
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interfacial chemistry in multivalent aqueous batteries: fundamentals, challenges, and advances
Zhengyu
Ju†
,
Tianrui
Zheng†
,
Bowen
Zhang
and
Guihua
Yu
 *
*
Materials Science and Engineering Program and Walker Department of Mechanical Engineering, The University of Texas at Austin, Austin, TX 78712, USA. E-mail: ghyu@austin.utexas.edu
First published on 19th August 2024
Abstract
As one of the most promising electrochemical energy storage systems, aqueous batteries are attracting great interest due to their advantages of high safety, high sustainability, and low costs when compared with commercial lithium-ion batteries, showing great promise for grid-scale energy storage. This invited tutorial review aims to provide universal design principles to address the critical challenges at the electrode–electrolyte interfaces faced by various multivalent aqueous battery systems. Specifically, deposition regulation, ion flux homogenization, and solvation chemistry modulation are proposed as the key principles to tune the inter-component interactions in aqueous batteries, with corresponding interfacial design strategies and their underlying working mechanisms illustrated. In the end, we present a critical analysis on the remaining obstacles necessitated to overcome for the use of aqueous batteries under different practical conditions and provide future prospects towards further advancement of sustainable aqueous energy storage systems with high energy and long durability.
 Zhengyu Ju | Zhengyu Ju is a postdoctoral fellow at the University of Texas at Austin. He received his BS degree in Chemistry at the University of Science and Technology of China (USTC) in 2018. After that, he completed his PhD degree in Materials Science and Engineering at the University of Texas at Austin under the supervision of Prof. Guihua Yu in 2023. His research interests mainly focus on electrochemical energy storage with two-dimensional nanomaterials. |
 Tianrui Zheng | Tianrui Zheng is an exchange student at the University of Texas at Austin, where he worked under the supervision of Prof. Guihua Yu on the development of advanced energy storage systems, including lithium batteries and beyond. |
 Bowen Zhang | Bowen Zhang is a research associate of Materials Science and Engineering at the University of Texas at Austin. He received his BS degree from Henan University, Master degree from the Beijing University of Chemical Technology, and PhD degree in Materials Science at the Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences. His research has been focused on nanostructured inorganic materials and their applications in electrochemical energy storage and conversion. |
 Guihua Yu | Guihua Yu is a Temple Foundation Professor of Materials Science and Mechanical Engineering at the University of Texas at Austin. He received his B.S. degree in chemistry with the highest honor from USTC, PhD degree from Harvard University, followed by postdoc research at Stanford. His research has been focused on creative design and synthesis of functional nanostructured materials, particularly organic and hybrid nanomaterials, fundamental understanding of their chemical and physical properties, and developing nanoscience-enabled solutions to address global challenges in advanced energy, environment and sustainability technologies. |
Key learning points(1) The interfacial challenges encountered by multivalent aqueous metal batteries, along with their underlying mechanisms.(2) The overarching design principles tailored to address the relevant interfacial issues. (3) The practical strategies and techniques that translate design principles into tangible solutions. (4) The future prospects and potential avenues for the advancement of multivalent aqueous batteries. |
1. Introduction
Electrochemical energy storage systems, as promising alternatives to fossil fuels, have been developed and extensively used in our daily life over the past several decades, ranging from portable electronic devices to electric transportation and grid-scale energy storage.1,2 Alkaline batteries, a key class of primary batteries, were invented and have been used since the last century which mainly include nickel–cadmium batteries (Cd//NiOH), mercury batteries (Zn//HgO), and zinc–manganese batteries (Zn//MnO2).3 Among them, alkaline Zn//MnO2 batteries are the most commonly used ones, with Zn as the anode, MnO2 as the cathode, and aqueous KOH as the electrolyte. However, such aqueous batteries pose significant challenges on environmental friendliness and user safety as they contain a highly concentrated alkaline electrolyte and toxic heavy metals and therefore standard disposal and recycling of these batteries are necessarily required.4Lithium-ion batteries (LIBs), which are a class of secondary batteries, have attracted considerable attention owing to their high energy density, long cycling life (>1000 cycles), high voltage (−3.04 V for Li+/Li vs. standard hydrogen evolution (SHE)) and low self-discharge rate.5 Current commercial LIBs include graphite as the anode, lithium metal oxide as the cathode, and organic electrolyte containing lithium (Li) salts. LIBs can be considered as rocking-chair batteries, as Li ions travel back and forth between the anode and the cathode during battery operation. The Li-ion storage at the anode and the cathode follows the intercalation/deintercalation mechanism, which means that Li ions are inserted into the crystal structure of the cathode/anode material and occupy the specific sites inside. Such a highly reversible charge storage mechanism ensures the stability of the material structure after repeated cycling, making LIBs rechargeable for thousands of times.
Although current LIBs are regarded as the most promising battery system adopted in electric or hybrid vehicles to replace gasoline-powered vehicles,6 the use of the organic electrolyte and transition metal-based electrode materials increases significant concerns mounting over their charging capability, safety, cost effectiveness, and environmental friendliness.7–10 The increasing market price of lithium and cobalt sources of the cathodes over the past few years poses great challenges for the adoption of LIB packs (∼$150 per kW h for LIBs in 2023) in grid-scale energy storage.11 Furthermore, firing/explosion is possible for cell phones and electric vehicles when the LIBs inside experienced a puncture or a fast thermal runaway due to the use of flammable organic solvent-based electrolytes.12 Replacing the graphite anode with the Li metal anode due to its high specific and volumetric capacities (3860 mA h g−1 and 2061 mA h cm−3) could improve the energy density of the cell (Fig. 1a); however, a dendritic issue of the metal anode brings more safety problems.13 Although solid-state electrolytes have been proposed and developed to overcome the firing issue when a short circuit occurs, the low Li-ion conductivity in solid electrolytes and the poor physical contact at the solid electrolyte–electrode interfaces remain to be resolved for practical applications.14 The developments in post-Li battery systems, such as sodium (Na) and potassium (K) batteries, address the critical cost concern due to their abundant sources in the Earth's crust and large mining production (Fig. 1b), lowering the price for Na-ion batteries (NIB) to ∼$40 per kW h, while their energy densities decrease due to the larger ionic radius and heavier atomic mass of Na+ and K+ compared to those of Li+.15
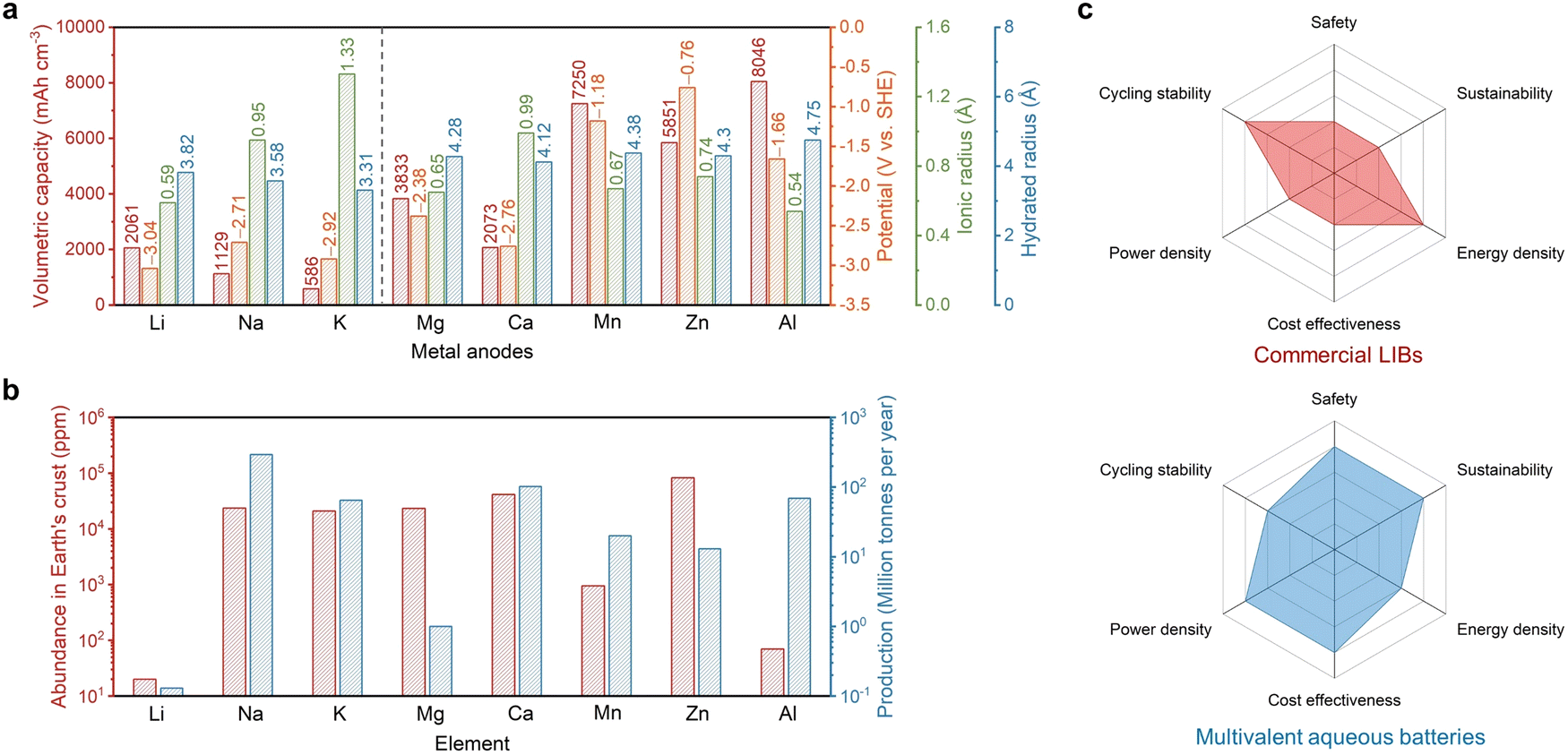 | ||
| Fig. 1 (a) Comparison of the metal anodes in terms of the volumetric capacity, redox potential, ionic radius, and hydrated radius. (b) Comparison of the element abundance in the Earth's crust and production. The Na, K, and Ca production is estimated from the corresponding main sources of NaCl, KCl, and CaCO3 from mining. Source: USGS mineral commodity summaries 2023. (c) Radar plots of the access of commercial LIBs and multivalent aqueous batteries regarding safety, cost effectiveness, sustainability, energy density, power density, and cycling stability. | ||
In pursuit of batteries with the characteristics of high safety and low cost for potential sustainable grid-scale energy storage, aqueous batteries stand out as a suitable choice.16 Aqueous batteries have several merits that outperform the commercial LIBs. First, the cost of electroactive materials and cell assembly can be significantly reduced due to the exclusion of expensive lithium/cobalt raw materials and oxygen- and water-free production lines. For example, the prices of Zn and Al are $2.7 per kg and $2.6 per kg, respectively, significantly cheaper than those of Li ($14 per kg) and Co ($27 per kg). Furthermore, the production and transportation of Zn/Al-based metal foils are much more cost-effective compared to water-sensitive LIB electrodes. Secondly, water-based electrolytes are more environmentally benign compared to volatile and flammable organic ester/ether-based electrolytes. Furthermore, recycling is required to prevent the improper processing and disposal of spent batteries which lead to contamination of the soil, water and air, and in such a process, the Zn recycling industry is well-established and provides about one-third of its demand, while Li recycling is more uncertain and in its infancy.17 In terms of cradle-to-gate environmental impacts, aqueous batteries (Zn-based) show a much smaller global warming indicator (45 CO2 equiv. per 1 kW h) compared to that of the LIBs (120 CO2 equiv. per 1 kW h), suggesting their positive effects on global warming reduction.18 Thirdly, ions can move faster in aqueous electrolytes compared to those in organic counterparts, which is promising for high-power applications requiring a large energy output within a short time. Last but not least, aqueous batteries are more bearable to battery pack deformation, puncture and electrolyte leaking. These advantages make aqueous batteries competitive alternatives to LIBs for clean and safe energy storage in the near future.
The charge carriers for aqueous batteries can be versatile including monovalent alkali metal ions (Li+, Na+, and K+), multivalent alkaline earth metal ions (Mg2+ and Ca2+) and transition or post-transition metal ions (Mn2+, Zn2+, Al3+, Fe2+, etc.). It should be noted that, for alkali metal-based batteries, metal anodes, which possess higher gravimetric/volumetric capacities compared to the traditional anode materials, cannot be used due to their low reduction potentials (−3.04 V for Li+/Li, −2.71 V for Na+/Na, and −2.92 V for K+/K), in which case a spontaneous reaction will occur between the metal anode and the water electrolyte. For alkaline earth metal ions with similarly low redox potentials, the reaction between the electrolyte and the metal anode is slower as hydroxide passivation layers (e.g., Mg(OH)2 and Ca(OH)2) would generate at the surface of the metal anode and prevent the further reaction. Transition or post-transition metals are typically more stable due to their relatively higher redox potentials (−1.18 V for Mn2+/Mn, −0.76 V for Zn2+/Zn, and −1.66 V for Al3+/Al), although corrosion and passivation could still occur at the metal surface. Due to the different redox potentials and reactivities of these multivalent metals, state-of-the-art aqueous Zn batteries (AZBs) can be stably cycled for tens of thousands of times,19,20 which significantly outperform commercial LIBs, while Mg-, Mn-, or Al-based batteries can typically be cycled for tens to hundreds of times suffering from severe side reactions.21,22 Apart from the stability issue in the aqueous environment, the working voltages of these multivalent metal-based batteries are lower than those of the typical organic LIBs. For example, the voltages of most of the AZBs are in the range of 1.0–1.5 V, resulting in their corresponding energy densities of 80–150 W h kg−1 in the current stage.23 Last but not least, it should be emphasized that after long-term cycling, the degradation at the metal anode side could generate a significant amount of hydrogen gas, which could seriously endanger the battery safety even in the aqueous system.
The overall comparison of commercial LIBs and multivalent aqueous batteries is shown in Fig. 1c. With the combined merits of low cost and simultaneously high safety, power, and sustainability, multivalent aqueous batteries are emerging as promising choices for large-scale electrochemical energy storage. Although at present, the energy density and cycle life of multivalent aqueous batteries have not shown significant advantages, the future optimization of battery components and interfaces can help overcome these two weaknesses. In the following, we discuss the main challenges at the interfaces in multivalent aqueous batteries, which are also one of the critical reasons that limit their energy density and electrochemical stability, as well as the design rationale to address these challenges. We believe that the interfacial design strategies in these aqueous battery systems are highly universal, which means that a successful solution in one battery case is likely to be applied to another system, thus promoting the development of the entire aqueous battery field to practical use.
2. Challenges at the electrode–electrolyte interfaces
2.1. Interfaces of the metal anode side
There are three main challenges at the interfaces of the metal anode side, including dendritic growth, the hydrogen evolution reaction (HER), and corrosion/passivation. According to the potential-pH diagrams of three commonly studied battery systems (Zn, Al, and Mn), it can be seen that the potential for hydrogen generation is always higher than that for metal deposition (Fig. 2a–c). The difference between these two potentials is small in the Zn potential-pH diagram (Fig. 2a) but huge in the Al potential-pH diagram (Fig. 2b), which means that, in the aqueous Al battery (AAB) case, the HER is prone to occur due to the lower potential required for Al3+ reduction. Another issue related to these diagrams is that hydroxides or oxides would generate causing surface passivation when the pH of the electrolyte is alkaline, neutral, or even mild acidic. This is due to the fact that, accompanying the HER, protons are consumed at the metal anode side leaving the local electrolyte pH increased. The passivation products are simple for the Zn and Al cases, but more complicated for Mn owing to the different reductive valence states of Mn.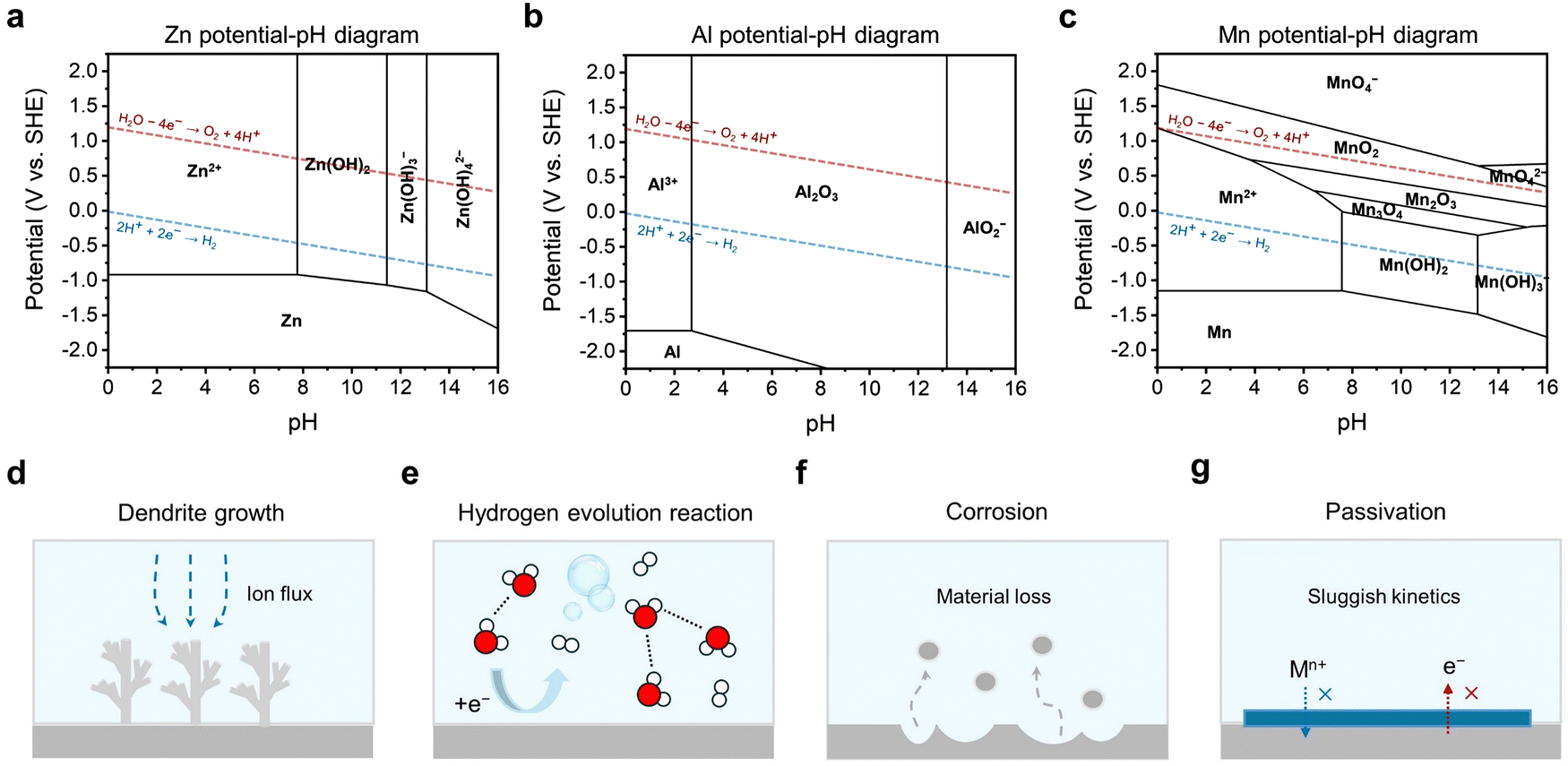 | ||
| Fig. 2 (a)–(c) Potential-pH diagrams of Zn (a), Al (b), and Mn (c). (d)–(g) Challenges at interfaces of the metal anode side including dendritic growth (d), the hydrogen evolution reaction (e), corrosion (f), and passivation (g). | ||
Firstly, the sharp, needle-like dendrites are likely to pierce the separator, coming in contact with the cathode, thus causing an internal short circuit. The internal short circuit results in immediate battery failure, and in some cases, even severe safety hazards such as thermal runaway, fire, and explosion. Even if a dendrite does not puncture through the separator thoroughly, the stress it imposes can locally deform the separator, disrupting the uniformity of ion flux and electric field distribution.31,32 Besides, a micro-short circuit can be another unwanted consequence, which poses a latent threat to the stable performance of a battery and even gives rise to catastrophic safety hazards.33–35 Secondly, upon stripping, due to weak adhesion, dendrites can be detached from the substrate and lead to ‘dead’ metal deposits. ‘Dead’ metal deposits are in the form of isolated islands floating in the electrolyte, which rarely participate in electrochemical reactions, thus causing capacity loss.36–38 Thirdly, the specific surface area of a dendrite is significantly larger compared to a flat and planar deposit. This increased surface area exacerbates undesirable side reactions and compromises the stability of the battery.39
Dendritic growth can be attributed to multiple factors. Two main factors, namely the electric field and the concentration gradient, are often studied and referred to.40–42 The onset of dendritic growth is usually associated with the uneven contour of the metal anode surface where small protrusions induce the tip effect. The tip effect describes the phenomenon of an elevated charge density and enhanced electric field near the tip region with a large curvature. Therefore, the small protrusions can alter the local electric field distribution and attract more metal ions to be preferentially deposited, thereby resulting in self-amplified rampant dendritic growth. Additionally, irregular nuclei as the product of uneven nucleation can also be potential dendrite formation sites. After initiation, dendritic growth predominantly takes place in the following plating process where the as-formed nuclei grow larger spontaneously. At this stage, in addition to the locally enhanced electric field, a local concentration gradient also plays a key role in dendritic growth: a large extent of local ion depletion limits ion diffusion and accelerates dendritic growth.
| 2H2O + 2e− → H2 (g) + 2OH− (aq) |
where E0 is the standard potential, E is the potential under specific conditions, a is the activity, and pH2/p0 is the partial pressure of hydrogen gas, which is equal to 1 in a standard atmosphere. R, T, and F here are the thermodynamic constant, temperature, and Faraday constant, respectively. It can be noted that the HER potential is related to the pH of the electrolyte. In practical cases, it is also related to the overpotential for driving the reaction to occur.
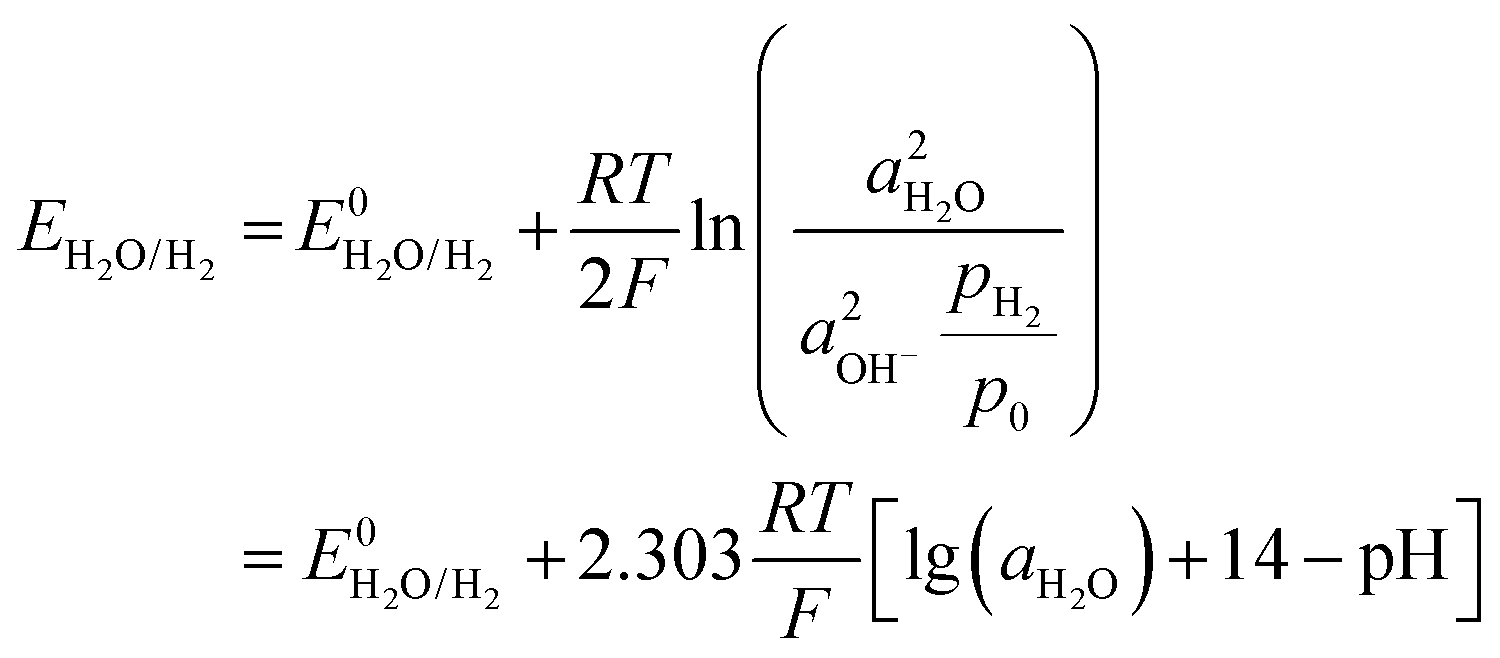
In common aqueous metal battery systems such as AZBs, AABs, aqueous Mn batteries and aqueous Mg batteries, the metal anodes are all prone to the HER. This is because the standard reduction potentials of these metals are all below the potential of the SHE, with −0.76 V for Zn2+/Zn, −1.66 V for Al3+/Al, −1.18 V for Mn2+/Mn, and −2.38 V for Mg2+/Mg (Fig. 2e). Although, in real cases, the conditions may well shift away from the standard ones, the standard reduction potentials still serve as an indicator that reflects the susceptibility of the metal anodes to the HER. Specifically, the lower the standard reduction potential, the more likely is for the HER to occur.
The HER is an undesirable side reaction that should be minimized. Firstly, the generation of H2 builds up an internal pressure of a battery which can potentially lead to bulge, expansion and deformation, or even leakage, burst and explosion, especially in the cases of pouch cells.43–45 Secondly, H2 bubble formation exacerbates the uneven surface contour of the metal anode, influencing the deposition behaviors and further deteriorating the battery stability.46 Thirdly, the HER is a competitive reaction with metal plating, which results in a narrow electrochemical window. This limits the working voltage and the energy output of a battery, reducing the energy density and specific energy. Last but not least, the HER induces a series of parasitic side effects that are detrimental to the battery performance. The parasitic side effects are mainly corrosion and passivation, which will be specified in the next section.
Passivation, associated with the HER and corrosion, arises from the by-product generated by those two reactions. Specifically, in the context of an aqueous metal battery, passivation, as a challenge, should denote the formation of a layer on the metal anode that is extremely low in both electronic and ionic conductivities (Fig. 2g). Comparatively, this passivation layer differs from other layers. While layers with high electronic conductivity can serve as electrodeposition substrates and layers with low electronic conductivity but high ionic conductivity can serve as solid–electrolyte interphases (SEIs) or interlayers through which metal ions pass and deposit beneath, passivation layers block both electrons and ion flow, potentially resulting in an open-circuit condition where electrochemical reactions cease to proceed anymore. Typical examples include Al(OH)3 or Al2O3 layers in AABs, MgO, Mg(OH)2, or MgCO3 layers in aqueous Mg batteries and Mn(OH)2 or other complex Mn oxides (Mn3O4, Mn2O3, and MnO2) in aqueous Mn batteries. These passivation layers, if in a dense and compact form, can passivate the anode and greatly impede the normal electrochemical reactions.51,52 A ZHS layer in an AZB, although often unfavorable for its randomly loose and porous morphologies that obstruct uniform deposition, is actually found to be ionically conductive and hence cannot be strictly counted as a passivation layer.50,53
2.2. Interfaces of the cathode side
The main issue at the interfaces of the cathode side is the material dissolution. It usually occurs in V- and Mn-based oxides. MnO2 is a cathode material commonly used in various aqueous batteries, and in a discharging process, it can be reduced to Mn3+. Mn3+ is not chemically stable in the aqueous electrolyte due to the Jahn-Teller effect, resulting in its spontaneous disproportionation reaction to soluble Mn2+ and insoluble MnO2:54| MnO2 (s) + e− + 4H+ (aq) → Mn3+ (s) + 2H2O |
| 2Mn3+ (s) + 2H2O → Mn2+ (aq) + MnO2 (s) + 4H+ (aq) |
This overall reaction consumes protons near the cathode side, leveling the pH up and increasing the Mn2+ concentration in the electrolyte. The transformation to soluble Mn2+ may be the main reason for the rapid depletion and capacity decay of cathode materials. Furthermore, the phase instability of the Mn-based host with the [MnO6] octahedral basic unit during cycling can lead to the transition to a layered structure and subsequent structural collapse, resulting in additional capacity loss.55
For V-based cathode materials such as V2O5, they also confront the severe dissolution issue in the aqueous electrolyte with a large number of free water molecules. The generation of the soluble species of VO2(OH)2− occurs in the mild acidic to neutral environment (pH: 4–8) due to the high polarity of the water molecules:56
| V2O5 (s) + 3H2O → 2VO2(OH)2− (aq) + 2H+ (aq) |
The dissolved VO2(OH)2− can further react with the active cations such as Zn2+ in the electrolyte and produce Zn3V2O7(OH)2·2H2O, leading to considerable capacity degradation as a result of the material instability.
For organic cathode materials mainly dependent on the carboxyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) as the redox center, it is also possible to form the soluble (C–O−) chemicals during reduction dissolved in the aqueous electrolyte.
O) as the redox center, it is also possible to form the soluble (C–O−) chemicals during reduction dissolved in the aqueous electrolyte.
Other challenges faced by the cathode materials include the formation of the by-products (e.g., ZHS) and the nature of the poor electrical conductivity of these popular cathode materials (e.g., 10−5–10−6 S cm−1 for MnO2 and 10−2–10−3 S cm−1 for V2O5). Potential interface/interphase engineering strategies utilized in overcoming these challenges can be categorized as artificial interlayer design and cathode–electrolyte interphase (CEI) construction. As the main research focus on multivalent aqueous batteries is at the metal anode side, we will focus on the design rationale at the anode–electrolyte interface and supplement the cathode–electrolyte interface designs in the relevant sections.
Given the aforementioned challenges at the interfaces in multivalent aqueous battery systems, here we outline three interfacial design concepts, including regulating metal deposition behavior, homogenizing ion flux, and tuning ion solvation chemistry (Fig. 3), to alter the electrochemical behavior at multiple interfaces/interphases and thus control the dendrite morphology, suppress the side reactions, and enlarge the electrochemical stability window of the aqueous batteries.
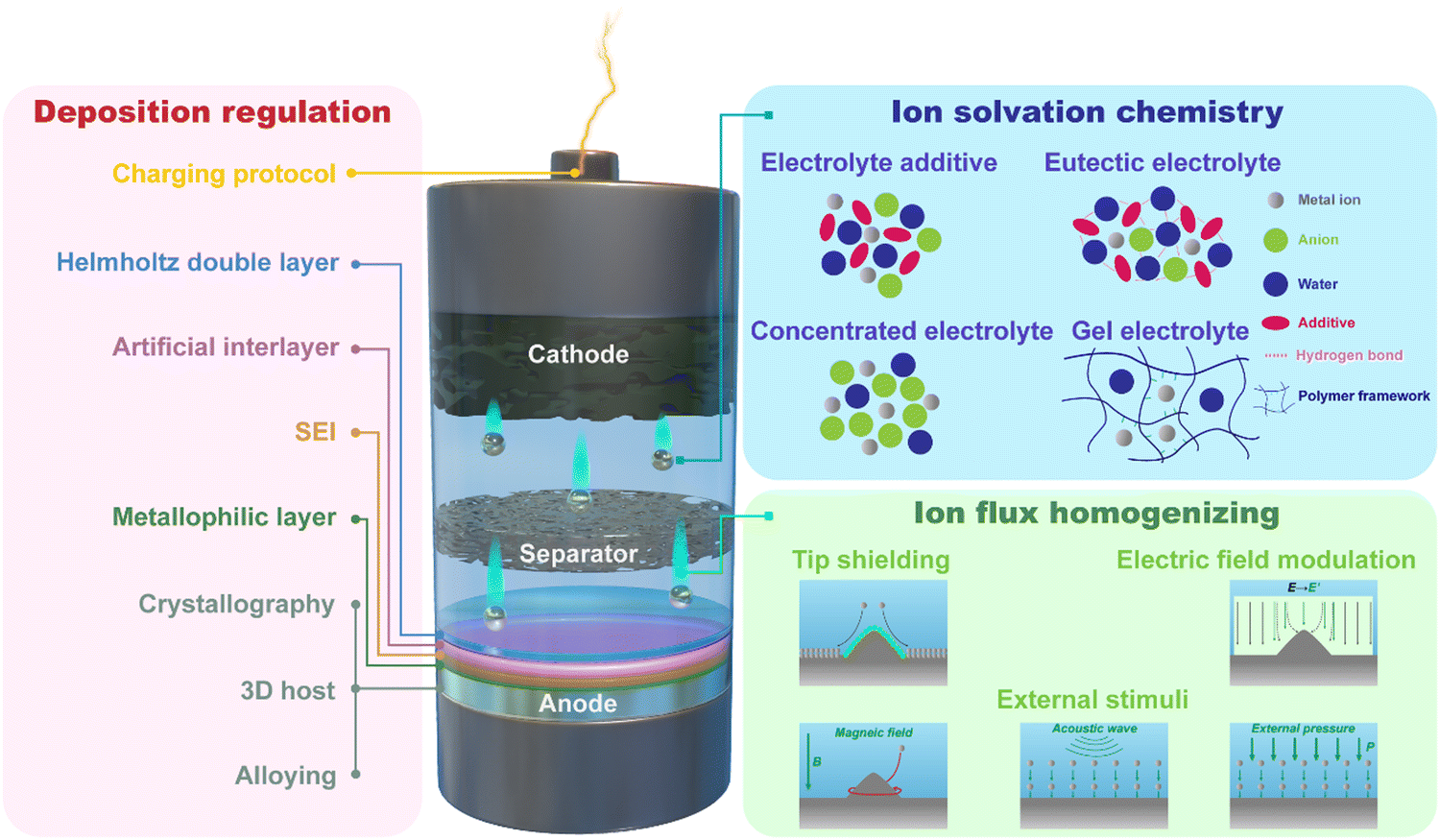 | ||
| Fig. 3 Schematic illustration of key components of the multivalent aqueous batteries and related strategies to address the main challenges at the interface/interphase via regulating metal deposition, tuning ion solvation chemistry, and homogenizing ion flux. | ||
3. Plating/stripping behavior regulation
3.1. Nucleation enhancement
Nucleation refers to the formation of a new solid phase on anode surfaces when ions transport from the cathode to the anode upon charging. In general, there are two types of nucleation, namely the homogeneous one and the heterogenous one. The case of metal deposition in aqueous batteries belongs to the latter since metal anodes serve as nucleation surfaces and provide nucleation sites. Energy barriers exist for both homogeneous nucleation and heterogenous nucleation, which can be characterized by ΔG(r), the Gibbs free energy change as a function of the radius of the nucleus (Fig. 4a). For homogeneous nucleation:where r is the radius of the nucleus, Δg is the negative free energy difference per unit volume between the stable and metastable phases, and σ is the surface tension between the nucleus and the electrolyte. The maximal ΔG(r) represents the energy barrier for nucleation, which is denoted as ΔG*. ΔG* can be expressed as follows:
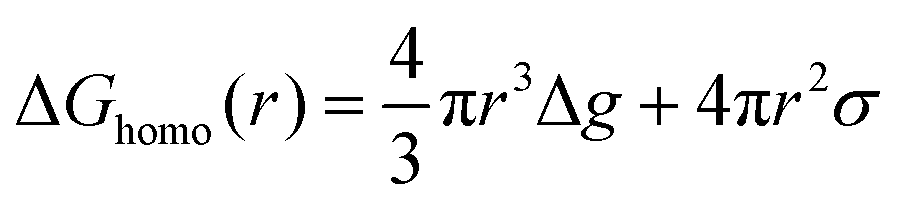
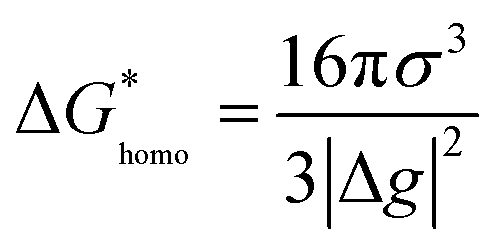
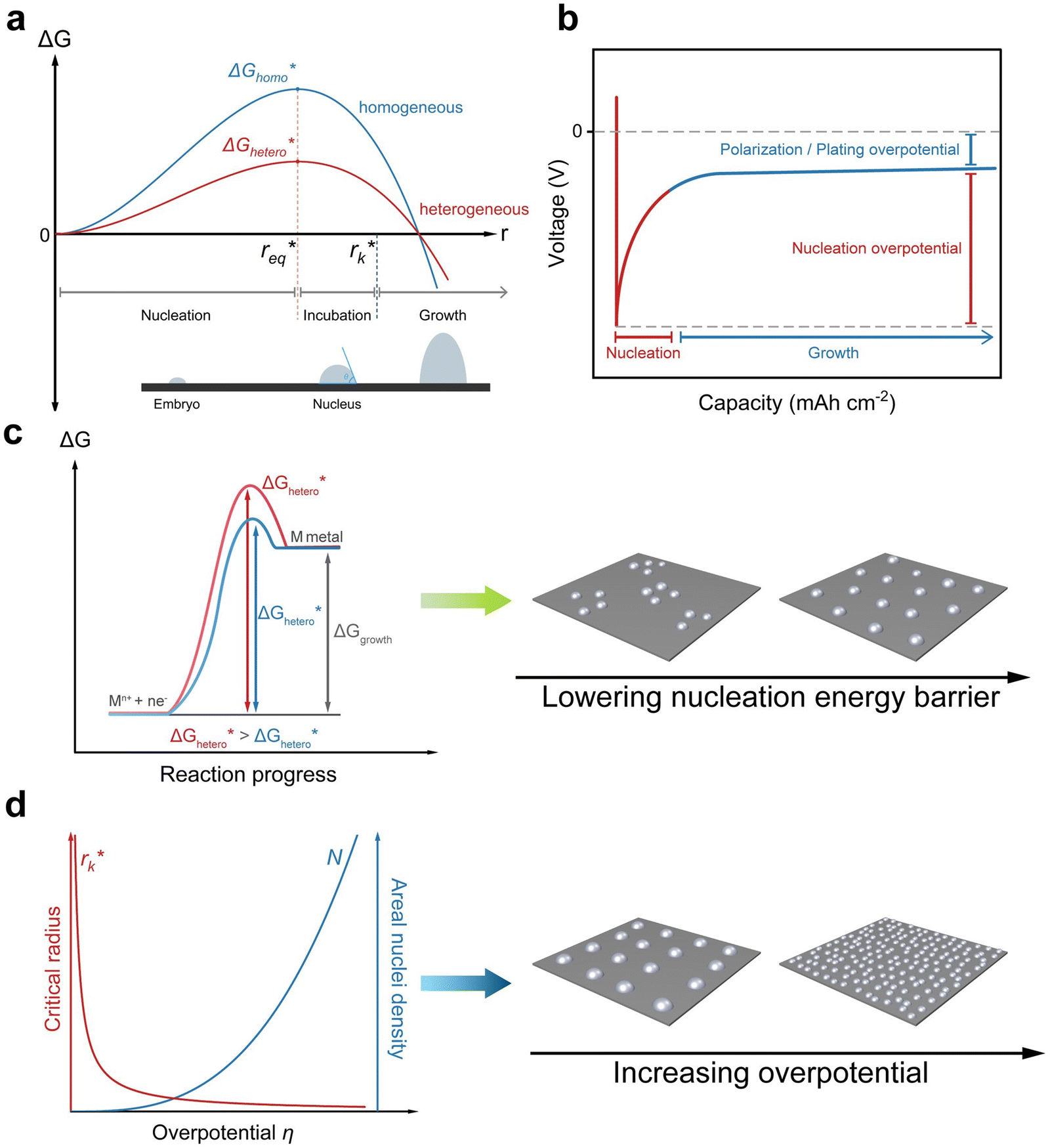 | ||
Fig. 4 Illustration of the nucleation mechanism and the related influencing factors. (a) Schematic diagram of Gibbs free energy to radius for homogeneous and heterogeneous nucleation, along with critical radii and the three stages of nucleation. (b) Voltage profiles dependent on plating capacity showing the regions for nucleation and growth. (c) The effect of heterogeneous nucleation Gibbs free energy 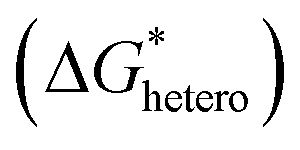 on the nucleation behavior. (d) The effect of overpotential (η) on the nucleation behavior. on the nucleation behavior. (d) The effect of overpotential (η) on the nucleation behavior. | ||
For heterogeneous nucleation, ΔG(r) is given by:
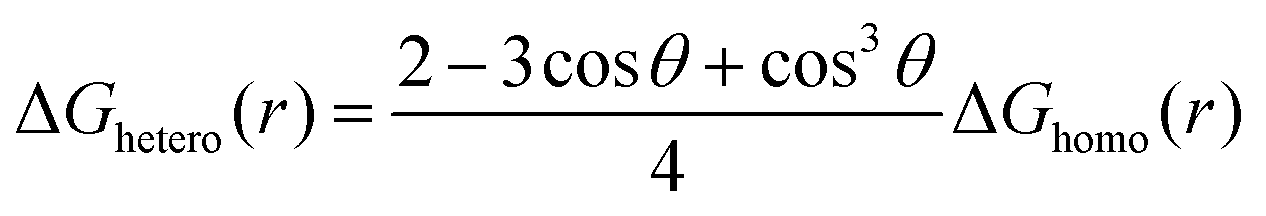
where θ is the contact angle between the nucleus and the surface. ΔG* corresponds to the critical radius of the nucleus, denoted as r* (when the overpotential η = 0), which is given by:
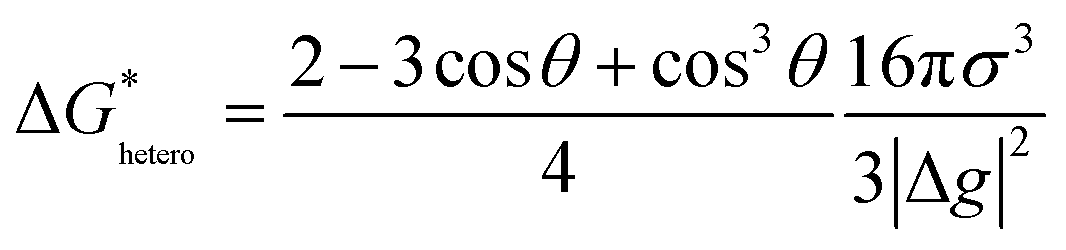
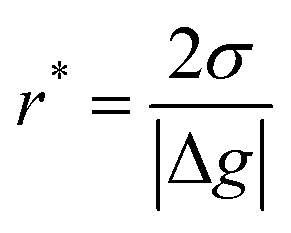
Considering the overpotential, the thermodynamic critical radius 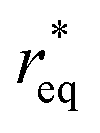 is given by:
is given by:
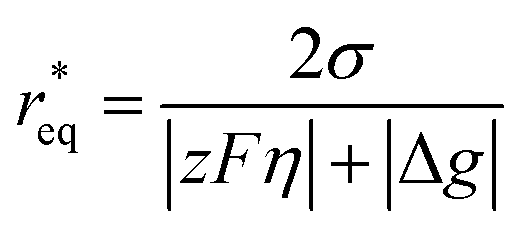
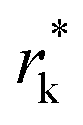 is given by:
is given by: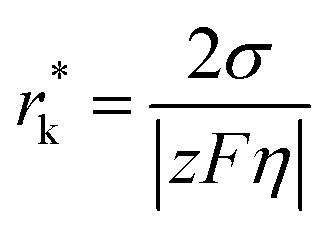
The critical overpotential for nucleation η0 can be given by:
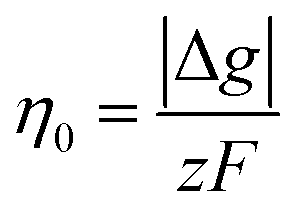
Kinetically, a nucleation and growth process can be divided into five regimes: the nucleation suppression regime, long incubation time regime, short incubation time regime, early growth regime, and late growth regime. In the nucleation suppression regime, the radii of the nuclei are below 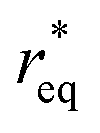 and the driving energy for nucleation is lower than
and the driving energy for nucleation is lower than 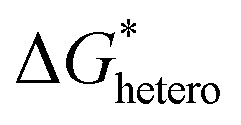 ; hence, the embryos are thermodynamically unstable. With a relatively low η (η < 2η0), when the radii of the nuclei become larger than
; hence, the embryos are thermodynamically unstable. With a relatively low η (η < 2η0), when the radii of the nuclei become larger than 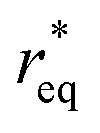 but lower than
but lower than 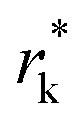 , the metastable embryos coarsen due to the Gibbs–Thomson interactions, which corresponds to the long incubation time regime. When η is relatively large (η > 2η0),
, the metastable embryos coarsen due to the Gibbs–Thomson interactions, which corresponds to the long incubation time regime. When η is relatively large (η > 2η0), 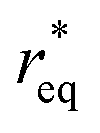 and
and 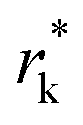 are rather close to each other, leading to accelerated growth of embryos, which is identified as the short incubation time regime. In the late growth regime, the growth of nuclei is thermodynamically favorable, and the evolution of the deposit is dominated by morphological instabilities and localized electric fields.40,57 The process of electrodeposition can be characterized by a voltage–capacity curve, where nucleation first takes place with a relatively high nucleation overpotential, and after reaching the critical sizes, nuclei grow with a polarization/plating overpotential (Fig. 4b).
are rather close to each other, leading to accelerated growth of embryos, which is identified as the short incubation time regime. In the late growth regime, the growth of nuclei is thermodynamically favorable, and the evolution of the deposit is dominated by morphological instabilities and localized electric fields.40,57 The process of electrodeposition can be characterized by a voltage–capacity curve, where nucleation first takes place with a relatively high nucleation overpotential, and after reaching the critical sizes, nuclei grow with a polarization/plating overpotential (Fig. 4b).
In practice, most studies consider low overpotential at a certain current density as a sign of desired nucleation, while there are also some studies that report that high overpotential can lead to stable cycling as well.58–60 Considering that it is possible to control the overpotential manually by controlling parameters such as current density, in general, the guiding principle for uniform nucleation mainly focus on lowering the nucleation energy barrier 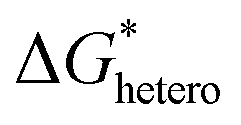 . By lowering the nucleation energy barrier, the nucleation Gibbs free energy becomes closer to the growth Gibbs free energy, hence enabling simultaneous nucleation and growth, which leads to uniform deposition rather than advantageous growth over new nucleation which is likely to result in dendrites (Fig. 4c).61 In addition, a lower nucleation energy barrier results in more nucleation sites, which favors dense and uniform nucleation (Fig. 4d).
. By lowering the nucleation energy barrier, the nucleation Gibbs free energy becomes closer to the growth Gibbs free energy, hence enabling simultaneous nucleation and growth, which leads to uniform deposition rather than advantageous growth over new nucleation which is likely to result in dendrites (Fig. 4c).61 In addition, a lower nucleation energy barrier results in more nucleation sites, which favors dense and uniform nucleation (Fig. 4d).
Based on this principle, the three strategies, including alloying, metallophilic layers and 3D host design, can be employed for interface and interphase engineering. In addition, a strategically charging protocol can also assist uniform nucleation and growth as a generic method without the specific context of specific chemistries.
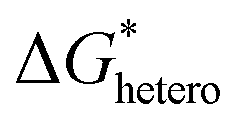 . Alloying elements with high binding energy also increase the number density of potential nucleation sites and favor uniform nucleation.61 The rational selection of the appropriate alloying elements can be based on the density functional theory (DFT) calculation that reveals the binding energy of a certain element with the metal atom to be deposited, and further experimental screening that selects out the alloy with low nucleation overpotential and high cycling stability (Fig. 5a).62
. Alloying elements with high binding energy also increase the number density of potential nucleation sites and favor uniform nucleation.61 The rational selection of the appropriate alloying elements can be based on the density functional theory (DFT) calculation that reveals the binding energy of a certain element with the metal atom to be deposited, and further experimental screening that selects out the alloy with low nucleation overpotential and high cycling stability (Fig. 5a).62
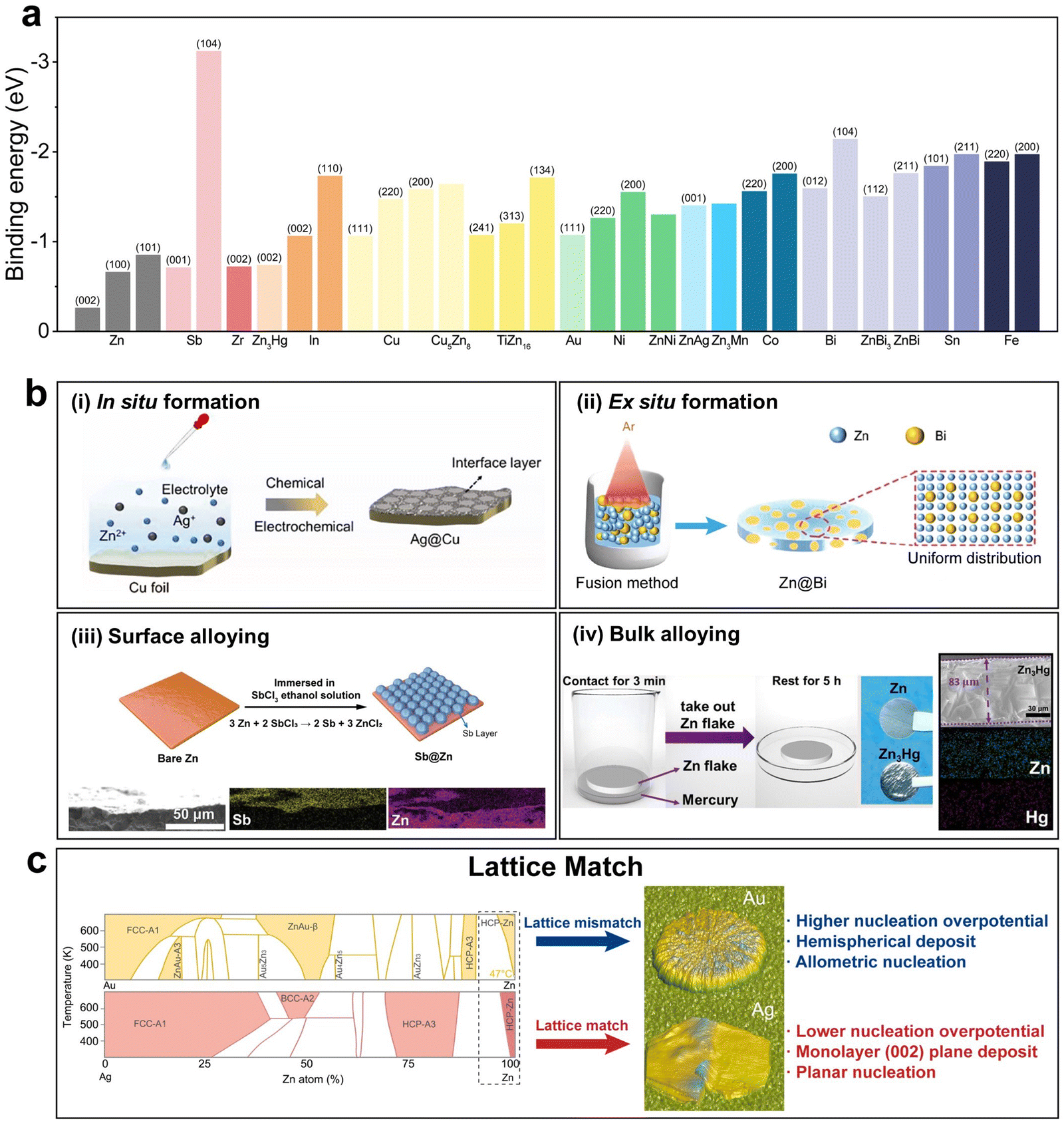 | ||
| Fig. 5 Alloying. (a) Binding energy of different metals or alloys with Zn.27,61–72 (b) Schematic illustration of different alloying strategies: (i) in situ formation of the Ag@Cu alloy. Reproduced with permission from ref. 73 Copyright 2023, Elsevier. (ii) Ex situ formation of the Zn@Bi alloy. Reproduced with permission from ref. 74 Copyright 2023, Wiley-VCH. (iii) Surface alloying of Sb@Zn. Reproduced with permission from ref. 63 Copyright 2022, Wiley-VCH. (iv) Bulk alloying of Zn3Hg. Reproduced with permission from ref. 72 Copyright 2022, Elsevier. (c) Schematic illustration of the lattice matching effect. Reproduced with permission from ref. 69 Copyright 2024, Wiley-VCH. | ||
There are various methods employed to obtain Zn alloys. Based on the formation and the alloy structure, these methods can be generally categorized into four groups: in situ formation, ex situ formation, surface alloying and bulk alloying. Zhang et al. introduced the AgNO3 additive in the conventional aqueous ZnSO4 electrolyte, enabling the in situ formation of a compact Ag layer via the replacement reaction between Ag+ and Cu.73 The Ag layer acted as zincophilic sites that lowered the nucleation energy barrier, beneficial for nucleation and growth. Du et al. utilized the fusion method to ex situ synthesize a Zn@Bi alloy anode.74 The Zn@Bi alloy managed to guide uniform nucleation and deposition of zinc due to its zincophilicity as well as to inhibit the HER and corrosion. Hong et al. constructed a uniform and robust metallic Sb layer on the surface of bare zinc via the replacement reaction between Sb3+ and Zn.63 The Sb@Zn anode provides abundant zincophilic sites for Zn nucleation and also acts as a protective layer against the HER. Tao et al. prepared a zinc-amalgam Zn3Hg alloy anode which represented bulk alloying.72 Not only was the binding ability between the Zn3Hg (002) surface and the Zn atom stronger than that between the Zn (101) surface and the Zn atom at all possible sites, the Zn3Hg anode also displayed a unique synergistic zinc plating/stripping and alloying/dealloying reaction mechanism. Besides the cases above, there are a number of other studies with alloying element selections such as Sn,75 Ag,69,70,76 Bi,62 Cu,67,77 Zr,65 Mn,27 In,66,78,79 Ti,61 Ni,71 and Ga–In80 based on the general guiding principle that contribute to uniform nucleation based on the basic principle of lowering the nucleation energy barrier for uniform nucleation.
Apart from the perspective of binding energy, a lattice match between the deposited metal and the substrate should also be considered, as was stated by Zhou et al., who reported that although Au had a higher binding energy with Zn than that with Ag, the lattice mismatch between the Zn@Au alloy and Zn led to higher overpotential and a less preferable nucleation morphology (Fig. 5d).69 Moreover, the lattice mismatch between the Zn@Au alloy and Zn resulted in allometric nucleation and hemispherical deposits, while the Zn@Ag alloy guaranteed favorable planar nucleation and (002) crystal plane dominated deposition.
It is noteworthy that the alloying method is extensively utilized not only in AZBs but also in AABs.81–84 However, the key objectives behind its use vary significantly. In AZBs, alloying vastly revolves around nucleation enhancement through the high binding energy of alloying elements, while in AABs, where controlling rampant side reactions is a critical concern, alloying primarily serves to inhibit these undesirable side reactions. The rational selection of the Al-based alloy can simultaneously achieve low nucleation energy barriers and inhibit side reactions. Ran et al. reported eutectic aluminum–copper (E-Al82Cu18) alloy lamellar heterostructures as anode active materials, which realized dendrite-free Al deposition by using periodic galvanic couplings of alternating monometallic α-Al and intermetallic Al2Cu nanometric lamellas.85 Wang et al. utilized a Ga-metal anode for reversible alloying/dealloying reactions between metallic Ga and Al, rendering high interfacial stability and low nucleation overpotential.86 Such a design principle can be further extended to other aqueous battery systems, e.g., Mg-metal batteries.87
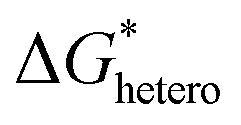 . The construction of a metallophilic layer changes the nucleation sites from the original metal anode to the top surface of the metallophilic layer. Therefore, nucleation behaviors are dependent on the properties of the metallophilic layer rather than the original metal anode and can then be regulated via the design of the metallophilic layer. A metallophilic layer is similar to the case of surface alloying mentioned above, but with more varieties in terms of the material selection. The appropriate materials should bear high adsorption energy with the metal atoms to be deposited to enhance nucleation. Additionally, to guarantee deposition on top of the metallophilic layer, the layer should have sufficient electrical conductivity.
. The construction of a metallophilic layer changes the nucleation sites from the original metal anode to the top surface of the metallophilic layer. Therefore, nucleation behaviors are dependent on the properties of the metallophilic layer rather than the original metal anode and can then be regulated via the design of the metallophilic layer. A metallophilic layer is similar to the case of surface alloying mentioned above, but with more varieties in terms of the material selection. The appropriate materials should bear high adsorption energy with the metal atoms to be deposited to enhance nucleation. Additionally, to guarantee deposition on top of the metallophilic layer, the layer should have sufficient electrical conductivity.
In AZBs, a large number of zincophilic layer designs have been reported. Based on the requirement of zincophilicity and electrical conductivity, carbon materials are suitable candidates, among which graphene and its derivatives have often been selected. It should be noted that the pronounced zincophilicity of graphene derivatives mainly stems from heteroatoms and functional groups including carbonyl, carboxyl, hydroxyl, epoxide, pyrrolic N, pyridinic N, graphitic N, sulfonate, etc., while the underemphasized zincophilicity of graphene itself stems from defects such as breakages, wrinkles and edges (Fig. 6a).88–91 Foroozan et al. grew monolayer graphene on the Cu substrate via chemical vapor deposition (CVD) as the electrodeposition substrate (Fig. 6b).92 The highly defective nature of CVD-grown graphene facilitated the creation of sites with low nucleation overpotential owing to the high binding energy involved. The lattice match between graphene and Zn favored the preferential formation of (002)-facet-oriented planar nucleation. N-doped graphene was introduced as a zincophilic layer by Xie et al. and characterized by both ex situ X-ray absorption spectroscopy and in situ Raman spectroscopy.91 It was revealed that Zn2+ was bonded with pyridine sites to form Zn–N bonds, which further induced the spacious nucleation of zinc (Fig. 6c). Besides the utilization of carbon materials themselves, Wang et al. assisted the zincophilic but poorly conductive Na3V2(PO4)3 (NVP) with conductive carbon materials such as activated carbon (AC), carbon nanotubes (CNT), and carbon black (CB) by exploiting their good electrical conductivity (Fig. 6d).93 The carbon coated Na3V2(PO4)3 proved to be an effective zincophilic layer on the zinc metal that induced uniform nucleation and deposition.
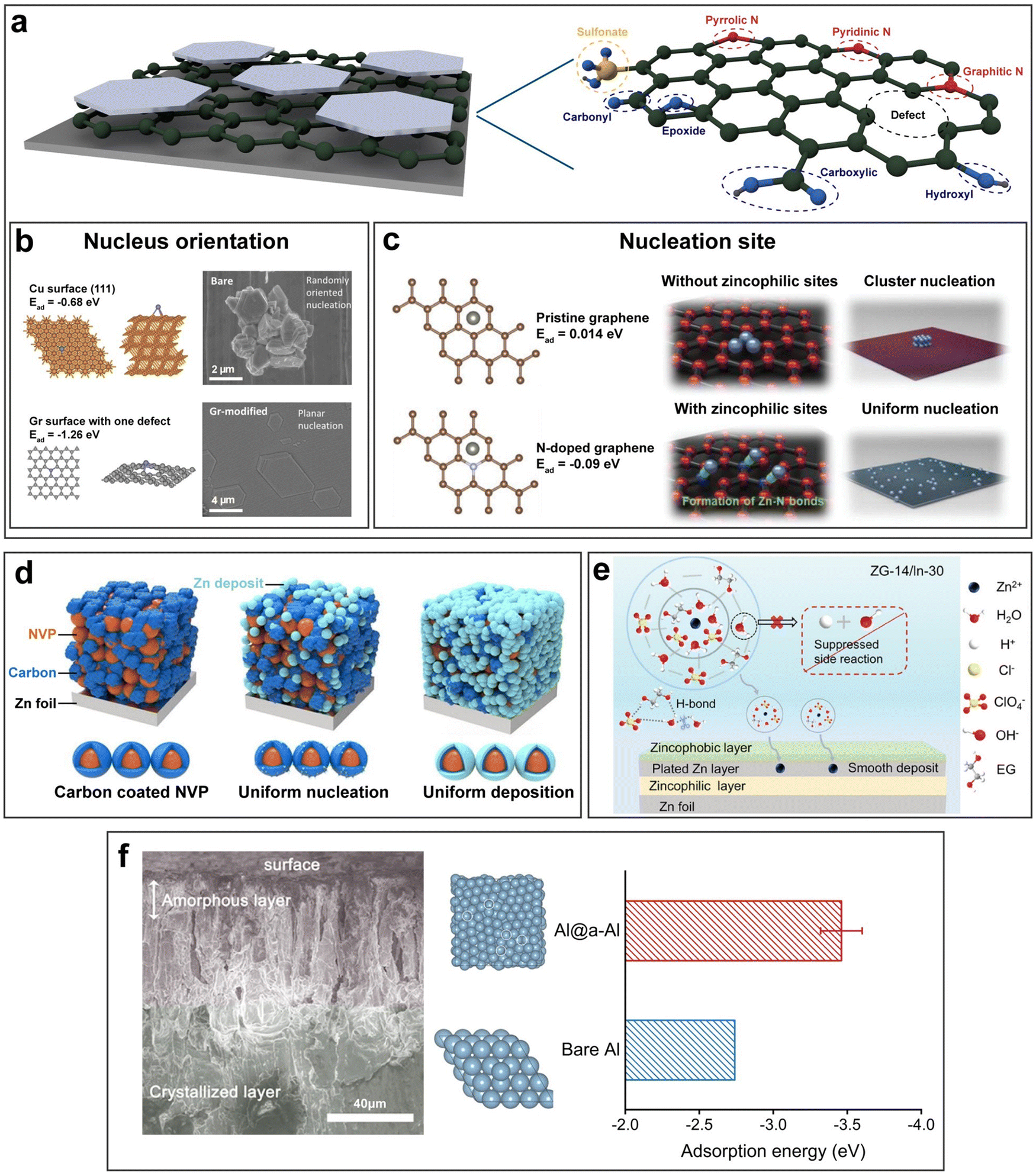 | ||
| Fig. 6 Metallophilic layers. (a) Schematic illustration of a graphene-based zincophilic layer. (b) and (c) The effect of a graphene-based zincophilic layer on the nucleus orientation (b) and nucleation site (c). Reproduced with permission from ref. 92 Copyright 2019, American Chemical Society. Reproduced with permission from ref. 91 Copyright 2021, Wiley-VCH. (d) Schematic illustration of a carbon-coated NVP zincophilic layer. Reproduced with permission from ref. 93 Copyright 2024, Elsevier. (e) Schematic of a tri-layer interface structure in a hybrid hydrated eutectic system. Reproduced with permission from ref. 94 Copyright 2024, Wiley-VCH. (f) Cross-sectional SEM images of the amorphous and crystalized Al layers and the corresponding binding energy. Reproduced with permission from ref. 95 Copyright 2022, American Chemical Society. | ||
However, the potential problem faced by a metallophilic layer is that although it can effectively enhance nucleation and initial growth, it will be fully covered by the newly deposited metal after further plating. In this case, new nucleation and growth take place on the surface of the metal that is already deposited, and their behaviors are no longer dependent on the initial metallophilic layer. Moreover, the direct exposure of a newly deposited metal to water can lead to the HER and corrosion that impairs cycling stability. Therefore, it is unlikely that a metallophilic layer itself is able to consistently regulate the deposition behavior throughout the whole plating process. To tackle such a problem, a more delicate and multifunctional multilayer structure was achieved by utilizing a novel hydrated eutectic electrolyte (HEE) composed of Zn(ClO4)2·6H2O, ethylene glycol, and InCl3 solution.94 The HEE in situ formed a zincophilic layer through the replacement reaction between In3+ and Zn on the Zn anode surface (Fig. 6e). Meanwhile, on top of the plated Zn, a zincophobic layer was also formed which acted as a barrier to prevent water penetration on the surface of the Zn anode, hence inhibiting side reactions.
For AABs, Yan et al. designed an amorphous Al layer on top of the crystallized one (Fig. 6f).95 It was found that the amorphous metals lacking the long-range ordered atomic arrangement exhibited elevated reactivity than the crystalline ones with long-range lattice periodicity, thereby demonstrating a higher adsorption energy and a lower Al nucleation energy barrier. Apart from nucleation enhancement, the reduced nucleation energy barrier also promoted competitive Al deposition over the HER, thereby minimizing side reactions. Meng et al. constructed an ultrathin platinum aluminophilic interface layer (AIL) on Ti for reversible Al plating/stripping.96 Due to the stronger binding energy between the Pt(111) plane and the Al atom compared to that of the Ti(101) and Al(111) planes, the Al nucleation barrier and nucleation overpotential could be lowered with such an AIL. For aqueous Sn batteries, Xu et al. employed Cu as a stannophile layer due to the high adsorption energy of Sn atoms on the Cu (111) plane.97 A low nucleation overpotential of 3.4 mV was thus achieved, and uniform nucleation as well as uniform deposition were realized.
To confirm the relationship between the nucleation site density and the deposition behavior, a brief theoretical analysis can be conducted. It is generally known that an electrodeposition process goes from the formation of nuclei at nucleation sites to the growth of nuclei through the continuous reduction of metal ions in the electrolyte.98,99 Assuming that nucleation sites are randomly distributed with number density N0, nucleation and growth can be described by the following first-order linear equation:
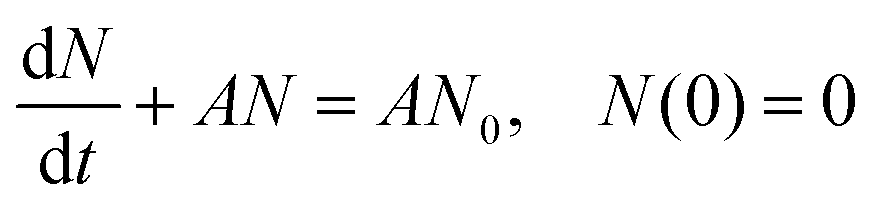
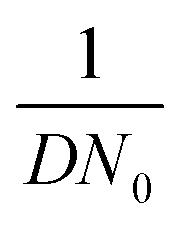 and charge transfer
and charge transfer 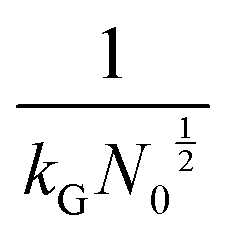 (D and kG denote the diffusion coefficient and the charge transfer rate constant, respectively), three different growth regimes can be distinguished: kinetic control
(D and kG denote the diffusion coefficient and the charge transfer rate constant, respectively), three different growth regimes can be distinguished: kinetic control 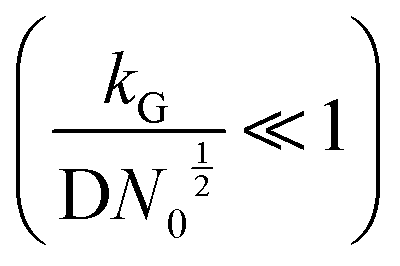 , mixed kinetic-diffusion control
, mixed kinetic-diffusion control 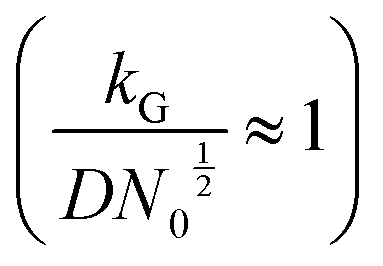 , and diffusion control
, and diffusion control 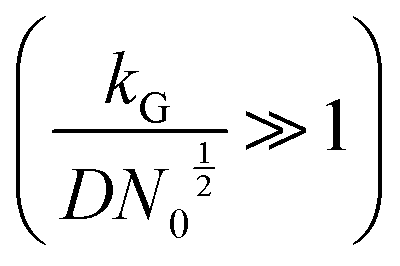 .102 With kG and D fixed, larger N0 results in lower
.102 With kG and D fixed, larger N0 results in lower 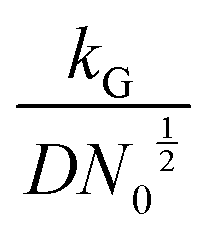 and favors the transition to the kinetic control growth mode. While the diffusion controlled growth is generally associated with dendrite formation, the kinetic control growth mode favors the uniform deposition.102–105 Therefore, increasing the nucleation site density is an effective approach to achieve uniform nucleation and deposition.
and favors the transition to the kinetic control growth mode. While the diffusion controlled growth is generally associated with dendrite formation, the kinetic control growth mode favors the uniform deposition.102–105 Therefore, increasing the nucleation site density is an effective approach to achieve uniform nucleation and deposition.
Apart from the advantage of augmenting the nucleation site density, 3D hosts possess the following merits as well: (i) a large surface area lowers the local current density, (ii) 3D geometric structures offer sufficient buffer space to accommodate volume changes, (iii) 3D scaffolds provide mechanical and chemical stabilities during electrochemical cycling, (iv) the unhampered pathways for ion transport lead to fast kinetics, and (v) the 3D structures are capable of rendering local electric field homogeneity. Importantly, 3D hosts should necessarily be accompanied with metallophilic modification, since the absence of metallophilic nucleation sites results in top-oriented nucleation,106–109 and in comparison, 3D hosts with metallophilic modification enable uniform nucleation on the inner surface of the whole structures, thus inducing uniform deposition (Fig. 7a).
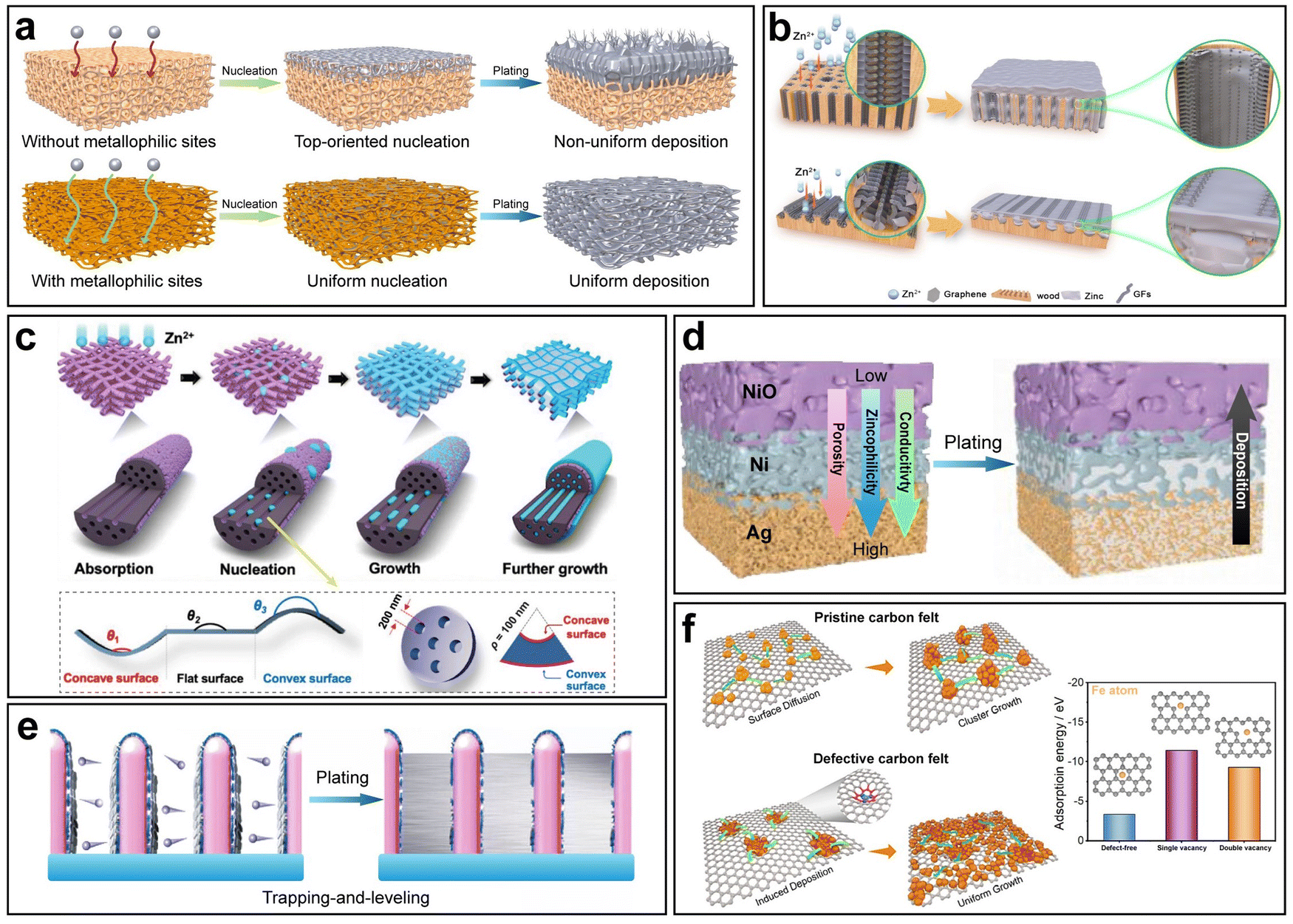 | ||
| Fig. 7 3D host design. (a) Schematic illustration of different metal deposition behaviors on 3D hosts with/without metallophilic sites. (b) Schematic illustration of two 3D hierarchically structured graphene matrices with different channel orientations. Reproduced with permission from ref. 110 Copyright 2023, Springer Nature. (c) Schematic illustration of lotus root-like multichannel carbon nanofibers. Reproduced with permission from ref. 111 Copyright 2024, Wiley-VCH. (d) Schematic illustration of Zn deposition on a 3D host with a triple gradient. Reproduced with permission from ref. 112 Copyright 2023, Wiley-VCH. (e) Schematic illustration of trapping-and-leveling deposition on arrays of hierarchical zincophilic nanorods. Reproduced with permission from ref. 67 Copyright 2024, Wiley-VCH. (f) Schematic illustration of Fe deposition behaviors on pristine and defective carbon felts and the corresponding adsorption energy. Reproduced with permission from ref. 113 Copyright 2024, Elsevier. | ||
In AZBs, a vast array of 3D host designs featuring diverse 3D geometric structures has been reported, and most of them adopt zincophilic modification so as to fully exploit the advantages of 3D hosts. Zhou et al. reported a composite anode based on porous Cu foam with wires inside having a ZnO–Zn–CuZn5–Cu multilevel lamellar structure.114 The CuZn5 layer acted as zincophilic sites to modulate Zn nucleation to achieve uniform deposition. Although many other studies have also reported Cu-based 3D hosts, it was pointed out that the prevailing 3D Cu frameworks fell short of energy density due to their inferior density and areal loading.115 In response, Shi et al. developed a novel lightweight 3D Cu architecture.115 A cross-linked polymer scaffold was 3D printed, followed by Cu metallization. Based on the pattern customizability enabled by 3D printing, geometrical optimization was conducted, and the optimized 3D Cu structure was able to reduce around 80% of weight and Cu consumption.
3D scaffolds based on other matrix materials with low densities, high electrical conductivities and sufficient mechanical strengths have also been widely investigated. Carbon materials, fitting all the criteria, are a common choice. Mu et al. developed 3D hierarchical graphene matrices consisting of N-doped graphene nanofiber clusters anchored on modified multichannel carbon converted from biomass wood (Fig. 7b).110 The N-doped graphene enhanced the binding between the matrices and Zn atoms. In addition, two geometrical patterns, namely matrices of a longitudinal direction and a radial direction were compared, and it was found that the latter achieved more uniform deposition due to the shorter ion diffusion pathways and more open space for Zn plating. Yu et al. fabricated a 3D framework constructed from lotus root-like multichannel Zn/N-doped carbon nanofibers decorated with Zn/N-doped carbon nanocage layers (Fig. 7c).111 The effect of host geometry on Zn nucleation was specifically investigated. According to the dependence of the nucleation energy barrier 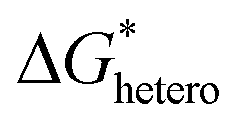 on the wetting angle θ between the substrate and Zn embryos, it can be deduced that a concave surface requires smaller embryos to reach the critical size than a flat or convex one, which lowers the nucleation barrier and promotes nucleation at lower overpotential.116 Because of this, along with the zincophilic N-doped graphene decoration, nucleation and early deposition preferentially took place in the inner side of a channel due to its concaveness compared to the outer surface, hence being confined inside the channels and leading to little overall volume expansion. In addition to the commonly adopted Cu and carbon materials, some other materials have also been utilized as the matrices for 3D hosts, such as stainless steel117 and Ti.118
on the wetting angle θ between the substrate and Zn embryos, it can be deduced that a concave surface requires smaller embryos to reach the critical size than a flat or convex one, which lowers the nucleation barrier and promotes nucleation at lower overpotential.116 Because of this, along with the zincophilic N-doped graphene decoration, nucleation and early deposition preferentially took place in the inner side of a channel due to its concaveness compared to the outer surface, hence being confined inside the channels and leading to little overall volume expansion. In addition to the commonly adopted Cu and carbon materials, some other materials have also been utilized as the matrices for 3D hosts, such as stainless steel117 and Ti.118
Beyond materials selections, some studies specify specific strategies unique to the 3D host design. One most commonly investigated strategy is the gradient design. As mentioned before, without sufficient metallophilic sites, especially without metallophilic sites at the bottom, nucleation is highly likely to take place at the top and then result in uneven deposition. However, if the distribution of metallophilic sites is uniform along the thickness direction, it is still likely that nonuniform deposition predominantly occurred at the top under conditions of high current density.44,119–121 To tackle this problem, the construction of a metallophilic gradient along the height direction is a solution. The upper part of a 3D host should exhibit lower metallophilicity, whereas the lower part should show higher metallophilicity. This design ensures nucleation and growth from the bottom to the top, which to the largest extent guarantees the spatially controllable deposition, accommodates the volume change and prevents the possible short circuit caused by top dendritic growth.122
In practice, control of the metallophilic gradient can be achieved by controlling the gradient distribution of metallophilic species. Lai et al. designed a 3D carbon fiber skeleton film with gradient zincophilicity and reversed gradient hydrophilicity.120 The two gradients were simultaneously realized by the gradient distribution of Cu nanoparticles. The abundance of Cu nanoparticles at the bottom resulted in regionally higher zincophilicity due to the highly zincophilic nature of Cu and lower hydrophilicity due to the high surface energy of ultrafine Cu nanoparticles. Such a dual-gradient design not only ensured the uniform bottom-up deposition, but also gave rise to a favorable effect that the top surface was accessible to the aqueous electrolyte while water was gradually repelled from top to bottom to effectively suppress side reactions. Moreover, Gao et al. proposed a more delicate strategy to realize 3D hosts with triple gradients of electrical conductivity, zincophilicity, and porosity (Fig. 7d).112 Ag was used as the bottom layer for its high conductivity and zincophilicity, NiO was used as the top layer for its semiconductivity and poor zincophilicity, and Ni was used as the middle layer for its moderate conductivity and zincophilicity between Ag and NiO. In addition, the porosity gradient was constructed by tuning the pore size inside each layer. The three gradients synergistically facilitated the smooth transport of the Zn2+ flux, optimized local charge transport dynamics at the bottom of the anode and promoted the desired bottom-up deposition behavior.
Another featured strategy in the 3D host design is ‘trapping-and-leveling’. As a proof of concept, arrays of core–shell Cu5Zn8 nanorods coated with N-doped carbon layers were constructed on Cu foam (Fig. 7e).67 Nucleation was guided by the zincophilic sites and preferentially took place within the interspace among the nanorods. Further high-capacity Zn plating could be tightly confined within the interspace, following a ‘trapping-and-leveling’ deposition process.
Despite all the advantages, a major challenge faced by a 3D host lies in the intensified side reactions due to the largely increased surface area. Therefore, protective methods should be applied accordingly to guarantee long stable cycling. It was reported that the Zn–Cu alloy itself could have a decent anti-corrosion effect due to the inactiveness of Cu.123 Apart from this, the ZnO outer layer could effectively inhibit the HER as well, achieving the negligible and stable HER with only 1.8 mmol h−1 cm−2 compared to 2.6–3.5 mmol h−1 cm−2 of the bare Zn anode.114 In fact, most of the studies on 3D hosts have taken into consideration the suppression of side reactions: the metallophilic materials can simultaneously present sufficient protection. Specifically, a strategy based on the 3D host design was proposed by Han et al. to target side reaction inhibition.124 The Cu nanorod arrays with hydrophobic long-chain alkyl groups in stearic acid provided ordered channels with both zincophilic and hydrophobic properties, introducing the ‘lotus effect’ to control water adsorption energy and thus fortify the anode against water-induced side reactions.
In AABs and aqueous Mg batteries, however, the utilization of 3D anode hosts is rarely reported, primarily due to the challenge of intensified side reactions as previously mentioned. Al and Mg possess lower reduction potentials than Zn, rendering them more vulnerable to the HER. Such susceptibility to the HER represents a critical concern that overshadows the advantages addressed by the design of 3D hosts.
In other aqueous battery systems, the 3D host design can be seen as well, such as in aqueous Fe metal batteries.113,125 Song et al. designed a modified carbon felt with abundant carbon defects that increased the affinity with Fe atoms (Fig. 7f).113 As a result, Fe could nucleate uniformly on defective carbon fibers and evolved into uniform deposits on a modified carbon felt, while nucleus clusters were present on pristine carbon fibers, leading to mossy deposition morphologies.
The 3D host design is widely investigated and applied in AZBs, while less common in some other aqueous battery systems, especially AABs and aqueous Mg batteries. The design strategies of a 3D host such as materials selection, gradient design, and side-reaction inhibition, however, are universal in various battery systems. With these strategies utilized, the 3D host design can exhibit excellent efficacy not only in nucleation enhancement, but also in the following deposit growth, thereby benefiting uniform deposition and prolonged life span of batteries.
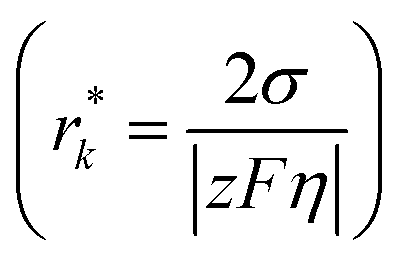 , and the areal nuclei density is proportional to the cube of overpotential (N ∝ η3). The resulting desired morphology of the initial nuclei appears as dense and fine particles that cover up the majority of the surface of the metal anode.126 High overpotential, however, is likely to result in nonuniform deposition and dendritic growth, the underlying mechanisms of which will be discussed in the following section.
, and the areal nuclei density is proportional to the cube of overpotential (N ∝ η3). The resulting desired morphology of the initial nuclei appears as dense and fine particles that cover up the majority of the surface of the metal anode.126 High overpotential, however, is likely to result in nonuniform deposition and dendritic growth, the underlying mechanisms of which will be discussed in the following section.
Based on the theoretical analysis above, a strategical charging protocol can be proposed: a large overpotential is applied for a short period of time at the beginning to promote uniform nucleation, followed by a relaxation step with zero current to offset the local concentration gradient. Afterwards, a low overpotential is applied in the further plating process (Fig. 8a). In practice, the control of overpotential can be achieved by controlling the current density. The relationship between the overpotential η and the current density J is given by: η ∝ log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) J, which is a simplification of the Butler–Volmer equation when η is relatively low (Fig. 8b).127 Hou et al. further experimentally verified this relationship.128 The three steps in the proposed charging protocol can be described as follows: high current density pulsed charging, relaxation and low current density charging.
J, which is a simplification of the Butler–Volmer equation when η is relatively low (Fig. 8b).127 Hou et al. further experimentally verified this relationship.128 The three steps in the proposed charging protocol can be described as follows: high current density pulsed charging, relaxation and low current density charging.
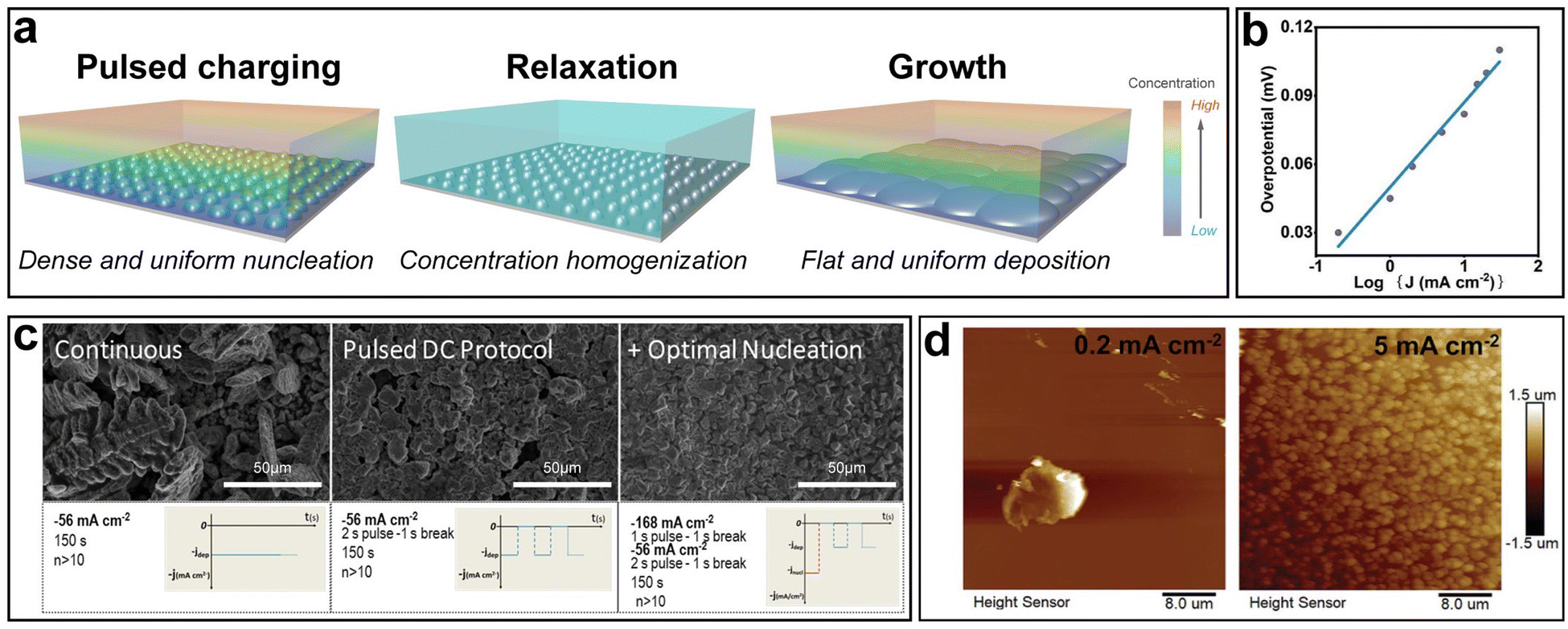 | ||
| Fig. 8 Strategical charging protocols. (a) Schematic of the process of an ideal pulsed charging protocol. (b) The dependence of overpotential on the current density. (c) Three different charging protocols and their effects on Zn nucleation. Reproduced with permission from ref. 129 Copyright 2017, American Chemical Society. (d) AFM images of Zn nuclei at different current densities. Reproduced with permission from ref. 128 Copyright 2022, Wiley-VCH. | ||
Garcia et al. utilized such a charging protocol in commercially available Ni–Zn batteries to optimize the Zn electroplating behavior.129 Three different charging protocols were compared (Fig. 8c). It was revealed that the continuous charging protocol resulted in dendrites, and the pulsed direct current (DC) protocol without nucleation optimization, despite dendrite prevention, resulted in nonuniform deposition as discontinuities such as empty gaps could be observed in the Zn film. The optimal nucleation charging protocol, where a short nucleation pulse at a high current density was introduced before the 2s-pulse-1s-break DC protocol for further growth, led to an obviously smoother deposition without visible discontinuities and dendritic growth. The role of the initial pulsed charging at a high current density was to promote dense and fine nucleation, while the role of the following breaks was to eliminate the local concentration gradient, which was critical in dendrite suppression.130,131 To better understand the effects of the initial pulsed charging on nucleation optimization, Hou et al. characterized the morphologies of Zn nuclei at different current densities via atomic force microscopy (AFM) (Fig. 8d).128 With the same deposition amount, the electrode surface was fully covered by fine Zn nuclei at a high current density of 5 mA cm−2, while at a low current density of 0.02 mA cm−2, a sparse and large nucleus was observed. Furthermore, Zn‖Zn symmetric cells with and without an initial high current density were cycled at 1 mA cm−2/1 mA h cm−2. It was demonstrated that the cell subjected to an initial high current density could maintain ultra-stable operation for 2500 h, a significant improvement over its counterpart which lasted only 300 h.
Ideally, applying an optimized charging protocol is a universal approach towards nucleation enhancement and desirable deposition morphologies that could work in all battery systems, such as AZBs, AABs and other aqueous ones. In practice, the potential parasitic effects of pulsed current density should be investigated in a specific system so as to minimize its detrimental impact.
3.2. Growth regulation
After the size of a nucleus reaches the critical radius and continues to grow,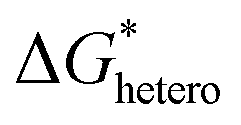 decreases and becomes negative. Since then, the thermodynamically favorable growth takes place spontaneously. A nucleation process typically occurs much more rapidly than the subsequent growth process. Consequently, while interfacial changes—such as the local concentration, electric field, and ion flux—may be negligible due to the relative instantaneity of nucleation, they become significant factors influencing the evolution of the electrodeposit over time during the growth stage. Thus, while the heterogeneous nucleation model suffices to describe the nucleation stage, those more complicated factors gain importance in shaping the electrodeposit's progression during growth. Therefore, it is crucial to first understand the growth process through theoretical analyses.
decreases and becomes negative. Since then, the thermodynamically favorable growth takes place spontaneously. A nucleation process typically occurs much more rapidly than the subsequent growth process. Consequently, while interfacial changes—such as the local concentration, electric field, and ion flux—may be negligible due to the relative instantaneity of nucleation, they become significant factors influencing the evolution of the electrodeposit over time during the growth stage. Thus, while the heterogeneous nucleation model suffices to describe the nucleation stage, those more complicated factors gain importance in shaping the electrodeposit's progression during growth. Therefore, it is crucial to first understand the growth process through theoretical analyses.
A common model used to describe a growth process is Chazalviel's space charge model.132–134 In a dilute electrolyte, when initial electrodeposition begins, metal cations start to diffuse towards the anode. At this stage, no significant accumulation of ions near the electrode is present, and the electric field is relatively weak. As metal cations approach the anode surface, they undergo a reduction reaction to form a solid deposit. The consumption of metal cations can be compensated by those extracted from the cathode. The anions, however, cannot be transferred to or from the electrodes. Thus, anions will be piled up near the anode and depleted near the cathode due to the limited mobility, which creates a space charge region. The presence of the space charge region alters the distribution of the electric field, the current density, and the ion flux. As a result of the space charge region, ramified growth of the metallic electrodeposit occurs.134 In a symmetric cell configuration with a small interelectrode distance, based on the space charge model, the concentration gradient at the anode surface 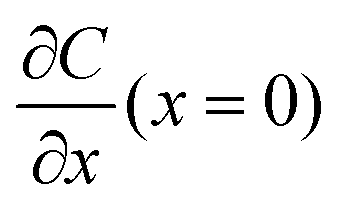 can be given by:
can be given by:
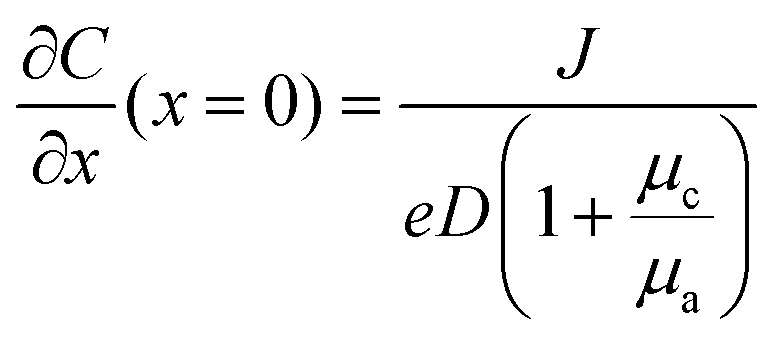
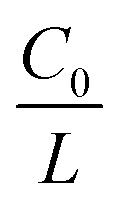 represents the initial average concentration, where L is the interelectrode distance and C0 is the initial concentration of the electrolyte. If the concentration gradient at the anode surface is lower than two times of the initial average concentration, or
represents the initial average concentration, where L is the interelectrode distance and C0 is the initial concentration of the electrolyte. If the concentration gradient at the anode surface is lower than two times of the initial average concentration, or 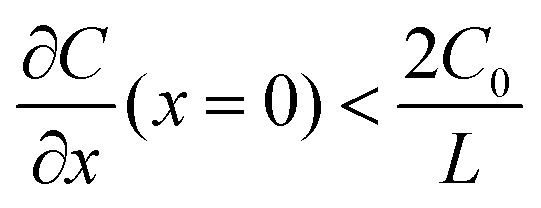 , the ionic concentration gradient at the negative electrode evolves to a steady state with a constant concentration gradient throughout the electrolyte and a stationary electric potential, leading to a smooth electrodeposit morphology.136 While if the concentration gradient at the anode surface is greater than two times of the initial average concentration, or
, the ionic concentration gradient at the negative electrode evolves to a steady state with a constant concentration gradient throughout the electrolyte and a stationary electric potential, leading to a smooth electrodeposit morphology.136 While if the concentration gradient at the anode surface is greater than two times of the initial average concentration, or  , the concentration will drop to zero at the negative electrode and the electric potential will eventually diverge (Sand's behavior) both at a time τ, which is called Sand's time.137 The Sand's time (τ) is given by:
, the concentration will drop to zero at the negative electrode and the electric potential will eventually diverge (Sand's behavior) both at a time τ, which is called Sand's time.137 The Sand's time (τ) is given by:and in which
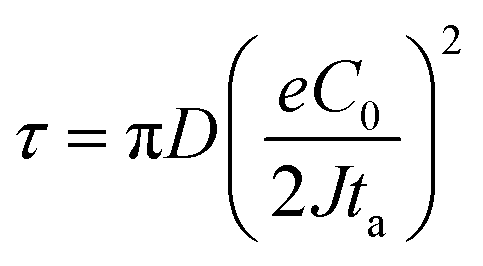
where ta is the anionic transference number and tc is the cationic transference number. A transference number is referred to as the ratio of the electric current carried by a certain ionic species to the total current carried by all ionic species. According to the space charge model, when Sand's time is reached, the ion concentration in the vicinity of the negative electrode drops to zero, resulting in the onset of dendritic growth.134
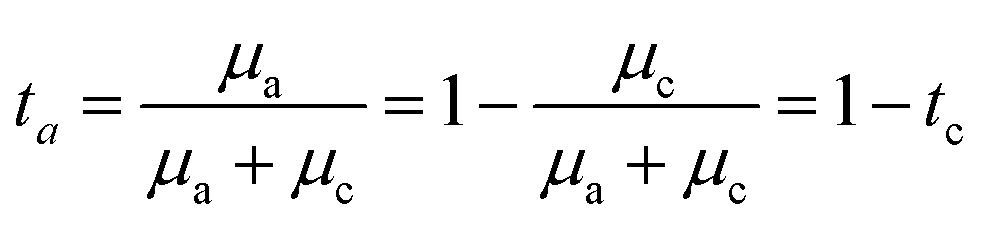
In addition, considering the dependence of 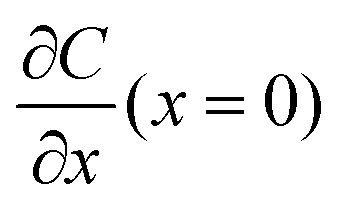 on J and the critical value of
on J and the critical value of 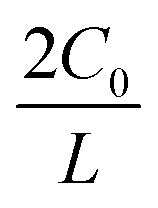 , the limiting current density J* can be given by:
, the limiting current density J* can be given by:
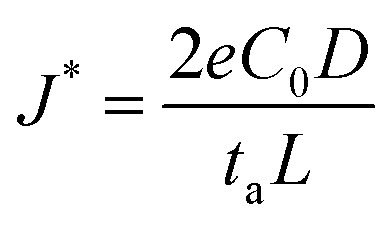
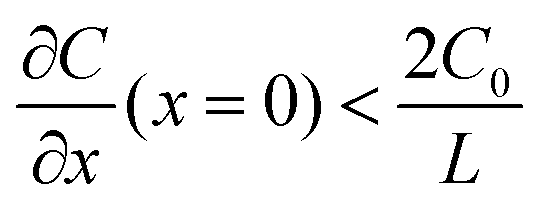 , a constant concentration gradient can be obtained. When J > J*, Sand's behavior occurs and induces dendritic growth.
, a constant concentration gradient can be obtained. When J > J*, Sand's behavior occurs and induces dendritic growth.
Based on the analysis above, it can be inferred that prolonging Sand's time τ delays the onset of dendritic growth and increasing the limiting current density J* suppresses the formation of dendrites. These can be achieved by decreasing the anionic transference number ta (or increasing the cationic transference number tc), increasing the diffusion coefficient D, increasing the initial electrolyte concentration C0, or lowering the effective current density J. This provides insight into the design principles for interface and interphase engineering targeting dendrite-free growth during an electroplating process.
In addition to being dendrite-free, an ideal electrodeposit morphology should also encompass traits such as being flat-surfaced, compact, and dense, thus minimizing the destructive effect on the separator and preventing the formation of ‘dead metal’ deposits. Regarding the morphologies of electrodeposits, it is widely reported that the second Damköhler number DaII, which governs the growth stability, can be used as an important indicator.138–142 DaII quantifies the relative rates of the electrode reaction and diffusion, and can be expressed as follows:
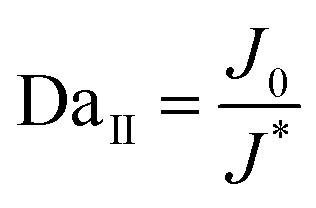
| J0 = nFCk0 |
Given the dependence of J*on the diffusion coefficient D and the dependence of J0 on the reaction coefficient k, a nondimensional electrochemical Damköhler number Da can be given in a simple form as follows:143
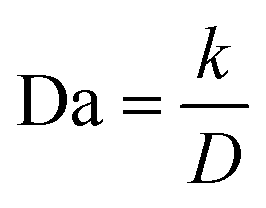
In short, in order to achieve the uniform deposition, a low Da is favored, which means that a lower reaction coefficient k, a lower exchange current density J0, and a higher diffusion coefficient are desirable.144 The requirement of a higher D aligns with the previous analysis on the space charge model and Sand's time.
Accordingly, three interface and interphase engineering strategies, including crystallographic manipulation, Helmholtz double layer regulation, and artificial interlayer and SEI design will be discussed based on the analyses of diffusion and deposition. Moreover, it is necessary to account for side reactions, which can significantly impact the morphologies of the electrodeposit substrates. Side reactions may alter both the original anode surface and the as-deposited metal surface, thereby influencing the deposition behavior. Thereby, when discussing the three aspects mentioned above, their effects in inhibiting side reactions will also be included.
In aqueous Fe metal batteries, for instance, it was reported that by adding ZnI2, Zn-doped Fe was obtained and it preferentially formed homogeneous small particles with the (110) plane parallel to the substrate, favoring uniform deposition.145
For aqueous Sn batteries, it was found that the Sn(OH)62−/Sn electrode naturally displayed smooth and dendrite-free deposition morphologies.146 The underlying reason was the intrinsic low surface-energy anisotropy of Sn. Sn adopts a body-centered tetragonal crystal structure and exhibits relatively minor surface energy differences between its various crystal planes (γ(102) = 0.023 ≈ γ(100) = γ(101) = γ(112) = 0.024 ≈ γ(201) = γ(210) = 0.025 ≈ γ(211) = γ(111) = 0.026, eV Å−2), which naturally results in the isotropic crystal growth mechanism during Sn electrodeposition.
For Al electrodeposition, the Al deposits tend to form (111) crystal facets during plating due to its lowest surface energy (0.96 J m−2), accounting for an equilibrium area fraction of 0.57 determined by the Wulff construction.147 The second significant facet is (100), with a surface energy of 1.05 J m−2 and an equilibrium area fraction of 0.17. Using a poor current collector such as stainless steel, the Al deposits tend to form large particles with a size of micrometers or classic dendritic morphologies at high current densities.148 Enhancing the interfacial interaction between the Al and the carbon fiber substrate could effectively downsize the Al deposits and achieve uniform deposition with enhanced (200) facet relative intensity.
Conversely, in AZBs, the more pronounced crystal plane-surface energy anisotropy of Zn is a contributing factor to dendrite formation. Consequently, a significant portion of research efforts in this field is dedicated to manipulating the crystallography of Zn. Therefore, the discussion on crystallographic manipulation herein will primarily focus on Zn.
Zn belongs to the P63/mmc space group and has a hexagonal closely packed (hcp) crystal structure. The three most common crystal planes referred to in the growth process of Zn electrodeposition are (002), (100) and (101) planes. The respective selective growth of the three crystal planes has all been reported. Without specific manipulation, random growth which involves all the three crystal planes is present, showing an undesired mossy and dendritic morphology. In comparison, the selective growth of a single crystal plane, regardless of which one among the three, yields an ordered, compact, and dendrite-free morphology. However, the determination of the most favorable crystal plane remains in suspense.
The (002) crystal plane garners the most attention in research, often being assumed as the most desirable crystal plane.149–154 The selective growth of the (002) plane showcases a planar structure parallel to the substrate with a hexagonal geometry, hence naturally favoring a flat and dendrite-free surface contour (Fig. 9a–c). The selective growth of the (100) and (101) planes, on the other hand, showcases a vertically aligned structure and a slightly tilted structure, respectively. Their highly ordered structures exhibit no dendrite growth either.
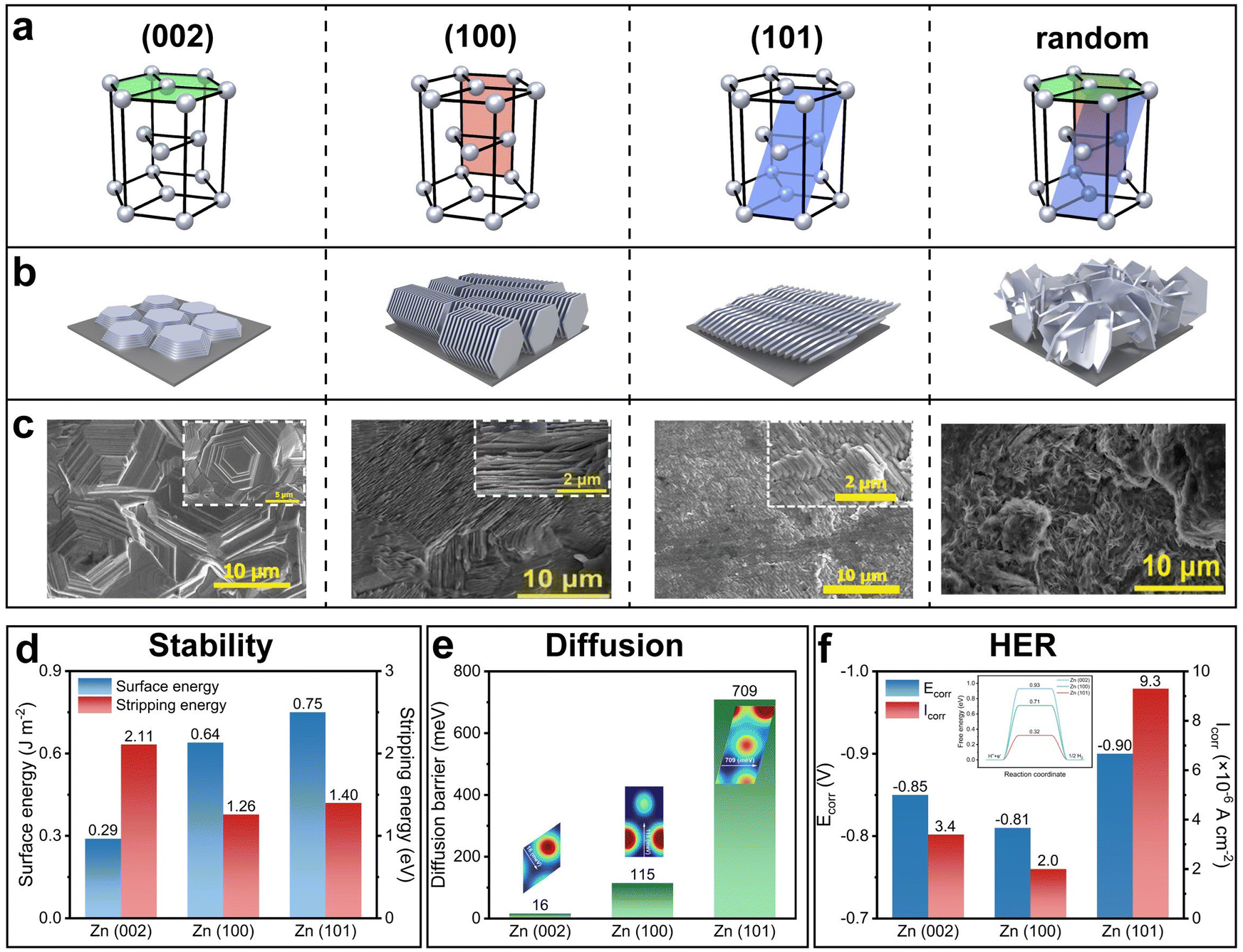 | ||
| Fig. 9 Zn crystal planes and their properties. (a)–(c) Schematic of the four common crystal planes of Zn (a) and their corresponding deposition orientations (b) and representative deposition morphologies (c). Reproduced with permission from ref. 155–157 Copyright 2023 and 2024, Wiley-VCH. (d) Surface energy and stripping energy of the (002), (100), and (101) crystal planes. Reproduced with permission from ref. 158 Copyright 2024, American Chemical Society. (e) Diffusion energy barrier of these crystal planes. (f) Corrosion potential, corrosion current density and HER energy barrier (inset) of these crystal planes. Reproduced with permission from ref. 159 Copyright 2024, National Academy of Sciences. | ||
In addition to the direct visual examination of the morphologies, other assessments of the three crystal planes in terms of their stability, the surface atom self-diffusion barrier and corrosion resistance ability have also been investigated. The (002) plane possesses the lowest surface energy and the highest stripping energy, indicating its superior stability (Fig. 9d).158,160 The (002) plane also exhibits the lowest surface atom self-diffusion barrier, suggesting a high diffusion coefficient at the surface and consequently a low propensity for dendrite formation,159 as supported by the previous analysis of the second Damköhler number DaII (Fig. 9e). For corrosion resistance, although the (002) plane has the highest energy barrier for the HER,158,161 the experimental results suggest that the (100) plane is least prone to corrosion, evidenced by its most positive corrosion potential Ecorr and lowest corrosion current density Icorr (a more positive Ecorr indicates a lower tendency to corrode, and a lower Icorr indicates a slower rate of corrosion) (Fig. 9f).159 This is attributed to the fact that the (100) plane has the lowest electrochemical active surface area: the electrochemical active surface area of the three crystal planes is in the order of (100) < (002) < (101).159
Although many of the studies emphasize that the selective growth of the (002) plane leads to superior cycling stability, some others argue otherwise.156,157,162 Despite the controversy, a clear conclusion that can be drawn from all the crystallography-related studies is that the selective growth of any of the three crystal planes outperforms random growth without specific crystallographic manipulation in terms of cycling stability. Therefore, the following discussion will focus on the detailed methods to control crystallography: substrate texturing, lattice match, selective shielding, and current density regulation.
3.2.1.1. Substrate texturing. Substrate texturing is a direct method to manipulate crystallography. Usually, commercially available Zn foil or Cu foil shows random polycrystalline orientations. If such foil is used as the electrodeposit substrate, the crystallographic orientation of the deposit is random. Through texturing, the initial randomly textured electrodeposit substrate can be transformed into a single-textured or even monocrystalline substrate.
Annealing, recrystallization, and mechanical rolling are effective ways to fabricate single-textured or monocrystalline substrates. Mechanical rolling is a straightforward and scalable way to obtain (002)-textured Zn substrates (Fig. 10a).157,163,164 Rolling creates severe plastic deformation and induces a slip that occurs preferentially at close-packed planes along the close-packed directions, such as the (002) plane along the 〈110〉 direction (Fig. 10b). The slip mechanism yields a characteristic texture of (002).164–166
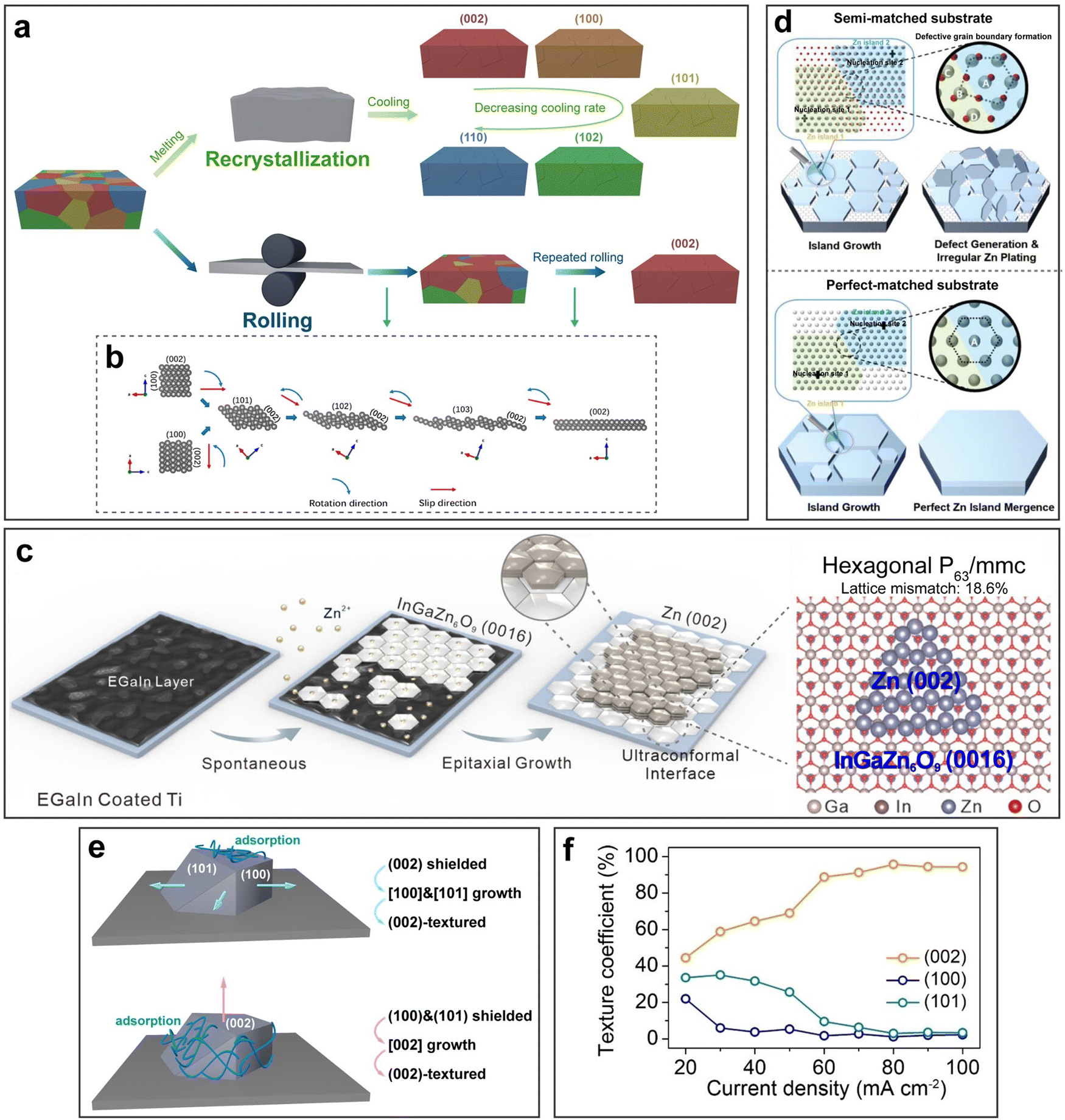 | ||
| Fig. 10 Crystallographic manipulation. (a) Schematic illustration of substrate texturing through recrystallization and rolling. (b) Atomic structure evolution showing the texturing mechanism of rolling. Reproduced with permission from ref. 165 Copyright 2022, American Chemical Society. (c) Schematic illustration of Zn deposition on EGaIn coated Ti and the atomic-scale matching of InGaZn6O9 and Zn crystal planes. Reproduced with permission from ref. 167 Copyright 2023, Wiley-VCH. (d) Schematic illustration of different growth behaviors on semi-matched/perfect-matched substrates. Reproduced with permission from ref. 168 Copyright 2022, Wiley-VCH. (e) Schematic of two possible selective shielding mechanisms. (f) The effect of the current density on the texture content. Reproduced with permission from ref. 160 Copyright 2023, Wiley-VCH. | ||
Compared to mechanical rolling, annealing and recrystallization can fabricate a wider range of single-textured substrates. Song et al. and Liu et al. fabricated single (002) and (101) textured Zn substrates via annealing, respectively.157,169 Chen et al. adopted a fast melting-solidification approach which circumvented the influences of initial textures, grain sizes, and residual stress to transform randomly textured Zn foils with micrometer grain sizes to single (002)-textured ones with millimeter grain sizes.170 Furthermore, Ren et al. revealed that the orientation of Zn could be precisely controlled by the Wulff construction prediction.159 They first melted the polycrystalline Zn foils and then carried out the recrystallization process by cooling the samples. Different cooling rates were selected based on the fact that a higher cooling rate benefited faster growth of facets with a lower surface energy. As a result, a library of monocrystalline Zn foils with (002), (100), (101), (102), and (110) planes were obtained.
Notably, in both mechanical rolling and heat treatment, residual stress exists inside the metal anode, which can result in crystallization stress accumulation during deposition and cause nonuniform deposition. Therefore, a stress-prerelease process can be adopted for metal anodes to reach sufficient strain capacity and for crystallization stress relaxation during deposition.171
Substrate texturing can also be achieved via electrolytic treatment of substrates, such as electro-plating and electro-stripping. Highly preferential crystal orientations can be achieved by introducing crystal facet selectors as additives into the electrolytes during electrolysis.172,173 For instance, Chen et al. constructed (002)-textured Zn by introducing KNO3 in the electrolyte.174 KNO3 could preferentially adsorb on the (001) and (101) planes, lowered their stripping energy, accelerated their dissolution upon electro-stripping, and hence rendered Zn with a dominant (002) exposure. In turn, without the use of crystal facet selectors, Niu et al. demonstrated that based on the highest stripping energy of the Zn (002) plane, applying an external anodic oxidizing electric field could prioritize the stripping of (100) and (101) planes, thus resulting in preferential exposure of the (002) plane.158
Last but not least, selective etching can be employed for substrate texturing as well.175 Liu et al. fabricated (002)-textured Zn foils by treating the original Zn foils in H3PO4 solutions.176 Since the (002) plane possessed lower reactivity to protons, a (002)-textured surface would be left after etching.
3.2.1.2. Lattice match. A lattice match between the electrodeposition substrate and a certain crystal plane of Zn can induce Zn deposition with highly preferential crystallographic orientations. This approach is mainly used to induce the growth of the (002)-textured Zn deposit. Graphene is commonly adopted for its rather low lattice mismatch with the Zn (002) plane.177 Zn and graphene shared the congruent hexagonal lattice structures, and their in-plane lattice constants are fairly close, with that of Zn being 266 pm and that of graphene being 246 pm, resulting in a low lattice mismatch of mere 7.4%.92,178 Therefore, epitaxial growth is favored for the formation of the (002)-textured Zn deposit. An even lower lattice mismatch of 1.6% was found between Cu (111) and Zn (002) by Su et al., who mass-produced single-crystal Cu (111) foils from commercial ones through a temperature-gradient-annealing route.179 The ultralow lattice mismatch enabled unprecedentedly flattened epitaxial lateral growth of Zn (002) and thereby guaranteed stable cycling of symmetric cells under an extreme condition of 50 mA cm−2/1 mA h cm−2. By introducing the eutectic Ga–In alloy (EGaIn) on the Zn anode, Zou et al. found out that the exposed (0016) plane of the spontaneously formed epitaxial interface InGaZn6O9 matched well with the (002) plane of Zn, inducing horizontal and dense Zn deposition (Fig. 10c).167
Notably, Pu et al. emphasized the significant role of defect-free single-crystal Zn in suppressing defect generation during electrodeposition.168 Due to the scarcity of grain boundaries, the sole exposure of the (002) plane and the perfect homoepitaxial lattice matching (Fig. 10d), the defect-free single-crystal Zn metal anode enabled reversible cycling at an unprecedented 200 mA cm−2/8 mA h cm−2, and the cycling could be maintained for over 1200 cycles at 50 mA cm−2/4 mA h cm−2. Besides, other materials including Zn–Ag alloys69,180 and ZnSe150 have also been reported to be compatible with the Zn (002) lattice and hence able to induce (002)-textured deposition.
3.2.1.3. Selective shielding. During the plating process, selective shielding blocks the growth of certain crystal planes while promoting the dominant growth along a certain direction, thus resulting in single-textured deposits. The shielding effect is enabled by electrolyte additives that selectively adsorb on certain crystal planes, hindering the deposition of ions onto those planes and thus suppressing their growth. According to the Bravais law, the orientation of a crystal plane is determined by the growth rates of all crystal planes.59,181,182 For instance, if the Zn (002) plane is adsorbed on and shielded by an electrolyte additive, its growth rate is greatly reduced. Meanwhile, the (001) and (101) planes remain exposed, with their growth rates unaffected (Fig. 10e). In this sense, due to the significantly higher growth rates of the (001) and (101) planes, growth along the [001] and [101] directions is dominant, which leads to the epitaxial growth of the (002) plane. Consequently, the (002)-textured deposition is realized in this case. Based on this theory, several studies have adopted electrolyte additives with the highest adsorption energy with the Zn (002) plane to obtain (002)-textured Zn deposits.59,133,151,181–183 Nonetheless, there are also studies that argue otherwise.156,162,184–186 For example, Zhang et al. stated that the additive they applied was preferentially adsorbed on the Zn (100) and (101) planes, forcing the deposition onto the (002) plane, thus inducing the preferential growth of the (002) plane and forming a flat (002) texture.185
3.2.1.4. Current density regulation. Current density is an important independent variable that determines the texture of the deposits. Yuan et al. used Cu as substrates and investigated the transformation of the textures of as-deposited Zn as the current density increases from 20 to 80 mA cm−2.155 It was revealed that the increasing current density resulted in a gradually increase of the (002) texture content and a decrease of the (100) and (101) texture contents (Fig. 10f). Similar studies were conducted to study a broader range of current densities.126,160,187 It was found that at a relatively low current density, nucleation and growth of electrodeposits were highly dependent on textures of the substrates, and textureless substrates could lead to mossy and dendritic morphologies. At a medium-high current density, the dependence of nucleation and growth behaviors on the substrates was weakened, and desirable (002)-textured Zn deposits could be formed at a suitable current density. When the current density is further elevated, however, the rampant growth, the diffusion limitation, the intensified space charge effect and electroconvection result in undesired dendritic morphologies.160,187
It should be noted that although primarily studied and applied in AZBs, crystallographic manipulation is a generic strategy in all aqueous metal batteries where the anisotropy of crystal planes, especially the surface energy difference of crystal planes in a metal, is significant. In this regard, although crystallographic manipulation may not be critical for aqueous Sn metal batteries due to the surface energy similarity of the crystal planes as mentioned above, in aqueous Mg batteries and aqueous Al batteries, crystallographic manipulation can be rather important considering that the surface energy differences of crystal planes in HCP Mg and face-centered-cubic (FCC) Al are relatively large.147 Studies on crystallographic manipulation in these two systems can be carried out on the premise that the major side-reaction problems of Mg and Al are solved.
 | ||
| Fig. 11 Helmholtz double layer regulation. (a) Schematic illustration of a Helmholtz double layer. (b) Distributions of different molecules near the Zn surface showing dynamic adsorption of maltose. Reproduced with permission from ref. 188 Copyright 2024, Elsevier. (c) Simulated electrolyte structures with and without the Py additive (cyan-blue: carbon; red: oxygen; yellow: sulfur; gray: zinc; blue: nitrogen; and white: hydrogen). Reproduced with permission from ref. 189 Copyright 2023, Wiley-VCH. (d) Schematic illustration of the trade-off between H2O-rich and H2O-poor electric double layers. Reproduced with permission from ref. 190 Copyright 2024, the Royal Society of Chemistry. (e) Regulation of the desolvation energy barrier by a ZnO layer. Reproduced with permission from ref. 191 Copyright 2020, the Royal Society of Chemistry. | ||
Although the HDL structure is often shrouded in oblivion, its significant influence on the electrochemical reaction, or to be more specific, the growth behavior of metal deposits, can by no means be overlooked. Firstly, the desolvation process at the OHP significantly impacts the transfer kinetics of the entire battery, as the energy barrier for the desolvation of the metal ion in the HDL is the primary contributor to the charge transfer migration barrier in the whole battery.192 Secondly, the presence of abundant water molecules in the IHP can lead to the undesired HER, especially when the redox potential of the metal anode is low. Given these critical factors, a fundamental principle for regulating the HDL emerges: minimizing the desolvation energy barrier and controlling the amount of water molecules in the IHP or creating a water-deficient HDL.
HDL regulation is primarily reported in AZBs, which is mostly achieved through the introduction of electrolyte additives. To obtain a water-deficient HDL, an electrolyte additive must be able to enter the IHP with high coverage on the Zn anode surface and expel the original water molecules. Additives such as polar organic molecules and positively or negatively charged ions may be drawn into the IHP driven by electrostatic forces.60,193,194 More importantly, electrolyte additives with high adsorption energy on Zn can overcome electrostatic repulsion to enter the IHP, be adsorbed on the Zn anode and replace the original water molecules to create a water-deficient HDL and thereby suppress the HER. For instance, Liu et al. introduced a trace amount of maltose as the additive to dynamically regulate the HDL.188 The maltose could be preferentially adsorbed onto the Zn anode, provided the IHP with a low-level water content and SO42− concentration and hence suppressed side reactions (Fig. 11b). It was also revealed that the adsorption of maltose followed a dynamic pattern, based on the MD simulations conducted on three states: a non-electrochemical cycling state (NECS) where the voltage is 0, a discharge state where the voltage is +100 mV, and a charge state where the voltage is −100 mV. At the charge state, adsorption of a high amount of maltose was observed, and the molecules near the Zn anode were dominantly maltose, with only a small proportion of water or SO42−. At the other two states, more maltose molecules tended to self-aggregate due to the strong intermolecular hydrogen bonding, and although the majority of the maltose molecules did not appear at the nearest surface of the Zn anode, the contents of H2O and SO42− in the IHP were still greatly reduced due to the steric hindrance effect of maltose. In fact, the HER is naturally inhibited near the anode in the discharge process as the electrons are flowing away from Zn, so the protective effect of electrolyte additives at this state is less pronounced compared to the NECS and the charging state.
Besides the sole adsorption of electrolyte additives, another approach is to bring the electrolyte additive molecules into the HDL by the solvation–desolvation effect. Luo et al. reported a universal high donor number (DN) additive pyridine (Py) for HDL regulation (Fig. 11c).189 Additives with a high DN, such as methanol (MeOH) and dimethyl sulfoxide (DMSO), have stronger interactions with metal ions.195,196 As a result, the metal ions coordinated with the electrolyte additive molecules could first enter the HDL, and then, after desolvation, the free electrolyte additive molecules can expel the water molecules. Furthermore, MD simulations visualized the effect of the Py additive in creating a water-deficient IHP.
Although most studies regard a water-poor HDL as favorable, a critical viewpoint was raised by Qi et al., stating that an excessively water-poor HDL was likely to cause undesired dead Zn formation.190 They added different amounts of aspartame (APM) additives to obtain a series of HDL structures ranging from water-rich to water-poor and revealed the trade-off between water-rich and water-poor HDLs (Fig. 11d). While a water-rich HDL caused rampant water-induced side reactions, nonuniform deposition and dendritic growth, an overly water-poor HDL deteriorated the electrode–solvent contact and impeded the solvation of Zn2+ with water during stripping, thus causing undesirable dead Zn formation. Therefore, a moderate water-poor HDL should be adopted by tuning the amount of the electrolyte additive.
When considering the reduction of the desolvation energy barrier, however, a dilemma arises with electrolyte additives. If an additive exhibits a high DN and thus possesses strong solvation capabilities with metal ions, it inherently requires a higher energy consumption for desolvation. Conversely, if an additive exhibits low solvation abilities, although the desolvation energy would decrease when metal ions were coordinated with it, the metal ions tend to prefer coordination with water due to the additive's inferior solvation power. Therefore, the desolvation energy barrier may not be lowered from the perspective of the tuning solvation structure. An alternative way is to introduce an electrolyte additive that is capable of undermining the original water hydrogen bonding networks. For instance, Cl− can bind water molecules via weak hydrogen bonds and thus lowers the desolvation energy barrier.197
Electrolyte additives aside, Xie et al. proposed novel HDL regulation that lowers the desolvation energy barrier by Zn anode modification.191 It was demonstrated that the 3D nanoporous ZnO coating on the Zn anode could lower the energy barrier of Zn2+ inserted into the modified anode almost eight times compared to bare Zn, and this led to electrostatic attraction toward Zn2+ rather than solvated Zn2+, thereby reducing the desolvation energy barrier in the HDL from 77.2 kJ mol−1 to 51.0 kJ mol−1 (Fig. 11e). The same regulation on the desolvation energy barrier can be realized by an ultrathin ZnS SEI layer as well.198
 | ||
| Fig. 12 In situ artificial interlayer design. (a) Schematic illustration showing interfacial challenges, different diffusion modes and the functions of an ideal interlayer. (b) Schematic illustration of the ‘anchor-capture’ mechanism. Reproduced with permission from ref. 199 Copyright 2023, Springer Nature. (c) Schematic illustration of an artificial interlayer of a polymer network. Reproduced with permission from ref. 200 Copyright 2023, Wiley-VCH. (d) Schematic illustration of a dynamic corrosion inhibitor and its mechanism. Reproduced with permission from ref. 201 Copyright 2022, Elsevier. (e) Schematic illustration of a proton filter. Reproduced with permission from ref. 95 Copyright 2022, American Chemical Society. | ||
The multifunctionality of such a layer raises certain requirements on the adsorbed species involved. The suitable molecule should contain a part with high binding energy with the metal anode for firm adsorption and another part with high binding energy with the metal ion to capture the ion and promote desolvation. A synergistic “anchor-capture” process can be used to describe the basic function of a typical adsorption layer. Such an “anchor-capture” mechanism was illustrated by Luo et al. using the amino acid glycine (Gly) as an electrolyte additive (Fig. 12b).199 The N atom of the amino group anchored the Gly molecule onto the Zn surface, while the strong nucleophilicity of the carboxyl group captured Zn2+. It was further demonstrated that the coexistence of both amino and carboxyl groups enabled Gly to stabilize the Zn metal anode via the “anchor-capture” mechanism, while, as a comparison, N-acetylglycine (Ac-Gly), which only contained a carboxyl group, and glycinamide hydrochloride (Gly-NH2), which only contained an amino group, both failed to stabilize the anode. Based on such a mechanism, other molecules with multiple polar functional groups, such as glutamine,202 tannic acid,203 and α-D-glucose204 could also construct organic interlayers on the metal anode.
Moreover, the anchoring of some specific additive molecules onto the metal anode, together with the steric hindrance, can contribute to long-term water isolation protection. Such a molecule usually has one end with a polar functional group that adheres to the metal anode and a hydrophobic long chain that repels water, such as perfluorooctanoic acid (PFOA).205 In PFOA, where C–F bonds are abundant, the substantially negatively charged terminal F atoms not only repel water molecules, but also generate strong ion–dipole interactions with positively charged Zn2+, thereby facilitating rapid ion transport among molecular chains forming a “Zn2+ pump”.
In addition, polymeric materials are frequently employed to in situ build multifunctional interlayers. The cross-linking and interpenetrating features have the potential to establish a protective layer that shields water and a percolating ion-conducting network that facilitates rapid and homogeneous ion diffusion. For instance, Meng et al. engineered a polyzwitterion protective layer (PZIL) by polymerizing 2-methacryloyloxyethyl phosphorylcholine (MPC) in carboxymethyl chitosan (CMCS), where the choline groups in MPC preferentially adsorbed on the Zn anode to circumvent side reactions and the phosphate groups chelated with Zn2+ to promote ion transport (Fig. 12c).200 Along with the Hofmeister effect between ZnSO4 and CMCS, a polymer network with abundant Zn2+ transport channels was thus formed. Besides the simple introduction of polymeric materials, Wang et al. revealed the important role of the orientation control of the functional groups in polymeric materials to optimize ion flux homogenization and ion transport kinetics improvement.206
Apart from polymers, self-assembly of a supramolecular framework is another approach to construct a multifunctional interlayer. For instance, Fu et al. developed a supramolecular organic framework through self-assembly based on H-bonds and π–π stacking interactions between melamine (ME) and cyanic acid (CA).207 This SOF exhibited triple functions: the in-plane H-bond networks firmly trapped water molecules to assist Zn2+ desolvation and prevent interfacial side reactions, the plentiful polar groups strongly interacted with Zn2+ and improved the ion transport kinetics, and the π–π stacked space-layout structure induced flat and smooth (002)-textured Zn deposition. The other supramolecular framework design includes self-assembled tannic acid/Zn2+ complex interlayers203 and 3D continuous spindle-shaped mesoporous chitosan frameworks.208
Notably, for AABs, water resistance is largely highlighted over other functions in terms of interlayer design, as water-induced side reactions are a major problem plaguing the batteries’ stability. Choi et al. designed a polyvinyl alcohol (PVA) interlayer on the Al metal anode by introducing PVA into the electrolyte for an aqueous Al–air battery (Fig. 12d).201 A dynamic corrosion inhibiting effect was featured as the hydroxyl group-rich PVA could be adsorbed on the Al surface to suppress corrosion in an open-circuit state, and detached upon discharging, leaving the Al surface exposed for a smooth stripping process. When adsorbed onto the Al surface, hydroxyl groups in PVA interacted with the surrounding water molecules by forming hydrogen bonds, intervening in the reaction between Al and water and therefore inhibiting the HER and corrosion. It was shown that the corrosion rate of Al in an open-circuit state with PVA was around one-fifth of that without PVA, displaying an inhibition efficiency of approximately 80%.
In aqueous Al metal batteries where a mildly acidic Al2(SO4)3 solution was used as the electrolyte, Yan et al. developed a proton-filtering polymer film as a functional interlayer (Fig. 12e).95 Poly(vinylidene difluoride) (PVDF) and Nafion were coated onto the Al metal anode as the polymer film.209,210 The polymer film served as a proton filter that not only rendered water molecules less accessible to the Al anode, but also prevented the contact with active protons via the electrostatic interactions between the polymer matrix and protons. The resultant CV curve of the Al anode with a proton-filtering polymer film showed no distinguishable hydrogen evolution peak in a symmetric cell configuration, indicating that an ultra-stable environment was enabled. Tao et al. reported the use of polyethylene glycol (PEG) as an electrolyte additive.211 During galvanostatic charging, polymerization of PEG led to the formation of a protective interlayer on the Al metal anode. This interlayer could effectively mitigate corrosion under acidic conditions with pH < 4 and render a flat anode surface for uniform plating/stripping.
Despite the spontaneous in situ interlayer formation featured by adsorption or self-assembly, critical requirements of the electrolyte additives involved may limit the materials selection. In comparison, ex situ methods such as spin-coating, magnetron sputtering and binder-coating enable the use of a wider range of materials, including cyanoacrylate adhesive (502 glue),212 commercial weighing paper,213 molecular sieves,214 TiO2,215 and porphyrin-Zn (ZnTBPP).216 Notably, the ZnTBPP-based interlayer designed by Chen et al. suppressed the HER in a novel way of interface redox potential modulation (Fig. 13a).216 Electrons at the Zn interface tended to migrate into the ZnTBPP molecules, forming a charge transfer layer. Furthermore, the electron cloud density of ZnTBPP was largely delocalized, which led to the charge depletion of the Zn interface and an increased redox potential of the Zn anode, hence suppressing the reduction of H+ into H2. The electron delocalization and electron withdrawing effect in ZnTBPP was attributed to two factors: the large π bond in the structural center, and the electron withdrawing –Br functional groups. To illustrate the importance of –Br groups, ZnTMPP (meso-tetratolylporphyrin-Zn) with electron-donating groups –CH3 replacing the –Br in ZnTBPP was used for comparison. It was shown that the electron cloud density in ZnTMPP was localized, and charge accumulation was observed on the Zn interface. As a result, ZnTBPP@Zn showed a higher HER energy barrier than ZnTMPP@Zn, and ZnTMPP@Zn still exhibited a higher HER energy barrier than the bare Zn due to electron delocalization by the large π bond.
 | ||
| Fig. 13 Ex situ artificial interlayer design. (a) Schematic illustration of interfacial redox potential modulation enabled by an artificial interlayer. Reproduced with permission from ref. 216 Copyright 2023, Wiley-VCH. (b) Schematic illustration of a COF design for an artificial interlayer. Reproduced with permission from ref. 217 Copyright 2024, Wiley-VCH. (c) Schematic illustration of a MOF design for an artificial interlayer. Reproduced with permission from ref. 153 Copyright 2023, Wiley-VCH. (d) Schematic illustration of a “capture-desolvation decoupling” design. Reproduced with permission from ref. 218 Copyright 2024, American Chemical Society. | ||
In addition to the previously mentioned materials for artificial interlayers, metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) are rather common choices, primarily due to their inherent tunable pores. For instance, Guo et al. designed a series of anhydride-based COFs with abundant zincophilic groups and precise tuning of hopping sites for Zn2+ transfer.217 The special ABC stacking of the designed COFs created specific zig-zag sites along the 1D channel that promoted faster Zn2+ diffusion than AA and AB stacking modes, as confirmed by the MD simulation on the activation energy of Zn2+ migration (Fig. 13b). In turn, Zhang et al. utilized an ethylenediaminetetraacetic acid (EDTA) grafted MOF as the artificial interlayer.153 EDTA served as tentacles that strongly coordinated with Zn2+, thus capturing the ions and facilitating prompt desolvation (Fig. 13c). The MOF matrix offered directional channels for ion conduction and improve Zn2+ diffusion kinetics. As a result, the MOF-modified substrate displayed a uniform (002)-textured deposition.
Critically, although most of the reports on artificial interlayer design claim the improved metal ion capture and desolvation behaviors, these two processes are actually coupled and incompatible, as the hydrophobic functional groups favoring repulsion of H2O and desolvation of the H2O-coordinated metal ion are actually adverse to the capture of metal ions. Moreover, the ion transport pathways in most artificial interlayers are highly tortuous, which may impair the diffusion kinetics. In response, Wang et al. proposed a tandem chemistry strategy to decouple the capture and desolvation processes and simultaneously boost the two within a sophisticatedly designed Janus mesopore accelerator.218 The accelerator had performative and aligned channels inside, with the upper inner surface decorated with hydrophilic –OH groups and the lower inner surface decorated with hydrophobic –F groups (Fig. 13d). Zn(H2O)62+ clusters could be easily captured at the upper –OH-rich region, and subsequently go through effective desolvation at the lower –F-rich region.
The concept of the artificial interlayer can be further employed in addressing the challenges for cathode materials. The key functions of the artificial layer are to inhibit the material dissolution and promote the electron transport at the cathode–electrolyte interface. Shuai et al. reported using an atomic layer-deposited ZnO layer to suppress the vanadium dissolution and side reactions.219 Interfacial coating with conductive polymers and carbon materials can be utilized to enhance the interfacial charge transport. Huang et al. designed a phytic acid/polypyrrole (PA/PPy) organic layer onto the Ca0.24V2O5·H2O cathode, which demonstrated synergistic positive effects of boosting the electronic conduction, facilitating the Zn2+ desolvation at the interface, and inhibiting the vanadium dissolution.220
3.2.4.1. SEI design rationales. The basic requirement for an ideal SEI should be low electronic conductivity, high ionic conductivity, water resistance and dendrite suppression (Fig. 14a).
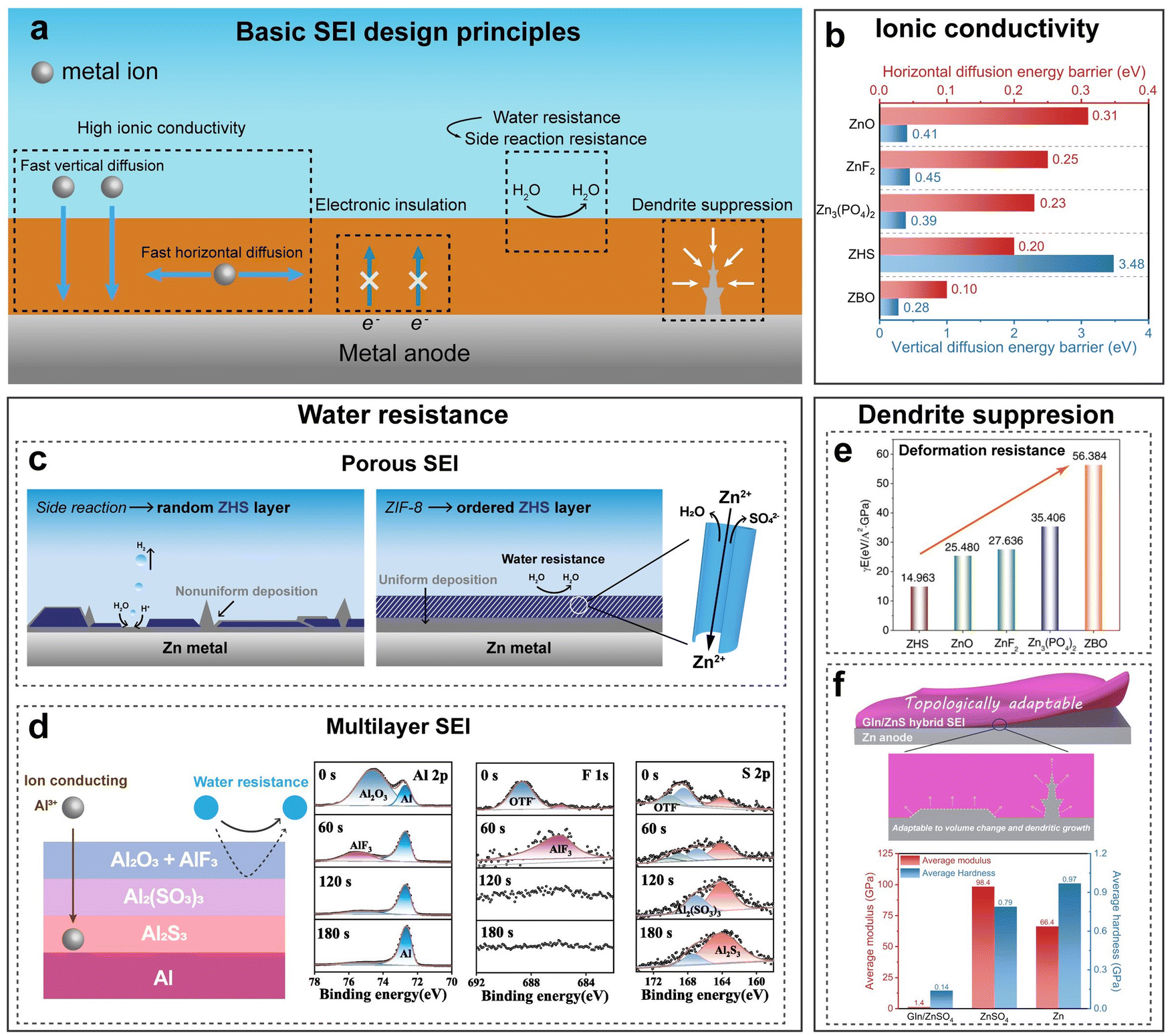 | ||
| Fig. 14 SEI design rationales. (a) Schematic illustration of an ideal SEI. (b) Ionic conductivity comparison of several SEI material candidates. Reproduced with permission from ref. 222 Copyright 2023, Wiley-VCH. (c) Schematic illustration of a porous ZHS SEI for water resistance. Reproduced with permission from ref. 223 Copyright 2024, the Royal Society of Chemistry. (d) Schematic illustration of a multilayer SEI for water resistance. Reproduced with permission from ref. 224 Copyright 2024, Elsevier. (e) Dendrite suppression ability comparison of several SEI candidates. Reproduced with permission from ref. 222 Copyright 2023, Wiley-VCH. (f) Schematic illustration of a topologically adaptable SEI design. Reproduced with permission from ref. 225 Copyright 2024, American Chemical Society. | ||
Sufficiently low electronic conductivity guarantees that deposition occurs below an SEI, rather than on top of it—otherwise an SEI would function as a substrate or a metallophilic layer as mentioned before. High electronic conductivity also leads to intensified interfacial redox side reactions, dendrite formation and potential dead metal deposit formation inside the SEI. On the other hand, high ionic conductivity ensures fast vertical and horizontal ionic diffusion, low polarization, uniform nucleation, and uniform growth. The positive effect of high ionic conductivity in dendrite-free deposition aligns with the previous discussion on the second Damköhler number DaII, as high ionic conductivity promotes fast diffusion and hence lowering DaII.
The common inorganic SEI compositions in AZBs including ZnF2,169,226–235 Zn3(PO4)2,169,176,228,232,236–239 ZnS,169,225,226,231,232,234,240,241 ZnCO3,227,231,234,242–244 ZnSO3,226,231,242 ZnO,226,241,244,245 Zn4(OH)6SO4·nH2O,50,246,247 ZnSe,150,169,248etc. all satisfy the requirement of decent ionic conductivity and electronic insulation. Wang et al. performed a rational screening of SEIs which included the dimensions of both electronic conductivity and ionic conductivity.222 In terms of ionic conductivity, the vertical diffusion through an SEI and the diffusion on the surface on an SEI were both considered, as the former influenced the transportation rate of Zn2+ while the latter affected the local distribution of Zn2+ (Fig. 14b). As a proof of concept, the two parameters above were calculated and compared between Zn3(BO3)2 (ZBO) and other common SEI materials, revealing the superior ionic conductivity of ZBO.
Besides the intrinsic electronic conductivity and ionic conductivity of an SEI itself, the thickness factor should also be considered. A thick SEI increases ionic resistance and may result in sluggish ionic diffusion.238,249 A thin SEI not only favors fast ionic diffusion but also may influence the electronic resistance. Specifically, when the thickness of an SEI goes below the 1 nm threshold, the electron tunneling effect becomes significant.250,251 Therefore, the thickness of an SEI should be preferably thin, but at least of a nanometer scale or greater. In AZBs, the reported thicknesses of SEIs usually fall in the range of 100 nm to 1 μm,150,222,228,232,238,249 with some ultrathin ones of tens of nanometers226,233,242 and some thick coatings of several micrometers.239,252 It should be noted that the thicknesses of the in situ formed SEIs are hardly controllable, but usually much thinner, while ex situ methods such as coating or hydrothermal reactions offer effective ways to control the SEI thicknesses.150,228
An ideal SEI should also prevent exposure of the metal anode to water so as to inhibit side reactions. This can be achieved by the design of dense SEIs, or alternatively, porous but functional SEIs. In AZBs, zinc hydroxide sulfate (ZHS), or Zn4(OH)6SO4·nH2O, is a by-product often considered harmful due to its micrometer sizes and random stacking that forms a loose layer on the Zn anode, which not only fails to protect the Zn anode from water, but also results in nonuniform deposition. However, its low electronic conductivity and high ionic conductivity, combined with its spontaneous formation, makes it an SEI candidate. To transform ZHS into a suitable SEI, Wang et al. realized in situ conformal growth of ZHS with the dominating (001) facet on (002)-textured Zn.50 The formed ZHS layer could serve as a dense, robust and uniform SEI which simultaneously regulated deposition and suppressed side reactions. In comparison, Xiang et al. constructed a porous ZHS layer derived from a ZIF-8 membrane (Fig. 14c).223 Inheriting the porous feature and well-ordered structure from ZIF-8, the ZHS layer provided ion transport channels which facilitated swift Zn2+ diffusion that favored uniform deposition and effective desolvation that inhibited side reactions. In AABs, a multilayer SEI was designed by Sun et al., which ensured effective isolation of water from the Al metal anode (Fig. 14d).224 The top layer of the SEI mainly contained AlF3, with a minor amount of Al2O3, and the subsequent layers contained Al2(SO3)3 and Al2S3, all derived from the Al(OTf)3-containing solution, as was characterized by the XPS spectra. Apart from water isolation, the SEI layer was also reported to be ionically conductive, which improved Al3+ transport kinetics and facilitated uniform deposition.
Last but not least, an ideal SEI should be capable of suppressing dendritic growth so as to guarantee safe and stable cycling. A tough and stiff SEI with high mechanical strength deformation resistance is usually considered the optimal choice. Wang et al. proposed the product of interfacial energy (γ) and Young's modulus (E) of the SEI as an indicator of dendrite suppression ability.222 A higher γE indicates more effective dendrite suppression, and a rational screening of γE showed that ZBO had superior dendrite suppression ability (Fig. 14e).222 Nevertheless, some pointed out that SEIs with high stiffness and hardness which could hardly adapt to the volume change of the Zn anode during cycling were susceptible to cracking.133,253,254 Therefore, Yan et al. introduced L-glutamine (Gln) as an additive to in situ produce a hybrid SEI consisting of ZnS and Gln-related species (Fig. 14f).225 This hybrid SEI possessed a rather low modulus and hardness, as well as good shape recovery capability, thus enabling topological adaptation to volume fluctuations during plating/stripping.
3.2.4.2. SEI design strategies. In addition to the fundamental SEI design principles outlined above, numerous studies have identified advantageous SEIs with additional desirable traits and featuring strategic fabrication methods. In situ construction of SEIs through the introduction of electrolyte additives is a commonly employed strategy.229,248 The widely reported ZnF2-containing SEIs can be produced by fluorine-containing species, such as Zn(OTf)2,232,242,246 Zn(TFSI)2,228,233,255 Zn(BF4)2,230etc, and similarly, Zn3(PO4)2-containing SEIs can also be produced in situ by electrolyte additives such as Zn(H2PO4)2,238 triethyl phosphate (TEP),232 tris(2,2,2-trifluoroethyl)-phosphate (TFEP),228etc. Specifically, a novel fluorine-containing zinc salt Zn(DFTFSI)2, which featured an asymmetric anion group, bi-(difluoromethanesulfonyl)(trifluoromethanesulfonyl)imide (DFTFSI−), was proposed by Chen et al. (Fig. 15a).235 DFTFSI− was demonstrated to be more effective in forming a robust ZnF2 SEI compared to TFSI−, due to the easy decomposition of DFTFSI− stemming from its asymmetric structure and the presence of an electrochemically labile CF2H moiety. Apart from anion-derived SEIs, some cations can also participate in the formation of SEIs.234,242,255 For instance, Cao et al. combined Zn(OTf)2 with trimethylethyl ammonium trifluoromethanesulfonate (Me3EtNOTf) and utilized the synergistic reactions between OTf− and Me3Et+ to synthesize poly-species SEI containing ZnF2, ZnCO3 and ZnSO3 (Fig. 15b).242
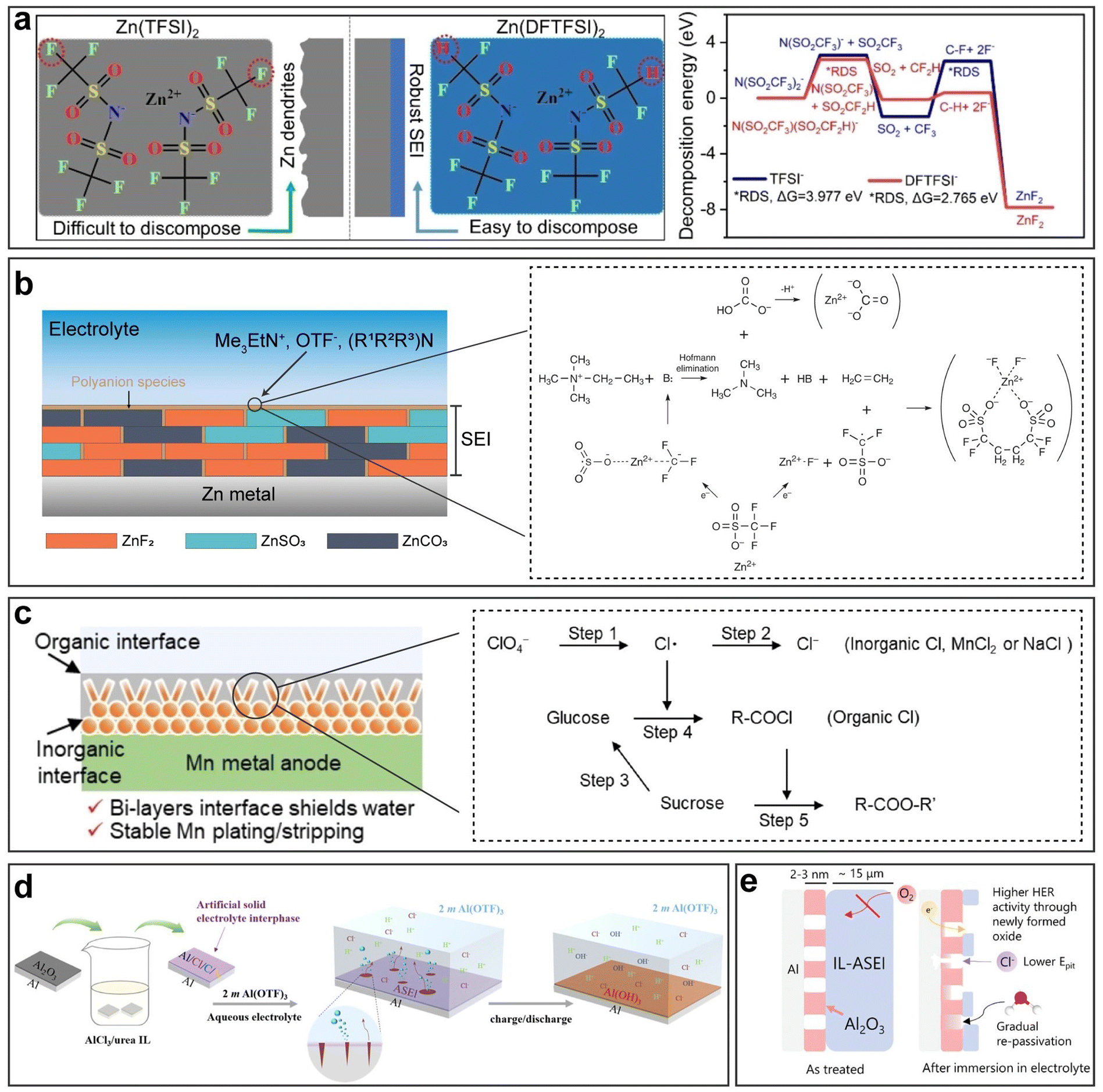 | ||
| Fig. 15 SEI design strategies. (a) Comparison of SEI formation processes based on two Zn salts. Reproduced with permission from ref. 235 Copyright 2024, Wiley-VCH. (b) Schematic illustration of a hybrid SEI and its formation mechanism. Reproduced with permission from ref. 242 Copyright 2021, Springer Nature. (c) Schematic illustration of an organic–inorganic SEI in aqueous Mn metal batteries. Reproduced with permission from ref. 256 Copyright 2022, Wiley-VCH. (d) Schematic illustration of the working mechanism of an AlCl3-containing SEI. Reproduced with permission from ref. 257 Copyright 2023, American Chemical Society. (e) Schematic illustration of the failure mechanism of an AlCl3-containing SEI. Reproduced with permission from ref. 258 Copyright 2021, Wiley-VCH. | ||
Organic–inorganic or polymer–inorganic hybrid SEIs represent a prevalent SEI design approach. In a hybrid SEI, the organic or polymeric components serve as a matrix that contributes to flexibility and ensures full coverage to repel water ingress, while the inorganic components are embedded in the matrix to provide sufficient mechanical strength and high ionic conductivity. Liu et al. in situ built an endogenous organic–inorganic hybrid SEI with the organic outer layer containing N–H and –CH3 and the inorganic inner layer consisting of ZnO and ZnCO3.244 The stable SEI with the complementary advantages of high ionic conductivity, sufficient structural integrity and toughness guaranteed a long lifespan for over 5500 hours at 3 mA cm−2/3 mA h cm−2. A similar design strategy was reported in aqueous manganese metal batteries by Yang et al. (Fig. 15c).256 The inorganic interface containing the Mn1.77Sn alloy and Mn2SnO4 was constructed through the replacement reaction between Mn and SnCl2. The inorganic interface had two layers: the upper layer containing vertically aligned flakes guided uniform ion diffusion and the lower layer consisting of dense particles physically isolated the Mn metal anode from water. The organic interface was formed by the esterification reaction between sucrose and NaClO4 and served as a dense protective layer against water.
Self-healing is another design feature of the SEI.247,248,259–261 The self-healing ability can be fulfilled by introduction of electrolyte additives which actively react with the exposed metal anode when the as-formed SEI cracks, thus regenerating new SEI layers to persistently guard the SEI integrity. Such electrolyte additives mimic the ‘early warning defense’ function of dendritic cells in the biological immunization system.259 Based on this philosophy, Zhang et al. proposed the multifunctional lithium bis-(oxalate)borate (LiBOB), which could not only derive an SEI containing ZnC2O4·2H2O with high ionic conductivity, but also preferentially adsorb towards and react with the freshly exposed Zn to repair the damaged SEI when defects or cracks of the previous SEI were present.259 Similarly, other chemistries with SeO2 repairing ZnSe-containing SEIs248 and sodium glycerophosphate C3H7Na2O6P repairing Zn3(PO4)2-containing SEIs260 have also been investigated. However, it should be noted that the lifespan of such a repairing effect is potentially limited by the amount of the electrolyte additive. Once the electrolyte additive is depleted, a damaged SEI can no longer be healed, and the metal anode is exposed to water unprotected. In response to this problem, Wu et al. designed a PVDF-based composite layer containing Zn(TFSI)2 which could continuously integrate the naturally generated by-product ZHS into a compact SEI film, thus achieving prolonged self-healability.247 To further enhance the reversibility of self-healing, Zhang et al. introduced nanosheet-like graphitic carbon nitride quantum dots (C3N4QDs) as additives to construct a dynamic and self-repairing SEI.261 Upon plating, layered C3N4QDs assembled into an SEI from its colloidal building blocks, exhibiting the ion-sieving effect of the single sheets to assure Zn2+ conduction while simultaneously suppressing the water-induced side reactions. Upon stripping, the as-formed protective C3N4QD SEI spontaneously redispersed into the electrolyte.
In AABs, the role of SEIs primarily revolves around inhibiting side reactions and passivation which pose primary challenges to an Al metal anode. Passivation in AABs is caused by the spontaneous formation of an Al2O3 or Al(OH)3 layer, which is neither electronically conductive nor ionically conductive. The removal of such a passivating layer and the subsequent construction of an effective SEI is the guiding principle. Zhao et al. proposed an artificial SEI formed on Al in contact with the AlCl3-[EMIm]Cl ionic liquid (IL)–eutectic electrolyte (denoted as IL-ASEI).262 The IL-ASEI was mainly composed of AlCl3 and exhibited high ionic conductivity. It was declared that the IL-ASEI was the tightly bound, IL-enriched film and capable of eroding the passivating Al2O3 oxide layer as well as protecting the Al anode against subsequent formation of oxide. Some other studies have also reported treatment of Al metals with AlCl3-based eutectic solutions or ionic liquids.263–265
To further investigate the efficacy of an SEI obtained by such treatment, Li et al. built an AlCl3-containing IL-ASEI by soaking Al in AlCl3/urea IL (Fig. 15d).257 They then revealed that the IL-ASEI actually promoted the corrosion of Al by providing Cl−, rather than facilitating the transport of Al3+. The remaining Cl− within the as-formed ASEI induced pitting corrosion and low-conductivity Al(OH)3 was generated as a by-product on the exposed Al surface during plating/stripping. Furthermore, H2 production was observed and the pits on the Al anode surface after cycling were attributed to H2. The detrimental effect of Cl− was further confirmed by Dong et al. (Fig. 15e).258 They illustrated that the AlCl3 in an IL-ASEI dissolved instantaneously from the initial SEI when immersed into the aqueous electrolyte. The resulting Cl− effectively lowered the pitting potential and promoted pitting corrosion. The broken SEI, in turn, led to gradual re-passivation. In conclusion, Cl-containing SEIs or the equivalent addition of Cl-containing salts to the electrolyte cannot be considered a sensible way to improve the Al electrode performance. In response, Sun et al. obtained a Cl-free, AlF3-rich SEI by the reactions between Al and OTf−.224 Besides abundant AlF3 in the outer layer, this SEI also contained Al2S3, Al2(SO3)3 and Al2O3. Coupled with a co-solvent acetonitrile which reduced the reactivity of water, the anode with the AlF3-rich SEI was largely immune to corrosion and passivation. However, the study on the SEI design in AABs is still insufficient, and the inferior cyclability of current AALBs remains an unresolved problem that demands the construction of a stable SEI that can effectively inhibit the HER, suppress corrosion and circumvent passivation simultaneously.
Besides the SEI design at the anode side, the construction of a CEI can further help overcome the cathode dissolution issue and prolong the full-cell cycling stability. For example, Guo et al. introduced PEG as a polymer additive into the electrolyte, efficiently suppressing the water activity through hydrogen bonding and material dissolution for stable sodium superionic conductor (NASICON) cathodes.266 Zhang et al. proposed an in situ artificial CEI strategy by introducing strontium ions into the vanadium oxide layers as a sacrifice guest.267 The strontium ions could precipitate onto the cathode surface once getting out of the vanadium-based cathode, forming a CEI that could suppress the vanadium dissolution.
4. Ion flux homogenization
The poor reversibility of a metal anode in an aqueous battery, apart from corrosion and side-reactions, mainly stems from nonuniform deposition due to the tip effect. The tips may come from the uneven surface of the pristine metal anode or the formation of protrusions during deposition. Such tips can distort the local electric field and attract more metal ions to deposit onto them or near them, thus causing dendritic growth. Considering obtaining an absolutely smooth and even surface is not practical, tips will inevitably form during deposition. In order to prevent the evolution from tips to dendrite, ion flux homogenizing strategies are applied to offset the tip effect and guide relatively uniform deposition in the presence of tips. Three approaches are often adopted to homogenize ion flux, which are electrostatic shielding, electric field modulation, and external stimuli.4.1. Tip shielding
Tip shielding aims to prevent the dendritic growth of protruding regions on the metal anode. With tip shielding, instead of being drawn to the tips and then forming dendritic deposits, the metal ions are driven towards flat regions and form even deposition. In order to achieve this function, the key to tip shielding is the response to the existence and evolution of protrusions.Such a response can be based on the mechanical pressure applied to a separator by the tips. Dong et al. designed a mesoporous ferroelectric polymer membrane as the separator, which could generate an internal piezoelectric field (EPZ) when compressed by protrusions (Fig. 16a).268 When the positive polarization (PPZ) side faced the metal anode surface, the local electric potential in the electrolyte (VEL) near the protrusions was increased due to the presence of a piezoelectric potential (VPZ), which was capable of repelling Zn2+ from migrating towards the tips. Notably, these effects selectively acted on the protruding regions, leaving the flat regions unaffected.
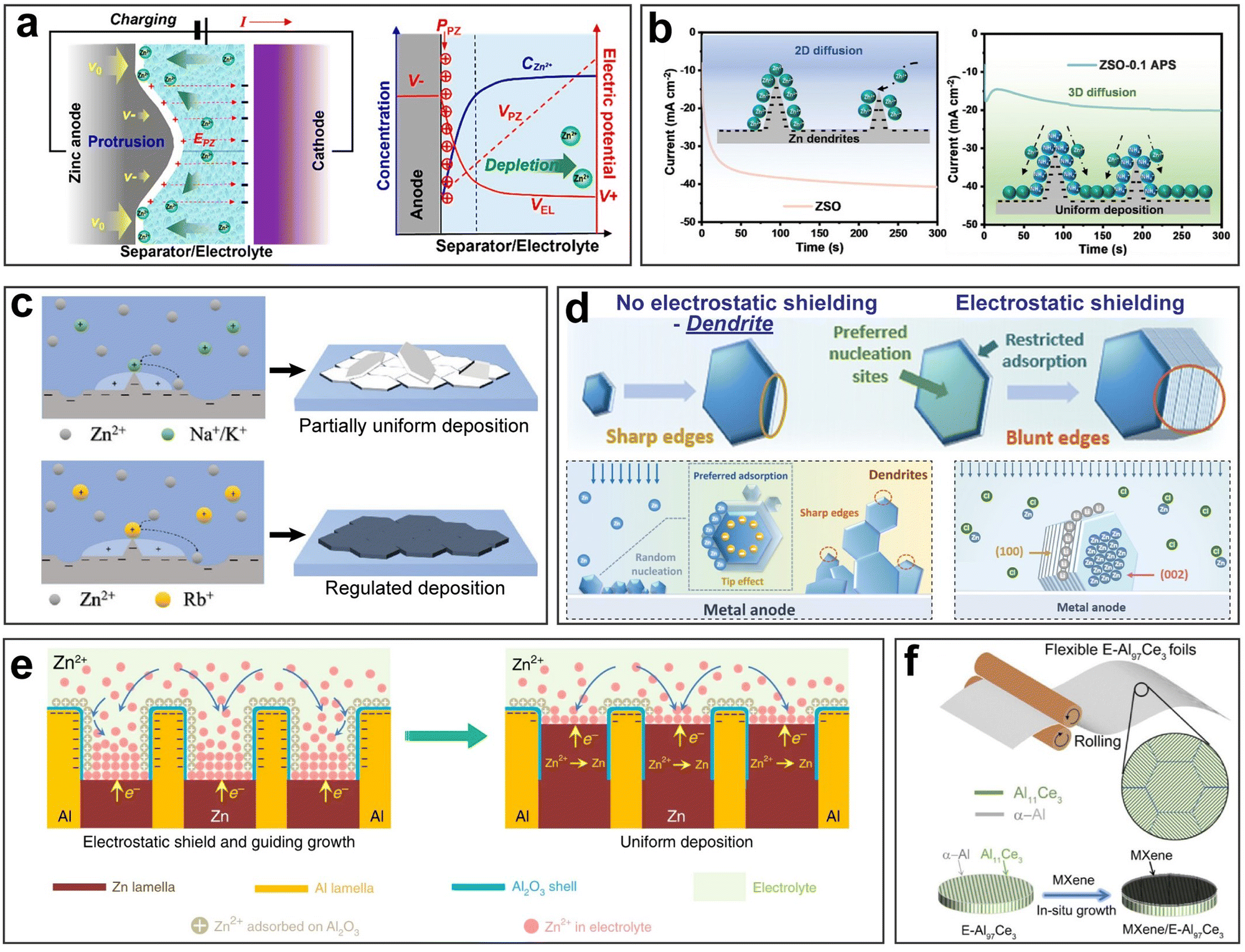 | ||
| Fig. 16 Tip shielding. (a) Schematic illustration of tip shielding enabled by a piezoelectric separator. Reproduced with permission from ref. 268 Copyright 2024, American Chemical Society. (b) Chronoamperometry curves showing 2D/3D diffusion with/without electrostatic shielding. Reproduced with permission from ref. 269 Copyright 2024, Wiley-VCH. (c) Schematic illustration of electrostatic shielding effects with different cations. Reproduced with permission from ref. 270 Copyright 2023, Wiley-VCH. (d) The influence of electrostatic shielding on crystallography. Reproduced with permission from ref. 271 Copyright 2024, Wiley-VCH. (e) Schematic illustration of electrostatic shielding in the eutectic Zn/Al alloy with a lamellar structure. Reproduced with permission from ref. 272 Copyright 2020, Springer Nature. (f) Schematic illustration of electrostatic shielding in the lamella-nanostructured Al/Ce alloy. Reproduced with permission from ref. 273 Copyright 2023, Wiley-VCH. | ||
Other than the shielding mechanism above, tip shielding is most often accomplished through electrostatic shielding, wherein specific electrolyte additives are introduced. These additives can selectively adhere to the protrusions, repelling metal ions towards flat regions for deposition, thereby impeding further dendritic growth of the protrusions. For example, Li et al. introduced ammonium persulfate (NH4)2S2O8 as an electrolyte additive, where NH4+ could preferentially adsorb on the protrusions due to the electron-rich nature of the tips, thus forming an electrostatic shield that guided Zn2+ towards the flat areas through electrostatic repulsion.269 It was revealed by CA curves that the electrostatic shielding could effectively restrain the 2D diffusion process which was associated with nonuniform deposition and promote the favorable 3D diffusion (Fig. 16b).
Common cations used for electrostatic shielding include Al3+,274 Na+,275–277 Sn2+,278 K+,279 and NH4+,269,280 with their radii being 0.53 Å, 1.02 Å, 1.2 Å, 1.38 Å and 1.48 Å, respectively. Zhang et al. compared the effects of cation radii on electrostatic shielding (Fig. 16c).270 Rb+, with a larger radius of 1.52 Å, was demonstrated to be more effective in electrostatic shielding because Rb+ was more efficient to occupy the tips and could induce a larger electrostatic shield, thereby prompting the lateral deposition of Zn2+ to form a well-regulated planar deposition morphology. On the other hand, Cheng et al. used Zr4+ as the cation for electrostatic shielding and stated that a higher valence could enhance the electrostatic shielding effect, and the further decreased Debye length also favored the Zn close-packed deposition.281 Other rare-earth cations with high valences such as Ce3+, La3+ and Y3+ have also been investigated as cations for electrostatic shielding.197,282
Specifically, using LiCl as the additive, Yuan et al. delved into electrostatic shielding from a crystallographic perspective (Fig. 16d).271 The rather small radius of Li+ (only 0.60 nm) took the electrostatic shielding effect to a crystallographic level, where sharp edges of Zn crystals served as tips. Li+ could effectively adsorb to the edges and tips of the existing Zn deposits due to the tip effect, thereby inhibiting secondary Zn nucleation along the (100) edges and encouraging deposition onto (002) facets instead, leading to a desirable dense and block-like deposition morphology. In addition to cationic electrostatic shielding, diethyl ether has also been reported to be an effective electrolyte additive for electrostatic shielding.154
Apart from electrolyte additives, electrostatic shielding can be achieved by the anode structure design as well. For instance, Wang et al. adopted a eutectic Al-Zn alloy with an alternating nano-lamellar structure as the anode, in which the insulating Al2O3 shell inhibited electron transfer from Al to Zn2+ and therefore prevented Zn deposition on the tips of Al/Al2O3 lamellas, enabling an electrostatic shield that guided uniform Zn deposition onto the Zn lamellas (Fig. 16e).272 The same strategy was also reported by Ran et al., who used a eutectic Ce–Al alloy (E-Al97Ce3) in situ grafted with uniform ultrathin MXene as the anode in AABs (Fig. 16f).273 Due to the high reactivity of Al and Ce, passivating and insulative oxide shells were in situ formed on Al11Ce3 nano-lamellas, which generated positive electrostatic shields that enabled directional Al deposition along the 2D channels of Al lamellas and hence guaranteed dendrite-free deposition.
4.2. Electric field modulation
A nonuniform electric field caused by protrusions or other contour irregularities leads to a nonuniform ion flux and eventually nonuniform deposition. In response to this, electric field modulation is adopted to offset the nonuniform electric field or guide a uniform electric field near the anode surface to ensure uniform deposition.Electric field modulation can be realized by electrode design. For instance, Ren et al. developed a uniform concave surface geometry on the Zn anode with nanometer-thick ZnO coating (ZnO–Zn) (Fig. 17a).245 Top-view SEM images showed that the surface of the bare Zn was rough and bumpy, full of irregular sharp edges, while that of ZnO–Zn displayed relatively smooth bowl-like concave morphologies. Correspondingly, the simulation demonstrated that the electric field in the ZnO–Zn model was uniform, precluding the sites with high current density due to the tip effect in the bare Zn model. Similarly, surface patterning that endowed the anode surface with regular microchannels was investigated by Wang et al. and proven to be effective in regulating the electric field distribution and homogenizing ion flux.66 Besides surface contour, more studies report the 3D electrode design for the uniform electric field distribution. Zhang et al. printed the 3D Ni–Zn anode with a highly ordered multi-channel lattice structure (3D Ni–Zn) (Fig. 17b).283 The simulations on the current density distribution from side views highlighted the significantly high local current density due to the tip effect in 2D Ni–Zn, while the relatively uniform electric field distribution in 3D Ni–Zn facilitated homogeneous deposition into the 3D microchannels. Various other 3D electrode designs, such as carbon networks and carbon cages, along with some cases mentioned before, all can contribute to electric field modulation.284
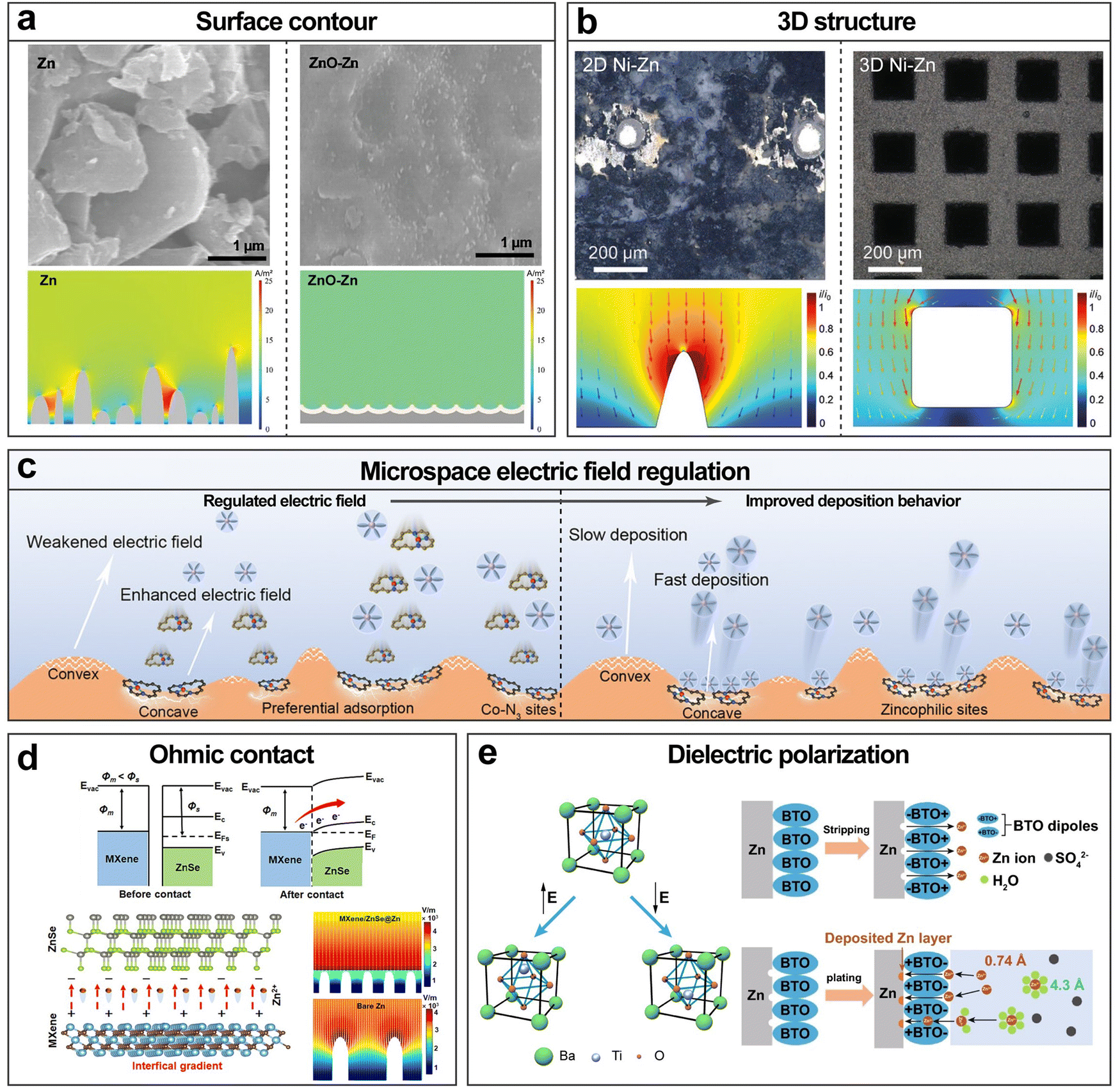 | ||
| Fig. 17 Electric field modulation. (a) Top-view SEM images and simulative electrical fields of Zn and ZnO–Zn. Reproduced with permission from ref. 245 Copyright 2023, Wiley-VCH. (b) Top-view SEM images and simulative models of the localized electric field for 2D/3D Ni–Zn. Reproduced with permission from ref. 283 Copyright 2021, Wiley-VCH. (c) Schematic illustration of microspace electric field regulation enabled by CoSA/C. Reproduced with permission from ref. 285 Copyright 2024, Wiley-VCH. (d) Schematic illustration of electric field modulation by MXene/ZnSe@Zn. (Evac, EF, Ec, Ev, and Φ represent the vacuum energy, Fermi level, conduction band, valence band, and work function, respectively). Reproduced with permission from ref. 286 Copyright 2024, Elsevier. (e) Schematic illustration of electric field modulation enabled by the BTO interlayer. Reproduced with permission from ref. 287 Copyright 2021, Springer Nature. | ||
A functional interlayer is also often utilized for electric field modulation. Interlayers composed of carbon networks288 or CNTs289 have been reported to redistribute the ion flux and homogenize the electric field. The Cu7Te4 interlayer proposed by Li et al. presented an ‘intercalation–deposition’ mechanism where Zn2+ was first inserted into Cu7Te4 and then uniformly deposited on Zn at successive low potential, leading to a more uniform distribution of the current density and ion concentration.290 Apart from these, the following cases featuring unique mechanisms associated with the electric field will be focused on.
An ingenious dynamic microspace electric field regulation strategy was proposed by Fan et al. by introducing trace addition of Co single atom anchored carbon (CoSA/C) (Fig. 17c).285 The empty 3d orbitals of Co accepted the lone-pair electrons of the nitrogen molecule, which induced the charge redistribution from the atomic-level and led to space charge polarization and localization, hence influencing the microspace electric field distribution. Upon charging, zincophilic micron-sized CoSA/C sheets could selectively adsorb on the spacious concave regions rather than the narrow convex protrusions. Such discriminative adsorption behavior, also referred to as the super filling phenomenon,291 combined with the charge polarization effect of CoSA/C, contributed to the enhanced electric field and current density on the concave surface. As a result, the electric field distribution could be regulated adaptively. The occurrence of self-amplifying protrusion growth induced by the tip effect was significantly limited and restrained, leading to flat and dense deposition. Consequently, the symmetric cells with the CoSA/C additive exhibited stable plating/stripping behaviors for over 1600 hours at 10 mA cm−2/2.5 mA h cm−2.
Ohmic contact can be employed as it creates a built-in electric field (BIEF), rearranges the energy bands, enhances the electrical conductivity, and therefore modulates the electric field and ion flux distribution. Ren et al. constructed a gradient heterostructure interlayer consisting of conductive MXene nanosheets and semiconductive ZnSe nanoparticles on the Zn anode surface (MXene/ZnSe) (Fig. 17d).286 Density functional theory (DFT) calculations showed that ZnSe exhibited a bandgap of around 1.156 eV near the Fermi level, while the MXene/ZnSe heterostructure displayed no bandgap, and its density of state (DOS) intensity was higher than that of ZnSe. Furthermore, energy band diagrams of metallic MXene and semiconductive ZnSe before and after contact illustrated that electrons transferred from MXene to ZnSe and formed a BIFE. The BIFE led to the charge redistribution and thus rendered the electric field distribution on the MXene/ZnSe@Zn surface uniform, as confirmed by simulations. Similarly, Ohmic contact interfaces could also be built by CeO2.292
Maxwell–Wagner polarization is another mechanism often considered for electric field modulation for its ability to create spatial charge separation and therefore contribute to the uniform electric field and homogeneous ion flux as well as HER inhibition.293,294 Maxwell–Wagner polarization refers to the charge separation phenomenon at the interfaces of two materials with different relaxation times 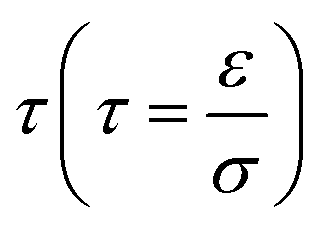 , where ε represents the dielectric constant and σ represents the electrical conductivity, and a pronounced Maxwell–Wagner polarization is achieved with a large relaxation time difference. Considering one of the materials is the metal anode with high electrical conductivity and a low dielectric constant, the interlayer should consist of materials with low electrical conductivity and a high dielectric constant.295 For example, Zhang et al. built an interlayer containing defect-rich HfO2−x polycrystals, which had a high dielectric constant of over 25 and a low electrical conductivity of 3.4 × 10−6 S m−1.296 In addition, the defect rich HfO2−x possessed strong zincophilicity due to the oxygen valence sites. As a result, the interlayer could not only induce Maxwell–Wagner polarization to regulate the ion flux, but also efficiently capture Zn2+ to improve the ion migration kinetics. Other materials with the low electrical conductivity and high dielectric constant including ZrO2,294,295,297 Si3N4298 and Nb2O5299 have also been reported to modulate the electric field via Maxwell–Wagner polarization.
, where ε represents the dielectric constant and σ represents the electrical conductivity, and a pronounced Maxwell–Wagner polarization is achieved with a large relaxation time difference. Considering one of the materials is the metal anode with high electrical conductivity and a low dielectric constant, the interlayer should consist of materials with low electrical conductivity and a high dielectric constant.295 For example, Zhang et al. built an interlayer containing defect-rich HfO2−x polycrystals, which had a high dielectric constant of over 25 and a low electrical conductivity of 3.4 × 10−6 S m−1.296 In addition, the defect rich HfO2−x possessed strong zincophilicity due to the oxygen valence sites. As a result, the interlayer could not only induce Maxwell–Wagner polarization to regulate the ion flux, but also efficiently capture Zn2+ to improve the ion migration kinetics. Other materials with the low electrical conductivity and high dielectric constant including ZrO2,294,295,297 Si3N4298 and Nb2O5299 have also been reported to modulate the electric field via Maxwell–Wagner polarization.
Another dielectric material, BaTiO3, due to its high dielectric constant and switchable polarization under external electric field, can be integrated in an interlayer for electric field modulation.287,300 Wu et al. constructed a BaTiO3 coating on the Zn anode (Fig. 17e).287 Upon charging, the corresponding external electric field induced deviation of Ti4+ from its original central position, causing polarization. The same phenomenon occurred upon discharge, but with an opposite polarization direction. As a result, a directional electric field could be built near the Zn anode surface. In addition, The BaTiO3 layer physically provided uniform ion pathways. The two mechanisms combined, the BaTiO3 layer significantly facilitated the uniform electric field distribution and homogenous ion flux, resulting in desirable smooth and flat deposition.
4.3. External stimuli
In addition to the material-specific ion flux homogenizing approaches, some universal measures, namely external stimuli, can be widely employed. Three external stimuli, such as magnetic fields, acoustic waves and external pressure, have been commonly studied in various battery systems301–307 and in aqueous batteries as well.Applying a vertical magnetic field can homogenize ion flux and alleviate dendritic growth.308–310 In the early stage of dendrite formation, metal ions are drawn towards the tips due to the tip effect. That is to say, a horizontal velocity component is present, in addition to the vertical velocity component drifting the ions from the cathode side to the anode side. A vertical magnetic field, in this case, generates the Lorentz force that acts on the metal ions being drawn to the tips. The direction of the Lorentz force is perpendicular to the plane consisting of the original vertical and horizontal velocities, inducing a new velocity that is perpendicular to both the original vertical and horizontal velocities. On the other hand, the magnetic field leaves the ions which are not subjected to the tip effect intact, as they exhibit no horizontal velocity components and hence are not subjected to any Lorentz force. The resulting trajectories of the metal ions subjected to both the tip effect and the Lorentz force are spiral lines revolving around the tips. The swirling motions of the metal ions can be referred to as the magnetohydrodynamic effect (Fig. 18a).310 With this effect, the metal ions that should be directly drawn to and deposited on the tips now are deposited in the regions near the protrusions, forming a relatively round deposit rather than a sharp dendrite. Such a mechanism can effectively mitigate the problem of dendrite formation and improve the reversibility of an aqueous metal battery.
 | ||
| Fig. 18 External stimuli. (a) (i) The effects of the external magnetic field on ion transport and deposition. (ii) Simulation of ion trajectories with/without the external magnetic field. Reproduced with permission from ref. 310 Copyright 2022, the Royal Society of Chemistry. (b) The effect of the acoustic wave on the ion transport and deposition. Reproduced with permission from ref. 311 Copyright 2024, Elsevier. (c) (i) Experimental results showing the effects of external pressures on deposition morphologies. Reproduced with permission from ref. 312 Copyright 2024, the Royal Society of Chemistry. (ii) The effects of the external pressure field on ion transport kinetics. Reproduced with permission from ref. 313 Copyright 2024, Wiley-VCH. | ||
Acoustic waves, or to be more specific, ultrasonic waves can induce dual effects that favor uniform deposition (Fig. 18b).311 Firstly, ultrasonic waves with a relatively low frequency (20–40 kHz) induce cavitation effects in electrolytes.314–316 Implosion of cavitation bubbles can release enough energy to both increase the local flow velocity and destruct the dendritic structure. Secondly, ultrasonic waves with a rather high frequency (100 MHz) generated by a surface acoustic wave device are able to generate acoustic streaming.301,317,318 The acoustic streaming can efficiently replenish ions in the boundary layer. At a high flow rate, a convective flow can even overtake ionic diffusion as the dominating ion transport and replenishment mechanism. Such effects can translate to the increasing diffusion coefficient substantially, which aligns with the previous analysis on the space charge model and Sand's time, significantly mitigating dendrite formation.
Finally, applying an external pressure is another plausible external approach for dendrite suppression. Experimental results suggested that as the applied stacking pressure increased from 100 kPa to 600 kPa, both the deposited Zn thickness and the porosity of the deposits decreased, along with top-view SEM images displaying denser and flatter deposition morphologies. Besides, Zn–Ti cells displayed higher Coulombic efficiency (CE) values with increasing pressure, and the average CE reached 88.9%, 91.2%, 94.3%, and 95.9% at 100, 200, 400, and 600 kPa, respectively. This indicated that applying the external pressure was capable of alleviating side reactions and preventing the dead Zn formation (Fig. 18c).312 Another study applying the reactive molecular dynamics (RMD) simulation with a reactive force field on the Zn electrode and the aqueous ZnSO4 electrolyte revealed that the addition of moderate pressure led to improved diffusion and reduction kinetics, regulating electrode crystallographic orientation towards the planar (002) texture, as well as a smaller solvation sheath with lower water coordination numbers, all of which favored dendrite suppression and uniform deposition.313
5. Solvation chemistry modulation
Often, multivalent ions are surrounded by the water molecules or ligands with counter-charged sites via the coordination interaction in the aqueous electrolyte. For those directly attached to the metal ions as the primary solvation shell, it will have the formula of [M(H2O)x(ligand)y]n+ where x + y is equal to 6 for most elements in periods 3 and 4 of the periodic table. The solvated structure of the ions is dependent on the interaction between the ligands (molecules, anions, etc.) and the metal ions. A strong ligand could repel the water molecules from the primary solvation shell, while a poor ligand would leave the solvated ion to fully hydrate as [M(H2O)6]n+. In the electrochemical deposition process, the solvated ions diffuse and approach the metal anode along the electric field, and then are desolvated at the surface for the following reduction reaction with charge transfer. In such a process, side reactions, such as the HER, could occur if the coordinated water number is high, while the desolvation kinetics would be slow if the metal ions are highly solvated by the ligands. In this part, the rational selection of ligands (anions from salts, organic additives, and polymer chains) and their functions in shaping the primary solvation shell and affecting the redox kinetics and side reactions will be introduced in multiple aqueous multivalent batteries.5.1. Deep eutectic electrolytes
The solvation chemistry of the metal cations can be controlled by adjusting the compositions and molecular chemical moieties of the electrolytes. Deep eutectic electrolytes, mainly consisting of hydrogen bond donor/acceptor pairs or Lewis acid/base pairs, have attracted broad attention in various electrochemical energy storage systems due to their good thermal and chemical stability, high tunability, and low vapor pressure.319,320 The hydrogen bond donors or Lewis bases interact with the metal cations, which repels the water molecules from their inner solvation sheath and suppresses the HER and passivation at the metal anode.321 One thing that should be noted is that there is a balance between the proportions of the solvated anions and water molecules, as too many solvated anions would decrease the ionic conductivity resulting in poor kinetics. The eutectic electrolytes can be mainly categorized into three classes: cosolvent eutectic electrolytes, hydrated eutectic electrolytes, and salt-additive eutectic electrolytes. Cosolvent eutectic electrolytes typically use organic solvents which are miscible in water and with a high donor number to change the first solvation shell of the metal ions. Commonly used solvents include dimethylformamide (DMF), dimethyl carbonate (DMC), DMSO, dimethylacetamide (DMA), dioxolane (DOL), Ch3CN, etc. For hydrated eutectic electrolytes, coordination agents, usually organic salts, are utilized to bond with metal ions and restrict the majority of water molecules inside the solvation structure. Although the water content is highly limited in such electrolytes, they could retain fluidity with a ‘water-in-eutectic’ status. The concept of salt-additive eutectic electrolytes is similar to the highly concentrated electrolytes, in which case, salt additives are added to the high-concentration electrolytes to further increase the salt concentration and decrease the hydrogen bonds formed with water molecules.For AZBs, Chen et al. reported a comprehensive study on the Zn(OTf)2 electrolytes with ethylene glycol (eg).322 The optimized binding configurations of different electrolytes were calculated by DFT calculations (Fig. 19a). Different from the normal solvation structure of [Zn(H2O)6]2+, the deep eutectic solvent (DES) showed the coordination with anions of OTf− and eg. When the water content in the DES increased to form hydrated DESs (HDES), the proportion of the coordinated anions decreased while the coordinated water increased. It should be noted that the anion-involved solvation structure in the DES is due to the weaker solvation capacity of eg compared with H2O, which results in low ionic conductivity and low binding energy. Simultaneous good ionic conductivity and low binding energy and overpotential could be realized in the electrolyte with a moderate water content, HDES30. The proportions of free anions (FA), loose ion pairs (LIP), and intimate ion pairs (IIP) in these HDES electrolytes were revealed by Raman spectroscopy (Fig. 19b). Among them, HDES30 showed a dramatically decreased LIP (Zn2+-OTf−) ratio, increased FA ratio, and moderate strong-H bond ratio, indicating a significantly improved ion dissociation degree and limited free water molecules (Fig. 19c). Wang et al. introduced a polar solvent, sulfolane, to constrain water molecules inside the reverse micelle nanodomains of the eutectic electrolyte, which limited the proton transport and suppress the HER.323 The sulfolane molecules not only form abundant hydrogen bonding with the water molecules through their strongly polar and hydrophilic group (O = S = O), but also coordinate with Zn2+ strongly forming the sulfolane-rich solvation shell.324 Other widely reported organic molecules used in eutectic electrolytes include acetamide,325,326 ethylene glycol,94,230 succinonitrile (SN),327 tetramethylene sulfone,328 urea,329,330etc.
 | ||
| Fig. 19 Deep eutectic electrolytes. (a)–(c) Coordination structures and average binding energies (a), Raman spectra (b), and proportions of the strong H-bond, free anion, and loose ion pair (c) of HDES50, HDES40, HDES30, HDES10, and DES, respectively. Reproduced with permission from ref. 322 Copyright, 2023, the Royal of Chemical Society. (d) Photographs of Al(ClO4)3·9H2O with SN in different ratios. (e) Solvated structure of the [Al(SN)5(H2O)]3+ complex. Reproduced with permission from ref. 331 Copyright, 2022, Wiley-VCH. (f) Photograph of the hydrated eutectic electrolyte made by Mg(NO3)2·6H2O and acetamide with a molar ratio of 1:8. (g and h) DSC (g) and FT-IR (h) curves of the electrolytes with different molar ratios. (i) RDF plots of Mg2+–O and the corresponding coordination environments of the Mg2+ ion in the electrolyte with a molar ratio of 1:8. Reproduced with permission from ref. 332 Copyright 2022, the Royal Society of Chemistry. | ||
The design rationales of eutectic electrolytes that explored in AZBs can be further employed in various multivalent aqueous battery systems as they share the similar solvation chemistry and mechanism. Meng et al. reported a new hydrated eutectic electrolyte with Al(ClO4)3·9H2O and a neutral ligand (SN) for AABs.331 It was found that Al(ClO4)3·9H2O is the best compared with other Al-based salts to form the hydrated eutectic electrolyte due to the strongest intermolecular interaction between Al(ClO4)3·9H2O and SN, which retained the liquid state even with a salt:ligand molar ratio of 1:20 (Fig. 19d). The coordinated SN (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N⋯Al3+) could be detected by Raman spectroscopy, distinguished from the free –C⋯N, with the highest coordination number of ∼5 when Al:SN is equal to 1:16 and the coordinated structure of [Al(SN)5(H2O)]3+ (Fig. 19e). The use of acetamide was also widely investigated in AABs. Chu et al. reported to use AlCl3/acetamide as the low-cost deep eutectic solvent for the Al–S batteries.333Via the coordination reaction, Al–Cl bond cleavage occurred and AlCl2+-coordinated cations and AlmCln− (AlCl4−, Al2Cl7−, etc.) anions formed.334 These newly formed anions could serve as the active species to help the reversible Al plating/stripping. Later, Chu et al. found that the use of a fluorine-substituted acetamide additive enabled an F-rich interphase layer which could further stabilize the Al plating/stripping.335 A large number of urea-like organic molecules, such as urea,263,336,337 methylurea,338 and thiourea,339 were reported for the formation of the deep eutectic electrolytes with Al salts. A hybrid Fe–Al liquid battery was reported by Zhang et al. using the AlCl3/urea deep eutectic electrolyte, which changed the coordination environment and the solvated structure to [AlCl2(urea)2]+ rather than Al2Cl7−.336 Luo et al. achieved a less-water-solvated structure, [Al(AN)2(TEP)(OTf)2(H2O)]3+, by using a combination of triethyl phosphate (TEP) and acetonitrile (AN) to prepare the hydrated eutectic electrolyte, which could inhibit the side reactions, lower the freezing point, and expand the electrochemical stability window.340
N⋯Al3+) could be detected by Raman spectroscopy, distinguished from the free –C⋯N, with the highest coordination number of ∼5 when Al:SN is equal to 1:16 and the coordinated structure of [Al(SN)5(H2O)]3+ (Fig. 19e). The use of acetamide was also widely investigated in AABs. Chu et al. reported to use AlCl3/acetamide as the low-cost deep eutectic solvent for the Al–S batteries.333Via the coordination reaction, Al–Cl bond cleavage occurred and AlCl2+-coordinated cations and AlmCln− (AlCl4−, Al2Cl7−, etc.) anions formed.334 These newly formed anions could serve as the active species to help the reversible Al plating/stripping. Later, Chu et al. found that the use of a fluorine-substituted acetamide additive enabled an F-rich interphase layer which could further stabilize the Al plating/stripping.335 A large number of urea-like organic molecules, such as urea,263,336,337 methylurea,338 and thiourea,339 were reported for the formation of the deep eutectic electrolytes with Al salts. A hybrid Fe–Al liquid battery was reported by Zhang et al. using the AlCl3/urea deep eutectic electrolyte, which changed the coordination environment and the solvated structure to [AlCl2(urea)2]+ rather than Al2Cl7−.336 Luo et al. achieved a less-water-solvated structure, [Al(AN)2(TEP)(OTf)2(H2O)]3+, by using a combination of triethyl phosphate (TEP) and acetonitrile (AN) to prepare the hydrated eutectic electrolyte, which could inhibit the side reactions, lower the freezing point, and expand the electrochemical stability window.340
A hydrated eutectic electrolyte with acetamide and Mg(NO3)2·6H2O was used for aqueous Mg-ion batteries by Zhu et al.332 The electrolyte remained in a liquid state with a Mg2+:acetamide molar ratio of 1:8 (Fig. 19f). Differential scanning calorimetry (DSC) curves revealed that high liquid temperatures (Tl) could be achieved when the molar ratio was too high or too low (Fig. 19g). Furthermore, the lowest melting point (Tm) at −76 °C could be obtained at a molar ratio of 1:8 due to the charge delocalization between the components. The bonding properties of the hydrated eutectic electrolyte could be further investigated by Fourier-transform infrared (FT-IR) spectroscopy, which suggested the break of hydrogen bonding in water clusters when increasing the acetamide (Fig. 19h). With the MD simulation, it showed that a low acetamide content (1:2) resulted in a well percolated hydrogen bonding network, while a high acetamide content (1:12) caused sluggish ionic transport due to the acetamide cluster (Fig. 19i). A ternary deep eutectic Mg-ion electrolyte was later reported by Song et al. using the mixture of Mg(Cl)2·6H2O, acetamide, and urea with the high ionic conductivity and enlarged working voltage.341
It can be seen that, in various aqueous battery systems, hydrated eutectic electrolytes are a promising design direction to alter the solvation chemistry of metal ions. More importantly, the introduced organic molecules, such as urea and acetamide, are universally effective in these cases. With a well-controlled solvated state, using eutectic electrolytes can suppress the side reactions, improve the ionic conductivity, and extend the electrochemical stability window simultaneously.
5.2. Organic polymer/chelating additives
Introducing electrolyte additives is a feasible and economical approach to control the dendrite growth, suppress the surface passivation, form stable protective SEI layers, and alleviate the dissolution of cathode materials in various aqueous battery systems.342,343 Inorganic additives, such as soluble salts and acids, are typically added to play the electrostatic shielding effect on suppressing the dendrites,344,345 involve in the electrochemical reduction to prevent the anode surface from the HER,346,347 and hinder the passivation layer formation by increasing the electrolyte acidity.348,349 Organic additives that are soluble in water can be categorized into surfactants, polymers, chelating agents, and organic salts. Surfactants, such as SDS350,351 and SDBS,352 show a strong tendency to enable the chemical adsorption on the metal surface and then alter its surface energy due to the interactions between the polar ends of the surfactant and the metal surface. The covering surfactants can homogenize the surface energy even with dendrite tips and its tail's hydrophobic nature can sterically hinder the approaching ions and redistribute the local ionic flux. Polymers with the strong polar ends (e.g., –NH2 and –OH) can also be absorbed onto the metal surface for tuning the interfacial properties.353–355 Furthermore, the interaction between the polymers and cations is able to reconstruct the solvation sheath as the chelating agents, which would be our focus in this section.356Jin et al. reported the in situ formation of nanometer-thick ion–oligomer interphases on a Zn metal anode for the chemical, morphological, and hydrodynamic instabilities.138 The structures of ion–oligomer complexes in aqueous electrolytes formed between Zn2+–halide ion complexes and PEG oligomer (300 Da) were studied with different zinc halide salts. The initial ion–oligomer complexes in aqueous media vary substantially as revealed by Raman vibrational spectroscopy, as shown in Fig. 20a. Octahedral [Zn(H2O)6]2+ and tetrahedral [ZnCl2(H2O)2] are the main ion states in the ZnSO4 and ZnCl2 electrolytes, respectively. In the ZnI2 electrolyte, besides the stable complexes [ZnI4]2−, several unstable complexes [ZnI3]−, [ZnI2] and [ZnI]+ also exist in a large proportion, while in the ZnBr2 electrolyte, [ZnBr2(H2O)2], [ZnBr4]2−, [ZnBr3]−, [ZnBr2] and [ZnBr]+ coexist. Such complexity suggests that the solvation structure of the Zn2+ in electrolytes is highly related to the steric structures and electronegativity of the anions. It is noted that in the case of ZnI2, no water molecules are involved in the complex formation, which is probably due to the lower electronegativity and higher polarization of iodine atoms. PEG with different molecular weights has also been reported effective in solvation regulation in other electrolyte systems.357–359 The chelating agents, such as 2-bis(2-hydroxyethyl)amino2-(hydroxymethyl)-1,3-propanediol (BIS-TRIS),360 trisodium citrate,361 triethanolamine,362 ethylenediaminetetraacetic acid (EDTA),363 and glucose,364 were reported to tune the solvation structures. Sun et al. introduced a small amount of glucose (10 mM) in the 1 M ZnSO4 electrolyte as the chelating agent to achieve highly reversible Zn plating/stripping.364 In contrast to the coordination structure of Zn2+ with six water molecules in the ZnSO4 electrolyte, one glucose molecule can enter the primary solvation shell to replace one water molecule there thus changing the solvation structure. Meng et al. proposed the use of the equilibrium constant of the complexation reaction (K), which affects the corrosion current density and nucleation overpotential, to evaluate the properties of the chelating agents to stabilize the anodes by comparing several Zn–ligand complexes.210 EDTA, with a superhigh K, was found to be the most effective chelating agent due to the highest energy barrier of Zn-EDTA for producing the Zn4SO4(OH)6 corrosion product in the ZnSO4 electrolyte.
 | ||
| Fig. 20 Organic polymer chelating additives. (a) Bonding between PEG and Zn ion complexes with electrolytes of ZnSO4, ZnCl2, ZnI2, and ZnBr2 with the 5 wt% of PEG oligomer. Reproduced with permission from ref. 138 Copyright 2022, Springer Nature. (b) Zn2+-solvation structure in the ZnSO4-glucose system by the MD simulation. Reproduced with permission from ref. 364 Copyright 2021, Wiley-VCH. (c) Schematic of Al3+-solvated structures with and without glucose addition. (d) LSV profiles in 4 M NaOH electrolytes with different glucose concentrations. Reproduced with permission from ref. 365 Copyright 2022, Elsevier. (e) The simulated geometrical structure of 0.8 m Mg(TFSI)2-85%PEG-15%H2O. (f) The g(r) and corresponding n(r) of Mg2+–H2O/PEG in 0.8 m Mg(TFSI)2-85%PEG-15%H2O. Reproduced with permission from ref. 366 Copyright 2022, Wiley-VCH. | ||
Wang et al. demonstrated the molecular crowding effect of the glucose-containing electrolyte in aqueous Al metal–air batteries.365 Different from the [Al(H2O)6]3+ structure in the normal electrolyte, free water molecules were found surrounded and confined by glucose molecules via the hydrogen bonds after introducing glucose (Fig. 20c). The potential for hydrogen evolution in the 4.5 M glucose electrolyte was −0.13 V more negative compared to the 0 M glucose electrolyte, which indicates the suppression of the hydrogen evolution reaction (Fig. 20d). Zhao et al. used polar pyridine-3-carboxylicacid (PCA) as the additive in the Al(OTf)3 electrolyte to alter the Al3+-solvation structure and restrain the corrosion and water activity.367 It was found that one PCA molecule can enter the primary solvation shell and replace one bounded H2O molecule and the adjacent Al3+ cations are connected by O, which differs significantly in the Al-centered solvated structure of [Al(H2O)6(OTf-H2O)3] in the electrolyte without the PCA. Such a solvation structure makes water decomposition thermodynamically unfavorable and enlarges the electrochemical stability window.
For AMIBs, Fu et al. reported an aqueous Mg-ion electrolyte with PEG and low-concentration salt of Mg(TFSI)2 with a wide ESW of 3.7 V.366 It was reported in the Mg(TFSI)2 electrolyte that Mg2+ tends to coordinate with the oxygen atoms of TFSI− ions and water with an octahedral arrangement, [Mg(H2O)6]2+ and [Mg(TFSI)3(H2O)3]−.368 PEG could directly coordinate with the Mg2+ cations and TFSI− anions, and thereby influence the solvation structure of Mg2+ ions in the electrolyte (Fig. 20e). The simulated geometrical structure of the Mg(TFSI)2–H2O electrolyte with and without PEG both showed a peak located at 0.20 nm, corresponding to the first solvation shell of Mg2+ (Fig. 20f). With PEG, an additional peak at 0.19 nm could be observed, which represented the coordination of Mg2+-PEG. Furthermore, the decreased integrated coordination number (ICN) with PEG indicated a decrease of the Mg2+–H2O coordination, implying that 36% of the solvated water in the first shell was replaced by two PEG molecules. Sun et al. also revealed that in the MgCl2–H2O-PEG electrolyte, four water molecules were coordinated with Mg2+ rather than six coordinated molecules in the MgCl2–H2O electrolyte.369
Overall, the introduced organic polymers or chelating agents could alter the solvation structure of the metal ions in the electrolyte, as showcased in different battery systems, via the coordination with the cations and the hydrogen bonds formed with water molecules and anions. Besides the effects on solvation, these additives often play the additional roles of restricting the activity of water molecules and constructing an organic interlayer on the metal anode, which synergistically benefit for the suppression of the HER and the free of corrosion/passivation.
5.3. Highly concentrated electrolytes
The concentration of the electrolyte can further significantly affect the solvation structure at the molecular level due to the change of the local environment for coordination, as well as the activity of water molecules in the aqueous electrolyte. When the salt concentration increases in the electrolyte, the IIP between the multivalent cations and anions from the salt would form, leaving the water molecules less coordinated but restrained in the local place. Such highly concentrated electrolytes with lean water are also called water-in-salt electrolytes when the salt outcomes the solvent by weight and volume. The desolvation behavior of the intimate ion pairs in WIS electrolytes is different from that of the common hydrated ions, which would promote the uniform deposition at the interface/interphase by regulating the interfacial properties. Furthermore, with the restrained activities of the water molecules, the hydrogen evolution and passivation occurred at the metal surfaces can also be suppressed.The concept of the WIS electrolyte was first introduced in the aqueous lithium-ion battery to overcome the water splitting issue in 2015.370 Suo et al. proposed to use the 21 m LiTFSI to broaden the electrochemical window to ∼3.0 V due to the evolution of the Li+ primary solvation sheath. Such WIS electrolyte design was also utilized in battery systems beyond Li, such as Na.371 Due to the high solubility of LiTFSI in water (>20 m) at room temperature, LiTFSI is a promising additive to form the WIS electrolyte combining with other redox active salts for multivalent aqueous batteries. Wang et al. used a mixed highly concentrated Zn-ion electrolyte (HCZE), 20 m LiTFSI and 1 m Zn(TFSI)2 for reversible Zn deposition and enlarged electrochemical window.372 As shown in Fig. 21a, the deposited Zn showed a dense and dendrite-free morphology after 500 cycles and no passivation product ZnO can be observed from XRD patterns. The highly reversible Zn plating/stripping can be attributed to the unique solvation sheath structure of Zn2+ in the HCZE, where anions are accumulated at the vicinity of Zn2+ and form (Zn–TFSI)+, rather than (Zn(H2O)6)2+ in the common electrolyte. Such coordination structures were revealed by MD simulations, which showed that Zn2+ coordinates with six water molecules with 5 m LiTFSI, and the structure evolves to coordinate with six oxygen atoms from the TFSI− anions when LiTFSI increases to 20 m (Fig. 21b). A large number of highly concentrated electrolytes were explored with different salt combinations showing the properties of suppressing the water decomposition and expanding the electrochemical stable window. Zhao et al. designed the WIS electrolyte of 21 m LiTFSI and 0.5 m ZnSO4 demonstrating a high-voltage aqueous Zn/LiMn0.8Fe0.2PO4 battery (>1.8 V).373 Hu et al. used 21 m LiTFSI with 1 m Zn(OTf)2 to make an aqueous hybrid-ion Zn/V2O5 battery with an improved discharge voltage platform of 1.0 V.374 Wan et al. further used the same WIS electrolyte to achieve an operating voltage of ∼1.6 V.375 Considering the high cost of the TFSI salts, the potential for using other cheap and commonly used salts for preparing WIS electrolytes has also been widely explored. For example, 30 m ZnCl2 can work as the WIS electrolyte due to the coordinated [Zn(H2O)2Cl4]2− and [ZnCl4]2− ions for preventing hydrogen evolution.376 Also, 3.3 m ZnSO4, forming [Zn(H2O)62+(SO42−)] in the electrolyte, was reported to use with a MOF-based protection layer to synergistically suppress the water activity and reduce the corrosion of the Zn anode.377
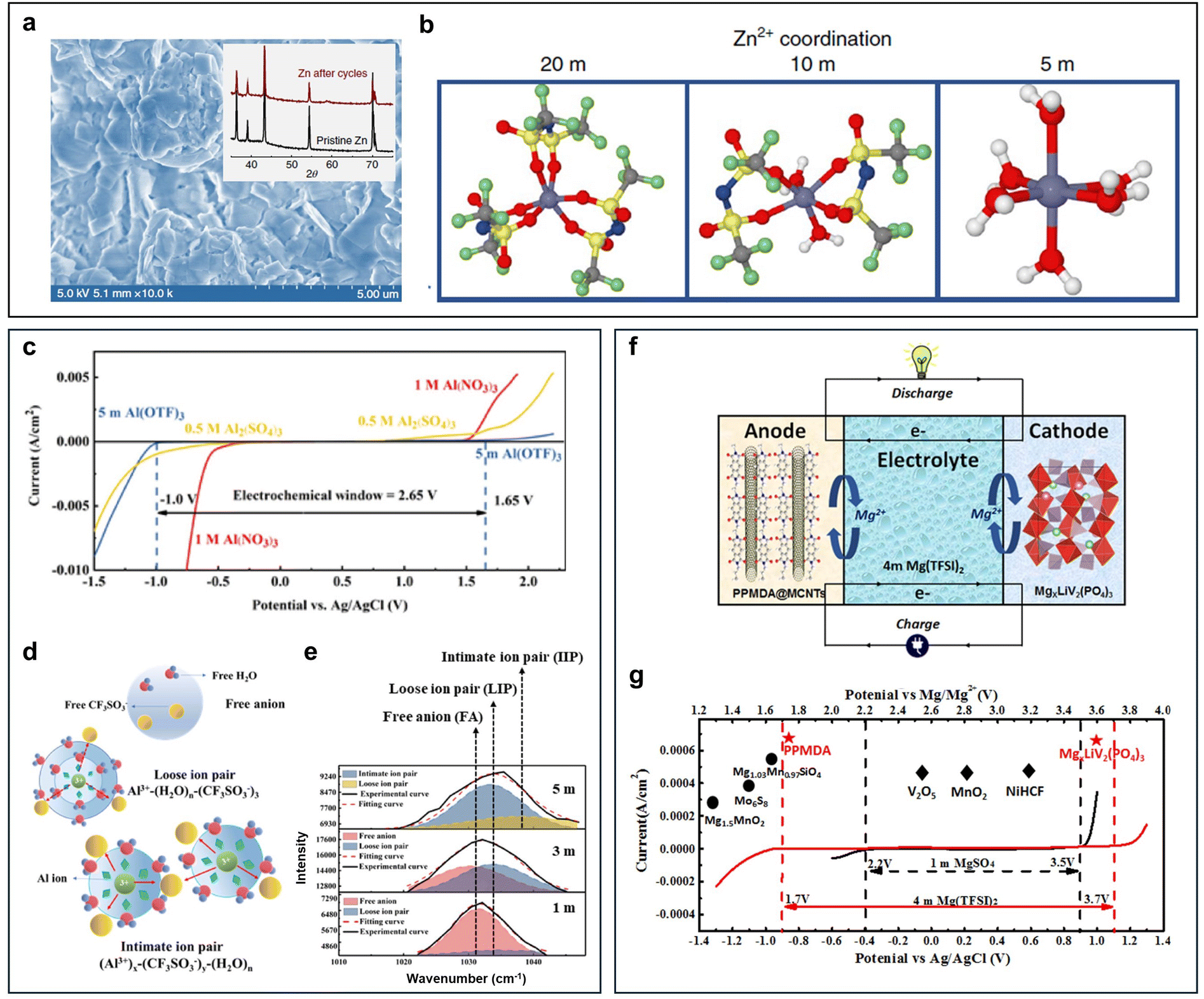 | ||
| Fig. 21 Water-in-salt electrolytes. (a) SEM image and XRD pattern (inset) of a Zn anode cycled in the HCZE. (b) Zn2+-solvation structures in the electrolytes with 1 m Zn(TFSI)2 + LiTFSI of 5 m, 10 m and 20 m, respectively. Reproduced with permission from ref. 372 Copyright 2018, Springer Nature. (c) The linear sweep voltammetry in the Al-WISE electrolyte in comparison to 1 M Al(NO3)3 and 0.5 M Al2(SO4)3 electrolytes. (d) Schematic diagram of the Al3+-solvation structures showing the free anion (FA), loose ion pair (LIP) and intimate ion pair (IIP). (e) Raman spectra of 1, 3, and 5 M Al(OTf)3. Reproduced with permission from ref. 378 Copyright 2019, American Chemical Society. (f) Schematic illustration of Mn-ion batteries with the electrolyte of 4 m Mg(TFSI)2. (g) Electrochemical stability window of the 4 m Mg(TFSI)2 aqueous electrolyte. Reproduced with permission from ref. 379 Copyright 2017, American Chemical Society. | ||
The WIS electrolyte design concept can be further used in Al-based aqueous battery systems. Compared with the redox potential of Zn/Zn2+ (−0.76 V vs. SHE), the redox potential for Al/Al3+ (−1.66 V vs. SHE) is much lower, which means that the hydrogen evolution issue at the metal anode side would be more severe. Zhou et al. reported that the 5 m Al(OTf)3 electrolyte, which is close to saturated, was effective to widen the electrochemical window as well as alleviate the dissolution issue of the Prussian blue cathode.378 As shown in Fig. 21c, compared with the electrolytes of 1 M Al (NO3)3 and 0.5 M Al2(SO4)3, with stability windows of 1.81 V and 1.40 V respectively, the WIS 5 m Al(OTf)3 electrolyte showed a broadened electrochemical window of 2.65 V. The suppressed water activity can be attributed to the formed LIP and intimate ion pairs in the WIS electrolyte due to the lack of water molecules to form two hydrated shells with Al3+, which is the case in dilute aqueous solutions (Fig. 21d).380 From the Raman spectra, in a dilute electrolyte of 1 m, the free anions (FA) are dominant species while an increasing proportion of the IIPs can be observed when the concentration of the electrolyte increases from 1 m to 5 m (Fig. 21e). However, Pastel et al. pointed out that the Al(OTf)3 electrolyte did not show obvious positive effects even with a concentration of 3.6 m, and the overpotential for hydrogen evolution was decreased in this case.51 It also should be noted that the Al(OTf)3–H2O system exhibited a much lower pH compared to the Li(OTf)–H2O system, which suggests that Al(OTf)3 could increase the proton activity and affect the cathode stability381 due to the existing hexa-aqua ion [Al(H2O)6]3+ although there are few unbound water molecules. Besides the use of Al(OTf)3, AlCl3 was selected as the salt due to its high solubility in aqueous solutions.28 With the AlCl3-based WIS electrolyte (4:1:1 of AlCl3·6H2O, MnSO4·6H2O and water in weight), a high discharge voltage of 1.9 V and a high discharge capacity of 285 mA h g−1 could be achieved for the MnO2/Al battery.382 Other advanced WIS electrolytes include the mixed salts, for example 1 M Al(OTf)3 and 15 M Li(OTf), which changes the Al3+-solvation sheath from the traditional octahedral Al(H2O)63+ configuration to mixed octahedral Al(OTf)x(H2O)6−x (x > 0) and tetrahedral Al(OTf)3OH− ion pairs, and thus extends the electrochemical stability window up to 4.35 V.383
The use of WIS electrolytes has also been exploited in other multivalent (e.g., Mg,379,384,385 and Ca386–388) aqueous batteries. The fundamental working principles of these WIS electrolytes are similar to those of the Zn and Al ones, which reshape the cation–solvation sheath structure and regulate the water bonding conditions. Wang et al. reported to achieve an enlarged electrochemical window with the WIS electrolyte of 4 m Mg(TFSI)2, which is noncorrosive and safe (Fig. 21f).379 Compared with the dilute 1 m MgSO4 electrolyte, whose electrochemical window is governed by the hydrogen and oxygen evolution potential, the WIS electrolyte exhibited a broadened window of 2.0 V (Fig. 21g), which is higher than most nonaqueous Mg-ion electrolytes.
Although there have been a decent number of studies on WIS electrolytes over the past decade, research thrusts can further be focused on developing novel WIS systems with multifunctional properties, for example, stabilizing the cathode materials during the electrochemical process. Also, exploring promising salts with large abundance and low cost is required to lower down the price for the electrolyte, making the WIS electrolyte-based aqueous batteries mass-produced for large-scale applications.
5.4. Hydrogel electrolytes
Hydrogel electrolytes have attracted broad attention in versatile electrochemical energy storage applications thanks to their high tunability in the surface functional groups and cross-linking status.389 The functional groups on the polymer chain determine its affinity with the water molecules in the aqueous electrolyte and the cross-linked polymer network constructs the pores with various sizes to hold water molecules inside. There are three main statuses for water molecules inside a hydrogel: free water, intermediate water, and bounded water.390 Free water molecules show a high degree of freedom, forming hydrogen bonds with the adjacent water molecules. These water molecules often have a high possibility to participate in the side reactions in the electrochemical process, e.g., the HER, causing detrimental effects on the cell cycle life. Bounded water are the water molecules at the vicinity of the polymer chain and forming hydrogen bonds with it and it is less likely for these water molecules to travel a long-range distance in a hydrogel electrolyte. By tuning the properties of the hydrogels, aqueous batteries with various features for different practical applications could be realized.Zwitterions, or ‘inner salts’, represent a unique class of small molecules in which cationic and anionic species are covalently bound, which typically exhibit high water retention abilities and solubilities. Polyzwitterions have been widely used in energy storage applications due to several advantages.391 Among them, the most important one is that, under the electric field, the cationic and anionic counterions on the polymer chains can be separated, which could facilitate the ion transport inside the channels due to the strong electrostatic interactions between the charges on the chains and cations in the electrolyte.392 Furthermore, the high polarity of the charged polymer chains generates a strong interaction between the hydrogel electrolyte and the metal anode. Fu et al. developed a supramolecular zwitterionic hydrogel electrolyte (SZHE) with cross-linked carbon nanofibers (CNF), polyacrylic acid (PAA), and zwitterionic betaine, which exhibited high ionic conductivity and self-healing properties due to the fast ion shuttles and high mechanical robustness of the hydrogel network (Fig. 22a).393 Mo et al. also constructed the fast ion migration channels via cross-linking [2-(methacryloy-loxy)ethyl]diethy-(3-sulopropyl) (MADES), a zwitterionic sulfobetaine monomer, in the cellulose nanofibril matrix (Fig. 22b).394 Separated ion migration channels for Zn2+ and SO42− ions could be achieved due to the zwitterionic groups on sulfobetaine chains in the zwitterionic sulfobetaine/cellulose (ZSC) hydrogel electrolyte.
 | ||
| Fig. 22 Hydrogel electrolytes. (a) Schematic illustration of the fabrication of SZHEs via the molecular engineering strategy. Reproduced with permission from ref. 393 Copyright 2023, the Royal Society of Chemistry. (b) Schematic of the ZSC gel electrolyte working under an external electric field. Reproduced with permission from ref. 394 Copyright 2020, Wiley-VCH. (c) Schematic of the ‘salting out’ for the PVA hydrogel in a mixture solvent of water and DMSO. Reproduced with permission from ref. 395 Copyright 2023, Wiley-VCH. (d) Linear voltammetry curves of the Ca(NO3)2 gel electrolyte and aqueous electrolytes with different concentrations. Reproduced with permission from ref. 396 Copyright 2021, Springer Nature. (e) Schematic of the electrolytes with and without PEG. (f) Photographs of the Al foil pristine and after cycling with and without PEG. Reproduced with permission from ref. 211 Copyright 2023, Wiley-VCH. | ||
Anti-freezing is a critically important feature for practical applications that could be realized via using the hydrogel electrolytes, as the water molecules bound to the polymer chain show a low degree of freedom and a lower temperature below the freezing point of water is required to freeze them. A poly(vinyl alcohol) (PVA)-based hydrogel electrolyte was fabricated via the integrated effects of co-nonsolvency and ‘salting-out’ with a DMSO/water mixed solvent (Fig. 22c).395 The PVA polymer chains were highly aggregated, forming the porous structure inside, and the hydrogen bonds among free water were disrupted, resulting in a high freezing tolerance of −77 °C. Shi et al. developed a Zn(BF4)2 and polyacrylamide (PAM)-based anti-freezing electrolyte by replacing the O–H⋯O bonding between water molecules with O–H⋯F due to the higher electronegativity of F atoms.397
Electrochemical stability windows can further be enlarged via rationally designing hydrogel electrolytes to reduce the free water content or generate a polymer electrolyte–metal interphase. A Ca2+-based hydrogel electrolyte with PVA was used for the protection of the SEI on the anode and suppressing both the HER and the OER for reversible Ca plating/stripping (Fig. 22d).396 For a 1 m Ca(NO3)2 solution, Ca2+ was solvated by six water molecules at the primary solvation sheath, while most of other water molecules freely formed hydrogen bonds, which would trigger the HER. When PVA was introduced in a saturated Ca(NO3)2, a polymer-like aggregation was exhibited, suggesting that water molecules were immobilized by the highly concentrated salt and the polymer chain. PEG was utilized as another skeleton for polymer electrolytes which could generate an Al electrolyte interface during charging (Fig. 22e).211 Such a protective layer not only inhibited the side reactions and avoided corrosion after repeated cycling (Fig. 22f), but also disrupted the hydrogen bond and increased the working temperature of the battery.
Generally speaking, hydrogel electrolytes are highly tunable by choosing different salts, polymer chains, and polymerization conditions for achieving various capabilities, such as high ionic conductivity, wide working temperature and voltage, and suppressed side reactions and corrosion. The fundamentals of the hydrogel electrolyte design are based on altering the interactions between the polymer and water and between the polymer and cations. Achieving aqueous electrolytes with multiple functions is highly promising through playing the interfacial chemistry among water, salts, and polymers.
6. Conclusions and outlook
To conclude, we explored the interfacial chemistry of multivalent aqueous batteries via the integration of fundamental working principles, dominating challenges at the interfaces, and rationale design strategies for regulating the interfacial properties. Specifically, aspects of regulating metal deposition, homogenizing ion flux, and tuning ion solvation chemistry were introduced to address the key interfacial problems at the metal anode and cathode, such as dendritic growth, side reactions, gas generation, and material dissolution. While considerable research has been focused on AZBs, other aqueous metal batteries have received comparatively less attention. Such disparity and imbalance present an opportunity to adapt and implement successful strategies developed for AZBs in other aqueous battery systems, as we have showed that the interfacial design strategies are actually highly universal in these multivalent aqueous battery systems. For example, alloying has been extensively studied and applied in AZBs, with its functions of nucleation enhancement as well as corrosion resistance both investigated. However, in AABs and other aqueous battery systems, alloying has not yet been fully studied. On one hand, alloying is a common and effective strategy in general metal corrosion prevention, the as-existing experience of which can be used as the reference for metal anode anti-corrosion. On the other hand, the nucleation enhancement effect of alloying has rarely been exploited in AABs and other aqueous battery systems. Similarly, other strategies such as metallophilic layers, crystallographic manipulation, tip shielding, electric field modulation, etc. can also be studied and applied in aqueous battery systems. It should also be noted that the metal anodes (e.g., Al, Mn, and Mg) are typically more susceptible to the HER and passivation/corrosion due to their intrinsic chemical and electrochemical properties. Therefore, employing the methodologies in Zn-based batteries in other metal-based batteries should carefully consider multiple aspects of anti-corrosion/passivation and anti-dendrite for achieving long battery lifetime.Despite the significant achievements in aqueous batteries, we summarize some key aspects that have not been well explored in current research, which would enable cost-effective, high-energy, long-duration multivalent batteries for various practical applications:
(1) High-voltage stability and wide-temperature properties. Other than the predominant emphasis on stability highlighted in numerous papers, it is imperative to consider several other dimensions when evaluating the performance of aqueous batteries. Two pivotal aspects warranting attention are high-voltage stability and wide-temperature performance, as they represent critical factors dominating the batteries’ stability and energy/power density and impeding the practical application of aqueous metal batteries. Current aqueous metal batteries are frequently plagued by side reactions, notably the electrolysis of water at high voltages. The constrained working voltage, typically around 1.5 V, is considerably lower than the average working voltage of lithium-ion batteries, which easily surpasses 3.5 V. The consequent low energy density and power density constitute significant drawbacks of aqueous metal batteries. Furthermore, the low-temperature performance of aqueous metal batteries is often compromised by diminished discharge capacity, reduced power density, and the susceptibility of electrolytes to freezing. Current state-of-the-art aqueous batteries with an anti-freezing electrolyte can operate at −85 °C398 and some others can work at −70 °C.399,400 However, the decrease in energy density is still inevitable due to the slow charge transport kinetics at such low temperatures. For high-temperature applications, the side reactions such as the HER will be more severe due to the high water molecule activity. With the advanced WIS electrolyte design, aqueous batteries can withstand the wide temperature range between −60 °C and +80 °C, while maintaining high stability and power density.401 However, more efforts are still required to advance the wide-temperature applications of various aqueous battery systems. The aforementioned limitations constrain the potential large-scale application scenarios of aqueous metal batteries and necessitate concerted efforts towards their resolution to propel advancements in the field.
(2) Discerning and understanding the underlying mechanisms. Despite numerous papers reporting research findings on aqueous metal batteries, many fail to delve into the fundamental mechanisms underlying the challenges that they aim to address or the rationale behind the proposed designs. Phenomenological evaluations merely answer whether a design is effective, without elucidating the underlying mechanisms or providing a logical and constructive framework. For example, in terms of electrolyte additives, interlayer design and SEI design, the choices of molecules and materials are miscellaneous. Although in general they may follow some overarching principles, the failure to sensibly discern different working mechanisms, the absence of systematic deduction and the lack of rational screening, to some extent, underscores the randomness of related materials selections. This calls for the unequivocal understanding of various mechanisms and in-depth comprehension via the integration of advanced characterization and simulation/modeling, instead of merely relying on electrochemical performance, to facilitate systematic and rational screening processes.
(3) Convincing and standardized characterization studies. In the calculation and experimental aspects, inconsistency exists among various studies. For instance, in DFT calculations, variations under simulation conditions lead to vastly fluctuating outcomes, such as reported surface energies of Zn and binding energies with alloying elements. This highlights the need for alternative experimental characterization to elucidate underlying mechanisms, as reliance solely on DFT calculations may lack persuasiveness. Similarly, the measurement of contact angles yields conflicting interpretations. While some take a decrease in contact angles as a sign of enhanced metallophilicity and improved electrolyte wettability that appears beneficial, some argue that a decreased contact angle indicates hydrophilicity which intensifies water-induced side reactions. Such a disagreement implies the intricacy of certain characterization methods, calling for rigorous interpretation and comprehensive analysis. Last but not least, there is a lack of a standardized baseline for the metal anode utilized in various studies. The stability performance of the metal anode may significantly depend on factors such as its initial surface morphology, composition, processing methods, and more. Therefore, the selection of characterization techniques should be standardized to ensure decisive and unambiguous insights into specific parameters or performances. For example, when evaluating corrosion resistance, priority should be given to techniques such as the Tafel plot, which provides critical information on corrosion potential and corrosion current density. Other characterization methods such as the theoretical calculation of hydrogen evolution energy barriers and the measurement of hydrogen generation rates could be auxiliary. In conclusion, convincing and standardized characterization studies, along with benchmarking studies, collaboration, and data sharing, are vital to enhancing viability, reproducibility, comparability, and scalability in electrochemical research.
(4) Exploring novel chemistries for cathodes. The current commonly used cathode materials in aqueous metal batteries are V- and Mn-based oxides, which suffer from the severe material dissolution issue, resulting in serious performance deterioration. Heteroatom doping has been utilized to optimize the interface of the cathode by means of forming a protective layer to suppress the dissolution. Other strategies include electrolyte modification such as the high-concentration design. However, there are not many simple, yet effective strategies developed so far to address such an issue. One possibility is to explore cathode materials with new chemistries beyond the insertion/extraction mechanism, and thus overcome the material dissolution. For example, organic materials, with the mechanism of the coordination reaction with the cations, could be potential cathodes for various aqueous batteries. The functional groups and steric structures of the organic materials could be highly tunable to meet the charge storage requirements for cations with different charges and hydrated sizes. Furthermore, the use of organic materials can take advantage of the ‘co-insertion’ of metal cations and protons to reach a high specific capacity and working voltage, and thus making the energy density of aqueous batteries more competitive.
(5) Artificial intelligence-guided fast screening and prediction. Traditional ‘trial-and-error’ processes require a vast number of tedious experiments, causing a dramatic waste of manpower and material resources. It is quite challenging especially when exploiting aqueous electrolytes, as their properties can be largely affected by the type of anions from the salts, electrolyte additives, and concentrations. Artificial intelligence or machine learning (AI/ML) has emerged as a powerful tool to unveil the material working/failure mechanisms and accelerate the discovery of battery materials and chemistries. The prerequisite of ML is the sufficient and solid dataset collected by simulations, advanced microscopies, and electrochemical characterization studies. Accordingly, ML algorithms that can learn complex correlations and patterns from the existing dataset provide a solution to the unresolved problems. The use of AI/ML would be very suitable for unlocking the solvation structure of metal ions with different anions, solvents, and ligands, given the large quantities of solvation structures that have been explored with MD simulations. Furthermore, with the dataset, it is promising that the AI/ML can help to predict the potential additives which can help regulate the solvation shell and guide the experimental research in a more time-efficient way.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported as part of the Center for Mesoscale Transport Properties, an Energy Frontier Research Center supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under award #DE-SC0012673.References
- Z. Yang, J. Zhang, M. C. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS
.
- J. B. Goodenough, Energy Environ. Sci., 2014, 7, 14–18 RSC
-
S. U. Falk and A. J. Salkind, Alkaline storage batteries, John Wiley & Sons, New York, 1969 Search PubMed
- M. Sun, X. Yang, D. Huisingh, R. Wang and Y. Wang, J. Cleaner Prod., 2015, 107, 775–785 CrossRef
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef
- C.-Y. Wang, T. Liu, X.-G. Yang, S. Ge, N. V. Stanley, E. S. Rountree, Y. Leng and B. D. McCarthy, Nature, 2022, 611, 485–490 CrossRef CAS PubMed
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed
- Z. Zhao, X. Liu, M. Zhang, L. Zhang, C. Zhang, X. Li and G. Yu, Chem. Soc. Rev., 2023, 52, 6031–6074 RSC
- Y. Ding, C. Zhang, L. Zhang, Y. Zhou and G. Yu, Chem. Soc. Rev., 2018, 47, 69–103 RSC
- Y. Huang and J. Li, Adv. Energy Mater., 2022, 12, 2202197 CrossRef CAS
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li, N. Tavajohi and B. Li, J. Energy Chem., 2021, 59, 83–99 CrossRef CAS
- D.-H. Liu, Z. Bai, M. Li, A. Yu, D. Luo, W. Liu, L. Yang, J. Lu, K. Amine and Z. Chen, Chem. Soc. Rev., 2020, 49, 5407–5445 RSC
- Y. Zheng, Y. Yao, J. Ou, M. Li, D. Luo, H. Dou, Z. Li, K. Amine, A. Yu and Z. Chen, Chem. Soc. Rev., 2020, 49, 8790–8839 RSC
- H. S. Hirsh, Y. Li, D. H. Tan, M. Zhang, E. Zhao and Y. S. Meng, Adv. Energy Mater., 2020, 10, 2001274 CrossRef CAS
- Z. Zhu, T. Jiang, M. Ali, Y. Meng, Y. Jin, Y. Cui and W. Chen, Chem. Rev., 2022, 122, 16610–16751 CrossRef CAS
- A. Innocenti, D. Bresser, J. Garche and S. Passerini, Nat. Commun., 2024, 15, 4068 CrossRef CAS PubMed
- Y. Li, Y. Li, Q. Liu, Y. Liu, T. Wang., M. Cui, Y. Ding, H. Li and G. Yu, Angew. Chem., Int. Ed., 2024, 63, e202318444 CrossRef CAS
- G. Li, Z. Zhao, S. Zhang, L. Sun, M. Li, J. A. Yuwono, J. Mao, J. Hao, J. Vongsvivut, L. Xing, C.-X. Zhao and Z. Guo, Nat. Commun., 2023, 14, 6526 CrossRef CAS PubMed
- X. Lu, Z. Liu, A. Amardeep, Z. Wu, L. Tao, K. Qu, H. Sun, Y. Liu and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202307475 CrossRef CAS PubMed
- J. Wang, T. Ghosh, Z. Ju, M.-F. Ng, G. Wu, G. Yang, X. Zhang, L. Zhang, A. D. Handoko, S. Kumar, C. Jiang, M. Wei, G. Yu and Z. W. Seh, Matter, 2024, 7, 1833–1847 CrossRef CAS
- X. Yang, C. Zhang, L. Chai, W. Zhang and Z. Li, Adv. Mater., 2022, 34, 2206099 CrossRef CAS
- P. Ruan, S. Liang, B. Lu, H. J. Fan and J. Zhou, Angew. Chem., 2022, 134, e202200598 CrossRef
- T. Wang, Y. Li, J. Zhang, K. Yan, P. Jaumaux, J. Yang, C. Wang, D. Shanmukaraj, B. Sun, M. Armand, Y. Cui and G. Wang, Nat. Commun., 2020, 11, 5429 CrossRef CAS
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS PubMed
- S. Belongia, X. Wang and X. Zhang, Adv. Funct. Mater., 2024, 34, 2302077 CrossRef CAS
- H. Tian, Z. Li, G. Feng, Z. Yang, D. Fox, M. Wang, H. Zhou, L. Zhai, A. Kushima, Y. Du, Z. Feng, X. Shan and Y. Yang, Nat. Commun., 2021, 12, 237 CrossRef CAS PubMed
- W. Pan, Y. Wang, Y. Zhang, H. Y. H. Kwok, M. Wu, X. Zhao and D. Y. Leung, J. Mater. Chem. A, 2019, 7, 17420–17425 RSC
- H. Wang, R. Tan, Z. Yang, Y. Feng, X. Duan and J. Ma, Adv. Energy Mater., 2021, 11, 2000962 CrossRef CAS
- X. Guo and G. He, J. Mater. Chem. A, 2023, 11, 11987–12001 RSC
- H. Wu, D. Zhuo, D. Kong and Y. Cui, Nat. Commun., 2014, 5, 5193 CrossRef CAS PubMed
- Z. Hao, Q. Zhao, J. Tang, Q. Zhang, J. Liu, Y. Jin and H. Wang, Mater. Horiz., 2021, 8, 12–32 RSC
-
D. H. Doughty, Vehicle battery safety roadmap guidance, National Renewable Energy Lab. (NREL), Golden, CO (United States), 2012 Search PubMed
- C. Hogrefe, T. Waldmann, M. Hölzle and M. Wohlfahrt-Mehrens, J. Power Sources, 2023, 556, 232391 CrossRef CAS
- L. Xue, W. Chen, Y. Hu, T. Lei, C. Yang, M. Zhou, X. Zhang, A. Hu, Y. Li, X. Wang and J. Xiong, Nano Energy, 2021, 79, 105481 CrossRef CAS
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M.-H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed
- Y. Xiang, M. Tao, G. Zhong, Z. Liang, G. Zheng, X. Huang, X. Liu, Y. Jin, N. Xu, M. Armand, J.-G. Zhang, K. Xu, R. Fu and Y. Yang, Sci. Adv., 2021, 7, eabj3423 CrossRef CAS PubMed
- F. Liu, R. Xu, Y. Wu, D. T. Boyle, A. Yang, J. Xu, Y. Zhu, Y. Ye, Z. Yu, Z. Zhang, X. Xiao, W. Huang, H. Wang, H. Chen and Y. Cui, Nature, 2021, 600, 659–663 CrossRef CAS PubMed
- C. Fan, W. Meng, D. Li and L. Jiang, Energy Storage Mater., 2023, 56, 468–477 CrossRef
- A. Jana and R. E. García, Nano Energy, 2017, 41, 552–565 CrossRef CAS
- A. Jana, S. I. Woo, K. Vikrant and R. E. García, Energy Environ. Sci., 2019, 12, 3595–3607 RSC
- W. Li, P. Luo, M. Chen, X. Lin, L. Du, H. Song, Y. Lu and Z. Cui, J. Mater. Chem. A, 2022, 10, 15161–15168 RSC
- J. Chen, W. Zhao, J. Jiang, X. Zhao, S. Zheng, Z. Pan and X. Yang, Energy Storage Mater., 2023, 59, 102767 CrossRef
- W. Du, E. H. Ang, Y. Yang, Y. Zhang, M. Ye and C. C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC
- Z. Cai, Y. Ou, J. Wang, R. Xiao, L. Fu, Z. Yuan, R. Zhan and Y. Sun, Energy Storage Mater., 2020, 27, 205–211 CrossRef
- P. Xiao, H. Li, J. Fu, C. Zeng, Y. Zhao, T. Zhai and H. Li, Energy Environ. Sci., 2022, 15, 1638–1646 RSC
- Z. Cai, J. Wang and Y. Sun, eScience, 2023, 3, 100093 CrossRef
- Y. Shi, Y. Chen, L. Shi, K. Wang, B. Wang, L. Li, Y. Ma, Y. Li, Z. Sun, W. Ali and S. Ding, Small, 2020, 16, 2000730 CrossRef CAS PubMed
- V. Verma, S. Kumar, W. Manalastas Jr and M. Srinivasan, ACS Energy Lett., 2021, 6, 1773–1785 CrossRef CAS
- J. Wang, B. Zhang, Z. Cai, R. Zhan, W. Wang, L. Fu, M. Wan, R. Xiao, Y. Ou, L. Wang, J. Jiang, Z. W. Seh, H. Li and Y. Sun, Sci. Bull., 2022, 67, 716–724 CrossRef CAS PubMed
- G. R. Pastel, Y. Chen, T. P. Pollard, M. A. Schroeder, M. E. Bowden, A. Zheng, N. T. Hahn, L. Ma, V. Murugesan, J. Ho, M. Garaga, O. Borodin, K. Mueller, S. Greenbaum and K. Xu, Energy Environ. Sci., 2022, 15, 2460–2469 RSC
- S. A. Rastabi, G. Razaz, M. Hummelgård, T. Carlberg, N. Blomquist, J. Örtegren and H. Olin, J. Power Sources, 2022, 523, 231066 CrossRef
- Z. Xiang, Y. Qiu, X. Guo, K. Qi, Z.-L. Xu and B. Y. Xia, Energy Environ. Sci., 2024, 17, 3409–3418 RSC
- H. Yaghoobnejad Asl and A. Manthiram, J. Am. Chem. Soc., 2020, 142, 21122–21130 CrossRef CAS PubMed
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed
- Y. Lu, T. Zhu, W. van den Bergh, M. Stefik and K. Huang, Angew. Chem., 2020, 132, 17152–17159 CrossRef
- D. R. Ely and R. E. García, J. Electrochem. Soc., 2013, 160, A662 CrossRef CAS
- J. L. Yang, J. Li, J. W. Zhao, K. Liu, P. Yang and H. J. Fan, Adv. Mater., 2022, 34, 2202382 CrossRef CAS PubMed
- T. Wei, H. Zhang, Y. Ren, L. E. Mo, Y. He, P. Tan, Y. Huang, Z. Li, D. Zhu and L. Hu, Adv. Funct. Mater., 2024, 2312506 CrossRef CAS
- Y. Wang, R. Zhao, M. Liu, J. Yang, A. Zhang, J. Yue, C. Wu and Y. Bai, Adv. Energy Mater., 2023, 13, 2302707 CrossRef CAS
- Y. Zhao, S. Guo, M. Chen, B. Lu, X. Zhang, S. Liang and J. Zhou, Nat. Commun., 2023, 14, 7080 CrossRef CAS PubMed
- R. Zhao, X. Dong, P. Liang, H. Li, T. Zhang, W. Zhou, B. Wang, Z. Yang, X. Wang, L. Wang, Z. Sun, F. Bu, Z. Zhao, W. Li, D. Zhao and D. Chao, Adv. Mater., 2023, 35, 2209288 CrossRef CAS PubMed
- L. Hong, L. Y. Wang, Y. Wang, X. Wu, W. Huang, Y. Zhou, K. X. Wang and J. S. Chen, Adv. Sci., 2022, 9, 2104866 CrossRef CAS PubMed
- X. Zheng, Z. Liu, J. Sun, R. Luo, K. Xu, M. Si, J. Kang, Y. Yuan, S. Liu, T. Ahmad, T. Jiang, N. Chen, M. Wang, Y. Xu, M. Chuai, Z. Zhu, Q. Peng, Y. Meng, K. Zhang, W. Wang and W. Chen, Nat. Commun., 2023, 14, 76 CrossRef CAS PubMed
- T. Wang, Y. A. Wang, X. Wang, M. Chang, Y. Zhang, J. You, F. Hu and K. Zhu, Adv. Funct. Mater., 2024, 2314157 CrossRef CAS
- T. Wang, Q. Xi, K. Yao, Y. Liu, H. Fu, V. S. Kavarthapu, J. K. Lee, S. Tang, D. Fattakhova-Rohlfing, W. Ai and J. S. Yu, Nano-Micro Lett., 2024, 16, 112 CrossRef CAS PubMed
- C. Tian, H. Wang, L. Xie, Y. Zhong and Y. Hu, Adv. Energy Mater., 2024, 2400276 CrossRef CAS
- Q. Zhao, Y. Wang, W. Liu, X. Liu, H. Wang, H. Yu, Y. Chen and L. Chen, Adv. Mater. Interfaces, 2022, 9, 2102254 CrossRef CAS
- X. Zhou, B. Wen, Y. Cai, X. Chen, L. Li, Q. Zhao, S. L. Chou and F. Li, Angew. Chem., Int. Ed., 2024, e202402342 CAS
- Y. Zhang, J. D. Howe, S. Ben-Yoseph, Y. Wu and N. Liu, ACS Energy Lett., 2021, 6, 404–412 CrossRef CAS
- P. Cao, J. Tang, A. Wei, Q. Bai, Q. Meng, S. Fan, H. Ye, Y. Zhou, X. Zhou and J. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 48855–48864 CrossRef CAS PubMed
- H. Tao, Z. Hou, L. Zhang, X. Yang and L.-Z. Fan, Chem. Eng. J., 2022, 450, 138048 CrossRef CAS
- X. Zhang, H. Luo, Y. Guo, C. Xu, Y. Deng, Z. Deng, Y. Zhang, H. Wu, W. Cai and Y. Zhang, Chem. Eng. J., 2023, 457, 141305 CrossRef CAS
- Y. Du, Y. Feng, R. Li, Z. Peng, X. Yao, S. Duan, S. Liu, S. C. Jun, J. Zhu, L. Dai, Q. Yang, L. Wang and Z. He, Small, 2024, 20, 2307848 CrossRef CAS PubMed
- J. Li, Z. Liu, S. Han, P. Zhou, B. Lu, J. Zhou, Z. Zeng, Z. Chen and J. Zhou, Nano-Micro Lett., 2023, 15, 237 CrossRef CAS PubMed
- Q. Lu, C. Liu, Y. Du, X. Wang, L. Ding, A. Omar and D. Mikhailova, ACS Appl. Mater. Interfaces, 2021, 13, 16869–16875 CrossRef CAS PubMed
- B. Liu, S. Wang, Z. Wang, H. Lei, Z. Chen and W. Mai, Small, 2020, 16, 2001323 CrossRef CAS PubMed
- J. C. Wu, X. Shen, H. Zhou, X. Li, H. Gao, J. Ge, T. Xu and H. Zhou, Small, 2024, 20, 2308541 CrossRef CAS PubMed
- J. Yin, Y. Wang, Y. Zhu, J. Jin, C. Chen, Y. Yuan, Z. Bayhan, N. Salah, N. A. Alhebshi, W. Zhang, U. Schwingenschlögl and H. N. Alshareef, Nano Energy, 2022, 99, 107331 CrossRef CAS
- C. Liu, Z. Luo, W. Deng, W. Wei, L. Chen, A. Pan, J. Ma, C. Wang, L. Zhu, L. Xie, X.-Y. Cao, J. Hu, G. Zou, H. Hou and X. Ji, ACS Energy Lett., 2021, 6, 675–683 CrossRef CAS
- H. Yu, C. Lv, C. Yan and G. Yu, Small Methods, 2023, 2300758 Search PubMed
- Z. Sun, H. Lu, L. Fan, Q. Hong, J. Leng and C. Chen, J. Electrochem. Soc., 2015, 162, A2116 CrossRef CAS
- M. Nestoridi, D. Pletcher, R. J. Wood, S. Wang, R. L. Jones, K. R. Stokes and I. Wilcock, J. Power Sources, 2008, 178, 445–455 CrossRef CAS
- C. Yan, C. Lv, L. Wang, W. Cui, L. Zhang, K. N. Dinh, H. Tan, C. Wu, T. Wu, Y. Ren, J. Chen, Z. Liu, M. Srinivasan, X. Rui, Q. Yan and G. Yu, J. Am. Chem. Soc., 2020, 142, 15295–15304 CrossRef CAS PubMed
- Q. Ran, H. Shi, H. Meng, S.-P. Zeng, W.-B. Wan, W. Zhang, Z. Wen, X.-Y. Lang and Q. Jiang, Nat. Commun., 2022, 13, 576 CrossRef CAS PubMed
- J. Wang, H. Jiao, W.-L. Song, M. Wang, J. Tu, Z. Tang and H. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 15063–15070 CrossRef CAS PubMed
- J. Niu, Z. Zhang and D. Aurbach, Adv. Energy Mater., 2020, 10, 2000697 CrossRef CAS
- Y. Zou, Y. Wu, W. Wei, C. Qiao, M. Lu, Y. Su, W. Guo, X. Yang, Y. Song, M. Tian, S. Dou, Z. Liu and J. Sun, Adv. Mater., 2024, 2313775 CrossRef CAS PubMed
- Y. Zou, C. Qiao, Y. Su, M. Lu, X. Yang, S. Dou and J. Sun, Appl. Phys. Lett., 2023, 123, 223903 CrossRef CAS
- J. Zhou, M. Xie, F. Wu, Y. Mei, Y. Hao, R. Huang, G. Wei, A. Liu, L. Li and R. Chen, Adv. Mater., 2021, 33, 2101649 CrossRef CAS PubMed
- F. Xie, H. Li, X. Wang, X. Zhi, D. Chao, K. Davey and S. Z. Qiao, Adv. Energy Mater., 2021, 11, 2003419 CrossRef CAS
- T. Foroozan, V. Yurkiv, S. Sharifi-Asl, R. Rojaee, F. Mashayek and R. Shahbazian-Yassar, ACS Appl. Mater. Interfaces, 2019, 11, 44077–44089 CrossRef CAS PubMed
- S. Wang, L. Hu, X. Li, D. Qiu, S. Qiu, Q. Zhou, W. Deng, X. Lu, Z. Yang, M. Qiu and Y. Yu, J. Energy Chem., 2024, 91, 203–212 CrossRef CAS
- J. Wan, R. Wang, Z. Liu, S. Zhang, J. Hao, J. Mao, H. Li, D. Chao, L. Zhang and C. Zhang, Adv. Mater., 2024, 36, 2310623 CrossRef CAS PubMed
- C. Yan, C. Lv, B.-E. Jia, L. Zhong, X. Cao, X. Guo, H. Liu, W. Xu, D. Liu, L. Yang, J. Liu, H. H. Hng, W. Chen, L. Song, S. Li, Z. Liu, Q. Yan and G. Yu, J. Am. Chem. Soc., 2022, 144, 11444–11455 CrossRef CAS PubMed
- Y. Meng, M. Wang, K. Li, Z. Zhu, Z. Liu, T. Jiang, X. Zheng, K. Zhang, W. Wang, Q. Peng, Z. Xie, Y. Wang and W. Chen, Nano Lett., 2023, 23, 2295–2303 CrossRef CAS PubMed
- D. Xu, J. Xie, L. Zhou, F. Yang, Y. Wang, Z. Yang, F. Wang, H. Zhang and X. Lu, Small, 2023, 19, 2301931 CrossRef CAS PubMed
-
A. Milchev, Electrocrystallization: fundamentals of nucleation and growth, Springer Science & Business Media, 2002 Search PubMed
-
E. B. Budevski, G. T. Staikov and W. J. Lorenz, Electrochemical phase formation and growth: an introduction to the initial stages of metal deposition, John Wiley & Sons, 2008 Search PubMed
- P. Altimari and F. Pagnanelli, Electrochim. Acta, 2016, 206, 116–126 CrossRef CAS
- S. Park, H. J. Jin and Y. S. Yun, Adv. Mater., 2020, 32, 2002193 CrossRef CAS PubMed
- L. Guo and P. C. Searson, Electrochim. Acta, 2010, 55, 4086–4091 CrossRef CAS
- S. Kaniyankandy, J. Nuwad, C. Thinaharan, G. Dey and C. Pillai, Nanotechnology, 2007, 18, 125610 CrossRef
- G. Li, Z. Liu, D. Wang, X. He, S. Liu, Y. Gao, A. AlZahrani, S. H. Kim, L.-Q. Chen and D. Wang, Adv. Energy Mater., 2019, 9, 1900704 CrossRef
- Y. Han, Y. Jie, F. Huang, Y. Chen, Z. Lei, G. Zhang, X. Ren, L. Qin, R. Cao and S. Jiao, Adv. Funct. Mater., 2019, 29, 1904629 CrossRef CAS
- Y. Zhang, M. Yao, T. Wang, H. Wu and Y. Zhang, Angew. Chem., Int. Ed., 2024, e202403399 CAS
- C. Wang, M. Liu, M. Thijs, F. G. Ooms, S. Ganapathy and M. Wagemaker, Nat. Commun., 2021, 12, 6536 CrossRef CAS PubMed
- C. Wu, L. Xue, A. Wang, L. Guo, W. Tang, S. X. Dou and C. Peng, J. Power Sources, 2024, 605, 234517 CrossRef CAS
- J. H. Kwak, S. Shin, Y. Jeoun, Y. Lee, S. Yu, Y. S. Yun, Y.-E. Sung, S.-H. Yu and H.-D. Lim, J. Power Sources, 2022, 541, 231724 CrossRef CAS
- Y. Mu, Z. Li, B.-K. Wu, H. Huang, F. Wu, Y. Chu, L. Zou, M. Yang, J. He, L. Ye, M. Han, T. Zhao and L. Zeng, Nat. Commun., 2023, 14, 4205 CrossRef CAS PubMed
- H. Yu, H. Yao, Y. Zheng, D. Liu, J. S. Chen, Y. Guo, N. W. Li and L. Yu, Adv. Funct. Mater., 2024, 34, 2311038 CrossRef CAS
- Y. Gao, Q. Cao, J. Pu, X. Zhao, G. Fu, J. Chen, Y. Wang and C. Guan, Adv. Mater., 2023, 35, 2207573 CrossRef CAS PubMed
- Y. Song, H. Yan, Z. Cong, J. Yang, Y. Li and A. Tang, Chem. Eng. J., 2024, 487, 150592 CrossRef CAS
- J. Zhou, F. Wu, Y. Mei, Y. Hao, L. Li, M. Xie and R. Chen, Adv. Mater., 2022, 34, 2200782 CrossRef CAS PubMed
- S. Shi, D. Zhou, Y. Jiang, F. Cheng, J. Sun, Q. Guo, Y. Luo, Y. Chen and W. Liu, Adv. Funct. Mater., 2024, 2312664 CrossRef CAS
- Z. Song, Z. Zhang, A. Du, S. Dong, G. Li and G. Cui, Adv. Mater., 2021, 33, 2100224 CrossRef CAS PubMed
- Q. Zhao, W. Liu, Y. Chen and L. Chen, Chem. Eng. J., 2022, 450, 137979 CrossRef CAS
- Y. An, Y. Tian, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2021, 15, 11828–11842 CrossRef CAS PubMed
- Y. Wang, J. Li, X. Li, H. Jin, W. Ali, Z. Song and S. Ding, J. Mater. Chem. A, 2022, 10, 699–706 RSC
- S. Lai, Y. Yuan, M. Zhu, S. Yin, Y. Huang, S. Guo and W. Yan, Small, 2024, 20, 2307026 CrossRef CAS PubMed
- M. Zhang, Y. Deng, Y. Yan, H. Mei, L. Cheng and L. Zhang, Energy Storage Mater., 2024, 65, 103156 CrossRef
- H. He, L. Zeng, D. Luo, J. He, X. Li, Z. Guo and C. Zhang, Adv. Mater., 2023, 35, 2211498 CrossRef CAS PubMed
- L. Zhou, F. Yang, S. Zeng, X. Gao, X. Liu, X. Cao, P. Yu and X. Lu, Adv. Funct. Mater., 2022, 32, 2110829 CrossRef CAS
- L. Han, Y. Guo, F. Ning, X. Liu, J. Yi, Q. Luo, B. Qu, J. Yue, Y. Lu and Q. Li, Adv. Mater., 2024, 36, 2308086 CrossRef CAS PubMed
- Y. Zhang, Y. Tian, Z. Wang, C. Wei, C. Liu, Y. An, B. Xi, S. Xiong and J. Feng, Chem. Eng. J., 2023, 458, 141388 CrossRef CAS
- Z. Liu, J. Liu, X. Xiao, Z. Zheng, X. Zhong, Q. Fu, S. Wang and G. Zhou, Adv. Mater., 2024, 2404140 CrossRef CAS PubMed
-
V. S. Bagotsky, Fundamentals of electrochemistry, John Wiley & Sons, 2005 Search PubMed
- Z. Hou, Y. Gao, R. Zhou and B. Zhang, Adv. Funct. Mater., 2022, 32, 2107584 CrossRef CAS
- G. Garcia, E. Ventosa and W. Schuhmann, ACS Appl. Mater. Interfaces, 2017, 9, 18691–18698 CrossRef CAS PubMed
- T. Dhara, A. Aryanfar, A. Ghosh, U. Ghosh, P. Mukherjee and S. DasGupta, J. Phys. Chem. C, 2023, 127, 4407–4415 CrossRef CAS
- T. Dhara, U. U. Ghosh, A. Ghosh, B. S. Vishnugopi, P. P. Mukherjee and S. DasGupta, Langmuir, 2022, 38, 4879–4886 CrossRef CAS PubMed
- X. Guan, A. Wang, S. Liu, G. Li, F. Liang, Y. W. Yang, X. Liu and J. Luo, Small, 2018, 14, 1801423 CrossRef PubMed
- L. Lin, H. Zheng, Q. Luo, J. Lin, L. Wang, Q. Xie, D. L. Peng and J. Lu, Adv. Funct. Mater., 2024, 2315201 CrossRef CAS
- J.-N. Chazalviel, Phys. Rev. A, 1990, 42, 7355 CrossRef CAS PubMed
- C. Brissot, M. Rosso, J.-N. Chazalviel and S. Lascaud, J. Power Sources, 1999, 81, 925–929 CrossRef
- P. G. Bruce and C. A. Vincent, J. Electroanal. Chem. Interf. Electrochem., 1987, 225, 1–17 CrossRef CAS
- H. J. Sand, Lond. Edinb. Dubl. Phil. Mag., 1901, 1, 45–79 CrossRef CAS
- S. Jin, J. Yin, X. Gao, A. Sharma, P. Chen, S. Hong, Q. Zhao, J. Zheng, Y. Deng, Y. L. Joo and L. A. Archer, Nat. Commun., 2022, 13, 2283 CrossRef CAS PubMed
- R. A. Enrique, S. DeWitt and K. Thornton, MRS Commun., 2017, 7, 658–663 CrossRef CAS
-
H. S. Fogler, Elements of chemical reaction engineering, Pearson Educacion, 1999 Search PubMed
- S. Jin, X. Gao, S. Hong, Y. Deng, P. Chen, R. Yang, Y. L. Joo and L. A. Archer, Joule, 2024, 8, 746–763 CrossRef CAS
- J. Zheng and L. A. Archer, Sci. Adv., 2021, 7, eabe0219 CrossRef CAS PubMed
- R. Davidson, A. Verma, D. Santos, F. Hao, C. Fincher, S. Xiang, J. Van Buskirk, K. Xie, M. Pharr, P. P. Mukherjee and S. Banerjee, ACS Energy Lett., 2018, 4, 375–376 CrossRef
- Z. Yu, Q. Wang, Y. Li, F. Zhang, X. Ma, X. Zhang, Y. Wang, J. Huang and Y. Xia, Joule, 2024, 8, 1063–1079 CrossRef CAS
- W. Wu, X. Yang, K. Wang, C. Li, X. Zhang, H.-Y. Shi, X.-X. Liu and X. Sun, Chem. Eng. J., 2022, 432, 134389 CrossRef CAS
- Y. Yao, Z. Wang, Z. Li and Y. C. Lu, Adv. Mater., 2021, 33, 2008095 CrossRef CAS PubMed
- K. S. Nagy, S. Kazemiabnavi, K. Thornton and D. J. Siegel, ACS Appl. Mater. Interfaces, 2019, 11, 7954–7964 CrossRef CAS PubMed
- J. Zheng, D. C. Bock, T. Tang, Q. Zhao, J. Yin, K. R. Tallman, G. Wheeler, X. Liu, Y. Deng, S. Jin, A. C. Marschilok, E. S. Takeuchi, K. J. Takeuchi and L. A. Archer, Nat. Energy, 2021, 6, 398–406 CrossRef CAS
- Z. Zhao, R. Wang, C. Peng, W. Chen, T. Wu, B. Hu, W. Weng, Y. Yao, J. Zeng, Z. Chen, P. Liu, Y. Liu, G. Li, J. Guo, H. Lu and Z. Guo, Nat. Commun., 2021, 12, 6606 CrossRef CAS PubMed
- X. Yang, C. Li, Z. Sun, S. Yang, Z. Shi, R. Huang, B. Liu, S. Li, Y. Wu, M. Wang, Y. Su, S. Dou and J. Sun, Adv. Mater., 2021, 33, 2105951 CrossRef CAS PubMed
- Z. Jiao, X. Cai, X. Wang, Y. Li, Z. Bie and W. Song, Adv. Energy Mater., 2023, 13, 2302676 CrossRef CAS
- C. Yang, P. Woottapanit, Y. Yue, S. Geng, J. Cao, X. Zhang, G. He and J. Qin, Small, 2024, 2311203 CrossRef CAS PubMed
- R. Zhang, Y. Feng, Y. Ni, B. Zhong, M. Peng, T. Sun, S. Chen, H. Wang, Z. Tao and K. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304503 CrossRef CAS PubMed
- Z. Shi, M. Yang, Y. Ren, Y. Wang, J. Guo, J. Yin, F. Lai, W. Zhang, S. Chen, H. N. Alshareef and T. Liu, ACS Nano, 2023, 17, 21893–21904 CrossRef PubMed
- W. Yuan, X. Nie, G. Ma, M. Liu, Y. Wang, S. Shen and N. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202218386 CrossRef CAS PubMed
- Q. Zhu, G. Sun, S. Qiao, D. Wang, Z. Cui, W. Zhang and J. Liu, Adv. Mater., 2024, 36, 2308577 CrossRef CAS PubMed
- Z. Liu, Z. Guo, L. Fan, C. Zhao, A. Chen, M. Wang, M. Li, X. Lu, J. Zhang, Y. Zhang and N. Zhang, Adv. Mater., 2024, 36, 2305988 CrossRef CAS PubMed
- Y. Niu, L. Chang, Q. Sun, Y. Liu, W. Nie, T. Duan, X. Lu and H. Cheng, ACS Appl. Mater. Interfaces, 2024, 16, 6988–6997 CrossRef CAS PubMed
- L. Ren, Z. Hu, C. Peng, L. Zhang, N. Wang, F. Wang, Y. Xia, S. Zhang, E. Hu and J. Luo, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2309981121 CrossRef CAS PubMed
- J. Zhang, W. Huang, L. Li, C. Chang, K. Yang, L. Gao and X. Pu, Adv. Mater., 2023, 35, 2300073 CrossRef CAS PubMed
- X. Liu, Y. Guo, F. Ning, Y. Liu, S. Shi, Q. Li, J. Zhang, S. Lu and J. Yi, Nano-Micro Lett., 2024, 16, 111 CrossRef CAS PubMed
- Z. Cheng, K. Wang, J. Fu, F. Mo, P. Lu, J. Gao, D. Ho, B. Li and H. Hu, Adv. Energy Mater., 2024, 2304003 CrossRef CAS
- M. Zhou, S. Guo, J. Li, X. Luo, Z. Liu, T. Zhang, X. Cao, M. Long, B. Lu, A. Pan, G. Fang, J. Zhou and S. Liang, Adv. Mater., 2021, 33, 2100187 CrossRef CAS PubMed
- J. Zheng, Y. Deng, J. Yin, T. Tang, R. Garcia-Mendez, Q. Zhao and L. A. Archer, Adv. Mater., 2022, 34, 2106867 CrossRef CAS PubMed
- Z. Chen, J. Zhao, Q. He, M. Li, S. Feng, Y. Wang, D. Yuan, J. Chen, H. N. Alshareef and Y. Ma, ACS Energy Lett., 2022, 7, 3564–3571 CrossRef CAS
- Y. Wang and J. Huang, Mater. Chem. Phys., 2003, 81, 11–26 CrossRef CAS
- J. Zou, Z. Zeng, C. Wang, X. Zhu, J. Zhang, H. Lan, L. Li, Y. Yu, H. Wang, X. Zhu, Y. Zhang, M. Zeng and L. Fu, Small Struct., 2023, 4, 2200194 CrossRef CAS
- S. D. Pu, C. Gong, Y. T. Tang, Z. Ning, J. Liu, S. Zhang, Y. Yuan, D. Melvin, S. Yang, L. Pi, J.-J. Marie, B. Hu, M. Jenkins, Z. Li, B. Liu, S. C. E. Tsang, T. J. Marrow, R. C. Reed, X. Gao, P. G. Bruce and A. W. Robertson, Adv. Mater., 2022, 34, 2202552 CrossRef CAS PubMed
- X. Song, L. Bai, C. Wang, D. Wang, K. Xu, J. Dong, Y. Li, Q. Shen and J. Yang, ACS Nano, 2023, 17, 15113–15124 CrossRef CAS PubMed
- Z. Chen, Q. Wu, X. Han, C. Wang, J. Chen, T. Hu, Q. He, X. Zhu, D. Yuan, J. Chen, Y. Zhang, L. Yang, Y. Ma and J. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202401507 CrossRef CAS PubMed
- X. Wu, W. Yuan, Y. Wu, C. Wang, L. Xue, Y. Zhou, X. Zhang, S. Jiang, B. Zhao, Y. Chen, C. Yang, L. Ding, Y. Tang and X. Ding, Adv. Energy Mater., 2024, 2304204 CrossRef CAS
- X. Zhang, J. Li, Y. Liu, B. Lu, S. Liang and J. Zhou, Nat. Commun., 2024, 15, 2735 CrossRef CAS PubMed
- D. Yuan, J. Zhao, H. Ren, Y. Chen, R. Chua, E. T. J. Jie, Y. Cai, E. Edison, W. Manalastas Jr, M. W. Wong and M. Srinivasan, Angew. Chem., 2021, 133, 7289–7295 CrossRef
- X. Chen, Z. Zhai, T. Yu, X. Liang, R. Huang, F. Wang and S. Yin, Small, 2024, 2401386 CrossRef PubMed
- F. Li, D. Ma, K. Ouyang, M. Yang, J. Qiu, J. Feng, Y. Wang, H. Mi, S. Sun, L. Sun, C. He and P. Zhang, Adv. Energy Mater., 2023, 13, 2204365 CrossRef CAS
- Y. Liu, J. Hu, Q. Lu, M. Hantusch, H. Zhang, Z. Qu, H. Tang, H. Dong, O. G. Schmidt, R. Holze and M. Zhu, Energy Storage Mater., 2022, 47, 98–104 CrossRef
- M. Xi, Z. Liu, W. Wang, Z. Qi, R. Sheng, J. Ding, Y. Huang and Z. Guo, Energy Environ. Sci., 2024, 17, 3168–3178 RSC
- J. Cao, D. Zhang, C. Gu, X. Wang, S. Wang, X. Zhang, J. Qin and Z. S. Wu, Adv. Energy Mater., 2021, 11, 2101299 CrossRef CAS
- Y. Su, B. Chen, Y. Sun, Z. Xue, Y. Zou, D. Yang, L. Sun, X. Yang, C. Li, Y. Yang, X. Song, W. Guo, S. Dou, D. Chao, Z. Liu and J. Sun, Adv. Mater., 2023, 35, 2301410 CrossRef CAS PubMed
- H. Lu, Q. Jin, X. Jiang, Z. M. Dang, D. Zhang and Y. Jin, Small, 2022, 18, 2200131 CrossRef CAS PubMed
- M. Qiu, P. Sun, Y. Wang, L. Ma, C. Zhi and W. Mai, Angew. Chem., Int. Ed., 2022, 61, e202210979 CrossRef CAS PubMed
- T. Wei, Y. Ren, Y. Wang, L. E. Mo, Z. Li, H. Zhang, L. Hu and G. Cao, ACS Nano, 2023, 17, 3765–3775 CrossRef CAS PubMed
- Z. Shen, J. Mao, G. Yu, W. Zhang, S. Mao, W. Zhong, H. Cheng, J. Guo, J. Zhang and Y. Lu, Angew. Chem., Int. Ed., 2023, 62, e202218452 CrossRef CAS PubMed
- Y. Lin, Z. Mai, H. Liang, Y. Li, G. Yang and C. Wang, Energy Environ. Sci., 2023, 16, 687–697 RSC
- H. Zhang, Y. Zhong, J. Li, Y. Liao, J. Zeng, Y. Shen, L. Yuan, Z. Li and Y. Huang, Adv. Energy Mater., 2023, 13, 2203254 CrossRef CAS
- W. Zhang, Q. Zhao, Y. Hou, Z. Shen, L. Fan, S. Zhou, Y. Lu and L. A. Archer, Sci. Adv., 2021, 7, eabl3752 CrossRef CAS PubMed
- Z. Cai, J. Wang, Z. Lu, R. Zhan, Y. Ou, L. Wang, M. Dahbi, J. Alami, J. Lu, K. Amine and Y. Sun, Angew. Chem., Int. Ed., 2022, 61, e202116560 CrossRef CAS PubMed
- Y. Liu, B. Xie, Q. Hu, R. Zhao, Q. Zheng, X. Huang, S. Deng, Y. Huo, J. Zhao, B. Xu and D. Lin, Energy Storage Mater., 2024, 66, 103202 CrossRef
- J. Luo, L. Xu, Y. Zhou, T. Yan, Y. Shao, D. Yang, L. Zhang, Z. Xia, T. Wang, L. Zhang, T. Cheng and Y. Shao, Angew. Chem., 2023, 135, e202302302 CrossRef
- K. Qi, P. Liang, S. Wei, H. Ao, X. Ding, S. Chen, Z. Fan, C. Wang, L. Song, X. Wu, C. Wu and Y. Zhu, Energy Environ. Sci., 2024, 17, 2566–2575 RSC
- X. Xie, S. Liang, J. Gao, S. Guo, J. Guo, C. Wang, G. Xu, X. Wu, G. Chen and J. Zhou, Energy Environ. Sci., 2020, 13, 503–510 RSC
- C. Yan, H.-R. Li, X. Chen, X.-Q. Zhang, X.-B. Cheng, R. Xu, J.-Q. Huang and Q. Zhang, J. Am. Chem. Soc., 2019, 141, 9422–9429 CrossRef CAS PubMed
- X. Nie, L. Miao, W. Yuan, G. Ma, S. Di, Y. Wang, S. Shen and N. Zhang, Adv. Funct. Mater., 2022, 32, 2203905 CrossRef CAS
- J. Fu, Q. Yin, H. Yao and H. Hu, Energy Storage Mater., 2023, 59, 102774 CrossRef
- T. Wang, Z. Tian, Z. You, Z. Li, H. Cheng, W. Li, Y. Yang, Y. Zhou, Q. Zhong and Y. Lai, Energy Storage Mater., 2022, 45, 24–32 CrossRef
- J. Hao, L. Yuan, C. Ye, D. Chao, K. Davey, Z. Guo and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 7366–7375 CrossRef CAS PubMed
- Y. Chen, S. Zhou, J. Li, J. Kang, S. Lin, C. Han, H. Duan, S. Liang and A. Pan, Adv. Energy Mater., 2024, 2400398 CrossRef CAS
- Y. Chen, Z. Deng, Y. Sun, Y. Li, H. Zhang, G. Li, H. Zeng and X. Wang, Nano-Micro Lett., 2024, 16, 96 CrossRef CAS PubMed
- Z. Luo, Y. Xia, S. Chen, X. Wu, R. Zeng, X. Zhang, H. Pan, M. Yan, T. Shi, K. Tao, B. B. Xu and Y. Jiang, Nano-Micro Lett., 2023, 15, 205 CrossRef CAS PubMed
- Z. Meng, Y. Jiao and P. Wu, Angew. Chem., 2023, 135, e202307271 CrossRef
- S.-R. Choi, K.-M. Kim and J.-G. Kim, J. Mol. Liq., 2022, 365, 120104 CrossRef CAS
- J. Yin, M. Li, X. Feng, T. Cui, J. Chen, F. Li, M. Wang, Y. Cheng, S. Ding, X. Xu and J. Wang, J. Mater. Chem. A, 2024, 12, 1543–1550 RSC
- Z. Zheng, D. Ren, Y. Li, F. Kang, X. Li, X. Peng and L. Dong, Adv. Funct. Mater., 2024, 2312855 CrossRef CAS
- F. Bu, Y. Gao, W. Zhao, Q. Cao, Y. Deng, J. Chen, J. Pu, J. Yang, Y. Wang, N. Yang, T. Meng, X. Liu and C. Guan, Angew. Chem., 2024, 136, e202318496 CrossRef
- F. Zhao, Z. Jing, X. Guo, J. Li, H. Dong, Y. Tan, L. Liu, Y. Zhou, R. Owen, P. R. Shearing, D. J. L. Brett, G. He and I. P. Parkin, Energy Storage Mater., 2022, 53, 638–645 CrossRef
- J. Wang, Y. Yu, R. Chen, H. Yang, W. Zhang, Y. Miao, T. Liu, J. Huang and G. He, Adv. Sci., 2024, 2402821 CrossRef CAS PubMed
- M. Fu, Q. Zhao, K. Long, Q. Li, G. C. Kuang, L. Zhou, W. Wei, X. Ji, L. Chen and Y. Chen, Adv. Funct. Mater., 2024, 34, 2311680 CrossRef CAS
- R. Wang, W. Wang, M. Sun, Y. Hu and G. Wang, Angew. Chem., Int. Ed., 2024, e202317154 CAS
- S. Liu, T. Qian, M. Wang, H. Ji, X. Shen, C. Wang and C. Yan, Nat. Catal., 2021, 4, 322–331 CrossRef CAS
- R. Meng, H. Li, Z. Lu, C. Zhang, Z. Wang, Y. Liu, W. Wang, G. Ling, F. Kang and Q. H. Yang, Adv. Mater., 2022, 34, 2200677 CrossRef CAS PubMed
- R. Tao, H. Fu, C. Gao, L. Fan, E. Xie, W. Lyu, J. Zhou and B. Lu, Adv. Funct. Mater., 2023, 33, 2303072 CrossRef CAS
- Z. Cao, X. Zhu, D. Xu, P. Dong, M. O. L. Chee, X. Li, K. Zhu, M. Ye and J. Shen, Energy Storage Mater., 2021, 36, 132–138 CrossRef
- Y. Guo, W. Cai, Y. Lin, Y. Zhang, S. Luo, K. Huang, H. Wu and Y. Zhang, Energy Storage Mater., 2022, 50, 580–588 CrossRef
- J. Zhu, Z. Bie, X. Cai, Z. Jiao, Z. Wang, J. Tao, W. Song and H. J. Fan, Adv. Mater., 2022, 34, 2207209 CrossRef CAS PubMed
- R. Zhao, Y. Yang, G. Liu, R. Zhu, J. Huang, Z. Chen, Z. Gao, X. Chen and L. Qie, Adv. Funct. Mater., 2021, 31, 2001867 CrossRef CAS
- J. Chen, J. Xiong, M. Ye, Z. Wen, Y. Zhang, Y. Tang, X. Liu and C. C. Li, Adv. Funct. Mater., 2023, 2312564 Search PubMed
- C. Guo, X. Huang, J. Huang, X. Tian, Y. Chen, W. Feng, J. Zhou, Q. Li, Y. Chen, S.-L. Li and Y.-Q. Lan, Angew. Chem., 2024, e202403918 CAS
- L. Wang, B. Zhang, W. Zhou, Z. Zhao, X. Liu, R. Zhao, Z. Sun, H. Li, X. Wang, T. Zhang, H. Jin, W. Li, A. Elzatahry, Y. Hassan, H. J. Fan, D. Zhao and D. Chao, J. Am. Chem. Soc., 2024, 146, 6199–6208 CrossRef CAS PubMed
- B. Shuai, C. Zhou, Y. Pi and X. Xu, ACS Appl. Energy Mater., 2022, 5, 6139–6145 CrossRef CAS
- H. Huang, X. Xia, J. Yun, C. Huang, D. Li, B. Chen, Z. Yang and W. Zhang, Energy Storage Mater., 2022, 52, 473–484 CrossRef
- C. Villevieille, Adv. Mater. Interfaces, 2022, 9, 2101865 CrossRef CAS
- D. Wang, H. Liu, D. Lv, C. Wang, J. Yang and Y. Qian, Adv. Mater., 2023, 35, 2207908 CrossRef CAS PubMed
- Z. Xiang, Y. Qiu, X. Guo, K. Qi, Z.-L. Xu and B. Y. Xia, Energy Environ. Sci., 2024, 17, 3409–3418 RSC
- Q. Sun, Y. Pang, H. Pan, W. Zhang and Z. Li, Chem. Eng. J., 2024, 489, 151296 CrossRef CAS
- T. Yan, S. Liu, J. Li, M. Tao, J. Liang, L. Du, Z. Cui and H. Song, ACS Nano, 2024, 18, 3752–3762 CrossRef CAS PubMed
- C. Chang, S. Hu, T. Li, F. Zeng, D. Wang, S. Guo, M. Xu, G. Liang, Y. Tang, H. Li, C. Han and H.-M. Cheng, Energy Environ. Sci., 2024, 17, 680–694 RSC
- M. Wu, X. Wang, F. Zhang, Q. Xiang, Y. Li and J. Guo, Energy Environ. Sci., 2024, 17, 619–629 RSC
- L. Cao, D. Li, T. Deng, Q. Li and C. Wang, Angew. Chem., 2020, 132, 19454–19458 CrossRef
- C. Li, A. Shyamsunder, A. G. Hoane, D. M. Long, C. Y. Kwok, P. G. Kotula, K. R. Zavadil, A. A. Gewirth and L. F. Nazar, Joule, 2022, 6, 1103–1120 CrossRef CAS
- D. Han, C. Cui, K. Zhang, Z. Wang, J. Gao, Y. Guo, Z. Zhang, S. Wu, L. Yin, Z. Weng, F. Kang and Q.-H. Yang, Nat. Sustainability, 2022, 5, 205–213 CrossRef
- Y. Yang, G. Qu, H. Wei, Z. Wei, C. Liu, Y. Lin, X. Li, C. Han, C. Zhi and H. Li, Adv. Energy Mater., 2023, 13, 2203729 CrossRef CAS
- S. Di, X. Nie, G. Ma, W. Yuan, Y. Wang, Y. Liu, S. Shen and N. Zhang, Energy Storage Mater., 2021, 43, 375–382 CrossRef
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef PubMed
- J. Weng, W. Zhu, K. Yu, J. Luo, M. Chen, L. Li, Y. Zhuang, K. Xia, Z. Lu, Y. Hu, C. Yang, M. Wu and Z. Zou, Adv. Funct. Mater., 2024, 2314347 CrossRef CAS
- S. Chen, S. Li, L. Ma, Y. Ying, Z. Wu, H. Huang and C. Zhi, Angew. Chem., Int. Ed., 2024, e202319125 CAS
- M. Zhu, H. Wang, H. Wang, C. Li, D. Chen, K. Wang, Z. Bai, S. Chen, Y. Zhang and Y. Tang, Angew. Chem., Int. Ed., 2024, 63, e202316904 CrossRef CAS PubMed
- H. Cheng, S. Zhang, W. Guo, Q. Wu, Z. Shen, L. Wang, W. Zhong, D. Li, B. Zhang, C. Liu, Y. Wang and Y. Lu, Adv. Sci., 2024, 11, 2307052 CrossRef CAS PubMed
- X. Zeng, J. Mao, J. Hao, J. Liu, S. Liu, Z. Wang, Y. Wang, S. Zhang, T. Zheng, J. Liu, P. Rao and Z. Guo, Adv. Mater., 2021, 33, 2007416 CrossRef CAS PubMed
- S. Zhang, M. Ye, Y. Zhang, Y. Tang, X. Liu and C. C. Li, Adv. Funct. Mater., 2023, 33, 2208230 CrossRef CAS
- J. Hao, B. Li, X. Li, X. Zeng, S. Zhang, F. Yang, S. Liu, D. Li, C. Wu and Z. Guo, Adv. Mater., 2020, 32, 2003021 CrossRef CAS PubMed
- X. Liu, H. Wang, X. Fan, Q. Wang, J. Liu, W. Xu, Z. Wu, J. Wan, C. Zhong and W. Hu, Energy Storage Mater., 2023, 58, 311–321 CrossRef
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Hou, K. Gaskell, J. T. Fourkas, X.-Q. Yang, K. Xu, O. Borodin and C. Wang, Nat. Nanotechnol., 2021, 16, 902–910 CrossRef CAS PubMed
- W. Li, W. Kong, W. Liu, S. Xu, H. Zhu, S. Liu, W. Yu and Z. Wen, Energy Storage Mater., 2024, 65, 103103 CrossRef
- X. Liu, X. Wang, J. Yao, J. Li, Y. Gan, Z. Wu, J. Zheng, W. Hao, L. Lv, L. Tao, J. Zhang, H. Wang, X. Ji, H. Wan and H. Wang, Adv. Energy Mater., 2024, 14, 2400090 CrossRef CAS
- Q. Ren, X. Tang, K. He, C. Zhang, W. Wang, Y. Guo, Z. Zhu, X. Xiao, S. Wang, J. Lu and Y. Yuan, Adv. Funct. Mater., 2023, 2312220 Search PubMed
- R. Chen, W. Zhang, C. Guan, Y. Zhou, I. Gilmore, H. Tang, Z. Zhang, H. Dong, Y. Dai, Z. Du, X. Gao, W. Zong, Y. Xu, P. Jiang, J. Liu, F. Zhao, J. Li, X. Wang and G. He, Angew. Chem., Int. Ed., 2024, e202401987 CAS
- W. Wu, Y. Deng and G. Chen, Chin. Chem. Lett., 2023, 34, 108424 CrossRef CAS
- C. Huang, X. Zhao, Y. Hao, Y. Yang, Y. Qian, G. Chang, Y. Zhang, Q. Tang, A. Hu and X. Chen, Adv. Funct. Mater., 2022, 32, 2112091 CrossRef CAS
- J. Han, H. Euchner, M. Kuenzel, S. M. Hosseini, A. Gross, A. Varzi and S. Passerini, ACS Energy Lett., 2021, 6, 3063–3071 CrossRef CAS
- H. Adenusi, G. A. Chass, S. Passerini, K. V. Tian and G. Chen, Adv. Energy Mater., 2023, 13, 2203307 CrossRef CAS
- H. Wu, H. Jia, C. Wang, J. G. Zhang and W. Xu, Adv. Energy Mater., 2021, 11, 2003092 CrossRef CAS
- Y. Song, Y. Liu, S. Luo, Y. Yang, F. Chen, M. Wang, L. Guo, S. Chen and Z. Wei, Adv. Funct. Mater., 2024, 2316070 CrossRef CAS
- X. Liang, X. Chen, Z. Zhai, R. Huang, T. Yu and S. Yin, Chem. Eng. J., 2024, 480, 148040 CrossRef CAS
- H. Du, R. Zhao, Y. Yang, Z. Liu, L. Qie and Y. Huang, Angew. Chem., 2022, 134, e202114789 CrossRef
- G. R. Pastel, T. P. Pollard, Q. Liu, S. Lavan, Q. Zhu, R. Jiang, L. Ma, J. Connell, O. Borodin, M. A. Schroeder, Z. Zhang and K. Xu, Joule, 2024, 8, 1050–1062 CrossRef CAS
- Q. Yang, X. Qu, H. Cui, X. He, Y. Shao, Y. Zhang, X. Guo, A. Chen, Z. Chen, R. Zhang, D. Kong, Z. Shi, J. Liu, J. Qiu and C. Zhi, Angew. Chem., Int. Ed., 2022, 61, e202206471 CrossRef CAS PubMed
- C. Li, Z. Lv, H. Du, L. Zhao, J. Yao, Y. Han, H. Chen, G. Zhang and Y. Bian, ACS Appl. Mater. Interfaces, 2023, 15, 50166–50173 CrossRef CAS PubMed
- T. Dong, K. L. Ng, Y. Wang, O. Voznyy and G. Azimi, Adv. Energy Mater., 2021, 11, 2100077 CrossRef CAS
- Z. Zhang, Y. Zhang, M. Ye, Z. Wen, Y. Tang, X. Liu and C. C. Li, Angew. Chem., Int. Ed., 2023, 62, e202311032 CrossRef CAS PubMed
- J. Hao, L. Yuan, Y. Zhu, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2022, 34, 2206963 CrossRef CAS PubMed
- W. Zhang, M. Dong, K. Jiang, D. Yang, X. Tan, S. Zhai, R. Feng, N. Chen, G. King, H. Zhang, H. Zeng, H. Li, M. Antonietti and Z. Li, Nat. Commun., 2022, 13, 5348 CrossRef CAS PubMed
- Q. Zhao, M. J. Zachman, W. I. Al Sadat, J. Zheng, L. F. Kourkoutis and L. Archer, Sci. Adv., 2018, 4, eaau8131 CrossRef CAS PubMed
- S. Kumar, T. Salim, V. Verma, W. Manalastas Jr and M. Srinivasan, Chem. Eng. J., 2022, 435, 134742 CrossRef CAS
- S. Kumar, V. Verma, H. Arora, W. Manalastas Jr and M. Srinivasan, ACS Appl. Energy Mater., 2020, 3, 8627–8635 CrossRef CAS
- Y. Li, L. Liu, Y. Lu, R. Shi, Y. Ma, Z. Yan, K. Zhang and J. Chen, Adv. Funct. Mater., 2021, 31, 2102063 CrossRef CAS
- G. Guo, X. Tan, K. Wang and H. Zhang, ChemSusChem, 2022, 15, e202200313 CrossRef CAS PubMed
- L. Zhang, B. Zhang, J. Hu, J. Liu, L. Miao and J. Jiang, Small Methods, 2021, 5, 2100094 CrossRef CAS PubMed
- Y. Dong, W. Liu, C. Carlos, Z. Zhang, J. Li, F. Pan, J. Sui and X. Wang, Nano Lett., 2024, 24, 4785–4792 CAS
- H. Li, L. Yang, S. Zhou, J. Li, Y. Chen, X. Meng, D. Xu, C. Han, H. Duan and A. Pan, Adv. Funct. Mater., 2024, 2313859 CrossRef CAS
- X. Zhang, J. Chen, H. Cao, X. Huang, Y. Liu, Y. Chen, Y. Huo, D. Lin, Q. Zheng and K. H. Lam, Small, 2023, 19, 2303906 CrossRef CAS PubMed
- Y. Yuan, S. D. Pu, M. A. Pérez-Osorio, Z. Li, S. Zhang, S. Yang, B. Liu, C. Gong, A. S. Menon, L. F. Piper, X. Gao, P. G. Bruce and A. W. Robertson, Adv. Mater., 2024, 36, 2307708 CrossRef CAS PubMed
- S.-B. Wang, Q. Ran, R.-Q. Yao, H. Shi, Z. Wen, M. Zhao, X.-Y. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef CAS PubMed
- Q. Ran, S. P. Zeng, M. H. Zhu, W. B. Wan, H. Meng, H. Shi, Z. Wen, X. Y. Lang and Q. Jiang, Adv. Funct. Mater., 2023, 33, 2211271 CrossRef CAS
- Y. Zeng, Z. Pei, Y. Guo, D. Luan, X. Gu and X. W. Lou, Angew. Chem., Int. Ed., 2023, 62, e202312145 CrossRef CAS PubMed
- X. Xu, X. Feng, M. Li, J. Yin, F. Li, J. Chen, W. Shi, Y. Cheng and J. Wang, Chem. Eng. J., 2023, 478, 147313 CrossRef CAS
- H. Tang, N. Hu, L. Ma, H. Weng, D. Huang, J. Zhu, H. Yang, Z. Chen, X. Yin, J. Xu and H. He, Adv. Funct. Mater., 2024, 2402484 CrossRef
- Y. Xu, J. Zhu, J. Feng, Y. Wang, X. Wu, P. Ma, X. Zhang, G. Wang and X. Yan, Energy Storage Mater., 2021, 38, 299–308 CrossRef
- Z. Liu, J. Ren, F. Wang, X. Liu, Q. Zhang, J. Liu, P. Kaghazchi, D. Ma, Z. Chi and L. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 27085–27095 CrossRef CAS PubMed
- K. Han, Z. Wang, F. An, Y. Liu, X. Qu, J. Xue and P. Li, ACS Appl. Mater. Interfaces, 2022, 14, 4316–4325 CrossRef CAS PubMed
- C. Lin, X. Yang, P. Xiong, H. Lin, L. He, Q. Yao, M. Wei, Q. Qian, Q. Chen and L. Zeng, Adv. Sci., 2022, 9, 2201433 CrossRef CAS PubMed
- Y. Cheng, Y. Jiao and P. Wu, Energy Environ. Sci., 2023, 16, 4561–4571 RSC
- Y. Ding, X. Zhang, T. Wang, B. Lu, Z. Zeng, Y. Tang, J. Zhou and S. Liang, Energy Storage Mater., 2023, 62, 102949 CrossRef
- G. Zhang, X. Zhang, H. Liu, J. Li, Y. Chen and H. Duan, Adv. Energy Mater., 2021, 11, 2003927 CrossRef CAS
- H. Zhang, L. Yang, H. Wang, B. Cui, J. Wang, X. Han and W. Hu, Adv. Funct. Mater., 2024, 34, 2312469 CrossRef CAS
- W. Fan, P. Li, J. Shi, J. Chen, W. Tian, H. Wang, J. Wu and G. Yu, Adv. Mater., 2024, 36, 2307219 CrossRef CAS PubMed
- Y. Ren, B. Wang, S. Chang, H. Wu, Y. Ma, F. Li, C. Wang, J. Gu, Y. Yang, S. Tang and X. Meng, Chem. Eng. J., 2024, 482, 148917 CrossRef CAS
- K. Wu, J. Yi, X. Liu, Y. Sun, J. Cui, Y. Xie, Y. Liu, Y. Xia and J. Zhang, Nano-Micro Lett., 2021, 13, 79 CrossRef PubMed
- Y. Li, D. Zhao, J. Cheng, Y. Lei, Z. Zhang, W. Zhang and Q. Zhu, Chem. Eng. J., 2023, 452, 139264 CrossRef CAS
- Y. Zhou, J. Xia, J. Di, Z. Sun, L. Zhao, L. Li, Y. Wu, L. Dong, X. Wang and Q. Li, Adv. Energy Mater., 2023, 13, 2203165 CrossRef CAS
- W. Li, Y. Ma, H. Shi, K. Jiang and D. Wang, Adv. Funct. Mater., 2022, 32, 2205602 CrossRef CAS
- Q. Wang, C. Yang, J. Yang, K. Wu, C. Hu, J. Lu, W. Liu, X. Sun, J. Qiu and H. Zhou, Adv. Mater., 2019, 31, 1903248 CrossRef CAS PubMed
- H. Liu, J. G. Wang, W. Hua, H. Sun, Y. Huyan, S. Tian, Z. Hou, J. Yang, C. Wei and F. Kang, Adv. Sci., 2021, 8, 2102612 CrossRef CAS PubMed
- T. Prodromakis and C. Papavassiliou, Appl. Surf. Sci., 2009, 255, 6989–6994 CrossRef CAS
- J. Cao, D. Zhang, C. Gu, X. Zhang, M. Okhawilai, S. Wang, J. Han, J. Qin and Y. Huang, Nano Energy, 2021, 89, 106322 CrossRef CAS
- P. Liang, J. Yi, X. Liu, K. Wu, Z. Wang, J. Cui, Y. Liu, Y. Wang, Y. Xia and J. Zhang, Adv. Funct. Mater., 2020, 30, 1908528 CrossRef CAS
- K. Zhang, C. Li, J. Liu, S. Zhang, M. Wang and L. Wang, Small, 2024, 20, 2306406 CrossRef CAS PubMed
- Y. Liu, T. Guo, Q. Liu, F. Xiong, M. Huang, Y. An, J. Wang, Q. An, C. Liu and L. Mai, Mater. Today Energy, 2022, 28, 101056 CrossRef CAS
- S. Zhou, Y. Wang, H. Lu, Y. Zhang, C. Fu, I. Usman, Z. Liu, M. Feng, G. Fang, X. Cao, S. Liang and A. Pan, Adv. Funct. Mater., 2021, 31, 2104361 CrossRef CAS
- S. So, Y. N. Ahn, J. Ko, I. T. Kim and J. Hur, Energy Storage Mater., 2022, 52, 40–51 CrossRef
- Q. Zong, B. Lv, C. Liu, Y. Yu, Q. Kang, D. Li, Z. Zhu, D. Tao, J. Zhang, J. Wang, Q. Zhang and G. Cao, ACS Energy Lett., 2023, 8, 2886–2896 CrossRef CAS
- A. Huang, H. Liu, O. Manor, P. Liu and J. Friend, Adv. Mater., 2020, 32, 1907516 CrossRef CAS PubMed
- A. Wang, Q. Deng, L. Deng, X. Guan and J. Luo, Adv. Funct. Mater., 2019, 29, 1902630 CrossRef
- Y.-S. Hong, N. Li, H. Chen, P. Wang, W.-L. Song and D. Fang, Energy Storage Mater., 2018, 11, 118–126 CrossRef
- K. Shen, Z. Wang, X. Bi, Y. Ying, D. Zhang, C. Jin, G. Hou, H. Cao, L. Wu, G. Zheng, Y. Tang, X. Tao and J. Lu, Adv. Energy Mater., 2019, 9, 1900260 CrossRef
- Z. Zhao, J. Zhao, Z. Hu, J. Li, J. Li, Y. Zhang, C. Wang and G. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 RSC
- W. Wang and Y. C. Lu, SusMat, 2023, 3, 146–159 CrossRef CAS
- C. M. Costa, K. J. Merazzo, R. Gonçalves, C. Amos and S. Lanceros-Méndez, iScience, 2021, 24, 102691 CrossRef CAS PubMed
- A. Sheelam, D. L. Glasco and J. G. Bell, Chem. Eng. J., 2023, 461, 141900 CrossRef CAS
- Q. Chen, C. Jiang, M. Chen, J. Zhang, G. Hou and Y. Tang, Surf. Interfaces, 2022, 31, 101972 CrossRef CAS
- P. Liang, Q. Li, L. Chen, Z. Tang, Z. Li, Y. Wang, Y. Tang, C. Han, Z. Lan, C. Zhi and H. Li, J. Mater. Chem. A, 2022, 10, 11971–11979 RSC
- Y. Zhang, H. Dong, R. Yang, H. He, G. He and F. Cegla, Electrochem. Commun., 2024, 162, 107700 CrossRef CAS
- F. Zhang, Z. Cai, R. Liu, Y. Sun and H. Pan, J. Mater. Chem. A, 2024, 12, 10341–10348 RSC
- Y. Li, C. B. Musgrave III, M. Y. Yang, M. M. Kim, K. Zhang, M. Tamtaji, Y. Cai, T. W. Tang, J. Wang, B. Yuan, W. A. Goddard III and Z. Luo, Adv. Energy Mater., 2024, 14, 2303047 CrossRef CAS
- M.-L. Doche, J.-Y. Hihn, A. Mandroyan, R. Viennet and F. Touyeras, Ultrason. Sonochem., 2003, 10, 357–362 CrossRef CAS PubMed
- H. Huang, P. Liu, Q. Ma, Z. Tang, M. Wang and J. Hu, Ultrason. Sonochem., 2022, 88, 106104 CrossRef CAS PubMed
- J. Klima, Ultrasonics, 2011, 51, 202–209 CrossRef CAS PubMed
- L. Y. Yeo and J. R. Friend, Annu. Rev. Fluid Mech., 2014, 46, 379–406 CrossRef
- A. Huang, H. Liu, P. Liu and J. Friend, Adv. Energy Sustainability Res., 2023, 4, 2200112 CrossRef CAS
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2020, 121, 1232–1285 CrossRef PubMed
- C. Zhang, L. Zhang and G. Yu, Acc. Chem. Res., 2020, 53, 1648–1659 CrossRef CAS PubMed
- X. Lu, E. J. Hansen, G. He and J. Liu, Small, 2022, 18, 2200550 CrossRef CAS PubMed
- R. Chen, C. Zhang, J. Li, Z. Du, F. Guo, W. Zhang, Y. Dai, W. Zong, X. Gao, J. Zhu, Y. Zhao, X. Wang and G. He, Energy Environ. Sci., 2023, 16, 2540–2549 RSC
- Y. Wang, T. Wang, S. Bu, J. Zhu, Y. Wang, R. Zhang, H. Hong, W. Zhang, J. Fan and C. Zhi, Nat. Commun., 2023, 14, 1828 CrossRef CAS PubMed
- C. Li, R. Kingsbury, A. S. Thind, A. Shyamsunder, T. T. Fister, R. F. Klie, K. A. Persson and L. F. Nazar, Nat. Commun., 2023, 14, 3067 CrossRef CAS PubMed
- J. Shi, T. Sun, J. Bao, S. Zheng, H. Du, L. Li, X. Yuan, T. Ma and Z. Tao, Adv. Funct. Mater., 2021, 31, 2102035 CrossRef CAS
- L. Su, F. Lu, Y. Li, X. Li, L. Chen, Y. Gao, L. Zheng and X. Gao, ChemSusChem, 2023, 16, e202300285 CrossRef CAS PubMed
- W. Yang, X. Du, J. Zhao, Z. Chen, J. Li, J. Xie, Y. Zhang, Z. Cui, Q. Kong, Z. Zhao, C. Wang, Q. Zhang and G. Cui, Joule, 2020, 4, 1557–1574 CrossRef CAS
- Y. Zhong, X. Xie, Z. Zeng, B. Lu, G. Chen and J. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202310577 CrossRef CAS PubMed
- Y. Xie, J. Huang, T. Kong, X. Zhou, K. Wu, X. Liu, J. Yi, L. Xing and Y. Xia, Energy Storage Mater., 2023, 56, 218–226 CrossRef
- X. Feng, P. Li, J. Yin, Z. Gan, Y. Gao, M. Li, Y. Cheng, X. Xu, Y. Su and S. Ding, ACS Energy Lett., 2023, 8, 1192–1200 CrossRef CAS
- P. Meng, J. Huang, Z. Yang, F. Wang, T. Lv, J. Zhang, C. Fu and W. Xiao, Adv. Mater., 2022, 34, 2106511 CrossRef CAS PubMed
- Y. Zhu, X. Guo, Y. Lei, W. Wang, A.-H. Emwas, Y. Yuan, Y. He and H. N. Alshareef, Energy Environ. Sci., 2022, 15, 1282–1292 RSC
- W. Chu, X. Zhang, J. Wang, S. Zhao, S. Liu and H. Yu, Energy Storage Mater., 2019, 22, 418–423 CrossRef
- F. Coleman, G. Srinivasan and M. Swadźba-Kwaśny, Angew. Chem., Int. Ed., 2013, 52, 12582–12586 CrossRef CAS PubMed
- W. Chu, X. Zhang, S. Zhao, M. Tang, S. Li, S. Liu and H. Yu, Adv. Funct. Mater., 2024, 34, 2305194 CrossRef CAS
- L. Zhang, C. Zhang, Y. Ding, K. Ramirez-Meyers and G. Yu, Joule, 2017, 1, 623–633 CrossRef CAS
- C. Zhang, Y. Ding, L. Zhang, X. Wang, Y. Zhao, X. Zhang and G. Yu, Angew. Chem., Int. Ed., 2017, 56, 7454–7459 CrossRef CAS PubMed
- P. Meng, J. Huang, Z. Yang, M. Jiang, Y. Wang, W. Zhang, J. Zhang, B. Sun and C. Fu, Nano-Micro Lett., 2023, 15, 188 CrossRef CAS PubMed
- S.-C. Wu, Y. Ai, Y.-Z. Chen, K. Wang, T.-Y. Yang, H.-J. Liao, T.-Y. Su, S.-Y. Tang, C.-W. Chen, D. C. Wu, Y.-C. Wang, A. Manikandan, Y.-C. Shih, L. Lee and Y.-L. Chueh, ACS Appl. Mater. Interfaces, 2020, 12, 27064–27073 CrossRef CAS PubMed
- X. Luo, R. Wang, L. Zhang, Z. Liu, H. Li, J. Mao, S. Zhang, J. Hao, T. Zhou and C. Zhang, ACS Nano, 2024, 18, 12981–12993 CrossRef CAS PubMed
- X. Song, Y. Ge, H. Xu, S. Bao, L. Wang, X. Xue, Q. Yu, Y. Xing, Z. Wu, K. Xie, T. Zhu, P. Zhang, Y. Liu, Z. Wang, Z. Tie, J. Ma and Z. Jin, J. Am. Chem. Soc., 2024, 146, 7018–7028 CrossRef CAS PubMed
- Y. Geng, L. Pan, Z. Peng, Z. Sun, H. Lin, C. Mao, L. Wang, L. Dai, H. Liu, K. Pan, X. Wu, Q. Zhang and Z. He, Energy Storage Mater., 2022, 51, 733–755 CrossRef
- S. Guo, L. Qin, T. Zhang, M. Zhou, J. Zhou, G. Fang and S. Liang, Energy Storage Mater., 2021, 34, 545–562 CrossRef
- X. Guo, Z. Zhang, J. Li, N. Luo, G.-L. Chai, T. S. Miller, F. Lai, P. Shearing, D. J. Brett, D. Han, Z. Weng, G. He and I. P. Parkin, ACS Energy Lett., 2021, 6, 395–403 CrossRef CAS
- G. Chang, S. Liu, Y. Fu, X. Hao, W. Jin, X. Ji and J. Hu, Adv. Mater. Interfaces, 2019, 6, 1901358 CrossRef CAS
- J. Gao, Y. Li, Z. Yan, Q. Liu, Y. Gao, C. Chen, B. Ma, Y. Song and E. Wang, J. Power Sources, 2019, 412, 63–70 CrossRef CAS
- M. Wang, Y. Meng, Y. Xu, N. Chen, M. Chuai, Y. Yuan, J. Sun, Z. Liu, X. Zheng, Z. Zhang, D. Li and W. Chen, Energy Environ. Sci., 2023, 16, 5284–5293 RSC
- Z. Hu, Y. Guo, H. Jin, H. Ji and L.-J. Wan, Chem. Commun., 2020, 56, 2023–2026 RSC
- H. Lv, S. Yang, C. Li, C. Han, Y. Tang, X. Li, W. Wang, H. Li and C. Zhi, Energy Storage Mater., 2021, 39, 412–418 CrossRef
- Z. Hou, X. Zhang, X. Li, Y. Zhu, J. Liang and Y. Qian, J. Mater. Chem. A, 2017, 5, 730–738 RSC
- K. E. Sun, T. K. Hoang, T. N. L. Doan, Y. Yu, X. Zhu, Y. Tian and P. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 9681–9687 CrossRef CAS PubMed
- J. Hao, J. Long, B. Li, X. Li, S. Zhang, F. Yang, X. Zeng, Z. Yang, W. K. Pang and Z. Guo, Adv. Funct. Mater., 2019, 29, 1903605 CrossRef
- Y. Jin, K. S. Han, Y. Shao, M. L. Sushko, J. Xiao, H. Pan and J. Liu, Adv. Funct. Mater., 2020, 30, 2003932 CrossRef CAS
- A. B. Hashemi, G. Kasiri and F. La Mantia, Electrochim. Acta, 2017, 258, 703–708 CrossRef
- M. Yan, N. Dong, X. Zhao, Y. Sun and H. Pan, ACS Energy Lett., 2021, 6, 3236–3243 CrossRef CAS
- X. Li, X. Wang, L. Ma and W. Huang, Adv. Energy Mater., 2022, 12, 2202068 CrossRef CAS
- Y. Wu, Z. Zhu, D. Shen, L. Chen, T. Song, T. Kang, Z. Tong, Y. Tang, H. Wang and C. S. Lee, Energy Storage Mater., 2022, 45, 1084–1091 CrossRef
- Z. Cao, X. Zhu, S. Gao, D. Xu, Z. Wang, Z. Ye, L. Wang, B. Chen, L. Li, M. Ye and J. Shen, Small, 2022, 18, 2103345 CrossRef CAS PubMed
- C. Li, R. Kingsbury, L. Zhou, A. Shyamsunder, K. A. Persson and L. F. Nazar, ACS Energy Lett., 2022, 7, 533–540 CrossRef CAS
- M. Luo, C. Wang, H. Lu, Y. Lu, B. B. Xu, W. Sun, H. Pan, M. Yan and Y. Jiang, Energy Storage Mater., 2021, 41, 515–521 CrossRef
- N. Wang, S. Zhai, Y. Ma, X. Tan, K. Jiang, W. Zhong, W. Zhang, N. Chen, W. Chen, S. Li, G. Han and Z. Li, Energy Storage Mater., 2021, 43, 585–594 CrossRef
- Y. Lin, Y. Hu, S. Zhang, Z. Xu, T. Feng, H. Zhou and M. Wu, CrystEngComm, 2022, 24, 7950–7961 RSC
- K. Xie, K. Ren, C. Sun, S. Yang, M. Tong, S. Yang, Z. Liu and Q. Wang, ACS Appl. Energy Mater., 2022, 5, 4170–4178 CrossRef CAS
- P. Sun, L. Ma, W. Zhou, M. Qiu, Z. Wang, D. Chao and W. Mai, Angew. Chem., 2021, 133, 18395–18403 CrossRef
- T. Wang, H. Cheng, Z. Tian, Z. Li, Z. Lin, Z. You, Y. Lu, Y. Zhu, W. Li, Y. Yang, Q. Zhong and Y. Lai, Energy Storage Mater., 2022, 53, 371–380 CrossRef
- Q. Fu, X. Wu, X. Luo, S. Indris, A. Sarapulova, M. Bauer, Z. Wang, M. Knapp, H. Ehrenberg, Y. Wei and S. Dsoke, Adv. Funct. Mater., 2022, 32, 2110674 CrossRef CAS
- Z. Zhao, Z. Zhang, T. Xu, W. Wang, B. Wang and X. Yu, J. Am. Chem. Soc., 2024, 146, 2257–2266 CrossRef CAS PubMed
- T. R. Kartha and B. S. Mallik, J. Chem. Eng. Data, 2021, 66, 1543–1554 CrossRef CAS
- C. Sun, G. Huang, C. Xu, L. Li, A. Tang, X. Zhou, J. Wang and F. Pan, Energy Storage Mater., 2024, 66, 103197 CrossRef
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed
- L. Suo, O. Borodin, Y. Wang, X. Rong, W. Sun, X. Fan, S. Xu, M. A. Schroeder, A. V. Cresce, F. Wang, C. Yang, Y.-S. Hu, K. Xu and C. Wang, Adv. Energy Mater., 2017, 7, 1701189 CrossRef
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed
- J. Zhao, Y. Li, X. Peng, S. Dong, J. Ma, G. Cui and L. Chen, Electrochem. Commun., 2016, 69, 6–10 CrossRef CAS
- P. Hu, M. Yan, T. Zhu, X. Wang, X. Wei, J. Li, L. Zhou, Z. Li, L. Chen and L. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 42717–42722 CrossRef CAS PubMed
- F. Wan, Y. Zhang, L. Zhang, D. Liu, C. Wang, L. Song, Z. Niu and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 7062–7067 CrossRef CAS PubMed
- C. Zhang, J. Holoubek, X. Wu, A. Daniyar, L. Zhu, C. Chen, D. P. Leonard, I. A. Rodríguez-Pérez, J.-X. Jiang, C. Fang and X. Ji, Chem. Commun., 2018, 54, 14097–14099 RSC
- H. Yang, Z. Chang, Y. Qiao, H. Deng, X. Mu, P. He and H. Zhou, Angew. Chem., 2020, 132, 9463–9467 CrossRef
- A. Zhou, L. Jiang, J. Yue, Y. Tong, Q. Zhang, Z. Lin, B. Liu, C. Wu, L. Suo, Y.-S. Hu, H. Li and L. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 41356–41362 CrossRef CAS PubMed
- F. Wang, X. Fan, T. Gao, W. Sun, Z. Ma, C. Yang, F. Han, K. Xu and C. Wang, ACS Central Sci., 2017, 3, 1121–1128 CrossRef CAS PubMed
- I. Persson, Pure Appl. Chem., 2010, 82, 1901–1917 CAS
- C. Wu, S. Gu, Q. Zhang, Y. Bai, M. Li, Y. Yuan, H. Wang, X. Liu, Y. Yuan, N. Zhu, F. Wu, H. Li, G. Lin and J. Lu, Nat. Commun., 2019, 10, 73 CrossRef CAS PubMed
- W. Pan, J. Mao, Y. Wang, X. Zhao, K. W. Leong, S. Luo, Y. Chen and D. Y. Leung, Small Methods, 2021, 5, 2100491 CrossRef CAS PubMed
- Y. Gao, Y. Li, H. Yang, L. Zheng, Y. Bai and C. Wu, J. Energy Chem., 2022, 67, 613–620 CrossRef CAS
- K. W. Leong, W. Pan, Y. Wang, S. Luo, X. Zhao and D. Y. Leung, ACS Energy Lett., 2022, 7, 2657–2666 CrossRef CAS
- K. W. Leong, W. Pan, X. Yi, S. Luo, X. Zhao, Y. Zhang, Y. Wang, J. Mao, Y. Chen, J. Xuan, H. Wang and D. Y. Leung, Sci. Adv., 2023, 9, eadh1181 CrossRef CAS PubMed
- Z. Tong, T. Kang, Y. Wan, R. Yang, Y. Wu, D. Shen, S. Liu, Y. Tang and C. S. Lee, Adv. Funct. Mater., 2021, 31, 2104639 CrossRef CAS
- M. Adil, A. Ghosh and S. Mitra, ACS Appl. Mater. Interfaces, 2022, 14, 25501–25515 CrossRef CAS PubMed
- F. Qiao, J. Wang, R. Yu, Y. Pi, M. Huang, L. Cui, Z. Liu and Q. An, Small Methods, 2024, 8, 2300865 CrossRef CAS PubMed
- Y. Guo, J. Bae, Z. Fang, P. Li, F. Zhao and G. Yu, Chem. Rev., 2020, 120, 7642–7707 CrossRef CAS PubMed
- X. Zhou, F. Zhao, Y. Guo, B. Rosenberger and G. Yu, Sci. Adv., 2019, 5, eaaw5484 CrossRef CAS PubMed
- S. Jin, P.-Y. Chen, Y. Qiu, Z. Zhang, S. Hong, Y. L. Joo, R. Yang and L. A. Archer, J. Am. Chem. Soc., 2022, 144, 19344–19352 CrossRef CAS PubMed
- X. Peng, H. Liu, Q. Yin, J. Wu, P. Chen, G. Zhang, G. Liu, C. Wu and Y. Xie, Nat. Commun., 2016, 7, 1–8 Search PubMed
- Q. Fu, S. Hao, X. Zhang, H. Zhao, F. Xu and J. Yang, Energy Environ. Sci., 2023, 16, 1291–1311 RSC
- F. Mo, Z. Chen, G. Liang, D. Wang, Y. Zhao, H. Li, B. Dong and C. Zhi, Adv. Energy Mater., 2020, 10, 2000035 CrossRef CAS
- Y. Yan, S. Duan, B. Liu, S. Wu, Y. Alsaid, B. Yao, S. Nandi, Y. Du, T. W. Wang, Y. Li and X. He, Adv. Mater., 2023, 35, 2211673 CrossRef CAS PubMed
- X. Tang, D. Zhou, B. Zhang, S. Wang, P. Li, H. Liu, X. Guo, P. Jaumaux, X. Gao, Y. Fu, C. Wang, C. Wang and G. Wang, Nat. Commun., 2021, 12, 2857 CrossRef CAS PubMed
- Y. Shi, R. Wang, S. Bi, M. Yang, L. Liu and Z. Niu, Adv. Funct. Mater., 2023, 33, 2214546 CrossRef CAS
- L. Jiang, S. Han, Y.-C. Hu, Y. Yang, Y. Lu, Y.-C. Lu, J. Zhao, L. Chen and Y.-S. Hu, Nat. Energy, 2024, 9, 839–848 CrossRef CAS
- Q. Zhang, Y. Ma, Y. Lu, L. Li, F. Wan, K. Zhang and J. Chen, Nat. Commun., 2020, 11, 4463 CrossRef CAS PubMed
- F. Yue, Z. Tie, S. Deng, S. Wang, M. Yang and Z. Niu, Angew. Chem., Int. Ed., 2021, 60, 13882–13886 CrossRef CAS PubMed
- C. Yang, J. Xia, C. Cui, T. P. Pollard, J. Vatamanu, A. Faraone, J. A. Dura, M. Tyagi, A. Kattan, E. Thimsen, J. Xu, W. Song, E. Hu, X. Ji, S. Hou, X. Zhang, M. S. Ding, S. Hwang, D. Su, Y. Ren, X.-Q. Yang, H. Wang, O. Borodin and C. Wang, Nat. Sustainability, 2023, 6, 325–335 CrossRef
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |