
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe promise of chiral electrocatalysis for efficient and sustainable energy conversion and storage: a comprehensive review of the CISS effect and future directions
Kyunghee
Chae†
 a,
Nur Aqlili Riana Che
Mohamad†
a,
Jeonghyeon
Kim
a,
Dong-Il
Won
a,
Zhiqun
Lin
*ab,
Jeongwon
Kim
*a and
Dong Ha
Kim
*a
a,
Nur Aqlili Riana Che
Mohamad†
a,
Jeonghyeon
Kim
a,
Dong-Il
Won
a,
Zhiqun
Lin
*ab,
Jeongwon
Kim
*a and
Dong Ha
Kim
*a
aDepartment of Chemistry and Nanoscience, Ewha Womans University, 52 Ewhayeodae-gil, Seodaemun-gu, Seoul 03760, Korea. E-mail: dhkim@ewha.ac.kr; kimgarden@ewha.ac.kr
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, Singapore 117585, Singapore. E-mail: z.lin@nus.edu.sg
First published on 19th August 2024
Abstract
The integration of chirality, specifically through the chirality-induced spin selectivity (CISS) effect, into electrocatalytic processes represents a pioneering approach for enhancing the efficiency of energy conversion and storage systems. This review delves into the burgeoning field of chiral electrocatalysis, elucidating the fundamental principles, historical development, theoretical underpinnings, and practical applications of the CISS effect across a spectrum of electrocatalytic reactions, including the oxygen evolution reaction (OER), oxygen reduction reaction (ORR), and hydrogen evolution reaction (HER). We explore the methodological advancements in inducing the CISS effect through structural and surface engineering and discuss various techniques for its measurement, from magnetic conductive atomic force microscopy (mc-AFM) to hydrogen peroxide titration. Furthermore, this review highlights the transformative potential of the CISS effect in addressing the key challenges of the NRR and CO2RR processes and in mitigating singlet oxygen formation in metal–air batteries, thereby improving their performance and durability. Through this comprehensive overview, we aim to underscore the significant role of incorporating chirality and spin polarization in advancing electrocatalytic technologies for sustainable energy applications.
 Kyunghee Chae | Kyunghee Chae obtained her BS from the Department of Chemistry and Nanoscience at Ewha Womans University in 2020. Currently, she is pursuing her PhD in the Department of Chemistry and Nanoscience at Ewha Womans University under the supervision of Prof. Dong Ha Kim. Her current research focuses on mechanistic studies of (photo)electrocatalytic water splitting and next-generation Li–air and Li–S batteries. |
 Nur Aqlili Riana Che Mohamad | Nur Aqlili Riana Che Mohamad received her MS degree in Catalysis in 2015 from Universiti Putra Malaysia, for the development of catalysts for biodiesel production. She worked as a research fellow in the Department of Chemical and Environmental Engineering of UPM (Malaysia). She is currently a PhD student in the Department of Chemistry and Nanoscience at Ewha Womans University under the supervision of Prof. Dong Ha Kim. Her research interests focus on the development of metal oxide catalysts for (photo)electrocatalytic water splitting and Li–air batteries. |
 Jeonghyeon Kim | Jeonghyeon Kim received her BS from the Department of Chemistry and Nanoscience at Ewha Womans University in 2023. Currently, she is pursuing her MS in the Department of Chemistry and Nanoscience at Ewha Womans University under the supervision of Prof. Dong Ha Kim. Her research interests focus on mechanistic studies of (photo)electrocatalysts for Li–air and Zn–air batteries. |
 Dong-Il Won | Dr Dong-Il Won obtained his BSc (2014) and MSc (2017) from Korea University. He received his PhD degree from the Department of Chemistry and Chemical Engineering at Inha University (2021). Since 2022, he has been working as a postdoctoral researcher in the Nanobio·Energy Material Center at Ewha Womans University. His research interests focus on the catalysis for renewable energy conversion and storage based on in situ/operando spectroscopic techniques. |
 Zhiqun Lin | Dr Zhiqun Lin is a professor in the Department of Chemical and Biomolecular Engineering at the National University of Singapore. His research interests include electrocatalysis, batteries, solar energy conversion, photocatalysis, semi-conductor organic–inorganic nanocomposites, multifunctional nanocrystals, conjugated polymers, block copolymers, hierarchical structure formation and assembly, and surface and interfacial properties. |
 Jeongwon Kim | Dr Jeongwon Kim received his PhD degree from the Department of Energy Engineering at the Ulsan National Institute of Science and Technology (UNIST). He is currently working as a research professor in the Nanobio·Energy Material Center at Ewha Womans University. His research interests lie in electrochemical or plasmonic-assisted catalysts based on transition metals aiming to develop efficient energy storage and conversion devices, such as metal–air batteries and polymer electrolyte membrane fuel cells, for hydrogen production and CO2 utilization. |
 Dong Ha Kim | Prof. Dong Ha Kim received his PhD degree from the Department of Fiber and Polymer Science at Seoul National University (2000). He worked as a postdoctoral researcher at the University of Massachusetts at Amherst and at the Max Planck Institute for Polymer Research. He assumed a faculty position in the Department of Chemistry and Nanoscience at Ewha Womans University (2006). His research interests include the development of hybrid nanostructures for energy storage and conversion, environmental remediation, memory devices, display devices, and theragnosis. |
1. Introduction
Electrochemistry presents a promising path for the sustainable conversion of molecules, leveraging electrons derived from renewable electricity.1–3 It holds a crucial position in propelling society towards a circular, sustainable, and equitable future. Consequently, substantial endeavors have been made to explore and harness diverse clean and sustainable energy sources, offering an optimistic avenue to markedly diminish reliance on conventional fossil fuels.4 Amidst the array of various sustainable electrochemical applications, the oxygen evolution reaction (OER) plays a pivotal role in various electrochemical energy conversion or storage systems, including water electrolyzers, electrochemical CO2 reduction systems, metal–air batteries, and fuel cells.5 Nevertheless, the OER remains a significant bottleneck because of the slow kinetics associated with its multi-electron and proton coupled electrochemical processes. This sluggish reaction requires a considerably higher energy input, manifesting as an increased overpotential, to overcome the kinetic barriers.6,7 In this regard, a large number of various strategies have been employed to improve the sluggish catalytic activities of the OER, to date. For example, while rutile ruthenium oxide (RuO2) demonstrates excellent OER catalytic activity in both acidic and alkaline electrolytes,8,9 it becomes highly unstable under high anodic potential, oxidizing to form RuO4 and dissolving in the solution.10 Iridium oxide (IrO2), another typical OER catalyst, also exhibits excellent OER performance; however, it suffers from a similar issue, as it oxidizes to form IrO3 and dissolves during the OER process.11 In addition to noble-metal-based electrocatalysts, non-noble-metal-based counterparts, such as transition metal electrocatalysts, have garnered considerable attention because of their outstanding catalytic efficacy. These catalysts feature versatile structures comprising transition metals exhibiting diverse coordination environments and oxidation states, enabling customizable oxygen evolution reaction (OER) behaviors. Moreover, incorporating diverse transition metals into a single metal oxide can elicit synergistic effects, offering an effective approach to enhance oxygen evolution reaction (OER) activity.12Compared to the conventional methods of screening metal element candidates, alternative approaches like magnetic manipulation are emerging as unconventional avenues to overcome the limitations delineated by the volcano plot within the framework of the Sabatier principle.2 This innovation leads to enhanced electrocatalytic efficiency and is gaining significant traction within the scientific community.13–15
In particular, R. Naaman and D. H. Waldeck's group first reported that the probability of electron transmission through chiral molecules depends on the electron spin, an effect that has been called chirality-induced spin selectivity (CISS).16,17 Since its initial discovery, the CISS effect has been explored across various practical applications. In this innovation, chiral electrocatalysts are employed for energy conversion applications such as water electrolysis, resulting in a notable enhancement in terms of energy efficiency compared to achiral electrocatalysts.16 These findings suggest that electrochemical reactions influenced by electron spin, which have been previously underestimated, hold potential as an effective approach to enhance sustainable electrochemical reaction efficiencies. The phenomenon of chirality-induced spin selectivity (CISS), which enables the manipulation of electron spin states in the absence of a magnetic field, is drawing significant interest because of its practicality, straightforwardness, and significant impact on the performance of electrochemical reactions.
Despite the promising advancements in chiral applications within electrochemistry, the field remains nascent, characterized by a scarcity of examples and limited methodologies for verifying electrocatalysts' chirality. This review endeavors to meticulously examine electrocatalysts' chirality and critically assess the influence of chiral nanostructures on electrochemical reactions, focusing on efficiency, catalytic activity, and mechanistic insights. Additionally, we propose rational design principles for chiral electrocatalysts and their implementation in sustainable energy conversion and storage solutions. We conclude by highlighting the challenges and prospective contributions of chiral nanomaterials to electrochemistry, underscoring the need for innovative research in this burgeoning domain (Scheme 1).
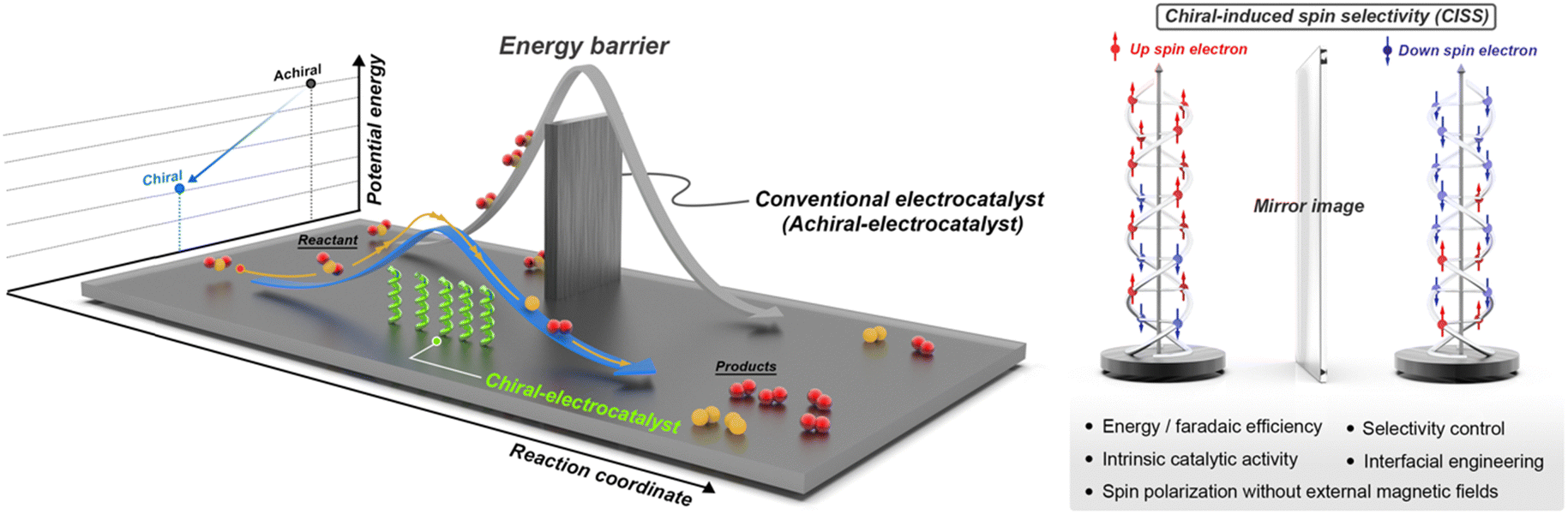 | ||
| Scheme 1 Schematic of activation energy barrier of chiral/achiral-electrocatalysts and the chirality-induced spin selectivity (CISS) effect in the transport of electrons with opposite spin (red and blue) through the chiral structure. | ||
2. Chirality induced spin selectivity effect
The concept of chirality-induced spin selectivity (CISS) has become a focal point of scientific inquiry, gaining substantial interest across the disciplines of physics, chemistry, and materials science. The CISS effect refers to the preferential electron transport of a particular spin orientation when interacting with chiral molecules or structures.18 Originally conceived as a theoretical notion, this effect has been validated by a growing body of experimental evidence, which has broad implications for multiple scientific and technological domains.19The importance of CISS cannot be overstated, given its potential applications in areas as diverse as spintronics, electronics, catalysis, and biotechnology.20 Spintronics, for instance, can benefit from the CISS effect through the development of spin filters, leading to more efficient data storage and transfer systems.21 In the field of electrocatalysis, CISS offers pathways for designing chiral catalysts with remarkable spin selectivity, thus providing new avenues for enantioselective transformations.22 This section aims to provide a comprehensive overview of the CISS effect, tracing its historical development, outlining the key theoretical principles, and summarizing the current state of experimental observations. Subsequent sections will delve into these aspects in detail to offer a thorough understanding of the CISS effect and its diverse applications.
2.1. Principle of the CISS effect
The chirality-induced spin selectivity (CISS) effect is a fascinating phenomenon that involves the interaction between the spin of an electron and the chirality of a molecule.16,17 When a chiral molecule participates in an electron transfer process, it can selectively promote or hinder the transfer of electrons based on their spin orientation. In other words, the CISS effect allows chiral molecules to act as spin filters, permitting only electrons with a specific spin orientation to pass through while blocking others.23,24 The CISS effect manifests when a chiral molecule participates in an electron transfer process, leading to preferential promotion or hindrance of electron transfer based on its spin orientation. This unique behavior allows chiral molecules to act as “spin filters,” selectively permitting only electrons with specific spin orientations to pass through while impeding others.18 To comprehend the underlying mechanisms of the CISS effect, several theoretical models have been proposed, unveiling the intriguing interplay between chirality and electron spin in chiral molecules. These models offer valuable insights into spin-selective phenomena occurring in chiral electrocatalysts and other spintronics applications.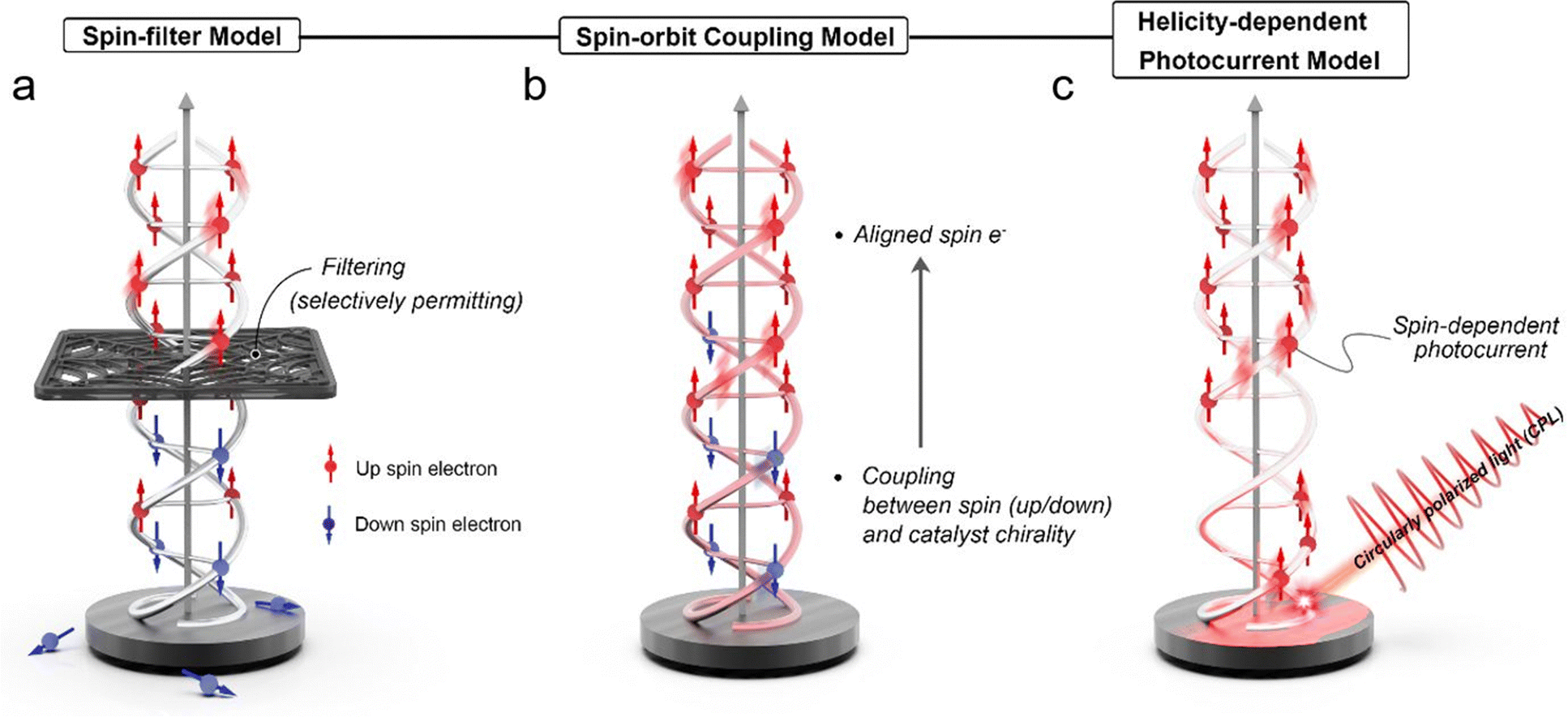 | ||
| Fig. 1 Schematic representation of electron transmission mechanisms through chiral-induced spin selectivity (CISS), illustrating three key models: (a) the spin-filter model, which demonstrates how electrons with specific spin states are selectively transmitted; (b) the spin–orbit coupling model, highlighting the interaction between an electron's spin and its orbital motion within the chiral structure; and (c) the helicity-dependent photocurrent model, which shows how the chirality of light influences the generation of photocurrents in materials with spin-selective properties. Each model is depicted to elucidate the fundamental processes contributing to spin selectivity in chiral materials. | ||
For instance, consider a hypothetical scenario involving a chiral electrocatalyst engaged in an electrochemical reaction. The chiral molecule, adorned with an asymmetric carbon center, exhibits two enantiomeric configurations—the left-handed (L-enantiomer) and the right-handed (D-enantiomer). In the context of an electrochemical process, the chiral electrocatalyst plays a pivotal role in mediating electron transfer during the reaction. As per the spin-filter model, electrons with specific spin orientations, such as spin-up electrons, encounter heightened and favourable coupling interactions with the chiral electrocatalyst.28 Consequently, their interaction with the chiral moiety exhibits increased efficacy, accelerating their transfer in the electrochemical reaction. Conversely, electrons with spin orientations opposite to the molecular chirality, exemplified by spin-down electrons, experience weaker coupling interactions, reducing their involvement in electron transfer processes, effectively resulting in spin selectivity.29,30
Zwang et al. further explored this concept by studying how a magnetic field could influence electron flow through native, hydrated double-stranded DNA. Their research highlighted a DNA-mediated CISS effect, where reversing the helical handedness of the DNA in the films created a diode-like spin-filtering response. This study underscores the potential of chiral molecular structures to influence electron dynamics significantly due to their inherent asymmetry.29
For example, consider a scenario involving a photoelectrochemical system, where a chiral molecule serves as a photoactive species, and circularly polarized light is used to excite electrons within the molecular entity. Within the framework of the spin–orbit coupling model, circularly polarized light plays a decisive role in influencing the spin polarization of photo-excited electrons within the chiral molecule. As a result of this intricate interplay, the spin state of the electron becomes intimately linked with the molecular chirality. The entwined spin-chirality correlation governs the electron's propensity to partake in charge transfer processes. Electrons featuring specific spin orientations, intricately interwoven with the molecular chirality, exhibit enhanced propensities for engaging in favourable charge transfer interactions with the chiral moiety. Conversely, electrons with spin orientations decoupled from the molecular chirality experience weaker coupling interactions with the chiral molecule, leading to a subdued involvement in electron transfer phenomena. The profound correlation between the electron's spin state and the molecular chirality bestows remarkable selective attributes in charge transfer processes. The judicious integration of chiral molecules and circularly polarized light provides an experimental platform for probing helicity-dependent variations in electron behavior and photocurrent, presenting tantalizing prospects for exploring spin-selective processes in photoelectrochemistry.
Steele et al. expanded this model by investigating the pronounced spin–orbit interactions in materials like carbon nanotubes, known for their non-trivial topological properties. Their research demonstrated that the spin–orbit coupling in carbon nanotubes varies with the chirality of the nanotube, significantly influencing the sign of the spin–orbit interaction. This finding suggests that different chiral configurations of nanotubes can distinctly affect electron behavior, emphasizing the critical role of molecular chirality in spin-related phenomena.32
For instance, consider a photoelectrochemical cell where a chiral photoactive material serves as the working electrode (Fig. 1c). When circularly polarized light illuminates the chiral material, it interacts differently with electrons of distinct spin states, contingent on their enantiomeric configurations. The circular polarization of light has notable implications for the spin polarization of the photo-excited electrons within the chiral material.20 The helicity of the incident light dictates the preferential excitation of electrons possessing specific spin orientations. As a corollary, the photoelectrochemical cell manifests helicity-dependent variations in photocurrent. Notably, the utilization of left-handed circularly polarized light may yield an augmented photocurrent compared to employing right-handed circularly polarized light, owing to the excitation of electrons with favoured spin orientations. The profound interplay between circularly polarized light, chiral molecules, and spin polarization forms the cornerstone of the helicity-dependent photocurrent model. Leveraging the controlled modulation of helicity allows for the manipulation of spin-dependent phenomena, enabling selective charge transfer processes in photoelectrochemical systems.
In summary, theoretical models—the spin-filter model, spin–orbit coupling model, and helicity-dependent photocurrent model—provide valuable insights into the underlying mechanisms of the CISS effect, shedding light on the intriguing interplay between chirality and electron spin in chiral molecules. Understanding these models is pivotal for designing efficient chiral electrocatalysts and exploring other spintronics applications. By harnessing the CISS effect, researchers can advance energy conversion and storage technologies, making significant strides toward enhanced efficiency and sustainability in the field of energy science and technology.
2.2. How to induce the CISS effect
The fabrication of nanomaterials with chiral configurations typically necessitates the introduction of a chiral bias to induce chiral features within the structures.37 This can be achieved through various means, including the incorporation of chiral inducers such as chiral ligands, chiral templates, magnetic fields, circularly polarized light, and macroscopic shear forces (Scheme 2). The methods can be broadly categorized into two types: structural and surface engineering. Structural engineering is a straightforward approach, often leveraging chiral templates and physical deposition strategies, while on the other hand, surface engineering methods involve the transference of chirality from chiral molecules to the nanostructures. This process disrupts the symmetry of the overall structures or induces specific morphologies, showcasing a more intricate and complex mechanism.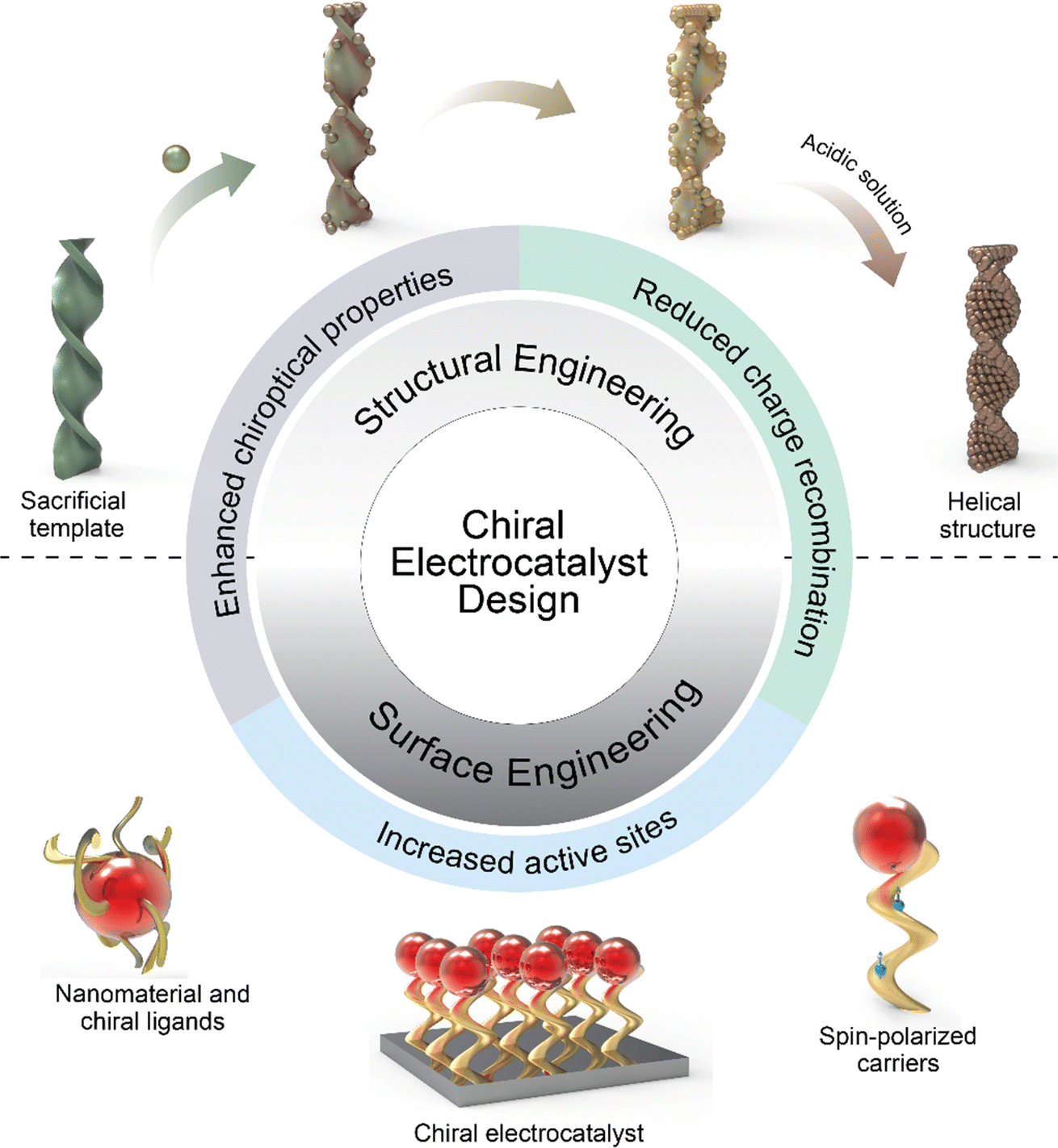 | ||
| Scheme 2 Schematic illustration of the fabrication process of nanomaterials with chiral configurations, employing two primary methodologies: structural and surface engineering. Structural engineering utilizes chiral templates and physical deposition; subsequent removal of the templates in the acidic solution yields chiral structures. Conversely, surface engineering transfers chirality from chiral molecules to nanomaterials through electrostatic interaction. Resultant chiral nanomaterials boast increased active sites, reduced charge recombination, and induce the CISS effect, enhancing their functional properties. | ||
2.2.1.1. Soft templates. Cellulose nanocrystals (CNCs) stand out as a prevalent and effective template for directing the spontaneous self-assembly of nanoparticles, resulting in the formation of optically active and highly ordered chiral nematic liquid crystal structures.38–40 CNCs are particularly appealing as templating agents due to their flexibility and highly crystalline and high-aspect-ratio rod-like nanocrystals, showing typical dimensions with widths in the range of ∼5–10 μm and lengths spanning from 50 to 300 nm.38 Moreover, the optical properties of CNC-templated chiral structures could be tuned by modifying the fabrication parameters of nanomaterials and CNC templates.45 A study reported by Liu et al. showed the concurrent dispersion and directed self-assembly of gold nanorods (GNRs) and CNCs to fabricate diverse mesostructured composite materials exhibiting distinct phases such as nematic-like, helicoidal cholesteric, and LC-isotropic phase-coexistence structures.46Fig. 2b and c show that at CNC concentrations below 3 wt%, both CNCs and GNRs were randomly oriented as validated by the absence of birefringence in the isotropic phase from polarized optical microscopy (POM). When the CNC concentration reached a critical concentration (Cc) of ∼3 wt%, an oriented ordered birefringence appeared as shown in Fig. 2e. The phenomenon stems from entropic steric interactions that mitigate the “excluded volume” while maximizing the translational entropy at the expense of the orientational component. In the nematic phase, translational entropy surpasses that of a disordered isotropic phase, stabilizing the ground-state nematic phase above the Cc.47 Lukach et al. also demonstrated the co-assembly between the CNCs and gold nanoparticles with different sizes, shapes, and compositions. Notably, their findings indicated that the size and surface charge of Au NPs did not exert a significant influence on the properties of CNCs. The concentration of Au NPs in the films, however, exhibited a pronounced impact on the degree of cholesteric (N*) order, as well as the extinction and circular dichroism (CD) properties of the films.33 Another conventional option is the utilization of a DNA-assisted template for the fabrication of chiral nanostructures with helical configurations.48 Kotov and colleagues presented an exemplary case involving the creation of heterogeneous chiral pyramids consisting of Au, Ag, and CdSe@ZnS quantum dots (QDs) positioned at the vertices of a tetrahedron.42 This intricate assembly process was facilitated through DNA hybridization across multiple steps. Notably, both R/S enantiomeric configurations displayed robust and complementary chiroptical signals, characterized by a significant g-factor of 1.9 × 10−2. This investigation underscored the promising potential of DNA for orchestrating the self-assembly of nanoparticles via partial hybridization.21
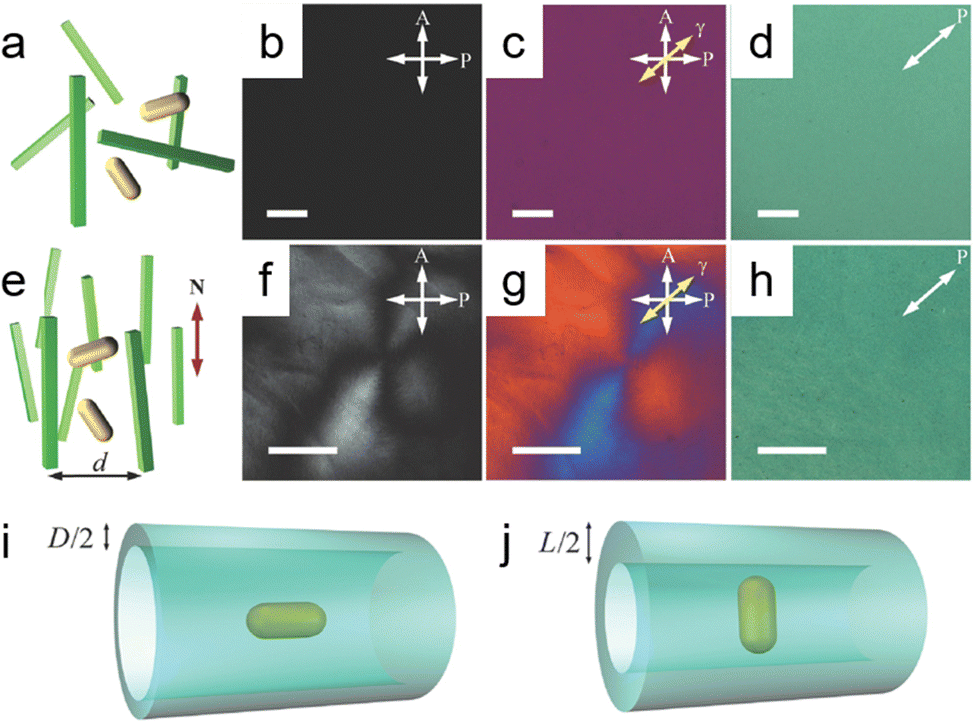 | ||
| Fig. 2 (a) Schematic illustrating randomly dispersed gold nanorods (GNRs) in a dilute green cellulose nanocrystal (CNC) dispersion. Polarized Optical Microscopy (POM) images show (b) without and (c) with a 530 nm retardation plate, indicating the slow axis (γ) with a yellow arrow. (d) A transmission optical microscopy image. (e) The diagram depicts gold nanorods (GNRs) dispersed randomly within nematic-like cellulose nanocrystals (CNCs), featuring a specified average distance between CNCs (d) and the local director (N). Additional POM images include (f) without and (g) with a full wavelength (530) nm retardation plate, highlighting the slow axis (γ) indicated by a yellow arrow and revealing a defect line within a schlieren texture. (h) Another transmission optical microscopy image is also presented. Illustrations of excluded volume of GNRs within the CNC host dispersion depict the effective confinement tube size when GNRs are (i) aligned parallel and (j) orthogonal to the director N. All images feature scale bars of 20 μm. Adapted with permission from ref. 46, Copyright 2014, John Wiley and Sons. | ||
2.2.1.2. Hard template. Chiral structures facilitated by flexible linkers often display modest interactions among adjacent components and limited stability. Conversely, chiral inorganic templates offer a more stable framework for imparting chirality to initially achiral building blocks attached to them.49 In recent decades, there has been a gradual advancement in the development of chiral SiO2 helices, serving as chiral inorganic hosts to create chiral composite materials with intricate 3D helical superstructures.50–52 These innovations hold promise for inducing novel chiral effects in guest components, such as plasmonic nanoparticles (NPs),53–55 perovskites56 and semiconductor nanocrystals.49 In a recent study, Correa-Duarte and collaborators demonstrated the fabrication of twisted SiO2@Au@TiO2 nanostructures through the electrostatic assembly of TiO2 nanoparticles onto SiO2@Au helices (Fig. 3a).43 The incorporation of TiO2 resulted in a further redshift of the optical signal emitted by the composite assemblies, attributed to changes in the surrounding dielectric medium. Additionally, this incorporation led to the Schottky barrier formation between Au and TiO2, enhancing the transfer of hot electrons from Au nanoparticles to TiO2 during polarization-sensitive photocatalytic processes (Fig. 3b). Apart from the adaptable silica helices, numerous other chiral materials act as sources of chirality. These include homochiral quantum dots (QDs),57 metal–organic frameworks (MOFs),58 and gold nanoparticles (Au NPs).59,60
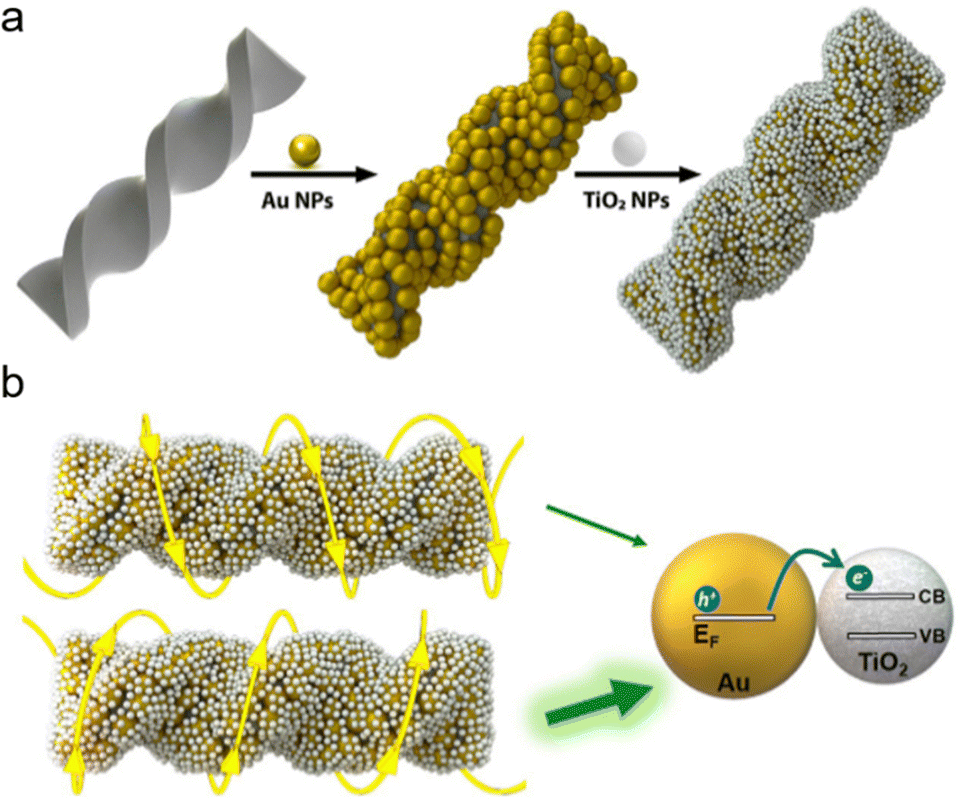 | ||
| Fig. 3 Schematics illustrating (a) the adsorption of gold (Au) and titanium dioxide (TiO2) nanoparticles (NPs) onto chiral silica (SiO2) nanoribbons and (b) the asymmetric interaction between a left-handed (L-handed) hybrid structure and circularly polarized light (CPL), either left (LCP) or right (RCP) circular polarization. Enhanced hot electron transfer occurs when the helicity of the NP assembly aligns with that of the CPL, facilitating optimal energy transfer from the metal's Fermi level to the semiconductor's conduction band. Adapted with permission from ref. 43, Copyright 2022, American Chemical Society. | ||
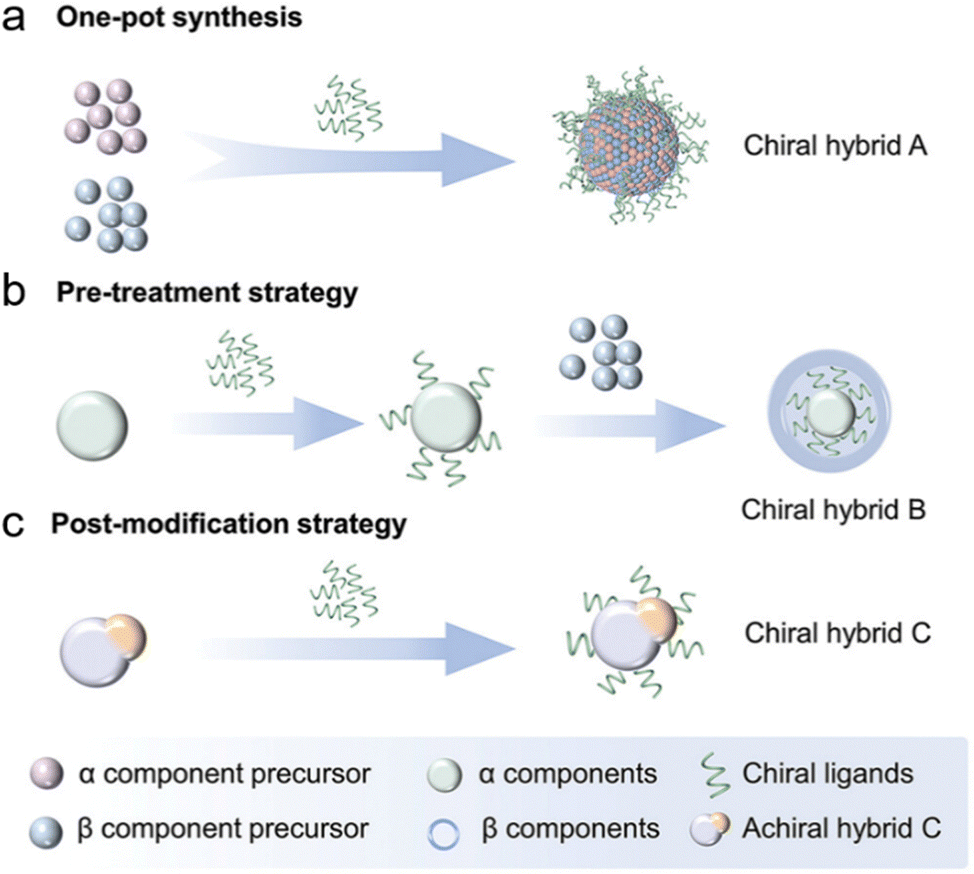 | ||
| Fig. 4 Schematic depiction of wet-chemical synthetic routes for creating plasmonic-based hybrid nanostructures using chiral ligands: (a) a one-pot synthesis method for fabricating multicomponent chiral structures by concurrently introducing metal precursors and chiral ligands into the reaction mixture. (b) A sequential methodology wherein an achiral component is first treated with chiral molecules, subsequently promoting the growth of an additional component. (c) A strategy involving the initial creation of achiral hybrid nanostructures, which are subsequently functionalized with chiral molecules via ligand exchange or electrostatic interactions to induce chirality. Adapted with permission from ref. 64, Copyright 2024, John Wiley and Sons. | ||
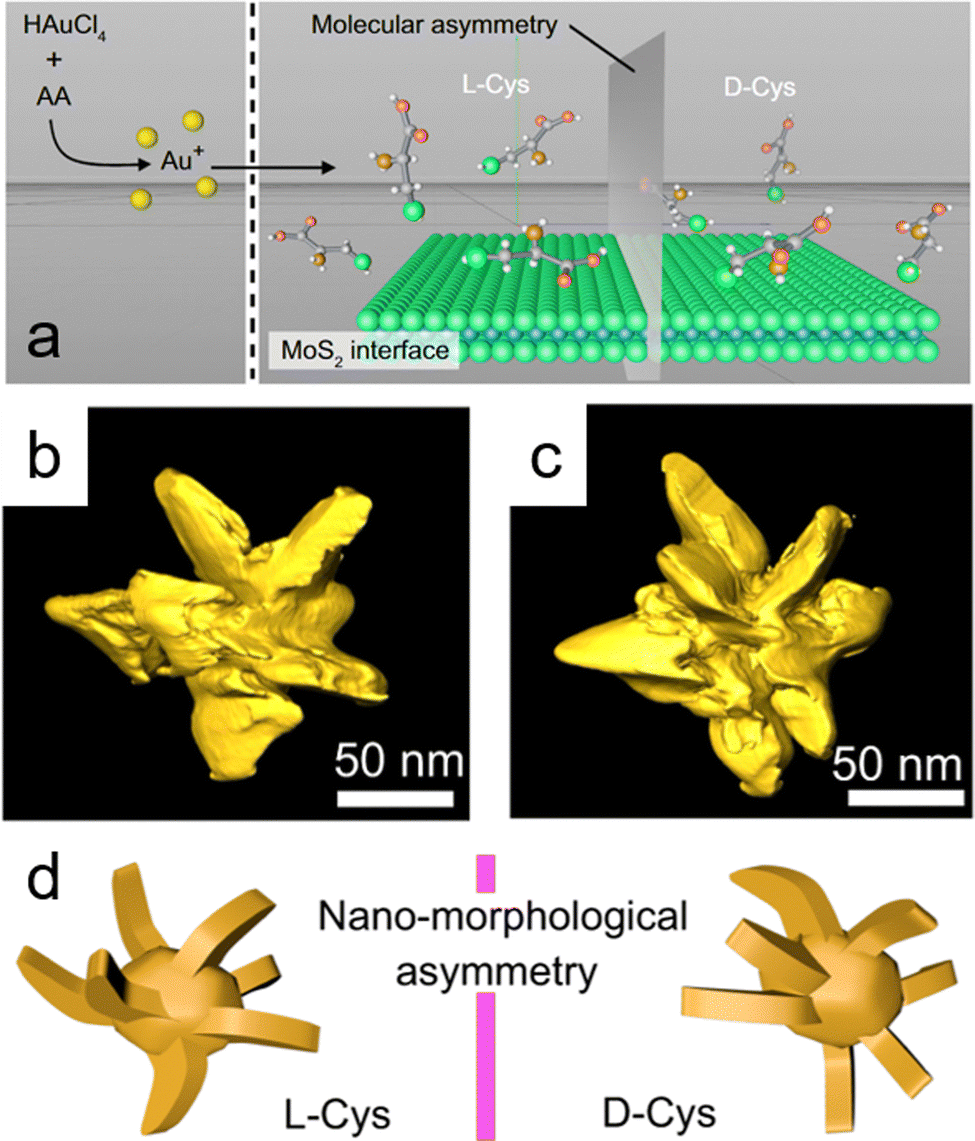 | ||
| Fig. 5 (a) Schematic illustration showing the growth of gold (Au) nanocrystals mediated by cysteine enantiomers in the presence of exfoliated molybdenum disulfide (MoS2) nanosheets; molybdenum (Mo) and sulfur (S) atoms are represented as blue and green spheres, respectively. (b) and (c) Transmission electron microscopy (TEM) tomography images of individual Au/MoS2 nanostructures. (d) Simulation of asymmetric morphology in synthesized Au/MoS2 nanostructures, derived from TEM images of nanostructures fabricated from L-cysteine (L-Cys) and D-cysteine (D-Cys). Adapted with permission from ref. 65, Copyright 2022, Springer Nature. | ||
An alternative approach for imparting chirality to metal or semiconductor surfaces involves the electrochemical coating of chiral molecules. Zhang et al. utilized this method to coat achiral Fe3O4 nanoparticles with four distinct chiral molecules—L-tryptophan, D-tryptophan, L-A3, and L-A11—resulting in the generation of chiral Fe3O4 nanoparticles through the adsorption of these chiral molecules.13 In a related study, Mtangi et al. explored two families of organic semiconductors, namely Zn porphyrins and tri(pyrid-2-yl)amine trisamide (TPyA), in both chiral (employing enantiomerically pure side chains) and achiral (utilizing achiral side chains) versions, coated onto indium tin oxide (ITO) substrates.68 Their findings revealed that manipulation of the side chains of the molecules allowed for control over the resulting helical supramolecular assemblies, enabling the formation of either a singular helical sense or a racemic mixture of both helical senses, respectively.68 In a recent development, Feng et al. synthesized a novel amorphous chiral tartaric acid–FeNi coordination polymer using an electrodeposition method.69 Following a conventional approach, the application of a negative potential induced the cathodic reduction of H2O and NO3−, leading to OH− formation and concurrent deprotonation of tartaric acid (TA). This promoted the coordination between Fe–Ni centers and TA ligands and simultaneous deposition. However, the resulting coordination polymer was found to be amorphous due to the rapid electrodeposition rate. The resulting chiral nanoparticles emerged through the ordered aggregation of smaller nanospheres with interparticle pores facilitated by coordination interactions.69
2.3. Measurement of the CISS effect
The evaluation of chiral purity is of utmost importance in the development of electrocatalysts, exerting a significant influence on both the inherent properties of electrocatalysts and the external environments impacting catalytic surfaces. Therefore, undertaking thorough and quantitative examination of CISS effects emerges as a pivotal element in the deliberate design and refinement of chiral-induced electrocatalysts. CISS analysis typically falls into two primary categories: direct detection measurements, which require the direct interaction of chiral structures with the electron spin state, and indirect detection measurements involving the quantitative analysis of products formed during electrochemical reactions.2.3.1.1. Low-energy photoelectron transmission (LEPET). Low-energy LEPET is the first indicator to observe the CISS effect and it involves studying electron transmission through chiral monolayers using photoemission techniques. It focuses on the dependence of electron transmission yield on the chirality of molecules and the light's handedness used for photoelectron ejection.16Fig. 6 illustrates the schematic setup of LEPET spectroscopy, designed to characterize electron energy distributions across chiral monolayers, omitting the spin of transmitted electrons. This method employs CPL to emit spin-polarized photoelectrons from the Au substrate.67 The experiment measures the quantum yield and the distribution of kinetic energy of photoelectrons in different molecular assemblies.21 In this work, Langmuir–Blodgett films of amino acids (L- or D-stearoyl lysine) were deposited on a polycrystalline gold substrate. It was found that a higher quantum yield was observed for a right-handed circularly polarized (RCP) light through L-stearoyl lysine assembly compared to R-stearoyl lysine assembly, despite identical film characteristics. The experimental results revealed a distinct correlation, wherein the quantum yield of photoelectrons traversing an L-stearoyl lysine assembly exhibited its peak magnitude under right-circularly polarized (RCP) light, followed by an intermediate level under linearly polarized light (with no discernible net spin polarization from the Au substrate), and the lowest efficacy under left-circularly polarized (LCP) light. Göhler and colleagues observed spin-selective electron transmission through self-assembled monolayers of double-stranded DNA on gold and measured the spin of transmitted electrons using a Mott polarimeter. To perform direct measurements, photoelectrons were guided through a 90° bender and transport optics to convert longitudinal spin polarization into transverse orientation for analysis. In the electron polarimeter, the scattering asymmetry induced by electron spin polarization was utilized, and transverse polarization was determined using the Sherman function. They found that by utilizing a Mott polarimeter to directly measure the spin of transmitted electrons, self-assembled monolayers of double-stranded DNA on gold exhibited spin polarizations exceeding 60% (room temperature), with spin-polarized photoelectrons detected even under unpolarized light excitation. This level of spin selectivity surpassed that achieved by other known spin filters. Moreover, the efficiency of spin filtration was found to be contingent upon the length and organization of the DNA within the monolayer.18 LEPET studies provided insights into the spin selectivity and transmission efficiency of photoelectrons, elucidating distinctions based on the chirality and helicity of the chiral molecules. This technique allowed for the examination of how the arrangement and geometric properties of chiral molecules impact the behaviour of photoelectrons, shedding light on their spin characteristics and transmission rates.
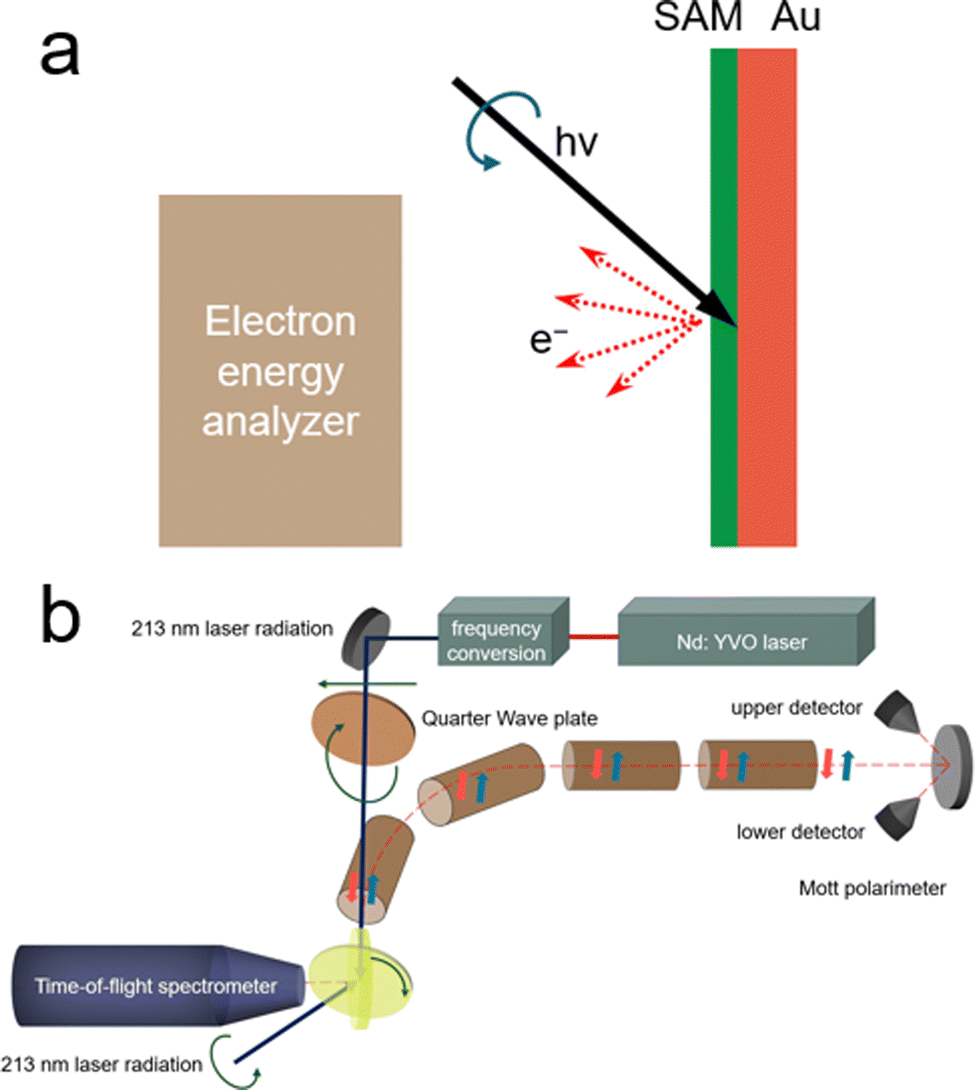 | ||
| Fig. 6 (a) Illustration of the LEPET (low energy photo-electron transmission) photoemission setup used to measure electron energy distributions across chiral monolayers, without the spin of transmitted electron determination. (b) Diagram depicting the experimental setup purposely designed to measure the spin polarization of photoelectrons. Adapted with permission from ref. 21, Copyright 2012, American Chemical Society. | ||
2.3.1.2. Magnetic conductive atomic force microscopy (mc-AFM). mc-AFM (magnetic-conductive atomic force microscopy) is a method developed by Naaman et al. and used to study the spin selectivity of chiral molecules at the nanoscale.70Fig. 7 shows the schematic of the mc-AFM setup used to measure the spin polarization. It probes the spin polarization of nanoscopic structures and analyses the effect of the interface between chiral materials and the ferromagnet used for spin analysis. mc-AFM measures the spin selectivity by comparing currents with different magnet configurations (up and down) at a specific voltage [(Iup/Idown)V], expressing spin polarization as a ratio or percentage {[(Iup − Idown)/(Iup + Idown)] × 100}. In this work, the supramolecular nanofibers are initially formed in a solution phase. The molecules are subsequently delicately transferred onto a gold-coated nickel surface (Au/Ni). This surface is magnetized perpendicular to its plane, with the north pole oriented either upwards or downwards. The electric potential is applied to ground the AFM tip, simultaneously adjusting the potential on the Au/Ni substrate. The studies were conducted on nanofiber-based materials assembled from three chiral and three achiral molecules. The effect of the magnetic field on spin polarization was significant for three chiral molecules studied. Fig. 7b and e shows that (S)-CBI-1 with 3,4,5-trialkoxyphenyl substituents exhibited a spin polarization of 40–50%. Conversely, the 3,5-dialkoxyphenyl-substituted (S)-CBI-3 displayed a notably larger disparity in currents upon magnetization switching of the Ni layer (Fig. 7c), resulting in a substantially heightened spin polarization of 85–90% at +3 V (Fig. 7f). Intrigued by the significant difference in spin polarization between (S)-CBI-1 and (S)-CBI-3, despite their strikingly similar molecular structures, further investigation extended to analyzing the spin polarization of nanofibers assembled from a tetra-amidated porphyrin with 3,5-dialkoxyphenyl wedges ((S)-Zn-P1). Remarkably, (S)-Zn-P1 exhibited a substantial 20
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 current ratio between magnet orientations (up and down) at +3 V (Fig. 7d and g).
:1 current ratio between magnet orientations (up and down) at +3 V (Fig. 7d and g).
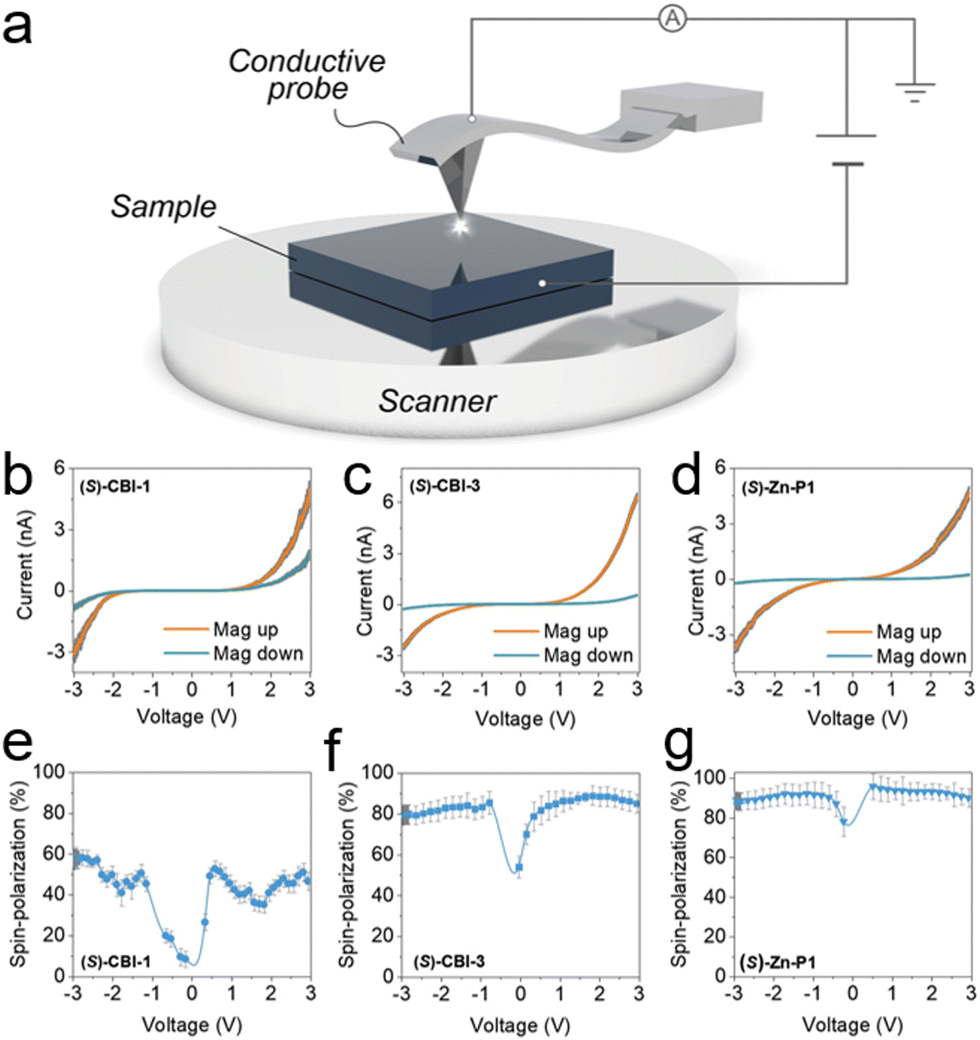 | ||
| Fig. 7 (a) Schematic of the magnetic-conductive atomic force microscopy (mc-AFM) setup employed to measure spin polarizations in supramolecular nanofibers. (b)–(d) (I–V) plots for supramolecular nanofibers of (S)-CBI-1, (S)-CBI-3, and (S)-Zn-P1, respectively, with the nickel film magnetized either with the north pole facing upwards and downwards (orange and cyan, respectively). (e)–(g) The spin polarization percentage ([(Iup − Idown)/(Iup + Idown)] × 100) of (S)-CBI-1 (e), (S)-CBI-3 (f), and (S)-Zn-P1 (g) with Iup and Idown representing the currents measured with the magnetic north pole oriented upwards and downwards, respectively. The mean standard error is depicted in gray. Adapted with permission from ref. 70, Copyright 2020, John Wiley and Sons. | ||
Another Naaman's work verified the spin selectivity of electron transmission through the Zn–porphyrin stacks.71 They demonstrated the correlation between the current's dependence on the orientation of the magnetic field at the mc-AFM tip and the chiral porphyrin stack. This observation confirms a clear preference for one spin orientation over its counterpart within the system. The ratio between the two spin currents is influenced by the 35% spin polarization of the tip, resulting in a 4:1 spin filtering in the molecular system. This means that only about 20% of electrons passing through the chiral molecular aggregates have the “wrong” spin. For achiral compounds, the current magnitude does not vary with the magnetic field orientation, indicating non-spin-specific conduction with an equal amount of right and left-handed helices. This contrasts with chiral porphyrins, which efficiently filter spins, consistent with previous findings on chiral molecules as effective spin filters.19,68
2.3.1.3. Circular dichroism (CD). Circular dichroism (CD) spectroscopy is a robust optical method utilized for the analysis of chiral materials and molecules. It enables the detection of enantioselective signals by measuring the disparate absorption of right and left circularly polarized light, typically achieved through polarization analysis of light transmitted through the sample of interest. Measurements conducted within the visible and ultraviolet regions of the electromagnetic spectrum observe electronic transitions. If the molecule under examination contains chiral molecules, one circularly polarized light (CPL) state will be absorbed more strongly than the other, resulting in a non-zero circular dichroism (CD) signal across the corresponding wavelengths. The CD signal can exhibit positivity or negativity, contingent upon whether left circularly polarized light (L-CPL) is absorbed to a greater degree than right circularly polarized light (R-CPL) (yielding a positive CD signal) or to a lesser extent (yielding a negative CD signal).
Moreover, previous research has revealed a strong correlation between spin polarization and observed changes in circular dichroism (CD) spectroscopy.70 The incorporation of both chiral and achiral building blocks results in an enhancement of the overall helicity within the stacks, wherein the chiral molecule dictates the supramolecular chirality of the helix while the achiral counterparts conform accordingly. The well-known “sergeants-and-soldiers” principle in chiral amplification experiments was investigated using (S)-coronene bisimide ((S)-CBI-3) and achiral-CBI-4, with measurements conducted using CD spectroscopy.72,73 As illustrated in Fig. 8a, the molar circular dichroism (Δε) demonstrates a non-linear relationship with the fraction of (S)-CBI-3 in the solution, affirming chiral amplification within the system. This amplification effect is further corroborated by the spin polarization of electron transport, which was measured using magnetic conductive atomic force microscopy (mc-AFM), as shown in Fig. 8e and f. Up to a 30% fraction of (S)-CBI-3, there is a notable nonlinear increase in spin polarization, which closely parallels the trend observed in the CD measurements. When the fraction of (S)-CBI-3 reaches and exceeds 50%, the magnitude of spin polarization approaches the levels observed in nanofibers assembled from enantiomerically pure (S)-CBI-3. This direct correlation between the changes in the CD signal and the spin polarization measured by mc-AFM, as a function of (S)-CBI-3 fraction, underscores the impact of chiral amplification on spin polarization. These findings suggest that the principles of chiral amplification can modulate the magnitude of spin polarization in helical nanofibers. Furthermore, the results indicate that the CISS effect probes the entire supramolecular chirality structure, rather than solely the number of stereocenters within the film. This study underscores the capability of the CISS effect to facilitate electrical measurements of chirality, demonstrating its potential to reveal the intricate relationship between molecular chirality and electron spin polarization.
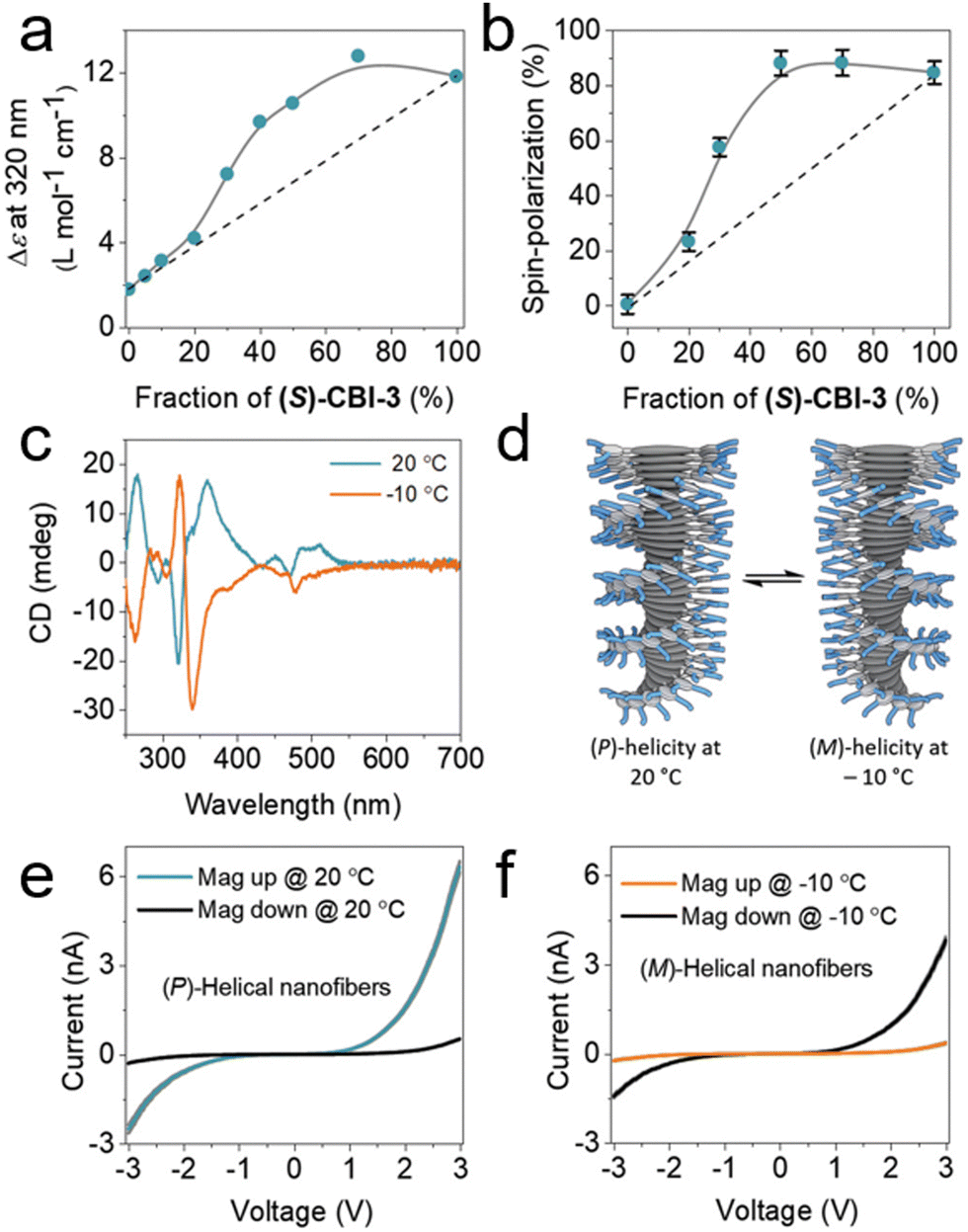 | ||
| Fig. 8 (a) The molar circular dichroism (Δε) evolution as a function of the proportion of the (S)-CBI-3 molecule with ac-CBI-4, in methylcyclohexane, MCH (50 × 10−6 M), at 20 °C. Circular dichroism spectroscopy shows a nonlinear Δε dependence on sergeant fraction, indicating chiral increment in solution-state assembly. (b) Spin polarization of (S)-CBI-3 constructed from a mixture of (S)-CBI-3 and ac-CBI-4 on Au/Ni substrates. The gray lines in (a) and (b) guide the eye while the dashed line signifies the presumed trend without increment of chirality. (c) Temperature-dependent CD spectra of (S)-CBI-3 in MCH (50 × 10−6 M). (d) Schematic showing the change in the helicity of supramolecular nanofibers with temperature shifts from +20 to −10 °C. (e) and (f) I–V curves of (S)-CBI-3 at 20 × 10−6 M on Au/Ni substrates at +20 (e) and −10 °C (f). All mc-AFM measurements were conducted at room temperature. Adapted with permission from ref. 70, Copyright 2020, John Wiley and Sons. | ||
2.3.1.4. Hall effect-based devices. The simplification of chirality measurement techniques can be achieved through the utilization of both Hall and Nanofloret-based devices, wherein the CISS effect enables electrical readout of chirality with high sensitivity even at low analyte concentrations. These methods are universal and do not necessitate specific recognition elements. In both device types, exchange interactions resulting from CISS induce notable transient spin-exchange forces between chiral molecules and magnetic surfaces.74 Hall measurements leverage changes in magnetization to investigate the chirality of adsorbed molecules. This is facilitated through a solid-state hybrid organic–inorganic device that relies on the Hall effect but functions without the need for an external magnetic field.75 The Hall effect is a long-known phenomenon, where a voltage is generated across a conductor transverse to the electric current when a magnetic field is applied perpendicular to the current flow.76 This effect serves as a fundamental tool in semiconductor studies, particularly for assessing properties such as charge carrier concentration and mobility.
Yossi Paltiel et al. demonstrated that the chiral monolayer acts as a spin filter, allowing for Hall effect observation without the need for an external magnetic field or a permanent magnet. In their experiment, molecules were chemisorbed onto the gold over-layer covering the conductive Ni channel, leading to a constant induced magnetic field. This setup enabled Hall measurements to be performed effectively. The resistance Rxy = Vy/Ix was determined, where Vy represents the potential measured between the Hall electrodes and Ix denotes the source-drain current. Notably, AHPA-L (α-helix polyalanine L) and AHPA-D (α-helix polyalanine D) molecules elicited magnetization in opposite directions, leading to opposing signs of Hall voltage, Vy, in both cases. Consequently, this results in a reversal of the sign of Rxy. The observed asymmetry between the measured values for the two enantiomers may arise from disparities in the current flow through the Hall channel due to inherent differences stemming from the fabrication process.77 During Anomalous Hall Effect (AHE) measurements, the device remained fixed to its substrate. A 4 mL volume of a 1 mM ethanolic solution containing molecules was drop-cast onto the device's surface and subsequently dried under inert conditions. This drop-casting technique preserves the integrity of the interconnections between the sample and the chip, thereby ensuring that any alterations in the Hall voltage readout solely reflect the influence of the added molecules. Additionally, it is pertinent to acknowledge that charge reorganization within the chiral molecule coincides with electron spin polarization.
To enhance the chirality signal and expand the spectrum probed by the Hall device, electrical gating was utilized.78 In an experimental configuration using a Hall device, the electrode integrated with a Hall circuit was positioned inside an electrochemical cell, with a voltage applied between it and a counter/gate electrode (see Fig. 9). This voltage induces an electric field across the molecular layer, prompting redistribution of electron clouds. When a chiral molecule film is present, it results in spin-polarized charging currents and generates a Hall voltage across the transverse electrodes. To evaluate the spin polarization associated with charge polarization, a single layer of these molecules adhered to the GaN surface of the Hall device/working electrode (see Fig. 9b). The GaN substrate was chosen for its extended spin lifetime and resistance to corrosion in aqueous solutions. Subsequently, the Hall device was immersed in a buffered electrolyte solution. Applying a voltage between the device and a gating electrode (G) induces an electrostatic field across the molecular film and the interior of an electrical double layer, causing charge polarization. If this polarization is accompanied by spin polarization, the Hall voltage increases. By employing the Hall device with electrical gating, electric-field-induced spin polarization was investigated in two chiral L-oligopeptides of varying lengths, a chiral D-oligopeptide and an achiral molecule serving as a control. This observation aligns with the known property of spin-filtering due to the CISS effect, contingent on current direction and chirality.
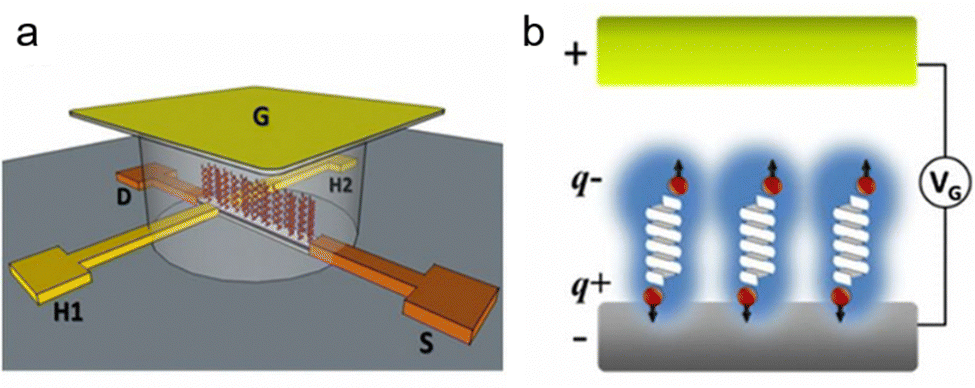 | ||
| Fig. 9 (a) Experimental setup diagram: a Hall device coated with an organic monolayer is immersed in a solution containing a graphene (G) electrode and an inert electrolyte. Applying an electric potential (VG) between the G electrode and the device induces polarization of the solution, generating an electric field that interacts with the adsorbed molecules. This leads to charge reorganization in the molecules (partial charges q+ and q−), inducing charge displacement at the device's surface and concurrent spin polarization, generating a magnetic field affecting electron flow between the source (S) and drain (D) electrodes. The resulting Hall potential (VH), arising from spin magnetization, is measured in relation to VG. (b) Diagram of spin polarization: an applied electric field (via VG) on a chiral molecule triggers charge reorganization, leading to spin polarization, illustrating the transformation from electrical to magnetic responses at the molecular level. Adapted with permission from ref. 78, Copyright 2017, National Academy of Sciences. | ||
2.3.1.5. Localized plasmon based chiral sensor. Localized surface plasmon resonance (LSPR) occurs when conduction electrons in a metallic nanoparticle oscillate coherently, typically when the nanoparticle's size is smaller than the wavelength of the illuminating light. LSPR is distinguished by an amplified electromagnetic field and confinement within the nanoparticle's dimensions. Research has demonstrated that covering a metallic nanoparticle with chiral molecules boosts the electronic circular dichroism signal and imparts chirality onto the LSPR mode.79 A recent report published by Amir Ziv and colleagues demonstrated that combining a chiral plasmonic metastructure with an electrode can produce a chiroptical response, directly translatable into an electrical signal readout (see Fig. 10).80 This device features a gold nanofloret (NF) hybrid structure, which is a combination of semiconductor and metal nanosystems. It consists of a SiGe nanowire tipped with an Au cap that contacts a counter electrode to form a nano-junction. When exposed to molecules, a metal–molecule–metal junction is formed. Chiral molecules adsorbed on the gold tip induce chirality in the localized plasmonic resonance at the electrode–tip junction, resulting in a specific current response depending on the molecule's chirality. Under illumination, the localized surface plasmon resonance (LSPR) of the gold nanocap, coupled to the SiGe nanowire, modulates the current flowing through the nanojunction. As shown in Fig. 10b, a voltage bias is applied across the suspended Au-NF, and the electrical current through the device is measured. Simultaneously, circularly polarized laser illumination is directed onto the sample using a microscope objective.
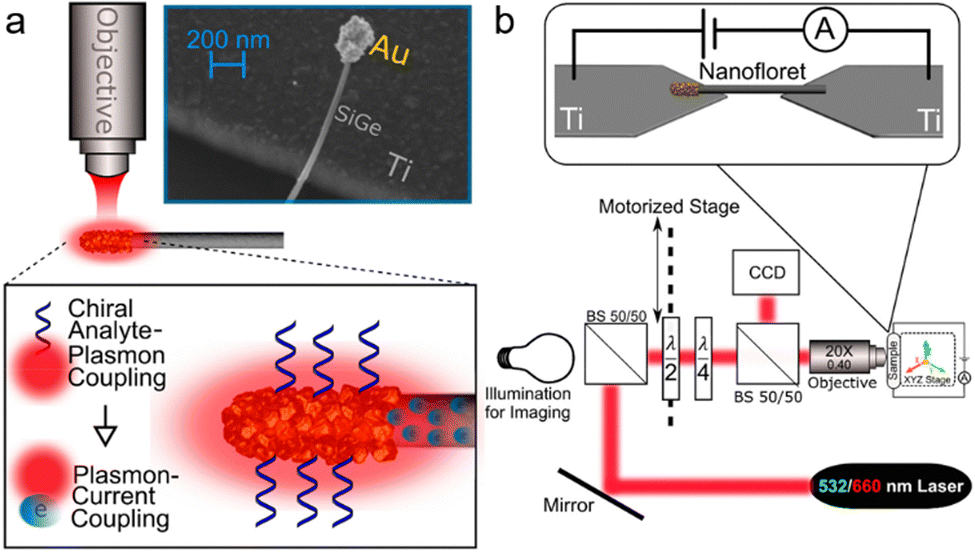 | ||
| Fig. 10 (a) Device measurement setup where an Au-nanofiber (Au-NF) junction is targeted by focused continuous-wave laser radiation, enhancing the interaction with a chiral substance linked to localized surface plasmons, which then influences the current through the Au-NF. Inset: SEM image illustrating the configuration of the Au-NF device, consisting of a semiconducting (SiGe) nanowire with an Au nanocap at its tip, suspended above a Ti macro-contact. The surface area of the cap is approximately 10−9 cm2. (b) Top-view schematic of the chirality sensor setup, where the Au-NF spans between two Ti contacts connected to a voltage source and an ammeter. Positioned on an XYZ stage, the sensor can be precisely aligned under a laser beam focused by a microscope objective. A quarter waveplate modifies the light to circular polarization, with its type (left or right) alternated by a half waveplate on a motorized stage. The setup is monitored using a CCD camera. Adapted with permission from ref. 80, Copyright 2021, American Chemical Society. | ||
Current–voltage (I(V)) measurements showed gap formation upon introducing an α-helix polyalanine (AHPA) solution, indicating monolayer formation at the Au–cap–Ti electrode gap. Before chiral molecules were added, illuminating the device with 532 nm left-circularly polarized (LCP) and right-circularly polarized (RCP) light triggered an inherent chiral response (see Fig. 11a). Due to the nonsymmetric shape and morphology of the NF gold cap, it inherently possesses chirality, as it does not exhibit symmetry under mirror reflection. Additionally, chiral metastructures can exhibit strong optical activity, with even individual metallic nanoparticles displaying optical activity due to imperfections in shape and crystal structure changes. As a result, the Au-NF chirality sensor behaves differently under LCP and RCP light even before adding chiral molecules. Upon exposure to the AHPA solution, an asymmetry in polarized excitation is observed, depending on the handedness of the AHPA (see Fig. 11b). This leads to changes in conductivity and photoresponse under different circular polarizations due to molecular adsorption. The relatively extended stabilization times are due to the lack of passivation at the NF junctions, causing surface states to change upon molecular adsorption. Therefore, the sensor's compact size and broad electrooptical response range facilitate the detection of ultra-low concentrations of chiral molecules using optical excitation and electrical readout.
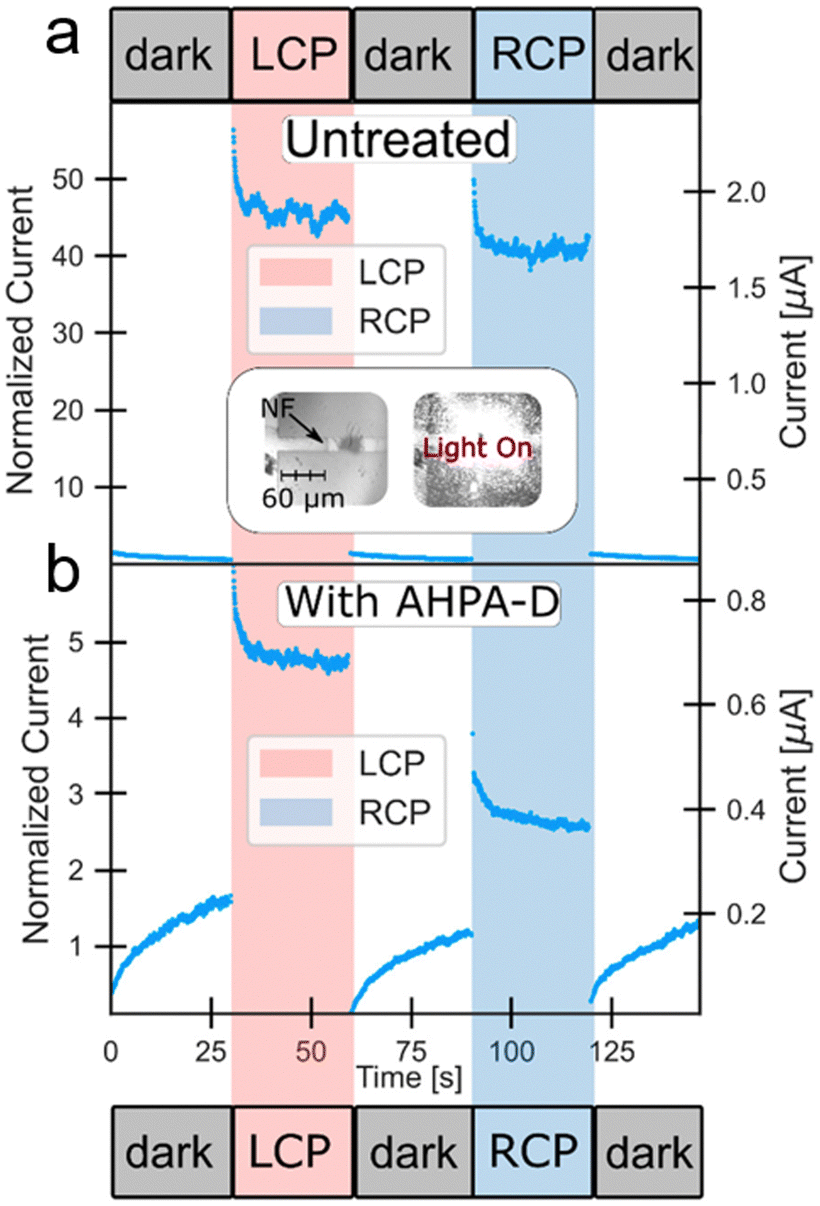 | ||
| Fig. 11 (a) Current over time recorded using the Au-NF chirality sensor at a fixed voltage of 1 V, comparing conditions with various circular polarizations and no illumination prior to molecule application via drop casting. Inset: optical images of the Au-NF junction, shown with and without illumination. (b) Current vs. time measurements for the Au-NF chirality sensor under a constant voltage, displaying responses to different circular polarizations and no illumination after molecules have been applied by drop casting. Adapted with permission from ref. 80, Copyright 2021, American Chemical Society. | ||
2.3.2.1. Titration of H2O2. To date, the most effective approach for realizing the CISS effect has primarily been within the domain of electrocatalysts deployed in the context of the oxygen evolution reaction (OER), serving as a representative system. The OER constitutes a critical bottleneck in electrocatalytic water splitting processes owing to its sluggish kinetics. This reaction necessitates a substantial overpotential to attain a requisite operating current, resulting in significant energy dissipation within water electrolyzers. In alkaline media, the proposed OER mechanism follows eqn (1)–(4), where * denotes the adsorption site:
| OH− + * → OH* + e− | (1) |
| OH* + OH− → O* + H2O + e− | (2) |
| O* + OH− → OOH* + e− | (3) |
| OOH* + OH− → O2 + H2O + e− + * | (4) |
The generation of hydrogen peroxide (H2O2) as a byproduct occurs through a two-electron pathway, as depicted in eqn (3) and (4), particularly under neutral or acidic conditions. While H2O2 and OH* serve as valuable products in other contexts, in this scenario, however, their presence impedes oxygen (O2) production consequently compromising the efficiency of water electrolysers.
| O* + OH− → OH* + e− | (4) |
| OH* + OH− → H2O2 + e− + * | (5) |
Bulk electrolysis conducted in the coulometry mode typically involves applying a constant voltage under neutral conditions, often in a solution like 0.1 M Na2SO4 aqueous solution. The presence of the H2O2 byproduct is commonly identified using a redox indicator such as o-tolidine or iron(II) chloride tetrahydrate. When reacting with o-tolidine, the integrated absorption peak around 436 nm signifies the generation of H2O2. In the case of iron(II) chloride tetrahydrate, a peak around 335 nm emerges due to the conversion of Fe2+ to Fe3+ induced by the presence of H2O2 in the electrolyte. A recent study conducted by the Masayuki Suda group showed the effective suppression of H2O2 formation through the utilization of a promising chiral van der Waals superlattice, namely, a chiral TiS2 crystal. The absorption peak at 437 nm in the electrolyte solution containing H2O2 exhibited heightened intensity when utilizing the TiS2_rac-PEA_PEG electrode, indicating a substantial yield of H2O2. Conversely, the absorption peaks nearly vanished in spectra obtained from electrodes employing TiS2_R-PEA_PEG and TiS2_S-PEA_PEG configurations. This phenomenon underscores the enhanced OER activity facilitated by the promotion of O2 generation and the suppression of H2O2 formation through the chirality inherent in TiS2_R-PEA_PEG and TiS2_S-PEA_PEG electrodes, thereby favouring the four-electron process.
3. Applications of the CISS effect in electrocatalysis
3.1. Oxygen evolution reaction
The oxygen evolution reaction (OER) represents a significant challenge in the realm of electrocatalytic water splitting, primarily due to its inherently sluggish kinetics.81 This reaction necessitates a substantial overpotential to achieve an adequate operating current, resulting in considerable energy losses in water electrolyzers.82,83 Enhancing the efficiency of the OER is a complex task, stemming from the intricate nature of its reaction mechanism and the often-limited stability of alloyed catalysts used in this process.In alkaline media, the oxygen evolution reaction (OER) is a sophisticated electrochemical process that entails the oxidation of water molecules to produce molecular oxygen, hydroxide ions, and electrons.84,85 This reaction is represented below (overall), occurring through a sequence of steps on the surface of a catalyst.86
| 4OH− → O2 + 2H2O + 4e− (overall) |
| OH− + * → *OH + e− | (1) |
| *OH → *O + H+ + e− | (2) |
| *O + OH− → *OOH | (3) |
| *OOH + OH− → O2 + H2O + e− | (4) |
In acidic media, the oxygen evolution reaction (OER) operates through distinct pathways involving either a two-electron or a four-electron process, each leading to the production of molecular oxygen.90–92 The four-electron process, which is more common and efficient, can be represented below (overall).
| 2H2O(l) → O2(g) + 4H+ + 4e− (overall) |
| H2O + * → *OH + H+ + e− | (5) |
| *OH → *O + H+ + e− | (6) |
| 2*O → *O–O* → O2 + 2* | (7) |
Conversely, the less common two-electron process, which typically leads to the formation of hydrogen peroxide (H2O2), is represented by 2H2O(l) → H2O2(l) + 2H+ + 2e−. In this process, the adsorbed water molecule is partially oxidized on the catalyst surface to form a hydroperoxide intermediate (*OOH), which then disassociates to release hydrogen peroxide.95
These mechanistic pathways in acidic media illustrate the complexity of the OER, with the four-electron process being generally more favorable due to its direct production of molecular oxygen and greater energy efficiency.96,97 The choice of catalyst plays a critical role in determining which pathway predominates, as well as the overall efficiency of the reaction, given the high overpotentials and harsh acidic conditions.98 Developing catalysts that favor the four-electron pathway while minimizing overpotential and maximizing stability remains a significant challenge in optimizing the OER for applications such as proton exchange membrane electrolyzers.99,100
In water electrolyzers, the efficiency of the OER can be compromised by the formation of hydrogen peroxide (H2O2), a byproduct that hinders the production of oxygen (O2).101,102 This issue arises because H2O2 is formed through a less efficient two-electron process, contrasting with the desired four-electron process that directly yields O2. The generation of H2O2 not only reduces the overall energy efficiency but also poses challenges due to its corrosive nature and potential to degrade electrolyzer components.101
A notable aspect of H2O2 is its electronic state – it only has a singlet state. In contrast, the more desirable O2 predominantly exists in a ground-state triplet form (3O2).103,104 This difference in electronic states provides an opportunity to use chiral molecules as spin-filters to manipulate the spin states of the intermediate radicals during the OER. By aligning the spins of these unpaired electrons, it's possible to favour the formation of triplet oxygen over singlet hydrogen peroxide.
In essence, the application of chiral molecules as spin-filters in water electrolyzers can significantly enhance the OER process. These chiral molecules can influence the spin orientation of intermediate radicals, thereby promoting the four-electron pathway that leads to the formation of 3O2. This approach effectively suppresses the less efficient two-electron pathway responsible for generating singlet H2O2. By reducing the production of H2O2, the overall efficiency of water splitting is improved, leading to more effective and cleaner generation of oxygen, which is a key goal in the development of sustainable energy systems.68
The introduction of the CISS effect into electrocatalytic water-splitting, pioneered by Naaman's group, represented a significant leap forward in the field.28 Their groundbreaking study involved using TiO2 films functionalized with organic linker molecules to attach CdSe nanoparticles (NP) as the anode—the site of electrochemical oxidation, as illustrated in Fig. 12a. The focus of their investigation was on evaluating the hydrogen evolution efficiency at the cathode, contrasting the effects of seven distinct organic molecules—three chiral and four achiral—when applied to the anode. Remarkably, the current gradients achieved with the chiral molecules at a voltage of 1.5 V significantly exceeded those of their achiral counterparts, a comparison depicted in Fig. 12b. Notably, all the achiral molecules used were considerably shorter than the chiral ones, with several exhibiting high levels of conjugation.105 Under normal circumstances, this would suggest better conductivity and higher current for the achiral molecules. However, the study found that despite their greater molecular lengths, the chiral molecules facilitated significantly lower threshold potentials for oxygen evolution, along with a corresponding increase in current, compared to the achiral molecules. The crucial factor contributing to this enhanced performance was the spin-dependent electron transfer from the CdSe NP through the chiral molecules to the TiO2 film, leading to an isotropic distribution of spin alignment.17 This specificity arises because chiral molecules efficiently transfer electrons featuring a particular spin state (either up or down). As a result, electrons remaining in the ground state have a spin orientation that is antiparallel to the transferred spin. Consequently, when an electron with a well-defined spin alignment is transferred from the CdSe NP, it leaves a hole with the same spin orientation. This uniformity in spin alignment means that electrons transferred from hole scavengers in the solution also possess the same spin alignment, resulting in all atoms having a consistent unpaired spin direction in the laboratory frame.28 Such uniformity in spin alignment promotes the formation of oxygen molecules with a higher cross-section. In contrast, without spin alignment as seen with achiral molecules, the formation of ground-state oxygen molecules requires the two oxygen atoms to be near, allowing strong exchange interactions between the spins for alignment. However, when spins are pre-aligned, as is the case with chiral molecules, the reaction can proceed even at greater distances.28 Naaman's group's seminal work thus provided a substantial foundation for subsequent research efforts focused on exploiting the CISS effect to enhance the OER process in electrocatalytic water-splitting applications, emphasizing the crucial role of spin alignment in improving reaction efficiency.
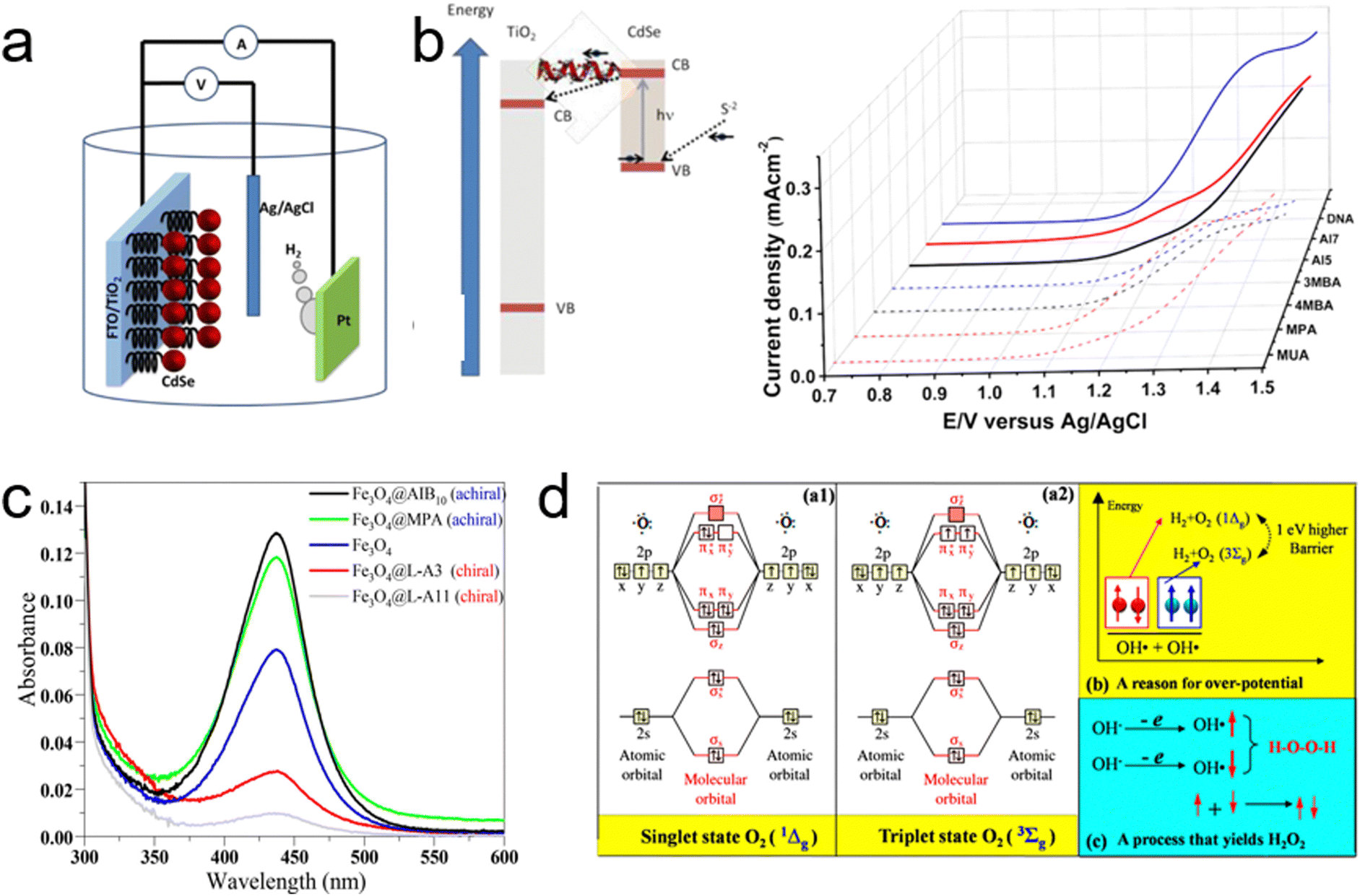 | ||
| Fig. 12 (a) Diagram of the photoelectrochemical cell used for water splitting, featuring CdSe nanoparticles (red) bound to TiO2 nanoparticles via chiral molecules, with TiO2 attached to the FTO conducting electrode (left) and a depiction of electron transfer between the S2− and TiO2 nanoparticles (right). (b) Current density versus potential for TiO2 electrodes coated with self-assembled monolayers of either achiral (dashed lines) or chiral (solid lines) molecules, measured against an Ag/AgCl electrode. Adapted with permission from ref. 28, Copyright 2015, American Chemical Society. (c) Visible absorption spectra of titration of a 0.1 M Na2SO4 electrolyte with o-tolidine, comparing bare Fe3O4, chiral-coated Fe3O4@L-A11 and Fe3O4@L-A3, and achiral-coated Fe3O4@MPA and Fe3O4@AIB10. (d) (a1) Illustration of a molecular orbital of a singlet-state oxygen molecule. (a2) Illustration of a molecular orbital of a triplet-state oxygen molecule. Adapted with permission from ref. 13, Copyright 2015, American Chemical Society. | ||
Building on their earlier work, Naaman's group further explored the application of the CISS effect in enhancing photoelectrochemical (PEC) water splitting, as reported in their subsequent study.13 In this research, they focused on enhancing the anode current, suppressing hydrogen peroxide production and reducing the overpotential. They achieved this by chemisorbing chiral molecules onto Fe3O4 nanoparticles deposited on a FTO anode. The introduction of chiral molecules induced chirality in the nanoparticles, making the electron transfer from the solution to the anode spin selective.17,21,27 This spin selectivity effectively suppressed hydrogen peroxide formation while enhancing the production of triplet oxygen. To empirically demonstrate the CISS effect's role, they analyzed the formation of H2O2 during water splitting with anodes coated with chiral versus achiral molecules. As evidenced in Fig. 12c, anodes coated with achiral Fe3O4 nanoparticles showed a significant absorption peak at 436 nm, indicative of H2O2 formation.106 In contrast, this peak was markedly reduced when using anodes coated with chiral Fe3O4 nanoparticles. This stark difference underscores the role of chiral nanoparticles in impeding H2O2 production. The mechanism behind this phenomenon lies in the spin-selective filtration by chiral Fe3O4 nanoparticles. When electrons transfer from OH−/H2O to the anode, these nanoparticles allow only electrons with a specific spin state to pass through. As a result, all generated ˙OH radicals have spins aligned parallel to each other, facilitating their interaction on a triplet potential surface to form ground-state triplet oxygen molecules (3Σg(3O2)) (Fig. 12d).68,107 Notably, the production of H2O2, which is spin-forbidden on this triplet potential surface, is significantly hindered. Conversely, when Fe3O4 nanoparticles are linked with achiral molecules, they do not filter the electrons’ spin state. This lack of spin selection results in ˙OH radicals with randomly oriented spins, some of which are aligned antiparallel to each other. These antiparallel-aligned radicals are more likely to interact on a singlet potential surface, leading to the formation of singlet hydrogen peroxide (1Δg(1O2)), which is energetically less favorable compared to the triplet ground state.
This uncontrolled oxidation process without spin restriction leads to a higher overpotential and promotes the formation of kinetically favourable H2O2. The accumulation of H2O2 on the electrodes can lead to poisoning, resulting in reduced anode current and compromised catalytic stability for water splitting.13
The CISS effect, arising from the movement of an electron's probability density within the chiral electrostatic field of molecules, generates an effective magnetic field that influences the electron's magnetic moment.21,108 While initial studies predominantly involved organic molecules in electron transmission, the transition to robust inorganic nanomaterials as spin filters opens new opportunities in electronics and electrocatalysis.13,28 A notable advancement in this direction was made by Waldeck's group, who first demonstrated the CISS effect with inorganic copper oxide (CuO) films, using this phenomenon to enhance selectivity in electrocatalytic water splitting.109 In their study, chiral CuO films were electrodeposited on a polycrystalline gold substrate from an electrolyte solution containing chiral complexes of Cu(II)–tartrate.31 The CD spectra reveal an approximate mirror symmetry between CuO films grown from aqueous solutions containing Cu–tartrate complexes of opposite chirality (Fig. 13a). The electrochemical cell equipped with the chiral CuO exhibited a higher current density compared to cells with achiral electrodes, corroborated by the data in Fig. 13c. This enhancement is attributed to the production of spin polarized OH radicals, which favor the generation of triplet oxygen over singlet oxygen and hydrogen peroxide. The study revealed that the onset potentials for oxygen production were lower for chiral CuO (1.38 V) than for achiral CuO (1.43 V), indicating a higher efficiency of the chiral CuO-coated anode in water splitting. The OER efficiency is also pH-dependent, with hydrogen peroxide often being a significant by-product. The generation of H2O2, as demonstrated with achiral electrodes (Fig. 13d), is notably reduced when using anodes composed of chiral CuO films, which also maintain lower overpotentials. Fig. 13e presents an energy scheme that rationalizes a mechanism based on the combination of surface-adsorbed OH radicals. Due to the CISS effect, charge transfer at the CuO surface results in spin polarization, thereby enhancing the production of the triplet oxygen species. While this rationale is presented in the context of recombining adsorbed OH intermediates, it can be extended to other mechanisms such as the oxide path and the metal peroxide path.110 The selectivity for oxygen production mirrors findings from earlier work using chiral molecules; however, in this study, an all-inorganic system (CuO) is employed. This system achieves a current density over 1000 times higher than that observed with organic linker molecule-coated electrodes (mA cm−2versus μA cm−2).28,109,111
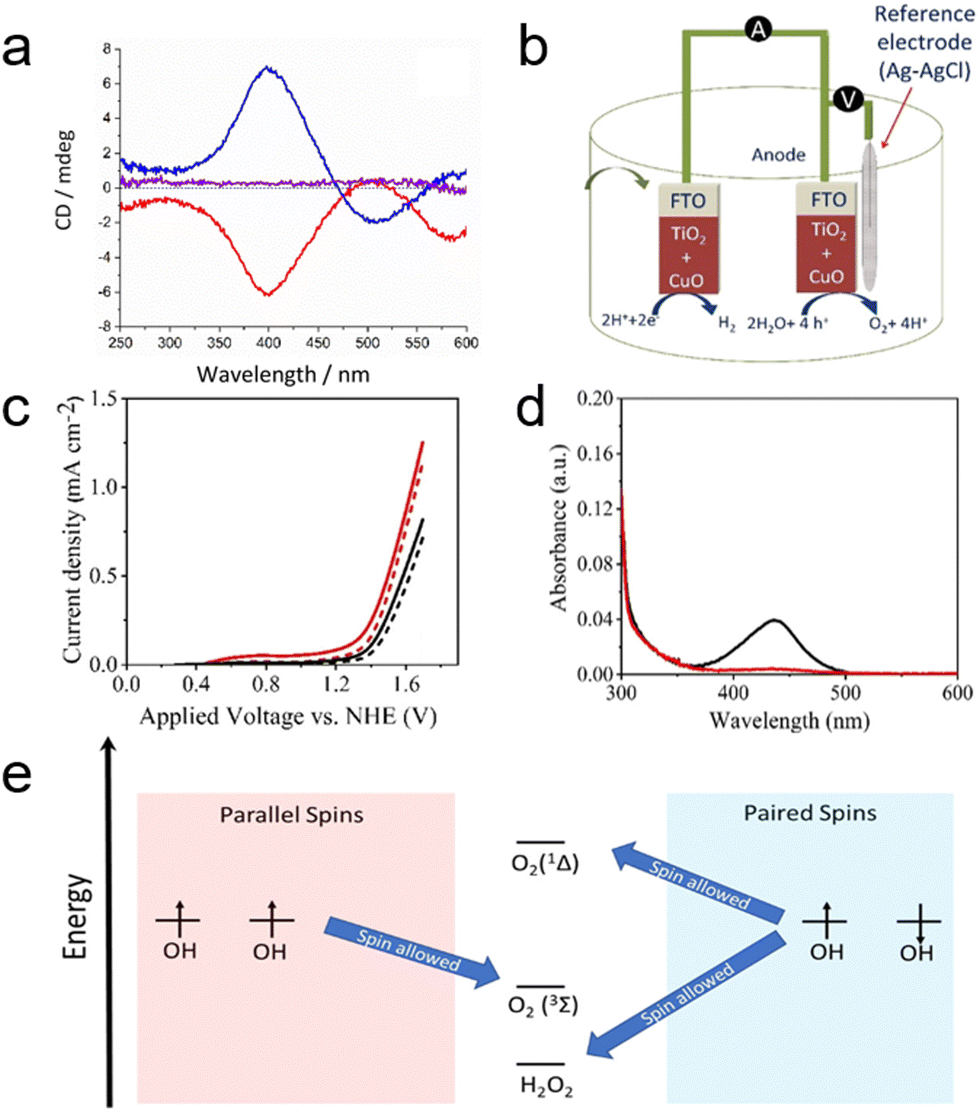 | ||
| Fig. 13 (a) Circular dichroism (CD) spectra showing 50 nm films of L-CuO (red), D-CuO (blue), and meso-CuO (purple). (b) Schematic of the electrochemical setup using electrochemically deposited CuO as both the cathode and anode. (c) Linear sweep voltammetry curves of chiral CuO (induced by L-tartaric acid, red line) and achiral CuO (induced by meso-tartaric acid, black line) electrodes in a pH 9 aqueous buffer solution under dark conditions. (d) Detection of H2O2. UV-vis absorption spectra of a 0.1 M Na2SO4 electrolyte post-titration with o-tolidine, illustrating results for chiral CuO (red line) and achiral CuO (black line). (e) Energy diagram depicting potential reaction products from hydroxyl groups on the CuO surface, highlighting the spin restriction upon recombination, which enhances selectivity against H2O2 formation when OH radical spins are aligned. Adapted with permission from ref. 109, Copyright 2019, American Chemical Society. | ||
Recently, Feng et al. explored the integration of the CISS effect with bimetallic Fe–Ni compounds, renowned for their OER catalytic efficiency, to create a chiral Fe–Ni electrocatalyst.69 They developed a chiral tartaric acid–FeNi coordination polymer (TA–FeNi CP) using an electrochemical deposition method, aiming to enhance the electrochemical OER process through the CISS effect. The chirality of tartaric acid was incorporated into the Fe–Ni sites via coordination interactions, as shown in Fig. 14a. This chiral D-TA–FeNi CP demonstrated a spin-polarized current with high spin polarization, a feature absent in its achiral DL-TA–FeNi CP counterpart. The combination of high spin polarization and good electrical conductivity rendered the chiral Fe–Ni electrocatalyst with outstanding OER activity and a remarkable long-term durability of 100 hours (Fig. 14b). They investigated the impact of chirality on the OER process by comparing the oxygen evolution activity and electrochemical behaviors of chiral and achiral TA–FeNi CP catalysts (Fig. 14c). Polarization curves showed that both chiral L- and D-TA–FeNi CP catalysts exhibited higher current densities for water oxidation than the achiral DL-FeNi CP, highlighting the positive influence of chirality on OER efficiency. The L-TA–FeNi CP catalyst, in particular, displayed low overpotentials of 205 mV and 280 mV to achieve current densities of 10 mA cm−2 and 100 mA cm−2, respectively, as illustrated in Fig. 14d. This research opens new prospects for leveraging spin control in electrochemistry to enhance oxygen evolution efficiency.
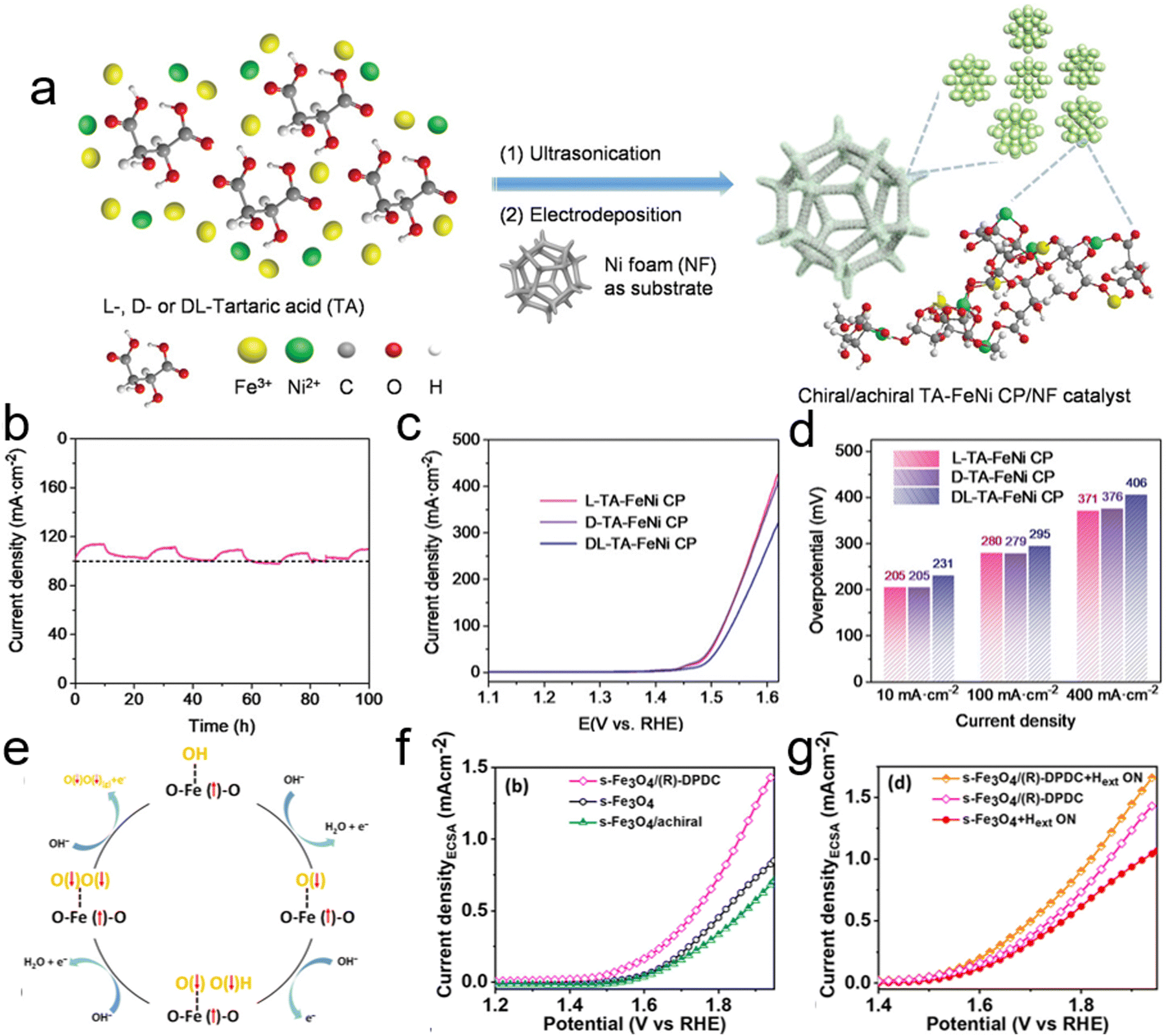 | ||
| Fig. 14 (a) Schematic showing the preparation of chiral (L- and D-) and achiral (DL-) TA–FeNi CP/NF catalysts. (b) Long-term durability test of the L-TA–FeNi CP electrocatalyst for the oxygen evolution reaction (OER), displaying periodic fluctuations in current density due to the cyclic accumulation and removal of O2 bubbles on the nanofiber (NF) during the OER. (c) Polarization curves adjusted for 80% iR compensation. (d) Comparison of overpotentials for different catalysts. Adapted with permission from ref. 69, Copyright 2023, John Wiley and Sons. (e) Schematic depicting the mechanism of the OER influenced by a magnetic field and spin selection dynamics. (f) Linear sweep voltammetry (LSV) polarization curves showing electrocatalytic activity. (g) LSV polarization curves for chiral Fe3O4 in the presence of an external magnetic field (Hext). Adapted with permission from ref. 112, Copyright 2023, American Chemical Society. | ||
Additionally, previous studies, including those conducted by Ren et al., have highlighted the potential of external magnetic fields in creating a spin-dependent environment conducive to improved OER processes.113 Further investigations have shown that combining magnetic fields with electrochemistry can more effectively facilitate spin transitions during the OER. An innovative approach involves using chiral molecules to induce spin polarization in ferrimagnetic and superparamagnetic electrocatalysts, thus enhancing the CISS effect. Nair et al. utilized ferrimagnetic and superparamagnetic Fe3O4 (f-Fe3O4 and s-Fe3O4, respectively) to provide a platform for studying the influence of magnetic properties on OER efficiency.112 These properties can be tuned without altering the chemical composition or structure of the catalysts, as depicted in Fig. 14e. By anchoring chiral molecules onto the surface of Fe3O4, they aimed to engineer total spin polarization in the catalysts through the combined impact of external magnetic fields and the CISS effect. In detail, building on their understanding of spin polarization in s-Fe3O4, they proposed a mechanism for the spin-polarized OER process. In this mechanism, Fe centers serve as the OER active sites in Fe3O4. Under an external magnetic field (Hext), the fixed spin direction at the Fe active center results in the generation of O(↓)− during the first electron transfer step, due to ferromagnetic exchange between the catalyst and the adsorbed oxygen species, following the spin angular momentum conservation principle. This process is followed by subsequent steps involving the formation of the triplet state intermediate O(↓)O(↓)H, ultimately leading to the favorable generation of triplet state O2. Their findings revealed a significant improvement, with an 89% increase in current density at 1.8 V vs. RHE and a low onset potential of 270 mV, as shown in Fig. 14f and g.
3.2. Oxygen reduction reaction
The oxygen reduction reaction (ORR) stands as a critical electrochemical process, underpinning the function and efficiency of various energy conversion systems, notably fuel cells and metal–air batteries.114 Integral to the energy landscape, especially in the context of sustainable and renewable energy sources, the role of the ORR in these devices is paramount in dictating their overall performance and efficiency.115 The mechanism of the ORR, while seemingly straightforward, involves complex electrochemical pathways that differ markedly depending on the medium–acidic or alkaline–in which the reaction occurs.116 This variability is particularly significant in terms of the byproducts generated, most notably hydrogen peroxide (H2O2), whose formation can influence not only the energy efficiency of the system but also its durability and operational stability.117 Understanding the nuances of the ORR mechanism in different environments is, therefore, essential for optimizing the design and functionality of fuel cells and related technologies, paving the way for more efficient, robust, and environmentally friendly energy solutions.In both acidic and alkaline media, the ORR is central to electrochemical energy conversion devices, with its mechanism varying significantly across these environments, particularly regarding the formation of hydrogen peroxide (H2O2) as a side product.118 In acidic media, the ORR typically follows a four-electron reduction pathway, forming water (Step (8)), but can also proceed through a less efficient two-electron pathway producing H2O2 (Step (9)).119
| O2 + 4H+ + 4e− → 2H2O | (8) |
| O2 + 2H+ + 2e− → H2O2 | (9) |
| O2 + 2H2O + 4e− → 4OH− | (10) |
| O2 + H2O + 2e− → H2O2 + OH− | (11) |
Sang et al. pioneered the hypothesis that the multi-electron reduction of diatomic oxygen (ORR) could be enhanced by utilizing spin-polarized electrons. They proposed that chiral biomolecules for inducing the CISS effect could be employed to boost ORR efficiency.121 To explore the impact of chirality on fuel cell materials, they focused on platinum (Pt), a quintessential electrocatalyst for oxygen reduction. Chiral Pt nanoparticles (NPs) were synthesized using L-cysteine, while their achiral counterparts were produced using a racemic mixture of L- and D-cysteine. The study revealed that chiral Pt NPs exhibit an onset potential of 0.92 V at −0.1 mA cm−2, modestly surpassing the commercial Pt/C by 30 mV and significantly outperforming the achiral Pt NPs by 80 mV (Fig. 15b and c).122 Furthermore, chiral Pt NPs also showed a higher half-wave potential (0.87 V) compared to achiral NPs (0.80 V) and Pt/C (0.84 V), suggesting that chirality enhances the ORR reaction rate, even in electrodes with high spin–orbit coupling.
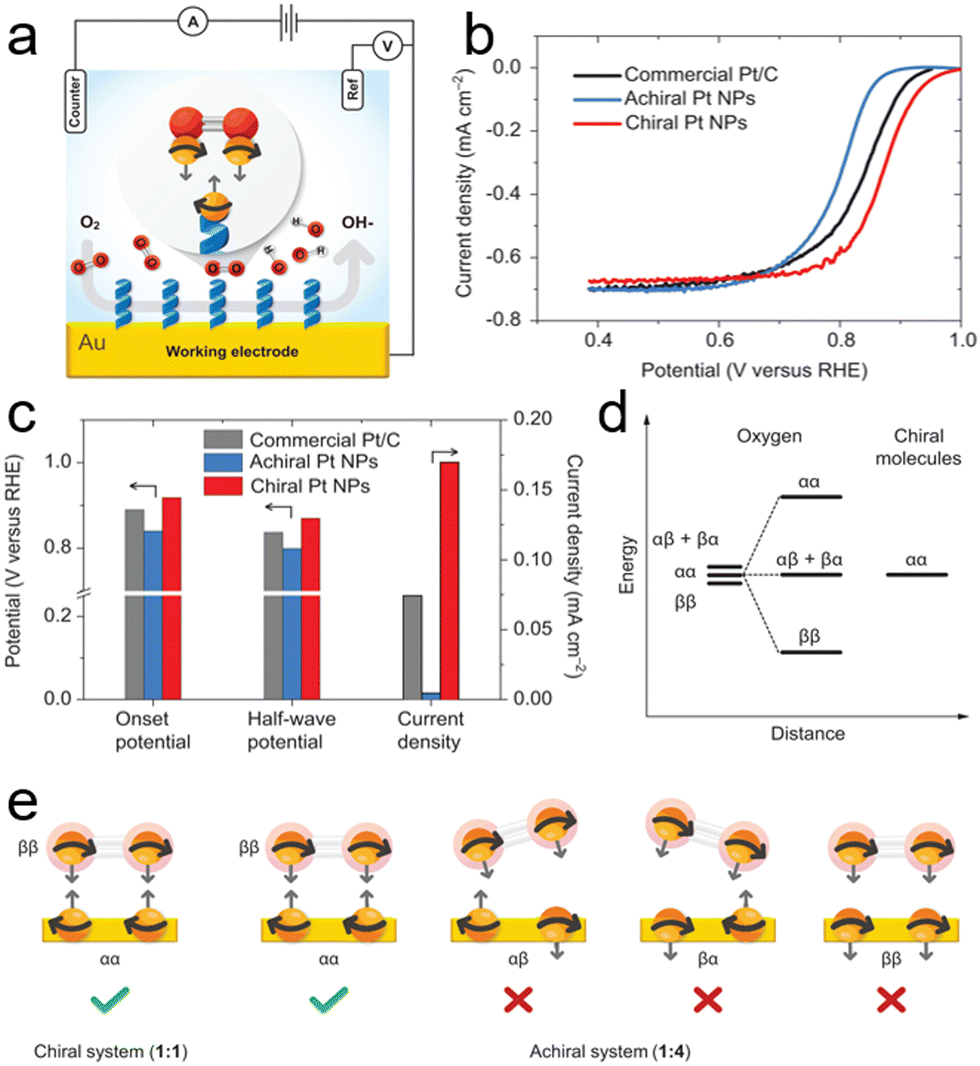 | ||
| Fig. 15 (a) Schematic of the electrochemical setup featuring a gold working electrode coated with a self-assembled monolayer (SAM) of either chiral or achiral molecules. (b) Rotating disk electrode (RDE) measurements showing ORR polarization curves for chiral and achiral Pt nanoparticles (NPs), alongside commercial Pt/C catalysts, in air-saturated 0.1 M KOH at 1500 rpm with a sweep rate of 5 mV s−1. (c) Comparative analysis of onset potentials (defined at −0.1 mA cm−2), half-wave potentials (E1/2), and current densities at 0.9 V (versus RHE). (d) Illustration of the spin state separation of triplet oxygen upon interaction with spin-polarized electrons from the chiral molecules. (e) Depiction of possible spin states in chiral (left) and achiral (right) systems. In achiral systems, the two electrons can adopt one of the four possible configurations, only one of which leads to reaction. In chiral systems, there is a single possible configuration where electrons are strongly coupled to the molecular frame, making it the only configuration conducive to reaction. Adapted with permission from ref. 121, Copyright 2022, National Academy of Sciences. | ||
The oxygen reduction process requires at least two electrons in the initial state to form either O22− or HO2−.123 The crucial factor is the projection of the O2 molecule's spin onto the chiral axis as it approaches the electrode. With three possible spin states for O2's unpaired electrons, αα, ββ, and 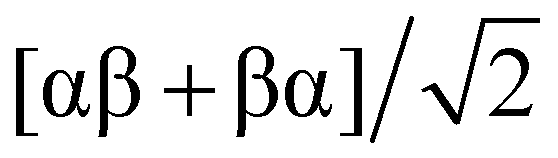 , chiral NPs ensure that the electrons injected from the chiral ligands have identical spin projections on the molecular axis, effectively lowering the reaction barrier due to entropic (spin statistics) and enthalpic (spin-exchange interaction) factors. As illustrated in Fig. 15d, the interaction of O2 with the chiral monolayer leads to a reduction in the enthalpic barrier and a more efficient spin injection into the oxygen system, facilitated by spin-exchange effects. This interaction aligns the spin-state of O2 with the chiral film, reducing the entropic contributions to the free energy barrier.123
, chiral NPs ensure that the electrons injected from the chiral ligands have identical spin projections on the molecular axis, effectively lowering the reaction barrier due to entropic (spin statistics) and enthalpic (spin-exchange interaction) factors. As illustrated in Fig. 15d, the interaction of O2 with the chiral monolayer leads to a reduction in the enthalpic barrier and a more efficient spin injection into the oxygen system, facilitated by spin-exchange effects. This interaction aligns the spin-state of O2 with the chiral film, reducing the entropic contributions to the free energy barrier.123
In contrast, the achiral film presents four potential spin states (αα, ββ, αβ, and βα), but only one of these enables efficient electron transfer to the oxygen, resulting in a lower reaction probability (Fig. 15e). Additionally, the lack of spin coupling in the achiral monolayers means they do not influence the spin states of the oxygen, failing to reduce the enthalpic barrier. This comparative analysis underscores the efficacy of chiral films in enhancing the ORR by manipulating electron spin states, as opposed to the limitations observed with achiral films.123
Recently, Scarpetta-Pizo et al. made a significant contribution to the field of electrocatalysis by demonstrating electron spin-dependent catalysis for the ORR using iron phthalocyanine (FePc), a well-known molecular catalyst for this reaction.124 In their innovative approach, they developed chiro-self-assembled FePc systems (CSAFePc) by self-assembling FePc onto a gold electrode surface using chiral peptides (L and D enantiomers). These chiral peptides served as spin filter axial ligands for FePc, significantly influencing the kinetics and thermodynamics of the ORR.
One of the key findings of their study was the impact of the peptides’ handedness and length on the ORR in CSAFePc. They observed that these factors could shift the onset potential up to 1.01 V vs. RHE in an alkaline medium, approaching the reversible potential of the O2/H2O couple, as illustrated in Fig. 16a. This indicates the substantial role of chirality and molecular structure in optimizing ORR catalysis.125
 | ||
| Fig. 16 (a) Linear sweep voltammetry (LSV) results for the oxygen reduction reaction (ORR) on chiro-self-assembled FePc systems (CSAFePcs): Au(111)/(1–4)L/FePc and Au(111)/(1–4)D/FePc systems, conducted in 0.1 M NaOH saturated with O2. The yellow line represents the unmodified Au(111) electrode, the blue line indicates Au(111)/FePc, the green line shows Au(111)/(1–4)D/FePc systems, and the red line shows Au(111)/(1–4)L/FePc. (b) General schematic of the ORR mechanism in chiro-self-assembled FePc systems (CSAFePcs). (c) Model of electronic spin states for the Fe–O2 adduct series, based on density functional theory (DFT) calculations, used to explain the chiral induced spin selectivity (CISS) effect observed in the differences in electrocatalytic activity for the ORR between enantiomeric peptide pairs in CSAFePc systems. In this model, α (yellow ball) and β (blue ball) represent the spin-up and spin-down states of the first electron transferred through the spin-filtering peptide-FePc–O2 circuit, leading to the charge transfer intermediary: peptide-FePc–O2− (rate-determining step). Adapted with permission from ref. 124, Copyright 2024, John Wiley and Sons. | ||
Scarpetta-Pizo et al. proposed a common rate-determining step (rds) for the ORR mechanism across the CSAFePcs, involving the formation of a peptide-FePc–O2 adduct with a concurrent electron transfer to form a charge-transfer intermediary (CTI) (Fig. 16b).126,127 This hypothesis was supported by a consistent Tafel slope observed across all CSAFePcs (≈−30 mV dec−1).124 A critical aspect of the ORR activation energy in this context is the spin inversion needed to transition the paramagnetic O2 from its spin-state (αα) to the diamagnetic states of the ORR products.67
Fig. 16c presents an electronic model explaining the spin-state dynamics of the L/D-FePc–O2 adduct series.128–130 The model, supported by computed Fe spin-states and spin-density changes, illustrates how the transferred electron spins interact in the first electron transfer for the ORR to form the CTI. Notably, the model reveals that the electronic effect in 3D systems favors a triplet-state in the Fe-center, conducive to a closed-shell singlet state in O2, which is consistent with the high ORR activity observed experimentally. In contrast, different electronic behaviors are noted for the 4L/D-FePc–O2 adducts, where both exhibit a total singlet-state, indicating a spin-polarized coupling between local triplet states of Fe and O2.
Thus, the CISS effect in the 1L/D-CSFePcs promotes one kind of electron spin-filtering, while in 2-4L/D-CSFePcs, it encourages the opposite.124 This nuanced understanding of electron spin dynamics, facilitated by the CISS effect, opens new avenues for enhancing ORR efficiency and selectivity through spin control.
3.3. Hydrogen evolution reaction
The hydrogen evolution reaction (HER) is a critical electrochemical process central to various green energy technologies, notably in water electrolysis and fuel cells, where it facilitates hydrogen gas production.131 This reaction occurs through a two-step mechanism that varies depending on whether it takes place in acidic or alkaline media.In acidic media, the HER begins with the Volmer step, where a proton (H*) from the electrolyte gains an electron (e−) from the electrode to form adsorbed hydrogen (Hads) on the surface of a catalyst, described by the reaction H+ + e− → Hads.132 This is followed by either the Heyrovsky step, where another proton reacts with Hads and an electron to release hydrogen gas (Hads + H+ + e− → H2), or the Tafel step, where two Hads atoms combine to form hydrogen gas (2Hads → H2).133
In alkaline media, the mechanism is similar but starts with the reduction of a water molecule instead of a proton. In the Volmer step, water (H2O) is reduced, releasing a hydroxide ion (OH−) and forming Hads (H2O + e− → Hads + OH−).2,134,135 The subsequent steps, Heyrovsky or Tafel, are akin to those in acidic media, involving the transformation of Hads into molecular hydrogen (Hads + H2O + e− → H2 + OH− for Heyrovsky and 2Hads → H2 for Tafel).136
While the application of the CISS effect in enhancing the HER has not been extensively explored, recent research conducted by Bhartiya et al. has shown promising results in this direction, albeit not directly through CISS but via the increased surface area provided by chirality.137 In their study, they utilized a chiral molecule-modified Au–Ni bilayer thin film electrode, coated with a self-assembled monolayer of chiral L-cysteine molecules (Ni/Au/Cys). As shown in Fig. 17a it can be inferred that the onset potential for the HER in the case of the Ni/Au/Cysteine sample (approximately 0.16 V) has been significantly reduced compared to bare Ni, Ni/Au, or Ni/Cys thin films. The absolute current density value (∼153 mA cm−2 at −0.6 V) achieved for the HER shows a dramatic 11-fold increase in cathodic current compared to the Ni/Au or Ni/Cys samples (Fig. 17b). Additionally, there is a notable reduction in overpotential, with the Ni/Au/Cys sample exhibiting a 320 mV and 240 mV decrease compared to Ni and Ni/Cys, respectively, to achieve a current density of 10 mA cm−2 (Fig. 17c).
 | ||
| Fig. 17 (a) Linear sweep voltammetry (LSV) curves, (b) current density at −0.6 V, (c) overpotential (η, in mV), and (d) Tafel slope measurements for the hydrogen evolution reaction (HER) for Ni, Ni/Cys (cysteine), Ni/Au, and Ni/Au/Cys films at pH 11 and room temperature, using an Ag wire as the reference, in 0.1 M KOH at a sweep rate of 10 mV s−1. Adapted with permission from ref. 137, Copyright 2022, Elsevier. | ||
The mechanism behind this enhanced HER activity is attributed to hydrogen adsorption on the large effective surface area, filled with electroactive sites, provided by the adsorbed chiral molecule on the Au thin film. The hydrogen binding sites are significantly increased due to the symmetrical orbital overlap between the 1s orbital of the hydrogen atom and the 3p orbitals of the sulfur atom in cysteine. This extensive hydrogen adsorption creates numerous sites for electron transfer, leading to a lower overpotential and a higher current density for the HER process.137 The research conducted by Bhartiya et al. opens new avenues for enhancing the HER through the strategic use of chiral molecules to increase the effective surface area of electrodes, highlighting the potential of chirality in electrocatalysis beyond the direct application of the CISS effect.
4. Future research directions
The chirality induced spin selectivity (CISS) effect introduces innovative pathways for electrocatalytic reactions, particularly those involving spin-sensitive intermediates.138 This phenomenon is notably significant in the realm of clean energy conversion, impacting key reactions such as the nitrogen reduction reaction (NRR) and the carbon dioxide reduction reaction (CO2RR). Furthermore, the CISS effect holds considerable promise in the energy storage sector, especially in the development and optimization of metal–air batteries. Through leveraging spin polarization, the CISS effect has the potential to enhance the efficiency and selectivity of these crucial electrocatalytic processes, offering new strategies for advancing sustainable energy technologies.While significant research has been conducted on enhancing catalytic activity through the application of magnetic fields, a major drawback of this approach is the requirement for external energy inputs. However, chiral electrocatalysts present a groundbreaking alternative by enabling the synthesis of intrinsically polarized catalysts. This intrinsic polarization allows for modifications in catalytic activity and selectivity without an external magnetic field. By shifting the focus from traditional magnetic field-based electrocatalyst enhancement to chiral electrocatalysts, a new research field is opened.139 This shift not only mitigates the dependency on external energy but also opens up novel avenues for research on enhancing electrocatalytic processes through chirality (Scheme 3).
 | ||
| Scheme 3 Depiction of spin polarization mechanisms in electrocatalysts, utilizing both magnetic fields and chiral structures, accompanied by a forward-looking analysis of potential applications in energy conversion and storage technologies. | ||
4.1. Energy conversion: NRR and CO2RR
The electrocatalytic NRR process is considered as one of the promising green technologies because it can be combined with renewable energy sources like wind and solar, presenting an environmentally friendly method for synthesizing ammonia. Yet, so far, several challenges have hindered the practical application of this process.140 The reaction mechanism itself is complex, necessitating multiple proton and electron transfers to activate the inert N2 molecule and convert it into NH3. The success of this conversion heavily relies on the capabilities of the electrocatalyst, which must efficiently adsorb and activate N2, facilitate the requisite electron transfer, and stabilize the nitrogen-containing intermediates that form during the reaction.144,145
Despite the apparent advantages, the electrocatalytic NRR faces significant obstacles, including low selectivity due to the competing hydrogen evolution reaction (HER),146 the high activation energy required to break N2's triple bond (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N),147 rapid deactivation of catalysts in aqueous environments,148 and the difficulty in identifying suitable electrocatalysts that offer high activity, selectivity, and stability. Addressing these challenges requires a holistic approach that encompasses the development of novel electrocatalysts through advancements in materials science, as well as a deep understanding of the reaction mechanisms to optimize conditions and suppress unwanted side reactions.142,149
N),147 rapid deactivation of catalysts in aqueous environments,148 and the difficulty in identifying suitable electrocatalysts that offer high activity, selectivity, and stability. Addressing these challenges requires a holistic approach that encompasses the development of novel electrocatalysts through advancements in materials science, as well as a deep understanding of the reaction mechanisms to optimize conditions and suppress unwanted side reactions.142,149
One of the groundbreaking aspects of using spin polarization in the NRR is its compatibility with various catalytic materials, including those that are not inherently magnetic. Advances in materials science and surface chemistry have enabled the design of catalysts that can induce spin polarization in adsorbed nitrogen molecules, thereby facilitating their reduction to ammonia. Moreover, this strategy opens new avenues for the development of catalysts that are selective for the NRR, offering a pathway to bypass the limitations of current catalysts that suffer from poor stability and low selectivity.149–151
Building on earlier work, Li et al. demonstrated that catalysts exhibiting a highly spin-polarized state could enhance N2 adsorption and activate its robust NN triple bond during the NRR.149 Extending this research, Zhang et al. revealed that highly spin-polarized Rh materials, combined with substrates like carbon nanotubes (CNTs), exhibit exceptional N2 adsorption and reduction capabilities due to the unique charge structure formed from Rh nanoparticles’ recombination with the substrate.150 Their findings indicated that the spin state and charge density on Rh atoms significantly influence N2 adsorption strength and the activation of the NN bond, with a high spin state facilitating electron transfer to the N2 molecule, thereby boosting NRR activity.
As illustrated in Fig. 18a, reducing Rh from bulk to clusters enhances the spin magnetic moment, indicating increased spin polarization, particularly at the edges/surfaces of Rh clusters. This size effect correlates with a high spin density and positive charge density at the Rh site, leading to strong N2 adsorption and effective NN bond weakening.152 Notably, spin densities at specific sites were found to be higher than others, promoting stronger N2 adsorption and further weakening of the NN bond.149 Free energy diagrams of potential intermediates during the NRR on Rh/graphene and bulk Rh surfaces show that certain pathways, particularly those at specific sites, are more energetically favourable, contributing to a lower overpotential and enhanced NRR electrocatalytic activity (Fig. 18b).150
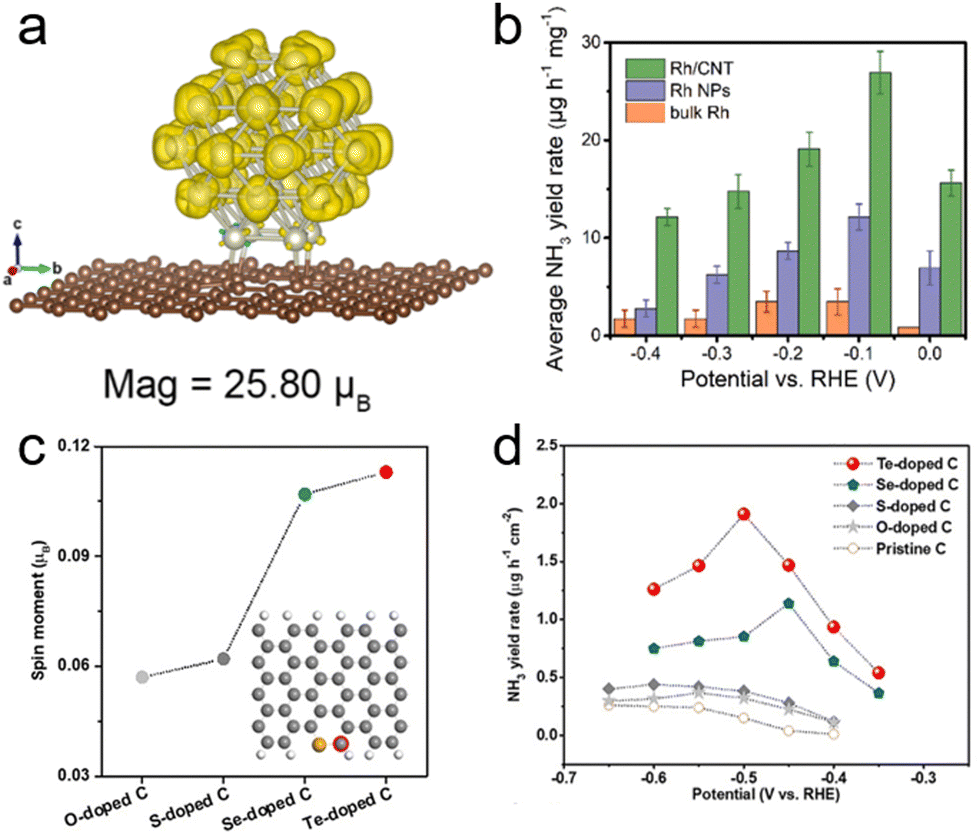 | ||
| Fig. 18 (a) Spin-resolved density visualization for Rh/graphene, with an iso-surface value set at 0.01 e Å−2. Areas of charge accumulation are highlighted in yellow. (b) Ammonia production rates using Rh/CNT, Rh nanoparticles, and bulk Rh as nitrogen reduction reaction (NRR) catalysts, compared across different potentials in phosphate-buffered saline (PBS). Adapted with permission from ref. 150, Copyright 2021, Elsevier. (c) Spin moment of a carbon atom adjacent to a dopant, shown in the inset. (d) Faradaic efficiency and ammonia yield rates for heteroatom-doped carbon catalysts at varying potentials. Adapted with permission from ref. 153, Copyright 2022, Springer Nature. | ||
Expanding on these findings, Yang et al. revealed that spin polarization notably advances the first protonation step to form NNH*, thereby augmenting the overall reaction efficiency.151 Their studies showed that heteroatom doping-induced charge accumulation promotes N2 adsorption on carbon atoms, while spin polarization enhances the potential-determining step of the first protonation to form NNH*.149Fig. 18c and d delineate the impact of the metal dopant on the spin moment of adjacent carbon atoms and its correlation with NH3 production.154 Furthermore, Cao et al. discovered a novel promotional effect in catalytic ammonia synthesis, confined to magnetic catalysts, which significantly lowers the activation energy, thus opening avenues for the discovery of new ammonia synthesis catalysts.153
The emerging research into chiral electrocatalysts for the NRR underscores a highly promising avenue in the field of electrocatalysis. Utilizing chiral molecules to induce spin polarization not only advances the development of more efficient and selective processes for ammonia synthesis but also represents a significant shift from traditional methods like the Haber–Bosch process, which is energy-intensive and environmentally taxing. The integration of the CISS effect in NRR catalysts leverages the unique properties of spin polarization to optimize electron interactions at the catalytic interface, enhancing both the adsorption and activation of nitrogen molecules.
These advancements highlight the pivotal role of spin polarization, influenced by the size and substrate effects, in lowering the Gibbs free energy for crucial steps in the NRR. The focus on harnessing the CISS effect to control and direct reaction pathways more effectively suggests a promising pathway for the development of groundbreaking ammonia synthesis technologies. This strategy not only improves the selectivity and activity of the catalysts under ambient conditions but also aligns with broader sustainability goals by enabling the use of renewable energy sources.
The potential of chiral electrocatalysts in the NRR has yet to be fully realized, inviting further exploration into how spin dynamics can be optimized to enhance electrocatalytic processes. By emphasizing spin polarization and integrating innovative chiral structures within catalyst designs, researchers open new avenues for advancing electrocatalytic strategies and achieving more efficient catalysts for ammonia synthesis. This optimistic outlook highlights the transformative potential of chiral electrocatalysts in the field, suggesting their crucial role in meeting global energy and environmental challenges.
Despite its promising outlook, the CO2RR is beset by several challenges that impede its practical application. The inherent stability of CO2 molecules renders the reaction kinetics sluggish, necessitating the development of catalysts capable of efficiently activating CO2.155 These catalysts must not only minimize the occurrence of the competing HER but also ensure high selectivity towards the desired reduction products. Achieving a high degree of selectivity, alongside maintaining catalyst activity and stability over prolonged operational periods, remains a formidable challenge. This is further complicated by the reaction's complexity, which involves multiple steps of proton and electron transfer processes, making the optimization of catalyst surface properties and reaction conditions critical for success.
In response to these challenges, a novel strategy leveraging spin polarization has recently emerged, offering a fresh perspective on enhancing CO2RR efficiency. This approach, rooted in the principles of spin chemistry and quantum mechanics, involves manipulating the spin states of electrons involved in the CO2 reduction process. Focusing on the conversion to formic acid, the importance of the singlet radical pair configuration ([CO2−˙↑⋯H˙↓]1) in the CO2RR has been highlighted over the triplet radical pair configuration ([CO2−˙↑⋯H˙↑]3).156,157 The application of a magnetic field has been shown to facilitate the transition of spin states from triplet to singlet, enhancing the CO2RR process.158 As shown in Fig. 19, Pan et al. have pioneered the use of Sn electrodes under the influence of a 0.9 T magnetic field to demonstrate the magneto current (MC) effect in the CO2RR, where an increase in reduction current, Faraday efficiency, and formic acid yield was observed.159 This magnetic field is posited to convert triplet states into singlet states, thereby boosting formic acid production. This seminal work suggests a novel pathway for enhancing the efficiency of the CO2RR (Fig. 19c).
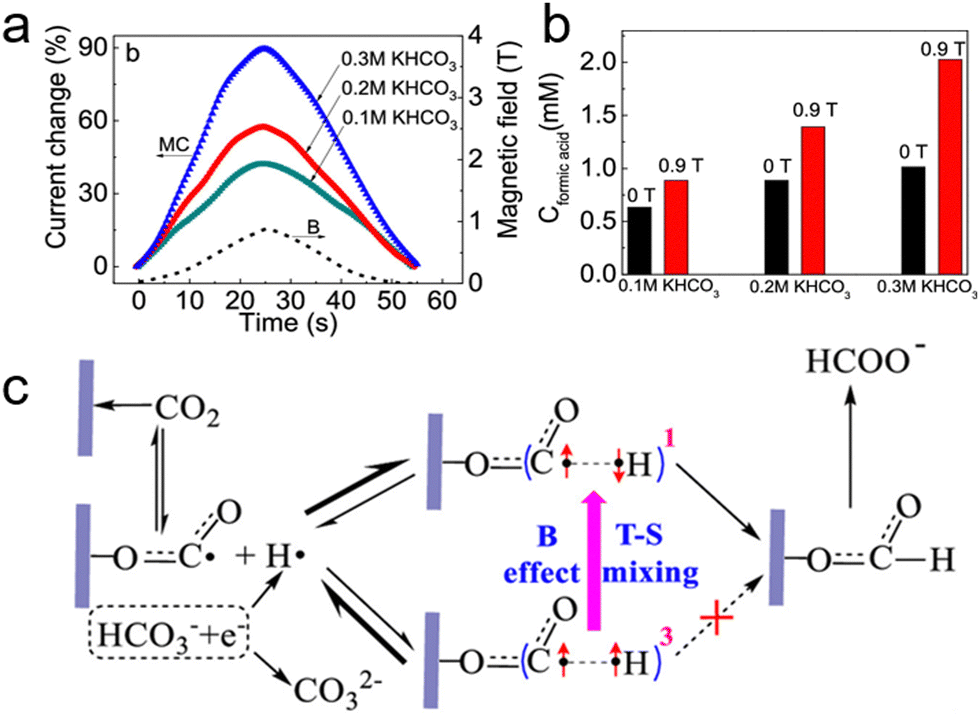 | ||
| Fig. 19 (a) Variation in current as a function of time under a triangular wave magnetic field at −1.7 V (vs. Ag/AgCl), at various concentrations of KHCO3 electrolyte saturated with CO2. (b) Dependence of formate production on the magnetic field at an applied electrode potential of −1.7 V (vs. Ag/AgCl) in 0.1, 0.2, and 0.3 M KHCO3 electrolytes. (c) Illustration of the proposed generation mechanism for magneto-current driven by electrocatalytic CO2 reduction. Adapted with permission from ref. 159, Copyright 2020, American Chemical Society. | ||
Beyond magnetic fields, the CISS effect offers an alternative for achieving efficient spin polarization, even in diamagnetic materials like Cu, which is known for converting CO2 into valuable hydrocarbon products with high faradaic efficiencies.160,161 The exploration of chiral CO2RR electrodes, which combine active catalysts with chiral molecules, could pave the way for development of electrodes that are highly selective and active toward the production of multi-carbon products. This development of hybrid electrodes utilizing the CISS effect represents a pivotal advancement in the selectivity and activity of electrocatalysts. By leveraging the intrinsic spin polarization properties introduced by chiral molecules, these electrodes promise to enhance the efficiency of CO2 utilization significantly. This approach could potentially transform the landscape of CO2 reduction technology, making it a more viable and efficient method for synthesizing hydrocarbon fuels and chemicals. Such advancements underscore the transformative potential of chiral electrocatalysts in improving the economic and environmental viability of CO2 reduction strategies, paving the way for more sustainable and efficient green chemistry technologies.
4.2. Energy conversion: metal–air batteries
Future energy storage requires the development of high-density and low-cost batteries.162,163 Metal–air batteries are some of the most promising battery chemistries that could satisfy these requirements.164 Central to their operation are two critical electrochemical reactions: the OER and the ORR, which play pivotal roles during the charge and discharge cycles, respectively.165 During the discharge phase, the ORR takes place at the cathode, where oxygen from the air is reduced, reacting with metal ions (such as lithium or sodium) to form metal peroxides, releasing energy in the process.166 This reaction is crucial for the battery's energy generation, as it directly influences the efficiency and capacity of the battery by determining how effectively oxygen is incorporated into the electrochemical process. Upon charging, the OER occurs, reversing the discharge process. Here, the metal peroxides formed during discharge are oxidized, releasing oxygen back into the atmosphere and regenerating the metal ions for the anode.167 The efficiency of the OER is vital for the rechargeability of metal–air batteries, impacting the energy required to recharge the battery and the longevity of the battery's life cycle.Metal–air batteries, across various chemistries, encounter several challenges that impact their efficiency and reversibility to different extents.168 A primary issue in all metal–O2 batteries is the occurrence of parasitic reactions at the positive electrode, significantly affecting the redox system's reversible operation and the overall battery performance. These issues are particularly prevalent in alkaline metal–O2 systems (involving metals like lithium and sodium), where a recurring pattern of degradation mechanisms has been identified. These mechanisms include: (a) nucleophilic attacks by reduced reactive oxygen species such as MeO2, O2−, MeO2−, and O22−,169,170 (b) hydrogen peroxide formation initiated by protons or water molecules,171,172 and (c) the heightened reactivity of singlet oxygen molecules produced through the oxidation of peroxides or the chemical disproportionation of superoxide species in the presence of weak Lewis acids.173,174 The generation and release of singlet oxygen in these systems have raised significant concerns, challenging the understanding of the reactions' thermodynamics and kinetics. Current insights into the reactive mechanisms associated with singlet oxygen, especially for metals other than lithium, remain incomplete and inadequately explained, highlighting a gap in the comprehensive understanding of these processes.175
In Li–O2 batteries, combating the detrimental effects of aggressive singlet oxygen (1O2) is crucial for enhancing performance and durability, as detailed in Fig. 20a.176 Two primary strategies emerge for mitigating 1O2: using traps and quenchers (Fig. 20b). Traps like 9,10-dimethylanthracene (DMA) engage in irreversible chemical reactions with 1O2, effectively capturing it, a process often monitored through UV-vis spectroscopy to assess the reduction of DMA by 1O2.177,178 Alternatively, quenchers deactivate 1O2 through a reversible physical process known as intersystem crossing (ISC), involving a transient charge transfer that facilitates the transition of 1O2 from its reactive excited state to a more stable triplet oxygen (3O2) state.179 This transition, highlighted in Fig. 20b, is characterized by a radiation less shift between electronic states of differing multiplicities, thereby diminishing the fluorescence intensity associated with 1O2 decay. Among quenchers, 1,4-diazabicyclo[2.2.2]octane (DABCO) stands out for its efficacy and is reported to reduce 1O2 byproducts by up to 70%. However, its narrow electrochemical potential window (2–3.6 V) limits its broader application in Li–O2 batteries.174 To circumvent this, derivatives known as “DABCOniums” have been developed to extend the usable potential window up to 4.2 V versus Li+/Li, although their quenching capability falls short compared to DABCO.180 This necessitates the exploration of quenchers combining an expansive potential window with robust quenching efficacy. Strategies under consideration include the incorporation of amines, sulfides,181 organometallic compounds,182 or antioxidant materials.183 Yet, enhancing quenching speed often compromises anodic stability, indicating a need for innovative solutions to balance these properties and effectively suppress singlet oxygen within Li–O2 batteries.184
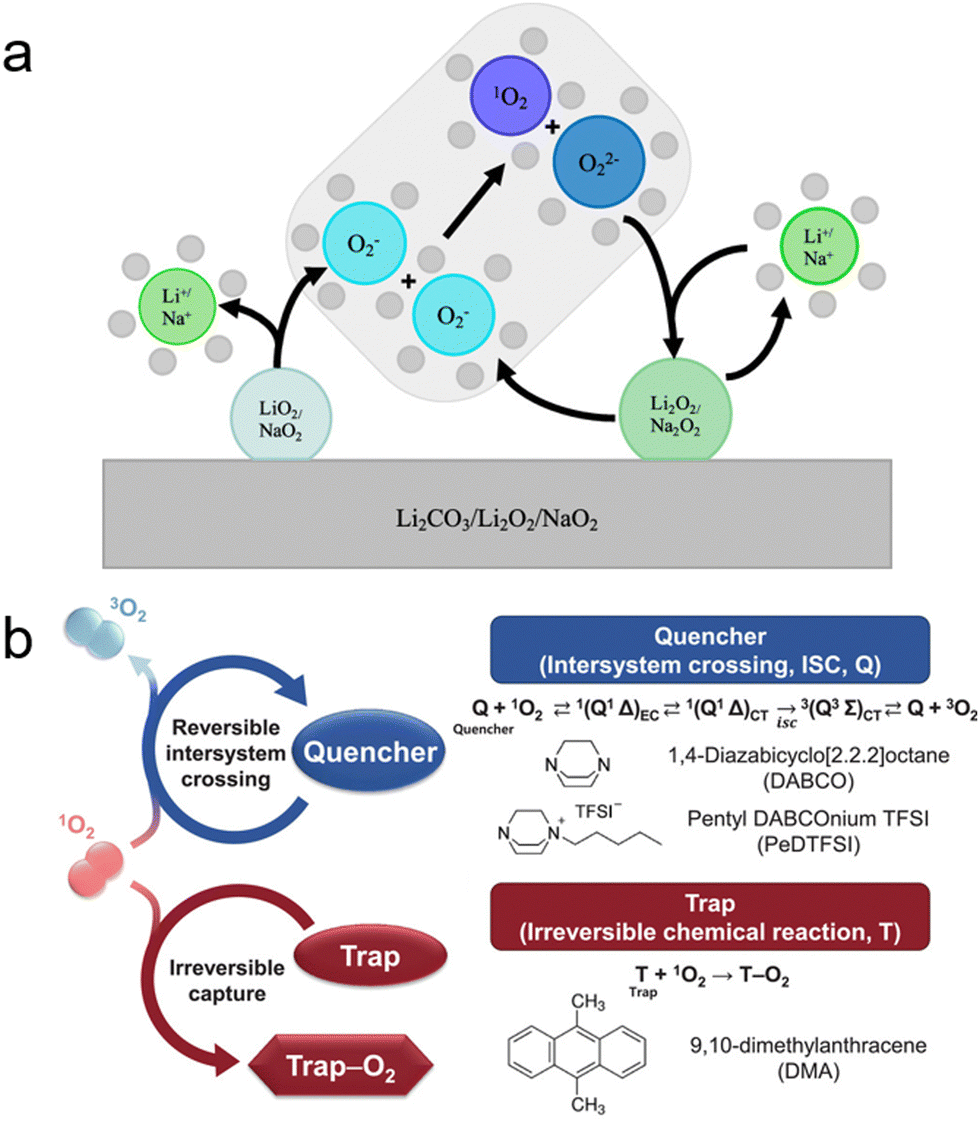 | ||
| Fig. 20 (a) Schematic diagram illustrating the role of singlet oxygen in metal–air batteries. Adapted with permission from ref. 168, Copyright 2020, American Chemical Society. (b) Schematic detailing the working principles of the quencher and the 1O2 trap in metal–air batteries, including reaction mechanisms of the quencher (represented by DABCO and DABCOnium) and the trap (represented by DMA). Adapted with permission from ref. 176, Copyright 2021, John Wiley and Sons. | ||
The application of magnetic fields as a means to energetically influence processes across physical spaces has garnered increased interest, particularly for its potential to enhance metal–air battery performance. This enhancement comes through the promotion of mass transfer, acceleration of charge transfer, and improvement of electrocatalytic capabilities, all achieved via the spin selectivity effect.185 Recent studies have shown that the application of an external magnetic field can markedly affect the spin selectivity observed in catalytic reactions. This includes the ability to interconvert two distinct spin states and facilitate the spin flip of reaction intermediates. The nature of the catalytic reaction's end-products is closely tied to these spin states, which can be dynamically altered in the presence of a magnetic field. Moreover, the spin states of intermediates, when in proximity to the electrocatalyst surface, can be modified by the applied magnetic field, leading to a preferential reaction pathway and thereby enhancing the chemical efficiency of the process.186 This relationship between catalytic activity, microstructure, and the spin effects introduced by electrochemistry underscores the profound impact of magnetic fields. By inducing spin selectivity, magnetic fields effectively navigate the reaction towards the most efficient pathway, either by restricting or permitting specific spin states, as illustrated in Fig. 21a.
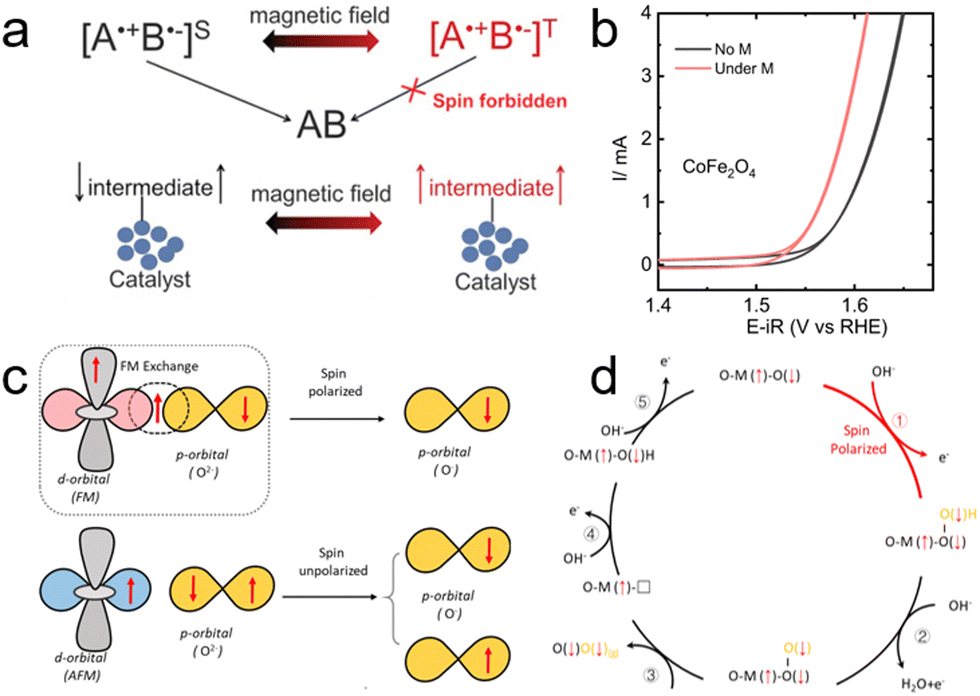 | ||
| Fig. 21 (a) Illustrations showing the effects of magnetic fields on the final product and the electrocatalytic reaction pathways. Adapted with permission from ref. 185, Copyright 2023, John Wiley and Sons. (b) Cyclic voltammetry (CV) curves of CoFe2O4 in O2-saturated 1 M KOH at a scan rate of 10 mV s−1, performed with and without an applied constant magnetic field. (c) Schematic of the spin-exchange mechanism in the oxygen evolution reaction (OER). This mechanism illustrates how the first electron transfer step is facilitated by spin polarization via ferromagnetic exchange (QSEI), leading to reduced electronic repulsion and a fixed spin direction for the adsorbed oxygen species. (d) Diagram detailing the spin-polarization mechanisms in the OER, focusing on the reaction step from O* + OH− to *OOH + e−. Adapted with permission from ref. 113, Copyright 2021, Springer Nature. | ||
Recent advancements underscore the significant impact of spin polarization on enhancing the efficiency of the OER, as detailed in a study published by Ren et al.113 This research highlights how electron spin polarization can markedly improve the catalytic activity associated with the OER (Fig. 21b). Specifically, oxygen produced during the OER possesses a paramagnetic triplet state, characterized by two unpaired electrons with parallel spins occupying the π orbital, in contrast to the diamagnetic singlet state of the reactants (OH−/H2O), where electrons are paired.181 By facilitating the alignment of electron spins in parallel, the efficiency of the OER can be significantly enhanced. Furthermore, the use of magnetic catalysts, which feature spin-polarized electrons, contributes to an improvement in OER dynamics (Fig. 21c and d).113 This enhancement is attributed to the quantum spin exchange mechanisms, offering a nuanced approach for optimizing the reaction pathway and efficiency.
In the realm of advancing battery technology, particularly for metal–air batteries, a series of studies have illuminated a path beyond the traditional use of magnetic fields for influencing reaction dynamics. The CISS effect emerges as a groundbreaking alternative, offering a nuanced approach for achieving efficient spin polarization. This innovative mechanism holds the potential to significantly mitigate the challenges posed by singlet oxygen, a notorious factor in the degradation and reduced efficiency of metal–air batteries. By leveraging the CISS effect, researchers envision a strategy that not only suppresses the detrimental impacts of singlet oxygen but also enhances the overall electrochemical performance of these batteries. This approach capitalizes on the intrinsic properties of chiral molecules to induce spin polarization, thereby steering electrocatalytic reactions towards greater efficiency and stability. The potential of the CISS effect in this context marks a promising frontier in the development of advanced battery systems, aiming to optimize energy storage solutions while addressing critical sustainability challenges.
5. Conclusions
The exploration of chirality and the CISS effect in electrocatalysis opens new horizons for enhancing the performance of critical electrochemical reactions fundamental to energy conversion and storage.187 Our review has systematically presented the CISS effect as a novel paradigm, capable of overcoming the inherent limitations of traditional electrocatalysis through the selective polarization of electron spins. By detailing the mechanisms through which the CISS effect can be induced and measured and by showcasing its application in key electrocatalytic processes such as the OER, ORR, and HER, this work lays the groundwork for future innovations in chiral electrocatalysis. Moreover, the potential of the CISS effect to revolutionize NRR and CO2RR processes, alongside its capability to enhance the efficiency and lifespan of metal–air batteries, marks a significant stride towards the development of more sustainable and efficient energy systems. As we look to the future, it is clear that the integration of the CISS effect into electrocatalytic designs not only offers a pathway to surmount existing challenges but also aligns with the broader scientific endeavor to harness renewable energy sources more effectively. The journey of incorporating chirality into electrocatalysis is just beginning, promising a future where energy technologies are both highly efficient and environmentally benign.Author contributions
D. H. Kim, J. Kim, and Z. Lin supervised the overall project. K. Chae and N. A. R. Che Mohamad drafted the outline and wrote the manuscript. J. Kim and D.-I. Won assisted in the development of a detailed strategy for organizing the manuscript. K. Chae and N. A. R. Che Mohamad contributed equally to this work. All the authors have read and approved the final manuscript.Data availability
No primary research results, software or codes have been included and no new data have been generated or analysed as part of this review. Further information about this review is available upon request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (2020R1A2C3003958), by the Basic Science Research Program (Priority Research Institute) through the NRF of Korea funded by the Ministry of Education (2021R1A6A1A10039823), and by the Korea Basic Science Institute (National Research Facilities and Equipment Center) grant funded by the Ministry of Education (2020R1A6C101B194). J. Kim acknowledges the financial support from the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. RS-2023-00212105). Z. Lin acknowledges the financial support by the Ministry of Education (MOE), Singapore, under its Academic Research Fund Tier 2 program (T2EP50123-0032) and Agency for Science, Technology and Research (A*STAR) under its RIE2025 MTC IRG program (M23M6c0106).Notes and references
- J. Greeley and N. M. Markovic, Energy Environ. Sci., 2012, 5, 9246–9256 RSC
.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed
- A. Raveendran, M. Chandran and R. Dhanusuraman, RSC Adv., 2023, 13, 3843–3876 RSC
- A. Marshall, B. Børresen, G. Hagen, M. Tsypkin and R. Tunold, Energy, 2007, 32, 431–436 CrossRef CAS
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed
- R. Frydendal, E. A. Paoli, B. P. Knudsen, B. Wickman, P. Malacrida, I. E. L. Stephens and I. Chorkendorff, ChemElectroChem, 2014, 1, 2075–2081 CrossRef CAS
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS
- R. Kötz, H. J. Lewerenz and S. Stucki, J. Electrochem. Soc., 1983, 130, 825 CrossRef
- E. Antolini, ACS Catal., 2014, 4, 1426–1440 CrossRef CAS
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef
- W. Zhang, K. Banerjee-Ghosh, F. Tassinari and R. Naaman, ACS Energy Lett., 2018, 3, 2308–2313 CrossRef CAS
- F. A. Garcés-Pineda, M. Blasco-Ahicart, D. Nieto-Castro, N. López and J. R. Galán-Mascarós, Nat. Energy, 2019, 4, 519–525 CrossRef
- X. Li, H. Liu, Z. Chen, Q. Wu, Z. Yu, M. Yang, X. Wang, Z. Cheng, Z. Fu and Y. Lu, Nat. Commun., 2019, 10, 1409 CrossRef PubMed
- K. Ray, S. P. Ananthavel, D. H. Waldeck and R. Naaman, Science, 1999, 283, 814–816 CrossRef CAS PubMed
- R. Naaman and D. H. Waldeck, Annu. Rev. Phys. Chem., 2015, 66, 263–281 CrossRef CAS PubMed
- B. Göhler, V. Hamelbeck, T. Z. Markus, M. Kettner, G. F. Hanne, Z. Vager, R. Naaman and H. Zacharias, Science, 2011, 331, 894–897 CrossRef PubMed
- Z. Xie, T. Z. Markus, S. R. Cohen, Z. Vager, R. Gutierrez and R. Naaman, Nano Lett., 2011, 11, 4652–4655 CrossRef CAS PubMed
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250–260 CrossRef CAS
- R. Naaman and D. H. Waldeck, J. Phys. Chem. Lett., 2012, 3, 2178–2187 CrossRef CAS PubMed
- Y. Liang, M. Lihter and M. Lingenfelder, Isr. J. Chem., 2022, 62, e202200052 CrossRef CAS
- S. Sanvito, Nat. Mater., 2011, 10, 484–485 CrossRef CAS PubMed
- M. Urdampilleta, S. Klyatskaya, J.-P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502–506 CrossRef CAS PubMed
- R. A. Rosenberg, D. Mishra and R. Naaman, Angew. Chem., Int. Ed., 2015, 54, 7295–7298 CrossRef CAS PubMed
- M. Kettner, B. Göhler, H. Zacharias, D. Mishra, V. Kiran, R. Naaman, D. H. Waldeck, S. Sęk, J. Pawłowski and J. Juhaniewicz, J. Phys. Chem. C, 2015, 119, 14542–14547 CrossRef CAS
- K. Michaeli, N. Kantor-Uriel, R. Naaman and D. H. Waldeck, Chem. Soc. Rev., 2016, 45, 6478–6487 RSC
- W. Mtangi, V. Kiran, C. Fontanesi and R. Naaman, J. Phys. Chem. Lett., 2015, 6, 4916–4922 CrossRef CAS PubMed
- T. J. Zwang, S. Hürlimann, M. G. Hill and J. K. Barton, J. Am. Chem. Soc., 2016, 138, 15551–15554 CrossRef CAS PubMed
- S. L. Kuzmin and W. W. Duley, Phys. Lett. A, 2014, 378, 1647–1650 CrossRef CAS
- R. Widmer, F.-J. Haug, P. Ruffieux, O. Gröning, M. Bielmann, P. Gröning and R. Fasel, J. Am. Chem. Soc., 2006, 128, 14103–14108 CrossRef CAS PubMed
- G. A. Steele, F. Pei, E. A. Laird, J. M. Jol, H. B. Meerwaldt and L. P. Kouwenhoven, Nat. Commun., 2013, 4, 1573 CrossRef CAS PubMed
- G. Koplovitz, D. Primc, O. Ben Dor, S. Yochelis, D. Rotem, D. Porath and Y. Paltiel, Adv. Mater., 2017, 29, 1606748 CrossRef PubMed
- O. Ben Dor, N. Morali, S. Yochelis, L. T. Baczewski and Y. Paltiel, Nano Lett., 2014, 14, 6042–6049 CrossRef CAS PubMed
- H. Al-Bustami, G. Koplovitz, D. Primc, S. Yochelis, E. Capua, D. Porath, R. Naaman and Y. Paltiel, Small, 2018, 14, 1801249 CrossRef PubMed
- B. P. Bloom, Y. Lu, T. Metzger, S. Yochelis, Y. Paltiel, C. Fontanesi, S. Mishra, F. Tassinari, R. Naaman and D. H. Waldeck, Phys. Chem. Chem. Phys., 2020, 22, 21570–21582 RSC
- Z. Bian, Y. Nakano, K. Miyata, I. Oya, M. Nobuoka, Y. Tsutsui, S. Seki and M. Suda, Adv. Mater., 2023, 35, 2306061 CrossRef CAS PubMed
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1900110 CrossRef CAS PubMed
- S. Zhao, Y. Yu, B. Zhang, P. Feng, C. Dang, M. Li, L. Zhao and L. Gao, Adv. Opt. Mater., 2021, 9, 2100907 CrossRef CAS
- T. Zhao, D. Meng, Z. Hu, W. Sun, Y. Ji, J. Han, X. Jin, X. Wu and P. Duan, Nat. Commun., 2023, 14, 81 CrossRef CAS PubMed
- X. Jin, J. Jiang and M. Liu, ACS Nano, 2016, 10, 11179–11186 CrossRef CAS PubMed
- W. Yan, L. Xu, C. Xu, W. Ma, H. Kuang, L. Wang and N. A. Kotov, J. Am. Chem. Soc., 2012, 134, 15114–15121 CrossRef CAS PubMed
- Y. Negrín-Montecelo, A. Movsesyan, J. Gao, S. Burger, Z. M. Wang, S. Nlate, E. Pouget, R. Oda, M. Comesaña-Hermo, A. O. Govorov and M. A. Correa-Duarte, J. Am. Chem. Soc., 2022, 144, 1663–1671 CrossRef PubMed
- M. Ramamurthy, P. Pachidis, B. M. Cote and V. E. Ferry, ACS Appl. Opt. Mater., 2023, 1, 491–499 CrossRef CAS
- X. Wu, C. Hao, J. Kumar, H. Kuang, N. A. Kotov, L. M. Liz-Marzán and C. Xu, Chem. Soc. Rev., 2018, 47, 4677–4696 RSC
- Q. Liu, M. G. Campbell, J. S. Evans and I. I. Smalyukh, Adv. Mater., 2014, 26, 7178–7184 CrossRef CAS PubMed
- L. Onsager, Ann. N. Y. Acad. Sci., 1949, 51, 627–659 CrossRef CAS
- W. Yan, L. Xu, W. Ma, L. Liu, L. Wang, H. Kuang and C. Xu, Small, 2014, 10, 4293–4297 CrossRef CAS PubMed
- P. Liu, Y. Battie, M. Decossas, S. Tan, E. Pouget, Y. Okazaki, T. Sagawa and R. Oda, ACS Nano, 2021, 15, 16411–16421 CrossRef CAS PubMed
- A. Guerrero-Martínez, J. L. Alonso-Gómez, B. Auguié, M. M. Cid and L. M. Liz-Marzán, Nano Today, 2011, 6, 381–400 CrossRef
- D. Vila-Liarte, N. A. Kotov and L. M. Liz-Marzán, Chem. Sci., 2022, 13, 595–610 RSC
- T. Delclos, C. Aimé, E. Pouget, A. Brizard, I. Huc, M.-H. Delville and R. Oda, Nano Lett., 2008, 8, 1929–1935 CrossRef CAS PubMed
- J. Cheng, G. Le Saux, J. Gao, T. Buffeteau, Y. Battie, P. Barois, V. Ponsinet, M.-H. Delville, O. Ersen, E. Pouget and R. Oda, ACS Nano, 2017, 11, 3806–3818 CrossRef CAS PubMed
- J. Gao, W. Wu, V. Lemaire, A. Carvalho, S. Nlate, T. Buffeteau, R. Oda, Y. Battie, M. Pauly and E. Pouget, ACS Nano, 2020, 14, 4111–4121 CrossRef CAS PubMed
- J. Xie, Y. Duan and S. Che, Adv. Funct. Mater., 2012, 22, 3784–3792 CrossRef CAS
- P. Liu, W. Chen, Y. Okazaki, Y. Battie, L. Brocard, M. Decossas, E. Pouget, P. Müller-Buschbaum, B. Kauffmann, S. Pathan, T. Sagawa and R. Oda, Nano Lett., 2020, 20, 8453–8460 CrossRef CAS PubMed
- Z. Zhu, J. Guo, W. Liu, Z. Li, B. Han, W. Zhang and Z. Tang, Angew. Chem., Int. Ed., 2013, 52, 13571–13575 CrossRef CAS PubMed
- O. Guselnikova, H. Lim, J. Na, M. Eguchi, H.-J. Kim, R. Elashnikov, P. Postnikov, V. Svorcik, O. Semyonov, E. Miliutina, O. Lyutakov and Y. Yamauchi, Biosens. Bioelectron., 2021, 180, 113109 CrossRef CAS PubMed
- L. Ohnoutek, N. H. Cho, A. W. Allen Murphy, H. Kim, D. M. Răsădean, G. D. Pantoş, K. T. Nam and V. K. Valev, Nano Lett., 2020, 20, 5792–5798 CrossRef CAS PubMed
- J. Zheng, C. Boukouvala, G. R. Lewis, Y. Ma, Y. Chen, E. Ringe, L. Shao, Z. Huang and J. Wang, Nat. Commun., 2023, 14, 3783 CrossRef PubMed
- H. Liu, A. E. Vladár, P.-P. Wang and M. Ouyang, J. Am. Chem. Soc., 2023, 145, 7495–7503 CrossRef CAS PubMed
- W. Liu, Z. Zhu, K. Deng, Z. Li, Y. Zhou, H. Qiu, Y. Gao, S. Che and Z. Tang, J. Am. Chem. Soc., 2013, 135, 9659–9664 CrossRef CAS PubMed
- H. Liu, Z. Li, Y. Yan, J. Zhao and Y. Wang, Nanoscale, 2019, 11, 21990–21998 RSC
- L. Tan, W. Fu, Q. Gao and P. Wang, Adv. Mater., 2024, 36, 2309033 CrossRef CAS PubMed
- B. L. Li, J. J. Luo, H. L. Zou, Q.-M. Zhang, L.-B. Zhao, H. Qian, H. Q. Luo, D. T. Leong and N. B. Li, Nat. Commun., 2022, 13, 7289 CrossRef CAS PubMed
- A. Vadakkayil, C. Clever, K. N. Kunzler, S. Tan, B. P. Bloom and D. H. Waldeck, Nat. Commun., 2023, 14, 1067 CrossRef CAS PubMed
- J. Fransson, J. Phys. Chem. Lett., 2019, 10, 7126–7132 CrossRef CAS PubMed
- W. Mtangi, F. Tassinari, K. Vankayala, A. Vargas Jentzsch, B. Adelizzi, A. R. A. Palmans, C. Fontanesi, E. W. Meijer and R. Naaman, J. Am. Chem. Soc., 2017, 139, 2794–2798 CrossRef CAS PubMed
- T. Feng, W. Chen, J. Xue, F. Cao, Z. Chen, J. Ye, C. Xiao and H. Lu, Adv. Funct. Mater., 2023, 33, 2215051 CrossRef CAS
- C. Kulkarni, A. K. Mondal, T. K. Das, G. Grinbom, F. Tassinari, M. F. J. Mabesoone, E. W. Meijer and R. Naaman, Adv. Mater., 2020, 32, 1904965 CrossRef CAS PubMed
- V. Kiran, S. P. Mathew, S. R. Cohen, I. Hernández Delgado, J. Lacour and R. Naaman, Adv. Mater., 2016, 28, 1957–1962 CrossRef CAS PubMed
- M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O’Leary and G. Willson, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef
- A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef CAS PubMed
- K. Banerjee-Ghosh, O. Ben Dor, F. Tassinari, E. Capua, S. Yochelis, A. Capua, S.-H. Yang, S. S. P. Parkin, S. Sarkar, L. Kronik, L. T. Baczewski, R. Naaman and Y. Paltiel, Science, 2018, 360, 1331–1334 CrossRef CAS PubMed
- M. Eckshtain-Levi, E. Capua, S. Refaely-Abramson, S. Sarkar, Y. Gavrilov, S. P. Mathew, Y. Paltiel, Y. Levy, L. Kronik and R. Naaman, Nat. Commun., 2016, 7, 10744 CrossRef CAS PubMed
- E. H. Hall, Am. J. Math., 1879, 2, 287–292 CrossRef
- O. Ben Dor, S. Yochelis, A. Radko, K. Vankayala, E. Capua, A. Capua, S.-H. Yang, L. T. Baczewski, S. S. P. Parkin, R. Naaman and Y. Paltiel, Nat. Commun., 2017, 8, 14567 CrossRef CAS PubMed
- A. Kumar, E. Capua, M. Kesharwani, J. Martin, E. Sitbon and D. Waldeck, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2474–2478 CrossRef CAS PubMed
- V. K. Valev, J. J. Baumberg, C. Sibilia and T. Verbiest, Adv. Mater., 2013, 25, 2517–2534 CrossRef CAS PubMed
- A. Ziv, O. Shoseyov, P. Karadan, B. P. Bloom, S. Goldring, T. Metzger, S. Yochelis, D. H. Waldeck, R. Yerushalmi and Y. Paltiel, Nano Lett., 2021, 21, 6496–6503 CrossRef CAS PubMed
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC
- J. R. Galan-Mascaros, Catal. Sci. Technol., 2020, 10, 1967–1974 RSC
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS
- M. Plevová, J. Hnát and K. Bouzek, J. Power Sources, 2021, 507, 230072 CrossRef
- X. Li, Z. Cheng and X. Wang, Electrochem. Energy Rev., 2021, 4, 136–145 CrossRef CAS
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC
- G. Bronoel and J. Reby, Electrochim. Acta, 1980, 25, 973–976 CrossRef CAS
- N. Yuan, Q. Jiang, J. Li and J. Tang, Arab. J. Chem., 2020, 13, 4294–4309 CrossRef CAS
- Y. Zhang, X. Zhu, G. Zhang, P. Shi and A.-L. Wang, J. Mater. Chem. A, 2021, 9, 5890–5914 RSC
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed
- A. F. Pedersen, M. Escudero-Escribano, B. Sebok, A. Bodin, E. Paoli, R. Frydendal, D. Friebel, I. E. L. Stephens, J. Rossmeisl, I. Chorkendorff and A. Nilsson, J. Phys. Chem. B, 2018, 122, 878–887 CrossRef CAS PubMed
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC
- J. An, Y. Feng, Q. Zhao, X. Wang, J. Liu and N. Li, Environ. Sci. Ecotech., 2022, 11, 100170 CAS
- J. Shan, Y. Zheng, B. Shi, K. Davey and S.-Z. Qiao, ACS Energy Lett., 2019, 4, 2719–2730 CrossRef CAS
- A. Zagalskaya and V. Alexandrov, ACS Catal., 2020, 10, 3650–3657 CrossRef CAS
- M. Wohlfahrt-Mehrens and J. Heitbaum, J. Electroanal. Chem. Interfacial Electrochem., 1987, 237, 251–260 CrossRef CAS
- K. A. Stoerzinger, O. Diaz-Morales, M. Kolb, R. R. Rao, R. Frydendal, L. Qiao, X. R. Wang, N. B. Halck, J. Rossmeisl, H. A. Hansen, T. Vegge, I. E. L. Stephens, M. T. M. Koper and Y. Shao-Horn, ACS Energy Lett., 2017, 2, 876–881 CrossRef CAS
- K. Macounova, M. Makarova and P. Krtil, Electrochem. Commun., 2009, 11, 1865–1868 CrossRef CAS
- R. Xu, H. Huang, W. Wang, L. Ding, Q. Lin, J. Li, Y. Zhang, Y. Han, J. Wang and X. Lu, J. Chem. Eng., 2023, 461, 141748 CrossRef CAS
- M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722–728 CrossRef CAS PubMed
- S. Chrétien and H. Metiu, J. Chem. Phys., 2008, 129, 074705 CrossRef PubMed
- E. Torun, C. M. Fang, G. A. de Wijs and R. A. de Groot, J. Phys. Chem. C, 2013, 117, 6353–6357 CrossRef CAS
- C. Nogues, S. R. Cohen, S. S. Daube and R. Naaman, Phys. Chem. Chem. Phys., 2004, 6, 4459–4466 RSC
- Z. Li, C. Kong and G. Lu, J. Phys. Chem. C, 2016, 120, 56–63 CrossRef CAS
- P. C. Mondal, W. Mtangi and C. Fontanesi, Small, Methods, 2018, 2, 1700313 Search PubMed
- E. Medina, L. A. González-Arraga, D. Finkelstein-Shapiro, B. Berche and V. Mujica, J. Chem. Phys., 2015, 142, 194308 CrossRef PubMed
- K. B. Ghosh, W. Zhang, F. Tassinari, Y. Mastai, O. Lidor-Shalev, R. Naaman, P. Möllers, D. Nürenberg, H. Zacharias, J. Wei, E. Wierzbinski and D. H. Waldeck, J. Phys. Chem. C, 2019, 123, 3024–3031 CrossRef CAS
- J. O. Bockris, J. Chem. Phys., 1956, 24, 817–827 CrossRef CAS
- F. Tassinari, K. Banerjee-Ghosh, F. Parenti, V. Kiran, A. Mucci and R. Naaman, J. Phys. Chem. C, 2017, 121, 15777–15783 CrossRef CAS
- A. N. Nair, S. Fernandez, M. Marcos-Hernández, D. R. Romo, S. R. Singamaneni, D. Villagran and S. T. Sreenivasan, Nano Lett., 2023, 23, 9042–9049 CrossRef CAS PubMed
- X. Ren, T. Wu, Y. Sun, Y. Li, G. Xian, X. Liu, C. Shen, J. Gracia, H.-J. Gao, H. Yang and Z. J. Xu, Nat. Commun., 2021, 12, 2608 CrossRef CAS PubMed
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed
- R. Ma, G. Lin, Y. Zhou, Q. Liu, T. Zhang, G. Shan, M. Yang and J. Wang, npj Comput. Mater., 2019, 5, 78 CrossRef
- C. H. Choi, M. W. Chung, H. C. Kwon, S. H. Park and S. I. Woo, J. Mater. Chem. A, 2013, 1, 3694–3699 RSC
- J.-M. You, M. S. Ahmed, H. S. Han, J. E. Choe, Z. Üstündağ and S. Jeon, J. Power Sources, 2015, 275, 73–79 CrossRef CAS
- Y. Sang, F. Tassinari, K. Santra, W. Zhang, C. Fontanesi, B. P. Bloom, D. H. Waldeck, J. Fransson and R. Naaman, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202650119 CrossRef CAS PubMed
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS
- D. Wang, X. Pan, P. Yang, R. Li, H. Xu, Y. Li, F. Meng, J. Zhang and M. An, ChemSusChem, 2021, 14, 33–55 CrossRef CAS PubMed
- L. Scarpetta-Pizo, R. Venegas, P. Barrías, K. Muñoz-Becerra, N. Vilches-Labbé, F. Mura, A. M. Méndez-Torres, R. Ramírez-Tagle, A. Toro-Labbé, S. Hevia, J. H. Zagal, R. Oñate, A. Aspée and I. Ponce, Angew. Chem., Int. Ed., 2024, 63, e202315146 CrossRef CAS PubMed
- J. H. Zagal and M. T. M. Koper, Angew. Chem., Int. Ed., 2016, 55, 14510–14521 CrossRef CAS PubMed
- C. Gutiérrez-Ceron, R. Oñate, J. H. Zagal, A. Pizarro, J. F. Silva, C. Castro-Castillo, M. C. Rezende, M. Flores, D. Cortés-Arriagada, A. Toro-Labbé, L. M. Campos, L. Venkataraman and I. Ponce, Electrochim. Acta, 2019, 327, 134996 CrossRef
- J. H. Zagal, M. Gulppi, M. Isaacs, G. Cárdenas-Jirón and M. J. S. Aguirre, Electrochim. Acta, 1998, 44, 1349–1357 CrossRef CAS
- C. Isvoranu, B. Wang, K. Schulte, E. Ataman, J. Knudsen, J. N. Andersen, M. L. Bocquet and J. Schnadt, J. Condens. Matter Phys., 2010, 22, 472002 CrossRef CAS PubMed
- Y. Wang, X. Li, X. Zheng and J. Yang, J. Chem. Phys., 2017, 147, 134701 CrossRef PubMed
- X. Zhang, C. Wolf, Y. Wang, H. Aubin, T. Bilgeri, P. Willke, A. J. Heinrich and T. Choi, Nat. Chem., 2022, 14, 59–65 CrossRef CAS PubMed
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed
- E. Skúlason, G. S. Karlberg, J. Rossmeisl, T. Bligaard, J. Greeley, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2007, 9, 3241–3250 RSC
- N. M. Markovića, S. T. Sarraf, H. A. Gasteiger and P. N. Ross, J. Chem. Soc., Faraday Trans., 1996, 92, 3719–3725 RSC
- S. Trasatti, J. Electroanal. Chem. Interf. Electrochem., 1972, 39, 163–184 CrossRef CAS
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 RSC
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed
- P. K. Bhartiya, M. Srivastava and D. Mishra, Int. J. Hydrogen Energy, 2022, 47, 42160–42170 CrossRef CAS
- B. P. Bloom, Y. Paltiel, R. Naaman and D. H. Waldeck, Chem. Rev., 2024, 124, 1950–1991 CrossRef CAS PubMed
- W. Xu, Y. Wu, W. Gu, D. Du, Y. Lin and C. Zhu, Chem. Soc. Rev., 2024, 53, 137–162 RSC
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed
- R. F. Service, Science, 2014, 345, 610 CrossRef CAS PubMed
- R. Zhao, H. Xie, L. Chang, X. Zhang, X. Zhu, X. Tong, T. Wang, Y. Luo, P. Wei, Z. Wang and X. Sun, J. Energy Chem., 2019, 1, 100011 Search PubMed
- Y. Bicer and I. Dincer, J. Electrochem. Soc., 2017, 164, H5036 CrossRef CAS
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS PubMed
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC
- Y. Abghoui and E. Skúlason, Catal. Today, 2017, 286, 69–77 CrossRef CAS
- X.-F. Li, Q.-K. Li, J. Cheng, L. Liu, Q. Yan, Y. Wu, X.-H. Zhang, Z.-Y. Wang, Q. Qiu and Y. Luo, J. Am. Chem. Soc., 2016, 138, 8706–8709 CrossRef CAS PubMed
- Y. Zhang, Q. Zhang, D.-X. Liu, Z. Wen, J.-X. Yao, M.-M. Shi, Y.-F. Zhu, J.-M. Yan and Q. Jiang, Appl. Catal., B, 2021, 298, 120592 CrossRef CAS
- Y. Yang, L. Zhang, Z. Hu, Y. Zheng, C. Tang, P. Chen, R. Wang, K. Qiu, J. Mao, T. Ling and S.-Z. Qiao, Angew. Chem., Int. Ed., 2020, 59, 4525–4531 CrossRef CAS PubMed
- P. Borthakur, P. K. Boruah, M. R. Das, M. M. Ibrahim, T. Altalhi, H. S. El-Sheshtawy, S. Szunerits, R. Boukherroub and M. A. Amin, ACS Appl. Energy Mater., 2021, 4, 1269–1285 CrossRef CAS
- A. Cao, V. J. Bukas, V. Shadravan, Z. Wang, H. Li, J. Kibsgaard, I. Chorkendorff and J. K. Nørskov, Nat. Commun., 2022, 13, 2382 CrossRef CAS PubMed
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed
- S. Kumar De, D.-I. Won, J. Kim and D. H. Kim, Chem. Soc. Rev., 2023, 52, 5744–5802 RSC
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed
- S. Li, J. Liu, Z. Yin, P. Ren, L. Lin, Y. Gong, C. Yang, X. Zheng, R. Cao, S. Yao, Y. Deng, X. Liu, L. Gu, W. Zhou, J. Zhu, X. Wen, B. Xu and D. Ma, ACS Catal., 2020, 10, 907–913 CrossRef CAS
- Y. Zhang, C. Liang, J. Wu, H. Liu, B. Zhang, Z. Jiang, S. Li and P. Xu, ACS Appl. Energy Mater., 2020, 3, 10303–10316 CrossRef CAS
- H. Pan, X. Jiang, X. Wang, Q. Wang, M. Wang and Y. Shen, J. Phys. Chem. Lett., 2020, 11, 48–53 CrossRef CAS PubMed
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC
- W. L. Fredericks, S. Sripad, G. C. Bower and V. Viswanathan, ACS Energy Lett., 2018, 3, 2989–2994 CrossRef CAS
- A. Manthiram, J. C. Knight, S.-T. Myung, S.-M. Oh and Y.-K. Sun, Adv. Energy Mater., 2016, 6, 1501010 CrossRef
- Y.-J. Lei, L. Zhao, W.-H. Lai, Z. Huang, B. Sun, P. Jaumaux, K. Sun, Y.-X. Wang and G. Wang, Chem. Soc. Rev., 2024, 53, 3829–3895 RSC
- T. Li, M. Huang, X. Bai and Y.-X. Wang, Prog. Nat. Sci.: Mater., 2023, 33, 151–171 CrossRef CAS
- S. Zago, L. C. Sacrpetta-Pizo, J. H. Zagal and S. Specchia, Electrochem. Energy Rev., 2024, 7, 1 CrossRef CAS
- L.-B. Liu, C. Yi, H.-C. Mi, S. L. Zhang, X.-Z. Fu, J.-L. Luo and S. Liu, Electrochem. Energy Rev., 2024, 7, 14 CrossRef CAS PubMed
- G. Houchins, V. Pande and V. Viswanathan, ACS Energy Lett., 2020, 5, 1893–1899 CrossRef CAS
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, T. Mori, R. Scheffler, A. Speidel, M. Sherwood and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 3043–3047 CrossRef CAS PubMed
- M. Carboni, A. G. Marrani, R. Spezia and S. Brutti, Chem. – Eur. J., 2016, 22, 17188–17203 CrossRef CAS PubMed
- A. Pierini, S. Brutti and E. Bodo, ChemPhysChem, 2020, 21, 2060–2067 CrossRef CAS PubMed
- E. Mourad, Y. K. Petit, R. Spezia, A. Samojlov, F. F. Summa, C. Prehal, C. Leypold, N. Mahne, C. Slugovc, O. Fontaine, S. Brutti and S. A. Freunberger, Energy Environ. Sci., 2019, 12, 2559–2568 RSC
- N. Mahne, S. E. Renfrew, B. D. McCloskey and S. A. Freunberger, Angew. Chem., Int. Ed., 2018, 57, 5529–5533 CrossRef CAS PubMed
- N. Mahne, B. Schafzahl, C. Leypold, M. Leypold, S. Grumm, A. Leitgeb, G. A. Strohmeier, M. Wilkening, O. Fontaine, D. Kramer, C. Slugovc, S. M. Borisov and S. A. Freunberger, Nat. Energy, 2017, 2, 17036 CrossRef CAS
- S. Koraltan, F. Slanovc, F. Bruckner, C. Nisoli, A. V. Chumak, O. V. Dobrovolskiy, C. Abert and D. Suess, npj Comput. Mater., 2021, 7, 125 CrossRef CAS
- H.-W. Lee, H. Kim, H.-G. Jung, Y.-K. Sun and W.-J. Kwak, Adv. Funct. Mater., 2021, 31, 2102442 CrossRef CAS
- N. Kitajima, S. Fukuzumi and Y. Ono, J. Phys. Chem., 1978, 82, 1505–1509 CrossRef CAS
- W. Fudickar and T. Linker, Angew. Chem., Int. Ed., 2018, 57, 12971–12975 CrossRef CAS PubMed
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed
- Y. K. Petit, C. Leypold, N. Mahne, E. Mourad, L. Schafzahl, C. Slugovc, S. M. Borisov and S. A. Freunberger, Angew. Chem., Int. Ed., 2019, 58, 6535–6539 CrossRef CAS PubMed
-
S. Beutner, B. Bloedorn, T. Hoffmann and H.-D. Martin, Singlet Oxygen, UV-A, and Ozone, Academic Press, 2000, vol. 319, pp. 226–241 Search PubMed
- E. J. Corey, M. M. Mehrotra and A. U. Khan, Science, 1987, 236, 68–69 CrossRef CAS PubMed
- K. C. Das and C. K. Das, Biochem. Biophys. Res. Commun., 2002, 295, 62–66 CrossRef CAS PubMed
- Z. Liang, Q. Zou, J. Xie and Y.-C. Lu, Energy Environ. Sci., 2020, 13, 2870–2877 RSC
- H. Wang, K. Wang, Y. Zuo, M. Wei, P. Pei, P. Zhang, Z. Chen and N. Shang, Adv. Funct. Mater., 2023, 33, 2210127 CrossRef CAS
- Z.-M. Liao, Y.-D. Li, J. Xu, J.-M. Zhang, K. Xia and D.-P. Yu, Nano Lett., 2006, 6, 1087–1091 CrossRef CAS PubMed
- H. J. Eckvahl, N. A. Tcyrulnikov, A. Chiesa, J. M. Bradley, R. M. Young, S. Carretta, M. D. Krzyaniak and M. R. Wasielewski, Science, 2023, 382, 197–201 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |