
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeted protein degradation directly engaging lysosomes or proteasomes†
Jiseong
Kim‡
 ab,
Insuk
Byun‡
ab,
Do Young
Kim‡
c,
Hyunhi
Joh
c,
Hak Joong
Kim
*c and
Min Jae
Lee
*abd
ab,
Insuk
Byun‡
ab,
Do Young
Kim‡
c,
Hyunhi
Joh
c,
Hak Joong
Kim
*c and
Min Jae
Lee
*abd
aDepartment of Biochemistry & Molecular Biology, Seoul National University College of Medicine, Seoul 03080, Korea. E-mail: minjlee@snu.ac.kr
bDepartment of Biomedical Sciences, Seoul National University Graduate School, Seoul 03080, Korea
cDepartment of Chemistry, College of Science, Korea University, Seoul 02841, Korea. E-mail: hakkim@korea.ac.kr
dDepartment of Pathology and Laboratory Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA
First published on 19th February 2024
Abstract
Targeted protein degradation (TPD) has been established as a viable alternative to attenuate the function of a specific protein of interest in both biological and clinical contexts. The unique TPD mode-of-action has allowed previously undruggable proteins to become feasible targets, expanding the landscape of “druggable” properties and “privileged” target proteins. As TPD continues to evolve, a range of innovative strategies, which do not depend on recruiting E3 ubiquitin ligases as in proteolysis-targeting chimeras (PROTACs), have emerged. Here, we present an overview of direct lysosome- and proteasome-engaging modalities and discuss their perspectives, advantages, and limitations. We outline the chemical composition, biochemical activity, and pharmaceutical characteristics of each degrader. These alternative TPD approaches not only complement the first generation of PROTACs for intracellular protein degradation but also offer unique strategies for targeting pathologic proteins located on the cell membrane and in the extracellular space.
 Jiseong Kim | Jiseong Kim graduated from Handong Global University (Korea) and received a BS in 2021. She then joined the group of Professor Min Jae Lee at Seoul National University College of Medicine and is currently in the PhD course. Her research interest is developing a new platform for targeted protein degradation that induces selective degradation of pathologic proteins. |
 Insuk Byun | Insuk Byun received his BS (2022) from Konkuk University, Seoul, Korea. He is currently pursuing his PhD degree under the supervision of Dr Min Jae Lee at Seoul National University College of Medicine. His research interests are developing anticancer drugs focusing on the ubiquitin-proteasome system and intracellular proteasome regulation. |
 Do Young Kim | Do Young Kim received his BS degree from Korea University in 2019 and completed his doctoral studies at the same university under the supervision of Dr Hak Joong Kim in 2024. His research interests have included repurposing antibacterial and anticancer drugs via developing an original drug delivery strategy. Currently, he is involved in the discovery of potent agents capable of inducing targeted degradation of secreted proteins relevant to cancer metastasis. |
 Hyunhi Joh | Hyunhi Joh graduated from the Department of Chemistry, Korea University and received a BS degree in 2023. She is currently studying under the supervision of Dr Hak Joong Kim at Korea University to pursue her master's degree. Her research interests include the discovery of novel targeted protein degradation agents and establishing a new screening platform based on the DNA-encoded library. |
 Hak Joong Kim | Hak Joong Kim is a professor in the Department of Chemistry at Korea University. He received his PhD degree under the guidance of Dr Hung-Wen Liu at the University of Texas at Austin and then joined Dr Peter G. Schultz at The Scripps Research Institute for postdoctoral research. His research interests include chemical biology and medicinal chemistry associated with infectious diseases and cancer, among which he currently focuses on the development of new platforms for targeted protein degradation. |
 Min Jae Lee | Min Jae Lee is a Professor of Biochemistry at Seoul National University College of Medicine in Korea. He received his BS in chemistry from SNU and his PhD in pharmaceutical sciences from University of Pittsburgh School of Pharmacy. His research interests are intracellular protein degradation through the ubiquitin-proteasome system and autophagy, its pathophysiological implications, and the development of pharmacological methods. He has authored >70 research papers with >5000 citations and was the recipient of the Research Excellent Prizes of the Korean Chemical Society (in 2016) and the Korean Society of Biochemistry and Molecular Biology (in 2019). |
Key learning points(1) A comprehensive overview of recent approaches in TPD, emphasizing ubiquitin-independent modalities from chemical and biological perspectives.(2) Chemical induction of autophagy-mediated degradation for elimination of intracellular proteins and organelles with pathological relevance. (3) Exploitation of lysosome-trafficking receptors for TPD of extracellular and membrane-associated proteins via the endosome–lysosome pathway. (4) Direct recruiting of the 26S proteasome to soluble proteins, a departure from the PROTAC technology based on target ubiquitination. (5) Comparative analysis on the advantages and limitations of ubiquitin-independent TPD technologies with a discussion on their therapeutic potentials. |
1. Introduction: targeted protein degradation
Targeted protein degradation (TPD) has emerged as a compelling pharmacological modality for selective removal of disease-associated proteins.1 In contrast to conventional small-molecule inhibitors or agonists/antagonists that typically require a high binding affinity with specific proteins, TPD can operate through comparably modest interactions between the targets (primarily proteins but cellular organelles as well) and binding molecules (encompassing chemical compounds and biologics).2,3 The first-generation protein degraders, such as PROteolysis-TArgeting Chimeras (PROTACs), are mainly synthetic heterobifunctional compounds that bind to a target protein on one end and recruit an E3 ubiquitin (Ub) ligase (or ligase complex) on the other end, connected by a diverse variety of linkers.4–7 The induced proximity between the hijacked E3 Ub ligase and “neo-substrate” in the cell leads to the forced polyubiquitination of the latter, resulting in its proteolysis by 26S proteasomes.8 Molecular glue degraders (MGDs) share a similar mode of action with PROTACs, chemically inducing a stable ternary complex with an E3 ligase and a target protein, have an analogous mode of action towards PROTACs.9–11 However, unlike PROTACs, MGDs lack the linker element and the capability to establish an intimate contact interface between two proteins. With their smaller size, MGDs can offer pharmacological advantages over PROTACs, albeit posing greater challenges for their rational chemical design.As a therapeutic strategy, specific removal of disease-associated proteins offers several advantages compared to conventional occupancy-driven drugs, which require stoichiometric target engagement.12,13 First, owing to their catalytic mode of action, PROTAC- and MGD-based drugs can exhibit high potency even at low cellular concentrations. Protein degraders can also overcome the drug resistance originating from somatic mutations or structural/functional changes in target proteins due to post-translational modifications (PTMs).14 PROTACs have enabled the targeting of scaffolding proteins and transcription factors that are therapeutically significant but usually lack well-defined binding pockets for conventional drugs, thereby expanding the range of druggable proteomes. Various PROTAC-based degraders have been developed, and their unique therapeutic potential has been demonstrated, with approximately 25 PROTAC drugs being under clinical trials and many more in the development pipeline.15–18
PROTACs, MGD, and many other early TPD approaches function by exploiting the Ub-proteasome system (UPS), the predominant proteolytic route within the cytoplasm and nucleus of eukaryotes.19 The UPS also plays a pivotal role in maintaining the quality control of cellular proteins. For instance, misfolded proteins, generated via genetic mutations, transcriptional/translational errors, or PTM-mediated structural changes, undergo rapid polyubiquitination and proteasomal degradation (mostly <10 min of an average half-life).20–22 The E3 Ub ligases play a critical role in determining substrate specificity by mediating a proximal interaction between the Ub-charged E2 Ub-conjugating enzymes and to-be-degraded substrates. The human genome encodes >600 E3 Ub ligases,23 but only a few have been employed for PROTACs; two E3s, CRBN and VHL, are responsible for ∼94% of all PROTACs (Please note that a detailed discussion of E3s in TPD is outside the scope of this review. Readers interested on this topic are encouraged to consult recent excellent reviews24,25).
Proteasomes are highly abundant (>200 nM) and stable (half-life >12 days) enzyme complexes present in most cell types,26–29 which makes them appropriate proteases for TPD. The 26S holoenzyme consists of two distinct and dissociable components—the 28-subunit catalytic complex (CP) and the 19-subunit regulatory complex (RP).30 While the cylindrical CP retains active proteolytic sites in its interior, the RP interacts with the polyubiquitin (polyUb) chains of target substrates through multiple Ub receptors, including PSMD2/Rpn1, PSMD4/Rpn10, and ADRM1/Rpn13.31 Upon binding, the RP triggers substrate unfolding, opens the “gate” at the substrate translocation channel, and translocates the substrates into the CP for proteolysis.32 The catalytic PSMB5/β5, PSMB6/β1, and PSMB7/β2 subunits in the CP are threonine proteases; the hydroxyl groups on their N-terminal threonine residues initiate multiple, simultaneous nucleophilic attacks on peptide bonds, leading to proteolysis in a processive manner.33
Proteasomes heterogeneously exist as a mixture of 20S (free CP as a stand-alone protease), 26S (singly-capped RP-CP), and 30S (doubly-capped RP2-CP) subfamilies within a cell. It is notable that free 20S proteasomes, which cannot recognise polyUb chains, account for more than half of the total proteasome.34,35 The equilibrium between Ub-dependent proteasome holoenzymes (26S and 30S) and Ub-independent 20S proteasomes appears to be dynamically adjusted in response to environmental changes. Although the detailed mechanism remains to be identified, the abundance of 26S/30S proteasomes available in target cells is expected to influence the overall degradation efficiency of various PROTACs, which can also be limited by the expression levels of hijacked E3 Ub ligases. When E3s are ubiquitously expressed, a TPD drug may exhibit “on-target, off-tumour” toxicity in healthy tissues.36,37 Overall, the mechanistic complexities involved in the UPS and the constant fluctuation of its proteolytic flux in diverse biological contexts imply that substrate polyubiquitination alone may not always be sufficient to trigger substrate degradation.38 The development of PROTACs and MGDs capable of forming ternary complexes with acceptable druggability and pharmacological profiles continues to heavily rely on empirical trial-and-error methods both in the chemical (synthesis) and biological (assay) process.
While these potential limitations do not preclude the viability of PROTACs and other TPD modalities exploiting the UPS, it is noteworthy that a number of alternative strategies have been explored to circumvent these issues. Within a cellular environment, the UPS is complemented by another central degradation system—the autophagy-lysosome system (hereafter referred to as autophagy).39 In contrast to the UPS, which operates continuously under physiological conditions, autophagy is primarily an inducible system that is activated only when cells are under relatively severe cellular stress, such as nutrient starvation, oxidative stress, and pathogen infection, thus providing metabolic intermediates and energy to cells and maintaining organelle homeostasis.40–42 Combined with lysosomes harbouring >60 hydrolytic enzymes, autophagy can also degrade non-proteinaceous entities, such as nucleic acids, intracellular pathogens, mitochondria, endoplasmic reticulum (ER), and lipid droplets.43,44 Consequently, integrating autophagic degradation with the concept of TPD has considerably broadened the range of druggable targets.
In autophagy, microtubule-associated protein 1 light chain 3 (LC3) plays a critical role in the regulation of autophagosome biogenesis, cargo recruitment, and lysosomal fusion.45 The biochemical cascade that involves the attachment of a phosphatidylethanolamine moiety to the C-terminal glycine of the cleaved LC3 (LC3-I) in autophagy resembles the E1-, E2-, and E3-mediated multistep Ub-conjugating system in the UPS.46,47 In contrast to the UPS that features hundreds of E3 Ub ligases, autophagy depends on only a handful of receptors connecting diverse cargoes to lipidated LC3 (LC3-II) on the phagophore membrane.46–48 Selective autophagy is known to be mediated by specific cargo receptors, such as SQSTM1/p62, NBR1, IPTN, NDP52, and TAX1BP1, all of which commonly feature an LC3-interacting region, a Ub-binding domain, and an oligomerisation domain. In non-selective, bulk autophagy, liquid–liquid phase separation, instead of specific cargo recognition, at the phagophore assembly site appears to be critical for importing substrates to the autophagosome.
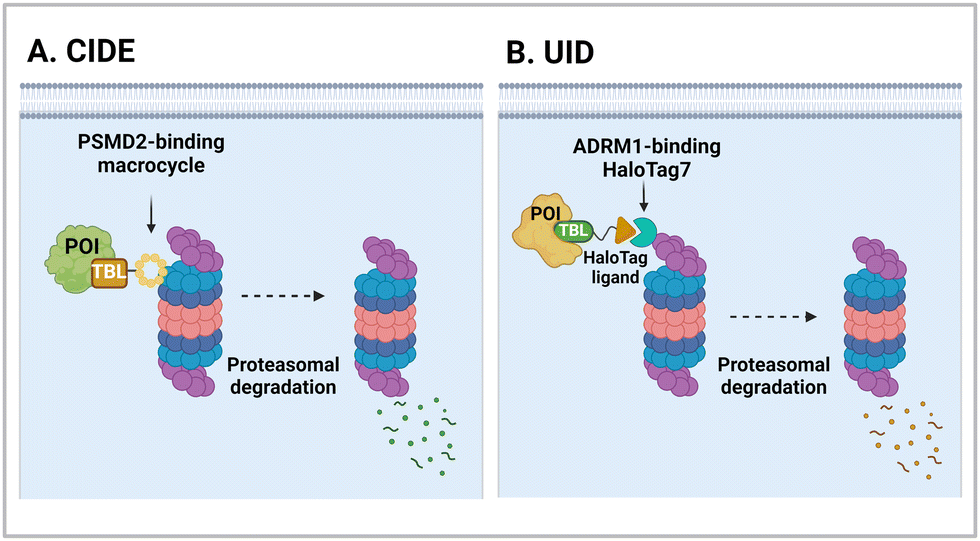
It has been proposed that TPD modalities that do not require the complex and energy-intensive ubiquitination process as in PROTACs could offer distinct advantages from both technical and therapeutic standpoints. Recently, innovative TPD tools, including AuTophagy-TEthering Compounds (ATTEC),49 LYsosome-TArgeting Chimeras (LYTAC),50 AUTOphagy-TArgeting Chimera (AUTOTAC),51 CytoKine receptor-TArgeting Chimera (KineTAC),52 Integrin-Facilitated Lysosomal Degradation (IFLD),53 and DENdronized DNA Chimera (DENTAC),54 have been developed to hijack endogenous lysosomes through lysosomal trafficking receptors, LC3-II on the growing phagophore, or oligomerised SQSTM1 (Fig. 1). These tools effectively degrade the intracellular, extracellular, and membrane proteins, protein aggregates, lipid droplets, and damaged organelles. Moreover, it has been shown that pathologic proteins can be directly recruited to the 26S proteasome and subjected to induced proteolysis using small molecules, such as the recently developed Chemical Inducers of DEgradation (CIDE)55 and Ub-Independent Degraders (UIDs).56 Here, we overview recent developments in these TPD technologies that directly engage with the lysosome or the proteasome and outline a comprehensive analysis of each technique, including its strengths and limitations from both chemical and biological perspectives. Please note that this review uses the official human gene/protein names, determined by the HUGO gene nomenclature committee (https://www.genenames.org/).
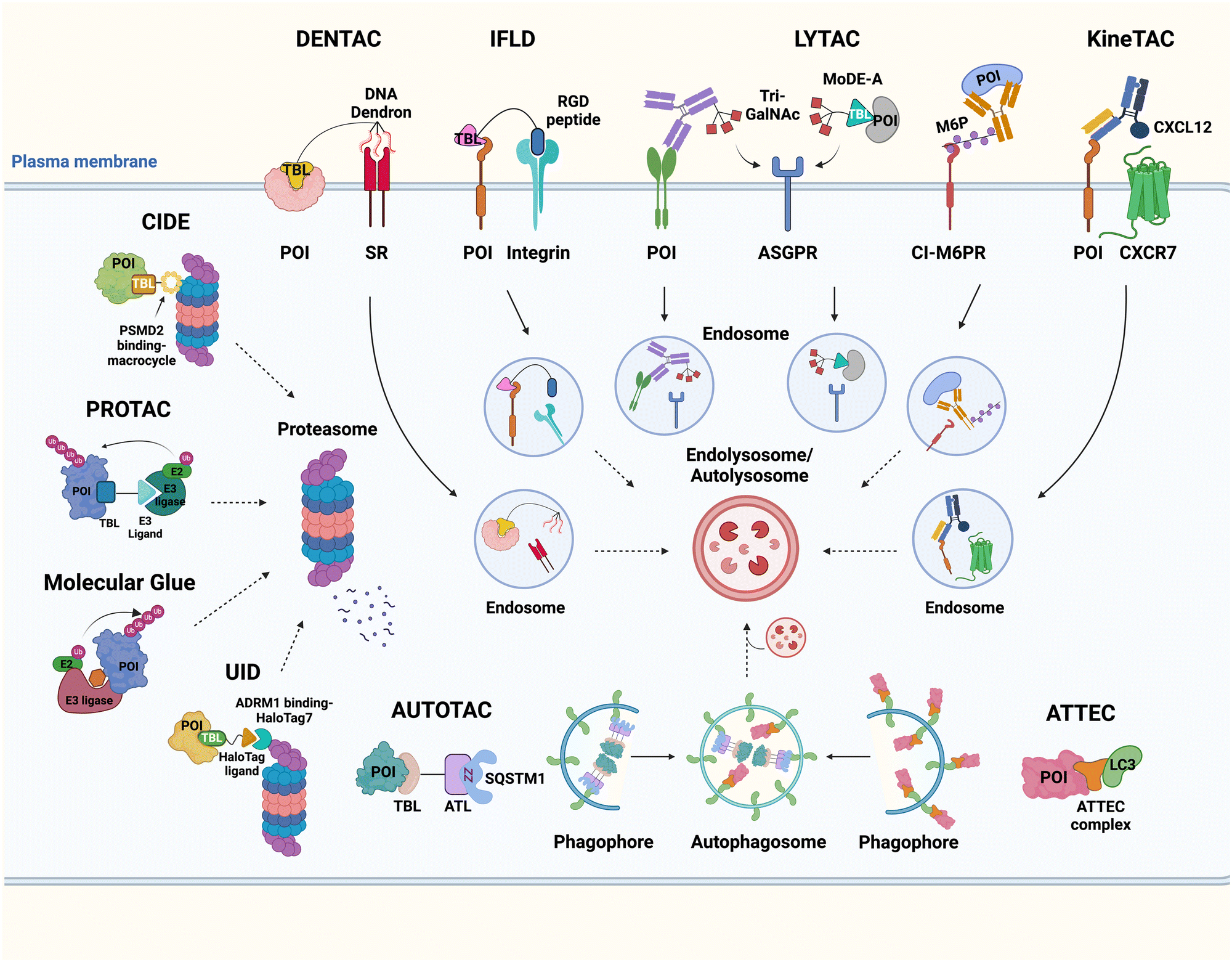 | ||
| Fig. 1 The illustration of Ub-independent TPD strategies. Arginine–glycine–aspartic acid (RGD); asialoglycoprotein receptor (ASGPR); autophagy-targeting ligand (ATL); autophagy-tethering compound (ATTEC); autophagy-targeting chimera (AUTOTAC); cation-independent mannose-6-phosphate receptor (CI-M6PR); chemical inducers of degradation (CIDE); cytokine receptor-targeting chimeras (KineTAC); C–X–C motif chemokine ligand 12 (CXCL12); C–X–C chemokine receptor type 7 (CXCR7); dendronized DNA chimera (DENTAC); integrin-facilitated lysosomal degradation (IFLD); lysosome-targeting chimera (LYTAC); mannose-6-phosphate (M6P); molecular degraders of extracellular proteins through the ASGPR (MoDE-A); a protein-of-interest (POI); proteolysis-targeting chimera (PROTAC); scavenger receptor (SR); target-binding ligand (TBL); triantenerray N-acetylgalactosamine (tri-GalNAc); ubiquitin-independent degrader (UID). | ||
2. Targeted degradation of intracellular proteins and organelles through the autophagy machinery
2.1. AuTophagy-TEthering compounds (ATTECs)
One of the initial Ub-independent TPD methodologies was designed to utilise LC3 ligands that directly interact with developing phagophores in the early stage of autophagy (Fig. 2(A)). As only a limited number of LC3 ligands were available, compared to the wide array of existing ligands for E3 enzymes in PROTACs, Li et al. screened chemical libraries in a high-throughput setting to identify compounds that could bind to LC3 and pathologic huntingtin (HTT) proteins simultaneously.49 The length of polyglutamine (polyQ) in HTT is known to be proportional to its aggregation propensity in Huntington's disease (HD).57 To selectively target the mutant HTT (mHTT) with long polyQ repeats such as HTT with 72 polyQ (HTT-Q72), they conducted a negative selection process to remove hit compounds interacting with non-pathological HTT-Q25. Interestingly, mHTT has been known to evade the UPS-mediated degradation, being preferentially cleared by autophagy,58–60 consistent with the prevailing notions about proteasomal degradation: (1) biased sequences, such as lengthy polyQ repeats, can impede proteasomal degradation of mHTT and (2) efficient degradation by the 26S proteasome necessitates a structurally disordered region in the substrate.61–63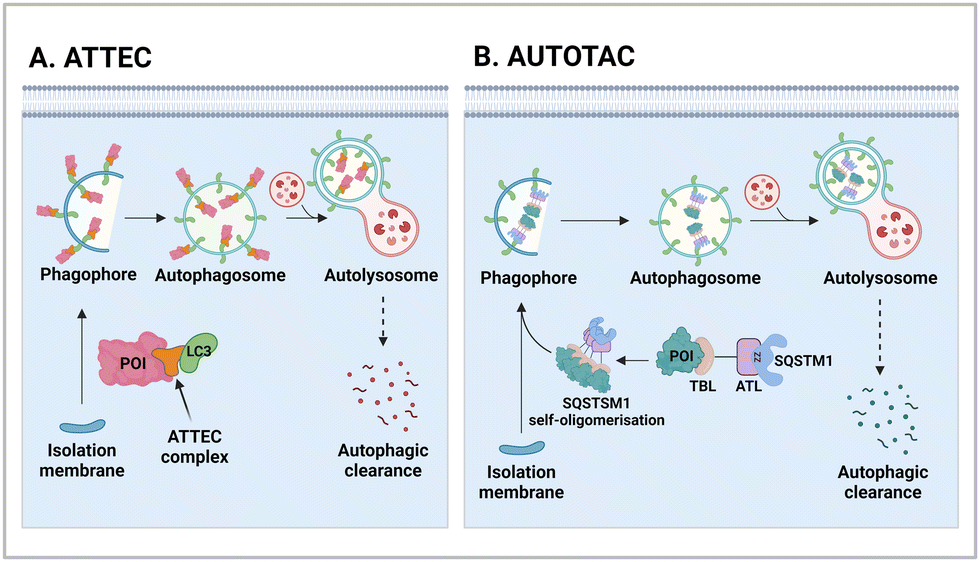 | ||
| Fig. 2 The mechanism of actions of autophagy-based degraders, such as autophagy-tethering compound (ATTEC; A) and autophagy-targeting chimera (AUTOTAC; B). While ATTECs engage with LC3, which is essential for autophagosome biogenesis and maturation, AUTOTACs interact with an autophagic receptor SQSTM1/p62, which undergoes self-oligomerization with cargoes to be directed to autophagosomes. ZZ; the ZZ domain in SQSTM1, which directly interacts with the AUTOTAC compounds. | ||
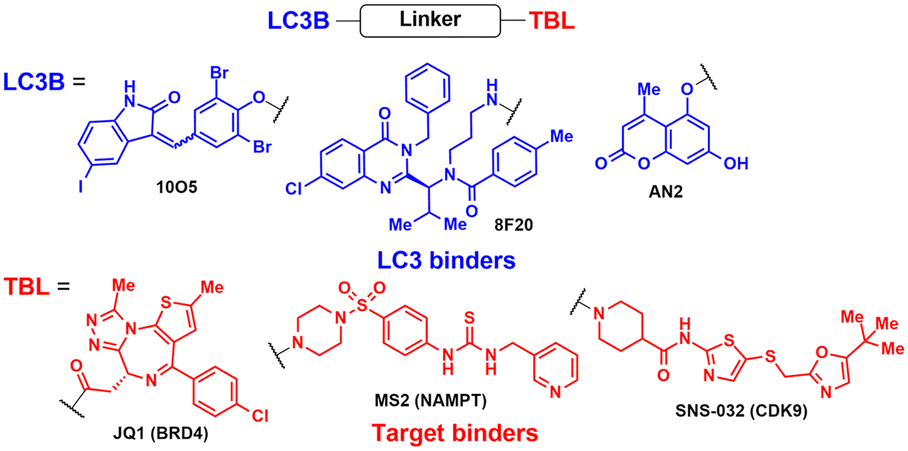
The lead ATTEC molecules, capable of binding to both LC3 and HTT-Q72, but not to HTT-Q25, possessed a lactam-based bicyclic structure with a halogen-substituted aryl group as a common structural motif (Table 1). Treatment with 100 nM of each ATTEC compound for 48 h potently decreased the levels of mHTT by 26–40% of maximal protein degradation (DMax) in primary cortical neurons isolated from a knock-in mouse expressing mHTT-Q140 (ESI,† Table S1; for quantitative values in experimental conditions and assay read-outs).49 In contrast, these compounds did not affect the levels of wild-type (wt) HTT-Q7. The analyses of global proteome in cultured cells and mice using mass spectrometry (MS) indicated that the levels of endogenous autophagic substrates, other than mHTT, remained largely comparable before and after the exposure of cells to ATTECs, demonstrating that the overall autophagic flux remained virtually unaffected (ESI,† Table S2).49
| TPD technology | Characteristics of degraders | Structure | Molecular weight | Target substrates | Ref. | |
|---|---|---|---|---|---|---|
| Chemicals or biologics | Target recognition by | |||||
| ATTEC | Chemicals | Ligands |
|
256–521 Da | mHTT, mutant ATXN3, polyQ-GFP | Z. Li (2019)49 |
|
641–1150 Da | BRD4, NAMPT, CDK9/cyclin T1 | J. Pei (2021),72 G. Dong (2022),73 and Y. Zeng (2023)74 | |||
|
745–1040 Da | Lipid droplets | Y. Fu (2021)67 | |||
|
683 Da (post-click) | Mito-chondria | M. Liu (2023)69 | |||
| TPD technology | Characteristics of degraders | Structure | Molecular weight | Target substrates | Ref. | |
|---|---|---|---|---|---|---|
| Chemicals or biologics | Target recognition by | |||||
| AUTOTAC | Chemicals | Ligands |
|
783–945 Da | ERβ, AR, MetAP2 | C. H. Ji (2022)51 |
|
625–817 Da | Aggregation prone proteins (desmin, tau, HTT, α-synuclein) | C. H. Ji (2022)51 and J. Lee (2023)82 | |||
| KineTAC | Biologics | Antibodies |

|
∼140 kDa | VEGF, TNF-α, PD-L1, HER2, EGFR, PD-1, CDCP1, TROP2 | K. Pance (2023)52 |
| TPD technology | Characteristics of degraders | Structure | Molecular weight | Target substrates | Ref. | |
|---|---|---|---|---|---|---|
| Chemicals or biologics | Target recognition by | |||||
| LYTAC | Biologics | Antibodies |
|
>500 kDa | mCherry, ApoE4, EGFR, CD-71, PD-L1 | S. M. Banik (2020)50 |
| Biologics | Antibodies |
|
∼152 kDa | EGFR, HER2 | X. Zhang (2022)119 | |
| Biologics or chemicals | Antibodies or ligands |
|
>160 kDa | Rabbit IgG, Mouse IgG, EGFR, HER2, integrin | G. Ahn (2021)124 and Y. Zhou (2021)127 | |
| Chemicals | Ligands |
|
1753–1788 Da | α-DNP antibody, MIF | D. F. Caianiello (2021)126 | |
| Biologics | Oligonucleotides |
|
∼33–47 kDa | Met, PTK-7, HER2 | Y. Miao (2021)129 and K. Hamada (2023)130 | |
| Biologics | Oligonucleotides |
|
∼17–21 kDa | PDGF, PTK-7 | Y. Wu (2023)131 | |
| TPD technology | Characteristics of degraders | Structure | Molecular weight (kDa) | Target substrates | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chemicals or biologics | Target recognition by | |||||||||||
| IFLD | Chemicals | Ligands |
|
1290 Da | mCherry, ApoE4, PD-L1 | J. Zheng (2022)53 | ||||||
| DENTAC | Chemicals | Oligonucleotides |
|
∼22–180 kDa | nucleolin, EGFR | C. Zhu (2023)54 | ||||||
| CIDE | Chemicals | Ligands |
|
2720 Da | BRD4 | C. Bashore (2022)55 | ||||||
| UID | Chemicals | Ligands |
|
838–926 Da | BRD2 | M. Balzarini (2023)56 | ||||||
Despite their bispecific binding properties, the sizes of these ATTECs were relatively small, being only 256–521 Da, similar to those of MGDs rather than PROTACs. Nevertheless, unlike MGDs, the majority of ATTECs displayed a “hook effect”, characterized by a diminishing effectiveness of a functional molecule beyond a certain dosage level. This phenomenon is frequently manifested in heterobivalent TPDs due to the formation of stable binary structures with either the target protein or E3 ligase at the oversaturated concentration range.49 At their effective concentrations, they significantly facilitated intracellular binding between mHTT and LC3. Moreover, ATTEC-dependent mHTT degradation was reversed either by lysosomal inhibitors (NH4Cl or chloroquine) or via ATG5 silencing (essential for autophagosome formation), suggesting that the autophagy-tethering effect of ATTEC was responsible for the targeted degradation of mHTT. Nevertheless, a deeper understanding is required to clarify the mechanism whereby these small molecules differentiate between wt HTT and mHTT. It is also notable that pathologic mHTT typically accumulates and aggregates in the nucleus,64,65 while autophagy occurs only in the cytoplasm.
ATTECs were also tested in fibroblasts and iPSC-derived neurons from patients with HD and healthy individuals. While the wt HTT levels in fibroblasts from healthy groups were not affected, a significant reduction in mHTT levels was apparent in HD fibroblasts.49 Additionally, the iPSC-derived neurons showed reduced mHTT levels when they were treated with the ATTEC compounds. The potential therapeutic effects of ATTECs have also been investigated in several in vivo models. In the inducible Drosophila model overexpressing mHTT-Q128, the flies treated with 10 μM ATTECs for six days showed significantly reduced mHTT levels and neurotoxicity as well as an extended lifespan.49 In the HdhQ7/Q140 mouse model, the mice injected daily with ATTECs for two weeks (intraperitoneal or ip, 0.5 mg kg−1 day−1) exhibited significant improvements in HD-related behavioural deficits.26 Both wt flies and mice displayed no apparent impacts in response to the ATTECs, which implicates the minimal off-target effects and the potential of this autophagy-mediated technology for the TPD of other aggregation-prone proteins.
The ATTEC platform has several similarities to AUtophagy-TArgeting Chimeras (AUTACs), both designed to mediate the autophagic degradation of target proteins. Arimoto et al. exploited a derivative of S-guanine, p-fluorobenzylguanine, as a stand-alone degradation tag inducing Lys63-linked polyUb chains on targets.66 AUTAC compounds linking this degradation tag and a substrate-targeting ligand could induce autophagic degradation of various proteins and mitochondria. While the degradation potential of the AUTAC platform has been demonstrated, the precise molecular mechanism underlying the action of S-guanine or its chemical analogue in generating Lys63-linked polyUb chains remains to be further elucidated.
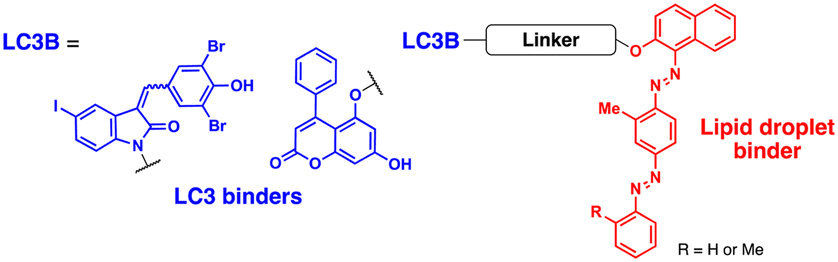
Fu et al. attempted to expand the scope of the ATTEC technology; they developed heterobivalent ATTECs comprising LC3- and lipid droplet (LD)-binders via linkers with varying sizes and types (molecular weights ranging from 745 to 1040 Da; Table 2).67 These LD-ATTECs effectively eliminated LDs in oleic acid-treated tumour cells and cultured adipocytes (at a concentration of 5 μM for 24 h) as well as in the liver of mouse models of hepatic lipidosis (ip injection, 30 mg kg−1 day−1, once a day for 2 weeks) in an autophagy-dependent manner.67 These results are one of the first demonstrations that non-proteinaceous biomolecules can be selectively cleared by Ub-independent TPD. The LD-ATTECs were also evaluated in mouse models of age-related macular degeneration (AMD),68 where a key pathological condition is the presence of drusen, the LD-containing deposits, in the retinal pigment epithelium. Intravitreal administration of LD-ATTEC in AMD model mice, once a week for 4 weeks, significantly reduced LD levels and delayed cytotoxicity and photoreceptor dysfunction in the retinal tissues.68 The LD-ATTEC study highlights the distinct advantage of autophagic degraders, particularly when compared to PROTACs, i.e., autophagic degraders can degrade not only soluble proteins but also aggregation-prone proteins and non-proteinaceous entities.
| TPD technology | Degradation mechanism | Target proteins | Ref. | |||
|---|---|---|---|---|---|---|
| Degradation initially mediated by | Hijacking cellular mechanisms | Targets degraded by | Target protein types | Substrates tested | ||
| ATTEC | LC3 binding ligands | Macroautophagy | Autophagy | Intracellular | mHTT, mutant ATXN3, polyQ-GFP, BRD4, NAMPT, CDK9/cyclin T1 | Z. Li (2019),49 J. Pei (2021),72 G. Dong (2022),73 and Y. Zeng (2023)74 |
| Intracellular organelle | Lipid droplets | Y. Fu (2021)67 | ||||
| Intracellular organelle | Mitochondria | M. Liu (2023)69 | ||||
| AUTOTAC | ZZ domain of SQSTM1/p62-binding ligands | SQSTM1/p62 oligomerization by ATL | Autophagy | Intracellular | ERβ, AR, MetAP2, aggregation-prone proteins (desmin, tau, HTT, α-synuclein) | C. H. Ji (2022),51 Y. Lee (2023),90 and J. Lee (2023)82 |
| Intracellular pathogen | Intracellular bacteria | |||||
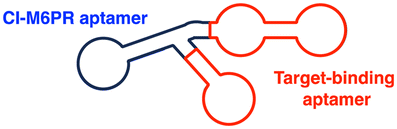
| LYTAC | M6Pn | M6Pn: CI-M6PR-mediated endocytosis | Lysosome | Extracellular | mCherry, ApoE4 | S. M. Banik (2020)50 and X. Zhang (2022)119 |
| Membrane | EGFR, HER2 CD-71, PD-L1 | |||||
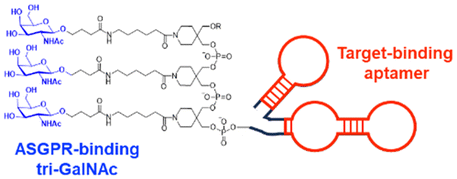
| tri-GalNAc | tri-GalNAc: ASGPR-mediated endocytosis | Extracellular | Mouse IgG, Rabbit IgG, α-DNP antibody, MIF | G. Ahn (2021),124 D. F. Caianiello (2021),126 and Y. Zhou (2021)127 | ||
| Membrane | EGFR, HER2, Integrin | |||||
| CI-M6PR binding aptamers | Aptamer: CI-M6PR-mediated endocytosis | Membrane | Met, PTK-7, HER2 | Y. Miao (2021)129 and K. Hamada (2023)130 | ||
| ASGPR binding aptamers | Aptamer: ASGPR-mediated endocytosis | Extracellular | PDGF | Y. Wu (2023)131 | ||
| Membrane | PTK-7 | |||||
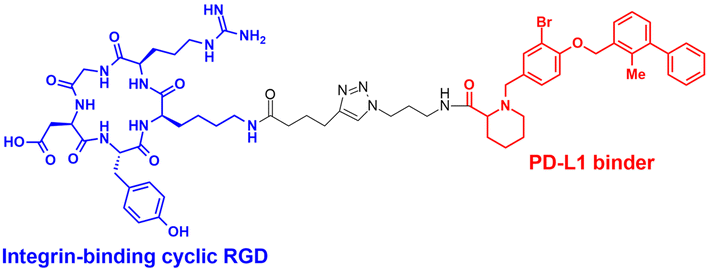
| IFLD | Integrin binding cyclic RGD peptides | RGD peptide: integrin-mediated endocytosis | Lysosome | Extracellular | mCherry, ApoE4, PD-L1 | J. Zheng (2022)53 |
| Membrane | PD-L1 | |||||
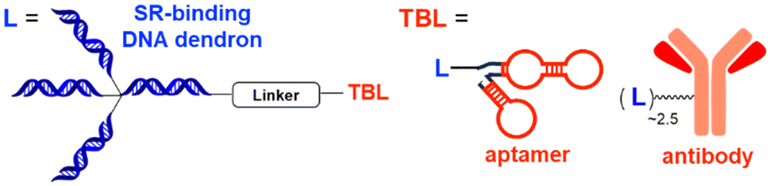
| DENTAC | Scavenger receptor binding DNA dendron | DNA dendron: SR-mediated endocytosis | Lysosome | Membrane | nucleolin, EGFR | C. Zhu (2023)54 |
| KineTAC | CXCL12 | CXCL12: CXCR7-mediated endocytosis | Lysosome | Extracellular | VEGF, TNF-α | K. Pance (2023)52 |
| Membrane | PD-L1, HER2, EGFR, PD-1, CDCP1, TROP2 | |||||
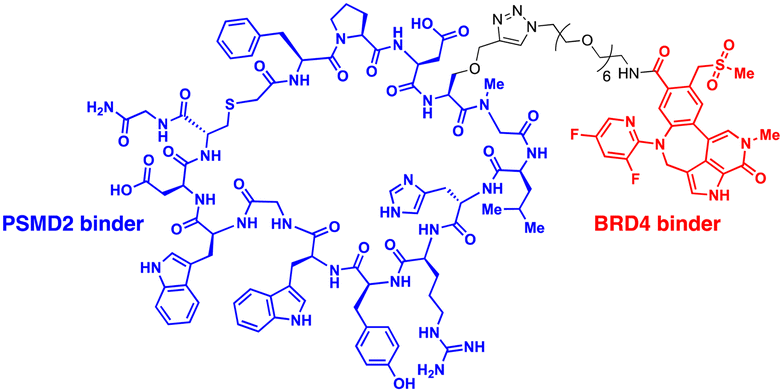
| CIDE | PSMD2 binding macrocycles | Proximity to AAA+ unfoldases | Proteasome | Intracellular | BRD4 | C. Bashore (2022)55 |
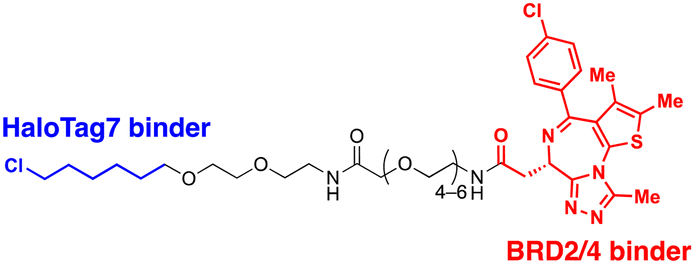
| UID | ADRM1-Halotag7 binding ligand | Proximity to 26S proteasomes | Proteasome | Intracellular | BRD2 | M. Balzarini (2023)56 |
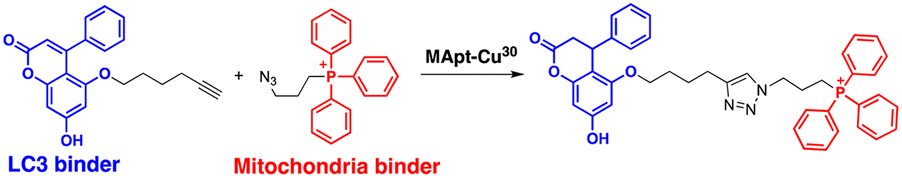
The range of potential ATTEC substrates has been broadened to include cellular organelles. The Qin group has developed mitochondria-targeting ATTECs, known as bio-ATTEC and mito-ATTEC, which combines an LC3 binder and the established mitochondrion-targeting motif, triphenylphosphonium cation.69,70 To minimise potential off-target effects, they utilised split ATTEC fragments with complementary click chemistry handles (azide and alkyne), allowing selective activation within the target cells. In bio-ATTECs, tissue specificity was achieved by employing cancer cell-specific surface antigen-binding aptamers. Specific aptamers, which are short single-stranded DNA or RNA nucleotides that fold into specific three-dimensional structures, are considered an effective alternative to antibodies and can be readily selected using an in vitro screening technique, called systematic evolution of ligands via exponential enrichment (SELEX).71 The authors observed mitochondrial depletion and autophagic tumour cell death after treatment with bio-ATTEC.69 It would be intriguing to find out which specific mitochondrial proteins are initially engaged with the bio-ATTEC degrader. In addition, more warheads and linkers should be assessed for further validation and generalisation of this modality.
Following the discovery of potent LC3 binders, numerous efforts have been made to harness autophagy for targeted intracellular protein degradation. Recent studies have reported several ATTECs targeting bromodomain-containing protein 4 (BRD4),72 nicotinamide phosphoribosyltransferase (NAMPT),73 and the cyclin-dependent kinase 9 (CDK9)/cyclin T1 complex.74 The optimised BRD4-ATTEC exhibited impressive degradation activity (DMax 92%, DC50 0.9 μM) in MDA-MB-231 breast cancer cells, which have the highest level of PD-L1, without apparent disturbance in cellular autophagy flux. The significantly reduced c-Myc expression, a key indicator of BRD4 inhibition, and potent anti-proliferative effects on various cancer cell lines following treatment with BRD4-ATTECs indicate that the LC3-engaging degraders could be a promising strategy for developing anti-cancer agents targeting BRD4. Zeng et al. made a noteworthy observation that CDK9-targeting ATTECs exhibit superior degradation and anti-proliferative activities compared to the corresponding CDK9-PROTACs, which share the same CDK9 binder.74,75 In addition, these ATTECs not only induced CDK9 degradation but also led to the degradation of CDK9-associated cyclin T1, which was not observed with the corresponding PROTAC. This collateral degradation implicates not only distinct mechanisms but also potentially different outcomes between these two approaches, although further comparative studies are necessary to fully grasp the respective strengths and limitations of each method. Nonetheless, the aforementioned results convincingly demonstrated the unique capability of the ATTEC platform to target a wide range of cellular components in a tissue-specific and autophagy-dependent manner.
2.2. AUTOphagy-TArgeting chimera (AUTOTAC)
Proteotoxic proteins, often found to be misfolded and aggregated, are known to disrupt normal cellular metabolism and, potentially, lead to cell death.76 The abnormal accumulation of proteotoxic proteins can arise from (1) self-oligomerisation acquired from mutations or PTMs (a protein-centric view), (2) compromised clearance due to dysfunctional UPS or autophagy (a proteolysis-centric view), or (3) a combination of both (a synergistic mechanism).77–79 Numerous neurodegenerative diseases, including HD, Alzheimer's (AD), and Parkinson's (PD) disease, feature protein aggregates as one of their key pathogenic hallmarks. Once considered undruggable, these proteins have gained considerable attention as targets of TPD drugs. Several studies have proposed that while the UPS is responsible for clearing the soluble forms of proteotoxic proteins, particularly during the early stage of neurodegeneration, autophagy-mediated protein degradation becomes critical as proteotoxic proteins begin to get fibrillised in neurons.80 To pharmacologically modulate this process, Ji et al. developed innovative autophagic degraders that exploit key components of the N-degron pathway (Fig. 2(B)).81,82In the N-degron pathway, formerly known as the “N-end rule” pathway, the in vivo half-life of a protein is primarily determined by its N-terminal amino acid.83,84 In the initial N-end rule, each amino acid within the genetic code was categorised into either “stabilizing” (including the start codon Met) or “destabilizing” (including positively charged Arg) residues for proteasomal degradation; however, it is now understood that all 20 amino acids can serve as N-degrons.83 One of the key biochemical processes that generates an N-degron substrate is the conjugation of Arg to the N-terminus of a protein.85 A set of ER-residing chaperons was reported to be N-terminally arginylated and retro-translocated to the cytosol, where the chaperons were bound to their new clients—cytosolic misfolded proteins. These complexes were then recognised by the autophagic adaptor SQSTM1 through its ZZ domain.86,87 This process involves the conformational change and self-oligomerisation of SQSTM1 for the delivery of cargoes (N-degrons) to autophagosomes.88,89 Therefore, N-terminal Arg functions not only as a binding site for E3 Ub ligases for ubiquitination but also as an activating ligand for SQSTM1 for activating autophagy.89–91 In the same context, SQSTM1 also has a dual role: as a prototype autophagic cargo adaptor and a novel N-recognin of destabilizing N-terminal residues.
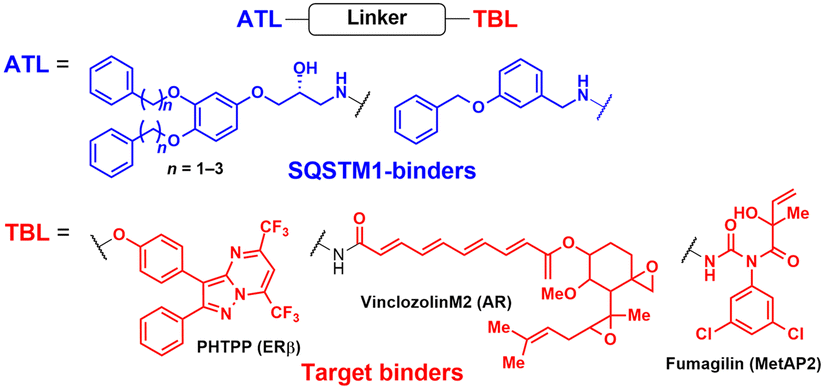
AUTOTAC degraders are bispecific molecules linking a target-binding ligand (TBL) and an autophagy-targeting ligand that binds to the ZZ domain of SQSTM1.51 Using 3D structural modelling and structure–activity relationship studies, Cha-Molstad et al. developed Arg-based SQSTM1 ligands featuring O-benzylated phenol or catechol moieties (Table 1).87 These compounds induced conformational activation of SQSTM1, transforming it into an autophagy-compatible oligomer. Therefore, these ZZ ligands essentially mimic the biochemical action of the “destabilizing” residues in the autophagic N-degron pathway. AUTOTAC degraders were then synthesized by conjugating the ZZ ligands to diverse TBLs, such as PHTPP (a nonsteroidal antagonist of the oestrogen receptor β [ERβ]), vinclozolin M2 (an androgen receptor [AR] blocker), and fumagillin (a ligand for methionine aminopeptidase 2 [MetAP2]) (Table 2).92–94 The effectiveness of these AUTOTAC compounds was demonstrated by achieving significant autophagic degradation of the cognate oncoproteins, such as ERβ, AR, and MetAP2, within the sub-micromolar ranges of half-degrading concentration values (DC50).43 Importantly, these activities were completely abolished by silencing either SQSTM1 or ATG5.51
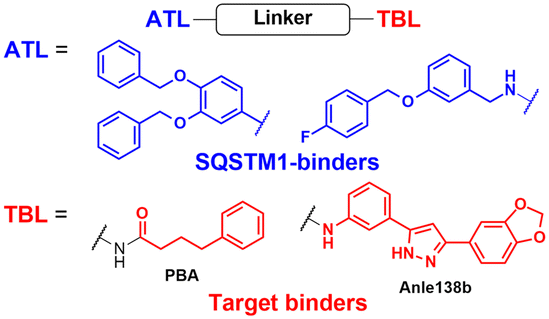
To assess whether these AUTOTAC degraders can degrade aggregated proteins, additional compounds were synthesized to incorporate TBLs that recognise exposed hydrophobic motifs, a common structural property of misfolded proteins. The following TBLs were conjugated to the ZZ ligands: (1) 4-phenylbutyric acid (PBA), an FDA-approved drug and a chemical chaperone that binds to hydrophobic residues, and (2) Anle138b, an inhibitor of α-synuclein aggregation.95–97 In these studies, the PBA and Anle138b moiety-conjugated AUTOTAC compounds (PBA-1105 and ATC161/Anle138b-F105) were effective in reducing the levels of mutant desmin, aggregation-prone tau-P301L, and mHTT, respectively.98–100 Remarkably, they did not affect the levels of non-pathologic counterparts such as wt desmin, tau, and HTT. The DC50 values for the mutant tau and HTT proteins ranged from 1–10 nM and 0.1–1 μM, respectively, and a hook effect was observed at high concentrations (ESI,† Table S3).43 For the in vivo evaluation, ZZ-PBA compounds were administered (ip injection, 20 or 50 mg kg−1, 3 times per week for 1 month) to transgenic mice overexpressing hTauP301L tagged with biomolecular fluorescence complementation (BiFC).101,102 Immunoblotting, immunostaining, and histochemical analyses showed reduced levels of RIPA-insoluble human hTauP301L and a decrease in BiFC fluorescence signal in the mouse brains.
The AUTOTAC platform was utilised to facilitate the autophagic clearance of intracellular bacteria and α-synuclein.82,90 As a part of their defence mechanisms, eukaryotic cells often rely on xenophagy, a type of selective autophagy, to capture intracellular pathogens, such as bacteria, protozoans, and viruses.103,104 However, these intracellular pathogens have also evolved to evade or even inhibit cellular xenophagy to survive within their host cells.105,106 The Kwon group discovered that treatment with SQSTM1-targeting ZZ ligands lacking TBL moieties was sufficient to induce the degradation of intracellular bacteria in cells and mice infected with Salmonella enterica and Mycobacterium tuberculosis.90 Recently, Lee et al. introduced new TBLs in the AUTOTAC toolkit, including baicalein (ATC162), a flavonoid compound that binds to α-synuclein, and resveratrol (ATC163), a phenol compound that binds to α-synuclein.82 In the PD mouse model (oral administration, 10 mg kg−1, 5 times per week, for 16 weeks), ATC161/Anle138b-F105 treatment reduced the number of α-synuclein aggregates in the brain, resulting in a significant delay in motor neuron degeneration. Similar to tau and α-synuclein, many target proteins tested with AUTOTAC degraders were either transiently or constitutively overexpressed. While it is a common practice to study proteotoxicity through transiently overexpressed proteins, a thorough evaluation of the overall efficacy for endogenous substrate proteins is crucial for a more accurate assessment of the therapeutic potential of the AUTOTAC technology. Furthermore, it also remains to be determined whether the AUTOTAC-induced oligomeric SQSTM1 captures off-target soluble proteins (potentially through phase separation)107 and organellar membrane proteins (via direct phagophore binding)108 at the proteomic level.
3. Targeted degradation of secreted and membrane proteins through the endolysosomal pathway
3.1. LYsosome-TArgeting chimeras (LYTACs)
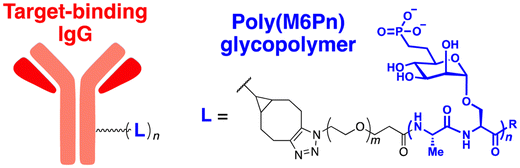
The initial non-Ub TPD degraders, including ATTECs and AUTOTACs, were designed for degrading intracellular proteins, although over one-third of protein-encoding genes produce secreted and membrane proteins.37,109 First developed by the Bertozzi's group, the LYTAC technology mediates the lysosomal degradation of these proteins.50 The original LYTAC molecules consisted of a substrate targeting antibody and a glycopolypeptide ligand for binding to the cation-independent mannose-6-phosphate (M6P) receptor (CI-M6PR; Fig. 3(A)). CI-M6PR is a plasma membrane-residing and endosome-targeting receptor that recognises N-glycans capped with M6P moieties.110 In addition, it functions as a decoy receptor, continuously shuttling M6P-modified cargoes from the plasma membrane to the endosomal compartments.111–114 In late endosomes and lysosomes, the low pH induces the dissociation of the endocytosed CI-M6PR and LYTAC-target complex and facilitates receptor recycling, while the target proteins are degraded in the lysosome.50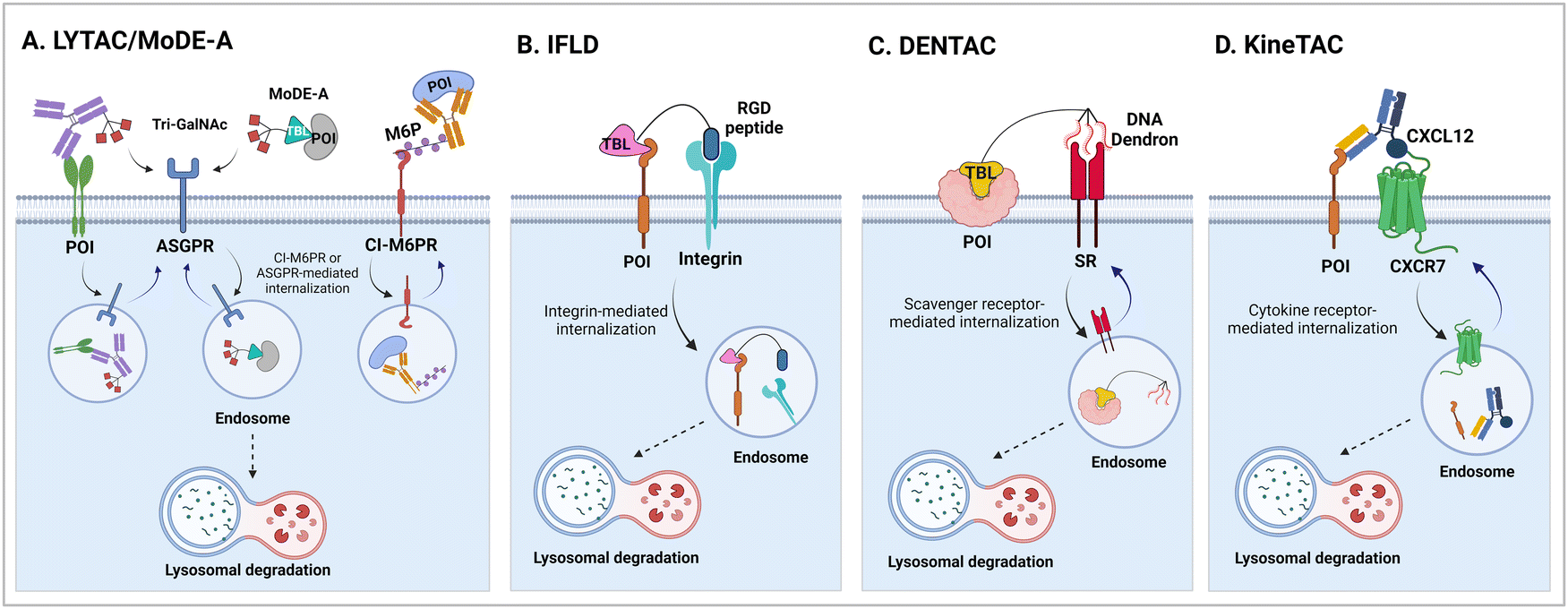 | ||
| Fig. 3 Lysosome-based degraders which utilise ligand-triggered receptor endocytosis, such as lysosome-targeting chimera (LYTAC; A), molecular degraders of extracellular proteins through the ASGPR (MoDE-A; A), integrin-facilitated lysosomal degradation (IFLD; B), dendronized DNA chimera (DENTAC; C), and cytokine receptor-targeting chimeras (KineTAC; D). Other similar platforms based on bifunctional antibodies, including TransTAC and EndoTag, have recently reported. | ||
To generate glycopolypeptides that are not only recognised by CI-M6PR but are also resistant to cellular phosphatase activity, Banik et al. synthesised N-carboxyanhydride (NCA)-derived Ser-Ala polypeptides (with various lengths) containing multiple serine-O-mannose-6-phosphonate moieties (M6Pn) (Table 1). The oxygen at the 6-position of mannose was replaced by the methylene linkage (CH2) to improve their metabolic stability.50 These M6Pn glycopolypeptides, unlike those containing mannose or N-acetylgalactosamine (GalNAc), successfully promoted the CI-M6PR-mediated uptake of cargo molecules by acidic endosomes and lysosomes in several cell lines.50 Using CRISPR/Cas9-knockout screening, it was found that IGF2R (insulin like growth factor 2 receptor, an alternative gene name for CI-M6PR) and several genes involved in protein neddylation and activation of the E3 ligase cullin-3 (CUL3) are essential for the cargo uptake and LYTAC recycling.115 These results provided not only an unbiased validation of the underlying molecular mechanism of LYTACs but also a potential indicator (e.g., neddylation of CUL3) to predict whether and to what extent LYTAC therapy will be effective in vivo.
To further validate this technology, the cellular uptake and degradation of apolipoprotein E4 (ApoE4), an extracellular protein that represents the most common genetic risk factor for AD, by a LYTAC molecule was tested (Table 2).116 First, a compound consisting of M6Pn-bearing glycopolypeptides conjugated to a goat anti-mouse IgG antibody (Ab-1) was developed. The conjugation involved random introduction of azide moieties onto multiple IgG lysine residues, followed by a copper-free, strain-promoted cycloaddition reaction with the bicyclononyne-bearing M6Pn ligand. Next, K562 lymphoblast cells were treated with 25 nM Ab-1 in the presence of both recombinant ApoE4 proteins and a mouse anti-ApoE4 antibody labelled with Alexa Fluor-647. The uptake of ApoE4 was increased by 13-fold following Ab-1 treatment. When leupeptin, an endolysosomal protease inhibitor, was co-treated with Ab-1 in the presence of mouse anti-ApoE4 antibodies, the intracellular levels of ApoE4 significantly increased, demonstrating the LYTAC-mediated lysosomal degradation of extracellular proteins. The LYTAC technology has also been utilised to directly target the epidermal growth factor receptor (EGFR) by conjugating the M6Pn glycopolypeptides to cetuximab, an EGFR-targeting antibody (Ab-2). Treatment with 10 nM Ab-2 for 24–48 h resulted in effective degradation of EGFR on the surface of breast and liver cancer cells, reaching a 61–83% DMax (ESI,† Table S4).50 Quantitative proteomic analysis demonstrated that this LYTAC-mediated EGFR degradation was more specific than the cetuximab treatment alone (ESI,† Table S2).
A similar LYTAC-mediated degradation was observed by targeting other membrane-bound proteins, such as transferrin receptor 1 (CD71) and programmed death-ligand 1 (PD-L1), using Ab-1 LYTAC (with a polyclonal anti-mouse IgG) co-treated with mouse anti-CD71 antibodies in EGFR-negative cells and Ab-3 LYTAC (with a monoclonal anti-PD-L1 antibody, atezolizumab) in PD-L1-positive cells, respectively.117,118 As observed previously,50 treatment with antibodies alone without glycopolypeptides did not have a significant effect on the target protein levels. Moreover, degradation was abolished when cells were treated with competing M6P or lysosome inhibitors, further confirming the mechanism of LYTAC action. Mice injected with the M6Pn-cetuximab conjugate (single injection, 5 mg kg−1) exhibited a two-phase (first rapid and then sustained) clearance of the LYTAC molecule.50 Since EGFR, CD71, and PD-L1 are all promising targets in cancer therapy, the LYTAC modality may represent a complementary or synergistic platform to conventional chemotherapy. However, the poor metabolic stability of these M6Pn-conjugated antibodies in vivo still needs to be improved to enhance their possible therapeutic benefits as anti-tumour agents.
Given the relatively long half-lives (>3 days) of cetuximab and atezolizumab, the rapid clearance of LYTACs in the circulatory system might result from the heteropolymorphic nature of the M6Pn glycopolypeptide ligand. The lack of site-specific conjugation of M6Pn to antibodies is also expected to contribute to this pharmacokinetic characteristic. More recently, Zhang et al. developed a chemoenzymatic method to prepare homogeneous CI-M6PR-targeting LYTACs.119 Based on their prior observation that simple glycans may possess high affinity for CI-M6PR with mannose-6-phosphate-α1,2-mannose disaccharide moieties, antibodies were modified with minimal M6P glycans (tri- or tetra-saccharides) instead of glycopolypeptides. The site-specific introduction of the M6P motif to the antibody was accomplished by using engineered endoglycosidases that function as a transglycosylation catalyst. This approach enabled the generation of homogeneous LYTACs that targeted EGFR and HER2, with their in vitro proteolytic activities being comparable to those achieved using polymeric M6Pn ligands. Nevertheless, whether the homogeneous structure of LYTAC antibodies improves their pharmacokinetic profiles in vivo remains to be determined.
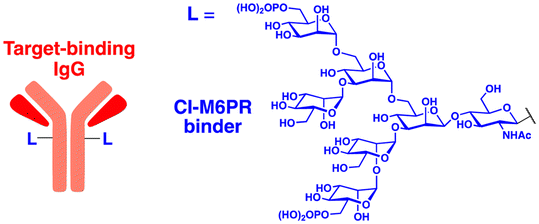
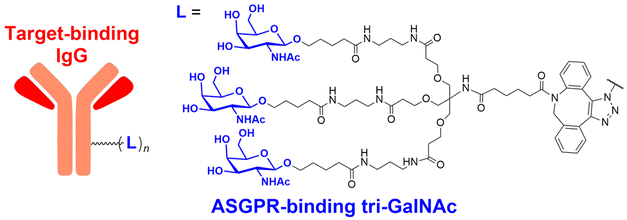
In their second LYTAC study, Bertozzi and co-workers used the asialoglycoprotein receptor (ASGPR), which is another endosome-targeting receptor exclusively expressed in the liver, as opposed to the ubiquitously expressed CI-M6PR.120,121 ASGPRs can recognise the galactose or GalNAc moieties on extracellular proteins and internalize them via clathrin-mediated endocytosis for lysosomal degradation.122 Specifically, a triantennary GalNAc (tri-GalNAc: 1.7 kDa) motif was exploited as an ASGPR-targeting ligand for LYTAC development, as it has been widely used as a vehicle for the targeted delivery of oligonucleotides into hepatocytes via ASGPRs.123 For proof of concept, tri-GalNAc was conjugated with a targeting ligand such as a synthetic polyspecific integrin-binding peptide (PIP: 3.4 kDa) as an alternative to antibodies (Fig. 3(A) and Table 1).124 The treatment of HEPG2 cells with tri-GalNAc-PIP resulted in a significantly facilitated degradation of cell-surface integrins and delayed cell proliferation. Similarly, the tri-GalNAc-cetuximab conjugate selectively degraded cell-surface EGFRs in HEP3B, HEPG2, and HUH7 cells (DC50 ≈ 1 nM), but not in non-hepatocellular carcinoma cell lines (ESI,† Table S4). Tri-GalNAc-pertuzumab conjugates also degraded ∼75% of total HER2 in HEPG2 cells, while pertuzumab alone reduced the HER2 levels by only ∼30%. Notably, these conjugates featured a single tri-GalNAc ligand, which was attached at specific locations utilising the SMARTag technology.125In vitro degradation assays revealed that the activity of the HER2-targeting LYTAC with a hinge modification was comparable to that of the non-specific counterpart, having more than 10 equivalents of the tri-GalNAc ligands. When homogeneous EGFR-targeting LYTACs were administered to mice for a week (ip injection, 5 mg kg−1, every 2 or 4 days), site-specific LYTAC conjugates showed sustained in vivo stability; by contrast, non-specific heterogeneous conjugates underwent rapid clearance. This observation may exemplify the significance of homogeneous antibody modifications in elevating metabolic stability in vivo.
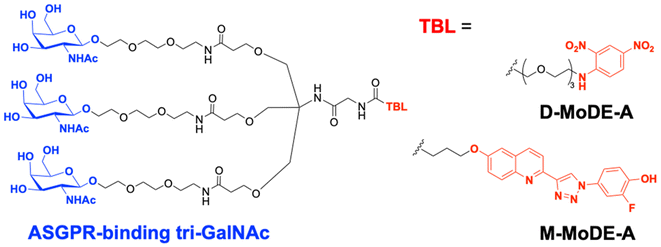
A key advantage of employing ASGPRs over CI-M6PR lies in their structural uniformity of the receptor-targeting motif, tri-N-GalNAc, and structural flexibility, which allows further functional optimisation. Almost simultaneously with Bertozzi's report on ASGPR-based LYTACs,124 Spiegel's and Tang's groups independently reported their TPD strategies, which also capitalized on the ASGPR and tri-GalNAc interactions (Table 1).126,127 Tang's design was based on the conjugation of tri-GalNAc to antibodies or antigen-binding fragments (Fabs), akin to Bertozzi's concept, whereas Spiegel et al. pursued a fully synthetic molecular approach akin to the tri-GalNAc-PIP concept, resulting in MoDE-A (MOlecular DEgraders of extracellular proteins through ASGPRs).126 They connected the ASGPR-binding tri-GalNAc motif to either a dinitrophenyl (DNP) group or the inhibitor of the cytokine macrophage migration inhibitory factor (MIF), which resulted in the formation of two distinct compounds—D-MoDE-A and M-MoDE-A (Fig. 3(A)). These derivatives effectively formed corresponding ternary complexes with ASGPR and their cognate targets, leading to the lysosomal degradation of α-DNP antibodies and MIFs, respectively. Moreover, M-MoDE-A effectively reduced the levels of exogenously administered human MIF in the sera of nude mice (ip injection, 1 mg kg−1 or 10 mg kg−1 M-MODE-A), although the in vivo effects of these modalities on endogenous protein targets still remain to be validated. Moreover, although these LYTACs are likely to be recycled without being co-degraded with their target proteins, the quantitative analysis of the in vivo DC50 and DMax would be informative for attaining a comprehensive understanding of their pharmacological property and therapeutic efficacy.
Aptamers have further broadened the scope of LYTAC-mediated degradation of extracellular and membrane-associated proteins.128 The following studies have been conducted to generate and utilise aptamer-conjugated LYTACs (also known as Apt-LYTACs): (1) bispecific DNA aptamers targeting both CI-M6PR and mesenchymal-epithelial transition receptor (Met) or protein tyrosine kinase-like 7 (PTK7),129 (2) bispecific DNA aptamers targeting CI-M6MR and HER2,130 and (3) tri-GalNAc-conjugated DNA aptamers targeting PTK7 or platelet-derived growth factors (PDGFs) (Table 2).131 Considering the rapid advancements in aptamer/carbohydrate chemistry and antibody engineering, the PIP-LYTAC, MoDE-A, and Apt-LYTAC molecules represent a novel class of efficient lysosome-based TPDs. Nevertheless, a more comprehensive understanding of lysosome homeostasis, especially under diverse disease conditions, appears to be imperative for these LYTAC molecules to achieve improved safety and effectiveness as therapeutics.
3.2. Related modalities: integrin-facilitated lysosomal degradation (IFLD) and DENdronised DNA chimera (DENTAC)
The LYTAC technology operates through ligand-triggered endocytosis and subsequent internalisation of target proteins to the lysosome. This opens up the potential for harnessing various analogous receptor–ligand pairs to drive the lysosomal degradation of extracellular and membrane-associated proteins. Integrins, which are known cell-to-cell adhesion receptors, are often overexpressed on various cancer cells and selectively interact with the Arg–Gly–Asp (RGD) peptides (MW = 619.7 Da). The Li group introduced the IFLD platform, wherein the RGD motif is linked to neo-substrate-targeting small molecules or antibodies (Fig. 3(B)).53 A conjugate composed of cyclic RGD and BMS-8, a small molecule PD-L1 binder, exhibited ∼70% degradation activity at a 25 nM concentration in MDA-MB-231 cells. In an in vivo melanoma xenograft mouse model (iv injection, 5 mg kg−1, every 2 days), these chimeric molecules led to significant tumour regression, accompanied by a notable decrease in PD-L1 levels in tumour tissues, as determined via immunohistochemistry analysis (ESI,† Table S5).Another approach utilised cell-surface scavenger receptors (SRs) that bind to anionic ligands. These receptors have been used to facilitate the intracellular delivery of polyanionic DNA nanomaterials through caveolae-mediated endocytosis.132,133 Zhu et al. developed DENTACs by combining dendritic DNAs with target-binding motifs, either antibodies or aptamers, to mediate the SR-dependent TPD (Fig. 3(C)).54 A specific DENTAC for nucleolin (NCL) was generated by conjugating poly-thymidine DNA dendrons to AS1411, a known NCL-binding DNA aptamer; it exhibited significant NCL degradation activity in A549 lung adenocarcinoma cells (with a DC50 of 25 nM and DMax of 60% at 50 nM). An in vivo study using an A549 mouse xenograft model (ip injection, 2 mg kg−1, every 2 days for 2 weeks) also showed significant regression of tumour size (∼76% reduction in weight) without apparent body weight loss (ESI,† Table S5). Recent reports in bioRxiv have suggested the potential of other naturally recycling receptors to redirect target proteins to lysosomal degradation, such as the transferrin receptor (TransTAC)134 and sortillin (EndoTag).135 While these findings are pending for evaluation, emergence of novel methods will broaden the array of lysosome-trafficking receptor and corresponding ligand pairs for alternative LYTAC modalities.
3.3. CytoKine receptor-TArgeting chimeras (KineTACs)
Specific antibodies can be discovered relatively rapidly using well-established techniques such as phage display or optofluidic systems. Numerous PROTAC degraders have been covalently linked to monoclonal antibodies to enhance their tissue-targeting ability, a concept largely resembling the antibody–drug conjugate system.136 For instance, the conjugation of trastuzumab to a BRD4-targeting PROTAC led to selective BRD4 degradation only in HER2-positive breast cancer cells.137 In the GlueTAC platform, a PD-L1 nanobody was cross-linked to proximity-reactive, non-canonical amino acids, which enabled covalent binding of the nanobody to PD-L1 receptors.138 The GlueTAC nanobodies also retained a cell-penetrating peptide and a lysosome-sorting sequence, facilitating their internalisation and the subsequent lysosomal degradation of PD-L1. Cotton et al. developed the first bispecific antibody-based TPD platform, called Antibody-based PROTAC (AbTAC). The AbTACs are capable of recognizing both a target membrane protein and the membrane-bound E3 Ub ligase RNF43.139 The AbTACs mediated target degradation by inducing polyubiquitination and subsequent trafficking of the cargo to the lysosome. By contrast, a similar bispecific antibody (PROTAB) recruiting zinc and ring finger 3 (ZNRF3), another cell-surface E3 ligase, has been demonstrated to degrade target substrates by 26S proteasomes.140 Recently, the Wells group expanded their TPD platforms from AbTACs to KineTACs (Fig. 3(D)),52 both of which share a common architecture of bispecific antibodies. Unlike AbTACs, which target only membrane proteins due to their reliance on membrane-bound RNF43, KineTACs are capable of triggering the lysosomal degradation of both membrane and extracellular proteins via the endolysosomal pathway, similar to the LYTAC technology.KineTACs achieved lysosome-mediated target degradation through cytokine-mediated endosome-targeting receptors. The knobs-into-holes method141 was adopted to generate recombinant antibodies with two distinct functional components—(1) human C–X–C motif chemokine ligand 12 (CXCL12) fused to the knob crystallizable fragment (Fc), which binds to the non-signalling chemokine receptor CXCR7, and (2) a Fab fused to the hole Fc domain (Table 1). KineTACs, similar to LYTACs, facilitated the lysosomal degradation of various proteins through the constant internalisation of CXCR7 with β-arrestin recruitment. The initial KineTAC bispecific antibody was generated to target PD-L1 by incorporating the Fab fragment of atezolizumab into the KineTAC scaffold. The treatment of different cancer cells with this CXCL12-atezolizumab chimera robustly reduced endogenous PD-L1 levels by ∼90%, which was a modestly higher reduction than that achieved by AbTAC (∼63% of DMax).139 In contrast, the atezolizumab Fab or CXCL12 isotype alone had little effect on PD-L1 levels. In addition to PD-L1, the KineTAC platform was applied to degrade other cell surface proteins, including HER2, EGFR, PD-1, CDCP1, and TROP2, by utilising trastuzumab, cetuximab, nivolumab, 4A06, and sacituzumab, respectively (Table 2).142–144 The landscape of the KineTAC targets has been further expanded to include soluble extracellular proteins, such as vascular endothelial growth factor (VEGF) and tumour necrosis factor α (TNFα). Specifically, chimeric CXCL12-targeting KineTAC antibodies, containing either bevacizumab or adalimumab, effectively induced the intracellular uptake and delivery of extracellular VEGF or TNFα to the lysosome; however, the degree of extracellular protein degradation by KineTACs remains to be assessed.
To evaluate the general applicability of KineTAC-mediated degradation, alternative chemokines (CXCL11 and vMIPII, both targeting the CXCR7 chemokine receptor) were incorporated into their functional repertoire. The results indicated that these variants achieved degradation efficiencies comparable to those of the original CXCL12-based KineTACs. KineTACs utilising the human interleukin-2 (IL-2) cytokine fused to nivolumab were also effective in degrading the cell surface PD-1 proteins (DMax of 86.7%) (ESI,† Table S6), which suggested that this modality might not be restricted to CXCR receptors but rather exhibit broad versatility across various cytokine receptor-mediated internalisation systems. Intravenously administered CXCL12-trastuzumab showed promising pharmacokinetic characteristics in mice, such as prolonged circulation time and reduced cross-reactivity. However, the therapeutic potential of KineTAC antibodies can be validated only when their ability to induce efficient target degradation is demonstrated in vivo. Owing to their relatively simple engineering and production processes, KineTACs offer a compelling technical advantage over functionally analogous LYTACs, which usually involve complex synthesis and conjugation chemistry. Moreover, KineTAC production can avoid concerns such as light chain-heavy chain mispairing, a common issue encountered in bispecific IgG production.141 Considering the flexibility in leveraging various cytokine-receptor pairs as well as the robust in vivo stability, it seems evident that the application of the KineTAC platform can rapidly expand the TPD of therapeutically relevant proteins.
4. TPD by direct recruitment of the 26S proteasome
4.1. Chemical inducers of DEgradation (CIDEs)
The 26S proteasome is the hybrid of an ancient unfolding-proteolysis machinery and a more recent polyUb-recognizing system, which are evolutionarily separated by billions of years. Chambered proteases in bacteria, such as ClpP, demonstrate remarkable structural and mechanistic similarities to the CP complex or 20S proteasome in archaea and eukaryotes.145 Since prokaryotes lack Ub, they employ specific peptide sequences, most notably the E. coli SsrA, which are recognised by the AAA+ ATPase ClpX.146 This AAA+ motor complex, arranged in a homohexameric ring, unfolds the substrates and translocates them through an axial channel into the catalytic chamber of ClpP complexes, which consist of two homoheptameric rings.147 In eukaryotic cells, six AAA+ ATPase subunits (PSMC1 to PSMC6 from six different genes) form a heterohexameric ring on the base of the RP contacting the PSMA heteroheptameric ring of the CP.148 The RP also plays a role in bringing in substrates to the CP using multiple Ub receptors, including a stoichiometric subunit PSMD2.31 It is largely accepted in the field that (1) Ub does not affect the thermodynamic stability of target proteins, (2) it only brings substrates into close proximity with 26S proteasomes, and (3) the direct recognition by the proteasome is usually sufficient for the substrates to be degraded in the CP.149 Under these presumptions, direct tethering of substrates to proteasomes through bifunctional ligands could be a viable strategy to induce TPD, instead of altering the global proteome.Bashore et al. in Genentech pioneered the direct proteasome targeting modality (referred to as CIDE) as an alternative TPD technology (Fig. 4(A)).55 Their initial breakthrough was the identification of ligands that could specifically bind to the Ub receptor PSMD2. By screening both 8- to 16-amino acid-long peptide phage libraries and 10- to 14-residue macrocyclic mRNA display libraries, one peptide (PP1) and three macrocycles (MC1 to MC3) with strong affinities (in a nanomolar range) to a recombinant PSMD2 protein were identified. Biotinylated versions of macrocycle MC1, but not its enantiomeric controls, effectively pulled down 26S proteasomes without compromising their structural integrity. Cryo-EM images with a 2.5 Å-resolution unveiled that MC1 binds to the solenoid domain and C-terminal region of PSMD2, which are proximal to the AAA+ unfoldase or the PSMC APTase ring. To further enhance the versatility of MC1, a click chemistry-based approach was utilised to synthesise various versions of MC1 linked to BETi, a BRD4 ligand/inhibitor,150 using PEG linkers with different lengths and conjugation sites (Table 1). These heterobifunctional MC1-BETi molecules were also able to strongly interact with PSMD2, forming a proper ternary complex with PSMD2 and BRD4 in vitro.
 | ||
| Fig. 4 Degraders directly engaging the 26S proteasome, such as chemical inducers of degradation (CIDE; A) and ubiquitin-independent degrader (UID; B). | ||
A fluorescence microscopy analysis revealed that CIDEs, despite their peptidic nature, can be internalized to the cytoplasm in an endocytosis-independent manner.151,152 In HEK293 cells, the most permeable MC1-BETi derivative (L-CIDE), but not its D-enantioisomer, triggered significant and dose-dependent BRD4 degradation. This phenomenon was effectively abolished by proteasome inhibitors. Although the DC50 of L-CIDE remained relatively high at a concentration of 0.73 μM (ESI,† Table S7), these results provided initial validation for the feasibility of TPD via the direct recruitment of the 26S proteasome. However, it remains uncertain whether the functionality of CIDE depends on or is affected by the presence of the polyUb chains on target substrates. This is implicated in the practicality of CIDE compounds in treating, for example, BETi-resistant cancer, where BRD4 is often poorly polyubiquitinated.153 More CIDE compounds targeting other endogenous proteins besides BRD4 need to be developed and assessed to understand the mechanistic basis of this TPD modality. If CIDE molecules could enable Ub-independent degradation simply by bringing proteins close to 26S proteasomes, these compounds could eliminate a wide range of cytosolic, nuclear, and membrane proteins, rendering CIDEs as more versatile TPD options than PROTACs.
4.2. ADRM1-mediated Ub-independent degraders (UIDs)
Recently, an alternative TPD strategy based on direct engagement of 26S proteasomes without involving polyUb chains was proposed by Kodadek et al.56 The UID system employs ADRM1, another Ub receptor on the 26S proteasome, located approximately 107 Å away from the PSMC1-6 ATPases on the RP base.31 Since a suitable ligand for ADRM1 did not exist, a cell line stably overexpressing the N-terminal segment of ADRM1 fused to HaloTag7 was used as a model system and a primary chloroalkane chain conjugated to a substrate-targeting ligand was tested as a cognate UID compound (Fig. 4(B)). The prototype UIDs targeting the bromo and extra-terminal domain (BET) family proteins were synthesised to have varying lengths of ethylene glycerol linkers between the chloroalkane group and a BET bromodomain ligand, JQ1. Significant degradation of BRD2 was observed only when the length of these linkers exceeded four ethylene glycerol units (Table 2). Neither a HaloTag7 ligand nor JQ1 alone triggered proteasomal recruitment or BRD2 degradation. Although both BRD2 and BRD4 could be brought into close proximity to ADRM1 through JQ1-chloroalkane conjugates, only BRD2, but not BRD4, underwent substantial degradation. Similarly, heterobivalent molecules (ByeTACs), consisting of an ADRM1 binder and JQ1, induced a Ub-independent, proteasomal degradation of BRD4 in a nanomolar concentration range.154 These findings need to be further validated but suggest that the recruitment of a target protein to the proteasome may be sufficient for effective degradation.In contrast to the CIDE study,55 which lacked detailed information regarding the involvement of substrate polyubiquitination, this study provided valuable insights into this question. In brief, the UID approach appeared to operate through a Ub-independent mechanism; while proteasome inhibitors significantly delayed Halo/UID-mediated TPD, E1 Ub activating enzyme inhibitors, which block the initial step of polyUb chain formation, did not affect the process.56 Proteasome-based TPD modalities, which essentially create “neo-proximity” between the 26S proteasome and substrates, hold promise for opening new opportunities in drug discovery. However, it should be noted that the proteasome operates through highly coordinated multistep mechanisms, with peptide bond cleavage being only the last step in the complex program of substrate manipulation. Therefore, additional studies are needed to further evaluate the potential changes in global proteostasis resulting from direct proteasome-targeting methods such as CIDE and UID, especially considering that the direct proteasome-targeting ligands may act as an antagonist of proteasomal degradation. Additionally, the effectiveness of these approaches as targeted protein degraders needs validation through animal model studies. Once such data become available, they will provide a more comprehensive biochemical framework for investigating the functionality, safety, and therapeutic applicability of these approaches.
5. Conclusions and perspectives
This tutorial review explores innovative technical platforms for degraders that do not rely on target ubiquitination and thus have considerably expanded the TPD target space (Fig. 1 and Table 1). The majority of TPD drugs currently undergoing clinical trials are PROTAC- and molecular glue-based degraders, utilising a very limited number of E3 Ub ligases.155 In addition, these approaches require precise spatial control to facilitate coordinated interactions among E2, E3, and the target protein and subsequent induction of substrate ubiquitination. Direct recruitment of targets to the 26S proteasome may present an alternative strategy that overcomes several hurdles of PROTAC-based therapies, such as limited E3 availability (only CRL4CRBN and CRL2VHL recruiters have entered the clinical trials) and drug resistance (due to somatic mutations of the E3 ligases in cancer cells).156–158 The TPD modalities that directly harness the lysosomal degradation system have the potential to target an even broader spectrum of substrates, including extracellular proteins (Table 2). It is worth noting that many human proteopathies occur outside cells, rather than within the intracellular space, rendering them inaccessible by conventional E3-recruiting TPD strategies.159 Our understanding of extracellular proteostasis is still limited. With the advancement in our knowledge of these mechanisms and pathological implications, leveraging lysosome/autophagy-based TPD approaches will fully unlock their therapeutic benefits.A potential concern regarding lysosome/autophagy-based TPDs is the risk of unintended proteolysis of off-target substrates because early endosomes and growing phagophores intrinsically engage with a number of proteins or organelles. Moreover, selective autophagy relies on only a few autophagic receptors embracing a wide variety of intracellular cargoes; in contrast, the UPS utilises approximately 600 E3 ligases, each with some level of specificity for distinct substrates.160 Thus, although these degraders are designed to meticulously discriminate their intended cargos from other cellular components, it seems crucial to evaluate any potential alterations in the turn-over rates of other endogenous substrates. For translating TPD technologies into therapeutic applications, a comprehensive global proteome analysis is crucial for a rigorous evaluation of potential on-site-off-targets as well as off-site-on-target effects.
Another important question is whether the action mechanism of lysosome-mediated TPD conforms to the event-driven pharmacology demonstrated by the catalytic and sub-stoichiometric mode of PROTACs or whether these lysosomal/autophagosomal degraders are irreversibly trapped in lysosomes along with their cargoes without recycling. From a biochemical standpoint, it is conceivable that degraders directly targeting 26S proteasomes (such as CIDEs, UIDs, and ByeTACs) are anticipated to be recycled similar to proteasomal Ub receptors and cellular Ub shuttle proteins. The small-molecule degraders targeting autophagosomes, such as ATTECs and AUTOTACs, may rely on their stability within the lysosomal compartments as well as their capability to escape from lysosomal degradation. The chemical modality likely plays a significant role in recyclability. While antibody- or peptide-based degraders are highly susceptible to various hydrolases and acidic environments within the lysosomes, small molecule degraders are more likely to maintain structural integrity under those conditions for catalytic functionality. Nonetheless, all these speculations lack sufficient evidence, warranting more comprehensive studies in that regard.
Apparently, it is more challenging to quantitatively assess the enzyme kinetics and potencies of lysosome/autophagy-mediated degraders in vitro, compared to those of PROTACs, because the former are engaged with more cellular components (both proteins and lipids) than the latter in exploiting relatively well-defined E3 enzymes and proteasomes. Moreover, given the current technical limitations in measuring autophagic flux as well as endpoint autophagic activity, finding a therapeutic window for these degraders in vivo appears more difficult than for conventional drugs or PROTACs. The lysosome or autophagy inducers are required in relatively high dosages to achieve sufficient potency, where the positive “cooperativity” in ternary complex formation cannot mitigate the hook effect. Moreover, their pharmacological characteristics can be altered as the total catalytic output of cellular autophagy keeps changing according to the metabolic states and is tightly balanced with that of the UPS.161–163 Lysosome/autophagy-targeting degraders, combined with in-depth mechanism studies into their biochemical and physiological effects, could offer a promising alternative to UPS-targeting TPDs. Lysosome/autophagy-targeting degraders, after in-depth mechanism studies of their biochemical and physiological effects, could offer a promising alternative to UPS-targeting TPDs.
More recently, Forte et al. developed heterobifunctional molecules which can recruit an E2 Ub-conjugation enzyme, such as UBE2D, and induce specific protein degradation.164 They also reported that the recruiters of a CUL4A adaptor protein DDB1 could be harnessed for effective TPD as well.165 While these alternative strategies are intriguing, their mechanistic principles closely resemble those of PROTACs utilising the cellular UPS system. Therefore, it remains to be determined whether these novel approaches can offer distinct advantages over the E3 recruitment strategy. Since the human genome encodes ∼40 E2 enzymes and E2s crosstalk with multiple E3 enzymes, E2-based degraders may target multiple substrates simultaneously. On the other hands, the direct guidance of disease-causing proteins to 26S proteasomes, as demonstrated by CIDE and UID technologies, stands out as a distinctive strategy, as it does not require polyubiquitination of target proteins. In the UPS-based proteolytic process, the delivery of polyubiquitinated substrates slated for proteasomal degradation solely relies on the ability of Ub receptors to aid their positioning in close proximity to the 26S proteasome. Thus, proteasome-recruiting compounds can essentially serve as faithful surrogates of polyUb tags. It is also possible that they may not require strict “cooperativity” to form a ternary complex, as is the case with E3-recruiting degraders. By bypassing the ubiquitination steps and allowing more flexible ternary conformations, these degraders are likely to trigger the target degradation more efficiently than PROTACs. Moreover, the ubiquitous and abundant expression of proteasomes (>106 molecules per cell)166 precludes many limitations of canonical PROTACs.
Considering that proteasomes are responsible for the proteolysis of at least 80% of cellular proteins (∼3 × 109 total proteins in a growing mammalian cell),167,168 an important question that arises regarding the direct 26S proteasome recruiters is whether these affect the normal function of proteasomes. Moreover, since the 26S proteasomes have many distinct conformational states for processing their polyubiquitinated substrates (at least seven conformations in mammals),169 the functionality of specific ligands may vary, for example, from the substrate-engaging states to degradation-competent states. The effectiveness of these modalities can also be compromised under various disease conditions, when an excess of substrates could “clog” the entry pore of the proteasome often observed in cancer and neurodegeneration. In such cases, the pharmacological enhancement of proteasomal activity, along with proteasome-recruiting degraders, could synergically reverse the adverse effects caused by excess pathologic proteins. We anticipate that more proteasome-recruiting degraders, which exploit various subunits in the RP and CP, will be developed in the future through AI-guided drug designs. Nevertheless, their effects on proteasome mechanics, interactions with other proteins, and the impacts on the global proteome should be rigorously assessed. Furthermore, a quantitative evaluation of the binding affinity between CIDE compounds and their cognate proteasome subunits would be essential for fine-tuning the degradation kinetics. This is also important because compounds with an excessively high binding affinity towards the 26S proteasome might impede substrate translocation into its catalytic chamber for proteolysis.
The mechanistic and quantitative comparisons (ESI,† Tables S1–S7) presented in this review may serve as a practical guide for the future development and evaluation of non-Ub-mediated TPD modalities. For successful translational applications, we believe that the following assessments should be the standard practice in developing lysosome/autophagy- or proteasome-targeting degraders: (1) measuring the extent to which the degraders bind to the components of proteolytic machinery in test tubes (biophysical assessment), (2) determining whether targeted degradation is completely compromised with genetic knockout or chemical inhibitors of key components in cultured cells (biochemical evaluation), and (3) assessing the level of specificity they can achieve in animal models (proteomic validation). In-depth investigations following proof-of-concept studies are crucial to prevent the premature use of unverified degraders in pre-clinical or clinical applications. The strategies are still evolving, and we anticipate encountering unprecedented challenges both in the mechanistic understanding and pharmacological application. However, establishing validated approaches directly targeting lysosomes or proteasomes seems to remain a feasible and important goal, diversifying the TPD strategies and offering an alternative route to address various human diseases. Classic biochemical studies on the UPS and autophagy have laid the foundation and provided the inspiration for the development of novel TPDs, a trend that will continue into the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Samsung Research Funding & Incubation Center of Samsung Electronics (SRFC-MA2202-06 to M. J. L.).References
- L. Zhao, J. Zhao, K. Zhong, A. Tong and D. Jia, Signal Transduction Targeted Ther., 2022, 7, 113 CrossRef CAS PubMed.
- X. Han, L. Zhao, W. Xiang, C. Qin, B. Miao, T. Xu, M. Wang, C. Y. Yang, K. Chinnaswamy, J. Stuckey and S. Wang, J. Med. Chem., 2019, 62, 11218–11231 CrossRef CAS PubMed.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Cell Chem. Biol., 2018, 25, 78–87 CrossRef CAS PubMed.
- J. L. Gustafson, T. K. Neklesa, C. S. Cox, A. G. Roth, D. L. Buckley, H. S. Tae, T. B. Sundberg, D. B. Stagg, J. Hines, D. P. McDonnell, J. D. Norris and C. M. Crews, Angew. Chem., Int. Ed., 2015, 54, 9659–9662 CrossRef CAS PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- A. Bricelj, C. Steinebach, R. Kuchta, M. Gütschow and I. Sosič, Front. Chem., 2021, 9, 707317 CrossRef CAS PubMed.
- L. Buetow and D. T. Huang, Nat. Rev. Mol. Cell Biol., 2016, 17, 626–642 CrossRef CAS PubMed.
- A. Ciechanover, A. Orian and A. L. Schwartz, BioEssays, 2000, 22, 442–451 CrossRef CAS PubMed.
- G. Dong, Y. Ding, S. He and C. Sheng, J. Med. Chem., 2021, 64, 10606–10620 CrossRef CAS PubMed.
- A. Domostegui, L. Nieto-Barrado, C. Perez-Lopez and C. Mayor-Ruiz, Chem. Soc. Rev., 2022, 51, 5498–5517 RSC.
- J. A. Dewey, C. Delalande, S. A. Azizi, V. Lu, D. Antonopoulos and G. Babnigg, J. Med. Chem., 2023, 66, 9278–9296 CrossRef CAS PubMed.
- S. An and L. Fu, EBioMedicine, 2018, 36, 553–562 CrossRef PubMed.
- J. C. Fuller, N. J. Burgoyne and R. M. Jackson, Drug Discovery Today, 2009, 14, 155–161 CrossRef CAS PubMed.
- M. Toure and C. M. Crews, Angew. Chem., Int. Ed., 2016, 55, 1966–1973 CrossRef CAS PubMed.
- T. Neklesa, L. B. Snyder, R. R. Willard, N. Vitale, J. Pizzano, D. A. Gordon, M. Bookbinder, J. Macaluso, H. Dong and C. Ferraro, J. Clin. Oncol., 2019, 37, 10.1200 Search PubMed.
- J. Flanagan, Y. Qian, S. Gough, M. Andreoli, M. Bookbinder, G. Cadelina, J. Bradley, E. Rousseau, R. Willard and J. Pizzano, Cancer Res., 2019, 79, P5-04-18 CrossRef.
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- D. A. Stevens, R. Ewesuedo, A. McDonald, S. Agarwal, P. Henrick, R. Perea, A. Gollerkeri and J. Gollob, J. Clin. Oncol., 2022, 40, TPS3170 CrossRef.
- E. M. Sontag, R. S. Samant and J. Frydman, Annu. Rev. Biochem., 2017, 86, 97–122 CrossRef CAS PubMed.
- A. Hershko, A. Ciechanover and A. Varshavsky, Nat. Med., 2000, 6, 1073–1081 CrossRef CAS PubMed.
- I. Amm, T. Sommer and D. H. Wolf, Biochim. Biophys. Acta, Mol. Cell Res., 2014, 1843, 182–196 CrossRef CAS PubMed.
- S. B. Qian, M. F. Princiotta, J. R. Bennink and J. W. Yewdell, J. Biol. Chem., 2006, 281, 392–400 CrossRef CAS PubMed.
- N. Zheng and N. Shabek, Annu. Rev. Biochem., 2017, 86, 129–157 CrossRef CAS PubMed.
- M. Schapira, M. F. Calabrese, A. N. Bullock and C. M. Crews, Nat. Rev. Drug Discovery, 2019, 18, 949–963 CrossRef CAS PubMed.
- A. Bricelj, C. Steinebach, R. Kuchta, M. Gutschow and I. Sosic, Front. Chem., 2021, 9, 707317 CrossRef CAS PubMed.
- S. Asano, Y. Fukuda, F. Beck, A. Aufderheide, F. Forster, R. Danev and W. Baumeister, Science, 2015, 347, 439–442 CrossRef CAS PubMed.
- C. G. Pack, H. Yukii, A. Toh-e, T. Kudo, H. Tsuchiya, A. Kaiho, E. Sakata, S. Murata, H. Yokosawa, Y. Sako, W. Baumeister, K. Tanaka and Y. Saeki, Nat. Commun., 2014, 5, 3396 CrossRef PubMed.
- W. H. Choi, Y. Yun, S. Park, J. H. Jeon, J. Lee, J. H. Lee, S. A. Yang, N. K. Kim, C. H. Jung, Y. T. Kwon, D. Han, S. M. Lim and M. J. Lee, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 19190–19200 CrossRef CAS PubMed.
- K. Tanaka and A. Ichihara, Biochem. Biophys. Res. Commun., 1989, 159, 1309–1315 CrossRef CAS PubMed.
- D. Finley, Annu. Rev. Biochem., 2009, 78, 477–513 CrossRef CAS PubMed.
- K. Martinez-Fonts, C. Davis, T. Tomita, S. Elsasser, A. R. Nager, Y. Shi, D. Finley and A. Matouschek, Nat. Commun., 2020, 11, 477 CrossRef CAS PubMed.
- J. Rabl, D. M. Smith, Y. Yu, S. C. Chang, A. L. Goldberg and Y. Cheng, Mol. Cell, 2008, 30, 360–368 CrossRef CAS PubMed.
- E. Seemuller, A. Lupas, D. Stock, J. Lowe, R. Huber and W. Baumeister, Science, 1995, 268, 579–582 CrossRef CAS PubMed.
- B. Fabre, T. Lambour, J. Delobel, F. Amalric, B. Monsarrat, O. Burlet-Schiltz and M. P. Bousquet-Dubouch, Mol. Cell. Proteomics, 2013, 12, 687–699 CrossRef CAS PubMed.
- B. Fabre, T. Lambour, L. Garrigues, M. Ducoux-Petit, F. Amalric, B. Monsarrat, O. Burlet-Schiltz and M. P. Bousquet-Dubouch, J. Proteome Res., 2014, 13, 3027–3037 CrossRef CAS PubMed.
- C. Zhao and F. J. Dekker, ACS Pharmacol. Transl. Sci., 2022, 5, 710–723 CrossRef CAS PubMed.
- M. Uhlen, L. Fagerberg, B. M. Hallstrom, C. Lindskog, P. Oksvold, A. Mardinoglu, A. Sivertsson, C. Kampf, E. Sjostedt, A. Asplund, I. Olsson, K. Edlund, E. Lundberg, S. Navani, C. A. Szigyarto, J. Odeberg, D. Djureinovic, J. O. Takanen, S. Hober, T. Alm, P. H. Edqvist, H. Berling, H. Tegel, J. Mulder, J. Rockberg, P. Nilsson, J. M. Schwenk, M. Hamsten, K. von Feilitzen, M. Forsberg, L. Persson, F. Johansson, M. Zwahlen, G. von Heijne, J. Nielsen and F. Ponten, Science, 2015, 347, 1260419 CrossRef PubMed.
- Y. Makaros, A. Raiff, R. T. Timms, A. R. Wagh, M. I. Gueta, A. Bekturova, J. Guez-Haddad, S. Brodsky, Y. Opatowsky, M. H. Glickman, S. J. Elledge and I. Koren, Mol. Cell, 2023, 83, 1921–1935 CrossRef CAS PubMed.
- D. C. Rubinsztein, P. Codogno and B. Levine, Nat. Rev. Drug Discovery, 2012, 11, 709–730 CrossRef CAS PubMed.
- G. E. Mortimore and C. M. Schworer, Nature, 1977, 270, 174–176 CrossRef CAS PubMed.
- G. Filomeni, D. De Zio and F. Cecconi, Cell Death Differ., 2015, 22, 377–388 CrossRef CAS PubMed.
- J. Huang and J. H. Brumell, Nat. Rev. Microbiol., 2014, 12, 101–114 CrossRef CAS PubMed.
- A. Stolz, A. Ernst and I. Dikic, Nat. Cell Biol., 2014, 16, 495–501 CrossRef CAS PubMed.
- H. Weidberg, E. Shvets and Z. Elazar, Annu. Rev. Biochem., 2011, 80, 125–156 CrossRef CAS PubMed.
- Y. K. Lee and J. A. Lee, BMB Rep., 2016, 49, 424–430 CrossRef CAS PubMed.
- V. I. Korolchuk, A. Mansilla, F. M. Menzies and D. C. Rubinsztein, Mol. Cell, 2009, 33, 517–527 CrossRef CAS PubMed.
- J. H. Lee, S. Park, E. Kim and M. J. Lee, Autophagy, 2019, 15, 726–728 CrossRef CAS PubMed.
- E. Adriaenssens, L. Ferrari and S. Martens, Curr. Biol., 2022, 32, R1357–R1371 CrossRef CAS PubMed.
- Z. Li, C. Wang, Z. Wang, C. Zhu, J. Li, T. Sha, L. Ma, C. Gao, Y. Yang, Y. Sun, J. Wang, X. Sun, C. Lu, M. Difiglia, Y. Mei, C. Ding, S. Luo, Y. Dang, Y. Ding, Y. Fei and B. Lu, Nature, 2019, 575, 203–209 CrossRef CAS PubMed.
- S. M. Banik, K. Pedram, S. Wisnovsky, G. Ahn, N. M. Riley and C. R. Bertozzi, Nature, 2020, 584, 291–297 CrossRef CAS PubMed.
- C. H. Ji, H. Y. Kim, M. J. Lee, A. J. Heo, D. Y. Park, S. Lim, S. Shin, S. Ganipisetti, W. S. Yang, C. A. Jung, K. Y. Kim, E. H. Jeong, S. H. Park, S. Bin Kim, S. J. Lee, J. E. Na, J. I. Kang, H. M. Chi, H. T. Kim, Y. K. Kim, B. Y. Kim and Y. T. Kwon, Nat. Commun., 2022, 13, 904 CrossRef CAS PubMed.
- K. Pance, J. A. Gramespacher, J. R. Byrnes, F. Salangsang, J. C. Serrano, A. D. Cotton, V. Steri and J. A. Wells, Nat. Biotechnol., 2023, 41, 273–281 CrossRef CAS PubMed.
- J. Zheng, W. He, J. Li, X. Feng, Y. Li, B. Cheng, Y. Zhou, M. Li, K. Liu, X. Shao, J. Zhang, H. Li, L. Chen and L. Fang, J. Am. Chem. Soc., 2022, 144, 21831–21836 CrossRef CAS PubMed.
- C. Zhu, W. Wang, Y. Wang, Y. Zhang and J. Li, Angew. Chem., Int. Ed., 2023, 62, e202300694 CrossRef CAS PubMed.
- C. Bashore, S. Prakash, M. C. Johnson, R. J. Conrad, I. A. Kekessie, S. J. Scales, N. Ishisoko, T. Kleinheinz, P. S. Liu, N. Popovych, A. T. Wecksler, L. Zhou, C. Tam, I. Zilberleyb, R. Srinivasan, R. A. Blake, A. Song, S. T. Staben, Y. Zhang, D. Arnott, W. J. Fairbrother, S. A. Foster, I. E. Wertz, C. Ciferri and E. C. Dueber, Nat. Chem. Biol., 2023, 19, 55–63 CrossRef CAS PubMed.
- M. Balzarini, W. Gui, I. M. Jayalath, B.-B. Schell, J. Tong and T. Kodadek, bioRxiv, 2023, preprint DOI:10.1101/2023.07.19.549534.
- E. J. Bennett, T. A. Shaler, B. Woodman, K. Y. Ryu, T. S. Zaitseva, C. H. Becker, G. P. Bates, H. Schulman and R. R. Kopito, Nature, 2007, 448, 704–708 CrossRef CAS PubMed.
- U. B. Pandey, Z. Nie, Y. Batlevi, B. A. McCray, G. P. Ritson, N. B. Nedelsky, S. L. Schwartz, N. A. DiProspero, M. A. Knight, O. Schuldiner, R. Padmanabhan, M. Hild, D. L. Berry, D. Garza, C. C. Hubbert, T. P. Yao, E. H. Baehrecke and J. P. Taylor, Nature, 2007, 447, 859–863 CrossRef CAS PubMed.
- S. Sarkar and D. C. Rubinsztein, FEBS J., 2008, 275, 4263–4270 CrossRef CAS PubMed.
- E. Kim, S. Park, J. H. Lee, J. Y. Mun, W. H. Choi, Y. Yun, J. Lee, J. H. Kim, M. J. Kang and M. J. Lee, Cell Rep., 2018, 24, 732–743 CrossRef CAS PubMed.
- S. Fishbain, T. Inobe, E. Israeli, S. Chavali, H. Yu, G. Kago, M. M. Babu and A. Matouschek, Nat. Struct. Mol. Biol., 2015, 22, 214–221 CrossRef CAS PubMed.
- M. A. Kalchman, R. K. Graham, G. Xia, H. B. Koide, J. G. Hodgson, K. C. Graham, Y. P. Goldberg, R. D. Gietz, C. M. Pickart and M. R. Hayden, J. Biol. Chem., 1996, 271, 19385–19394 CrossRef CAS PubMed.
- P. Venkatraman, R. Wetzel, M. Tanaka, N. Nukina and A. L. Goldberg, Mol. Cell, 2004, 14, 95–104 CrossRef CAS PubMed.
- F. Gasset-Rosa, C. Chillon-Marinas, A. Goginashvili, R. S. Atwal, J. W. Artates, R. Tabet, V. C. Wheeler, A. G. Bang, D. W. Cleveland and C. Lagier-Tourenne, Neuron, 2017, 94, 48–57 CrossRef CAS PubMed.
- H. Li, S. H. Li, Z. X. Yu, P. Shelbourne and X. J. Li, J. Neurosci., 2001, 21, 8473–8481 CrossRef CAS PubMed.
- D. Takahashi, J. Moriyama, T. Nakamura, E. Miki, E. Takahashi, A. Sato, T. Akaike, K. Itto-Nakama and H. Arimoto, Mol. Cell, 2019, 76, 797–810 CrossRef CAS PubMed.
- Y. Fu, N. Chen, Z. Wang, S. Luo, Y. Ding and B. Lu, Cell Res., 2021, 31, 965–979 CrossRef CAS PubMed.
- Y. Zhang, J. Huang, Y. Liang, J. Huang, Y. Fu, N. Chen, B. Lu and C. Zhao, Autophagy, 2023, 19, 2668–2681 CrossRef CAS PubMed.
- M. Liu, Z. Liu, G. Qin, J. Ren and X. Qu, Nano Lett., 2023, 23, 4965–4973 CrossRef CAS PubMed.
- Z. Liu, G. Qin, J. Yang, W. Wang, W. Zhang, B. Lu, J. Ren and X. Qu, Chem. Sci., 2023, 14, 11192–11202 RSC.
- M. Blind and M. Blank, Mol. Ther.–Nucleic Acids, 2015, 4, e223 CrossRef PubMed.
- J. Pei, X. Pan, A. Wang, W. Shuai, F. Bu, P. Tang, S. Zhang, Y. Zhang, G. Wang and L. Ouyang, Chem. Commun., 2021, 57, 13194–13197 RSC.
- G. Dong, Y. Wu, J. Cheng, L. Chen, R. Liu, Y. Ding, S. Wu, J. Ma and C. Sheng, J. Med. Chem., 2022, 65, 7619–7628 CrossRef CAS PubMed.
- Y. Zeng, J. Xiao, Y. Xu, F. Wei, L. Tian, Y. Gao, Y. Chen and Y. Hu, J. Med. Chem., 2023, 66, 12877–12893 CrossRef CAS PubMed.
- C. M. Olson, B. Jiang, M. A. Erb, Y. Liang, Z. M. Doctor, Z. Zhang, T. Zhang, N. Kwiatkowski, M. Boukhali, J. L. Green, W. Haas, T. Nomanbhoy, E. S. Fischer, R. A. Young, J. E. Bradner, G. E. Winter and N. S. Gray, Nat. Chem. Biol., 2018, 14, 163–170 CrossRef CAS PubMed.
- A. Folger and Y. Wang, Cells, 2021, 10, 2835 CrossRef CAS PubMed.
- H. Olzscha, Biol. Chem., 2019, 400, 895–915 CrossRef CAS PubMed.
- C. Alquezar, S. Arya and A. W. Kao, Front. Neurol., 2021, 11, 595532 CrossRef PubMed.
- D. A. T. Nijholt, L. De Kimpe, H. L. Elfrink, J. J. M. Hoozemans and W. Scheper, Curr. Med. Chem., 2011, 18, 2459–2476 CrossRef CAS PubMed.
- A. Ciechanover and Y. T. Kwon, Exp. Mol. Med., 2015, 47, e147 CrossRef CAS PubMed.
- C. H. Ji, M. J. Lee, H. Y. Kim, A. J. Heo, D. Y. Park, Y. K. Kim, B. Y. Kim and Y. T. Kwon, Autophagy, 2022, 18, 2259–2262 CrossRef CAS PubMed.
- J. Lee, K. W. Sung, E. J. Bae, D. Yoon, D. Kim, J. S. Lee, D. H. Park, D. Y. Park, S. R. Mun, S. C. Kwon, H. Y. Kim, J. O. Min, S. J. Lee, Y. H. Suh and Y. T. Kwon, Mol. Neurodegener., 2023, 18, 41 CrossRef CAS PubMed.
- A. Varshavsky, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 358–366 CrossRef CAS PubMed.
- Y. Jiang, J. Lee, J. H. Lee, J. W. Lee, J. H. Kim, W. H. Choi, Y. D. Yoo, H. Cha-Molstad, B. Y. Kim, Y. T. Kwon, S. A. Noh, K. P. Kim and M. J. Lee, Autophagy, 2016, 12, 2197–2212 CrossRef CAS PubMed.
- M. J. Lee, D. E. Kim, A. Zakrzewska, Y. D. Yoo, S. H. Kim, S. T. Kim, J. W. Seo, Y. S. Lee, G. W. Dorn, 2nd, U. Oh, B. Y. Kim and Y. T. Kwon, J. Biol. Chem., 2012, 287, 24043–24052 CrossRef CAS PubMed.
- H. Cha-Molstad, K. S. Sung, J. Hwang, K. A. Kim, J. E. Yu, Y. D. Yoo, J. M. Jang, D. H. Han, M. Molstad, J. G. Kim, Y. J. Lee, A. Zakrzewska, S. H. Kim, S. T. Kim, S. Y. Kim, H. G. Lee, N. K. Soung, J. S. Ahn, A. Ciechanover, B. Y. Kim and Y. T. Kwon, Nat. Cell Biol., 2015, 17, 917–929 CrossRef CAS PubMed.
- H. Cha-Molstad, J. E. Yu, Z. Feng, S. H. Lee, J. G. Kim, P. Yang, B. Han, K. W. Sung, Y. D. Yoo, J. Hwang, T. McGuire, S. M. Shim, H. D. Song, S. Ganipisetti, N. Wang, J. M. Jang, M. J. Lee, S. J. Kim, K. H. Lee, J. T. Hong, A. Ciechanover, I. Mook-Jung, K. P. Kim, X. Q. Xie, Y. T. Kwon and B. Y. Kim, Nat. Commun., 2017, 8, 102 CrossRef PubMed.
- A. J. Jakobi, S. T. Huber, S. A. Mortensen, S. W. Schultz, A. Palara, T. Kuhm, B. K. Shrestha, T. Lamark, W. J. H. Hagen, M. Wilmanns, T. Johansen, A. Brech and C. Sachse, Nat. Commun., 2020, 11, 440 CrossRef CAS PubMed.
- C. H. Ji, H. Y. Kim, A. J. Heo, S. H. Lee, M. J. Lee, S. B. Kim, G. Srinivasrao, S. R. Mun, H. Cha-Molstad, A. Ciechanover, C. Y. Choi, H. G. Lee, B. Y. Kim and Y. T. Kwon, Mol. Cell, 2019, 75, 1058–1072 CrossRef CAS PubMed.
- Y. J. Lee, J. K. Kim, C. H. Jung, Y. J. Kim, E. J. Jung, S. H. Lee, H. R. Choi, Y. S. Son, S. M. Shim, S. M. Jeon, J. H. Choe, S. H. Lee, J. Whang, K. C. Sohn, G. M. Hur, H. T. Kim, J. Yeom, E. K. Jo and Y. T. Kwon, Autophagy, 2022, 18, 2926–2945 CrossRef CAS PubMed.
- S. M. Shim, H. R. Choi, S. C. Kwon, H. Y. Kim, K. W. Sung, E. J. Jung, S. R. Mun, T. H. Bae, D. H. Kim, Y. S. Son, C. H. Jung, J. Lee, M. J. Lee, J. W. Park and Y. T. Kwon, Autophagy, 2023, 19, 1642–1661 CrossRef CAS PubMed.
- L. Xiao, M. Xiao, M. Zou and W. Xu, Int. J. Clin. Exp. Pathol., 2017, 10, 8569–8576 Search PubMed.
- J. M. Molina-Molina, A. Hillenweck, I. Jouanin, D. Zalko, J. P. Cravedi, M. F. Fernández, A. Pillon, J. C. Nicolas, N. Olea and P. Balaguer, Toxicol. Appl. Pharmacol., 2006, 216, 44–54 CrossRef CAS PubMed.
- E. C. Griffith, Z. Su, S. Niwayama, C. A. Ramsay, Y. H. Chang and J. O. Liu, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15183–15188 CrossRef CAS PubMed.
- L. Cortez and V. Sim, Prion, 2014, 8, 197–202 CrossRef CAS PubMed.
- L. Antonschmidt, D. Matthes, R. Dervişoğlu, B. Frieg, C. Dienemann, A. Leonov, E. Nimerovsky, V. Sant, S. Ryazanov, A. Giese, G. F. Schröder, S. Becker, B. L. de Groot, C. Griesinger and L. B. Andreas, Nat. Commun., 2022, 13, 5385 CrossRef CAS PubMed.
- D. Matthes, V. Gapsys, C. Griesinger and B. L. de Groot, ACS Chem. Neurosci., 2017, 8, 2791–2808 CrossRef CAS PubMed.
- J. Lewis, E. McGowan, J. Rockwood, H. Melrose, P. Nacharaju, M. Van Slegtenhorst, K. Gwinn-Hardy, M. Paul Murphy, M. Baker, X. Yu, K. Duff, J. Hardy, A. Corral, W. L. Lin, S. H. Yen, D. W. Dickson, P. Davies and M. Hutton, Nat. Genet., 2000, 25, 402–405 CrossRef CAS PubMed.
- E. F. Kuiper, E. P. de Mattos, L. B. Jardim, H. H. Kampinga and S. Bergink, Front. Neurosci., 2017, 11, 145 CAS.
- N. Kedia, K. Arhzaouy, S. K. Pittman, Y. Sun, M. Batchelor, C. C. Weihl and J. Bieschke, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16835–16840 CrossRef CAS PubMed.
- S. Shin, D. Kim, J. Y. Song, H. Jeong, S. J. Hyeon, N. W. Kowall, H. Ryu, A. N. Pae, S. Lim and Y. K. Kim, Prog. Neurobiol., 2020, 101782 CrossRef CAS PubMed.
- D. H. Han, H. K. Na, W. H. Choi, J. H. Lee, Y. K. Kim, C. Won, S. H. Lee, K. P. Kim, J. Kuret, D. H. Min and M. J. Lee, Nat. Commun., 2014, 5, 5633 CrossRef CAS PubMed.
- V. Sharma, S. Verma, E. Seranova, S. Sarkar and D. Kumar, Front. Cell Dev. Biol., 2018, 6, 147 CrossRef PubMed.
- Y. W. Wu and F. Li, Virulence, 2019, 10, 352–362 CrossRef CAS PubMed.
- J. M. Yuk, T. Yoshimori and E. K. Jo, Exp. Mol. Med., 2012, 44, 99–108 CrossRef CAS PubMed.
- E. Campoy and M. I. Colombo, Curr. Top. Microbiol. Immunol., 2009, 335, 227–250 CAS.
- N. N. Noda, Z. Wang and H. Zhang, J. Cell Biol., 2020, 219, e202004062 CrossRef CAS PubMed.
- T. Lamark and T. Johansen, Annu. Rev. Cell Dev. Biol., 2021, 37, 143–169 CrossRef CAS PubMed.
- S. Ghaemmaghami, W. K. Huh, K. Bower, R. W. Howson, A. Belle, N. Dephoure, E. K. O'Shea and J. S. Weissman, Nature, 2003, 425, 737–741 CrossRef CAS PubMed.
- P. Ghosh, N. M. Dahms and S. Kornfeld, Nat. Rev. Mol. Cell Biol., 2003, 4, 202–212 CrossRef CAS PubMed.
- E. Dalle Vedove, G. Costabile and O. M. Merkel, Adv. Healthcare Mater., 2018, 7, e1701398 CrossRef PubMed.
- D. B. Oh, BMB Rep., 2015, 48, 438–444 CrossRef CAS PubMed.
- L. Beljaars, G. Molema, B. Weert, H. Bonnema, P. Olinga, G. M. Groothuis, D. K. Meijer and K. Poelstra, Hepatology, 1999, 29, 1486–1493 CrossRef CAS PubMed.
- D. B. Berkowitz, G. Maiti, B. D. Charette, C. D. Dreis and R. G. MacDonald, Org. Lett., 2004, 6, 4921–4924 CrossRef CAS PubMed.
- G. Ahn, N. M. Riley, R. A. Kamber, S. Wisnovsky, S. Moncayo von Hase, M. C. Bassik, S. M. Banik and C. R. Bertozzi, Science, 2023, 382, eadf6249 CrossRef CAS PubMed.
- M. Safieh, A. D. Korczyn and D. M. Michaelson, BMC Med., 2019, 17, 64 CrossRef PubMed.
- M. L. Burr, C. E. Sparbier, Y. C. Chan, J. C. Williamson, K. Woods, P. A. Beavis, E. Y. N. Lam, M. A. Henderson, C. C. Bell, S. Stolzenburg, O. Gilan, S. Bloor, T. Noori, D. W. Morgens, M. C. Bassik, P. J. Neeson, A. Behren, P. K. Darcy, S. J. Dawson, I. Voskoboinik, J. A. Trapani, J. Cebon, P. J. Lehner and M. A. Dawson, Nature, 2017, 549, 101–105 CrossRef CAS PubMed.
- P. Huang, X. Xu, L. Wang, B. Zhu, X. Wang and J. Xia, J. Cell. Mol. Med., 2014, 18, 218–230 CrossRef CAS PubMed.
- X. Zhang, H. Liu, J. He, C. Ou, T. C. Donahue, M. M. Muthana, L. Su and L. X. Wang, ACS Chem. Biol., 2022, 17, 3013–3023 CrossRef CAS PubMed.
- T. S. Zimmermann, V. Karsten, A. Chan, J. Chiesa, M. Boyce, B. R. Bettencourt, R. Hutabarat, S. Nochur, A. Vaishnaw and J. Gollob, Mol. Ther., 2017, 25, 71–78 CrossRef CAS PubMed.
- R. J. Stockert, Physiol. Rev., 1995, 75, 591–609 CrossRef CAS PubMed.
- M. Spiess, Biochemistry, 1990, 29, 10009–10018 CrossRef CAS PubMed.
- M. Egli and M. Manoharan, Nucleic Acids Res., 2023, 51, 2529–2573 CrossRef CAS PubMed.
- G. Ahn, S. M. Banik, C. L. Miller, N. M. Riley, J. R. Cochran and C. R. Bertozzi, Nat. Chem. Biol., 2021, 17, 937–946 CrossRef CAS PubMed.
- D. Rabuka, J. S. Rush, G. W. de Hart, P. Wu and C. R. Bertozzi, Nat. Protoc., 2012, 7, 1052–1067 CrossRef CAS PubMed.
- D. F. Caianiello, M. Zhang, J. D. Ray, R. A. Howell, J. C. Swartzel, E. M. J. Branham, E. Chirkin, V. R. Sabbasani, A. Z. Gong, D. M. McDonald, V. Muthusamy and D. A. Spiegel, Nat. Chem. Biol., 2021, 17, 947–953 CrossRef CAS PubMed.
- Y. Zhou, P. Teng, N. T. Montgomery, X. Li and W. Tang, ACS Cent. Sci., 2021, 7, 499–506 CrossRef CAS PubMed.
- G. Kaur and I. Roy, Expert Opin. Invest. Drugs, 2008, 17, 43–60 CrossRef CAS PubMed.
- Y. Miao, Q. Gao, M. Mao, C. Zhang, L. Yang, Y. Yang and D. Han, Angew. Chem., Int. Ed., 2021, 60, 11267–11271 CrossRef CAS PubMed.
- K. Hamada, T. Hashimoto, R. Iwashita, Y. Yamada, Y. Kikkawa and M. Nomizu, Cell Rep. Phys. Sci., 2023, 4, 101296 CrossRef CAS.
- Y. Wu, B. Lin, Y. Lu, L. Li, K. Deng, S. Zhang, H. Zhang, C. Yang and Z. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202218106 CrossRef CAS PubMed.
- A. Lacroix and H. F. Sleiman, ACS Nano, 2021, 15, 3631–3645 CrossRef CAS PubMed.
- M. E. Distler, M. H. Teplensky, K. E. Bujold, C. D. Kusmierz, M. Evangelopoulos and C. A. Mirkin, J. Am. Chem. Soc., 2021, 143, 13513–13518 CrossRef CAS PubMed.
- D. Zhang, J. Duque-Jimenez, G. Brixi, F. Facchinetti, K. Rhee, W. W. Feng, P. A. Jänne and X. Zhou, bioRxiv, 2023, preprint DOI:10.1101/2023.08.10.552782.
- B. Huang, M. Abedi, G. Ahn, B. Coventry, I. Sappington, R. Wang, T. Schlichthaerle, J. Z. Zhang, Y. Wang, I. Goreshnik, C. W. Chiu, A. Chazin-Gray, S. Chan, S. Gerben, A. Murray, S. Wang, J. O’Neill, R. Yeh, A. Misquith, A. Wolf, L. M. Tomasovic, D. I. Piraner, M. J. D. Gonzalez, N. R. Bennett, P. Venkatesh, D. Satoe, M. Ahlrichs, C. Dobbins, W. Yang, X. Wang, D. Vafeados, R. Mout, S. Shivaei, L. Cao, L. Carter, L. Stewart, J. B. Spangler, G. J. L. Bernardes, K. T. Roybal, J. Per Greisen, X. Li, C. Bertozzi and D. Baker, bioRxiv, 2023, preprint DOI:10.1101/2023.08.19.553321.
- P. S. Dragovich, T. H. Pillow, R. A. Blake, J. D. Sadowsky, E. Adaligil, P. Adhikari, S. Bhakta, N. Blaquiere, J. Chen, J. Dela Cruz-Chuh, K. E. Gascoigne, S. J. Hartman, M. He, S. Kaufman, T. Kleinheinz, K. R. Kozak, L. Liu, L. Liu, Q. Liu, Y. Lu, F. Meng, M. M. Mulvihill, A. O’Donohue, R. K. Rowntree, L. R. Staben, S. T. Staben, J. Wai, J. Wang, B. Wei, C. Wilson, J. Xin, Z. Xu, H. Yao, D. Zhang, H. Zhang, H. Zhou and X. Zhu, J. Med. Chem., 2021, 64, 2534–2575 CrossRef CAS PubMed.
- M. A. Maneiro, N. Forte, M. M. Shchepinova, C. S. Kounde, V. Chudasama, J. R. Baker and E. W. Tate, ACS Chem. Biol., 2020, 15, 1306–1312 CrossRef CAS PubMed.
- H. Zhang, Y. Han, Y. Yang, F. Lin, K. Li, L. Kong, H. Liu, Y. Dang, J. Lin and P. R. Chen, J. Am. Chem. Soc., 2021, 143, 16377–16382 CrossRef CAS PubMed.
- A. D. Cotton, D. P. Nguyen, J. A. Gramespacher, I. B. Seiple and J. A. Wells, J. Am. Chem. Soc., 2021, 143, 593–598 CrossRef CAS PubMed.
- H. Marei, W. K. Tsai, Y. S. Kee, K. Ruiz, J. He, C. Cox, T. Sun, S. Penikalapati, P. Dwivedi, M. Choi, D. Kan, P. Saenz-Lopez, K. Dorighi, P. Zhang, Y. T. Kschonsak, N. Kljavin, D. Amin, I. Kim, A. G. Mancini, T. Nguyen, C. Wang, E. Janezic, A. Doan, E. Mai, H. Xi, C. Gu, M. Heinlein, B. Biehs, J. Wu, I. Lehoux, S. Harris, L. Comps-Agrar, D. Seshasayee, F. J. de Sauvage, M. Grimmer, J. Li, N. J. Agard and E. M. F. de Sousa, Nature, 2022, 610, 182–189 CrossRef CAS PubMed.
- J. B. Ridgway, L. G. Presta and P. Carter, Protein Eng., 1996, 9, 617–621 CrossRef CAS PubMed.
- A. H. Boekhout, J. H. Beijnen and J. H. Schellens, Oncologist, 2011, 16, 800–810 CrossRef CAS PubMed.
- S. Li, K. R. Schmitz, P. D. Jeffrey, J. J. Wiltzius, P. Kussie and K. M. Ferguson, Cancer Cell, 2005, 7, 301–311 CrossRef CAS PubMed.
- S. L. Topalian, F. S. Hodi, J. R. Brahmer, S. N. Gettinger, D. C. Smith, D. F. McDermott, J. D. Powderly, R. D. Carvajal, J. A. Sosman, M. B. Atkins, P. D. Leming, D. R. Spigel, S. J. Antonia, L. Horn, C. G. Drake, D. M. Pardoll, L. Chen, W. H. Sharfman, R. A. Anders, J. M. Taube, T. L. McMiller, H. Xu, A. J. Korman, M. Jure-Kunkel, S. Agrawal, D. McDonald, G. D. Kollia, A. Gupta, J. M. Wigginton and M. Sznol, N. Engl. J. Med., 2012, 366, 2443–2454 CrossRef CAS PubMed.
- C. M. Pickart and R. E. Cohen, Nat. Rev. Mol. Cell Biol., 2004, 5, 177–187 CrossRef CAS PubMed.
- A. W. Karzai, E. D. Roche and R. T. Sauer, Nat. Struct. Biol., 2000, 7, 449–455 CrossRef CAS PubMed.
- R. T. Sauer and T. A. Baker, Annu. Rev. Biochem., 2011, 80, 587–612 CrossRef CAS PubMed.
- S. Bar-Nun and M. H. Glickman, Biochim. Biophys. Acta, Mol. Cell Res., 2012, 1823, 67–82 CrossRef CAS PubMed.
- R. F. Kalejta and T. Shenk, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3263–3268 CrossRef CAS PubMed.
- B. Donati, E. Lorenzini and A. Ciarrocchi, Mol. Cancer, 2018, 17, 164 CrossRef CAS PubMed.
- H. S. Kruth, N. L. Jones, W. Huang, B. Zhao, I. Ishii, J. Chang, C. A. Combs, D. Malide and W. Y. Zhang, J. Biol. Chem., 2005, 280, 2352–2360 CrossRef CAS PubMed.
- S. D. Conner and S. L. Schmid, Nature, 2003, 422, 37–44 CrossRef CAS PubMed.
- N. Liu, R. Ling, X. Tang, Y. Yu, Y. Zhou and D. Chen, Front. Oncol., 2022, 12, 847701 CrossRef CAS PubMed.
- E. M. H. Ali, C. A. Loy and D. J. Trader, bioRxiv, 2024, preprint DOI:10.1101/2024.01.20.576376.
- A. Mullard, Nat. Rev. Drug Discovery, 2019, 18, 237–239 Search PubMed.
- L. Zhang, B. Riley-Gillis, P. Vijay and Y. Shen, Mol. Cancer Ther., 2019, 18, 1302–1311 CrossRef CAS PubMed.
- K. Yang, Y. Zhao, X. Nie, H. Wu, B. Wang, C. M. Almodovar-Rivera, H. Xie and W. Tang, Cell Chem. Biol., 2020, 27, 866–876 CrossRef CAS PubMed.
- C. Otto, S. Schmidt, C. Kastner, S. Denk, J. Kettler, N. Muller, C. T. Germer, E. Wolf, P. Gallant and A. Wiegering, Neoplasia, 2019, 21, 1110–1120 CrossRef CAS PubMed.
- A. R. Wyatt, J. J. Yerbury, S. Poon and M. R. Wilson, Curr. Med. Chem., 2009, 16, 2855–2866 CrossRef CAS PubMed.
- W. J. Liu, L. Ye, W. F. Huang, L. J. Guo, Z. G. Xu, H. L. Wu, C. Yang and H. F. Liu, Cell. Mol. Biol. Lett., 2016, 21, 29 CrossRef PubMed.
- W. K. Wu, C. H. Cho, C. W. Lee, Y. C. Wu, L. Yu, Z. J. Li, C. C. Wong, H. T. Li, L. Zhang, S. X. Ren, C. T. Che, K. Wu, D. Fan, J. Yu and J. J. Sung, Autophagy, 2010, 6, 228–238 CrossRef CAS PubMed.
- K. Zhu, K. Dunner, Jr. and D. J. McConkey, Oncogene, 2010, 29, 451–462 CrossRef CAS PubMed.
- S. Kageyama, Y. S. Sou, T. Uemura, S. Kametaka, T. Saito, R. Ishimura, T. Kouno, L. Bedford, R. J. Mayer, M. S. Lee, M. Yamamoto, S. Waguri, K. Tanaka and M. Komatsu, J. Biol. Chem., 2014, 289, 24944–24955 CrossRef CAS PubMed.
- N. Forte, D. Dovala, M. J. Hesse, J. M. McKenna, J. A. Tallarico, M. Schirle and D. K. Nomura, ACS Chem. Biol., 2023, 18, 897–904 CrossRef CAS PubMed.
- M. Meyers, S. Cismoski, A. Panidapu, B. Chie-Leon and D. K. Nomura, bioRxiv, 2023, preprint DOI:10.1101/2023.08.11.553046.
- E. V. Rusilowicz-Jones, S. Urbe and M. J. Clague, Mol. Cell, 2022, 82, 1414–1423 CrossRef CAS PubMed.
- J. Zhao, B. Zhai, S. P. Gygi and A. L. Goldberg, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15790–15797 CrossRef CAS PubMed.
- S. B. Cambridge, F. Gnad, C. Nguyen, J. L. Bermejo, M. Kruger and M. Mannw, J. Proteome Res., 2011, 10, 5275–5284 CrossRef CAS PubMed.
- Y. Dong, S. Zhang, Z. Wu, X. Li, W. L. Wang, Y. Zhu, S. Stoilova-McPhie, Y. Lu, D. Finley and Y. Mao, Nature, 2019, 565, 49–55 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cs00344b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |