
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe route for commercial photoelectrochemical water splitting: a review of large-area devices and key upscaling challenges
António
Vilanova
 ab,
Paula
Dias
a,
Tânia
Lopes
a and
Adélio
Mendes
*a
ab,
Paula
Dias
a,
Tânia
Lopes
a and
Adélio
Mendes
*a
aLEPABE – Laboratory for Process Engineering, Environment, Biotechnology and Energy, Faculty of Engineering, University of Porto, Rua Dr. Roberto Frias, 4200-465 Porto, Portugal. E-mail: mendes@fe.up.pt
bINL – International Iberian Nanotechnology Laboratory, Avenida Mestre José Veiga, 4715-330, Braga, Portugal
First published on 30th January 2024
Abstract
Green-hydrogen is considered a “key player” in the energy market for the upcoming decades. Among currently available hydrogen (H2) production processes, photoelectrochemical (PEC) water splitting has one of the lowest environmental impacts. However, it still presents prohibitively high production costs compared to more mature technologies, such as steam methane reforming. Therefore, the competitiveness of PEC water splitting must rely on its environmental and functional advantages, which are strongly linked to the reactor design, to the intrinsic properties of its components, and to their successful upscaling. This review gives special attention to the engineering aspects and categorizes PEC devices into four main types, according to the configuration of electrodes and strategies for gas separation: wired back-to-back, wireless back-to-back, wired side-by-side, and wired separated electrode membrane-free. Independently of the device architecture, the use of concentrated sunlight was found to be mandatory for achieving competitive green-H2 production. Additionally, feasible strategies for upscaling the key components of PEC devices, especially photoelectrodes, are urgently needed. In a pragmatic context, the way to move forward is to accept that PEC devices will operate close to their thermodynamic limits at large-scale, which requires a solid convergence between academics and industry. Research efforts must be redirected to: (i) build and demonstrate modular devices with a low-cost and highly recyclable embodiment; (ii) optimize thermal and power management; (iii) reduce ohmic losses; (iv) enhance the chemical stability towards a thousand hours; (v) couple solar concentrators with PEC devices; (vi) boost PEC-H2 production through the use of organic compounds; and (vii) reach consensual standardized methods for evaluating PEC devices, at both environmental and techno-economic levels. If these targets are not met in the next few years, the feasibility of PEC-H2 production and its acceptance by industry and by the general public will be seriously compromised.
 António Vilanova | António Vilanova obtained an MSc degree in environmental engineering, in 2014, from the Faculty of Engineering of the University of Porto (FEUP) and completed his PhD degree in 2021 at the University of Porto on Photoelectrochemical devices for solar H2 production. From 2021 to 2023 A. Vilanova worked as a research fellow in the NOA group at the International Iberian Nanotechnology Laboratory. A. Vilanova currently works as an innovation developer at Capwatt and he is an effective member of the Portuguese Engineers Association. Research topics of interest include renewable fuels production, PEC water splitting, energy storage systems, CFD, carbon footprint, LCA, and circular economy. |
 Paula Dias | Paula Dias is a chemical engineer and junior researcher at LEPABE-FEUP. She received her PhD degree in chemical and biological engineering from the University of Porto in 2016 on the development of innovative materials for Photoelectrochemical (PEC) water splitting. Her research activities aim at (photo)electrochemistry, namely PEC devices for solar energy harvesting, conversion and storage, redox flow cells, and hydrogen production through methane splitting technology. P. Dias has been involved in several research projects and, presently, (co-)leads ASAPFuels, TanPT, and 112CO2. Currently, P. Dias is an invited Professor for the Renewable Energies I course at the FEUP, and co-founder of Pixel Voltaic and Halius. |
 Tânia Lopes | Tânia Lopes, a chemical engineer and assistant researcher at LEPABE-FEUP, earned her PhD in Chemical and Biological Engineering from the University of Porto in 2014 on Photoelectrochemical (PEC) Cells for Solar Hydrogen Production. Her current research centers on PEC systems for solar energy harvesting, conversion and storage (experimental and theoretical studies, and scaling-up strategies); electrolysis and synthetic fuels production (renewable methanol); and thermal energy storage systems. She has been contributing to over 20 research projects (6 funded by the EU), and (co-)leads HopeH2, ASAPFuels and H2Driven. She is an invited Professor for the Renewable Energies III and Automated Laboratory Data Acquisition courses at the FEUP. |
 Adélio Mendes | Adélio Mendes is a Full Professor at the Chemical Engineering Department, Faculty of Engineering, University of Porto, and he leads the Energy, Processes, and Products group at the Laboratory for Process Engineering Environment Biotechnology and Energy. He is the author of more than 450 peer-reviewed articles and a textbook, the inventor or co-inventor of more than 35 patents, and the coordinator of a FET-Proactive EU project. Prof. Mendes has been awarded 17 notable distinctions, namely the Municipal Medal of Merit – Gold Grade (Porto, 2015), the Coimbra University Prize of 2016, and the Technological Innovation Award of the University of Porto (2017). |
1. Introduction
Renewable energy sources have been widely used by humankind for several millennia. In fact, until 250 years ago, hydro, wind, solar, and biomass were the only known and available energy sources.1 After the Industrial Revolution in the 18th century, human society witnessed unprecedented technological development, with fossil fuels in the spotlight of energy production.2 Energy demand never stopped growing, as the world population started experiencing significant growth, along with the improvement of living conditions.3,4 As a consequence, energy consumption resulted in a progressively higher negative impact on the environment.5,6 Emissions of greenhouse gases (GHGs) have continuously increased due to the consumption of fossil fuels, leading to an accelerated global warming phenomenon with severe consequences for the climate stability of planet Earth, resulting in extreme and unpredictable weather events.7,8 There is a global consensus to implement drastic changes in the way energy is harvested, stored and used, leading to an extensive reduction of GHG emissions aiming at keeping global warming below 1.5 °C relative to the pre-industrial times.9 As humankind enters into the 4th Industrial Revolution, such a goal represents a great challenge to the scientific community, industry, and governments.10,11Solar-powered technologies for producing chemical and electrochemical fuels are a clean, cost-competitive alternative to traditional fossil-fuel-based systems. Electrochemical solar hydrogen (H2) is particularly interesting since it allows the efficient harvest and conversion of abundant solar energy into a storable, transportable, environmentally friendly fuel. Green-H2 is a valuable industrial chemical feedstock, especially for producing ammonia and steel and for CO2 reduction into liquid synthetic fuels.12,13 It can also be used in medium- and high-temperature heating processes in the industry for which electricity is not effective, or converted downstream into electricity for stationary or mobile applications.14 These applications complement currently available technologies that capture solar energy and generate electricity (e.g. photovoltaics) or heat (e.g. solar-thermal systems).15 At present, the most relevant processes for generating solar-H2 are: (i) photovoltaic–electrolysis (PV–EC); (ii) photoelectrochemical (PEC) water splitting; (iii) photocatalysis; (iv) solar thermochemical processes; and (v) biological processes. Decoupled PV–EC is the available technology with the highest technological readiness level (TRL) due to the maturity of both photovoltaic (PV) and electrolyser (EC) technologies, which have been optimized for decades.15 However, PEC systems integrate light absorption and electrocatalysis in a single device and provide the opportunity to use thermal management for improving energy conversion efficiency.16,17 The conversion of solar energy into chemical energy can also be accomplished via photocatalytic water splitting using particulate semiconductors, which is considered a promising technology with tremendous potential for large-scale applications, not only due to the readiness level on the photocatalyst synthesis but also due to the simple reactor designs.18 Although the scope of the present review is not related to photocatalytic water splitting, one of the largest demonstrations of direct solar conversion into hydrogen is attributed to the research group of Domen, who successfully demonstrated a panel of 1 m2-size photocatalytic water-splitting working under natural sunlight with a solar-to-H2 (STH) conversion efficiency of 0.4%.19 The authors used a particulate RhCrOx/SrTiO3:Al photocatalyst in a one-step photoexcitation route.20
The successful implementation of PEC water splitting systems in the renewable energy market relies on their efficiency, durability, scalability, and cost-effectiveness. This route requires the optimization of device architectures for developing PEC systems that comply with the circular economy principles, comprising technical solutions already developed for high-performing laboratory cells.16,21 The history of modern photoelectrochemistry is based on discoveries made in the mid-20th century on semiconductor surfaces.22–24 Building upon these findings, notable advancements in photoelectrochemistry occurred in subsequent years, as is the case of the photoelectrocatalytic splitting of water. The original concept of PEC water splitting was first demonstrated by Boddy, in 1968,25 who used an n-type titanium dioxide (TiO2) semiconductor to drive the water splitting reaction upon UV-light illumination and applying an external bias potential. However, it was the pivotal work of Fujishima and Honda, in 1972,26 that boosted the research interest in the PEC water splitting field and different PEC devices and configurations have been studied since then. Thereafter, and over the last 50 years, PEC water splitting has been widely and exhaustively investigated. Unfortunately, the development and optimization of PEC devices addressing key upscaling challenges has received considerably less attention than materials development at lab-scale. Luckily, an ongoing change in paradigm is perceptible in the literature, with increasing relevance being given to the design and construction of large-area, or at least scalable, devices – Fig. 1. It is clear that the number of publications demonstrating large-area PEC devices almost doubled every five years for the past twenty-five years.
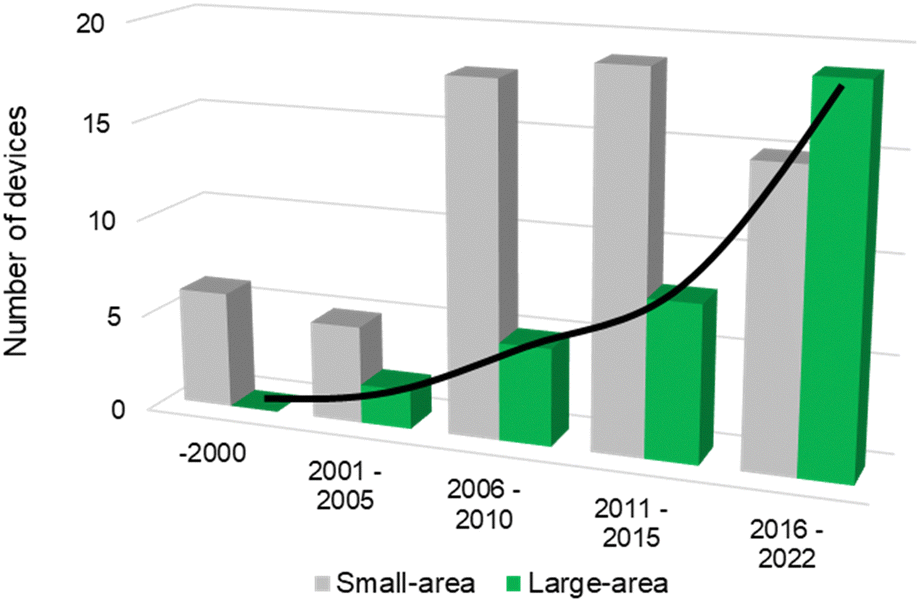 | ||
| Fig. 1 Histogram of reported PEC reactors over the last 25 years concerning the number of small-area devices (grey) and large-area devices with an illuminated area higher than 50 cm2 (green) – (searched keywords: “photoelectrochemical” + “water splitting” + “small area” + “large area” + “device”; database: Google Scholar, Scopus and SolarfuelsDB27). | ||
The STH conversion efficiency is a critical parameter to assess the competitiveness of PEC systems.28 The United States Department of Energy has set an efficiency target of 25% to reach market competitiveness of PEC water splitting systems.29 The highest reported STH efficiency of 19.3% was achieved using multijunction monolithic PEC cells incorporating III–V semiconductors, but the technology is still far from commercialization due to its high cost and complexity.30 Up to date, no single, earth-abundant and low-cost semiconductor material was identified to promote unbiased solar water splitting, delivering a high efficiency while operating stably on a timescale of years. Due to the high potential difference needed to drive the overall water splitting reaction (typically >1.6 V), a dual-absorber tandem configuration, in which the bottom and top materials absorb complementary regions of the solar spectrum, has been increasingly investigated to enhance the generated photopotential.28 To achieve STH efficiencies higher than 25%, pairing top absorbers with a bandgap range of 1.65–1.80 eV and bottom absorbers with a bandgap range of 0.95–1.15 eV is necessary.31 The PEC–PV device, which combines the photoelectrode paired in tandem with a PV cell, envisaged as the most feasible tandem approach, surpassing the efficiency of PV–EC systems when using the same PV technology in both cases. As demonstrated by Rothschild and Dotan, a tandem PEC–PV system can be more competitive than a PV–EC system (using the same PVs in both cases).32 By using a figure-of-merit (FOM) that considers the ratio between the STH efficiency of the complete system and the power conversion efficiency (PCE) of the PV module that drives the system, and then representing this FOM as a function of the fraction of the total power allocated to chemical power generation (assuming PV cells with a PCE of 20%, an electrolysis efficiency of 68.3% for the electrolysers and PEC cells operating at the same bias potentials and current densities), the authors identified several points to boost the efficiency of PEC–PV tandem systems, e.g. through the use of power management and light management strategies.32 Among the studied PEC–PV tandem configurations, special attention has been given to coupling a typical semiconductor in series with low-cost PV cells, including silicon (Si) PV solar cells, perovskite solar cells (PSCs), and dye-sensitized solar cells (DSSCs). As an example, tandem systems using a bismuth vanadate (BiVO4) photoanode were reported, displaying photocurrent densities close to the theoretical maximum limit.33–35 Recently, the remarkable 17.6% STH efficiency was reported for a low-cost PEC–PV tandem cell comprising perovskite–silicon photocathode absorber.36 These results show that there are ample opportunities for further improving the efficiency and reducing the costs of PEC systems, namely through the systematic optimization of each component individually. Along with performance indicators, the cost of produced H2 is a critical metric for assessing the viability and future commercialization of the PEC–PV water splitting technology – Fig. 1. Pinaud et al.37 estimated a levelized cost of H2 (LCOH) from PEC systems between 4.10 USD and 10.40 USD per kg of H2, considering an STH efficiency of 10%, a lifetime of 10 years, and panel costs between 153 USD m−2 and 316 USD m−2. Given an equivalent STH efficiency, Shaner et al.38 concluded that utilizing an EC stack and a PV module would result in a higher cost of dispensed H2, mainly due to the fact that transporting H2 from the PV panels to compression units would be less expensive than transporting and conditioning electricity from the panels to the electrolysers; still, the authors highlight that PEC systems still need great advancements, namely regarding the panel design and mounting to become truly competitive against grid-supplied PV–EC units.
As schematized in Fig. 2, a set of key requirements should be considered in fully integrated PEC–PV tandem systems. Apart from the performance of the photoabsorber materials and electrocatalysts, when commercialization is brought into sight, the PEC reactor itself, integrating all components, plays a crucial role in balancing the cost-effectiveness of such systems. It was established that an optimized PEC reactor must address the following requirements:39,40 (i) maximum absorption of solar energy by the photoelectrodes and/or PV components (also under concentrated sunlight); (ii) electrolyte recirculation with an optimized internal flow pattern, assuring efficient heat dissipation and gas bubble removal; (iii) optimum arrangement of electrodes to minimize overpotential losses (ionic and electronic); (iv) efficient separation of the evolved gases; (v) comprise photo and chemically stable components; (vi) construction using highly recyclable components; (vii) use of inexpensive materials and low-cost maintenance; and (viii) scalability and easy integration in an H2 production plant. A well-engineered PEC reactor that is suitable for large-scale PEC–PV systems must combine all these requirements.41
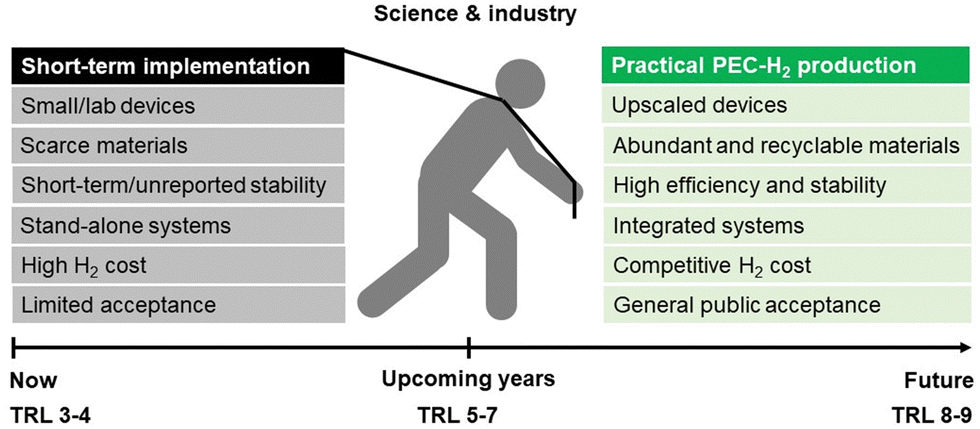 | ||
| Fig. 2 Holistic pathway for commercial PEC-H2 production and key challenges that must be addressed simultaneously by the scientific community and the industry. | ||
The following sections will address the progress on materials engineering for the main components of PEC systems, key design features of the main PEC device architectures, the challenges resulting from the use of concentrated sunlight and biomass derivatives, as well as the current status of PEC-H2 production based on environmental and economic metrics.
2. PEC water splitting components
The field of materials remains the most challenging for addressing the key challenges for commercial PEC water splitting, mainly because: (i) for long-term PEC operation, the photoelectrode (PE), the counter-electrode (CE) and the membrane need to be chemically stable in the presence of aqueous electrolytes and evolved gases, as well as continuous electrolyte recirculation, exposure to normal and concentrated sunlight; and (ii) each component of the device must incorporate Earth-abundant, non-toxic and low-cost materials, prepared by simple and scalable processes.42,43 This section reviews promising materials used in the main components of a PEC device, focusing on low-cost and scalable fabrication techniques and strategies for improving their performance.2.1. Photoelectrodes
PEC water splitting devices rely on photoactive electrodes, i.e. photoelectrodes, which have the ability to convert incident photons into electron–hole pairs (e−CB + h+VB) in the conduction and valence bands, respectively, when exposed to sunlight.44 This is only possible if photons’ energy (E), h·ν (h is the Planck's constant and ν is the frequency), is equal to or higher than the semiconductor's energy bandgap (EG); EG is given by the difference between the bottom of the conduction band (EC) and the top of the valence band (EV).45 When the semiconductor is in contact with an electrolyte whose redox energy (ERedox) differs from the Fermi level energy (EF), the equilibrium of the electrochemical potential is achieved by electron exchange at the interface between the semiconductor and the electrolyte. Electrons are transferred to the electrolyte, leaving in the semiconductor a depleted layer (space charge layer);44 the Fermi level is then shifted due to the formation of an electric double layer at the electrode–electrolyte interface.46 The depletion in electron concentration at the surface of the PE induces an electric field between the semiconductor's bulk and its surface, leading to the band bending phenomenon.44,47 For a given semiconductor/electrolyte combination, there is a unique potential at which the potential drop between the surface and the bulk is zero, and there is no space charge layer – the flatband potential. For a certain redox pair, the flatband potential corresponds to the redox potential of the electrolyte when there is no excess charge in the semiconductor and, consequently, no band bending.48 When a semiconductor, in equilibrium with an electrolyte, absorbs light, it has the capacity to change the band bending, since the electron and hole densities increase due to light absorption.44,47 One of the key aspects concerning the choice of a semiconductor is related to its band edge positions, which determine the upper limits of reduction and oxidation potentials for the photogenerated electrons and holes.44 The energy difference between the oxidizing hole and the reducing electron should be higher than the electrochemical potential required for water dissociation (>1.23 V).47,49 An EG of 1.6–2.0 eV, which covers the enthalpy of the reaction (1.48 V) and overpotentials, is considered to be the best compromise for a PE used in a PEC device.Given these assumptions, the standardized metric for evaluating the overall performance of a PEC system is STH efficiency (ηSTH), defined as the amount of chemical energy produced, i.e. the rate of hydrogen production (mmol of H2 s−1) against the solar energy input. The chemical energy produced is multiplied by the change in Gibbs free energy per mol of H2 (at 25 °C, ΔG = 237 kJ mol−1). The solar energy input is the incident illumination power density (Pin – 0.1 W cm−2, AM 1.5 G, considering standard PEC operating conditions) multiplied by the illuminated PE area (A, cm2). The H2 production rate can be directly measured by gas chromatography or mass spectrometry.
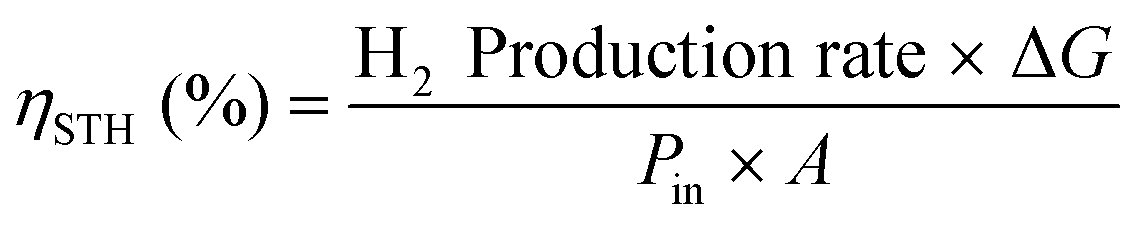 | (1) |
Additionally, ηSTH can also be determined using the relation that power is the product of potential, current, and the faradaic efficiency for H2 evolution (ηF) as follows:
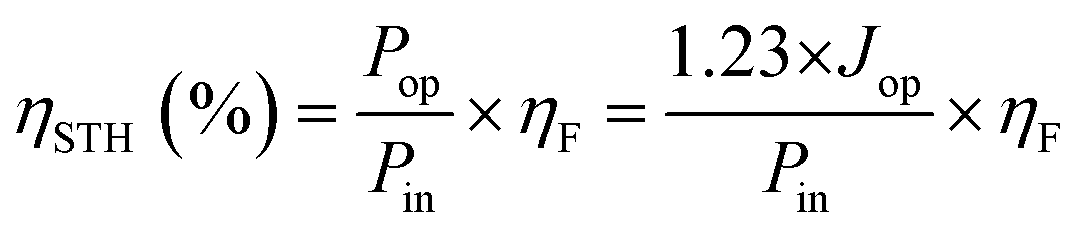 | (2) |
 | ||
| Fig. 3 Conventional PEC device: (a) schematic representation of a generic PEC device, assembled in a three-electrode configuration, including the relevant reactions of photo-oxidation of water using an n-type photoelectrode. (b) Simplified schematic of a potentiostat attached to a PEC cell using working electrode (WE), working sense (WS), counter-electrode (CE), and reference electrode (RE) leads. The working electrode is typically the photoelectrode used for studying any electrochemical processes. The potential within the cell is measured between RE and WS. The current flow through both potential-sensing electrodes is minimized (ideally zero). The CE completes the electrical circuit. Current (I) flows between CE and WE, while potential (E) is read between WE and WS. Energy diagram of a PEC cell based on: (c) a photoanode (n-type semiconductor) and a dark electrode (metal cathode) in the dark (left side) and under illumination (right side); (d) a photocathode (p-type semiconductor) and a dark electrode (metal cathode) under illumination; and (e) a tandem arrangement, which combines a photoanode (n-type semiconductor) and a photocathode (p-type semiconductor) under illumination. | ||
PEC devices designed for practical applications usually consist of a narrow reactor – the cell – where the electrolyte and both electrodes are inserted, typically assembled in separate compartments divided by an ion exchange membrane, which allows for charge balance while avoiding gas mixing. Front and/or back transparent windows allow sunlight to reach the surface of the photoactive materials.51 Still, when someone addresses the topic of materials within the field of PEC-H2 production, the component that instantly comes to mind is the photoabsorber material, i.e. the semiconductor or photoelectrode, as it causes the photon-to-H2 conversion.52 In fact, PEs are the key component that makes PEC cells more attractive compared to PV–EC systems, as they enable light absorption, charge generation and separation, hole, electron, ionic and molecular transport in a single device. Additionally, PEC devices can be operated at lower current densities, exhibiting lower overpotentials than commercial electrolysers.53,54
An ideal semiconductor for PEC water splitting should meet the following key criteria: (i) good visible light absorption and narrow bandgap energy between 1.8 eV and 2.4 eV; 1.8 eV corresponds to the theoretical minimum bandgap for water splitting of 1.23 eV plus the thermodynamic losses (0.3–0.4 eV) and overpotentials (0.4–0.6 eV) and 2.4 eV is the maximum bandgap value to achieve the target of 10% STH efficiency; (ii) suitable valence and conduction band edge positions, as the valence band should be below the redox potential for O2 evolution and the conduction band should be above the redox potential for H2 evolution; (iii) effective separation and transport of charge carriers, relying on both intrinsic characteristics (hole and electron mobility) and extrinsic properties (surface area, crystallinity, grain boundaries); (iv) high catalytic activity, essential for avoiding surface recombination and for decreasing the activation overpotentials; (v) chemical stability, which depends on the relative positions of the band edges and the respective decomposition potentials; and (vi) non-toxicity, Earth-abundance and low-cost fabrication.39,55 So far, there is no single semiconductor reported that meets all these requirements.
The materials used to prepare photoelectrodes for PEC water splitting can essentially be divided into the so-called “classic” and emerging semiconductor materials.56–58 Considering the short time window for making PEC water splitting competitive, conventional metal-oxide semiconductor materials should be preferable for optimizing large-area PEs, since they are easy to prepare and have already been deeply studied and demonstrated. Typically, transition metal-oxide PEs include TiO2, hematite (α-Fe2O3), tungsten trioxide (WO3), bismuth vanadate (BiVO4), and cuprous oxide (Cu2O); tantalum nitride (Ta3N5) and silicon-based PEs are also being highly reported. Among the emerging materials, organic, perovskite and kesterite semiconductors are three of the most studied.
Hematite (α-Fe2O3) is one of the most promising photoanodes for solar water splitting and has been widely studied since the late 1990s. This metal-oxide presents remarkable chemical stability, low toxicity, and low-cost.64 Also, it has a bandgap between 1.9 eV and 2.3 eV, which is nearly ideal for visible light harvesting, resulting in a maximum theoretical STH efficiency of ca. 16% (corresponding to 12.6 mA cm−2 at 1.23 VRHE).59 However, its use as a semiconductor for PEC-H2 generation entails some drawbacks, including: (i) slow mobility of holes (ca. 0.2 cm2 V−1 s−1), resulting in a very short diffusion length of 2–4 nm; (ii) poor surface water oxidation kinetics, leading to high surface recombination due to the accumulation of holes; and (iii) low absorption coefficient (on the order of 103 cm−1), requiring film thicknesses of at least 400–500 nm for full sunlight absorption.65,66 Among the various reported techniques to prepare α-Fe2O3 PEs, solution-based methods have raised particular interest. The resulting PEs show different nanostructured morphologies, like nanorods, nanowires or worm-like protuberances, all with a high surface area. The state-of-the-art α-Fe2O3 PE is shaped in nanowire arrays, prepared by a hydrothermal method, and is capable of delivering a record photocurrent density of 6 mA cm−2 at 1.23 VRHE (ca. 7% STH efficiency);67 this result corresponds to ca. 50% of the theoretical limit for this semiconductor. Still, nanostructured α-Fe2O3 presents stability challenges when compared with more compact structures, typically prepared by atomic layer deposition (ALD) or spray pyrolysis (SP). The most stable α-Fe2O3 PE reported to date is a 19 nm film prepared by SP, that is capable of generating 1.05 mA cm−2 at 1.45 VRHE over 1000 h, under continuous simulated sunlight conditions, in 1 M KOH.64 This preparation technique was successfully upscaled to a 50 cm2 PE that generated ca. 0.45 mA cm−2 at a bias potential of 1.6 V, over 1008 h under 1000 W m−2, and with electrolyte recirculation at 45 °C.43 Indeed, this review's authors have consistently demonstrated the successful scale-up of flat and nanostructured α-Fe2O3 PEs over the years. Their achievements include scaling up to 25 cm2 nanostructured hematite of porous nanocubes,68 but also compact photoelectrodes up to 50 cm2 and 100 cm2.43,69
Tungsten trioxide (WO3) is another n-type semiconductor that has been studied for PEC applications since the mid-1970s, presenting a good electron mobility (6 cm2 V−1 s−1) and a long charge carrier diffusion length (ca. 150 nm). Although this material has a bandgap of 2.7–2.8 eV, larger than α-Fe2O3, it is still capable of absorbing a reasonable fraction of the solar spectrum, with limited maximum theoretical STH efficiency (4.8%).70 In contrast to α-Fe2O3, it is stable only in aqueous acidic solutions (pH < 4), such as H2SO4, H3PO4, Na2SO4 or CH3SO3H. There are several reported techniques for preparing low-cost and scalable nanostructured WO3 PEs.71,72 Sputtering allows obtaining photoanodes with good crystallinity, low intergrain and lattice defects; however, this technique is quite expensive and not easily scalable. Nanoporous or worm-like structures can be obtained by electrochemical anodization of metallic tungsten foils; the as-prepared PEs are opaque, and the roughness of the foil affects the growth shape and direction.73 Electrodeposition allows the deposition of WO3 films on glass substrates, but these PEs usually present low current densities (ca. 0.35 mA cm−2 at 1.23 VRHE).74 Overall, sol–gel remains the most common chemical process used to prepare WO3 PEs; the resulting semiconductor layer is thick and highly porous. Reinhard et al. reported bare WO3 PEs, prepared via a sol–gel method, consisting of consecutive depositions of a colloidal precursor solution by doctor-blading, followed by thermal annealing, which displays ca. 3.5 mA cm−2 at 1.23 VRHE, remaining the state-of-the-art achievement.75
Bismuth vanadate is the most successfully demonstrated ternary metal oxide photoanode, being also a promising candidate for large-area PEC-H2 production. This material has a bandgap of 2.4 eV, resulting in a theoretical maximum photocurrent density of 7.4 mA cm−2 at 1.23 VRHE, corresponding to an STH efficiency of 9.1%. Similar to α-Fe2O3, the performance of BiVO4 photoanodes is mainly limited by the fast charge carrier recombination, due to the short hole diffusion length, which is estimated to be 70–100 nm; it is also limited by the slow water oxidation kinetics.76,77 To overcome these limitations, BiVO4 is often doped with Mo and W, which enhances the charge carrier-separation efficiency.78 Spray pyrolysis is a common technique used to fabricate BiVO4 photoanodes;77 Ahmet et al. reported BiVO4 photoanodes prepared by spray pyrolysis that achieved a photocurrent density of ca. 3.3 mA cm−2 at 1.23 VRHE, with an onset potential of 0.4 VRHE.79 BiVO4 photoanodes are often used in hetero-junctions, usually with WO3, allowing photocurrent densities higher than 6 mA cm−2 to be to achieved at 1.23 VRHE, where nanostructured WO3 is used as scaffold for the BiVO4 top layer.76 There are several reported techniques for fabricating these heterostructures. Porous WO3 structures (the scaffold) can be fabricated using colloidal templates, such as polystyrene spheres; the BiVO4 precursor solution is later impregnated on the template followed by a heat treatment, which allows the removal of the polystyrene and crystallizing the doped BiVO4.80,81 Another interesting approach consists of combining glancing angle deposition (GLAD) to fabricate WO3 nanorods, followed by electrochemical deposition of BiVO4, which already allowed a PE displaying a photocurrent density of 6.72 mA cm−2 to be fabricated at 1.23 VRHE (>90% of its theoretical limit).82 The most stable BiVO4 PE was demonstrated by Kuang et al.83 These authors reported a particulate Mo:BiVO4/Ni/Sn electrode that generated ca. 2.6 mA cm−2 at 0.6 VRHE over 1100 h, without noticeable performance losses after the complete test; stability measurements comprised eleven 100-h runs, with electrolyte replacement between any two runs and in situ self-regeneration of the catalyst. The photocorrosion inhibition was attributed to the specific high-temperature treatment; in situ self-generation of catalyst was assured by the thin Ni layer, which acted simultaneously as an ion source and electrical back contact.
From the family of oxynitrides, tantalum nitride (Ta3N5) has received special attention, due to its 2.1 eV direct bandgap and suitably positioned energy band edges for solar water splitting applications. Indeed, Ta3N5 is probably the most promising semiconductor among the ones reaching photocurrents close to the theoretical limit of STH efficiency under sunlight. Liu et al. reported a multi-step strategy for improving the performance of Ta3N5 grown on Ta metal foils, which yielded a maximum photocurrent density of ca. 12.1 mA cm−2 at 1.23 VRHE, corresponding to 94% of its theoretical performance limit; this is an unprecedented achievement within the PEC-H2 field.84 The application of Ni(OH)x/ferrihydrite (Fh) layers onto Ta3N5 proved to be efficient in mediating the interfacial charge transfer with Co and Ir catalysts, and a TiOx blocking layer for reducing the electron–hole recombination. Additionally, the Fh overlayer was also employed for protecting Ta3N5 against photocorrosion.85 Despite this impressive progress, Ta3N5 still faces two critical shortcomings: poor stability and low photopotential. Fortunately, different strategies have been successfully employed to tackle these issues. First, stability can be improved, e.g. using electrolyte solutions containing a ferrocyanide hole scavenger; still, the reported 30 h of stable performance remains far from real application needs.86 On the other hand, the low photopotential can be overcome by using tandem arrangements, which have the potential to generate sufficient photopotential to achieve water splitting with high STH efficiency. The most efficient Ta3N5 PEs were prepared on opaque Ta foils, which limits the assembling options to front illumination only. To be used as front-side PE (back illumination), semi-transparent Ta3N5 has to be fabricated on transparent conductive oxide (TCO) glass substrates, but they seem to reach lower photocurrents.87 Higashi et al. fabricated a Ta3N5 planar thin film grown on a transparent GaN/Al2O3 substrate and decorated with an ultrathin NiFeOx electrocatalyst layer, generating 7.4 mA cm−2 at 1.23 VRHE.88 When paired in tandem with CuInSe2 PV, an STH efficiency of 9% was obtained, which is the highest efficiency ever reported among metal-oxide/nitride-based PEC–PV tandem cells; unfortunately, this device showed only 2 h of stable operation.
Spinel ferrites (MFe2O4, M = Zn, Mg, Cu, Ca, etc.) have also been the subject of several research studies on PEC water splitting. These ternary metal oxides are made of abundant materials, present a suitable bandgap for light harvesting in the visible range, i.e. 1.4–2.1 eV, and have displayed long-term photostability. ZnFe2O4 (known as ZFO), with a bandgap range of 1.9–2.1 eV, enables broad absorption of visible light while presenting favourable chemical and morphological stability.89 This material arose as a substitute for typically less efficient PEs, being frequently used as a secondary overlayer on composite PEs, such as TiO2/ZFO or α-Fe2O3/ZFO, boosting the charge extraction in the primary oxide.90 However, as the main active photoanode material, the performance of ZFO has remained modest; among the best results, Xu et al. reported ZFO decorated AZO nanowires that generated a photocurrent of ca. 1.72 mA cm−2 at 1.23 VRHE in a neutral electrolyte (0.1 M Na2SO4).89
Cuprous oxide is the most studied photocathode for solar water splitting, being non-toxic, and abundant, presenting a bandgap of 2.0 eV, suitable for absorbing a significant part of the solar spectrum.91 Despite having a maximum theoretical STH efficiency of 18%, Cu2O has two main drawbacks that strongly limit its use for practical applications. First, this material is unstable under water-based solutions, as the reduction and oxidation potentials of monovalent copper oxide are located within the water splitting potentials, favouring photocorrosion.92 Stability can be significantly improved by applying protective overlayers, such as TiO2 deposited by ALD; 50 h of stable performance was reported by Tilley et al.93 Also, combining Cu2O with an n-type semiconductor with a more positive conduction band, which forms a p–n junction that promotes a faster transfer of electrons from Cu2O to the n-type semiconductor, allows improving both stability and efficiency. The second major drawback of Cu2O is related to its charge carrier diffusion length. Cu2O films typically are >1 µm thick to efficiently absorb sunlight; however, the electron diffusion length is limited to about 200 nm, which hinders the collection of photogenerated carriers. Accurate morphology control, based on advanced preparation techniques that allow obtaining nanostructured PEs, is the most attractive approach to address this challenge. A good example was implemented by Pan et al.,35 reporting Cu2O photocathodes able to deliver an unprecedented photocurrent density of 10 mA cm−2 over 100 h of continuous operation, which was a state-of-the-art result, but still far from the ultimate target STH efficiency of 25%, set by the Department of Energy.29
Silicon-based PEs, the most common semiconductors in the PV industry, have also been widely used for PEC water splitting; the bandgap of 1.1 eV matches well with the solar spectrum, presenting a theoretical maximum photocurrent of 44 mA cm−2 under 1-sun. However, bare Si PEs undergo fast oxidation when in contact with aqueous electrolytes. This challenge can be overcome by applying protective layers that passivate the surface states of Si. As an example, the alignment of TiO2's conduction band with both the hydrogen evolution reaction (HER) and with Si enables its use as a protective overlayer; additionally, TiO2 facilitates electron transfer from Si with negligible resistance.94 Inspired by the low-cost and high open-circuit potential difference displayed by amorphous Si (a-Si) solar cells, Lin et al. reported a Si photocathode coated with a TiO2 protection layer, and with Pt or Ni–Mo co-catalysts that generated 10 mA cm−2 at 0.6 VRHE in 0.5 M KHP (potassium hydrogen phthalate), for 12 h of continuous operation.95 In 2020, Fu et al. reported a bifacial Si photocathode coated with a semi-transparent 5 nm Pt layer that showed an unprecedented photocurrent density of 61.2 mA cm−2 at 0 VRHE under bifacial 2-sun illumination; this system enables harvesting sunlight both at the front and at the back sides, yielding ca. 57% of excess H2 when compared with monofacial PEC systems, presenting a stable performance over 370 h in 1 M H2SO4.96 Despite being largely used in first generation PV cells, it is important not to forget that metallic Si is listed as critical raw material (CRM), so its use must be greatly reduced in the next few years, including in PEC applications.
Fig. 4(a) shows the number of publications on some of the most studied semiconductors over the past decades, namely α-Fe2O3, BiVO4, WO3, Cu2O and Ta3N5. Fig. 4(b) shows the state-of-the-art of these PE materials, based on the three-pillar criteria for commercialization: scalability, stability, and efficiency. Hematite α-Fe2O3 has been the most studied material, but publications on BiVO4 have increased exponentially in the last 5-year period, followed by Cu2O, Ta3N5, and WO3. Nevertheless, it is clear that efficiency has received more attention among the published research studies. So far, α-Fe2O3 is the semiconductor material that shows the best compromise among the three key criteria, although there is still no α-Fe2O3 PE capable of fully address simultaneously these targets. Nonetheless, some published studies have shown the potential of this semiconductor material to achieve important halfway targets for more than one pillar criterion simultaneously, such as an active area of ≥50 cm2 and a stable performance for more than 1000 h.43 Overall, these results highlight the complexity of preparing large-area PEs that are simultaneously efficient and stable; it is important to emphasize that the process of discovering functional PE materials typically takes a minimum of 10 years.
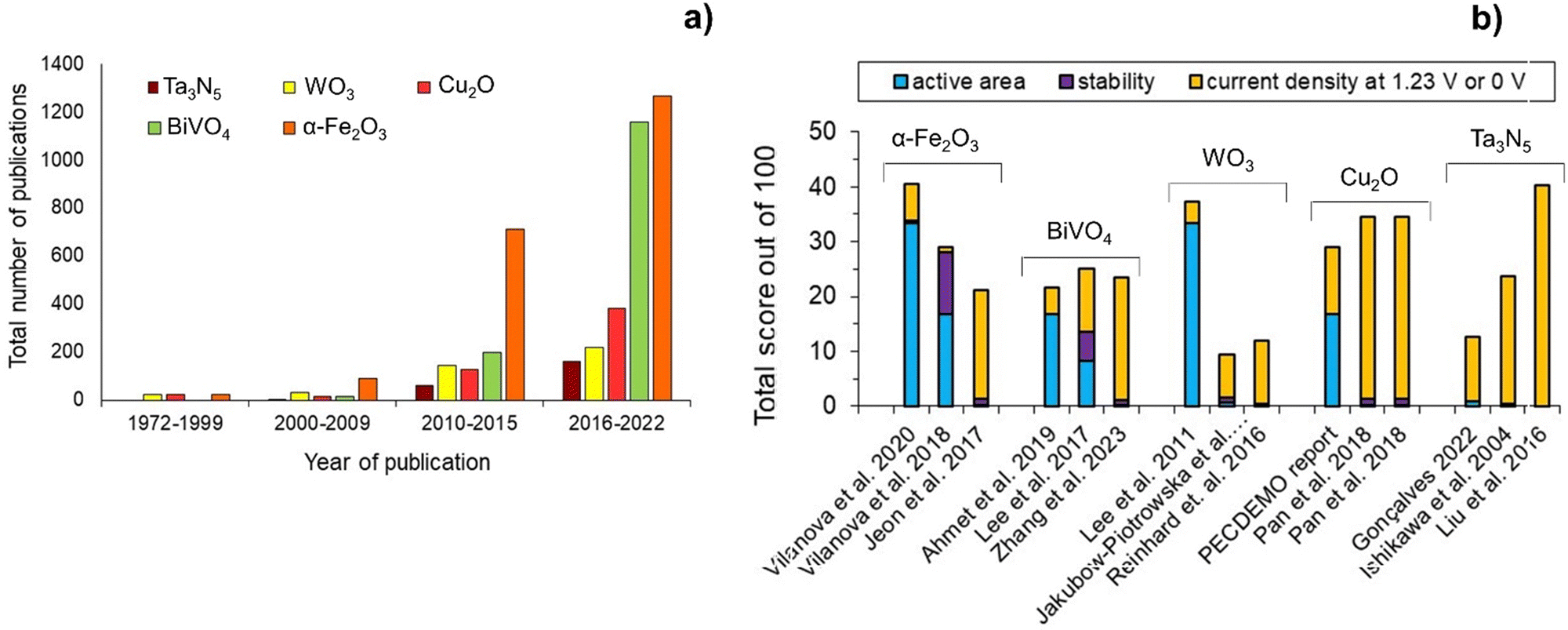 | ||
| Fig. 4 Overview of the most studied semiconductors for PEC water splitting: (a) number of publications over the past decades (database: Google scholar and SolarfuelsDB27); and (b) performance ranking of α-Fe2O3,43,67,97 BiVO4,79,98,99 WO3,75,100,101 Cu2O35,102 and Ta3N584,86,103 PEs based on active area, stability and current density generated at a reference potential of 1.23 V (except 0 V for Cu2O); normalization considered equal 33.3(3)% contribution for each criterion, delivering a total score out of 100, and considering the following targets: (i) active area of 100 cm2; (ii) 2920 h of stable performance (8 h over 365 days; max. 10% current loss); and (iii) current density of 10 mA cm−2. | ||
These advantages can be a differentiating aspect in PEC applications if successfully replicated. Nevertheless, the use of organic semiconductors in PEC devices has remained barely addressed, mainly due to their poor stability in aqueous solutions.106 Early organic PE materials included polypyrrole, polyacetylene, polythiophene derivatives, poly(ethylenedioxythiophene) (PEDOT) derivatives, polyaniline, polythiophene-based photocathodes, poly(thienothiophene) (PTT) and poly(dithienothiophene) (PDTT).107 Given the huge variety of organic semiconductors, they can essentially be grouped in dye-sensitized and thin films.105 Since the first demonstrations of efficient proton reduction using organic PEs, research has been focused on the buried-junction approach, especially on introducing an anti-corrosion layer, and on developing highly tunable non-fullerene acceptors and wide-bandgap donor polymers.108 Still far from the PEC performance of other semiconductor materials, the photocurrent density generated by organic photocathodes in PEC water-splitting devices has increased from a few µA cm−2 in acidic solutions at high negative potentials to over 100 µA cm−2 in neutral solutions at low/no applied potentials.107 Indeed, the rapid advances within the organic semiconductor field for solar water splitting have provided a vast number of high-performance donor polymers and non-fullerene acceptors with carefully tailored chemical structures, allowing them to reach high photocurrent and open circuit potential. As the band positions and bandgaps of organic semiconductors can be easily tuned, in multijunction devices, by combining two or more sub-cells with complementary bandgaps and connecting them in series, the total open circuit potential (EOC) can theoretically be sufficient to drive water splitting, resulting in a maximum STH efficiency of up to 30%, avoiding the need of coupling an external PV cell, which is often considered an essential requirement for PEC water splitting feasibility.109 Since water oxidation is the limiting half-reaction, and given that many organic materials are easily degraded upon oxygen exposure, organic photocathodes are considered more promising than organic photoanodes.110 Even so, high-performance and stable organic materials were already reported. As an example, Yu et al. used nickel foils, GaIn eutectic, and layered double hydroxides to passivate the photoactive layer of p-type polymers and n-type non-fullerene materials.111 These organic photoanodes generated 15.1 mA cm−2 at 1.23 VRHE and displayed an onset potential of 0.55 VRHE, resulting in an STH efficiency of 4.33%. These photoanodes were able to maintain 90% of the initial photocurrent for 10 h, when continuously operated at 1.3 VRHE. Zhang et al. have demonstrated a robust organic photocathode comprising a bulk heterojunction (BHJ) and an organic charge-selective layer.112 This optimized photocathode generated ca. 4 mA cm−2 at 0 VRHE, retaining 90% of the initial performance over 6 h. Combining the optimized BHJ photocathode with a benchmark BHJ photoanode, the authors then demonstrated a 2.4 cm2 organic PEC tandem cell for complete solar water splitting, capable of generating a photocurrent of 0.6 mA cm−2 in a two-electrode configuration, resulting in an STH efficiency of 0.3%. Finally, organic-based PEs include the so-called metal–organic framework-based materials (MOFs). These materials essentially comprise heterostructures used to fabricate thin films, where the MOF layer is used as a sacrificial template (Ti-based, Fe-based, Zn-based, and Co-based) or as a coating.113 An example of a MOF-based photoelectrode displaying good PEC performance was reported by Pan et al.; the authors used NiFe-metal–organic frameworks (NiFe-MOFs) as a protective layer and cocatalyst on oxygen-vacancy-rich BiVO4 (Ov-BiVO4), forming a core–shell structure that produced 5.3 mA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−2 at 1.23 VRHE.114
cm−2 at 1.23 VRHE.114
Since the early 2010s, metal halide perovskites have emerged as one of the most promising materials for low-cost and efficient third-generation PV cells.115 These materials comprise a polycrystalline structure with the chemical formula ABX3, with A referring to methylammonium (MA), formamidinium (FA), or cesium (Cs), B referring to Pb or Sn, and X referring to the halide atom, i.e. I−, Br−, or Cl−, or combinations of them.3 As photoabsorber materials for PV applications, perovskites exhibit remarkable optoelectronic properties, namely high absorption coefficients, high electron and hole mobilities and diffusion lengths, superior structural defect tolerance and shallow point defects, low surface recombination rate and favorable grain boundaries, which have led to an unprecedented increase in efficiency, from 3.8% in 2009 to the present 26.1%.116 Like in PV applications, perovskite materials, in this case perovskite oxides with the chemical structure ABO3, became highly attractive for PEC applications, offering better optoelectronic properties than simple oxides, mainly related to superior structural flexibility, high electrocatalytic activity, better sunlight absorption capability and simultaneous control of bandgaps and band edges.117 For practical PEC water splitting, perovskite materials can be used as the main photoelectrode, i.e., in contact with the electrolyte solution, or used in a tandem arrangement. In fact, Karuturi et al. achieved a remarkable STH efficiency of 17.6% for self-driven solar water splitting by pairing a semitransparent perovskite with a high bandgap (1.75 eV) with a p+nn+-Si/Ti/Pt photocathode capable of generating a current density of 39.7 mA cm−2 at 0 VRHE and an onset potential of 590 mV.36 The Reisner group has also been developing interesting solutions that use perovskites for solar water splitting, either as main PE or as dual absorbers in tandem arrangements.118,119 One of the most relevant contributions was reported in 2022, where the authors successfully demonstrated the fabrication of scalable lightweight artificial leaves made of lead halide perovskite photocathodes deposited onto indium tin oxide-coated polyethylene terephthalate, comprising thin, flexible substrates and carbonaceous protection layers. This lightweight perovskite–BiVO4 PEC device achieved an STH efficiency of 0.58% and its scalability was demonstrated using a 100cm2 stand-alone device, which sustained stable performance for 24h.120 Still, to be used as the main photoelectrode in conventional PEC device architectures, perovskite oxides have to be stable to the highly demanding operating conditions, namely to water exposure, temperature, and mechanical erosion promoted by gas bubbles evolution and electrolyte recirculation.121 Possible ways to overcome such adversities include the use of thin overlayers and encapsulation. An example of the successful application of a thin protective overlayer was demonstrated by Liu et al.122 The authors have reported porous perovskite photoanodes capable of being operated in standard PEC conditions, i.e. in contact with an aqueous electrolyte. The BiFeO3 photoanodes, covered with an ultra-thin TiO2 overlayer (5–6 nm) and a cobalt oxide/oxyhydroxide co-catalyst, generated 0.16 mA cm−2 at 1.23 VRHE in 1 M NaOH, achieving an onset potential of 0.6 VRHE. On the other hand, Chen et al. have successfully demonstrated encapsulated perovskite-based PEs for solar water splitting. In this case, the device layout consisted of two monolithically encapsulated perovskite (FA0.80MA0.15Cs0.05PbI2.55Br0.45) solar cells, covered with CoP and FeNi(OH)x co-catalysts, working as a photocathode and a photoanode, respectively. The CoP-based photocathode generated 12.4 mA cm−2 at 0 V, whereas the FeNi(OH)x-based photoanode generated 11 mA cm−2 at 1.23 VRHE, resulting in an unassisted overall STH efficiency of 8.54%. The encapsulation was made using a thin polyisobutylene (PIB) layer and a cover glass, enabling these electrodes to be in direct contact with the electrolyte (0.5 M NaOH) and presenting a stable performance over 13 h of continuous operation.123 Another remarkable achievement using perovskite PEs for PEC water splitting was reported by Daboczi et al. The authors reported a CsPbBr3-based photoanode that displayed a 0.4 VRHE onset potential and generated 8 mA cm−2 at 1.23 VRHE, being capable of retaining 100% of its stabilized photocurrent density after 100 h of continuous operation. Enhanced charge transfer was attributed to the phase engineering of CsPbBr3 and to the NiFeOOH catalyst applied on a protective graphite sheet.124
Kesterite (Cu2ZnSn(S,Se)4, CZTS) materials are another class of emerging materials for PEC applications.63 Kesterites have been primarily studied for PV applications, in part because they are abundant, low-cost, and non-toxic. The interest in CZTS materials for PEC-H2 production has arisen due to the capability of changing the bandgap energy from 1.0 eV to 1.5 eV – or even higher - depending on the used elements, chemical reactivity, and the very high coefficient of light absorption, among others. As a p-type semiconductor with suitable optoelectronic properties (absorption coefficient of ca. 104 cm−1 and energy bandgap of ca. 1.4–1.5 eV), CZTS can be used as a photocathode for H2 evolution.125 Yokoyama et al. first demonstrated the application of CZTS as a photocathode in 2010.126 Surface modifications, i.e. CdS and TiO2 layers, were implemented to accelerate charge transfer and to protect the lower layer from corrosion in the electrolyte solution. Ros et al. were able to demonstrate the feasibility of CZTS/Se PEs in highly acidic conditions (pH < 1), with no detected degradation, by applying a protective TiO2 layer deposited by ALD, achieving a current density of 10 mA cm−2 at 0 VRHE.127 However, kesterite-based PEC systems still lack adequate demonstration in high-performing and upscaled devices, mainly due to limitations arising from (1) performance and stability problems; (2) misalignment of energy bands and interface defects; (3) the lack of compatible surface catalysts; and (4) the complexity of kesterite-based architectures for high-performing and stable devices. Similar to PV applications, the performance of CZTS highly depends on the buffer layer, as it forms a p–n junction that facilitates the separation of photo-induced electron–hole pairs by the built-in electric field of the junction. Therefore, in CZTS PEs, having a charge-selective contact layer is essential for the efficient extraction of photoexcited charge carriers. Among many major sources of interfacial recombination in these materials, the energy band misalignment and recombination through interfacial defects can be pointed as the major ones. Also, elemental interdiffusion at the interface affects the carrier transport. Charge-separating contacts with favorable energy band-alignment using interface passivation strategies can be a possible solution, as it has already demonstrated promising results in PV applications. For that, the conduction band position of buffer layers (e.g. CdS, Zn(O,S) and TiO2) can be tuned for better band matching. Other buffer layer parameters should also be optimized for optimum transparency and conductivity, charge carrier lifetime, and extraction. Surface passivation strategies include chemical and thermal treatments of the kesterite/buffer layer. Doping has also been proven to be an effective strategy for improving the PEC performance of CZTS PEs. As an example, Si-doping has been demonstrated to improve the generated photocurrent by 2.39 times, achieving ca. 5.57 mA cm−2 at −0.41 VRHE in a CZTSiS/CdS/ZnO PE.128
Morphology control has been widely used for increasing the photocurrent density generated at lower potentials; it overcomes the trade-off between enhancing the optical absorption and shortening the diffusion length of minority carriers, while improving the specific surface area, essential to drive the redox reactions.130 Indeed, many different preparation techniques have been described for obtaining semiconductors with specific morphologies.50,131,132 On the one hand, nanoparticles, dendrites, and mesoporous structures suffer from recombination and poor transport across the semiconductor, especially due to grain boundaries. Nanostructures such as worm-like protrusions, nanowires, nanotubes and nanorods, which have high aspect ratio and large surface area, enhance charge carrier collection by minimizing recombination at grain boundaries; in nanostructures with small diameters, the distance needed for the holes to diffuse to the semiconductor–electrolyte interface is shortened, thus mitigating the bulk recombination.66 Typically, thick and porous morphologies have a higher surface area and are capable of generating higher photocurrent densities; however, they usually present low stability and the resulting photoanodes are opaque (which is a drawback when multi-layer approaches or tandem arrangements are envisioned). Also, thicker semiconductor layers have increasing recombination problems.50 On the other hand, compact planar films, usually relatively thin (<40 nm), present high transmittance and assure the full coverage of the underneath conductive substrate, being therefore remarkably stable.133 However, due to the small specific interfacial area, these morphologies present lower performance, typically photocurrent densities <1.5 mA cm−2 at 1.23 VRHE.64,134
The incorporation of doping agents in the semiconductor lattice introduces additional majority carriers and creates steeper potential gradients (thinner space-charge layers), therefore improving charge separation and reducing the electron–hole recombination rate.135,136 Still, doping can decrease the particle size and crystallinity of the hosting semiconductor, increasing bulk recombination and forcing charge carriers to travel longer distances to reach the current collector.137 There are different reported strategies for incorporating dopants in the semiconductor lattice.138,139 Intrinsic doping is the simplest approach, such as via high-temperature annealing of semiconductors prepared over FTO glass substrates (normally over 650 °C), which enables the migration of Sn from the FTO layer to the main lattice of the semiconductor.140,141 Other strategies usually comprise the addition of the doping agent to the precursor. However, there is no standardized procedure for choosing the more suitable doping agent, or the optimal doping level, since the beneficial effect of each doping agent is highly dependent on both the intrinsic properties of the semiconductor and on its morphology. As an example, the addition of Ti as a dopant to α-Fe2O3 precursors must be carried with utmost precision since slightly too high concentrations may lead to the complete vanishing of the photoactivity. In the specific case of α-Fe2O3, several doping agents can be used to enhance its performance, namely Si, Ti, Sn, Nb, Zr, Pt, Mn, Zn and Ni; from these dopants, Sn is more effective for thin planar films, while Ti plays a more significant role on porous nanostructures.142 For highly doped semiconductors, one possibility is to introduce a gradient in the dopant concentration across the film; as an example, doping BiVO4 with a gradient of tungsten atoms allows the creation of a distributed n+ n− homojunction with a corresponding electric field that enhances charge separation.78
The surface of the semiconductor, in contact with the electrolyte, requires special attention when improving the photocurrent density generated at lower potentials. In a semiconductor–liquid junction, the rate of charge transfer at the interface relates to the rate of carrier generation and separation in the semiconductor, which makes surface states critical to form the depletion region that influences charge transport. Therefore, surface states located on truncated sites may act as charge-trapping spots, recombination centres and Fermi level pinning states, being highly detrimental to the overall performance. Chemical etching is a simple and effective technique for improving the oxygen evolution reaction (OER), promoting the interfacial charge transfer, and inhibiting surface recombination, by changing the chemical state of the semiconductor surface. A good example of this strategy was reported by Cao et al. for α-Fe2O3 PEs;143 a synergetic cathodic shift of 100 mV in the photocurrent onset potential and an increase of the photocurrent density from 1.2 mA cm−2 to 1.6 mA cm−2 measured at 1.23 VRHE were demonstrated and attributed to surface corrosion promoted by acid treatment over 90 min (with an aqueous solution of HCl). Although no obvious change was observed in the surface morphology, electrochemical impedance spectroscopy (EIS) analyses showed that the interface charge transfer rate increased after surface corrosion, indicating that back-reaction losses were mostly suppressed.
The deposition of co-catalysts is another surface treatment that allows reducing surface states, enhancing the performance of a PE, mainly at more cathodic potentials. Co-catalyst materials can play different roles at the semiconductor–liquid interface, as they can: (i) facilitate the bond-making and bond-breaking reactions; (ii) passivate recombination sites; (iii) tune the band structure energetics; and (iv) act as a protective layer against corrosion.144 Catalysts can be applied as an impermeable layer, turning the semiconductor–liquid junction into a semiconductor–catalyst junction. Therefore, the band bending is no longer governed by the electrolyte redox potential but becomes dependent on the work function difference at the semiconductor–catalyst interface. The band edge positions can then change depending on the chosen catalyst, which may allow overcoming unfavourable band edge positions. Depending on the type of application, co-catalysts are usually divided into three main categories: (i) noble metals; (ii) transition metals; and (iii) non-metals and metalloids. Among all, noble metals such as iridium and ruthenium for the OER and platinum for the HER are among the best-performing catalysts; however, these are critical raw materials.55,145,146 For minimizing their use, the surface-to-bulk atomic ratio can be increased without compromising efficiency and with substantial gains in terms of metal utilization. Dias et al.140 reported the use of IrO2/RuO2 as an OER catalyst, deposited on α-Fe2O3 thin films, to reduce overpotentials and improve photopotential. These authors achieved a 140% increase in photocurrent density at 1.23 VRHE, compared with a bare α-Fe2O3 PE and a final photopotential of 1.20 V, the highest value reported for α-Fe2O3-based PEs. Due to the scarcity, and high cost of Ir-based electrocatalysts, several new approaches have been conducted aiming to lower this noble metal content without impairing performance, such as dispersing Ir oxides on highly conductive, stable, and with large specific surface area supports.147 This strategy was mainly reported for water electrolysis but it can be used for PEC water splitting as well. Non-noble metal oxides such as antimony-doped tin oxide (ATO),148 indium tin oxide (ITO),149 and fluorine-doped tin oxide (FTO)150 are examples of these supports. Alternatively, transition metals and metalloids have been studied as co-catalysts in PEC systems. Despite presenting a slightly lower activity than noble metal-based catalysts, these elements are cheaper and more abundant, being a more sustainable approach for industrial applications. Over the past decade, co-catalysts such as cobalt phosphate (Co–Pi), Fe- or Ni-oxides have been widely studied for passivating surface states on different semiconductors.151 As an example, Co-based catalysts can be used to assist photogenerated holes to move away from recombination sites, located at the surface;152 however, Co-based materials bring special environmental concerns, which can significantly hinder their applicability. Nickel oxides (NiOx) have received particular attention due to their remarkable stability and functionality.153 The use of NiOx overlayers on BiVO4 photoanodes promotes an efficient charge transfer to the surface, thus reducing charge recombination and improving OER kinetics. In a study by Zhang et al., NiOx-modified/Mo:BiVO4 photoanodes generated a photocurrent density of 2.44 mA cm−2 at 1.23 VRHE, which is a 2-fold increase compared to uncatalyzed samples.153 Some co-catalysts can also act as a protective overlayer, when applied as a thin uniform coating, acting as a physical barrier against severe reaction conditions.154,155 An example of this approach is the use of a TiO2 overlayer on Cu2O and PEs.93 Nevertheless, the protective effect can only be accomplished if the catalyst coating is highly stable, and transparent, to avoid compromising light-harvesting by the semiconductor layer.94 When catalyst films are not applied uniformly, the STH efficiency can be hindered, since most catalysts have a high extinction coefficient (opaque) or are electrochromic under electrolysis potentials;156 as so, the optimal thickness of a uniform catalyst overlayer should be of few nanometres. Alternatively, catalysts can be applied as random or regular arrays with low geometric filling fractions, which reduces the negative effect of opacity.
The application of co-catalysts or other surface treatments plays a crucial role in the stability of the photoelectrode in contact with an electrolyte solution, a process that is known to be the competition between anodic/cathodic dissociation and redox reaction, controlled by thermodynamic and kinetic parameters, respectively.147,157 The selection of a suitable co-catalyst and surface treatment requires a set of experimental studies to conclude on the products of HER/OER photo(corrosion) and other secondary reactions.155 This issue was reviewed in detail by Ros et al. The authors concluded that the application of thin metallic films with a few nanometers thick (enough to be transparent) that could act both as catalysts and as a protective layer, was not an effective solution.63 Indeed, for protecting short bandgap photoabsorbers, such as Si, CZTS or similar materials, protective layers of several tens of nanometers thick are required. For that, TiO2 and NiO have been the most studied materials, the former acting as a HER/OER catalyst and the later as an OER catalyst. Still, the authors highlight that for both TiO2 and NiO, the deposition technique, and the intermittent polarization during long-term operation can hinder their electronic structure, therefore compromising stability.
Despite some performance limitations, TCO-glass remains the most used substrate for solar water-splitting electrodes. TCOs present unique advantages, such as (i) a combination of optical transparency with electrical conductivity; (ii) tunable energy bandgap (2.8–4.2 eV); and (iii) chemical and mechanical stability.161,162 As a result, TCOs have the advantages of metals typically used for electrical contacts, while avoiding their main drawbacks, i.e. opacity and corrosion issues.163,164 Among TCOs, ITO is the most broadly studied and is used in multiple optoelectronic applications. However, ITO lacks temperature and chemical stability, which compromises several preparation steps of the PE downstream, and since indium is a scarce and toxic element, its use for PEC devices has been limited.165 FTO-coated glass substrates became the best alternative to ITO for PEC applications, presenting a wide energy bandgap (ca. 3.6 eV), low-cost production, thermal stability, chemical inertness, low sheet resistance and high optical transmittance (usually series sheet resistance ≤15 Ω □−1 and transmittance ≥80% at 550 nm).166,167 Also, FTO thin films can be easily prepared by simple and low-cost methods, such as screen printing, doctor blade, and spray pyrolysis168–171 Due to their versatility, the preparation of FTO glass substrates by spray pyrolysis allows for full optimization and customization for PEC applications. By varying the sprayed volume and the [F]/[Sn] atomic ratio and using a figure of merit to evaluate the optoelectronic performance of FTO substrates, Pinheiro et al. were able to tune the optoelectronic properties of these substrates for PV applications, achieving PCE improvements of 8.9% in DSSCs and 4.7% in PSCs.172 Hence, when upscaling of PEs is foreseen, developing custom-made large-area FTO-glass substrates is essential to optimize the optoelectronic performance of the resulting PEs.
Although FTO-glass substrates have remained the preferable substrate for the preparation of PEC photoelectrodes, they also present non-negligible limitations. First, when practical mounting issues arise, such as screwing, sealing, and sudden temperature variations (especially when the PE is used simultaneously as a PEC cell window), the risk of cracking becomes high. Furthermore, the use of rigid substrates greatly limits the use of roll-to-roll manufacturing techniques, which have been widely used in large-scale production processes. Aiming to overcome these limitations, some studies reported the preparation of flexible PEs for solar water splitting, i.e., the deposition of semiconductor materials over flexible substrates. Hou et al. reported an innovative ZnO-based flexible PE prepared on micropatterned polyurethane acrylate (PUA) fabricated by a mold transfer process.173 A thin layer of ITO was deposited on the polyurethane acrylate substrate to assure electrical conductivity, and subsequently, one dimensional ZnO nanorods (NRs) arrays were prepared by a facile seed layer and hydrothermal method. These PEs were able to generate 0.52 mA cm−2 at 1.23 VRHE. In a different approach, Thakur et al. demonstrated a flexible photoanode prepared on micro-porous laboratory filter paper decorated with polypyrrole, which displayed a bandgap of 1.98 eV, yielding 9.5 mA cm−2 at 1.23 VRHE.174
2.2. Counter-electrodes
The classic configuration of a PEC device (Fig. 3(a)), where only one of the electrodes is photoactive, comprises a semiconductor operated as a working electrode (WE) and a “dark electrode”, the so-called counter-electrode (CE), where the complementary HER or OER takes place.175 As so, the CE plays an equally relevant role in the overall STH efficiency, and an inappropriate selection of materials or design criteria can significantly hinder the performance of a PEC device, as it must address some key features: (i) high surface area, at least the double than that of the PE, and good catalytic activity (to prevent undesirable photocurrent limitations originated by the overpotentials at the counter-electrode); (ii) good stability on strong acidic or alkaline media; (iii) ductility for enabling different assembling strategies, avoiding inhomogeneous pathways of charge carriers and parasitic ionic paths in the electrolyte; and finally (iv) non-toxicity, abundance and low-cost. Unfortunately, there are few available materials that meet all the aforementioned requirements, which makes some noble metals the obvious choice for lab-scale experiments. As a result, Pt has been used as a CE in small PEC devices for decades, mainly due to its low overpotential (ca. 0.1 V) and high chemical stability over a wide range of electrolytes and pH values. Almost like a standardized procedure, using a spiral-shaped Pt next to the PE became the most reported configuration among PEC characterization setups. Despite being commercially available in multiple configurations, the price and scarceness of Pt make it non-viable for large-area applications.176 Platinized meshes appeared as an alternative for larger devices, usually consisting of Pt-coated titanium or niobium meshes, having a higher surface area, while preserving the stability and low overpotential features.39,43 Even so, the amount of Pt incorporated in these electrodes, as well as other critical raw materials (CRMs), like Ti or Nb, make them suitable only for pilot-scale demonstration devices.Research efforts have been made to develop highly efficient and stable non-noble transition-metal-based and metal-free electrodes for water splitting applications. Liu et al. proposed an efficient flexible binder-free electrode for the HER, showing a low overpotential, compared with Pt, and notable electrocatalytic performance.177 The authors reported the electrophoretic-deposition of MoSe2 nanosheets on carbon cloth, which is a low-cost textile with high electric conductivity, large surface area and excellent flexibility and strength, which facilitates its integration in PEC devices. The prepared electrodes show a large number of exposed active edges due to the nanostructured MoSe2 sheets and the large surface area of the 3D-carbon cloth support, displaying remarkable catalytic activity. Binder-free catalysts to be used in the counter-electrode half-reaction own clear advantages as the catalyst is solely formed by the aggregation or self-supporting nature of the active material, improving: (i) activity, as the absence of binders allows the reactants to reach the active site easily, (ii) stability, as some binders can degrade or react with the active components of the catalyst under harsh operating conditions, (iii) mass transport within the catalyst structure, and (iv) selectivity towards the desired products. However, some binder-free catalysts/counter-electrodes may have a fragile structure, being more susceptible to physical damage, which can sometimes make it difficult to shape counter-electrodes to specific reactor designs and catalyst recovery. However, for a given catalytic process, binders may provide additional stability or tailor the catalyst properties.178 Similar approaches are being pursued using half-membrane electrode assemblies (H-MEA), where the separate evolution of O2 and H2 gases is assured by bonding the CE to an ion-exchange membrane. When MEAs are used, i.e. catalyst-coated membranes (CCM), the direct coating can be made on commercially available membranes (i.e. Aemion, Sustainion, Piperion, Nafion) or substrates, e.g. carbon cloth or carbon paper. Usually, a catalyst-ink is applied by spray-coating or doctor-blading, followed by thermal and pressure-finishing, with subsequent characterization, for assessing mechanical stability, morphology, and conductivity.179 MEAs can be prepared with nanoparticles of non-noble metals, such as Ni-nanoparticles, sprayed onto a hydrophilic carbon-based current collector particle support, which is normally bonded to an ion-exchange membrane using an ionomer glue. The shape of the electrode can be adapted to any reactor design and has the advantage of enabling the operation with a vapour phase on the HER side. An example of a carbon-based electrode was reported by Wang et al.,180 where 3-D carbon paper/tubes/cobalt–sulphide sheets were fabricated and tested as electrodes for the HER and OER. These electrodes presented high activity and stability for both cases, which was attributed to the increased exposure of active sites, improved electron transport, and good release of gaseous products, with demonstrated continuous water electrolysis over 2 h, presenting an overpotential of only 64 mV comparatively to Pt/C electrodes. Noble-metal free transition metal chalcogenide catalyst systems, i.e. (Fe,Ni)9X8 and NiX with X = S, Se, Te, and pentlandites (Fe4.5Ni4.5S8), have shown quite promising results when used as CEs in water electrolysis systems.181,182 Solution-based bottom-up approaches (precipitation, solvothermal) can be implemented for the synthesis of small-particle size seleno- and telluro-pentlandites. Still, the variation of chalcogenide/pentlandite composition as well as particle size distribution should be optimized for each PEC system. Fig. 5 shows schematically the incorporation of an MEA in two different PEC device architectures.
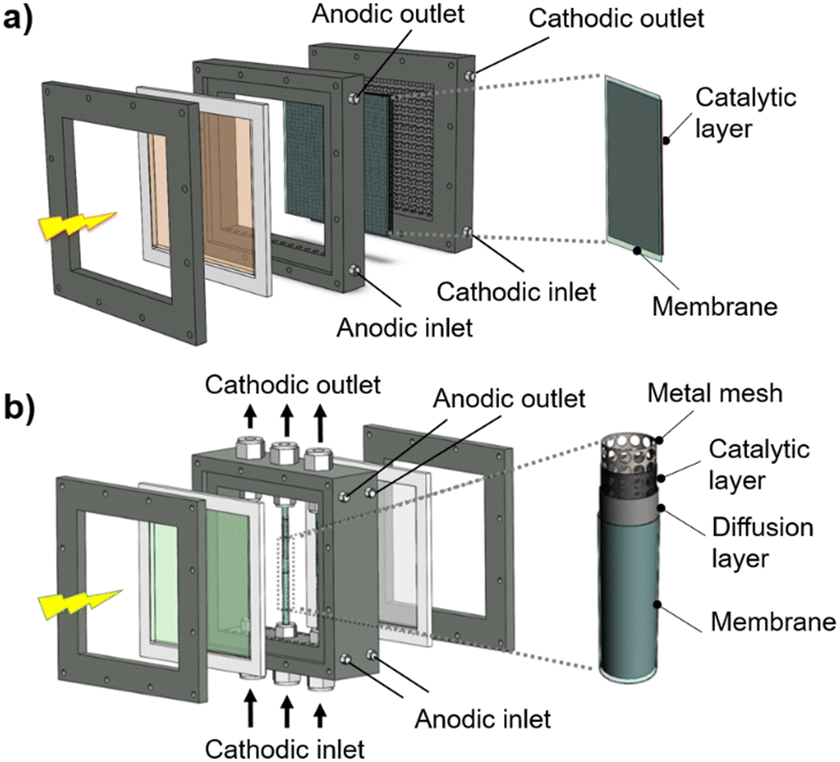 | ||
| Fig. 5 Schematic representation of MEAs comprising catalyst-coated membranes assembled in different PEC device architectures: (a) back-to-back configuration; and (b) side-by-side configuration. | ||
2.3. Anode/cathode separation materials and designs
Typical PEC devices require the incorporation of an internal separator to prevent gas mixing, avoiding the need for further processing downstream and assuring safer and more efficient operation. In current PEC systems, and considering the operation at ≤10 mA cm−2, the membrane can represent up to 17% of the total cost of the device, more than that of the electrodes.38 Indeed, as demonstrated by Grimm et al., the membrane is the most expensive component of a PEC module.183 Ion-exchange membranes, i.e. cation or anion exchange membranes (CEMs, AEMs), are common choices, being also widely applied in electrolysers, fuel cells and redox flow batteries.184–186 Whatever the choice for the membrane, inserting a selective porous separator in a PEC cell represents an additional resistance to species transport between electrodes due to its intrinsic properties (conductivity and permeability to the evolved gases) and due to its influence on the device design, i.e. the location of the membrane, in a large-area device, influences directly the distance between the electrodes. As an example, Vilanova et al. demonstrated that increasing the distance between two Pt–Ti meshes from 1 cm to 9 cm resulted in an increase of the ohmic losses from 73 mV to 297 mV, at a current density of 8.1 mA cm−2 (simulating 12% STH efficiency) in 1.0 M KOH.43 In another work, Berger et al. concluded that, among the main properties of membranes, the permeability to gases is the one that has the greatest influence on the device configuration.187 Apart from safety-related problems, gas crossover in a PEC device leads to an obvious decrease in net H2 collection, therefore requiring the operation at higher current densities to collect the same amount of H2. Finally, depending on the arrangement of PEs, namely in tandem configurations, membranes may need to address other critical requirements, such as high transmittance to sunlight.Considering water electrolysis in acidic media, perfluorinated sulfonic acid (PFSA) polymers, such as DuPont's Nafion, are the most commonly used.188 Despite being commercially available for large-area applications, CEMs still have a high cost per unit area (0.25–0.85 USD cm−2). Similar to CEMs, AEMs comprise fixed positive charges, which are compensated by mobile anions, with costs ranging from 0.16 USD cm−2 to 2.5 USD cm−2. Typical AEMs include reinforced PET or PEEK, which have been reported in several PEC devices. Poly(norbornene)-based resin membranes are among the most expensive; they present an innovative ionomer structure that contains a poly(norbornene) backbone with quaternary ammonium functional groups, with an integrated reinforcement layer that provides enhanced mechanical properties.
In contrast to conventional monopolar membranes, bipolar membranes (BPMs) were not designed for ion transport across them. These polymeric membranes comprise a negatively charged cation-exchange layer and a positively charged anion-exchange layer. BPMs promote the electro-dissociation of water into protons and hydroxide ions at the bipolar junction, without any gas formation; still, their use in PEC devices has been barely assessed.189 Even so, BPMs can be an interesting approach for PEC devices that comprise photocathodes and photoanodes, allowing them to pair two different electrolytes suitable for their respective oxidation and reduction reactions. Moreover, using strong acidic and basic electrolytes at the cathode and anode sides, respectively, reduces the overall losses in a BPM-based cell and the induced pH gradient may tune the redox potentials for a better alignment of the PE band edges. In this arrangement, H+ ions can pass through the cation exchange layer, while OH− can pass through the anion exchange layer; the quantity of generated protons and hydroxides at the BPM matches the consumption at the electrodes, maintaining a constant pH difference across the membrane. The total bias over the cell is not increased nor decreased by the water dissociation in the BPM, since the potential difference required to dissociate water over the membrane causes an equal shift in redox potentials for the HER and OER, due to the pH difference between electrodes. Vermaas et al. demonstrated for the first time photoassisted water splitting performed in a PEC device with an acidic cathodic compartment and a neutral or alkaline anodic compartment separated by a BPM.190 The use of a BPM allowed for maintaining the pH gradients and reducing the needed bias potential. PEC water splitting was promoted by a Pt cathode immersed in 1 M H2SO4 (pH 0), and by a BiVO4 photoanode, immersed in 1 M KOH (pH 14), resulting in a current density of 2 mA cm−2 at 1.3 V; the authors reported that a minor fraction of other ions was transported through the membrane due to imperfect selectivity.
Aiming at eliminating the resistance imposed by selective internal separator membrane-free PEC devices and integrated PEC (IPEC) systems have been proposed and demonstrated. However, these approaches often result in higher ohmic losses, due to the larger electrode spacing, or in additional and unnecessary complexity, due to the need of placing extra parts or integrating convoluted electrolyte paths to prevent gas mixing. Nevertheless, for small-scale applications, such as standard lab measurements, membrane-less devices are widely used, such as the Cappuccino PEC cell.191 Here, given the small illuminated area (ca. 0.5 cm2), the amount of evolved gases does not represent any safety concern, though the energy performance becomes affected.175 Landman et al. proposed an innovative membrane-free PEC design where H2 and O2 are generated in separated cells, and a solid-state redox system mediates the ion exchange between the anode and cathode, both immersed in alkaline aqueous solutions. The auxiliary electrodes (AEs) were a NiOOH/Ni(OH)2 redox couple, which had the possibility of being cycled multiple times; electrons are transferred between these AEs through metal wires.192 This approach presents some advantages over the typical approach that comprises a physical gas separator: (i) H2 can be produced in a centralized generator that can be placed at the end-user's location; (ii) gas mixing inside the PEC reactor is totally eliminated; and (iii) the O2 evolution compartment does not need to be sealed, thus avoiding piping costs. Still, for large-area applications, membrane-free systems are far from implementation, mainly because the auxiliary electrodes require frequent replacement and the whole system needs further experimental validation.
3. Upscaling of PEC water splitting
Despite being investigated for over 50 years, PEC-H2 production is still far from commercialization, with few demonstrated large-area devices. Although remarkable progress has been made in materials for water splitting, research work is still mostly focused on lab-scale experiments.193–196 Nevertheless, for PEC water splitting to become a competitive green-H2 production technology, it is required that research devices are designed to be scalable. Moreover, it is being recognized that the PEC device design plays an equally important role as the photoelectrode during the upscaling process. Device architectures with an illuminated area of at least 50 cm2 can be considered as a first step towards large-scale applications, given the limitations imposed by some manufacturing equipment and by the electronic resistance of some substrates, namely transparent conductive oxides (TCOs). Indeed, some authors who have addressed the challenge of upscaling photoelectrodes (PEs) for PEC water splitting have considered 50 cm2 as a benchmark for labelling their devices as “large-area”.43,79,197,198 Alternatively, scalability can be achieved by coupling module units of smaller dimensions. Therefore, materials engineering and fabrication techniques must be cleverly optimized envisaging upscaling, while preserving the performance of state-of-the-art materials and assuring a low-cost assembly towards higher TRL.The following subsections address relevant strategies and fabrication techniques for the preparation of efficient large-area PEs, as well as PEC device architectures suitable for upscaling.
3.1. Engineering solutions for large-scale photoelectrodes
The upscaling of efficient PEs remains one of the main challenges for the industrial implementation of PEC-H2 technology. Although nanostructured materials have been greatly optimized at the lab-scale (usually ≤5 cm2),67,140 upscaling top-performing PEs towards the expected dimensions of commercial PEC panels (≥1 m2) without significant losses is a demanding task.199,200 Indeed, Hankin et al.201 demonstrated that the performance of PEC devices is greatly affected by the geometry of the electrodes, especially for PEs with the smallest dimension >0.1 m. Table 1 summarizes common challenges during the upscaling of PEs and provides a comparative overview of the advantages and challenges of using smaller and larger PEs in PEC devices. – easy to implement/achieve;
– easy to implement/achieve;  possible to achieve, but with severe constraints/limitations;
possible to achieve, but with severe constraints/limitations;  – very difficult to implement/achieve
– very difficult to implement/achieve
| Lab-scale (≤5 cm2) | Multi-PE (≥25 cm2) | Large PEs (≥50 cm2) | PEs with metal lines (≥50 cm2) | Modular device (≥100 cm2) | PEC panel (≥1 m2) | |
|---|---|---|---|---|---|---|
| Schematics of the PE(s) |

|
|

|

|
|
|
| Preparation |

|

|

|

|

|

|
| Testing |

|

|

|

|

|

|
| Efficiency |

|

|

|

|

|

|
| Stability |

|

|

|

|

|

|
| Active area |

|

|

|

|

|

|
| Published device for demonstration (example) |

|

|

|

|

|

|
As mentioned before, FTO-glass stands as the most widely used PE substrate in PEC water splitting.202,203 These substrates are resistant to corrosion by commonly used electrolyte solutions and allow pairing of two photoabsorbers in series, which is a requirement for PEC–PV tandem systems, often considered the most feasible arrangement for reaching the STH efficiency targets.77,195 However, compared to metallic substrates, glass-coated substrates have a relatively high sheet resistance (usually ranging from 7–15 Ω □−1). Some promising strategies for overcoming this drawback have been demonstrated over the past decade – Fig. 6. The use of conductive lines across the substrate seems to be an obvious and easy approach for reducing the electronic path until the current collection point.204 Lee et al. successfully demonstrated a 131 cm2 WO3 PE prepared on an FTO glass substrate with an embedded silver grid – Fig. 6(a); this PE remains the largest prepared on a glass substrate containing metal lines.100 Silver current collector grid lines (width × height: 0.6 mm × 10 µm) were prepared by screen-printing a silver paste on the FTO substrates, followed by annealing at 180 °C. This substrate presented a resistance of ca. 1 Ω between two points 5 cm apart, considerably lower than the standard specification of the substrate, which is 10 Ω for the same distance (10 Ω □−1). WO3 was later deposited between the silver lines by screen printing and sintered at 500 °C. Finally, the silver lines were encapsulated with a non-conductive and non-corrosive epoxy resin. This 131 cm2 PE generated a current density of 1.18 mA cm−2 at 1.23 VRHE. In 2019, Ahmet et al. reported a 50 cm2 BiVO4 PE prepared on an FTO glass with electrodeposited Ni lines that allowed achieving a current density of 1.5 mA cm−2 at 1.23 VRHE – Fig. 6(b).79 This work followed a different approach to applying metal lines in a large-area glass substrate; the photoactive material was deposited over the whole substrate, including exposed FTO and Ni lines. These lines (7 cm long, 2 mm wide) were electrochemically deposited in a pre-treated and masked FTO glass, followed by the deposition of a 10 nm thick tin oxide (SnO2) layer and the 200 nm thick BiVO4 layer.
 | ||
| Fig. 6 Demonstrated strategies for overcoming the ohmic losses in large FTO glass substrates: (a) metal lines deposited on TCO glass substrates protected by epoxy resin; (b) metal lines deposited on TCO glass substrates and protected by the semiconductor, applied directly over the metal lines; (c) metal lines applied on ablated glass, covered by TCO, prior to the deposition of the semiconductor; (d) multi-PE, comprising several small-size PEs externally connected in parallel. | ||
Despite the current density generated by these large-area PEs (Fig. 6(a) and (b)) was less than half of the current density generated by equivalent small-sized PEs, the use of metal lines demonstrated to be an effective approach to overcome the electronic resistance across the substrate. However, the stability of these metal lines remains a massive challenge, since they need to be protected against corrosion, which is hard to achieve by encapsulation, such as using an epoxy resin, or covering the substrate with photoactive materials.21,205 Some innovative approaches have been proposed for overcoming most of the limitations imposed by the application of metal lines in conductive glass substrates. The ETCO (embedded TCO) – Fig. 6(c) – basically consists of applying metal lines beneath the TCO layer, inside ablated channels. This approach was reported by Ivanou et al.206 for a 6.5 × 6.5 cm2 FTO glass substrate. ETCO presented full stability and suppressive behaviour for recombination in DSSCs, allowing an increase in the cross-sectional area of current collectors and enabling the use of metals with good specific conductivity. However, this concept still lacks a further demonstration for PEC applications, i.e. with recirculation of corrosive electrolytes under uninterrupted solar illumination. The use of metal lines in large-area PEs, with its associated problems, can be overcome by simply connecting small PEs in parallel – Fig. 6(d), fulfilling large-area requirements while avoiding corrosion-related problems, as demonstrated by Vilanova et al.207 The authors developed a multi-electrode support with 5 × 10 cm2 that can be assembled as a PEC cell window. Through the connection of eight PEs in parallel using this support, a slightly higher photocurrent density and photopotential were reported, compared with a single 50 cm2 α-Fe2O3 PE. Apart from the significant performance losses imposed by the substrates, the successful demonstration of upscaled PEs is also related to the preparation technique of the semiconductor. Given the obstacles presented by upscaling of PEs, “classic” metal oxide PEs have been the subject of relevant studies aiming at increasing the active area without significant losses in performance while improving their stability under standard operating conditions. Table 2 provides an overview of the current status of large-area PEC water splitting.
| Top absorber [bottom or side absorber] | Electrolyte/pH | Area (cm2) | Current density (mA cm−2)/ηSTH (%) | Stability (h) | PEC architecture | Year | Ref. |
|---|---|---|---|---|---|---|---|
| FTO/WO3/Mo–BiVO4/Co–Pi | 0.5 M Na2SO4 + 0.1 M KH2PO4/pH 7 | 25 | 2.20@1.23 V | 1 | Type I | 2018 | Yao et al.208 |
| FTO/Fe2O3 | 1.0 M KOH/pH 13.6 | 26 | 1.11@1.45 V | — | Type III | 2018 | Vilanova et al.207 |
| LaTiO2N | 0.1 M Na2SO4 + 1 M NaOH/pH 13.4 | 40 | 0.56@1.23 V | — | Type I | 2019 | Dilger et al.209 |
| W/WO3 | 3 M MSA acid/pH 0.14 | 49 | 0.90@1.45 V | — | Type I | 2014 | Lopes et al.39 |
| FTO/Fe2O3 [2 × SHJ Si] | 1.0 M KOH/pH 13.6 | 50 | 1.05@1.45 V | 1008 | Type III | 2017 | Vilanova et al.43 |
| FTO/Ag–Pt/W:BiVO4/CoPi [2 × SHJ Si] | 0.1 M KPi buffer/pH 7 | 50 | 1.50@1.23 V/ 2.1 | — | Type III | 2019 | Ahmet et al.79 |
| FTO/Cu grids/Cu2O/AZO/Ga2O3/RuOx | 0.5 M Na2SO4 + 0.1 M KPi/pH 5 | 50.4 | 3.70@0 V | — | Type I | 2016 | PECDEMO102 |
| FTO/Fe2O3 | 1.0 M KOH/pH 13.6 | 51 | 0.47@1.45 V | — | Type I | 2014 | Lopes et al.39 |
| FTO/Au grids/NiFe–BiVO4 | 0.1 M H3BO3+ Na2B4O7·10H2O/pH 8.4 | 54.3 | 2.55@1.23 V | 5 | Type I | 2020 | Huang et al.210 |
| 2 series-connected a-Si:H/µc-Si:H tandem PV cells | 1.0 M KOH/pH 13.6 | 64 | —/3.9 | 40 | Type II | 2016 | Turan et al.21 |
| FTO/Fe2O3 [c-Si] | 1.0 M KOH/pH 13.6 | 100 | 0.56@1.84 V/0.71 | 8 | Type IV | 2020 | Landman et al.211 |
| FTO/WO3 [2 × SHJ Si] | 0.5 M H2SO4/pH 0.3 | 131 | 1.18@1.23 V/ 2 | — | Type II | 2011 | Lee et al.100 |
| FTO/Ti–Fe2O3 | 1.0 M KOH/pH 13.6 | 200 | 2.00@1.45 V | 48 | Type III | 2019 | Vilanova et al.97 |
| Mo–BiVO4/CoPi [Si PV] | 0.1 M KPi buffer/pH 7 | 16000 |
— | — | Type I | 2017 | Tolod et al.199 |
3.2. Brief overview of scalable fabrication techniques
Deposition techniques can be divided into chemical and physical, as well as into dry and wet. Dry techniques include processes such as thermal and plasma-assisted atomic layer deposition (ALD), sputtering and evaporation, while wet techniques include the application of inks/pastes, solution-based methods or chemical sprays with subsequent thermal processing.212 Most lab equipment is optimized and calibrated to prepare highly efficient PEs only for small-area applications;156,213 record-performing PEs usually have few square centimetres.39,175 Moreover, even when the preparation techniques allow the use of large-area substrates, it is difficult to ensure uniform layers across the entire PE area. Ahmet et al. assessed this problem by dividing their 50 cm2 BiVO4 PE into four equal parcels of 12.5 cm2;79 each of them was tested individually. The authors concluded that the variation between the samples remained below 13%. However, even when the prepared material is uniform, the performance deviation between small and large samples is often attributed to the preparation technique. As an example, Lee et al. reported that the shape of the metal grid applied on the 131 cm2 WO3 PE did not allow using the 5 multi-step deposition procedure optimized for smaller samples, which do not have a metal grid.100 Consequently, instead of presenting an optimal thickness of 25 µm, large WO3 PEs were prepared via a one-step deposition procedure, presenting a thickness of 5 µm. Table 3 presents a summary of comparison between technologies suitable for preparing large-area PEs.42| Synthesis | Techniques | Characteristics | ||||
|---|---|---|---|---|---|---|
| Waste | Time-consuming | Quality | Cost | Comments | ||
| Physical | Spin coating (wet) | ●●○ | ●○○ | ●○○ | ○○○ | Limited for multilayers |
| Evaporation | ○○○ | ●●○ | ●○○ | ●●○ | Low melting point only | |
| Magnetron sputtering | ○○○ | ●●● | ●●● | ●●● | Expensive equipment | |
| Chemical | ALD | ○○○ | ●●● | ●●● | ●●● | Limited chemistries; expensive equipment |
| CVD | ●●● | ●●○ | ●○○ | ●●○ | High contamination | |
| Plasma ALD/CVD | ○○○ | ●●○ | ●●● | ●●● | Expensive equipment | |
| Spray pyrolysis (wet) | ○○○ | ●●○ | ●●○ | ●○○ | Ideal for oxides | |
| Ink jet (wet) | ●○○ | ○○○ | ●○○ | ●○○ | Ideal for patterned areas | |
| Electro/electroless plating (wet) | ●●● | ●○○ | ●○○ | ○○○ | Ideal for metals | |
| Sol–gel/slurry (wet) | ●○○ | ●○○ | ○○○ | ○○○ | Low reproducibility | |
| Solution-based methods (hydrothermal) | ●○○ | ●●○ | ●●○ | ○○○ | Low-cost, limited quality control | |
When a preferable preparation technique does not allow achieving a uniform semiconductor deposition across the substrate, having a multi-PE window could be an interesting approach, as it allows working with small PEs prepared using state-of-the-art techniques/equipment. Even so, this multi-electrode window needs further optimization, especially due to the increased complexity brought by electrical connections and the associated costs.207 As described by the authors, this window limits the implementation of tandem arrangements, since the electrical connections made at the back of the cell block part of the incident light. The research group of Lee has achieved a significant breakthrough in large-scale PE manufacturing using inkjet printing.214 The proof-of-concept of fully inkjet-printed α-Fe2O3 layers on an FTO glass substrate was demonstrated with a 20 × 25 array and photo-illumination area of 500 cm2; the authors also reported the formulations for preparing other semiconductors. However, they did not provide any information regarding the performance of large-area inkjet-printed PEs or on economic and environmental analyses.
3.3. Device architectures for practical applications
Among the various architectures used in PEC devices, two main groups of planar architectures can be identified, based on the implemented strategy for separating the evolved H2 and O2: (i) PEC reactors comprising an internal mechanism to separate the evolved gases; and (ii) membrane-free systems comprising separated cells.42 Considering the strategy for connecting the electrodes, it is possible to divide planar PEC device architectures into four main types: the back-to-back design, where the electrodes can be connected wired or wireless – type I and type II, respectively; the side-by-side design – type III; and the membrane-less dual reactor device – type IV. Types I, II and III use a membrane separator for dividing the anode from the cathode inserted in a single container. Fig. 7 shows schematically these four main PEC device architectures. Table 4 presents more detailed information regarding PEC reactors of each type, designed and implemented for large-scale solar H2 products. The back-to-back PEC design containing the electrodes in a wired configuration (type I) is the most studied architecture for both lab- and large-scale applications. This design is quite similar to the well-known alkaline electrolysers (AEC), comprising a reservoir for the electrolyte wherein the electrodes are immersed and physically separated by an ion-exchange membrane. Over the past decade, different approaches were reported for scaling up this family of devices: (i) in 2011, a 100 cm2 “stacked” PEC reactor was reported for the first time by the Imperial College of London;215,216 (ii) in 2014 UPORTO developed the 100 cm2 “PortoCell”, which was further improved in 2016;39,217 (iii) in 2015, the European Union (EU) project ArtipHyction developed the largest PEC prototype to date, with an irradiated surface area of 1.6 m2;218 and (iv) in 2019, EPFL patented an integrated PEC (IPEC) device, whose concept was validated experimentally using a close electronic integration between the III–V-based photoabsorber (4 cm2) and the IrRuOx-Pt-based electrocatalyst.17 Holmes-Gentle et al. reported an STH efficiency of >20% at a H2 production rate of >2.0kW (>0.8gmin−1),219 landmark results with PV-grade PEs, such as Si-perovskite cells. Although great efforts have been pursued to successfully upscale type I PEC cells, the arrangement of electrodes for a PEC–PV tandem configuration has always been a limiting factor for the efficient utilization of the solar spectrum, and also required an optimized electrolyte flow pattern for efficient dissipation of heat and evolved gases. The upgraded version of the “PortoCell” was the first PEC design that included an optimized flow pattern and simultaneously addressed ohmic losses across the electrolyte reservoir; still, this design presents some disadvantages under real operating conditions.217 On the other hand, the ArtipHyction prototype was the first device that validated the transfer from lab-scale cells to pilot-scale units; however, its viability was compromised by the low overall PEC performance: STH conversion efficiency of ca. 2% with a 5% decrease over 1000 h of continuous operation.218
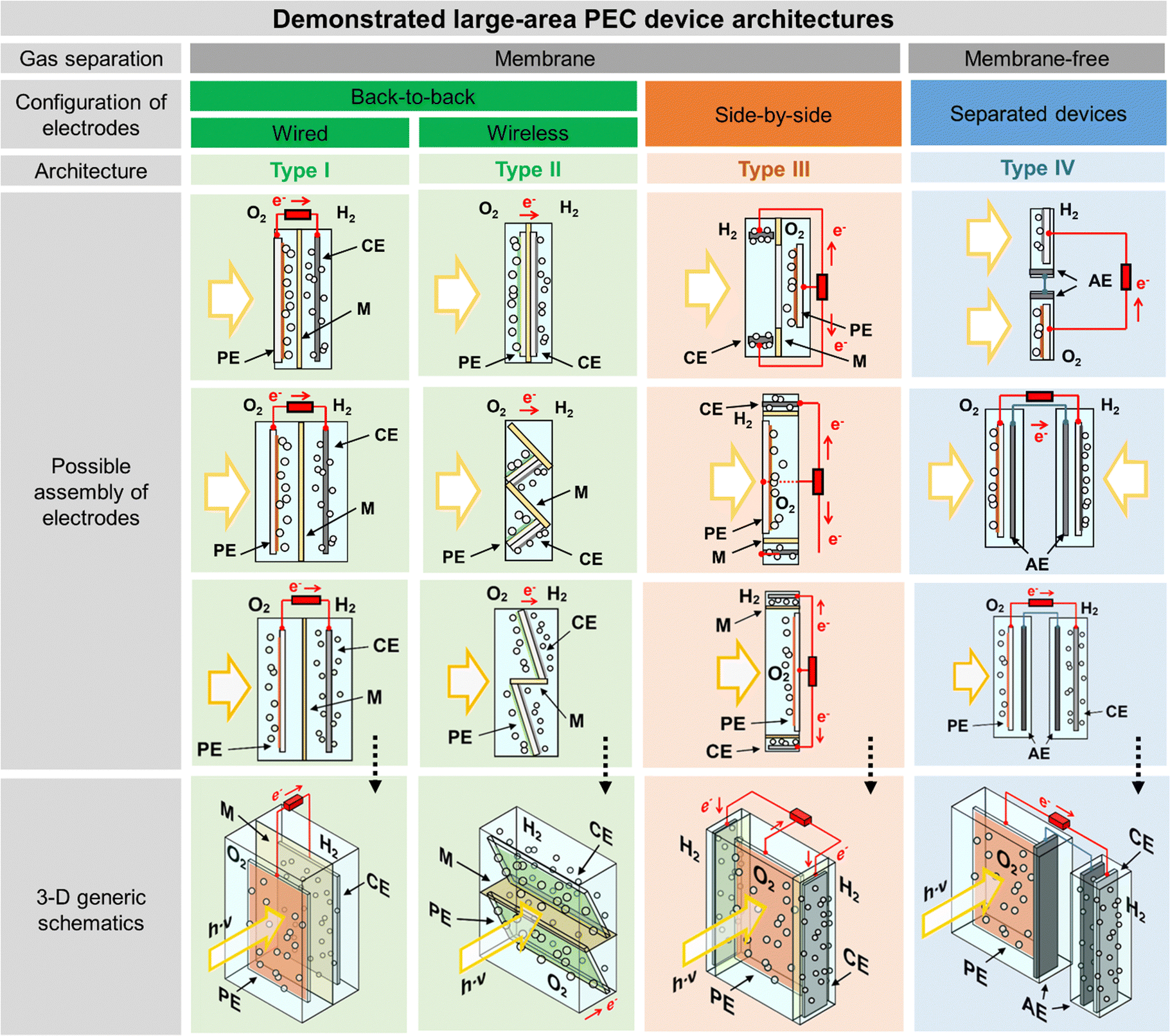 | ||
| Fig. 7 Schematic representation of the four main planar PEC device architectures. PE – photoelectrode; CE – counter-electrode; M – membrane; AE – auxiliary electrode. | ||
 – successfully implemented/optimized;
– successfully implemented/optimized;  – not assessed;
– not assessed;  – non-optimized or not possible to implement
– non-optimized or not possible to implement
| Year | Author designation | Design type (according to Fig. 7) | Photoactive area (cm2) | Design features | General indicators | Ref. |
|---|---|---|---|---|---|---|
| 2011 | Stacked PEC reactor | Type I | • Demonstrated: |
 Gas separation: Nafion membrane Gas separation: Nafion membrane |
• PE: Si-doped α-Fe2O3 | 215,216 |
| – (10 × 10) cm2 | ||||||
 PV integration: a PV cell can be placed outside the PEC reactor or immersed in the electrolyte using protective materials PV integration: a PV cell can be placed outside the PEC reactor or immersed in the electrolyte using protective materials |
• Performance: 2.5 mA cm−2@0.7 VHgO|Hg in 1 M NaOH under white (Xe) lamp irradiation (35 W m−2) | |||||
 Electrolyte feeding: not optimized for continuous operation Electrolyte feeding: not optimized for continuous operation |
• Stability: not assessed. | |||||
 Configuration of electrodes: not suitable for tandem arrangements (photoanode//HER metal catalyst) Configuration of electrodes: not suitable for tandem arrangements (photoanode//HER metal catalyst) |
||||||
 Concentrated sunlight: not assessed. Concentrated sunlight: not assessed. |
||||||
| 2014–2016 | PortoCell | Type I | • Demonstrated: |
 Gas separation: Teflon® or ion exchange membranes; gas crossover was observed when the device was operated in a tilted position Gas separation: Teflon® or ion exchange membranes; gas crossover was observed when the device was operated in a tilted position |
• PE: α-Fe2O3 | 39,217,220 |
| – 2014 version: |
 PV integration: PV cell placed behind the PE PV integration: PV cell placed behind the PE |
• Performance: 0.5 mA cm−2@1.45 VRHE in 1 M NaOH under AM 1.5G illumination (1000 W m−2); STH efficiency not presented for tandem configuration | ||||
| (10 × 10) cm2 |
 Electrolyte feeding: fully optimized in the 2nd device version (2016) Electrolyte feeding: fully optimized in the 2nd device version (2016) |
• Stability: not assessed. | ||||
| – 2016 version: |
 Configuration of electrodes: not ideal for tandem arrangements (photoanode//photocathode – dual side illumination using a dual mirror system; photoanode//HER metal catalyst; and photocathode//OER metal catalyst) Configuration of electrodes: not ideal for tandem arrangements (photoanode//photocathode – dual side illumination using a dual mirror system; photoanode//HER metal catalyst; and photocathode//OER metal catalyst) |
|||||
| (7.2 × 7.2) cm2 |
 Concentrated sunlight: assessed in 2016 (2nd device version – the front window slightly overheated; further adjustments are necessary). Concentrated sunlight: assessed in 2016 (2nd device version – the front window slightly overheated; further adjustments are necessary). |
|||||
| • Prototype: | ||||||
| – 4 PEC cell modules of | ||||||
| (7 × 7) cm2 | ||||||
| 2014–2016 | EMPA PEC reactor – BioPEC | Type III | • Demonstrated: |
 Gas separation mechanism was not specified Gas separation mechanism was not specified |
• PE: α-Fe2O3 photoanode | 221–223 |
| – (10 × 10) cm2 |
 PV connection: a PV cell can be placed outside the PEC reactor or immersed in the electrolyte using protective materials or tandem PEs PV connection: a PV cell can be placed outside the PEC reactor or immersed in the electrolyte using protective materials or tandem PEs |
• Performance | ||||
 Electrolyte feeding system was not investigated Electrolyte feeding system was not investigated |
0.45 mA cm−2@1.45 VRHE in 1 M KOH under AM 1.5G illumination (1000 W m−2) | |||||
 Configuration of electrodes suitable for tandem arrangements (photoanode⊥HER metal catalyst and photocathode⊥OER metal catalyst) Configuration of electrodes suitable for tandem arrangements (photoanode⊥HER metal catalyst and photocathode⊥OER metal catalyst) |
• Stability | |||||
 Concentration sunlight: not assessed. Concentration sunlight: not assessed. |
70 min running time | |||||
| 2015 | ArtipHyction prototype | Type I | • Demonstrated: |
 Gas separation: Nafion membrane Gas separation: Nafion membrane |
• PE: BiVO4 photoanode | 218 |
| – 1.6 m2, composed of 20 strings of 5 PEC cell modules of (58.0 × 12.4) cm2 |
 PV integration: PV cells were placed outside the PEC reactor (the overall exposed illuminated area was increased; the final configuration is not a real tandem system) PV integration: PV cells were placed outside the PEC reactor (the overall exposed illuminated area was increased; the final configuration is not a real tandem system) |
• Performance: 2% STH efficiency | ||||
 Electrolyte feeding: assessed Electrolyte feeding: assessed |
• Stability: Not clearly described. | |||||
 Configuration of electrodes: not suitable for tandem arrangements (photoanode//HER metal catalyst and photocathode//OER metal catalyst) Configuration of electrodes: not suitable for tandem arrangements (photoanode//HER metal catalyst and photocathode//OER metal catalyst) |
||||||
 Concentrated sunlight: not assessed. Concentrated sunlight: not assessed. |
||||||
| 2015 | Louvered cell | Type II | • Demonstrated: |
 Gas separation: Nafion membrane (only works in acidic media due to electrode constraints) Gas separation: Nafion membrane (only works in acidic media due to electrode constraints) |
• Performance: 0.24% STH efficiency | 224–226 |
| – 2 PEC cell modules of (1.6 × 4.4) cm2 – possible to increase |
 PV integration: PV immersed in the electrolyte (tandem PE) PV integration: PV immersed in the electrolyte (tandem PE) |
• Stability: 20 h. | ||||
 Electrolyte feeding: assessed through multi-physics modelling Electrolyte feeding: assessed through multi-physics modelling |
||||||
 Configuration of electrodes: not ideal for tandem arrangements (photoanode//photocathode) Configuration of electrodes: not ideal for tandem arrangements (photoanode//photocathode) |
||||||
 Concentrated sunlight: not assessed Concentrated sunlight: not assessed |
||||||
 Dual side illumination: no information concerning the system transparency. Dual side illumination: no information concerning the system transparency. |
||||||
| 2016–2020 | Membrane-free PEC cell for alkaline water electrolysis in separate H2 and O2 cells | Type IV | • Demonstrated: |
 Gas separation: H2 and O2 in separate cells Gas separation: H2 and O2 in separate cells |
• Electrodes: Ni foil and Pt-plated stainless-steel mesh | 69,192,227 |
| – 1 and 100 cm2 |
 PV integration: the PV cell can be placed outside the PEC reactor, or immersed in the electrolyte using protective coatings or tandem PEs PV integration: the PV cell can be placed outside the PEC reactor, or immersed in the electrolyte using protective coatings or tandem PEs |
• Performance | ||||
| • Forecasted: |
 Electrolyte feeding: not assessed Electrolyte feeding: not assessed |
(STH efficiency): | ||||
| – 5 × 9 array of (10 × 10) cm2 |
 Configuration of electrodes: suitable for tandem arrangements Configuration of electrodes: suitable for tandem arrangements |
1 cm2: 7.5% | ||||
 Concentrated sunlight: not assessed, but it was tested under natural sunlight in outdoor conditions. Concentrated sunlight: not assessed, but it was tested under natural sunlight in outdoor conditions. |
100 cm2: 0.68% | |||||
| • Stability: | ||||||
| 1 cm2: 1 h; 100 cm2: 83 h. | ||||||
| 2016–2019 | CoolPEC cell | Type III | • Demonstrated: |
 Gas separation: ion exchange membrane Gas separation: ion exchange membrane |
• PE: α-Fe2O3 and dual CoPi/W:BiVO4 | 43,79,217 |
| – 50 cm2 |
 PV integration: PV cell placed behind the PE, or immersed in the electrolyte using protective materials or tandem PEs PV integration: PV cell placed behind the PE, or immersed in the electrolyte using protective materials or tandem PEs |
• Performance | ||||
 Electrolyte feeding: fully optimized Electrolyte feeding: fully optimized |
(STH efficiency): | |||||
 Configuration of electrodes: ideal for tandem arrangements (photoanode⊥HER catalyst and photocathode⊥OER catalyst) Configuration of electrodes: ideal for tandem arrangements (photoanode⊥HER catalyst and photocathode⊥OER catalyst) |
α-Fe2O3: 1% | |||||
 Concentrated sunlight: assessed in the SoCRatus setup. Concentrated sunlight: assessed in the SoCRatus setup. |
W:BiVO4: 2% | |||||
| • Stability: | ||||||
| α-Fe2O3: 1008 h. | ||||||
| 2018–2020 | CoolPEC module | Type III | • Demonstrated: |
 Gas separation: ion exchange membrane Gas separation: ion exchange membrane |
• PE: 32 small α-Fe2O3 PEs or single 50 cm2 planar α-Fe2O3 PEs, connected in parallel | 97 |
| – 200 cm2, 1 × 4 array of 50 cm2 PEC cells |
 PV integration: If multi-electrode windows are used instead of standard 50 cm2 PEs, the possibility of placing a PV cell at the back of the PE is compromised. PV integration: If multi-electrode windows are used instead of standard 50 cm2 PEs, the possibility of placing a PV cell at the back of the PE is compromised. |
• Performance | ||||
 Electrolyte feeding: fully optimized Electrolyte feeding: fully optimized |
2 mA cm−2@1.45 V, 1 M KOH, ca. 6000 W m−2 | |||||
 Configuration of electrodes: ideal for tandem arrangements (photoanode⊥HER catalyst and photocathode⊥OER catalyst); Configuration of electrodes: ideal for tandem arrangements (photoanode⊥HER catalyst and photocathode⊥OER catalyst); |
• Stability | |||||
 Concentrated sunlight: assessed in the SoCRatus setup. Concentrated sunlight: assessed in the SoCRatus setup. |
4 days. | |||||
| 2019 | Integrated PEC (IPEC) device | Type I | • Demonstrated: |
 Gas separation: MEA (proton-exchange nanoporous solid conductor, Nafion 115 membrane coated with catalyst particles on each side, sandwiched between two gas diffusion layers and two bipolar flow plates) Gas separation: MEA (proton-exchange nanoporous solid conductor, Nafion 115 membrane coated with catalyst particles on each side, sandwiched between two gas diffusion layers and two bipolar flow plates) |
• PE: Concept validated with triple junction InGaP/InGaAs/Ge PV solar cell | 17 |
| – 4 cm2, 4 × 1 cm2 III–V PV cells |
 Electrolyte feeding: fully optimized Electrolyte feeding: fully optimized |
• Performance: 0.88 A cm−2, 17.2% STH efficiency, under ca. 474 kW m−2 | ||||
| – Possible to increase |
 Configuration of electrodes: may be suitable for tandem arrangements with a buried junction (electronic integration between the photoabsorber and electrocatalyst) Configuration of electrodes: may be suitable for tandem arrangements with a buried junction (electronic integration between the photoabsorber and electrocatalyst) |
• Stability: 2 h. | ||||
 Concentrated sunlight: assessed with thermal and mass transport optimization. Concentrated sunlight: assessed with thermal and mass transport optimization. |
Type II PEC reactors are rarely reported; an example of this architecture was successfully demonstrated in 2015 by Walczak and his co-authors224 with the development of the so-called Louvered cell. In the Louvered prototype, planar macroscale tandem PEs are installed at a certain angle in a chamber; the PE compartments are separated by an anion-exchange membrane.224,225 During the water splitting process, O2 and H2 are collected in different compartments of the cell, respectively – cf. Fig. 7.224,225 The overall efficiency of these PEC reactors is highly dependent on the performance of the tandem PEs but is equally important to ensure a proper ionic transport with low overpotentials.228
Within the type II architecture, integrated tandem monolithic PEs, where the top and bottom absorbers are combined into a single monolithic electrode, offer several practical benefits. Since the illumination path is confined through a single thin monolithic device, optical scattering, and parasitic absorption losses are mitigated. Also, due to the monolithic nature, ohmic losses are significantly minimized due to the low dispersed current density paths and tunneling between junctions.40 By using integrated tandem monolithic PEs, the PEC reactor design can be more easily narrowed, which is essential for reducing manufacturing costs and facilitating device installation. Still, monolithic wireless configurations have been less investigated than other electrode configurations, which is mainly due to the complexities and difficulties related to electrode fabrication and characterization. The characterization of a wireless monolithic device is particularly challenging, as it does not allow having any direct electrical wiring to a potentiostat. Therefore, the performance of the device can only be assessed by collecting the gaseous products, which is experimentally more difficult, as gas leakage, especially H2, is one of the most reported issues in lab-scale devices.229 Notwithstanding, monolithically integrated PEs offer unique advantages for achieving commercial PEC water splitting, due to the simplicity and compactness of the associated device. Ahmet et al. have demonstrated the use of low-cost and scalable deposition techniques to produce a planar and nanostructured tandem photoanode, consisting of n-Si/SiOx/TiO2/WO3/BiVO4/Fe(Ni)OOH and a Pt cathode as an H2 evolution catalyst, which provided a combined photopotential capable of unassisted solar water splitting, generating 0.3 mA cm−2 in 1.0 M KBi pH 9.3 buffer solution.230 Zhu et al. have also demonstrated a monolithic PEC device with simple engineering and low loading of catalysts through the integration of a new class of double perovskite cobaltites for the OER, reporting a 6.6% STH under 71 h of continuous illumination.231 Monolithic architectures can also be considered for the full integration of conventional PV electrodes in PEC devices, enabling to achieve remarkable STH efficiencies.21,37,57,232 Kistler et al. demonstrated a wireless monolithic vapor-fed PEC device assembled with a III–V triple-junction photovoltaic (PV) electrode embedded in a Nafion membrane, where the catalyst was applied by compression of catalyst-modified carbon electrodes against the front and back PV contacts.233 This device allowed for achieving an unprecedented 14% STH efficiency over 1000 h of cumulative device operation.
In a side-by-side design (type III), as the nomenclature suggests, CEs are placed side-by-side to the photoactive WE, perpendicular to the direction of the incident light; a membrane is placed between electrodes on both sides to avoid gas mixture – cf. Fig. 7. Therefore, the side-by-side architecture is considered more feasible for large-area tandem PEC–PV devices, since it comprises an open pathway for sunlight to reach the sublayers of photoactive electrodes. An example of a demonstrated type III device is the “CoolPEC” cell.43 This 50 cm2 device was optimized for continuous operation, including some key features: (i) the PE is simultaneously one window of the cell; (ii) uses a feeding manifold for the electrolyte, assuring efficient gas bubble detachment from the windows, low concentration polarization of the electrolyte and efficient heat dissipation (crucial when the cell is operated under concentrated sunlight); and (iii) a narrow, easy-assemble and cost-efficient embodiment. The “CoolPEC” was successfully demonstrated as a tandem PEC–PV device; when equipped with a 50 cm2 α-Fe2O3 photoanode displayed a constant current density of ca. 0.45 mA cm−2 at 1.6 V, under 1000 W m−2 and with constant electrolyte feeding at 45 °C, over 42 days (1008 h) of continuous operation; the extra bias potential was provided through the combination of two silicon heterojunction (SHJ) solar cells.43 This cell was also tested with a 50 cm2 dual CoPi/W:BiVO4 photoanode assembled in a tandem arrangement with 2-series connected SHJ cells, generating ca. 1.7 mA cm−2.79 In 2020, Vilanova et al. demonstrated a 200 cm2 modular device, comprising four 50 cm2 CoolPEC cells in a linear array; this device was tested outdoors under homogeneously concentrated solar radiation (up to 12.8 kW m−2) provided by a linear Fresnel reflector and achieved a maximum current density of ca. 4 mA cm−2.97,234 Although the reported STH efficiency for this architecture remains below 10%, this target can be achieved with more efficient PEs, such as GaInP/GaInAs tandem absorbers, which can achieve an STH efficiency of 16%.235
Type IV PEC devices consist of a membrane-free water splitting system comprising separated cells for the hydrogen and oxygen evolution, and auxiliary electrodes to ensure the charged species transport between the anode and cathode sites. This architecture, which uses a photoelectrode under alkaline conditions, was disclosed by Technion in 2015227 and reported by Landman et al. in 2017192 and in 2020.69 In the demonstrated device, Ni(OH)2/NiOOH electrodes, commonly used in rechargeable alkaline batteries, were assembled as auxiliary electrodes (AEs). During electrolysis, the NiOOH electrode is charged in the oxygen cell, while the Ni(OH)2 electrode is discharged in the H2 cell. The oxygen produced at the photoanode can be released to the atmosphere, eliminating the need for sealing the photopanels, while H2 is produced under unilluminated conditions, in sealed reactors, enabling a centralized production, and avoiding large piping for H2 collection. This concept was successfully demonstrated in small-area cells (6.03 cm2), achieving an STH efficiency of 7.5%.192 Subsequently, it was upscaled to 100 cm2 and comprising dual α-Fe2O3 photoanodes assembled in tandem arrangement with a Si–PV module, generating ca. 0.55 mA cm−2 over 10 cycles of 8.3 h. Type IV devices incorporate important cost reduction strategies since the use of membranes is avoided and piping is shortened; even so, the STH efficiency of large-area devices remains too low, and the need for regularly replacing the AEs is a serious drawback. Besides the device architectures considered in Fig. 7 and Table 4, other PEC designs can also be found in the literature.41,194,236 However, these designs are still at a conceptual stage, due to the lack of experimental validation, or because their scalability is not possible. For instance, a particle-suspension PEC system appears to be a simple and attractive design, with no need for large electrodes, wires and compartmentalization; however, no efficient gas separation method is available for this approach and the price of good-performing particles is prohibitively high.195,237 Some other reported architectures might prove promising in the near future, but the lack of experimental validation hinders a fair comparison with designs that were already experimentally assessed. Holmes-Gentle et al.41 demonstrated an innovative approach of a membrane-less PEC cell using multi-physics modelling, where the evolved gases are kept separated by hydrodynamic control; this conceptual design was engineered to minimize energy losses associated with the ionic transport and to avoid membrane costs. PEC designs that consider operation with water vapour have also been proposed, but are still in early stages of development.194,238 Several patents disclosing PEC reactors have also been filled by different groups/companies over the years; however, in most cases, only the conceptual design is reported, and few report large-area devices.239–241 Independently from the device architecture, a decision-tree approach is a very effective strategy for designing and optimizing a PEC device, as exemplified in Fig. 8.
 | ||
| Fig. 8 Example of a decision-tree used to develop a hypothetical and generic new PEC device. | ||
3.4. Assembling requirements for practical PEC devices
When the commercialization of a technology comes into sight, and particularly in the renewable energy market, discussing an economy of scale becomes inevitable. In this context, parameters such as the modularity and scalability of the devices, their CAPEX, EPC (engineering, procurement, and construction) efforts and, naturally, the cost per unit of mass of produced H2 turn out to be the differentiating factors when choosing between technology A or B. Although the performance of the device is directly related to the performance of the photoelectrodes, which translates into greater or lesser amounts of H2 produced by the device, the reactor embodiment assumes an extremely relevant role in the final cost of the technology. On a laboratory scale, the main objective is to build a reactor that is stable to a wide range of electrolytes and that is easily assembled and disassembled. Given these assumptions, most of the reported laboratory devices tend to incorporate expensive materials, such as PEEK,242 PMMA,233 and quartz.97 In an economy of scale, these materials will naturally have to be replaced by cheaper ones commonly used in commercial reactors, such as polyurethane or composite materials. Proper sealing of PEC reactors is also an extremely important aspect for commercialization, both to avoid leakage of corrosive or environmentally harmful liquids and to avoid gas leaks, especially H2. Considering PEC devices described in the literature, different sealing strategies have been used, including conventional rubber O-rings,39 silicone gaskets,233 and glues.207 Yet, commercial sealing solutions will most likely be similar to those used in fuel cells and electrolysers, such as elastomeric gaskets and stack plates. As so, research studies on new PEC devices should take these requirements into consideration and should aim at developing PEC reactors made of low-cost and stable materials already used on a commercial scale, incorporating practical sealing solutions that avoid liquid and gas leakage and do not compromise an easy assembly and replacement of the electrodes. Finally, bearing in mind, the growing environmental restrictions and circular economy directives, the embodiment of commercial PEC reactors should incorporate recyclable, reusable and/or biodegradable materials, such as composite materials or polyurethane.243–245 Incorporating these materials will be essential to lowering the technology's carbon footprint and ensuring fair green marketing.4. Use of concentrated sunlight and thermal management in PEC devices
The industrialization of PEC-H2 devices does not rely only on yielding high efficiencies, but also on ensuring a stable and safe operation and reducing production costs. Concentrating solar radiation is a clever strategy for meeting these targets; still, it brings additional challenges, namely related to the increased temperature load and the continuous operation with high current density values.17,156 Integrating solar concentrators in PEC systems requires further optimization following a symbiotic approach, i.e. PEC devices need to be optimized to be operated under concentrated sunlight and solar concentrators need to be redesigned to be compatible with several material restrictions of PEC devices. Also, the operation under an optimum concentration factor depends on the available solar irradiance (i.e. varies with the site location) and requires appropriate thermal management solutions for each PEC device architecture.4.1. Overview of concentrating solar technologies
At present, the use of solar concentrators in PEC systems remains almost unaddressed, mainly because large-scale PEC-H2 production is still at an early stage of development and optimization, with few demonstrated devices. The closest approach to this synergetic strategy at a high TRL is the use of solar concentrators in PV-electrolysers.246 Both large-scale alkaline electrolysers and PV cells have been optimized, demonstrated and used for industrial H2 production for many decades and, in more recent years, solar concentrators have been used to further enhance the power output of the PV component.247,248 Currently, there is no standardized nomenclature in the literature for grouping solar concentrators.249 Nevertheless, some authors grouped solar concentrators into different categories, usually depending on the operational characteristic that better distinguishes the purpose for which they are used. One option for categorizing solar concentrators considers the type of focal area, which can be linear or punctual.250 This criterion is directly related to the concentration factor and temperature achieved in the receiver. Linear concentrators provide a homogeneous light concentration along a planar surface or a line, allowing moderate to high-temperature values in the receiver to be reached, usually, ranging from 100 °C to 500 °C.249,251 On the other hand, punctual concentrators focus sunlight in a single point, allowing very high temperature values in the receiver to be achieved, usually above 500 °C. Another option for categorizing solar concentrators considers the way the solar radiation is focused on the receiver, being reflective or refractive.252 Caron et al. suggested another categorization for solar concentrators suitable for planar PEC devices, based on the type of motion (fixed, translational and rotational) and on the type of tracking (stationary, one-axis and two-axis tracking).253Fig. 9 represents schematically the main types of solar concentrators. PEC devices and solar concentrators can be coupled via two main assembly arrangements: (i) externally, in separate structures, focusing sunlight only in the PV or PV/PEC components; and (ii) integrated in the device itself, i.e. comprised in the glass windows.254,255Table 5 qualifies the compatibility of each solar concentrator depicted in Fig. 9 with the PV and PEC technologies, based on multiple categorization criteria. | ||
| Fig. 9 Schematic representation of the different types of solar concentrators: (a) parabolic trough collector (PTC); (b) linear Fresnel reflector (LFR); (c) compound parabolic concentrator (CPC); (d) luminescent solar concentrator (LSC); (e) parabolic dish concentrator (PDC); (f) Fresnel lens (FL); and (g) convex lens (CL). The focal area in the receiver is highlighted in red. | ||
| Categorization | Solar concentrators (abbreviation) | |||||||
|---|---|---|---|---|---|---|---|---|
| PTC (a) | LFR (b) | CPC (c) | LSC (d) | PDC (e) | FL (f) | CL (g) | ||
| *NA – non-applicable.**NR – non-relevant. | ||||||||
| Working principle | Reflective | ● | ● | ● | ● | ● | ○ | ○ |
| Refractive | ○ | ○ | ○ | ● | ○ | ● | ● | |
| Focal area | Linear | ● | ● | ● | ● | ○ | ○ | ○ |
| Punctual | ○ | ○ | ○ | ○ | ● | ● | ● | |
| Receiver | Planar | ○ | ● | ○ | ● | ● | ● | ● |
| Cylindrical | ● | ● | ● | ○ | ● | ○ | ○ | |
| Compatibility | PV | ○ | ● | ○ | ● | ○ | ● | ● |
| PEC | ● | ● | ● | ● | ○ | ● | ● | |
| Association with PV/PEC | External | ● | ● | ● | ○ | ● | ○ | ○ |
| Integrated | ○ | ○ | ○ | ● | ○ | ● | ● | |
| Main application | Fluid heating | ● | ● | ● | ○ | ○ | ○ | ○ |
| Power generation | ○ | ● | ○ | ○ | ● | ○ | ○ | |
| Glass/windows | ○ | ○ | ○ | ● | ○ | ● | ● | |
| Coupling with PV | ○ | ● | ○ | ○ | ○ | ● | ● | |
| Defining operating parameter | Fluid temperature (°C) | 350–400; up to 650 | 100–300; up to 600 | 80–250 | NA* | NR** | NR** | NR** |
| Concentration factor (x) | NR** | 30–80 | NR** | up to 100 | up to 3000 | 60–260; up to 720 | 500; up to 900 | |
Reflective concentrators with linear focus are the most common type of solar concentrators, including parabolic trough collectors (PTC), linear Fresnel reflectors (LFR), and compound parabolic concentrators (CPCs). Being studied since the early 1910s, parabolic trough collectors (PTC, Fig. 9(a)) are the most used linear solar concentrators, mainly due to their simple design and low-cost.251 These concentrators work on a single axis and have a linear receiver. Basically, curved mirrors reflect sunlight towards a dark-coated tube (absorber), which is located just above the mirror and contains the recirculating working fluid.256 Linear Fresnel reflectors (Fig. 9(b)) are used since the mid-1960s and, like parabolic troughs, operate with a single-axis mirror.257 These concentrators consist essentially of different arrays of mirrors that focus the solar radiation in a linear central receiver, providing concentration ratios between 30 and 80. The solar receiver is located on the focal line, defined by the tracking mirrors. These mirrors are positioned at different angles and the tracking system is installed at each line of mirrors to concentrate sunlight on the fixed receiver.258
Compound parabolic concentrators (CPCs, Fig. 9(c)) are also a mature technology and have been used commercially for a few decades, namely for PV applications, since they enable capturing the diffuse solar radiation together with the direct component. Among CPCs, the most studied and efficient type is the 2-D CPC trough, which allows collecting and concentrating all the rays within a specified acceptance. Some novel features have been added to this type of solar concentrator, namely lens-walled structures, enabling a larger acceptance angle and better optical efficiency. Also, customized 3-D CPCs, ideal for PV applications, are a great improvement over traditional 2-D CPC troughs, achieving optical efficiencies higher than 90%, considering that the incident rays are smaller than the acceptance angle.259 Advantageous design features of the different reflective linear solar concentrators may be combined to produce better solar concentrators. The best example is the combination of LFRs with CPCs, where the receiver of the LFR incorporates a secondary reflector, usually based on a 2-D CPC.260 Due to the linear and cylindrical receiver configuration, reflective linear solar concentrators are mainly used for heating a thermal fluid or for steam generation; therefore, they are not usually coupled to water electrolysis devices. Nevertheless, these solar concentrators can be redesigned for coupling with PEC devices, including double-axis tracking systems and special supports for holding linear-array reactors. As a result, these concentrators can provide homogeneous concentrated sunlight in a rectangular focal plane (low to medium concentration range), with the advantage of having a small form factor.261 SoCRatus, developed by the German Aerospace Centre, is one of the best examples of an LFR suitable for PEC devices, having been already demonstrated for this purpose.97 This solar concentrator, equipped with 22 linear mirrors and a two-axis tracking system, concentrates sunlight in a rectangular flat focus of 250 × 10 cm2, ideal for holding linear-array reactors.43,262
Solar concentrators with a punctual focus can be considered the second big group of solar concentrating devices, including the reflective and refractive types. Parabolic dish concentrators (PDCs, Fig. 9(e)) are probably the most well-known punctual solar concentrators, presenting a simple design, based on a parabolic reflective dish, usually equipped with a dual-axis solar tracking system, being commonly used for power generation. The central receiver is located at the focal point,263 reaching concentration factors up to 3000. PV and PEC devices can be coupled with PDCs, but only if structural changes are made, namely placing the receiver out of the focal point, introducing secondary reflectors or using materials with lower reflectance in the dish.264,265 Still, by changing the optimal operating parameters of the PDC, the receiver will not get homogenous radiation, which can lead to the appearance of parasitic species transport paths and recombination losses.266 The concentration of solar radiation in a punctual focus area can also be achieved using Fresnel lenses (Fig. 9(e)) or convex lenses (Fig. 9(g)). This technical solution is mainly used in small devices or modular devices comprising small individual cells. In fact, these types of lenses have been used in concentrated photovoltaic electrolysers (CPVE) for a few decades.267 In this case, lenses are typically used to focus sunlight on a special tailored multi-junction solar cell, which is then connected to the anode of the electrolyser.268 Overall, both types of lenses can be coupled to PEC devices, directing the concentrated solar radiation to the photoelectrode, or integrated in the device itself, as a front window.269
Finally, luminescent solar concentrators (LSCs, Fig. 9(d)) are a distinct type of solar concentrating devices, with a different working principle comparatively to the other types. Typical LSCs consist of plastic optical waveguides doped with fluorophores, or glass windows coated with active layers of emissive materials, which can absorb direct and diffused sunlight and then re-emit it at longer wavelengths. Total internal reflection propagates the luminescence towards a receiver (normally a PV cell), placed on the opposite side.270 LSCs combine the advantage of reflective and refractive solar concentrators – collecting sunlight from a larger area and focusing onto a smaller area; as so, LSCs are ideal for windows.271 These devices have been pointed as a cost-effective alternative to optic-based solar concentration systems aiming at enhancing the performance of stand-alone solar cells and allowing their integration into existing or newly constructed buildings, i.e. semi-transparent PV windows capable of transforming energy-passive façades into large-area energy generation units, a particularly appealing strategy towards nearly zero-energy buildings.272,273 LSCs can be coupled to PEC units, replacing one of the transparent windows. Additionally, if the emission wavelength of the LSC emitters matches the spectral absorption peak of the photo-active electrode, the overall efficiency can be further increased.274,275
Concentrated photovoltaic electrolysers (CPVEs) are the closest available technology to future systems that combine solar concentrators with PEC devices. CPVEs have been investigated as a thriving way for low-cost and clean H2 production from sunlight, aiming at improving the overall performance of existing solar-powered electrolysers. Design guidelines have been presented for conceptual designs, based on performance modelling. Based on modelling calculations and LCA analysis, Dumortier et al.276 showed that the use of an integrated concentrated PV electrolyser allows reducing the GHG emissions of H2 production by up to seven times and increasing the energy yield ratio – the ratio between the energy generated during the lifetime of the equipment and the energy that they incorporate – by four times. A good example of a successfully demonstrated CPVE device was reported by Jia et al., who reported a CPVE device comprising a PEM electrolyser assembled in series with an InGaP/GaAs/GaInNAsSb triple-junction solar cell. This device was operated under concentrated sunlight, at ca. 42-suns, where the maximum power point of the PV was adjusted to the operating capacity of the electrolysers, achieving an STH efficiency of 30% over 48 h.277 Still, it is important to keep in mind that in CPVEs, solar concentrators and tracking systems will represent a significant part of the operational costs, which will affect the final H2 production cost. Fresnel and convex lenses are also often used in CPVE applications. Price et al.278 demonstrated that using high-efficiency micro PV cells embedded between a pair of plastic lens arrays allowed reaching a 200-fold increase in the power output comparatively to an unconcentrated cell, at an optical efficiency of 70%, for incidence angles of ca. 60°, at a fixed latitude tilt. This work demonstrates that high efficiencies (up to 40%) can be achieved by simply incorporating arrays of convex plastic lenses in the front window and convex mirrors at the back, resulting in a compact and inexpensive design.
Concentrated-PEC (CPEC) systems, coupling two technologies in a single unit, allow for reducing costs and further improving the overall efficiency, since concentrated sunlight is used by both the PV and the electrolyser components.279 Unfortunately, research work addressing the use of concentrated sunlight in PEC systems is strongly limited by the lack of commercial PEC devices, particularly with an illuminated area larger than 50 cm2.175,199 In a pioneer work by Segev et al.,280 two different types of α-Fe2O3 PEs, i.e. a 50 nm polycrystalline dense film and a 500 nm mesoporous layer with cauliflower structures, were subjected to simulated concentrated sunlight, ranging from 1 to 25 kW m−2. It was observed that the photocurrent produced by both 0.28 cm2 samples increased linearly with solar flux, while the photopotential scaled logarithmically with light intensity. An example of external coupling of a PEC device to a solar concentrator was demonstrated by Khan et al., which coupled a PEC reactor assembled with a triple-junction GaInP/GaInAs/Ge PV cell (0.25 cm2), and a Ni foil as an O2 evolution catalyst, with a Fresnel lens, kept at a focal distance of 5 cm, achieving an STH efficiency of 13%.281 Despite the STH efficiency surpassing that assumed as the commercialization target, i.e. 10%, the active area of 0.25 cm2 remains far from any commercial ambitions. An example of a large-area CPEC device was demonstrated by Vilanova et al.97 The authors reported a 200 cm2 PEC modular device designed and optimized for continuous operation under real concentrated sunlight that was coupled with a modified linear Fresnel reflector (LFR). When equipped with nanostructured α-Fe2O3 PEs, this device generated ca. 2.0 mA cm−2, below 14 kW m−2, corresponding to a hydrogen production of 5.5 × 10−5 gH2 h−1 cm−2, the highest reported for a large-area CPEC device; still, the STH efficiency values varied between 0.36% and 0.12%, which highlights the importance of the compromise between area and efficiency. Splitting the solar spectrum can be a promising strategy to further increase STH efficiency, particularly for combined CPV-PEC devices,.282,283 In the work of Bicer et al.,284 the use of light splitters allowed for achieving power conversion efficiencies of up to 15%, enabling the use of lower wavelengths by a Cu2O PE.
In general, CPEC devices allow for reaching higher photocurrent densities at lower applied potentials, but they still result from the combination of two separate units, a PEC device and a solar concentrator. Hence, concentrator-integrated PEC (CIPEC) systems can be the ultimate approach since all components are combined in a single unit.285 The CIPEC approach has been investigated for different PEC systems over the last few years. Zhang et al. reported a luminescent solar concentrator based on quantum dots (QDs), which, combined with TiO2 photoanodes, achieved a current density of ca. 16.0 mA cm−2 (at 0.9 VRHE), the highest value reported for PEC cells based on CdS/CdSe QDs; still, the stability was quite low, i.e. a 17% photocurrent decrease was recorded after 4 h, attributed to the self-oxidation of QDs induced by ultraviolet radiation.286 Another example of CIPEC technology was addressed by the SHINE-Nanotera consortium. This project aimed at developing a PEC device with integrated solar concentrator units for operation under medium-range solar concentration, i.e. from ca. 20 to 50.287 In this case, the solar concentrator embodied a self-tracking design, with an angular acceptance of 32° (±16°) and an effective concentration of up to 20; water microfluidics was used for thermal management.288 This smart system used a dichroic mirror to split the solar radiation, where the transmitted part (>750 nm) was absorbed by a black paraffin wax layer. Under solar radiation, paraffin melts and expands upwards, creating a coupling feature for the reflected light.289 Then, the focal spot changes and a different part of the actuator is activated, depending on the position of the sun throughout the day/season. Simpler CIPEC devices can be assembled by just replacing one window of a PEC reactor with Fresnel or convex lenses. The use of lenses with a short focus distance is compatible with planar PEC device architectures, providing moderate concentration factors, without the need for additional cooling systems.
At an industrial level, the use of concentrated sunlight for PEC-H2 production is essential to boost H2 production and thus easily overcome the claimed disadvantage that PEC systems generate lower current densities than their counterparts. Additionally, the dissipation of excess heat from increased solar load can be more easily achieved in PEC devices than in PVs, since PEC devices comprise electrolyte recirculation by default, which can be optimized for thermal management purposes. Overall, the upscaling of PEC devices for operation under concentrated sunlight must take advantage of the know-how acquired from mature technologies that use concentrated sunlight to drive the water electrolysis, like currently existing CPVE plants.247
4.2. Thermal management strategies
Any device that is designed to be operated outdoors, such as PV cells and PEC devices, must incorporate thermal management strategies since, throughout its lifetime, it will experience significant temperature fluctuations, whether related to the ambient temperature, which can vary greatly between seasons of the year, or even throughout the day (depending on the site location), or related to sun exposure. Even under 1-sun, photo(electro)chemical systems can reach working temperatures of ca. 60–90 °C. While excessive heating can reduce the device's lifetime, careful thermal management may improve the overall STH efficiency.290 Thermal management becomes even more important if coupling with concentrating solar technologies is foreseen, as the receiver area will experience a high thermal load. Taking the example of CPVEs, thermal management was pointed out as the most critical design feature for assuring the expected performance increase due to high photon flux.291 Also, thermal management can play a determinant role in the final H2 cost; it should represent at least 20% of the total power costs, being ascribed mainly to water supply for cooling purposes.The simplest and most effective strategy to assure proper thermal management in a PEC device is through the optimization of the electrolyte circulation for this purpose. Indeed, water cooling is the most widely used strategy to achieve proper thermal management in multiple applications. By tailor-making electrolyte feeding and recirculation, proper heat dissipation can be achieved, ensuring safe operating conditions that prevent the device from experiencing advanced degradation. As an example, Vilanova et al. used fluid mechanics simulation software to design the fluid body of a PEC reactor to optimize the electrolyte flow.43 Following a stepwise approach by changing the diameter and location of inlets and outlets, the authors developed an electrolyte inlet manifold incorporating several diffusers that allowed reaching an optimized electrolyte upward flow. When subjected to continuous recirculation at 0.5 L min−1, the PEC reactor with a photoelectrochemically active area of 200 cm2 was demonstrated to effectively maintain a desired operating temperature and dissipate excess heat under concentrated sunlight, at ca. 18-suns. Khan et al. have also demonstrated that electrolyte recirculation was sufficient to dissipate heat in a tailor-made PEC reactor operating under 42-suns.281
Apart from optimized electrolyte recirculation, there are many different thermal management strategies that can be used for PEC applications. Rashidi et al. have reviewed some of the most used thermal management strategies in fuel cells, electrolysers and supercapacitors.292 From the many different options, those that can be potentially interesting for new PEC devices are summarized in Table 6.
| Thermal management strategy | Brief description | Pros/benefits | Cons/challenges |
|---|---|---|---|
| Liquid cooling | The simplest and most used strategy. Consists of using the electrolyte flow inherent to a PEC device to cool it. The reactor is designed for having an electrolyte flow that assures proper heat dissipation. | – Highly effective | – Requires a tailor-made manifold design |
| – Simple integration | – May require a high electrolyte flow rate | ||
| – Also promotes an effective collection of evolved gases | – High simulation efforts | ||
| – Great versatility and adaptability to electrolytes and absorber materials | |||
| Nanofluid cooling | Use of nanofluids, consisting of liquids containing uniformly distributed nanoparticles. | – The required cooling area can be reduced by about 10% | – Nanoparticles increase the viscosity of the liquid |
| – Significant improvement of thermal conductivity and diffusivity | – Nanoparticles can cause light trapping and react with the electrolyte | ||
| – Can be an upgrade to standard liquid cooling | – Nanoparticles may represent additional challenges to the recirculation equipment, namely pumps | ||
| Evaporative cooling | Refers to phase-change cooling processes, including boiling and evaporation. Heat of vaporization is the basis of the cooling process. | – Decreased coolant flow rate | – Requires the use of a vapor-phase electrolyte |
| – Simpler and more compact reactor layout | – Two-phase flow instabilities | ||
| – Use of latent heat of phase change | – Not suitable for all photo absorber materials | ||
| – May increase degradation | |||
| Heat spreaders | Heat is first transferred by conduction and subsequently it is dissipated by forced or free convection. Heat spreaders act as heat sinks. Heat spreaders that have high thermal conductivity are employed near the feeding channel. | – In large stacks, it reduces the size of the cooling system. | – Complex design |
| – Low-cost graphite-based materials can be used as heat spreaders. | – The thermal conductivity of the cooling plates has to be very high | ||
| – Difficult integration in conventional PEC device architectures | |||
| Integrated thermoelectric generators (TEG) | An array of p- and n-type doped semiconductors are assembled electrically in series and thermally in parallel to form a thermoelectric generator. A steady temperature gradient is sustained via passive heating, by conveniently placing the commercial TEG between a hot PEC reactor and a room-temperature medium. | – Compact design | – Complex design |
| – Waste heat can be used for generating additional bias potential | – The energy levels required for overall water splitting limit the choice of materials to a small selection of wide bandgap semiconductors | ||
| – Allows integrating either single or tandem light absorbers with the TEG in a single optical light path | |||
| Heat pipes | Similar to a heat exchanger, heat pipes with a recirculating thermal fluid are used to promote heat exchange. | – Highly effective | – Complex design |
| – Thermal management can be performed in a separate unit, allowing for more accurate control | – As the heat pipes need to pass through the reactor, the pipe materials have to be resistant to the electrolyte | ||
| – Cooling can be optimized separately | – Depending on their location, the inclusion of heat pipes might disturb evolved gas collection and might cause shadowing | ||
| – Fluids with high thermal conductivity can be used, independently from the electrolyte properties. | – Passing pipes through the reactor can add sealing challenges | ||
| – Added maintenance efforts | |||
| Air cooling | An air flow is promoted perpendicular to the PEC reactor to remove excess heat. | – Thermal management can be performed in a separate unit, allowing for more accurate control | – Since air has poor thermal conductivity, a high flow rate must be used, increasing power consumption |
| – Cooling can be optimized separately | – Colling is promoted from the outside; if the reactor embodiment comprises materials with poor thermal conductivity, such as most polymers, dissipating heat from the inside of the reactor will be difficult | ||
| – No special material requirements are needed, namely corrosion resistance | – Added maintenance efforts |
It is important to note that operating under high sunlight concentration requires both thermal and power management solutions.287 Since the energy required for promoting the water electrolysis is much higher than the thermodynamic value related to the energy of the reaction, mainly due to high ohmic losses, associated with various components of the cell, i.e. the electrodes, the electrolyte, and the membrane, which increase linearly with the generated current but decrease with temperature, thermal management is essential for minimizing performance losses. Tembhurne et al. developed a 2-D multi-physics model to investigate and simulate CIPEC devices subjected to highly concentrated sunlight, working at high temperatures and currents.254 It was concluded that the performance decreased due to the faster degradation of light absorber materials and catalysts in two distinct zones, one corresponding to a low operating current density zone, mostly affected by temperature, and the other corresponding to a high operating current density zone, characterized by mass transport limitations, significantly affected by the maximum power point. The underlying degradation mechanisms can be controlled and counterbalanced with proper thermal and power management, by controlling the electrolyte flow and by installing a power control unit; the authors reported that a 12% degradation in STH efficiency could be compensated by increasing the mass flow rate of the electrolyte by seven times. Tembhurne et al. also demonstrated that the recirculation of the aqueous electrolyte solution allows the dissipation of heat from the illuminated semiconductor, preventing photopotential losses.17
One of the most interesting approaches for efficient thermal management in PEC water splitting under concentrated sunlight was disclosed by Pornrungroj et al.293 The authors reported an integrated thermoelectric (TE)-PEC design for overall water splitting under concentrated solar radiation, where single or tandem light absorbers and heat-harvesting thermoelectric generator (TEG) are assembled in a single optical light path. Moreover, waste heat can be used to generate additional photopotential to accomplish unassisted solar water splitting. A stable temperature can be achieved through passive heating, by placing the TEG between the heated PEC reactor and a room-temperature medium. The fundamental concept relies on assembling electrically in series p- and n-type doped semiconductors, but thermally in parallel to form a thermoelectric generator. The authors were able to report unassisted water splitting under 2-sun by wiring a BiVO4 PE to a thermoelectric element. Also, the photocurrent of a thermoelectric–perovskite–Fe2O3 system was enhanced 29.7 times under 5-sun. Still, the authors also reported that scalability might be a challenging issue to address for these systems, as in-plane parasitic effects were already noticeable in the reported 6 cm2 device.
Hence, commercial PEC-H2 production must consider the use of concentrating solar technologies and thermal management for increasing H2 production and decreasing performance losses. Therefore, full-scale PEC-H2 should comprise key elements, as follows: (i) an electrical power station; (ii) a water pre-treatment unit, including filtration systems, reverse osmosis, and a mixing system for adding the electrolyte; (iii) the PEC devices coupled with solar concentrators; (iv) gas–liquid separation systems, one for H2 and another for O2 (when O2 is considered a valuable by-product; the remaining liquid, should be sent back to the PEC units); and (v) an H2 purification system, including de-oxidizer and drying sub-systems.294Fig. 10 shows a schematic of a hypothetical CPEC-H2 production plant.
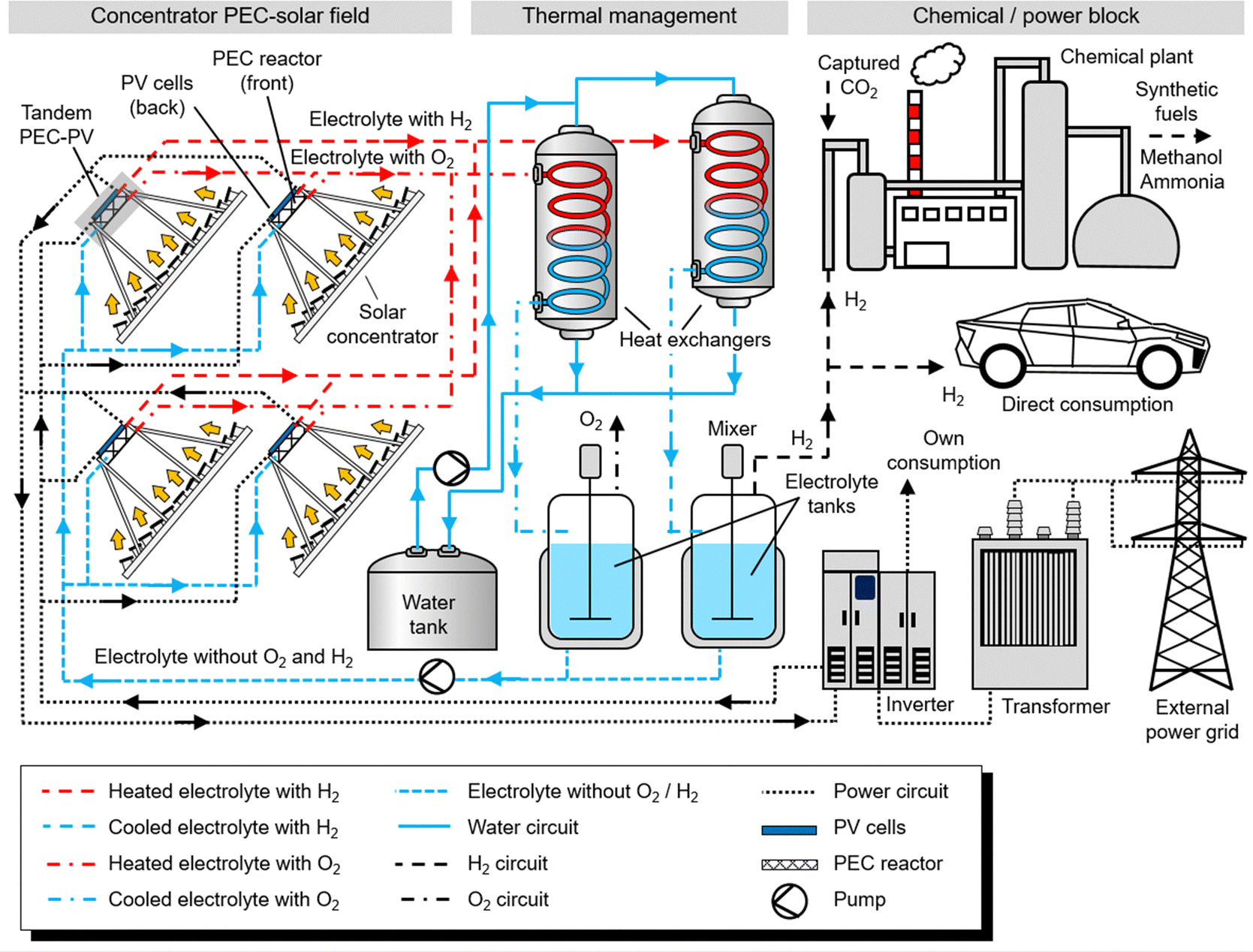 | ||
| Fig. 10 Schematic representation of a CPEC-H2 production plant, comprising LFRs coupled with tandem PEC–PV devices and an external thermal management system. | ||
5. Use of biomass derivatives in PEC-H2 production
The use of biomass derivatives has been pointed out as a clever approach to boost H2 generation in PEC devices, allowing them to overcome thermodynamic and kinetic constraints of conventional PEC water splitting systems and to fulfil circular economy requirements. In these systems, the Gibbs free energy is smaller than that for the water splitting, as the redox potentials of the biomass derivatives are much more negative than that of water, indicating that they are more easily oxidized, making this process more efficient from a thermodynamic point of view.295 The operating principles of PEC-H2 production from biomass derivatives and water consist of (i) generation of electron–hole pairs; (ii) movement of electrons from the PE to the CE through an external circuit; (iii) water reduction at the cathode; and lastly (iv) oxidation of biomass by the photogenerated holes to form simpler organic intermediates and CO2.296 The last step (iv) is equivalent to the donation of electrons from the biomass to the PE to consume the photogenerated holes. Overall, the fundamental mechanism of PEC-H2 production from biomass-derived oxygenates (denoted as CxHyOz) and water can be simplified as follows:| CxHyOz + (2x − 2z)H2O → xCO2 + (2x − 2z + (y/2))H2 | (3) |
In this case, the performance of the PEC cell is projected to rely on the efficient separation of charge within the PE, on the biomass capability to exchange electrons with the PE, and on ion transport through the electrolyte. Therefore, the addition of organic molecules to the electrolyte started to be explored in the PEC-H2 field, since it boosts electron transfer due to the smaller activation overpotentials.297 For PEC devices that use biomass derivatives, H2 production strongly depends on the properties of the PE, which should fulfil similar key requirements as those of conventional devices: (i) suitable band edge positions; (ii) strong light harvesting ability; (iii) efficient charge transport; (iv) chemical and photoelectrochemical stability; (v) high selectivity and tolerance to poison-species; (vi) low overpotentials for the reduction–oxidation reactions; and (vii) low-cost and abundance. Among the several materials studied for PEC-H2 using biomass derivatives and water, α-Fe2O3 has been pointed out as one of the most promising materials. This classical semiconductor meets several fundamental requirements, namely a suitable bandgap energy of 1.9–2.2 eV, ideal for solar spectrum harvest, appropriate valence band edge towards water and organic oxidation, and chemical stability in alkaline environments, apart from the well-known abundance, low-toxicity and low-cost. However, it comprises some detrimental features such as low optical absorption due to its indirect bandgap and high recombination rates leading to a short hole diffusion length (ca. 5 nm). As α-Fe2O3 is the benchmark in stability, but seems to be limited in performance, in the pathway for a highly efficient biomass-driven PEC-H2 generation device, spinel complex ferrites based on non-CRMs, such as ZnFe2O4 and CuFe2O4, have also been studied – Fig. 11(a). Both zinc and copper ferrite compounds have favourable bandgap values of ca. 2.0 eV and 1.9 eV, respectively, and they can generate higher photocurrent densities under back-side illumination, ideal in a near-zero-gap design. Nonetheless, high recombination rates and low carrier diffusion lengths are critical challenges that have been difficult to overcome. Therefore, upscaled systems should consider clever design features, such as multilayer tailored arrangements, passivation tunnelling, as well as innovative contact architectures, assuring field effect passivation, suitable band alignment, and improved oxidation and reduction processes.
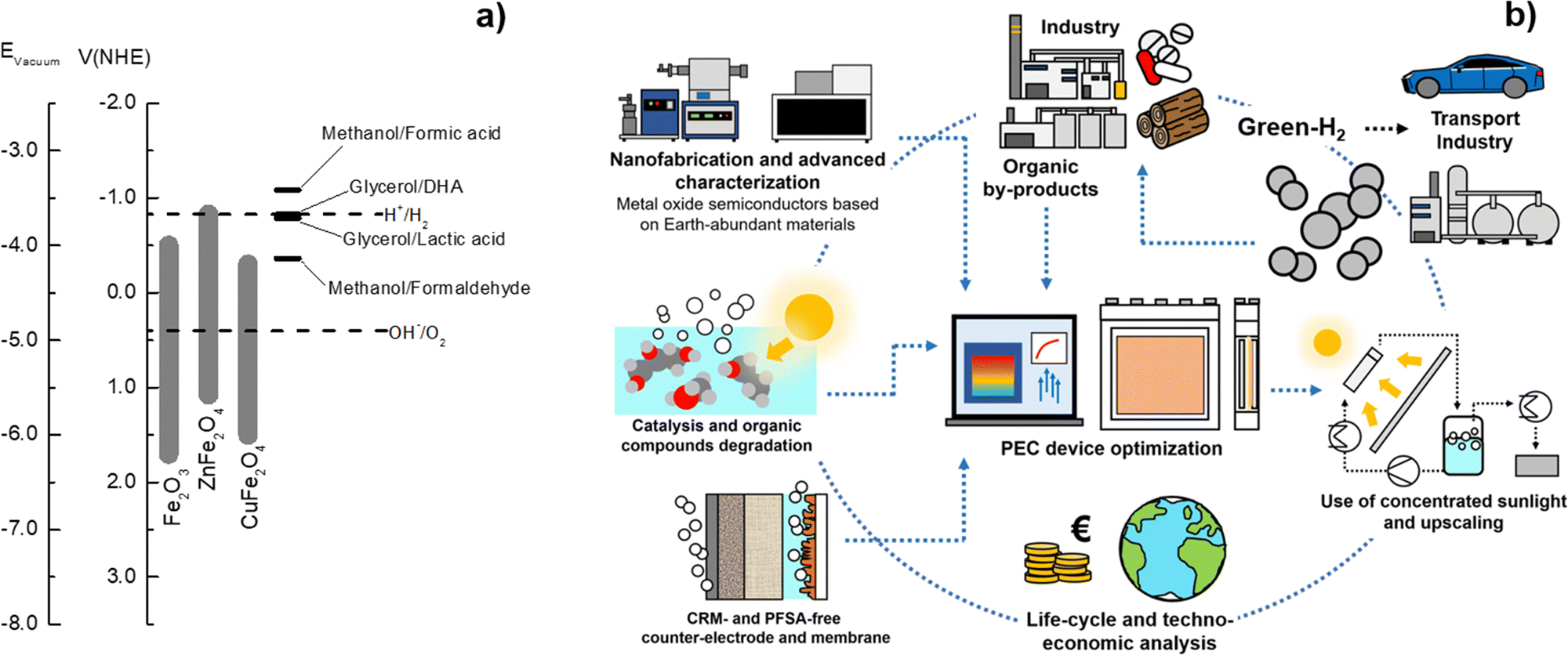 | ||
| Fig. 11 The use of biomass derivatives for PEC-H2 production: (a) band alignment of some promising metal oxide semiconductors for PEC-H2 production intermediated by some industrial organic by-products; and (b) added-value of PEC-H2 production from biomass derivatives and water to existing supply chains, using industrial by-products, CRM-free electrodes and concentrated sunlight. | ||
From an environmental point of view, these systems are more competitive than traditional ones, as several biomass by-products, such as methanol and glycerol, are regarded as renewables.298 Moreover, transforming wastewater and sunlight into green-H2 breaks the high verticalization of the H2 market by directly turning an energy consumer into a prosumer. Glycerol is an important by-product of biodiesel and soap production. Particularly in the biodiesel industry, the largest residue is washing wastewater.299 Biomass-derived biodiesel waste streams can contain glycerol at ca. 50% of purity, along with methanol and organic acids. Also, conventional crops used for glycerol production present a high environmental impact mainly arising from significant land use, especially in the cases of palm oil, soybean oil rapeseed oil and sunflower oil.297 N-type spinel oxide PEs based on MCo2O4 have shown promise, like CuCo2O4;300 still, as Co is listed as CRM, CuFeOx PEs could be more interesting for this application. However, another study on glycerol oxidation on bare catalyst-free α-Fe2O3 photoanodes did not result in the production of formate but originated a variety of products, indicating selectivity challenges.301 Methanol (MeOH) is another common pollutant in the wastewater from wood pulp, paper, and pharmaceutical industries.302 Also, the conversion of CO2 into methanol, a process with particular interest for the upcoming years, usually produces large volumes of wastewater containing trace amounts of MeOH, but enough to prohibit its reuse for electrolysis or as a cooling fluid. Although MeOH is valuable, it is difficult to recover from dilute waste streams, becoming a cheap and valuable resource for PEC-H2 production. Bare α-Fe2O3 has been demonstrated to be active for both glycerol and methanol oxidation. Likewise, Zn and Cu ferrites comprise structural motifs that make them promising candidates for biomass oxidation, especially regarding their catalytic activity, as their valence band positions present sufficient thermodynamic driving force.
Nonetheless, using wastewater as an electrolyte in PEC devices is challenging, namely because of the harsh chemical environment created (like in the case of MeOH) and added viscosity and clogging (like in the case of glycerol). These challenges can be tackled by (i) using tailored catalyst layers, providing the surface of the PE with “self-cleaning” and anti-sticking capabilities, (ii) optimizing fluid flow, preventing stagnant zones and facilitating the removal of organic deposits, and (iii) using concentrated sunlight, favouring the degradation of organics, both by intensifying photocatalysis and by increasing the thermal load.303Fig. 11(b) schematically shows the combination of strategies that should be implemented to achieve high-performing PEC-H2 production from biomass derivatives and water. As concluded by Huang et al., apart from understanding photocatalytic mechanisms and kinetics at the fundamental level and developing new advanced materials, designing novel photo-reactors able to optimize mass transfer, photon transfer, light utilization, and ready for upscaling is of utmost importance.304 Moreover, the authors also concluded that optimizing the reactor operating conditions, including photocatalyst loading, light intensity, pressure, pH (liquid phase), reactant feed ratio, etc. is equally important for efficient PEC-H2 production from biomass-derived products. This vision is accompanied by Luo et al., as the authors highlight that the rational design of semiconductor-cocatalyst systems based on Earth-abundant elements is essential for assuring high efficiency and selectivity for a given by-product used as feedstock.297
Complementarily, the system as a whole requires a tailored optimization, where reaction parameters should be carefully chosen, namely temperature, feedstock concentration, electrode distance and electrolyte flow. Also, to improve the environmental performance of these systems, the development of a mature market for oxidation products is essential. Another interesting approach has been explored by the research group of Reisner. The authors have been demonstrating the materialization of different circular economy approaches by combining waste photodegradation with solar fuel production in PEC devices.305,306 In this case, particulate photocatalysts are often considered, which is not the focus of this review. Nevertheless, Bhattacharjee et al. have successfully used perovskite PEs for this purpose.118 Additionally, the authors have also addressed one of the most important environmental problems of the present, which is the contamination of water resources with plastics;307 here, plastics can be seen as a raw material for boosting H2 production in PEC devices. As an example, the authors reported a perovskite-based PEC device capable of simultaneously producing H2 and reforming soluble biomass and plastic derived, using a novel Cu30Pd70 oxidation catalyst, achieving 60–90% product selectivity and sustaining unassisted photocurrents of 4–9 mA cm−2 for plastic, biomass, and glycerol conversion, in either a two-compartment or integrated “artificial leaf” configuration.
6. Overview of techno-economic analysis and life-cycle assessment on H2 production
Selecting the appropriate H2-production technology for industrial applications is a massive challenge. At present, green-H2 is produced mainly using mature technologies, such as conventional electrolysers, which are often operated with electricity from the grid. There are few available wide comparative studies on H2 production methods that combine life cycle and techno-economic indicators, which are essential for understanding how PEC-H2 production performs from an all-inclusive point of view. Indeed, for PEC-H2 production, which is at an early stage of development, adopting a holistic approach is vital for improving its competitiveness, regarding both environmental and economic metrics.Life cycle assessment (LCA) is the most widely used tool for assessing the environmental impacts attributed to a product or a service, comprising three major stages: (i) goal and scope definition, including the selection of life cycle boundaries; (ii) inventory data collection on material and energy flows, where emissions and consumption of resources are identified and quantified; and (iii) estimation of the environmental impacts of each life cycle step, normalization of the impact category indicators and grouping by weighting or significance.308 The most common type of LCA is the cradle-to-grave approach, which considers all steps of the life cycle, from the extraction of raw materials until the disposal/end-of-life. For energy systems, due to their complexity, a cradle-to-gate approach is more effective, usually including only the production and distribution stages. Particularly for PEC-H2, since commercial large-scale PEC devices are not available yet, LCA studies are based mainly on assumptions and extrapolations from existing devices, which were developed for laboratory purposes. This can result in values with no practical significance and are unfairly detrimental to the PEC water splitting technology. Sathre et al. performed a close approach to what could be an ideal prospective life-cycle inventory for PEC water splitting devices.232,309 Based on the detailed inventory, the authors were able to calculate relevant techno-economic indicators: large-scale PEC-H2 production with a 10% STH efficiency and a life span of 20 years (considering 1 GW per year, equivalent to 610 ton of H2 per day) would have an energy payback time (EPBT) of 8.1 years, an energy return on energy invested (ERoEI) of 1.7, and a life-cycle primary energy balance of 5.0 × 1017 J, considering 40 years of projected service life. If the STH efficiency is increased to 20%, the EPBT can be further reduced to 3 years.232 Although Sathre's study focused on net-energy parameters, it was possible to identify the most relevant components of the life cycle inventory: (i) fabrication of active materials; (ii) production of the materials used in the embodiment; (iii) assembly of the cell; (iv) assembly of the module; (v) operation of the module (including auxiliary equipment); and (vi) end-of-life. The STH efficiency and the life span of the PEC cells remain critical parameters, since they affect the net energy balance, whereas the balance of system (BOS) parameters, such as the construction and operation of liquid and gas infrastructures, present a lesser significance. However, the most limiting factor of comparative analyses based on LCA studies is the multiplicity of reported impact categories and the lack of standardization in the way results are presented. Following, the comparative outlook presented here considers only the global warming potential (GWP, kgCO2eq. kgH2−1) and acidification potential (AP, gSO2eq. kgH2−1) as categories of environmental impacts, since they are the most addressed indicators; the functional unit is 1 kg of generated H2.
Similar to environmental performance, assessing the economic viability of H2 production methods is crucial for having a holistic view of their competitiveness, and especially for understanding how competitive PEC-H2 is against the other options.37,38 Due to the lack of standardization of results and ambiguities in the definition of variables and criteria for quantifying economic performance, the payback time and the levelized cost of hydrogen (LCOH) were the economic indicators selected for the comparative analysis presented here. In short, the payback time indicates the duration for which a facility needs to be operated to produce an amount of H2 equivalent to the energy required for its manufacturing, construction, and decommissioning; it is calculated as the ratio between the fixed energy input and the annual net energy output under full-scale steady-state operation.232 This parameter is not suitable for classification as a life-cycle metric, since it does not consider the energy generated after reaching the payback time; therefore, the service life of the facility is not included in the calculations. The LCOH usually refers to the ratio between the net present value of the total cost of constructing and operating an H2 plant and the total H2 production over its lifetime. As so, it can be assumed as the total cost needed to bring an H2 production facility to a commercially operational status, combining individual purchase costs with installation factors, indirect costs (i.e. engineering, construction, contingency costs, taxes, etc.) and maintenance during the lifetime of the facility.
Finally, it is essential to choose the H2-production technologies that should be compared with PEC water splitting. At present, there is no specific taxonomy for grouping H2 production methods, but one can assume a logical categorization depending on the main energy input, being classified as fossil fuel- or renewable-based.310,311 Fossil fuel-based technologies are more mature, widely developed and account for almost 95% of current H2 production.312 Among them, steam methane reforming (SMR) and underground coal gasification (UCG) are the most widespread; others include hydrocarbon pyrolysis and autothermal reforming. Despite accounting for only 5% of current H2 production, renewable-based technologies are more diverse but essentially comprise (i) water splitting based technologies, which include electrolysers (alkaline – AEC, proton exchange membrane – PEM, and solid oxide electrolysis cells – SOEC, supplied with electricity from the grid or from wind, hydro or solar-PV plants), photoelectrochemical (PEC) water splitting and high-temperature electrolysis (HTE), and (ii) bio- and thermochemical processes, where biomass and organic matter are degraded, generating H2, which can be assisted or not by solar radiation (direct and indirect bio-photolysis, photo and dark fermentation).313 A new way of classifying H2 production methods has emerged in recent years, consisting basically of a colour code.314 This colour categorization started when H2 production based exclusively on renewable sources became unanimously recognized as “green hydrogen”. However, given the diversity of H2 production technologies, there was a need to create a colour code that could also encompass other technologies, distinguishing the working principle, the primary resources, and the generated emissions. Even so, this colour code is not unanimous and, depending on the literature, some colours might have different definitions, thus making the classifications imprecise. For instance, yellow-H2 can easily be found as referring to PV-electrolysis or grid-fed electrolysis. Fundamentally, there are three colours that are widely used and accepted, and whose definition is not controversial: (i) grey, produced by steam methane reforming, partial oxidation, or autothermal reforming; (ii) blue, produced from fossil fuels, but with carbon capture, utilization, and storage; and (iii) green, produced by water electrolysis using exclusively electricity from renewable sources. Fig. 12 schematizes the adopted taxonomy for H2 production based on renewability and on the H2 colour code. Although the colour code considered in Fig. 12 might seem something trivial or of little importance, the truth is that it has become extremely relevant in the H2 market, since this classification has been directly influencing the value of products made from H2, such as synthetic fuels, ammonia, methanol, among others. Furthermore, the inconsistency and dynamics of the CO2 market315,316 have played a crucial role in the H2 price, boosting the choice for processes that are based on renewable sources and/or have a neutral or very low carbon footprint.317 Given the evident trend of increasing taxes and penalties for the emission of greenhouse gases (translated into tons of CO2 eq.), this has become the present determining criterion for choosing a H2 production technology. In fact, presently the economic competitiveness of H2 production processes relies more on the cost of CO2 emissions rather than on the cost of raw materials.318,319 Furthermore, this discussion often leads to existentialist questions such as “how green electrolysis really is ?” If one considers only the H2 production process, water electrolysis does not generate CO2 emissions, but if one considers, for instance, the life cycle of PV panels, the CO2 balance may not be so green. On the other hand, methane decomposition results in the production of turquoise H2 which, if based on natural gas, can have a neutral CO2 balance, but if biomethane is used, the CO2 balance can be negative. Therefore, it becomes imperative to assess the environmental performance of a certain H2 production process based on its life cycle, particularly regarding its global warming potential, as it will strongly influence the LCOH.
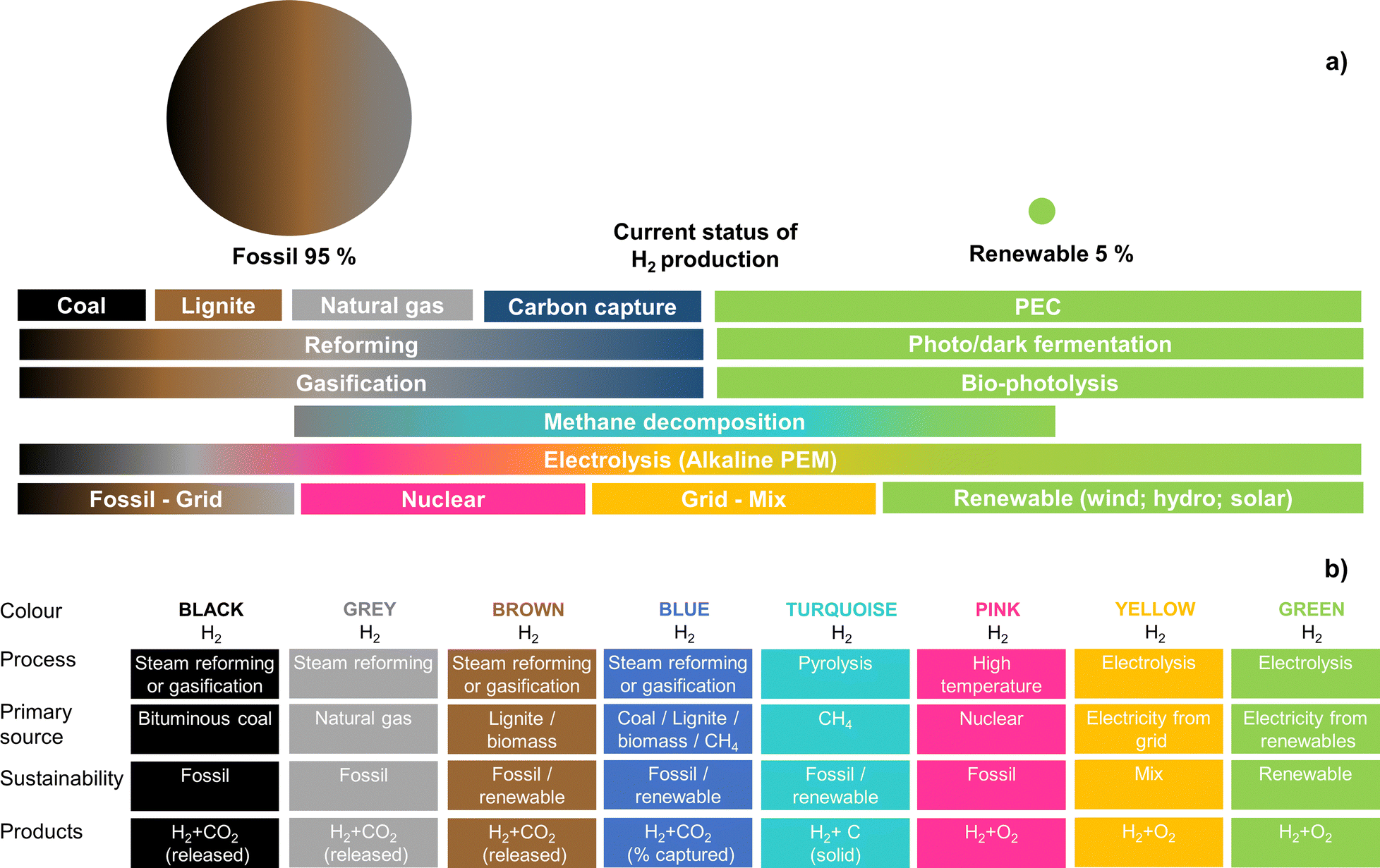 | ||
| Fig. 12 Grouping and categorization of H2 production methods: (a) based on renewability; and (b) based on colour code. | ||
For the present comparative overview, a brief description of each H2 production method, as well as its pros and cons, benefits and challenges, is summarized in Table 7. Only H2 production technologies for which it was possible to collect enough data to perform a reasonable comparison were considered, as detailed in Table 8. The comparative outlook of environmental impact categories and production costs of the selected H2 production method is presented in Fig. 13.
| Technology/abbreviation | Brief description | Pros/benefits | Cons/challenges |
|---|---|---|---|
| Underground coal gasification/UCG320 | Still, one of the most used processes to produce H2. Coal is subjected to high temperature and pressure conditions in the presence of a controlled amount of O2 (or steam), being broken down into syngas, i.e. carbon monoxide (CO), hydrogen (H2), and methane (CH4). | – One of the most mature technologies for H2 production | – High emissions of GHG |
| – Low payback time | – High acidification potential | ||
| – Low water footprint | – As it uses a fossil fuel, the generated H2 is not renewable | ||
| – Cleaner and more effective than coal combustion (if power generation is considered) | – If carbon capture is not considered, the high carbon taxes may undermine cost-effectiveness | ||
| Steam methane reforming/SMR321,322 | Methane is first catalytically reformed at high temperatures, producing carbon monoxide and a syngas mixture. H2 is further produced through the water–gas shift reaction. | – Currently the most common and cost-effective method for H2 production. | – High emission of GHG |
| – High capital costs | |||
| – Coke deposition during the reaction. | |||
| Biomass gasification/BG323 | One of the main routes for the thermochemical conversion of biomass, occurring at higher temperatures than pyrolysis (700–1200 °C), in the presence of an oxidizing agent, originating mainly gaseous products (syngas, i.e. H2 and CO). | – Abundant, renewable, and cheap feedstock. | – Formation of tar aerosols and ash (leading to slagging, fouling and agglomeration) |
| – It is a renewable process | – As the process generates CO and/or CO2, the resulting H2 is considered brown-H2. | ||
| – Promotes forest cleaning, reducing the forest fuel and preventing wildfires | – Expensive separation and purification units | ||
| – Possibility of also using municipal waste in the process. | – Not suitable for intermittent operation | ||
| – Substrate pre-treatment is crucial | |||
| – It can generate undesirable subproducts | |||
| Biogas reforming/BGR324 | Similar to fossil fuel-based SMR, but biogas is used as a renewable resource for producing H2 (usually 55–65% CH4, and 30–45% CO2). | – Biogas is a renewable source of methane | – Feedstock quality |
| – Coupling with commercial digesters, landfills, or wastewater treatment plants. | – Carbon capture and storage | ||
| – Low tolerance for impurities. | |||
| Steam methane reforming with carbon capture and storage/SMR-CCS | The same technology as conventional SMR, but with a carbon capture and storage unit downstream. | – Despite using fossil fuels, the whole carbon balance can be almost zero | – Increased capital cost |
| – Mature process | – It is still non-renewable | ||
| – Carbon capture is quite expensive | |||
| Methane splitting/MS325 | Methane decomposition, also known as methane pyrolysis or methane cracking, is an endothermic process (that occurs typically at 700–1100 °C) where CH4 is decomposed in the presence of a catalyst, generating H2 and solid carbon. | – H2 is easily purified | – Still at low TRL; few commercial solutions available |
| – If bio-methane is used, the carbon balance might be negative | – Innovative catalysts and reactors are needed for the reaction to occur at lower temperature | ||
| – Absence of GHG emissions | |||
| High-temperature electrolysis/HTE326 | Water splitting occurs at high temperatures (700–1000 °C) and the primary source (nuclear, concentrated solar power) generates both thermal and electrical inputs. Solid oxide electrolysis cells (SOECs) are a commonly used process. | – Coupling to existing nuclear power plants allowing large-scale production | – Few chemically stable materials for high temperature and highly reducing/oxidizing environment. |
| – Free of GHG emissions. | – It carries all the well-known problems of nuclear energy, such as safety issues, hazardous waste handling, and the discussion of whether it is renewable or not. | ||
| – Combined heat and gas generation | |||
| Alkaline electrolysis via electricity from the grid/AEC-grid327 | Electric current is provided to two electrodes immersed in a liquid electrolyte, usually concentrated KOH, resulting in the production of H2 at the cathode and O2 at the anode. The two electrodes are separated by a selective gas diaphragm. | – Mature and reliable technology | – High capital cost |
| – Already optimized for large-scale operation | – Corrosive operational environment | ||
| – Required constant power supply, preventing intermittent operation. | – Operation at high potential (1.8–2.4 V) | ||
| – Not necessarily green | |||
| – Need for ultrapure water (leading to the formation of a concentrated wastewater stream) | |||
| Photovoltaic electrolysis/PV–EC322,328 | Solar panels generate electricity, feeding an electrolyser (PEM or alkaline) with the appropriate potential to split water. | – Comprises two mature technologies already optimized for large-scale operation | – Two separate devices are required to drive water splitting (PV unit and electrolyser) |
| – The main energy source is renewable and clean. | – Single-bandgap PV cell | ||
| – Considerable land footprint | |||
| – Intermittence of sunlight | |||
| Wind electrolysis/Wind-EC329 | Wind power plants (grid-connected or autonomous) feed the electrolyser (directly or through batteries). | – The main energy source is renewable | – Intermittence of wind |
| – Comprises two mature technologies already optimized for large-scale operation | – High capital cost | ||
| – Aesthetic impact and noise. | |||
| Photoelectrochemical/PEC44,97 | The electrodes are in contact with an electrolyte in a single device where, at least, one is photoactive. H2 and O2 can be produced in the same compartment (divided by an ionic exchange membrane) or in physically separate compartments. | – Possibility of using concentrated sunlight | – Still at an early development stage |
| – Low-cost materials | – Scalability and stability of PEs | ||
| – Operation at low temperature | – Operation in a corrosive environment (strong alkaline or acidic media). | ||
| – Operation at low potentials | |||
| – Plenty of room for optimization. |
| GWP | AP | EPBT | LCOH | Efficiency | |
|---|---|---|---|---|---|
| kgCO2eq. kgH2−1 |
gSO2eq. kgH2−1 |
Years | $ kgH2−1 | % | |
| UCG | 18.0330–333 | 34.1330,331 | 6.9334 | 0.9330 | 72330,335 |
| SMR | 16.614,336–343 | 16.614,337,339,341–343 | 4.6344 | 1.5329,337,342,345–347 | 65.5348,349 |
| BG | 4.3326,350–352 | 21.2330,351–354 | 4.8350 | 3.5330,355–357 | 45.0330,350 |
| BGR | 7.6337–340 | 4.9345,358,359 | 8.0360 | 3.6337,347,361 | 44.3347 |
| SMR + CCS | 2.3311,330,362 | 16.8353 | 7.5363 | 1.6311,330,362,364 | 65.8330 |
| MS | 5.4365–367 | 14.7365 | n.a. | 2.4325,366–368 | 58.0369 |
| HTE | 1.4326,339,342,351,352,370 | 3.5339,342,351,352,359,370 | 10371 | 4.5330,342,372,373 | 82.0330,374,375 |
| AEC-grid | 24.6341,376–379 | 1.9330,353,376 | 3.1380 | 7.2327,342,346,352,357,372 | 74.2341,352 |
| PV–EC | 3.414,337,342,351,352,381 | 9.014,337,342,351,352 | 10.0382 | 7.5279,330,337,373 | 12.3277,383 |
| Wind-EC | 0.914,337,339,341,342,352,381,384 | 3.714,337,339,341,342,352 | 6.4385,386 | 5.1329,337,342,347,386,387 | 31.0347,358 |
| PEC | 1.029,330,343,351,352 | 2.0330,343,351,352 | 5.4232,309,388 | 6.929,330,352,373,389 | 16.184,232 |
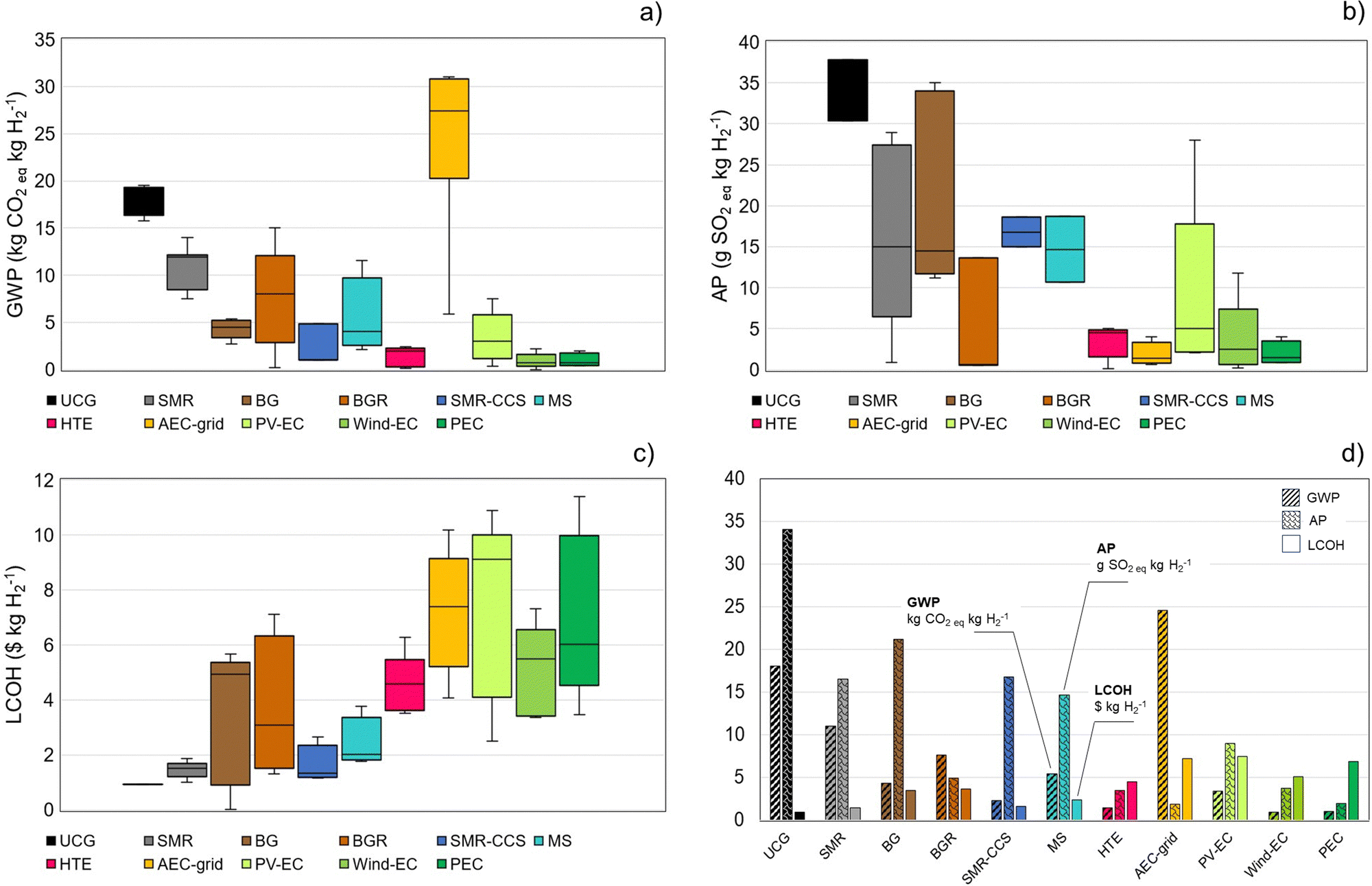 | ||
| Fig. 13 Comparison of H2 production processes based on LCA indicators and costs: (a) global warming potential (GWP); (b) acidification potential (AP); (c) levelized cost of hydrogen (LCOH); and (d) global comparison between the previous indicators. | ||
The obtained results clearly show that UCG is the H2 production technology with the worst environmental performance, with a GWP of 18.0 kgCO2eq. kgH2−1 and an AP of 34.1 gSO2eq. kgH2−1. Nevertheless, AEC grid presented the highest GWP, with a total of 24.6 kgCO2eq. kgH2−1. It is important to note that this GWP value comes with a significant error, which is due to the highly variable nature of electricity from the grid; in many countries, the grid is supplied by fossil-fuel-powered plants, which significantly hinders the environmental performance of AECs.342,376 On the other hand, wind-electrolysis presented the lowest GWP contribution, with 1.0 kgCO2eq. kgH2−1, while PEC water splitting had the lowest associated AP, with 1.3 gSO2eq. kgH2−1. Overall, PEC water splitting is the H2 production method with the lowest environmental impact, which is consistent with other comparative studies.390 As an example, Karaca et al. performed a cradle-to-gate LCA study considering PEC-H2 production based on an 870 cm2 PEC reactor that generated 3.15 mA cm−2.343 The results obtained for PEC-H2 production were compared with other three H2 production processes, namely SMR, PV–EC, and wind-driven PEM water electrolysis. It was concluded that PEC water splitting is the most environmentally friendly technology, namely considering the GWP and AP impact categories. This study also reveals that the PEC reactor and the membrane are the components with greater contribution to the total environmental impact, which confirms the importance of using non-toxic, abundant, and highly recyclable materials in device design.
As solar-based AEC is the currently available industrial technology that is closer to PEC-H2 technology, a parallel comparison between LCA studies can provide key information on which components and which lifecycle stages have a higher contribution to the environmental impacts of future PEC-H2 plants. In an LCA study conducted by Palmer et al. on PV–electrolysis, the authors found that most of the normalized environmental impact of the complete system came from the PV panels, mainly related to the production of metallic silicon.391 Hence, PEC devices can be competitive, as they are not so dependent on metallic silicon and can incorporate photo-absorbing electrodes that are made from Earth-abundant materials and do not require such energy-intensive manufacturing processes. In another LCA study on alkaline water electrolysis, Koj et al. concluded that the anode and the cathode have the highest contribution to the acidification potential (AP), while the infrastructure and the electrodes contribute equally to the GWP.376 This reinforces the need for choosing non-toxic, abundant, and chemically stable materials for PEC water splitting, produced via clean and non-complex fabrication techniques. It was also found that operating an AEC-H2 plant for 5 years has an impact on AP and GWP that is 9 times higher than the construction stage; as so, optimizing the operating parameters, such as maximum solar harvesting and thermal and power management, becomes critical for reducing the environmental impact of future PEC-H2 production plants. In terms of economic performance, SMR remains the most competitive option, with a payback time of 4.6 years and a production cost of 1.5 $ kgH2−1, although AEC-grid has the lowest payback time, averaging only 3.1 years. However, it is important to note that these results are based on studies that do not consider sudden events that can strongly affect the price of fossil fuels and, particularly, the price of natural gas; as an example, in a timeline of only a few months during 2022, the price of natural gas increased by more than 200%, reaching values as high as 315 € MWh−1, which can ruin the competitiveness of SMR.392 In this scenario, and based only on the cost of thermodynamic energy, SMR would produce H2 at a cost of 21 USD kg−1, and renewable-based H2 production would become much more competitive. Nevertheless, and based on a “so-called standard scenario”, AEC-grid achieves the lowest payback time; indeed, it is the first H2 production technology, studied over two centuries. On the other hand, HTE has the highest payback time, mainly because it is commonly associated with nuclear power plants, which require a large initial investment. This comparative analysis also shows that PEC water splitting generates H2 at one of the highest costs, averaging 6.9 $ kgH2−1. This result can be attributed to its early development stage, resulting in considerable expenses for producing and replacing PEs (which still have a low lifespan) and for manufacturing PEC panels (which still require the use of expensive and scarce materials, i.e. highly resistant polymers, noble metals).
After quantifying the environmental and economic performance of each H2 production method, normalization was made using the following equation:351
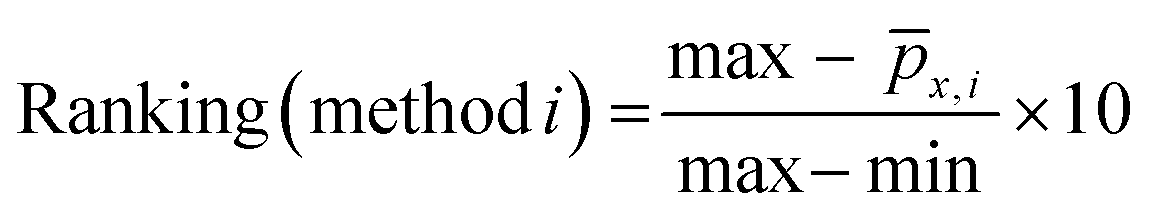 | (4) |
![[p with combining macron]](https://www.rsc.org/images/entities/i_char_0070_0304.gif) x,i is the average value of parameter x of the H2 production method i; ranking equal to 0 translates to the poorest performance and 10 indicates the best performance (zero-cost and zero-emissions). The normalized results are presented in Fig. 14. PEC water splitting remains a promising option for sustainable H2 production, scoring a remarkable environmental performance, but lacking economic arguments. It is important to note that the use of biomass derivatives as feedstock for PEC-H2 production can significantly improve the environmental performance comparatively to conventional PEC systems, as the oxidation of biomass shortens the pathway towards carbon neutrality and circular economy, also being quite interesting from an economic perspective.
x,i is the average value of parameter x of the H2 production method i; ranking equal to 0 translates to the poorest performance and 10 indicates the best performance (zero-cost and zero-emissions). The normalized results are presented in Fig. 14. PEC water splitting remains a promising option for sustainable H2 production, scoring a remarkable environmental performance, but lacking economic arguments. It is important to note that the use of biomass derivatives as feedstock for PEC-H2 production can significantly improve the environmental performance comparatively to conventional PEC systems, as the oxidation of biomass shortens the pathway towards carbon neutrality and circular economy, also being quite interesting from an economic perspective.
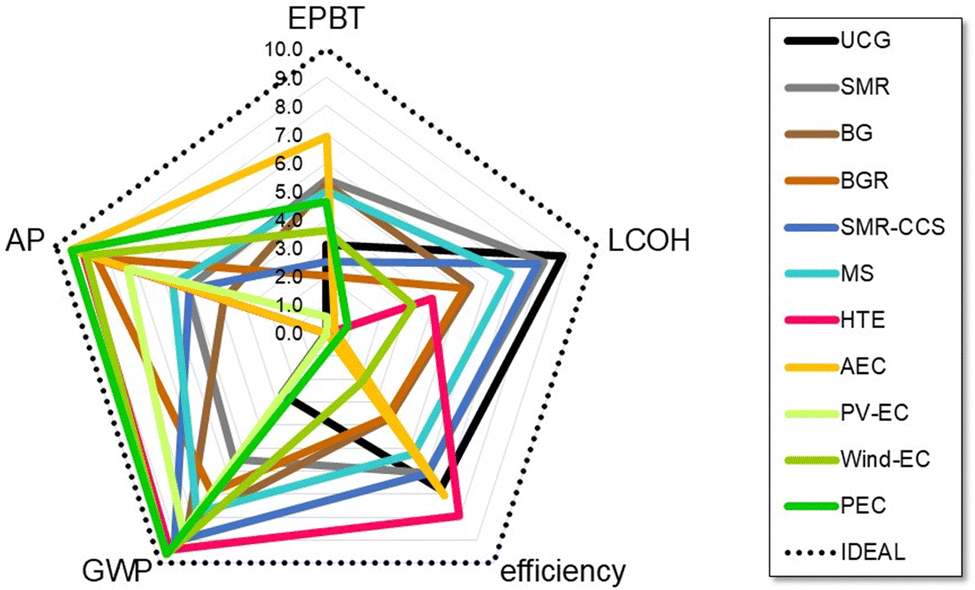 | ||
| Fig. 14 Overall performance comparison of H2 production processes. | ||
On the other hand, more mature and widespread technologies, such as UCG and SMR, are financially safer, and more appealing to investors and H2 consumers. So far, steam methane reforming with carbon capture and storage is the H2 production technology that assures the best compromise between environment and economics. Nevertheless, due to the geographically dispersed availability of primary renewable energy sources, i.e. wind, solar, and water resources, each case has to be analyzed separately, independently from the environmental and economic ranking; as an example, one can assume that wind-powered electrolysis would be a preferable option over PV-electrolysis for Nordic countries, such as Denmark, and solar-power electrolysis would be recommended for desert regions, such as North Africa or the Arabian peninsula. Overall, water electrolysis technologies offer more room for achieving a better environmental performance whereas fossil fuel-based technologies allow for a better economic performance.352,390 However, the margin for reducing the environmental impact of fossil fuel-based technologies is very small, while the margin for improving the economic performance of renewable-based H2 production processes is huge, which represents a vital endorsement for future investments in green-H2 production.
7. Conclusions, closing remarks and future opportunities
PEC water splitting celebrated its 50th anniversary in 2022. Over 50 years, and side-by-side with a never-before-seen development in materials and technologies, PEC water splitting has been the subject of numerous publications and the forerunner of several discoveries and advancements in semiconductors, catalysts, nanostructures, and many others. Almost inexplicably, PEC-H2 never left the lab and remains at a low TRL, still far from commercialization. Fortunately, the current global situation became a unique opportunity for the development of technologies that promote decarbonization, with a particular focus on the production of green fuels, where green-H2 has clearly assumed the status of a hot topic. Indeed, the third decade of the 21st century witnessed sudden events that greatly affected the energy market dynamics, with a subsequent brutal impact on energy prices. World-leading countries started to question if the resulting economic turmoil could muffle their willingness to implement key changes in the energy market, endangering ambitious carbon neutrality goals, namely through green-H2 production.393 By 2022, fossil fuels supplied ca. 95% of the global H2 production, more specifically 48% produced from natural gas, 30% from oil, and 18% from coal, which undermines the “green” labeling that H2 is ambitioning.312,394 Surprisingly, many private and government institutions interpreted the current situation as a great opportunity to implement drastic changes in the energy market, attributing to green-H2 a prominent position in the financial packages to be distributed.395 For example, in the European Green Deal, green-H2 is a priority for achieving carbon neutrality by 2050. It is mentioned that the large-scale deployment of green-H2 is mandatory for reducing GHG emissions towards 55% by 2030, requiring research and innovation efforts, manufacturing capabilities for supplying H2 to large consumers, and especially, it is necessary to accelerate the development of promising technologies at low TRL, such as PEC water splitting.396,397 For instance, within the European context, the latest update of the Renewable Energy Directive (RED III) stipulates that the utilization of Renewable Fuels of Non-Biological Origin (RFNBOs), where renewable gaseous H2 is included, will result in a carbon credit bonus for meeting the European targets mandated by the directive. As such, low-carbon fuels, particularly those whose production relies on green-H2, are currently witnessing unparalleled and distinctive valorisation, which is translated into a unique opportunity for PEC water splitting, as the technology is expected to have one of the lowest carbon footprints among H2 production technologies. Sustainable energy production based on renewable sources and diversified technologies is essential to avoid sudden and steep fluctuations in energy prices due to unpredictable events, especially in countries where fossil fuel imports account for a considerable market share.398There is no doubt that green-H2 has gained the reputation of “key player” in future energy systems; still, a critical question remains: would PEC water splitting be able to compete with other green-H2 production technologies? The simple answer is yes. Although in terms of thermodynamics and kinetics, the best PEC-H2 systems can only hope to match the best PV–EC;399 the differentiating competitive factor relies on the environmental and functional advantages of PEC water splitting, as PEC devices benefit from the integration of light absorbers with electrolysis technologies in a single unit, with no peripheral electronics.32 LCA studies show that, among currently available H2 production processes, PEC water splitting has the lowest environmental impact, despite coal gasification and steam methane reforming remaining the more financially appealing options to investors and H2 consumers, due to their economy of scale and maturity. This reinforces that scalability is one of the most critical challenges for turning PEC water splitting economically viable in the upcoming years.15,57,277 With state-of-the-art photoelectrodes, to reach a similar H2 output as PV–EC, the active area of PEC devices must be at least 50 times larger, which means in the range of a few hundred centimeters.15 Therefore, the commercial viability of PEC-H2 production strongly relies on the successful upscaling of energy-efficient PEs,183 namely on achieving the benchmark values of the three-pillar criteria for commercialization, i.e. an active area of 100 cm2, 2920 h of stable performance (8 h over 365 days with a decrease of max. 10% of the initial current density) and a current density of 10 mA cm−2. Complementarily to the successful upscaling of efficient PEs, using concentrated sunlight is a simple and clever way to boost H2 production. For that, solar concentrators must be compatible with PEC device restrictions, and PEC devices must be compatible with the specifications of solar concentrators. No matter the concentrating solar technology, adopting thermal management strategies is essential to avoid performance losses due to the added thermal load. Finally, the competitiveness of PEC water splitting can be further enhanced using biomass derivatives, which allows overcoming thermodynamic and kinetic constraints of conventional systems and meeting circular economy requirements, by using wastewater as an electrolyte.
During the upcoming years, the field of PEC-H2 production shall experience an unparalleled economic and research stimulus, which will allow for bridging lab-scale research with commercial interests. The best way forward is to accept, at first, that PEC devices will need to operate very close to their fundamental thermodynamic limits, at large scale.188,400 This will require a solid convergence between academics and industry and research efforts should be directed to: (i) build and demonstrate large-scale PEC devices assembled in a highly recyclable embodiment, incorporating clever design features and CRM-free electrodes and catalysts; (ii) demonstrate high STH efficiency values for large-area PEs that are in the same range as the PV cells; (iii) develop preparation techniques for large-area PEs that can be easily implemented on an industrial scale; (iv) enhance the chemical stability of PEs targeting tens of thousands of hours; (v) investigate and develop new approaches for coupling solar concentrators with PEC devices; (vi) find suitable organic compounds for PEC-H2 production systems, with the corresponding ideal PEs, catalysts and membranes; and (vii) reach consensual standardized methods for evaluating PEC devices, based on environmental and techno-economic metrics.
If these targets are not met in the next decade, the feasibility of PEC-H2 production will be seriously compromised and the topic could tend to become another subject without practical application for society. Nevertheless, investment in research on PEC devices should move forward, especially given the quest for decarbonization. Moreover, PEC devices are finding other applications beyond water splitting, such as in solar redox flow cells.68,401 Overall, the incorporation of PEC-H2 production into the energy market should be a gradual process and should not aim at dethroning existing technologies, but aspire instead to be part of a symbiotic integration (Fig. 15).
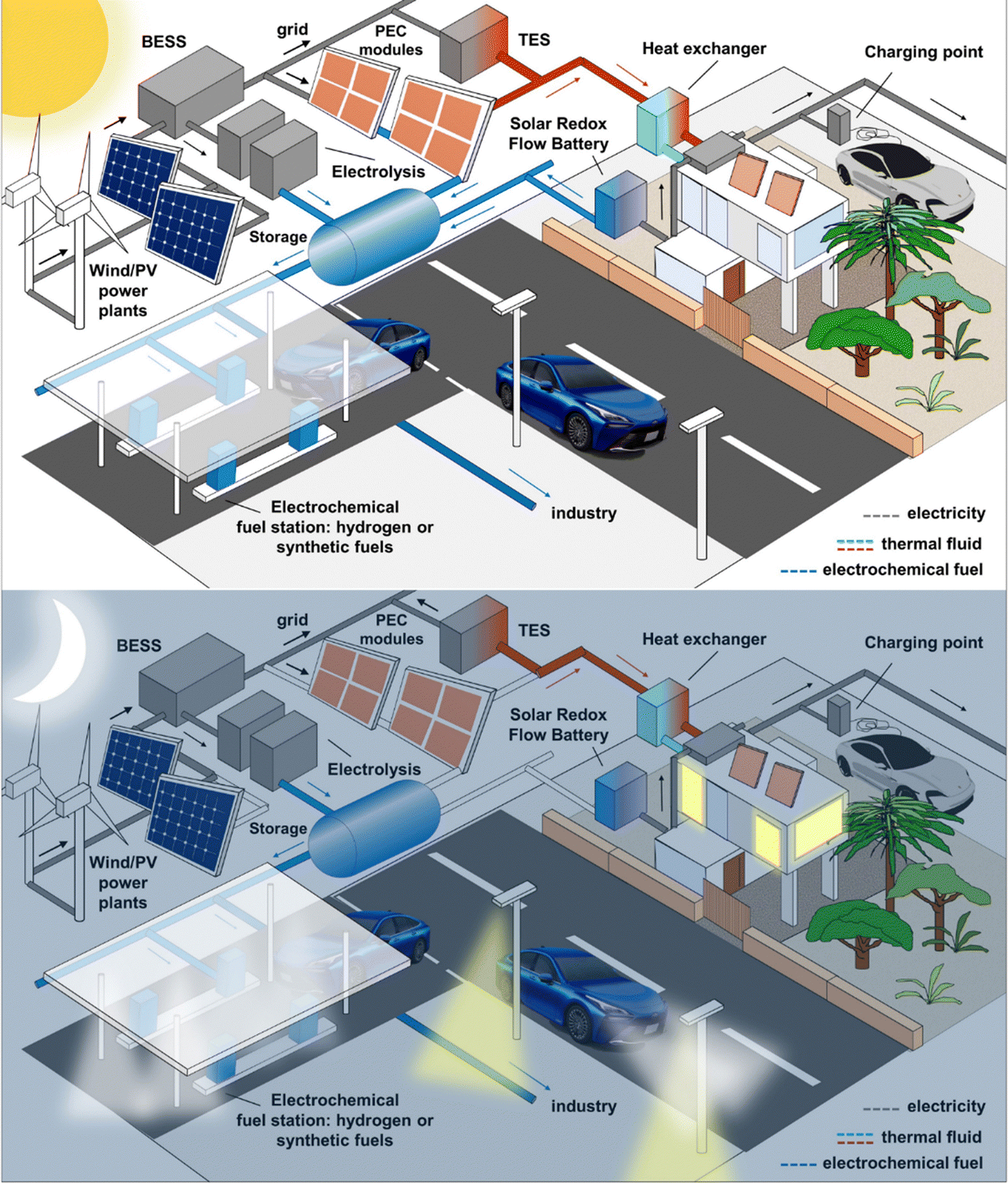 | ||
| Fig. 15 Hypothetical scenario of the integration of PEC-H2 production in the energy grid, comprising the electrical, thermal and fuel flows for day and night periods. | ||
Abbreviation
| AE | Auxiliary electrode |
| AEC | Alkaline electrolysis |
| AEM | Anion exchange membrane |
| ALD | Atomic layer deposition |
| AP | Acidification potential |
| ATO | Antimony-doped tin oxide |
| AZO | Aluminium-doped zinc oxide |
| BESS | Battery energy storage system |
| BG | Biomass gasification |
| BGR | Biogas reforming |
| BHJ | Bulk heterojunction |
| BIPV | Building-integrated photovoltaic |
| BOS | Balance of system |
| BPM | Bipolar membrane |
| CAPEX | Capital Expenditure |
| CCM | Catalyst-coated membranes |
| CCS | Carbon capture and storage |
| CE | Counter-electrode |
| CEM | Cation exchange membrane |
| CIPEC | Concentrator-integrated photoelectrochemical |
| CL | Convex lenses |
| CPC | Compound parabolic concentrator |
| CPEC | Concentrated-photoelectrochemical |
| CPVE | Concentrated photovoltaic electrolysis |
| CRM | Critical raw material |
| CZTS | Copper zinc tin sulfide |
| DLR | German Aerospace Centre (Deutsches Zentrum für Luft- und Raumfahrt) |
| DSSC | Dye-sensitized solar cell |
| EC | Electrolysis/electrolyser |
| EIS | Electrochemical impedance spectroscopy |
| EPBT | Energy payback time |
| ERoEI | Energy return on energy invested |
| ETCO | Embedded transparent conductive oxide |
| EU | European Union |
| FL | Fresnel lenses |
| FTO | Fluorine-doped tin oxide |
| FOM | Figure-of-merit |
| GHG | Greenhouse gases |
| GLAD | Glancing angle deposition |
| GWP | Global warming potential |
| HER | Hydrogen evolution reaction |
| HTE | High-temperature electrolysis |
| IPEC | Integrated photoelectrochemical |
| ITO | Indium tin oxide |
| LCA | Life cycle assessment |
| LCOH | Levelized cost of hydrogen |
| LFR | Linear Fresnel reflector |
| LSC | Luminescent solar concentrator |
| MEA | Membrane electrode assembly |
| MOFs | Metal–organic framework-based materials |
| MS | Methane splitting |
| OER | Oxygen evolution reaction |
| PCE | Power conversion efficiency |
| PDC | Parabolic dish concentrator |
| PE | Photoelectrode |
| PEC | Photoelectrochemical |
| PEEK | Polyether ether ketone |
| PEM | Polymer electrolyte membrane |
| PET | Polyethylene |
| PFSA | Perfluorinated sulfonic acid |
| PMMA | Poly(methyl 2-methylpropenoate) |
| PS | Polystyrene |
| PSC | Perovskite solar cell |
| PTC | Parabolic trough collector |
| PV | Photovoltaic |
| PV-EC | Photovoltaic-electrolysis |
| QD | Quantum dot |
| RE | Reference electrode |
| RFNBO | Renewable fuel from non-biological origin |
| RHE | Reversible hydrogen electrode |
| SMR | Steam methane reforming |
| SOEC | Solid oxide electrolyser cell |
| SP | Spray pyrolysis |
| STH | Solar-to-hydrogen |
| TCO | Transparent conductive oxide |
| TE | Thermoelectric |
| TES | Thermal energy storage |
| TEG | Thermoelectric generator |
| TRL | Technological readiness level |
| UPORTO | University of Porto |
| UCG | Underground coal gasification |
| US | United States |
| UV | Ultraviolet |
| WE | Working electrode |
| WS | Working sense |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. Vilanova, P. Dias and T. Lopes are grateful to the Portuguese Foundation for Science and Technology (FCT) for funding (References SFRH/BD/121039/2016, CEECIND/02862/2018 and CEECIND/02385/2021, respectively). The research leading to these results has received funding from: (i) projects PTDC/EQU-EQU/30760/2017 – HopeH2 and PTDC/EQU-EQU/4225/2021 – ASAPFuels, funded by FEDER, through COMPETE2020 and by national funds, through FCT; (ii) project Baterias 2030, with the reference POCI-01-0247-FEDER-046109, co-funded by Operational Programme for Competitiveness and Internationalisation (COMPETE 2020), under the Portugal 2020 Partnership Agreement, through the European Regional Development Fund (ERDF); (iii) “H2 Driven Green Agenda”, nr. C644923817-00000037, investment project nr. 50, financed by the Recovery and Resilience Plan (PRR) and by the European Union – NextGeneration EU; and (iv) LA/P/0045/2020 (ALiCE), UIDB/00511/2020 and UIDP/00511/2020 (LEPABE), funded by national funds through FCT/MCTES (PIDDAC).References
- C. S. Lai, Y. Jia, L. L. Lai, Z. Xu, M. D. McCulloch and K. P. Wong, Renewable Sustainable Energy Rev., 2017, 78, 439–451 CrossRef.
- N. Abas, A. Kalair and N. Khan, Futures, 2015, 69, 31–49 CrossRef.
- I. Mesquita, L. Andrade and A. Mendes, Renewable Sustainable Energy Rev., 2018, 82, 2471–2489 CrossRef CAS.
- M. González-Eguino, Renewable Sustainable Energy Rev., 2015, 47, 377–385 CrossRef.
- M. A. Khan, M. Z. Khan, K. Zaman and L. Naz, Renewable Sustainable Energy Rev., 2014, 29, 336–344 CrossRef.
- A. Vaona, Renewable Energy, 2016, 86, 354–359 CrossRef.
- K. Zaman and M. Abd-el Moemen, Renewable Sustainable Energy Rev., 2017, 74, 1119–1130 CrossRef.
- G. Nicoletti, N. Arcuri, G. Nicoletti and R. Bruno, Energy Convers. Manage., 2015, 89, 205–213 CrossRef.
- G. P. Peters, R. M. Andrew, J. G. Canadell, S. Fuss, R. B. Jackson, J. I. Korsbakken, C. Le Quéré and N. Nakicenovic, Nat. Clim. Change, 2017, 2, 118–122 CrossRef.
- H. J. Schellnhuber, S. Rahmstorf and R. Winkelmann, Nat. Clim. Change, 2016, 6, 649–653 CrossRef.
- M. Lee, J. Yun, A. Pyka, D. Won, F. Kodama, G. Schiuma, H. Park, J. Jeon, K. Park and K. Jung, J. Open Innov.: Technol. Mark. Complex., 2018, 4, 21 CrossRef.
- F. Yilmaz, M. T. Balta and R. Selbaş, Renewable Sustainable Energy Rev., 2016, 56, 171–178 CrossRef CAS.
- F. Zhang, P. Zhao, M. Niu and J. Maddy, Int. J. Hydrogen Energy, 2016, 41, 14535–14552 CrossRef CAS.
- F. Suleman, I. Dincer and M. Agelin-Chaab, Int. J. Hydrogen Energy, 2015, 40, 6976–6987 CrossRef CAS.
- S. Ardo, D. F. Rivas, M. A. Modestino, V. S. Greiving, F. F. Abdi, E. A. Llado, V. Artero, K. Ayers, C. Battaglia and J.-P. Becker, Energy Environ. Sci., 2018, 11, 2768–2783 RSC.
- W. Yang, R. R. Prabhakar, J. Tan, S. D. Tilley and J. Moon, Chem. Soc. Rev., 2019, 48, 4979–5015 RSC.
- S. Tembhurne, F. Nandjou and S. Haussener, Nat. Energy, 2019, 4, 399 CrossRef CAS.
- D. M. Fabian, S. Hu, N. Singh, F. A. Houle, T. Hisatomi, K. Domen, F. E. Osterloh and S. Ardo, Energy Environ. Sci., 2015, 8, 2825–2850 RSC.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome and M. Katayama, Joule, 2018, 2, 509–520 CrossRef CAS.
- T. Takata and K. Domen, ACS Energy Lett., 2019, 4, 542–549 CrossRef CAS.
- B. Turan, J.-P. Becker, F. Urbain, F. Finger, U. Rau and S. Haas, Nat. Commun., 2016, 7, 12681 CrossRef PubMed.
- W. H. Brattain and J. Bardeen, Bell Syst. Tech. J., 1953, 32, 1–41 CrossRef.
- C. Garrett and W. H. Brattain, Phys. Rev., 1955, 99, 376 CrossRef.
- H. Gerischer, J. Electrochem. Soc., 1966, 113, 1174 CrossRef CAS.
- P. Boddy, J. Electrochem. Soc., 1968, 115, 199 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- EPFL, SolarFuelsDB, https://solarfuelsdb.epfl.ch/, (accessed July, 2023).
- K. T. Fountaine, H. J. Lewerenz and H. A. Atwater, Nat. Commun., 2016, 7, 13706 CrossRef CAS PubMed.
- Technical Targets for Hydrogen Production from Photoelectrochemical Water Splitting, https://www.energy.gov/eere/fuelcells/doe-technical-targets-hydrogen-production-photoelectrochemical-water-splitting, (accessed July 2023, 2023).
- W.-H. Cheng, M. H. Richter, M. M. May, J. Ohlmann, D. Lackner, F. Dimroth, T. Hannappel, H. A. Atwater and H.-J. Lewerenz, ACS Energy Lett., 2018, 3, 1795–1800 CrossRef CAS.
- S. Hu, C. Xiang, S. Haussener, A. D. Berger and N. S. Lewis, Energy Environ. Sci., 2013, 6, 2984–2993 RSC.
- A. Rothschild and H. Dotan, ACS Energy Lett., 2017, 2, 45–51 CrossRef CAS.
- J. H. Kim, J.-W. Jang, Y. H. Jo, F. F. Abdi, Y. H. Lee, R. van de Krol and J. S. Lee, Nat. Commun., 2016, 7, 13380 CrossRef CAS PubMed.
- X. Shi, H. Jeong, S. J. Oh, M. Ma, K. Zhang, J. Kwon, I. T. Choi, I. Y. Choi, H. K. Kim, J. K. Kim and J. H. Park, Nat. Commun., 2016, 7, 11943 CrossRef CAS PubMed.
- L. Pan, J. H. Kim, M. T. Mayer, M.-K. Son, A. Ummadisingu, J. S. Lee, A. Hagfeldt, J. Luo and M. Grätzel, Nat. Catal., 2018, 1, 412–420 CrossRef CAS.
- S. K. Karuturi, H. Shen, A. Sharma, F. J. Beck, P. Varadhan, T. Duong, P. R. Narangari, D. Zhang, Y. Wan, J.-H. He, H. H. Tan, C. Jagadish and K. Catchpole, Adv. Energy Mater., 2020, 10, 2000772 CrossRef CAS.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum and S. Ardo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- M. R. Shaner, H. A. Atwater, N. S. Lewis and E. W. McFarland, Energy Environ. Sci., 2016, 9, 2354–2371 RSC.
- T. Lopes, P. Dias, L. Andrade and A. Mendes, Sol. Energy Mater. Sol. Cells, 2014, 128, 399–410 CrossRef CAS.
- S. Haussener, C. Xiang, J. M. Spurgeon, S. Ardo, N. S. Lewis and A. Z. Weber, Energy Environ. Sci., 2012, 5, 9922–9935 RSC.
- I. Holmes-Gentle, F. Hoffmann, C. A. Mesa and K. Hellgardt, Sustainable Energy Fuels, 2017, 1, 1184–1198 RSC.
- M. T. Spitler, M. A. Modestino, T. G. Deutsch, C. X. Xiang, J. R. Durrant, D. V. Esposito, S. Haussener, S. Maldonado, I. D. Sharp, B. A. Parkinson, D. S. Ginley, F. A. Houle, T. Hannappel, N. R. Neale, D. G. Nocera and P. C. McIntyre, Sustainable Energy Fuels, 2020, 4, 985–995 RSC.
- A. Vilanova, T. Lopes, C. Spenke, M. Wullenkord and A. Mendes, Energy Storage Mater., 2018, 13, 175–188 CrossRef.
- R. van de Krol, in Photoelectrochemical Hydrogen Production, ed. R. van de Krol and M. Grätzel, Springer Science + Business Media, New York, USA, 2012, vol. VIII, ch. 2, pp. 13–67 Search PubMed.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 RSC.
- N. Papageorgiou, Coord. Chem. Rev., 2004, 248, 1421–1446 CrossRef CAS.
- F. Le Formal, On the Morphology and Interfaces of Nanostructured Hematite Photoanodes for Solar-Driven Water Splitting, PhD thesis, EPFL, Lausanne, Switzerland, 2011, p. 216 DOI:10.5075/epfl-thesis-5180.
- H. Dotan, N. Mathews, T. Hisatomi, M. Grätzel and A. Rothschild, J. Phys. Chem. Lett., 2014, 5, 3330–3334 CrossRef CAS PubMed.
- C. Ilkay, Solar photoelectrolysis of water with translucent nano-structured hematite photoanodes, PhD thesis, EPFL, Lausanne, Switzerland, 2007, p. 200 DOI:10.5075/epfl-thesis-3914.
- A. G. Tamirat, J. Rick, A. A. Dubale, W.-N. Su and B.-J. Hwang, Nanoscale Horiz., 2016, 1, 243–267 RSC.
- P. Zhai, S. Haussener, J. Ager, R. Sathre, K. Walczak, J. Greenblatt and T. McKone, Energy Environ. Sci., 2013, 6, 2380–2389 RSC.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- F. A. S. Moranchell, J. M. S. Pineda, J. N. H. Pérez, U. S. Silva-Rivera, C. A. C. Escobedo and R. d G. G. Huerta, Int. J. Hydrogen Energy, 2020, 45, 13683–13692 CrossRef.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- C. Jiang, S. J. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- K. Sivula and R. Van De Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- J. H. Kim, D. Hansora, P. Sharma, J.-W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- S. K. Saraswat, D. D. Rodene and R. B. Gupta, Renewable Sustainable Energy Rev., 2018, 89, 228–248 CrossRef CAS.
- S. Shen, S. A. Lindley, X. Chen and J. Z. Zhang, Energy Environ. Sci., 2016, 9, 2744–2775 RSC.
- D. Wang, X.-T. Zhang, P.-P. Sun, S. Lu, L.-L. Wang, Y.-A. Wei and Y.-C. Liu, Int. J. Hydrogen Energy, 2014, 39(28), 16212–16219 CrossRef CAS.
- M. M. Momeni and Y. Ghayeb, J. Alloys Compd., 2015, 637, 393–400 CrossRef CAS.
- X. Li, P. S. Bassi, P. P. Boix, Y. Fang and L. H. Wong, ACS Appl. Mater. Interfaces, 2015, 7, 16960–16966 CrossRef CAS PubMed.
- C. Ros, T. Andreu and J. R. Morante, J. Mater. Chem. A, 2020, 8, 10625–10669 RSC.
- P. Dias, A. Vilanova, T. Lopes, L. Andrade and A. Mendes, Nano Energy, 2016, 23, 70–79 CrossRef CAS.
- I. S. Cho, H. S. Han, M. Logar, J. Park and X. Zheng, Adv. Energy Mater., 2016, 6, 1501840 CrossRef.
- K. Sivula, F. Le Formal and M. Gratzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- T. H. Jeon, G.-h Moon, H. Park and W. Choi, Nano Energy, 2017, 39, 211–218 CrossRef CAS.
- T. da Silva Lopes, P. Dias, R. Monteiro, A. Vilanova, D. Ivanou and A. Mendes, Adv. Energy Mater., 2021, 2102893 Search PubMed.
- A. Landman, R. Halabi, P. Dias, H. Dotan, A. Mehlmann, G. E. Shter, M. Halabi, O. Naseraldeen, A. Mendes, G. S. Grader and A. Rothschild, Joule, 2020, 4, 448–471 CrossRef CAS.
- A. Apolinário, T. Lopes, C. D. Costa, J. P. Araújo and A. Mendes, ACS Appl. Energy Mater., 2019, 2, 1040–1050 CrossRef.
- Y. Wang, W. Tian, C. Chen, W. Xu and L. Li, Adv. Funct. Mater., 2019, 29, 1809036 CrossRef.
- T. Zhu, M. N. Chong and E. S. Chan, ChemSusChem, 2014, 7, 2974–2997 CrossRef CAS PubMed.
- A. Tacca, L. Meda, G. Marra, A. Savoini, S. Caramori, V. Cristino, C. A. Bignozzi, V. G. Pedro, P. P. Boix and S. Gimenez, Chem. Phys. Chem., 2012, 13, 3025–3034 CrossRef CAS PubMed.
- W. Kwong, H. Qiu, A. Nakaruk, P. Koshy and C. Sorrell, Energy Procedia, 2013, 34, 617–626 CrossRef CAS.
- S. Reinhard, F. Rechberger and M. Niederberger, ChemPlusChem, 2016, 81, 935 CrossRef CAS PubMed.
- Y. Pihosh, I. Turkevych, K. Mawatari, J. Uemura, Y. Kazoe, S. Kosar, K. Makita, T. Sugaya, T. Matsui and D. Fujita, Sci. Rep., 2015, 5, 11141 CrossRef PubMed.
- F. F. Abdi, L. Han, A. H. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 CrossRef PubMed.
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 CrossRef PubMed.
- I. Y. Ahmet, Y. Ma, J.-W. Jang, T. Henschel, B. Stannowski, T. Lopes, A. Vilanova, A. Mendes, F. Abdi and R. van de Krol, Sustainable Energy Fuels, 2019, 3, 2366–2379 RSC.
- K. Ouyang, S. Xie, P. Wang, J. Zhu and P. Zhan, Int. J. Hydrogen Energy, 2019, 44, 7288–7299 CrossRef CAS.
- M. Zhou, J. Bao, Y. Xu, J. Zhang, J. Xie, M. Guan, C. Wang, L. Wen, Y. Lei and Y. Xie, ACS Nano, 2014, 8, 7088–7098 CrossRef CAS PubMed.
- S. Kosar, Y. Pihosh, I. Turkevych, K. Mawatari, J. Uemura, Y. Kazoe, K. Makita, T. Sugaya, T. Matsui and D. Fujita, Jpn. J. Appl. Phys., 2016, 55, 04ES01 CrossRef.
- Y. Kuang, Q. Jia, G. Ma, T. Hisatomi, T. Minegishi, H. Nishiyama, M. Nakabayashi, N. Shibata, T. Yamada and A. Kudo, Nat. Energy, 2016, 2, 1–9 Search PubMed.
- G. Liu, S. Ye, P. Yan, F. Xiong, P. Fu, Z. Wang, Z. Chen, J. Shi and C. Li, Energy Environ. Sci., 2016, 9, 1327–1334 RSC.
- G. Liu, J. Shi, F. Zhang, Z. Chen, J. Han, C. Ding, S. Chen, Z. Wang, H. Han and C. Li, Angew. Chem., Int. Ed., 2014, 53, 7295–7299 CrossRef CAS PubMed.
- A. Ishikawa, T. Takata, J. N. Kondo, M. Hara and K. Domen, J. Phys. Chem. B, 2004, 108, 11049–11053 CrossRef CAS.
- M. Higashi, K. Domen and R. Abe, Energy Environ. Sci., 2011, 4, 4138–4147 RSC.
- T. Higashi, H. Nishiyama, V. Nandal, Y. Pihosh, Y. Kawase, R. Shoji, M. Nakabayashi, Y. Sasaki, N. Shibata and H. Matsuzaki, Energy Environ. Sci., 2022, 15, 4761–4775 RSC.
- Y.-F. Xu, H.-S. Rao, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Mater. Chem. A, 2016, 4, 5124–5129 RSC.
- D. H. Taffa, R. Dillert, A. C. Ulpe, K. C. Bauerfeind, T. Bredow, D. W. Bahnemann and M. Wark, J. Photonics Energy, 2016, 7, 012009 CrossRef.
- J. Azevedo, S. D. Tilley, M. Schreier, M. Stefik, C. Sousa, J. P. Araújo, A. Mendes, M. Grätzel and M. T. Mayer, Nano Energy, 2016, 24, 10–16 CrossRef CAS.
- J. Azevedo, L. Steier, P. Dias, M. Stefik, C. Sousa, J. Araujo, A. Mendes, M. Graetzel and S. Tilley, Energy Environ. Sci., 2014, 7, 4044–4052 RSC.
- S. D. Tilley, M. Schreier, J. Azevedo, M. Stefik and M. Graetzel, Adv. Funct. Mater., 2013, 24, 1616–3028 Search PubMed.
- D. Bae, B. Seger, P. C. Vesborg, O. Hansen and I. Chorkendorff, Chem. Soc. Rev., 2017, 46, 1933–1954 RSC.
- Y. Lin, C. Battaglia, M. Boccard, M. Hettick, Z. Yu, C. Ballif, J. W. Ager and A. Javey, Nano Lett., 2013, 13, 5615–5618 CrossRef CAS PubMed.
- H.-C. Fu, P. Varadhan, M.-L. Tsai, W. Li, Q. Ding, C.-H. Lin, M. Bonifazi, A. Fratalocchi, S. Jin and J.-H. He, Nano Energy, 2020, 70, 104478 CrossRef CAS.
- A. Vilanova, P. Dias, J. Azevedo, M. Wullenkord, C. Spenke, T. Lopes and A. Mendes, J. Power Sources, 2020, 454, 227890 CrossRef CAS.
- D. K. Lee and K.-S. Choi, Nat. Energy, 2018, 3, 53–60 CrossRef CAS.
- Z. Zhang, X. Huang, B. Zhang and Y. Bi, Energy Environ. Sci., 2022, 15, 2867–2873 RSC.
- W. J. Lee, P. S. Shinde, G. H. Go and E. Ramasamy, Int. J. Hydrogen Energy, 2011, 36, 5262–5270 CrossRef CAS.
- K. Jakubow-Piotrowska, D. Kurzydlowski, P. Wrobel and J. Augustynski, AACS Phys. Chem. Au, 2022, 2, 299–304 CrossRef CAS PubMed.
- Final Report Summary – PECDEMO (Photoelectrochemical Demonstrator Device for Solar Hydrogen Generation), 2017. Available online: https://cordis.europa.eu/project/id/621252/reporting (accessed on 3 August 2023).
- A. Gonçalves, Development of Tantalum Nitride Photoelectrodes, PhD thesis, UPORTO-FEUP, Porto, Portugal, 2022, p. 195, https://hdl.handle.net/10216/147462.
- F. Santos, J. Martins, J. Capitão, S. Emami, D. Ivanou and A. Mendes, ACS Appl. Energy Mater., 2022, 5, 7220–7229 CrossRef CAS PubMed.
- A. Sekar and K. Sivula, Chimia, 2021, 75, 169 CrossRef CAS PubMed.
- L. Steier and S. Holliday, J. Mater. Chem. A, 2018, 6, 21809–21826 RSC.
- S. Otep, T. Michinobu and Q. Zhang, Sol. RRL, 2020, 4, 1900395 CrossRef CAS.
- Q. Yin, K. Zhang, L. Zhang, J. Jia, X. Zhang, S. Pang, Q.-H. Xu, C. Duan, F. Huang and Y. Cao, J. Mater. Chem. A, 2019, 7, 12426–12433 RSC.
- T. Ameri, G. Dennler, C. Lungenschmied and C. J. Brabec, Energy Environ. Sci., 2009, 2, 347–363 RSC.
- L. Yao, A. Rahmanudin, N. Guijarro and K. Sivula, Adv. Energy Mater., 2018, 8, 1802585 CrossRef.
- J. M. Yu, J. Lee, Y. S. Kim, J. Song, J. Oh, S. M. Lee, M. Jeong, Y. Kim, J. H. Kwak and S. Cho, Nat. Commun., 2020, 11, 5509 CrossRef CAS PubMed.
- D. Zhang, H. H. Cho, J. H. Yum, M. Mensi and K. Sivula, Adv. Energy Mater., 2022, 12, 2202363 CrossRef CAS.
- X. Li, Z. Wang and L. Wang, Small Science, 2021, 1, 2000074 CrossRef CAS.
- J. B. Pan, B. H. Wang, J. B. Wang, H. Z. Ding, W. Zhou, X. Liu, J. R. Zhang, S. Shen, J. K. Guo and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 1433–1440 CrossRef CAS PubMed.
- A. S. Bati, Y. L. Zhong, P. L. Burn, M. K. Nazeeruddin, P. E. Shaw and M. Batmunkh, Commun. Mater., 2023, 4, 2 CrossRef CAS.
- H. Min, D. Y. Lee, J. Kim, G. Kim, K. S. Lee, J. Kim, M. J. Paik, Y. K. Kim, K. S. Kim and M. G. Kim, Nature, 2021, 598, 444–450 CrossRef CAS PubMed.
- W. Wang, M. Xu, X. Xu, W. Zhou and Z. Shao, Angew. Chem., Int. Ed., 2020, 59, 136–152 CrossRef CAS PubMed.
- S. Bhattacharjee, V. Andrei, C. Pornrungroj, M. Rahaman, C. M. Pichler and E. Reisner, Adv. Funct. Mater., 2022, 32, 2109313 CrossRef CAS.
- E. Edwardes Moore, V. Andrei, S. Zacarias, I. A. Pereira and E. Reisner, ACS Energy Lett., 2019, 5, 232–237 CrossRef PubMed.
- V. Andrei, G. M. Ucoski, C. Pornrungroj, C. Uswachoke, Q. Wang, D. S. Achilleos, H. Kasap, K. P. Sokol, R. A. Jagt and H. Lu, Nature, 2022, 608, 518–522 CrossRef CAS PubMed.
- A. Guerrero and J. Bisquert, Curr. Opin. Electrochem., 2017, 2, 144–147 CrossRef CAS.
- G. Liu, S. K. Karuturi, H. Chen, D. Wang, J. W. Ager, A. N. Simonov and A. Tricoli, Sol. Energy, 2020, 202, 198–203 CrossRef CAS.
- H. Chen, M. Zhang, T. Tran-Phu, R. Bo, L. Shi, I. Di Bernardo, J. Bing, J. Pan, S. Singh and J. Lipton-Duffin, Adv. Funct. Mater., 2021, 31, 2008245 CrossRef CAS.
- M. Daboczi, J. Cui, F. Temerov and S. Eslava, 2023.
- S. Oueslati, M. Pilvet, M. Grossberg, M. Kauk-Kuusik, J. Krustok and D. Meissner, Thin Solid Films, 2021, 739, 138981 CrossRef CAS.
- D. Yokoyama, T. Minegishi, K. Jimbo, T. Hisatomi, G. Ma, M. Katayama, J. Kubota, H. Katagiri and K. Domen, Appl. Phys. Express, 2010, 3, 101202 CrossRef.
- C. Ros, T. Andreu, S. Giraldo, V. Izquierdo-Roca, E. Saucedo and J. R. Morante, ACS Appl. Mater. Interfaces, 2018, 10, 13425–13433 CrossRef CAS PubMed.
- M. Vishwakarma, M. Kumar, M. Hendrickx, J. Hadermann, A. P. Singh, Y. Batra and B. Mehta, Adv. Mater. Interfaces, 2021, 8, 2002124 CrossRef CAS.
- Y. Park, K. J. McDonald and K.-S. Choi, Chem. Soc. Rev., 2013, 42, 2321–2337 RSC.
- P. Dias and A. Mendes, in Encyclopedia of Sustainability Science and Technology, ed. R. A. Meyers, Springer, New York, New York, NY, 2017, pp.1–52 DOI:10.1007/978-1-4939-2493-6_957-1.
- J. Zhou, S. Lin, Y. Chen and A. Gaskov, Appl. Surf. Sci., 2017, 403, 274–281 CrossRef CAS.
- Y. Kuang, Q. Jia, H. Nishiyama, T. Yamada, A. Kudo and K. Domen, Adv. Energy Mater., 2016, 6, 1501645 CrossRef.
- F. Le Formal, M. Grätzel and K. Sivula, Adv. Funct. Mater., 2010, 20, 1099–1107 CrossRef CAS.
- X. Zhao, J. Feng, S. Chen, Y. Huang, T. C. Sum and Z. Chen, Phys. Chem. Chem. Phys., 2017, 19, 1074–1082 RSC.
- M. J. Katz, S. C. Riha, N. C. Jeong, A. B. Martinson, O. K. Farha and J. T. Hupp, Coord. Chem. Rev., 2012, 256, 2521–2529 CrossRef CAS.
- Y. Ling, G. Wang, D. A. Wheeler, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 2119–2125 CrossRef CAS PubMed.
- I. Cesar, K. Sivula, A. Kay, R. Zboril and M. Grätzel, J. Phys. Chem. C, 2008, 113, 772–782 CrossRef.
- Y.-S. Hu, A. Kleiman-Shwarsctein, A. J. Forman, D. Hazen, J.-N. Park and E. W. McFarland, Chem. Mater., 2008, 20, 3803–3805 CrossRef CAS.
- J. H. Kim and J. S. Lee, Adv. Mater., 2019, 31, 1806938 CrossRef PubMed.
- P. Dias, L. Andrade and A. Mendes, Nano Energy, 2017, 38, 218–231 CrossRef CAS.
- M. Li, Y. Yang, Y. Ling, W. Qiu, F. Wang, T. Liu, Y. Song, X. Liu, P. Fang and Y. Tong, Nano Lett., 2017, 17, 2490–2495 CrossRef CAS PubMed.
- K. D. Malviya, H. Dotan, D. Shlenkevich, A. Tsyganok, H. Mor and A. Rothschild, J. Mater. Chem. A, 2016, 4, 3091–3099 RSC.
- D. Cao, W. Luo, J. Feng, X. Zhao, Z. Li and Z. Zou, Energy Environ. Sci., 2014, 7, 752–759 RSC.
- M. R. Nellist, F. A. Laskowski, F. Lin, T. J. Mills and S. W. Boettcher, Acc. Chem. Res., 2016, 49, 733–740 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- T. R. Hellstern, J. D. Benck, J. Kibsgaard, C. Hahn and T. F. Jaramillo, Adv. Energy Mater., 2016, 6, 1501758 CrossRef.
- H. Gerischer, J. Electroanal. Chem. Interfacial Electrochem., 1977, 82, 133–143 CrossRef CAS.
- B. Han, M. Risch, S. Belden, S. Lee, D. Bayer, E. Mutoro and Y. Shao-Horn, J. Electrochem. Soc., 2018, 165, F813 CrossRef CAS.
- L. Alibabaei, M. K. Brennaman, M. R. Norris, B. Kalanyan, W. Song, M. D. Losego, J. J. Concepcion, R. A. Binstead, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20008–20013 CrossRef CAS PubMed.
- H. S. Oh, H. N. Nong and P. Strasser, Adv. Funct. Mater., 2015, 25, 1074–1081 CrossRef CAS.
- J. Deng, Q. Zhang, K. Feng, H. Lan, J. Zhong, M. Chaker and D. Ma, ChemSusChem, 2018, 11, 3783–3789 CrossRef CAS PubMed.
- K. J. McDonald and K.-S. Choi, Chem. Mater., 2011, 23, 1686–1693 CrossRef CAS.
- M. Zhang, R. P. Antony, S. Y. Chiam, F. F. Abdi and L. H. Wong, ChemSusChem, 2019, 12, 2022–2028 CrossRef CAS PubMed.
- S. Hu, N. S. Lewis, J. W. Ager, J. Yang, J. R. McKone and N. C. Strandwitz, J. Phys. Chem. C, 2015, 119, 24201–24228 CrossRef CAS.
- T. Lopes, L. Andrade and A. Mendes, Sol. Energy Sci. Eng. Appl., 2013, 128, 399–410 Search PubMed.
- C. Xiang, A. Z. Weber, S. Ardo, A. Berger, Y. Chen, R. Coridan, K. T. Fountaine, S. Haussener, S. Hu and R. Liu, Angew. Chem., Int. Ed., 2016, 55, 12974–12988 CrossRef CAS PubMed.
- R. Memming, Semiconductor electrochemistry, John Wiley & Sons, Germany, 2nd edn, 2015, p. 484 Search PubMed.
- M. M. Momeni, M. Mahvari and Y. Ghayeb, J. Electroanal. Chem., 2019, 832, 7–23 CrossRef CAS.
- X. Lv, I. Rodriguez, C. Hu, J. Shang, P. H.-L. Sit, C. Ye, G. Oskam and W. Y. Teoh, Mater. Today Chem., 2019, 12, 7–15 CrossRef CAS.
- P. Dias, T. Lopes, L. Meda, L. Andrade and A. Mendes, Phys. Chem. Chem. Phys., 2016, 18, 5232–5243 RSC.
- S. D. Ponja, B. A. Williamson, S. Sathasivam, D. O. Scanlon, I. P. Parkin and C. J. Carmalt, J. Mater. Chem. C, 2018, 6, 7257–7266 RSC.
- N. Noor and I. P. Parkin, J. Mater. Chem. C, 2013, 1, 984–996 RSC.
- R. M. Pasquarelli, D. S. Ginley and R. O’Hayre, Chem. Soc. Rev., 2011, 40, 5406–5441 RSC.
- D.-w Choi, W. Maeng and J.-S. Park, Appl. Surf. Sci., 2014, 313, 585–590 CrossRef CAS.
- E. M. Bomhard, Environ. Toxicol. Pharmacol., 2018, 58, 250–258 CrossRef CAS PubMed.
- C. Khelifi, A. Attaf, A. Yahia and M. Dahnoun, Surf. Interfaces, 2019, 15, 244–249 CrossRef CAS.
- A. Way, J. Luke, A. D. Evans, Z. Li, J.-S. Kim, J. R. Durrant, H. K. Hin Lee and W. C. Tsoi, AIP Adv., 2019, 9, 085220 CrossRef.
- M. Asadzadeh, F. Tajabadi, D. Dastan, P. Sangpour, Z. Shi and N. Taghavinia, Ceram. Int., 2021, 47, 5487–5494 CrossRef CAS.
- I. Y. Bu, Ceram. Int., 2014, 40, 417–422 CrossRef CAS.
- A. Kumar, S. K. Swami and V. Dutta, J. Alloys Compd., 2014, 588, 546–550 CrossRef CAS.
- J. T. Wang, X. L. Shi, W. W. Liu, X. H. Zhong, J. N. Wang, L. Pyrah, K. D. Sanderson, P. M. Ramsey, M. Hirata and K. Tsuri, Sci. Rep., 2014, 4, 1–9 Search PubMed.
- X. Pinheiro, A. Vilanova, D. Mesquita, M. Monteiro, J. Eriksson, J. Barbosa, C. Matos, A. Oliveira, K. Oliveira and J. Capitão, Ceram. Int., 2022, 49(8), 13019–13030 CrossRef.
- T.-F. Hou, A. Shanmugasundaram, I. V. Bagal, S.-W. Ryu and D.-W. Lee, Mater. Sci. Semicond. Process., 2021, 121, 105445 CrossRef CAS.
- A. Thakur, P. Kumar, S. M. Thalluri, R. Sinha and P. Devi, Int. J. Hydrogen Energy, 2021, 46, 8444–8453 CrossRef CAS.
- T. Bosserez, J. Rongé, J. Van Humbeeck, S. Haussener and J. Martens, Oil Gas Sci. Technol., 2015, 70, 877–889 CrossRef CAS.
- E. Kemppainen, A. Bodin, B. Sebok, T. Pedersen, B. Seger, B. Mei, D. Bae, P. C. K. Vesborg, J. Halme and O. Hansen, Energy Environ. Sci., 2015, 8, 2991–2999 RSC.
- Y. Liu, L. Ren, Z. Zhang, X. Qi, H. Li and J. Zhong, Sci. Rep., 2016, 6, 22516 CrossRef CAS PubMed.
- Y. Liu, L. Ren, Z. Zhang, X. Qi, H. Li and J. Zhong, Sci. Rep., 2016, 6, 22516 CrossRef CAS PubMed.
- D. Siegmund, S. Metz, V. Peinecke, T. E. Warner, C. Cremers, A. Grevé, T. Smolinka, D. Segets and U.-P. Apfel, JACS Au, 2021, 1, 527–535 CrossRef CAS PubMed.
- J. Wang, H.-x Zhong, Z.-l Wang, F.-l Meng and X.-b Zhang, ACS Nano, 2016, 10, 2342–2348 CrossRef CAS PubMed.
- B. Konkena, K. Junge Puring, I. Sinev, S. Piontek, O. Khavryuchenko, J. P. Dürholt, R. Schmid, H. Tüysüz, M. Muhler and W. Schuhmann, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- D. Siegmund, N. Blanc, M. Smialkowski, K. Tschulik and U. P. Apfel, ChemElectroChem, 2020, 7, 1514–1527 CrossRef CAS.
- A. Grimm, W. A. de Jong and G. J. Kramer, Int. J. Hydrogen Energy, 2020, 45, 22545–22555 CrossRef CAS.
- K. Zhang, M. B. McDonald, I. E. Genina and P. T. Hammond, Chem. Mater., 2018, 30, 6420–6430 CrossRef CAS.
- S. Zhang, C. Yin, D. Xing, D. Yang and X. Jian, J. Membr. Sci., 2010, 363, 243–249 CrossRef CAS.
- D. Chen, M. A. Hickner, E. Agar and E. C. Kumbur, Electrochem. Commun., 2013, 26, 37–40 CrossRef CAS.
- A. Berger, R. Segalman and J. Newman, Energy Environ. Sci., 2014, 7, 1468–1476 RSC.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2013, 26, 407–414 CrossRef.
- N. M. Vargas-Barbosa, G. M. Geise, M. A. Hickner and T. E. Mallouk, ChemSusChem, 2014, 7, 3017–3020 CrossRef CAS PubMed.
- D. A. Vermaas, M. Sassenburg and W. A. Smith, J. Mater. Chem. A, 2015, 3, 19556–19562 RSC.
- T. Lopes, L. Andrade, H. A. Ribeiro and A. Mendes, Int. J. Hydrogen Energy, 2010, 35, 11601–11608 CrossRef CAS.
- A. Landman, H. Dotan, G. E. Shter, M. Wullenkord, A. Houaijia, A. Maljusch, G. S. Grader and A. Rothschild, Nat. Mater., 2017, 16, 646–651 CrossRef CAS PubMed.
- J. W. Ager Iii, M. Shaner, K. Walczak, I. D. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811–2824 RSC.
- M. A. Modestino and S. Haussener, Annu. Rev. Chem. Biomol. Eng., 2015, 6, 13–34 CrossRef CAS PubMed.
- T. J. Jacobsson, V. Fjallstrom, M. Edoff and T. Edvinsson, Energy Environ. Sci., 2014, 7, 2056–2070 RSC.
- S. Chu, W. Li, Y. Yan, T. Hamann, I. Shih, D. Wang and Z. Mi, Nano Futures, 2017, 1, 022001 CrossRef.
- M.-K. Son, Energies, 2021, 14, 7422 CrossRef CAS.
- Y. Chen, Y. Liu, F. Wang, X. Guan and L. Guo, J. Energy Chem., 2021, 61, 469–488 CrossRef CAS.
- K. R. Tolod, S. Hernández and N. Russo, Catalysts, 2017, 7, 13 CrossRef.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- A. Hankin, F. Bedoya-Lora, C. Ong, J. Alexander, F. Petter and G. Kelsall, Energy Environ. Sci., 2017, 10, 346–360 RSC.
- T. Lopes, L. Andrade, F. Le Formal, M. Gratzel, K. Sivula and A. Mendes, Phys. Chem. Chem. Phys., 2014, 16, 16515–16523 RSC.
- L. Han, F. F. Abdi, R. van de Krol, R. Liu, Z. Huang, H. J. Lewerenz, B. Dam, M. Zeman and A. H. Smets, ChemSusChem, 2014, 7, 2832–2838 CrossRef CAS PubMed.
- A. I. Pereira, J. Martins, C. J. Tavares, L. Andrade and A. Mendes, Appl. Surf. Sci., 2017, 423, 549–556 CrossRef CAS.
- S.-Y. Park, M.-g Kim, J. Jung, J. Heo, E. M. Hong, S. M. Choi, J.-Y. Lee, S. Cho, K. Hong and D. C. Lim, J. Power Sources, 2017, 341, 411–418 CrossRef CAS.
- D. Ivanou, J. Capitão, J. Maçaira, A. I. Pereira and A. Mendes, J. Electrochem. Soc., 2018, 165, H1040 CrossRef CAS.
- A. Vilanova, T. Lopes and A. Mendes, J. Power Sources, 2018, 398, 224–232 CrossRef CAS.
- X. Yao, D. Wang, X. Zhao, S. Ma, P. S. Bassi, G. Yang, W. Chen, Z. Chen and T. Sritharan, Energy Technol., 2018, 6, 100–109 CrossRef CAS.
- S. Dilger, M. Trottmann and S. Pokrant, ChemSusChem, 2019, 12, 1931–1938 CrossRef CAS PubMed.
- M. Huang, W. Lei, M. Wang, S. Zhao, C. Li, M. Wang and H. Zhu, J. Mater. Chem. A, 2020, 8, 3845–3850 RSC.
- A. Landman, R. Halabi, P. Dias, H. Dotan, A. Mehlmann, G. E. Shter, M. Halabi, O. Naseraldeen, A. Mendes and G. S. Grader, Joule, 2020, 4, 448–471 CrossRef CAS.
- C. Acar and I. Dincer, Int. J. Hydrogen Energy, 2016, 41, 7950–7959 CrossRef CAS.
- A. Eftekhari, V. J. Babu and S. Ramakrishna, Int. J. Hydrogen Energy, 2017, 42, 11078–11109 CrossRef CAS.
- D. Hansora, D. Cherian, R. Mehrotra, J.-W. Jang and J. S. Lee, Joule, 2023, 7, 884–919 CrossRef CAS.
- C. Carver, Z. Ulissi, C. Ong, S. Dennison, G. Kelsall and K. Hellgardt, Int. J. Hydrogen Energy, 2012, 37, 2911–2923 CrossRef CAS.
- C. K. Ong, PhD thesis, Imperial College, London, 2013.
- A. Vilanova, S. Miranda, A. Apolinário, L. Andrade, T. Lopes and A. Mendes, Public report on PEC device design University of Porto – Engineering Faculty, 2016.
- G. Saracco, Report on the study on the potential future penetration of the ArtipHyction technology in the devised or in spin-off fields, Instituto Politécnico de Turim, Artiphyction: Fully artificial photo-electrochemical device for low temperature hydrogen production, 2015.
- I. Holmes-Gentle, S. Tembhurne, C. Suter and S. Haussener, Nat. Energy, 2023, 1–11 Search PubMed.
- T. Lopes, Characterization and Phenomenological Modeling of Photoelectrochemical Cells for Hydrogen Production from Solar Energy, PhD thesis, UPORTO-FEUP, Porto, Portugal, 2014, https://hdl.handle.net/10216/99846 Search PubMed.
- J. Ihssen, A. Braun, G. Faccio, K. Gajda-Schrantz and L. Thony-Meyer, Curr. Protein Pept. Sci., 2014, 15, 374–384 CrossRef CAS PubMed.
- A. Braun, M. Diale, K. Maabong and R. Toth, Safe And Decentralised Hydrogen Fuel Production And Storage For Residential Building And Mobility Applications, 2016.
- A. Braun, M. M. Diale, K. D. Maabong and R. Toth, Safe and decentralised solar hydrogen fuel production and storage for residential building and mobility applications, 6th Int’l Disaster Risk Conf., 2016, pp. 102–105 Search PubMed.
- K. Walczak, Y. Chen, C. Karp, J. W. Beeman, M. Shaner, J. Spurgeon, I. D. Sharp, X. Amashukeli, W. West and J. Jin, ChemSusChem, 2015, 8, 544–551 CrossRef CAS PubMed.
- N. S. Lewis, Nat. Nanotechnol., 2016, 11, 1010–1019 CrossRef CAS PubMed.
- J. M. Spurgeon, M. G. Walter, J. Zhou, P. A. Kohl and N. S. Lewis, Energy Environ. Sci., 2011, 4, 1772–1780 RSC.
- A. Rothschild, G. Grader, G. Shter, A. Landman and H. Dotan, Methods and system for hydrogen production by water electrolysis, US Pat., US10487408B2, 2015 Search PubMed.
- S. Miranda, A. Vilanova, T. Lopes and A. Mendes, RSC Adv., 2017, 7, 29665–29671 RSC.
- C. Moon and B. Shin, Discov. Mater., 2022, 2, 5 CrossRef.
- I. Y. Ahmet, S. Berglund, A. Chemseddine, P. Bogdanoff, R. F. Präg, F. F. Abdi and R. van de Krol, Adv. Energy Sustainability Res., 2020, 1, 2000037 CrossRef.
- J. Zhu, J. B. Guđmundsdóttir, R. Strandbakke, K. G. Both, T. Aarholt, P. A. Carvalho, M. H. Sørby, I. J. Jensen, M. N. Guzik and T. Norby, ACS Appl. Mater. Interfaces, 2021, 13, 20313–20325 CrossRef CAS PubMed.
- R. Sathre, C. D. Scown, W. R. Morrow, J. C. Stevens, I. D. Sharp, J. W. Ager, K. Walczak, F. A. Houle and J. B. Greenblatt, Energy Environ. Sci., 2014, 7, 3264–3278 RSC.
- T. A. Kistler, M. Y. Um and P. Agbo, J. Electrochem. Soc., 2020, 167, 066502 CrossRef CAS.
- M. Wullenkord, C. Spenke, A. Vilanova, T. Lopes and A. Mendes, PECDEMO Project Deliverable Report D6.4: Public Report on Performance of the Large Area Prototype Array, 2017. Available online: https://archiveweb.epfl.ch/pecdemo.epfl.ch/files/content/sites/pecdemo/files/public%20files/PECDEMO%20Deliverable%206.4_DLR_UPorto.pdf (accessed on 10 August 2023).
- J. L. Young, M. A. Steiner, H. Döscher, R. M. France, J. A. Turner and T. G. Deutsch, Nat. Energy, 2017, 2, 17028 CrossRef CAS.
- E. L. Miller, R. E. Rocheleau and X. M. Deng, Int. J. Hydrogen Energy, 2003, 28, 615–623 CrossRef CAS.
- C. Xiang, A. Z. Weber, S. Ardo, A. Berger, Y. Chen, R. Coridan, K. T. Fountaine, S. Haussener, S. Hu and R. Liu, Angew. Chem., Int. Ed., 2016, 55, 12974 CrossRef CAS PubMed.
- C. Xiang, A. Z. Weber, S. Ardo, A. Berger, Y. Chen, R. Coridan, K. T. Fountaine, S. Haussener, S. Hu, R. Liu, N. S. Lewis, M. A. Modestino, M. M. Shaner, M. R. Singh, J. C. Stevens, K. Sun and K. Walczak, Angew. Chem., Int. Ed., 2016, 55, 12974–12988 CrossRef CAS PubMed.
- X. Deng and L. Xu, Integrated photoelectrochemical cell and system having a liquid electrolyte, WO2004050961A1, University of Toledo, Ohio, 2005 Search PubMed.
- X. Deng and L. Xu, Interconnected photoelectrochemical cell, US20080223439A1, Midwest Optoelectronics LLC and University of Toledo, Ohio, 2008 Search PubMed.
- M. Grätzel and J. Augustynski, Tandem Cell for Water Cleavage by Visible Light, WO0102624 (A1), EPFL, Switzerland, 2001 Search PubMed.
- L. Pan, Y. Liu, L. Yao, D. Ren, K. Sivula, M. Grätzel and A. Hagfeldt, Nat. Commun., 2020, 11, 318 CrossRef CAS PubMed.
- M. Cregut, M. Bedas, M.-J. Durand and G. Thouand, Biotechnol. Adv., 2013, 31, 1634–1647 CrossRef CAS PubMed.
- Z. Huaiwei and H. Xin, Resour., Conserv. Recycl., 2011, 55, 745–754 CrossRef.
- M. Y. Khalid, Z. U. Arif, W. Ahmed and H. Arshad, Sustainable Mater. Technol., 2022, 31, e00382 CrossRef CAS.
- G. Gahleitner, Int. J. Hydrogen Energy, 2013, 38, 2039–2061 CrossRef CAS.
- R. Boudries, Int. J. Hydrogen Energy, 2013, 38, 11507–11518 CrossRef CAS.
- A. Hussain, S. M. Arif and M. Aslam, Renewable Sustainable Energy Rev., 2017, 71, 12–28 CrossRef.
- M. S. Guney, Renewable Sustainable Energy Rev., 2016, 57, 776–785 CrossRef.
- D. Azzouzi, B. Boumeddane and A. Abene, Renewable Energy, 2017, 106, 111–121 CrossRef.
- A. Kumar, O. Prakash and A. Dube, Renewable Sustainable Energy Rev., 2017, 79, 304–307 CrossRef.
- A. Algarue, S. Mahmoud and R. Al-Dadah, Energy Procedia, 2014, 61, 2375–2378 CrossRef.
- S. Caron, M. Röger and M. Wullenkord, Energies, 2020, 13, 5196 CrossRef CAS.
- S. Tembhurne and S. Haussener, J. Electrochem. Soc., 2016, 163, H999–H1007 CrossRef CAS.
- R. Abbas, M. Montes, A. Rovira and J. Martínez-Val, Sol. Energy, 2016, 124, 198–215 CrossRef.
- V. Jebasingh and G. J. Herbert, Renewable Sustainable Energy Rev., 2016, 54, 1085–1091 CrossRef.
- H. Beltagy, D. Semmar, C. Lehaut and N. Said, Renewable Energy, 2017, 101, 782–793 CrossRef.
- G. Zhu, T. Wendelin, M. J. Wagner and C. Kutscher, Sol. Energy, 2014, 103, 639–652 CrossRef.
- Q. Xuan, G. Li, G. Pei, J. Ji, Y. Su and B. Zhao, Sol. Energy, 2017, 158, 808–818 CrossRef.
- E. Bellos, Therm. Sci. Eng. Prog., 2019, 10, 112–137 CrossRef.
- M. Wullenkord, C. Jung and C. Sattler, 2014, 45868, V001T02A034.
- M. Wullenkord and C. Jung, 2015, ISF-1, E: Po-4.
- J. P. Bijarniya, K. Sudhakar and P. Baredar, Renewable Sustainable Energy Rev., 2016, 63, 593–603 CrossRef.
- F. Dähler, M. Wild, R. Schäppi, P. Haueter, T. Cooper, P. Good, C. Larrea, M. Schmitz, P. Furler and A. Steinfeld, Sol. Energy, 2018, 170, 568–575 CrossRef.
- G. Muthu, S. Shanmugam and A. Veerappan, Energy Procedia, 2014, 54, 2–10 CrossRef CAS.
- L. Xiao, F.-W. Guo, S.-Y. Wu and Z.-L. Chen, Renewable Energy, 2020, 145, 878–892 CrossRef.
- D. Esteve, C. Ganibal, D. Steinmetz and A. Vialaron, Int. J. Hydrogen Energy, 1982, 7, 711–716 CrossRef CAS.
- A. Fallisch, L. Schellhase, J. Fresko, M. Zechmeister, M. Zedda, J. Ohlmann, L. Zielke, N. Paust and T. Smolinka, Int. J. Hydrogen Energy, 2017, 42, 13544–13553 CrossRef CAS.
- G. Peharz, F. Dimroth and U. Wittstadt, Int. J. Hydrogen Energy, 2007, 32, 3248–3252 CrossRef CAS.
- P. Moraitis, R. Schropp and W. Van Sark, Opt. Mater., 2018, 84, 636–645 CrossRef CAS.
- W. G. Van Sark, K. W. Barnham, L. H. Slooff, A. J. Chatten, A. Büchtemann, A. Meyer, S. J. McCormack, R. Koole, D. J. Farrell and R. Bose, Opt. Express, 2008, 16, 21773–21792 CrossRef CAS PubMed.
- M. G. Debije and P. P. Verbunt, Adv. Energy Mater., 2012, 2, 12–35 CrossRef CAS.
- M. D. Hughes, C. Maher, D.-A. Borca-Tasciuc, D. Polanco and D. Kaminski, Renewable Energy, 2013, 52, 266–272 CrossRef.
- G. Panzeri, E. Tatsi, G. Griffini and L. Magagnin, ACS Appl. Energy Mater., 2020, 3, 1665–1671 CrossRef CAS.
- G. Liu, B. Sun, H. Li, Y. Wang and H. Zhao, J. Mater. Chem. A, 2019, 7, 18529–18537 RSC.
- M. Dumortier and S. Haussener, Energy Environ. Sci., 2015, 8, 3069–3082 RSC.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS PubMed.
- J. S. Price, X. Sheng, B. M. Meulblok, J. A. Rogers and N. C. Giebink, Nat. Commun., 2015, 6, 1–8 CAS.
- C. A. Rodriguez, M. A. Modestino, D. Psaltis and C. Moser, Energy Environ. Sci., 2014, 7, 3828–3835 RSC.
- G. Segev, H. Dotan, K. D. Malviya, A. Kay, M. T. Mayer, M. Grätzel and A. Rothschild, Adv. Energy Mater., 2016, 6, 1500817 CrossRef.
- M. A. Khan, I. Al-Shankiti, A. Ziani, N. Wehbe and H. Idriss, Angew. Chem., 2020, 132, 14912–14918 CrossRef.
- A. Mojiri, R. Taylor, E. Thomsen and G. Rosengarten, Renewable Sustainable Energy Rev., 2013, 28, 654–663 CrossRef.
- M. A. Green and A. Ho-Baillie, Prog. Photovoltaics Res. Appl., 2010, 18, 42–47 CrossRef CAS.
- Y. Bicer, A. F. V. Sprotte and I. Dincer, Appl. Energy, 2017, 197, 169–182 CrossRef CAS.
- S. Tembhurne and S. Haussener, Sustainable Energy Fuels, 2019, 3, 1297–1306 RSC.
- H. Zhang, L. V. Besteiro, J. Liu, C. Wang, G. S. Selopal, Z. Chen, D. Barba, Z. M. Wang, H. Zhao and G. P. Lopinski, Nano Energy, 2020, 105416 Search PubMed.
- A. Leblebici, P. Mayor, M. Rajman and G. De Micheli, Nano-Tera. ch, Springer, 2019, pp.109–137 Search PubMed.
- V. Zagolla, D. Dominé, E. Tremblay and C. Moser, Opt. Express, 2014, 22, A1880–A1894 CrossRef PubMed.
- V. Zagolla, C. Moser and J.-E. Moser, Self-tracking solar concentrator device, US20150063751A1, 2017.
- Q. Wang, C. Pornrungroj, S. Linley and E. Reisner, Nat. Energy, 2022, 7, 13–24 CrossRef.
- M. E. Oruc, A. V. Desai, P. J. Kenis and R. G. Nuzzo, Appl. Energy, 2016, 164, 294–302 CrossRef CAS.
- S. Rashidi, N. Karimi, B. Sunden, K. C. Kim, A. G. Olabi and O. Mahian, Prog. Energy Combust. Sci., 2022, 88, 100966 CrossRef.
- C. Pornrungroj, V. Andrei and E. Reisner, J. Am. Chem. Soc., 2023, 145(25), 13709–13714 CrossRef CAS PubMed.
- R. Boudries, A. Khellaf, A. Aliane, L. Ihaddaden and F. Khida, Int. J. Hydrogen Energy, 2014, 39, 15188–15195 CrossRef CAS.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- X. Lu, S. Xie, H. Yang, Y. Tong and H. Ji, Chem. Soc. Rev., 2014, 43, 7581–7593 RSC.
- H. Luo, J. Barrio, N. Sunny, A. Li, L. Steier, N. Shah, I. E. Stephens and M. M. Titirici, Adv. Energy Mater., 2021, 11, 2101180 CrossRef CAS.
- N. Ibrahim, S. K. Kamarudin and L. Minggu, J. Power Sources, 2014, 259, 33–42 CrossRef CAS.
- J. Plácido and S. Capareda, Bioresour Bioprocess, 2016, 3, 1–12 CrossRef.
- X. Han, H. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. Qiu and S. Jin, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- N. Perini, C. Hessel, J. L. Bott-Neto, C. T. Pires, P. S. Fernandez and E. Sitta, J. Solid State Electrochem., 2021, 25, 1101–1110 CrossRef CAS.
- Y. Wang, G. Bu, X. Geng, Z. Zhu, P. Cui and Z. Liao, J. Cleaner Prod., 2019, 218, 212–224 CrossRef CAS.
- G. Ramis, E. Bahadori and I. Rossetti, Int. J. Hydrogen Energy, 2021, 46, 12105–12116 CrossRef CAS.
- C.-W. Huang, B.-S. Nguyen, J. C.-S. Wu and V.-H. Nguyen, Int. J. Hydrogen Energy, 2020, 45, 18144–18159 CrossRef CAS.
- D. Antón-García, E. Edwardes Moore, M. A. Bajada, A. Eisenschmidt, A. R. Oliveira, I. A. Pereira, J. Warnan and E. Reisner, Nat. Synth., 2022, 1, 77–86 CrossRef.
- C. Pulignani, C. A. Mesa, S. A. Hillman, T. Uekert, S. Giménez, J. R. Durrant and E. Reisner, Angew. Chem., Int. Ed., 2022, 61, e202211587 CrossRef CAS PubMed.
- G. G. N. Thushari and J. D. M. Senevirathna, Heliyon, 2020, 6, e04709 CrossRef CAS PubMed.
- M. Demertzi, R. P. Silva, B. Neto, A. C. Dias and L. Arroja, J. Cleaner Prod., 2016, 112, 1985–1994 CrossRef.
- R. Sathre, J. B. Greenblatt, K. Walczak, I. D. Sharp, J. C. Stevens, J. W. Ager and F. A. Houle, Energy Environ. Sci., 2016, 9, 803–819 RSC.
- P. Nikolaidis and A. Poullikkas, Renewable Sustainable Energy Rev., 2017, 67, 597–611 CrossRef CAS.
- M. Yu, K. Wang and H. Vredenburg, Int. J. Hydrogen Energy, 2021, 46, 21261–21273 CrossRef CAS.
- D. Girelen, E. Taibi and R. Miranda, Hydrogen: a renewable energy perspective Report 978-92-9260-151-5, International Renewable Energy Agency, 2019.
- I. Dincer, Int. J. Hydrogen Energy, 2012, 37, 1954–1971 CrossRef CAS.
- J. M. M. Arcos and D. M. Santos, Gases, 2023, 3, 25–46 CrossRef CAS.
- Y. Lovcha, A. Perez-Laborda and I. Sikora, Appl. Energy, 2022, 305, 117903 CrossRef CAS.
- M. Sato, R. Rafaty, R. Calel and M. Grubb, Wiley Interdiscip. Rev. Clim. Change, 2022, 13, e796 CrossRef.
- A. Ajanovic, M. Sayer and R. Haas, Int. J. Hydrogen Energy, 2022, 47, 24136–24154 CrossRef CAS.
- F. Pruvost, S. Cloete, C. A. del Pozo and A. Zaabout, Energy Convers. Manage., 2022, 274, 116458 CrossRef CAS.
- J. F. George, V. P. Müller, J. Winkler and M. Ragwitz, Energy Policy, 2022, 167, 113072 CrossRef CAS.
- A. Midilli, H. Kucuk, M. E. Topal, U. Akbulut and I. Dincer, Int. J. Hydrogen Energy, 2021, 46, 25385–25412 CrossRef CAS.
- A. Iulianelli, S. Liguori, J. Wilcox and A. Basile, Catal. Rev., 2016, 58, 1–35 CrossRef CAS.
- I. Dincer and C. Zamfirescu, Int. J. Hydrogen Energy, 2012, 37, 16266–16286 CrossRef CAS.
- L. Cao, K. Iris, X. Xiong, D. C. Tsang, S. Zhang, J. H. Clark, C. Hu, Y. H. Ng, J. Shang and Y. S. Ok, Environ. Res., 2020, 186, 109547 CrossRef CAS PubMed.
- O. Bereketidou and M. Goula, Catal. Today, 2012, 195, 93–100 CrossRef CAS.
- L. Alves, V. Pereira, T. Lagarteira and A. Mendes, Renewable Sustainable Energy Rev., 2021, 137, 110465 CrossRef CAS.
- M. R. Giraldi, J.-L. François and C. Martin-del-Campo, Int. J. Hydrogen Energy, 2015, 40, 4019–4033 CrossRef CAS.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrogen Energy, 2017, 42, 30470–30492 CrossRef CAS.
- T. L. Gibson and N. A. Kelly, Int. J. Hydrogen Energy, 2008, 33, 5931–5940 CrossRef CAS.
- B. Olateju, A. Kumar and M. Secanell, Int. J. Hydrogen Energy, 2016, 41, 8755–8776 CrossRef CAS.
- C. Acar and I. Dincer, Int. J. Hydrogen Energy, 2022, 47, 40118–40137 CrossRef CAS.
- D. Burchart, M. Gazda-Grzywacz, P. Grzywacz, P. Burmistrz and K. Zarębska, Energies, 2022, 16, 383 CrossRef.
- J. Li and W. Cheng, Int. J. Hydrogen Energy, 2020, 45, 27979–27993 CrossRef CAS.
- A. Verma and A. Kumar, Appl. Energy, 2015, 147, 556–568 CrossRef CAS.
- H. Liu, W. Guo and S. Liu, Energy, 2022, 258, 125001 CrossRef CAS.
- Y. Ju and C.-H. Lee, Energy Convers. Manage., 2017, 143, 123–136 CrossRef CAS.
- K. Bareiß, C. de la Rua, M. Möckl and T. Hamacher, Appl. Energy, 2019, 237, 862–872 CrossRef.
- C. Acar and I. Dincer, Int. J. Energy Res., 2015, 39(13), 1757–1768 CrossRef CAS.
- G. Di Marcoberardino, X. Liao, A. Dauriat, M. Binotti and G. Manzolini, Processes, 2019, 7, 86 CrossRef.
- A. Mehmeti, A. Angelis-Dimakis, G. Arampatzis, S. J. McPhail and S. Ulgiati, Environments, 2018, 5, 24 CrossRef.
- N. Hajjaji, S. Martinez, E. Trably, J.-P. Steyer and A. Helias, Int. J. Hydrogen Energy, 2016, 41, 6064–6075 CrossRef CAS.
- R. Bhandari, C. A. Trudewind and P. Zapp, J. Cleaner Prod., 2014, 85, 151–163 CrossRef CAS.
- M. Ji and J. Wang, Int. J. Hydrogen Energy, 2021, 46, 38612–38635 CrossRef CAS.
- A. E. Karaca and I. Dincer, Chemosphere, 2023, 337, 139367 CrossRef CAS PubMed.
- C. Yan, A. Yang, I.-L. Chien, W. Shen and J. Ren, Energy Convers. Manage., 2019, 199, 111963 CrossRef CAS.
- A. Valente, D. Iribarren, J.-L. Gálvez-Martos and J. Dufour, Sci. Total Environ., 2019, 650, 1465–1475 CrossRef CAS PubMed.
- B. Lee, H. Chae, N. H. Choi, C. Moon, S. Moon and H. Lim, Int. J. Hydrogen Energy, 2017, 42, 6462–6471 CrossRef CAS.
- A. Maljush, M. Wullenkord, E. Smirnova and C. Jung, Report on the results of life-cycle analysis, economic analysis and benchmarking, PECDEMO, 2017.
- N. Hajjaji, M.-N. Pons, A. Houas and V. Renaudin, Energy Policy, 2012, 42, 392–399 CrossRef CAS.
- A. Maljusch and M. Wullenkord, Advances in Photoelectrochemical Water Splitting, Royal Society of Chemistry, 2018, pp.266–284 Search PubMed.
- G. Li, S. Wang, J. Zhao, H. Qi, Z. Ma, P. Cui, Z. Zhu, J. Gao and Y. Wang, Energy, 2020, 117488 CrossRef.
- I. Dincer and C. Acar, Int. J. Hydrogen Energy, 2015, 40, 11094–11111 CrossRef CAS.
- R. S. El-Emam and H. Özcan, J. Cleaner Prod., 2019, 220, 593–609 CrossRef CAS.
- A. Valente, D. Iribarren and J. Dufour, Int. J. Hydrogen Energy, 2019, 44, 19426–19433 CrossRef CAS.
- D. Iribarren, A. Susmozas, F. Petrakopoulou and J. Dufour, J. Cleaner Prod., 2014, 69, 165–175 CrossRef CAS.
- M. Kumar, A. O. Oyedun and A. Kumar, Int. J. Hydrogen Energy, 2019, 44, 10384–10397 CrossRef CAS.
- H. R. Sara, B. Enrico, V. Mauro and N. Vincenzo, Energy Procedia, 2016, 101, 806–813 CrossRef.
- J. Yao, M. Kraussler, F. Benedikt and H. Hofbauer, Energy Convers. Manage., 2017, 145, 278–292 CrossRef CAS.
- E. Rillo, M. Gandiglio, A. Lanzini, S. Bobba, M. Santarelli and G. Blengini, Energy, 2017, 126, 585–602 CrossRef CAS.
- A. Patyk, T. M. Bachmann and A. Brisse, Int. J. Hydrogen Energy, 2013, 38, 3865–3880 CrossRef CAS.
- L. B. Braga, J. L. Silveira, M. E. Da Silva, C. E. Tuna, E. B. Machin and D. T. Pedroso, Renewable Sustainable Energy Rev., 2013, 28, 166–173 CrossRef CAS.
- Y. M. Camacho, S. Bensaid, G. Piras, M. Antonini and D. Fino, Clean Technol. Environ. Policy, 2017, 19, 1437–1447 CrossRef.
- M. Katebah and P. Linke, Clean. Eng. Technol., 2022, 10, 100552 CrossRef.
- H. Song, Y. Liu, H. Bian, M. Shen and X. Lin, Energy Convers. Manage., 2022, 258, 115513 CrossRef CAS.
- A. Al-Qahtani, B. Parkinson, K. Hellgardt, N. Shah and G. Guillen-Gosalbez, Appl. Energy, 2021, 281, 115958 CrossRef CAS.
- I. J. Okeke, B. A. Saville and H. L. MacLean, Int. J. Hydrogen Energy, 2023, 48, 12581–12599 CrossRef CAS.
- A. Oni, K. Anaya, T. Giwa, G. Di Lullo and A. Kumar, Energy Convers. Manage., 2022, 254, 115245 CrossRef CAS.
- F. Kerscher, A. Stary, S. Gleis, A. Ulrich, H. Klein and H. Spliethoff, Int. J. Hydrogen Energy, 2021, 46, 19897–19912 CrossRef CAS.
- S. Timmerberg, M. Kaltschmitt and M. Finkbeiner, Energy Convers. Manage.: X, 2020, 7, 100043 CAS.
- N. Sánchez-Bastardo, R. Schlögl and H. Ruland, Ind. Eng. Chem. Res., 2021, 60, 11855–11881 CrossRef.
- A. E. Karaca, I. Dincer and J. Gu, Int. J. Hydrogen Energy, 2020, 45, 22148–22159 CrossRef CAS.
- M. Sorrentino, A. Adamo and G. Nappi, Energies, 2019, 12, 3224 CrossRef CAS.
- R. Anghilante, D. Colomar, A. Brisse and M. Marrony, Int. J. Hydrogen Energy, 2018, 43, 20309–20322 CrossRef CAS.
- M. Lin and S. Haussener, Sol. Energy, 2017, 155, 1389–1402 CrossRef CAS.
- O. Posdziech, K. Schwarze and J. Brabandt, Int. J. Hydrogen Energy, 2019, 44, 19089–19101 CrossRef CAS.
- E. Giglio, A. Lanzini, M. Santarelli and P. Leone, J. Energy Storage, 2015, 1, 22–37 CrossRef.
- J. C. Koj, C. Wulf, A. Schreiber and P. Zapp, Energies, 2017, 10, 860 CrossRef.
- A. Valente, D. Iribarren and J. Dufour, Sci. Total Environ, 2020, 728, 138212 CrossRef CAS PubMed.
- N. Gerloff, ACS Sustainable Chem. Eng., 2021, 9, 10123–10141 CrossRef CAS.
- B. Lee, H.-S. Cho, H. Kim, D. Lim, W. Cho, C.-H. Kim and H. Lim, J. Environ. Chem. Eng., 2021, 9, 106349 CrossRef CAS.
- J. E. Mason and K. Zweibel, Int. J. Hydrogen Energy, 2007, 32, 2743–2763 CrossRef CAS.
- E. Cetinkaya, I. Dincer and G. F. Naterer, Int. J. Hydrogen Energy, 2012, 37, 2071–2080 CrossRef CAS.
- M. Fereidooni, A. Mostafaeipour, V. Kalantar and H. Goudarzi, Renewable Sustainable Energy Rev., 2018, 82, 415–423 CrossRef.
- M. Reuß, J. Reul, T. Grube, M. Langemann, S. Calnan, M. Robinius, R. Schlatmann, U. Rau and D. Stolten, Sustainable Energy Fuels, 2019, 3, 801–813 RSC.
- S. Ghandehariun and A. Kumar, Int. J. Hydrogen Energy, 2016, 41, 9696–9704 CrossRef CAS.
- T. Ayodele and J. Munda, Int. J. Hydrogen Energy, 2019, 44, 17669–17687 CrossRef CAS.
- R. Fang, Int. J. Hydrogen Energy, 2019, 44, 29399–29408 CrossRef CAS.
- M. Aguado, E. Ayerbe, C. Azcárate, R. Blanco, R. Garde, F. Mallor and D. M. Rivas, Int. J. Hydrogen Energy, 2009, 34, 2845–2854 CrossRef CAS.
- J. B. Greenblatt, Integrated Solar Fuel Generators, 2018, vol. 22, p.43 Search PubMed.
- L. Xu, X. Deng, A. Abken, X. Cao, W. Du, A. Vijh, W. Ingler, C. Chen, Q. Fan and R. Collins, Critical Research for Cost-Effective Photoelectrochemical Production of Hydrogen, Midwest Optoelectronics, LLC, Toledo, OH (United States), 2014 Search PubMed.
- C. Acar and I. Dincer, J. Cleaner Prod., 2019, 218, 835–849 CrossRef CAS.
- G. Palmer, A. Roberts, A. Hoadley, R. Dargaville and D. Honnery, Energy Environ. Sci., 2021, 14, 5113–5131 RSC.
- A. P. Ravikumar, M. Bazilian and M. E. Webber, Nat. Energy, 2022, 1–3 Search PubMed.
- I. Kougias, N. Taylor, G. Kakoulaki and A. Jäger-Waldau, Renewable Sustainable Energy Rev., 2021, 144, 111017 CrossRef CAS.
- S. Sadeghi, S. Ghandehariun and M. A. Rosen, Energy, 2020, 208, 118347 CrossRef CAS.
- J. Tian, L. Yu, R. Xue, S. Zhuang and Y. Shan, Appl. Energy, 2022, 307, 118205 CrossRef CAS PubMed.
- A hydrogen strategy for a climate-neutral Europe, European Commission, 2020. Available online: https://energy.ec.europa.eu/system/files/2020-07/hydrogen_strategy_0.pdf (accessed on 3 August 2023).
- I. Dincer and C. Acar, Int. J. Hydrogen Energy, 2018, 43, 8579–8599 CrossRef CAS.
- A. Gatto, Energy Res. Soc. Sci., 2022, 89, 102639 CrossRef.
- T. J. Jacobsson, V. Fjällström, M. Edoff and T. Edvinsson, Sol. Energy Mater. Sol. Cells, 2015, 134, 185–193 CrossRef CAS.
- T. J. Jacobsson, Energy Environ. Sci., 2018, 11, 1977–1979 RSC.
- W. Li, J. Zheng, B. Hu, H.-C. Fu, M. Hu, A. Veyssal, Y. Zhao, J.-H. He, T. L. Liu and A. Ho-Baillie, Nat. Mater., 2020, 19, 1326–1331 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |