the preceding one. As illustrated in Table 1 there is no discernible difference in stability between fcc and hcp hollow sites on Pt(111), with an energy difference of approximately 0.10 eV. These values are in close alignment with those observed in the terrace regions of stepped surfaces, which suggests the absence of substantial step–step interactions. Table 1 demonstrates that the differences in the energy required for the adsorption of an Au on Pt surfaces, Pt(332) and Pt(442), are negligible. We selected Pt(442) for further calculations as it has 33% fewer atoms.
| Site | Pt(111) | Pt(332) | Pt(442) | |
|---|---|---|---|---|
| 2 | Hfcc (T) | −3.72 | −3.88 | −3.84 |
| 3 | Hhcp (T) | −3.61 | −3.79 | −3.73 |
| 2 | Hfcc (S) | — | −4.14 | −4.11 |
| 3 | Hhcp (S) | — | −4.69 | −4.66 |
Conversely, the presence of steps alters the surface characteristics, resulting in a variation in the energy required for adsorption at different sites. Significant differences in Ead are observed when Au is adsorbed on the terrace or at the edge of the step, with the most stable adsorption site for an Au atom being the fcc gap on the step, which is approximately 0.80 eV more stable than on the terrace. Therefore, this adsorption site was selected for the remainder of our calculations involving Au atoms on Pt step surfaces.
In order to gain a more general and representative insight, the adsorption of a single Au atom on the step and of a complete row on Pt(442) are selected for comparison. This enables a detailed examination of the effects of complete and partial coverage of the step. Fig. 1c and d illustrate the most stable surfaces for the adsorption of a single Au atom (Pt(442)/Au@at) and a single row of Au atoms (Pt(442)/Au@nw), respectively, on Pt(442). It can be observed that the Au atoms are positioned preferentially within the step of the Pt surface (442), resulting in the formation of a new step exclusively composed of this metal in the case of the complete Au row. Consequently, the latter surface has been designated as “Au nanowire” on Pt(442), Pt(442)/Au@nw. This investigation focuses on the analysis of phase-separated systems in alloys, specifically examining a scenario comprising two distinguishable phases: a bulk platinum phase and a surface layer comprising gold atoms in contact with the Pt surface. The specific scenario in which Pt serves as a host and Au atoms act as surface impurities, as demonstrated by Nørskov and colleagues,44,45 illustrates a situation in which the segregation energy is such that the possibility of atom mixing is not thermodynamically feasible.
As a first insight into the electronic properties of these surfaces, the projected density of states (pDOS) of the d orbital is shown in Fig. 2. For surfaces without an Au atom, no significant difference is observed between Pt(111) surface atoms, Pt(442) terrace atoms and Pt(442) step atoms, apart from a subtle narrowing of the d-band in the latter. In the presence of an Au atom step, the Pt atoms adjacent to Au also show no significant change. However, a clear difference is observed between Pt and Au atoms. In Pt the projected density of states (pDOS) decays abruptly after the Fermi energy, whereas in Au it starts to decay about 3.5 eV beyond the Fermi energy. According to the d-band model,46,47 this suggests that Pt forms strong bonds with adsorbates, as the corresponding anti-bonding states appear above the Fermi energy and thus remain unoccupied. In contrast, Au forms weak bonds with adsorbates as the anti-bonding states appear below the Fermi energy and are therefore occupied by electrons. Taken together, these differences in the projected density of states underline the different ability of Pt and Au to interact with adsorbates, which may play a crucial role in the catalytic applications of these surfaces.
 | ||
| Fig. 2 Projected density of states for the 3d orbital of surface atoms in Pt(111), Pt(442) and Pt(442)/Au@nw. For (442) surfaces, terrace and step atoms are differentiated. Pt step* surface correspond to the Pt line of atoms next to the Au step. | ||
3.2 CO adsorption and diffusion
The principal objective of this study is to examine the impact of Au on the adsorption and diffusion of CO on the aforementioned surfaces. Previous experimental research has indicated that the oxidation of CO on Pt is influenced by the presence of adsorbed Au atoms.23,30,31,33 In order to gain this insight, several calculations were performed to determine the Ead of CO in different sites over surface. Fig. 3 illustrates the adsorption sites for CO molecules over Pt stepped surfaces, to observe the values on the Pt(111) monocrystalline surface in ESI,† kindly refer to Fig. S1 and Table S1 (ESI†). The circles, which are coloured differently, represent different environments of adsorption. The white circles symbolise terrace sites, while the grey ones represent sites over a step. The numbers in the figure indicate the different sites: (1) bridge, (2) hollow fcc, (3) hollow hcp and (4) top. The results of the Ead are presented in Table 2. | ||
| Fig. 3 Adsorption sites for CO molecules over Pt stepped surfaces. The circles, which are coloured differently, represent different environments of adsorption. The white circles symbolise terrace sites, while the grey ones represent sites over a step. The numerical values represent the distinct locations, as follows: (1) bridge, (2) hollow fcc, (3) hollow hcp, and (4) top. | ||
| Site | Pt(111) | Pt(442) | Pt(442)/Au@at | Pt(442)/Au@nw | |
|---|---|---|---|---|---|
| a The absence of values at some adsorption sites is a consequence of the relaxation of the molecule at those sites resulting in its adsorption at a nearby but different site. | |||||
| 1 | Bridge (T) | −2.40 | −2.30 | —a | −0.95(o) |
| 2 | Hfcc (T) | −2.49 | −2.25 | −1.37(n); | −2.13(f) |
| −2.06(f) | |||||
| 3 | Hhcp (T) | −2.45 | −2.30 | −1.63(n) | — |
| 4 | Top (T) | −2.28 | −2.07 | −1.23(o); | −0.94(o); |
| −1.45(n) | −1.06(n); | ||||
| −2.13(f) | |||||
| 1 | Bridge (S) | — | −2.53 | −2.22(f) | — |
| 2 | Hfcc (S) | — | −2.25 | — | — |
| 3 | Hhcp (S) | — | −1.91 | — | — |
| 4 | Top (S) | — | −2.58 | −2.22(f) | −1.92(f) |
Our findings indicate that Ead in Pt(111) and Pt(442) is comparable when considering the terrace region. However, markedly disparate are observed values when the adsorption occurs at the border of the step. Furthermore, the site over the terrace and behind the border step exhibit smaller Ead. The values indicate that the terrace sites of the step system used closely replicate the behaviour observed in defect-free systems, and additionally demonstrate a marked proclivity for CO adsorption at the summit of the step. The order of CO adsorption sites, from the most to least adsorbed, is as follows: top edge of step, terrace, bottom edge. The aforementioned observations align with the sequence of catalytic activity proposed by García and Koper in 2011.25
Adsorption energy calculations of CO and Au on Pt(111) showed that both species share the same preferential adsorption sites. As shown in Tables 1 and 2, the lowest Ead are observed in the hollow sites, both fcc and hcp structures, with similar values for each adsorbate. This suggests that the prior adsorption of one species could influence the availability of preferential sites for the other, thus affecting the distribution and stability of adsorbates on the Pt surface during catalytic processes. The Ead of CO in Pt(111) in the presence of adsorbed Au has been calculated and the results are presented in Tables S2, S3 and Fig. S2, S3 in ESI.† The results indicate that the presence of adsorbed Au induces a repulsive effect on the CO molecule, reducing its affinity for the Pt(111) surface.
As evinced in Table 2, the adsorption energies of CO on Pt(442) indicate that the step edge is the most favourable site for CO adsorption on Pt(442). As previously stated, the adsorption of Au on Pt(442) also exhibits a proclivity for adsorption at the step edge. Although both Au and CO display a preference for adsorption at the step edge, their respective behaviours exhibit notable differences. In contrast to CO, which adsorbs on top, moving the O atom as far away from the surface as possible, Au exhibits a different adsorption behaviour, adsorbing below the step and increasing its coordination number to reduce its surface energy. The observed preference for the adsorption site by both species to reduce the surface energy may be indicative of a competition between Au and CO for this site. Nevertheless, as the adsorption of Au occurs at an early stage, specifically during the synthesis of the catalyst and prior to exposure to CO, it is the adsorption of CO that is influenced by the presence of Au.23,30
In order to gain insight into this phenomenon, the Ead of CO at various sites, including those in close proximity (n) to, distant from (f) or situated on (o) the adsorbed Au atoms, has been examined in both the Pt(442)/Au@at and Pt(442)/Au@nw structures. It is observed that the adsorption of CO on the surface increases significantly when the distance to Au is larger than 3 Å. The observed energy values range from −1.9 eV to −2.2 eV, which is analogous to what has been observed on the monocrystalline Pt(111) surface (ESI†). Conversely, at distances closer than 2.5 Å from Au, the adsorption of CO is observed to weaken, with energy values between −1 eV and −1.5 eV. As evidenced by the Ead calculations presented in Table 1, a repulsive force exists between CO and Au, impeding CO adsorption in the vicinity of Au. Consequently, CO adsorption predominantly occurs in regions distant from Au, although CO adsorption on Au is not precluded. This finding is also corroborated by the electronic behaviour observed using pDOS, whereby Au forms relatively weak bonds with adsorbates, as discussed in the preceding section.
Although the existence of defects or adsorbed species can influence the adsorption of CO on the surface, this does not necessarily imply that the diffusion of CO is also affected. In their work, García and Koper25 proposed that the presence of steps alters local surface binding and diffusion of absorbed species as a result of a change in the electronic structure of the surface. To evaluate how the presence of steps and adsorbed Au affects CO diffusion, we have performed numerous NEB calculations along different paths in Pt(111), Pt(442), Pt(442)/Au@at and Pt(442)/Au@nw. Fig. 4 illustrates the results of a series of NEB calculations between adsorption sites, selected for the purpose of conducting a comprehensive investigation of the surface in question. The activation energies for each concatenation are shown in Table 3. The complete pathways were selected based on the direction of diffusion, which was determined to be either parallel or perpendicular to the step. In the event that the diffusion is parallel to the step and occurs on the terrace, the path is designated as (‖T). Conversely, if the diffusion occurs on the step, the nomenclature is (‖S). In the event that the diffusion is perpendicular to the step and the step does not have Au adsorbed, the nomenclature is (⊥). Conversely, if the diffusion is crossing an Au atom, the nomenclature is (⊥Au).
 | ||
| Fig. 4 Top view and energy variation of NEB paths for three different directions along Pt step surfaces. The purple line represents a path perpendicular to the step, while the orange and green lines represent parallel paths to the step. The orange lines are over the step, while the green lines are over the terrace. (a) Pt(442); (b) Pt(442)/Au@at; (c) Pt(442)/Au@nw. | ||
| Path | Pt(442) | Pt(442)/Au@at | Pt(442)/Au@nw |
|---|---|---|---|
| a The nomenclature of the pathways was selected based on the direction of diffusion, whether parallel (‖) or perpendicular (⊥) to the step. In the event that the diffusion is parallel, occurs on a terrace (‖T) or on the step (‖S), and is perpendicular to the step, and if it is crossing an Au atom (⊥Au) or not (⊥). | |||
| ‖Ta | 0.10 | 0.57 | 0.11 |
| ‖S | 0.21 | — | 0.14 |
| ⊥ | 0.74 | 0.86 | — |
| ⊥Au | — | 0.78 | 1.69 |
Regarding to Pt(111), it was observed that the diffusion of CO in any direction over the surfaces has at most an activation energy of 0.12 eV when it moves between hollow and bridge positions, Fig. S4 and Table S4 in ESI.† Furthermore, an elevated activation energy is evident when CO is compelled to ascend to a top site (Fig. S4 and Table S4 in ESI†). With respect to Pt(442), Table 3 demonstrates that the activation energy for CO diffusion in the terrace parallel to the step (T) and all Pt(111) passages exhibits a striking similarity, indicating a comparable behaviour of CO surface diffusion on the terrace and on the Pt(111) surface. It is noteworthy that the activation energies for all pathways are relatively low, below 0.3 eV, which suggests a favourable arrangement for surface diffusion.
For Pt(111), it can be observed (Fig. S4 and Table S4 in ESI†) that the activation energies are all lower than 0.25 eV, so diffusion in any direction is possible for this surface. Conversely, the activation barrier for CO diffusion from the terrace and perpendicular to the step edge (⊥) are higher, reaching almost 0.8 eV. This indicates that the presence of steps gives rise to anisotropic diffusive behaviour, whereby free diffusion occurs over the terrace but a significant barrier is encountered when crossing the steps. The activation barrier values for leaving the terrace and remaining on the edge are up to 0.4 eV, but the cost of going down or up the terrace is twice as much energy as it takes to reach the terrace. This anisotropic behaviour has been described by other authors48–50 and corresponds here to the intuition that if a molecule of CO is absorbed in one of the terrace sites, it can move freely to the step boundary, which is the preferred absorbed site. And once there, the molecule is available to react.
On the other hand, NEB calculations for the Pt(442)/Au@at and Pt(442)/Au@nw surfaces show a clear increase in the activation energy for CO diffusion when the path crosses a region close to the Au atoms. A similar trend is observed for Au-modified Pt(111) surfaces (Fig. S5, S6 and Tables S5, S6, ESI†). The activation energy for CO diffusion is not influenced by the presence of a single Au atom adsorbed on the step when the analysed path is distant from the Au atom. Nevertheless, it is evident that the CO initial linear trajectory is influenced by the presence of the Au atom, with the molecule exhibiting a repulsive force when attempting to diffuse in its proximity.
In contrast, when the step is completely covered by Au atoms, the activation energy for CO diffusion in the direction perpendicular to the step increases by almost 1 eV. This result shows the crucial role of Au atoms in CO diffusion over Pt surfaces, as the energies involved increase significantly. As previously suggested, this is an effect of Au–CO repulsion. It is worth noting that the diffusion in the terrace parallel to the step is not affected.
The activation energies permit the calculation of the rate constant51 for CO diffusion on these surfaces using the Arrhenius equation:
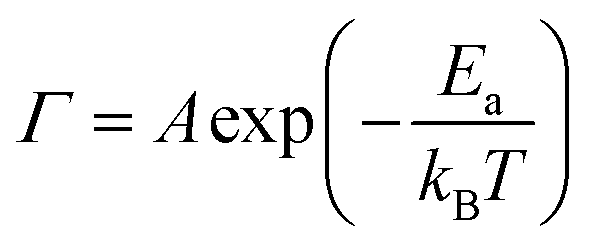 | (2) |
| Path | Pt(442) | Pt(442)/Au@at | Pt(442)/Au@nw |
|---|---|---|---|
| All values are in s−1. | |||
| ‖T | 2.09 × 1011 | 2.65 × 1003 | 1.45 × 1011 |
| ‖S | 2.96 × 1009 | — | 4.45 × 1010 |
| ⊥ | 3.70 × 1000 | 3.57 × 10−02 | — |
| ⊥Au | — | 7.87 × 10−01 | 4.06 × 10−16 |
Values of rate constant higher than 1.00 × 106 s−1 represents extremely rapid diffusion, where CO molecules move between adsorption sites almost continuously. Values around 1.00 s−1 reflect a slow diffusion, the energy barrier is not insurmountable but still significant enough to slow down the diffusion process. Finally, values below 1.00 × 10−6 s−1 indicate an almost complete suppression of CO diffusion for that path.
It is not computationally feasible to perform NEB calculations of all possible CO diffusion pathways across the surface. A slightly less costly method for the assessment of the diffusion pathways and potential energy barriers is through the use of a potential energy surface (PES), which identifies the lowest CO adsorption energy for a specific xy point on the metal surface.
Nevertheless, the application of these techniques to all the aforementioned surfaces entails a considerable computational expense. Consequently, we have elected to utilise the integrated methodology of CI-NEB and PES for the stepped Pt(442) system, which offers a more illustrative representation. The results are presented in Fig. 5, which comprises two rows. The top row depicts the atomic models, while the bottom row illustrates the corresponding PES maps, wherein the colours represent energy levels. The colour purple indicates a low-energy region, whereas the colour yellow represents a high-energy area. Conversely, the colour blue indicates a low-energy region, while the colour red represents a high-energy area. The scale on the right is expressed in electron-volts (eV).
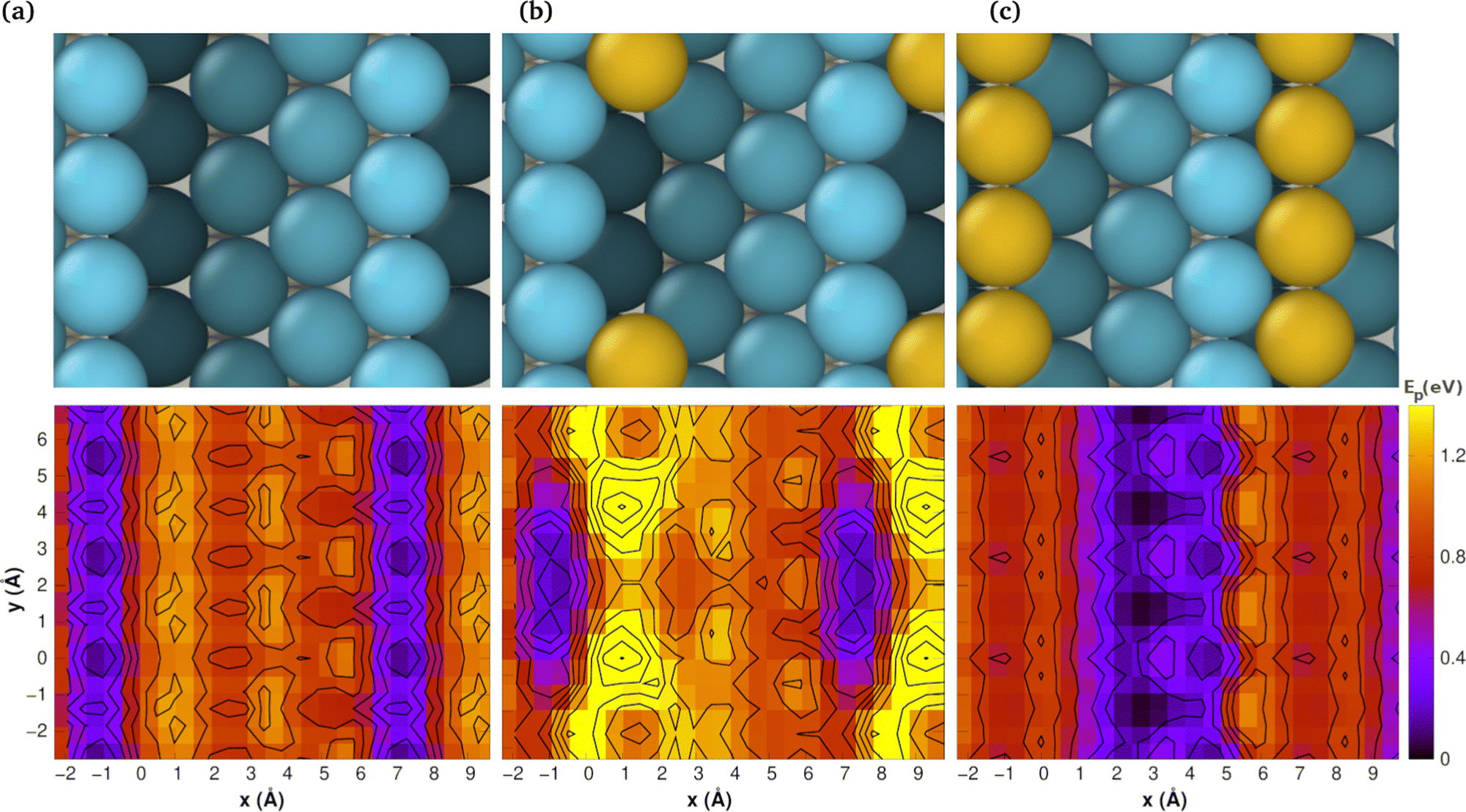 | ||
| Fig. 5 Top views and potential energy surfaces (PES) of (a) Pt(442), (b) Pt(442)Au@at and (c) Pt(442)Au@nw. The PES correspond to CO at several fixed xy position on the metal at their equilibrium height. The potential energy (Ep) is measured relative to the lowest adsorption energy for each surface. The contour lines represents energy differences of 0.2 eV. | ||
The most pertinent aspect of CO diffusion, as elucidated by the PES, is that diffusion in a direction perpendicular to the steps is markedly less probable than that in a direction parallel to the steps. In other words, CO encounters low-energy barriers when moving in a parallel direction to the edge and high barriers when moving across the steps.
In the case of Pt(442), the most favourable route for CO diffusion is along the top edge. Any alternative site is at least 0.8 eV higher in energy. This restriction is evidenced by the existence of a narrow, low-energy pathway within the PES.
In the case of a single Au atom at the step (Fig. 5b), the ease of CO diffusion along the top step is impeded by the presence of high-energy barriers, which are a consequence of the interactions between the CO molecule and the Au atom. This is demonstrated by the presence of yellow and red regions, which interrupt the blue stripe along the step.
Of interest is the observation that when a string of Au is covering the step (see Fig. 5c), the most facilitated route for CO diffusion shifts to the middle of the terrace, resulting in a markedly broader pathway. This is evidenced by the presence of a more extensive blue region in the centre of the PES map.
It is noteworthy that the PES maps exhibit periodic patterns, which correspond to the repeating structure of the surface. This conclusion is further supported by the experimental findings, which indicate a reduction in diffusion speed on Pt surfaces that have been modified with Au ad-atoms.23,30,33
4 Conclusions
Our studies of CO behaviour on Pt surfaces show significant differences between planar and stepped surfaces. On planar Pt surfaces, CO diffuses freely in all directions with low activation barriers. However, stepped surfaces exhibit anisotropic diffusion, where CO moves easily parallel to the steps but is hindered perpendicular to them, especially when crossing step edges.The presence of Au atoms has a significant impact on the adsorption and diffusion of CO. Although there was no discernible competition between Au and CO for adsorption sites, the preference of both for the step edges was evident. The proximity of Au atoms on the surface with CO adsorbed on Pt demonstrated a notable electrostatic repulsion, which affected its surface mobility. Even a single Au atom can impede the diffusion of CO along the step edges. The presence of Au nanowires on steps has been observed to shift the preferred CO diffusion path towards terrace centres, thereby altering the surface dynamics in comparison to those observed in pure Pt or surfaces with isolated Au atoms.
These effects of Au on CO behaviour explain the improved catalytic performance experimentally observed on Pt/Au surfaces. The Au atoms act as barriers, interfering with CO diffusion to and from active Pt sites. The present research offers a pioneering mechanistic understanding of the synergistic effects observed in bimetallic catalysts, thereby furnishing invaluable insights that can be employed in the design of more efficacious catalytic materials.
Author contributions
Ana Lucia Reviglio: investigation, validation, writing – review & editing. Paula S. Cappellari: conceptualization, investigation, writing – review & editing. German M. Soldano: methodology, investigation, writing – review & editing. Marcelo M. Mariscal: writing – review & editing. Gabriel A. Planes: conceptualization, writing – original draft, supervision.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank financial support from CONICET through Grant PIP 11220200101385CO, PIP 11220200102004CO, PIBAA 28720210100231CO, FONCyT PICT-A-2020-2943, PICT-A-2020-01436, PICT-I-2018-02221, PICT-I-2021-367, and SeCyT-UNC Program PAGE 1. A. L. R. thank CONICET for postdoctoral fellowship. Computational resources were provided by Centro de Computo de Alto Desempeño (CCAD-UNC), Universidad Nacional de Cordoba (https://ccad.unc.edu.ar/).Notes and references
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. Li, H. Wang, C. Priest, S. Li, P. Xu and G. Wu, Adv. Mater., 2021, 33, 2000381 CrossRef CAS.
- M. A. Abdelkareem, K. Elsaid, T. Wilberforce, M. Kamil, E. T. Sayed and A. Olabi, Sci. Total Environ, 2021, 752, 141803 CrossRef CAS.
- N. Dyantyi, J. Calderón Gómez, L. Mekuto, P. Bujlo and G. Pattrick, PEM Fuel Cells – Electrocatalysts: selectivity and utilization, Elsevier, 2022, ch. 3, pp. 55–70 Search PubMed.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: fundamentals and applications, John Wiley & Sons, 2022 Search PubMed.
- C. G. Zoski, Handbook of electrochemistry, Elsevier, 2007 Search PubMed.
- R. Adzic, N. Marinkovic, R. Adzic and N. Marinkovic, Platinum monolayer electrocatalysts, Springer, 2020 Search PubMed.
- Á. Cuesta and C. Gutiérrez, Catal. Electrochem., 2011, 339–373 Search PubMed.
- C. Farrell, C. Gardner and M. Ternan, J. Power Sources, 2007, 171, 282–293 CrossRef CAS.
- M. T. Darby, E. C. H. Sykes, A. Michaelides and M. Stamatakis, Top. Catal., 2018, 61, 428–438 CrossRef CAS PubMed.
- C. P. O’Brien and I. C. Lee, J. Phys. Chem. C, 2017, 121, 16864–16871 CrossRef.
- N. Shaari, S. K. Kamarudin, R. Bahru, S. H. Osman and N. A. I. Md Ishak, Int. J. Energy Res., 2021, 45, 6644–6688 CrossRef CAS.
- V. F. Valdés-López, T. Mason, P. R. Shearing and D. J. Brett, Prog. Energy Combust. Sci., 2020, 79, 100842 CrossRef.
- P. J. Sarma, C. L. Gardner, S. Chugh, A. Sharma and E. Kjeang, J. Power Sources, 2020, 468, 228352 CrossRef CAS.
- M. Subhramannia and V. K. Pillai, J. Mater. Chem., 2008, 18, 5858–5870 RSC.
- S. M. McClure and D. W. Goodman, Chem. Phys. Lett., 2009, 469, 1–13 CrossRef CAS.
- N. Lebedeva, M. Koper, E. Herrero, J. Feliu and R. Van Santen, J. Electroanal. Chem., 2000, 487, 37–44 CrossRef CAS.
- R. Antoniassi, J. Silva, T. Lopes, A. O. Neto and E. Spinacé, Int. J. Hydrogen Energy, 2017, 42, 28786–28796 CrossRef CAS.
- A. Ferre-Vilaplana, R. Gisbert and E. Herrero, Electrochim. Acta, 2014, 125, 666–673 CrossRef CAS.
- G. García and M. T. Koper, Phys. Chem. Chem. Phys., 2008, 10, 3802–3811 RSC.
- G. García, A. González-Orive, M. Roca-Ayats, O. Guillén-Villafuerte, G. Á. Planes, M. V. Martínez-Huerta, A. Hernández-Creus and E. Pastor, Int. J. Hydrogen Energy, 2016, 41, 19674–19683 CrossRef.
- Q. Xu, S. Liu, F. Longhin, G. Kastlunger, I. Chorkendorff and B. Seger, Adv. Mater., 2024, 36, 2306741 CrossRef CAS.
- D. Minudri, A. Y. Tesio, F. Fungo, R. E. Palacios, P. S. Cappellari, E. Pastor and G. A. Planes, J. Power Sources, 2021, 483, 229189 CrossRef CAS.
- M. Awad, T. Ohsaka and M. A. Kassem, J. Mol. Liq., 2021, 344, 117938 CrossRef CAS.
- G. García and M. T. Koper, ChemPhysChem, 2011, 12, 2064–2072 CrossRef.
- Z. A. Makrodimitri, D. J. M. Unruh and I. G. Economou, Phys. Chem. Chem. Phys., 2012, 14, 4133–4141 RSC.
- X. Ren, Q. Lv, L. Liu, B. Liu, Y. Wang, A. Liu and G. Wu, Sustainable Energy Fuels, 2020, 4, 15–30 RSC.
- J. Wang, C. Liu, S. Li, Y. Li, Q. Zhang, Q. Peng, S. T. John and Z. Wu, Chem. Eng. J., 2022, 428, 132558 CrossRef CAS.
- S. Wang, N. Kristian, S. Jiang and X. Wang, Nanotechnology, 2008, 20, 025605 CrossRef PubMed.
- P. S. Cappellari, G. García, J. Florez-Montano, C. A. Barbero, E. Pastor and G. A. Planes, J. Power Sources, 2015, 296, 290–297 CrossRef CAS.
- P. S. Cappellari, A. M. Baena-Moncada, R. Coneo-Rodríguez, M. S. Moreno, C. A. Barbero and G. A. Planes, Int. J. Hydrogen Energy, 2019, 44, 1967–1972 CrossRef CAS.
- R. Muralidharan, M. McIntosh and X. Li, Phys. Chem. Chem. Phys., 2013, 15, 9716–9725 RSC.
- G. Del-Giudice, A. Y. Tesio, P. S. Cappellari, R. E. Palacios and G. A. Planes, Electrochim. Acta, 2018, 270, 48–53 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, et al. , J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- K. Laasonen, A. Pasquarello, R. Car, C. Lee and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 10142 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef.
- K. F. Garrity, J. W. Bennett, K. M. Rabe and D. Vanderbilt, Comput. Mater. Sci., 2014, 81, 446–452 CrossRef CAS.
- V. Ásgeirsson, B. O. Birgisson, R. Bjornsson, U. Becker, F. Neese, C. Riplinger and H. Jónsson, J. Chem. Theory Comput., 2021, 17, 4929–4945 CrossRef.
- D. Sheppard, R. Terrell and G. Henkelman, J. Chem. Phys., 2008, 128, 134106 CrossRef PubMed.
- R. ShyamYadav, M. G. Gebru, H. Teller, A. Schechter and H. Kornweitz, J. Mater. Chem. A, 2024, 12, 3311–3322 RSC.
- S. Xue, P. Chaudhary, M. R. Nouri, E. Gubanova, B. Garlyyev, V. Alexandrov and A. S. Bandarenka, J. Am. Chem. Soc., 2024, 146, 3883–3889 CrossRef CAS PubMed.
- A. Boronat-González, E. Herrero and J. Feliu, J. Solid State Electrochem., 2020, 24, 2871–2881 CrossRef.
- C. Busó-Rogero, E. Herrero, J. Bandlow, A. Comas-Vives and T. Jacob, Phys. Chem. Chem. Phys., 2013, 15, 18671–18677 RSC.
- A. Christensen, A. V. Ruban, P. Stoltze, K. W. Jacobsen, H. L. Skriver, J. K. Nørskov and F. Besenbacher, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 5822–5834 CrossRef CAS.
- M. Pedersen, S. Helveg, A. Ruban, I. Stensgaard, E. Lægsgaard, J. Nørskov and F. Besenbacher, Surf. Sci., 1999, 426, 395–409 CrossRef CAS.
- E. Santos and W. Schmickler, ChemPhysChem, 2006, 7, 2282–2285 CrossRef CAS PubMed.
- E. Santos, P. Quaino and W. Schmickler, Phys. Chem. Chem. Phys., 2012, 14, 11224–11233 RSC.
- J. M. Piñeiros-Bastidas, S. V. Auras and L. B. Juurlink, Surf. Sci., 2023, 732, 122270 CrossRef.
- E. D. Williams, Surf. Sci., 1994, 299–300, 502–524 CrossRef CAS.
- E. Preuss, N. Freyer and H. P. Bonzel, Appl. Phys. A: Solids Surf., 1986, 41, 137–143 CrossRef.
- M. Mantina, Y. Wang, R. Arroyave, L. Q. Chen, Z. K. Liu and C. Wolverton, Phys. Rev. Lett., 2008, 100, 215901 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03618b |
| This journal is © the Owner Societies 2024 |