
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion⋯anion self-assembly under the control of σ- and π-hole bonds
Andrea
Pizzi
 ,
Arun
Dhaka
,
Roberta
Beccaria
and
Giuseppe
Resnati
*
,
Arun
Dhaka
,
Roberta
Beccaria
and
Giuseppe
Resnati
*
NFMLab, Department of Chemistry, Materials, Chemical Engineering “Giulio Natta”, Politecnico di Milano, via Mancinelli 7, I-20131 Milano, Italy. E-mail: giuseppe.resnati@polimi.it
First published on 13th June 2024
Abstract
The electrostatic attraction between charges of opposite signs and the repulsion between charges of the same sign are ubiquitous and influential phenomena in recognition and self-assembly processes. However, it has been recently revealed that specific attractive forces between ions with the same sign are relatively common. These forces can be strong enough to overcome the Coulomb repulsion between ions with the same sign, leading to the formation of stable anion⋯anion and cation⋯cation adducts. Hydroden bonds (HBs) are probably the best-known interaction that can effectively direct these counterintuitive assembly processes. In this review we discuss how σ-hole and π-hole bonds can break the paradigm of electrostatic repulsion between like-charges and effectively drive the self-assembly of anions into discrete as well as one-, two-, or three-dimensional adducts. σ-Hole and π-hole bonds are the attractive forces between regions of excess electron density in molecular entities (e.g., lone pairs or π bond orbitals) and regions of depleted electron density that are localized at the outer surface of bonded atoms opposite to the σ covalent bonds formed by atoms (σ-holes) and above and below the planar portions of molecular entities (π-holes). σ- and π-holes can be present on many different elements of the p and d block of the periodic table and the self-assembly processes driven by their presence can thus involve a wide diversity of mono- and di-anions. The formed homomeric and heteromeric adducts are typically stable in the solid phase and in polar solvents but metastable or unstable in the gas phase. The pivotal role of σ- and π-hole bonds in controlling anion⋯anion self-assembly is described in key biopharmacological systems and in molecular materials endowed with useful functional properties.
 Andrea Pizzi | Dr Andrea Pizzi is an Assistant Professor at Politecnico di Milano, in the Laboratory of Nanostructured Fluorinated Materials (NFMLab). His PhD thesis, under the supervision of Prof. Pierangelo Metrangolo, was focused on halogen bonding in peptides and proteins. His research interest is in the field of supramolecular chemistry based on σ-hole interactions, aimed at investigating the functioning of catalysts and obtaining new biomimetic materials. He is currently investigating the role of anion⋯anion interactions in proteins and small molecules in the solid state. |
 Arun Dhaka | Dr Arun Dhaka is a MSCA post-doctoral researcher at Politecnico di Milano, in the Laboratory of Nanostructured Fluorinated Materials (NFMLab). His Phd work on condensed matter was carried out under the supervision of Dr Marc Fourmiguè at Universitè de Rennes, France. His research interests broadly encompass the field of crystal engineering and molecular materials, with a focus on self-assembly processes driven under the control of σ-hole interactions, creating novel crystalline solids. He is currently investigating anion⋯anion interactions in molecular solids. |
 Roberta Beccaria | Roberta Beccaria is a PhD candidate in the Laboratory of Nanostructured Fluorinated Materials (NFMLab) at Politecnico di Milano. She obtained her Master's degree in Chemistry at the Università degli Studi di Torino in 2022 with a thesis on pharmaceutical cocrystals. Her areas of interest in research concern supramolecular chemistry and crystal engineering, with a particular focus on analytical techniques for assessing the formation of self-assembled systems mainly in the solid (X-ray analyses) but also in solution (NMR). |
 Giuseppe Resnati | Giuseppe Resnati is Professor of general chemistry for materials and nanotechnologies at the Politecnico di Milano. His interest in fluoroorganic chemistry prompted him to start the Laboratory of Nanostructured Fluorinated Materials (NFMLab) to investigate the asymmetric synthesis of fluorinated compounds and the chemistry of fluorous oxydizing agents. In the last decades, he focused on recognition and self-assembly processes driven by halogen, chalcogen, and pnictogen bonds as well as by analogous interactions involving a d block element as the electrophile. Anion⋯anion interactions are included in this latter area of interest. |
Key learning points1. The distribution of electron density at the surface of bonded atoms is anisotropic.2. In many polyatomic anions (e.g., IO4−, NO3−, PdCl42−, and AuCl4−), atoms having regions with depleted electron density (electrophilic sites) and excess electron density (nucleophilic sites) may both be present. 3. Attractive interactions between these electrophilic and nucleophilic sites can drive the formation of anion⋯anion adducts. 4. These adducts can exist as stable species in the solid form or in solution and occasionally also in the gas phase. 5. Such anion⋯anion interactions have significant impacts on compounds as relevant as ATP and nonlinear optical materials. |
1. Introduction
Molecular recognition and self-assembly are the holistic result of the balanced combination of a multitude of electronic and geometric features of interacting units. The electrostatic attraction between charges of opposite sign is one of the most common and influential effects which can bring molecular entities close together and drive their assembly into well-organized adducts.1 The repulsion between charges of the same sign (like-charges) is equally important and tends to separate anions from each other. The self-assembly of anions into discrete or infinite adducts can occur only if specific attractive forces are present and can successfully overcome the Coulombic anion⋯anion repulsion.2,3 It might thus be expected that these phenomena are exceptional, but it has been reported that a variety of natural anions, e.g., malonate,4 citrate,5 and carboxylate forms of amino acids,6 can aggregate and give rise to short anion⋯anion contacts.7 Indeed, in biological systems, the presence of anion⋯anion interactions is fairly common. Also, synthetic anions (e.g., hexacyanoferrate3,8 and nitroprusside anions9) self-assemble under different conditions.10 In this review, we will describe how σ-11,12 and π-hole13,14 interactions can effectively counteract the electrostatic repulsion between like charges; specifically, we will describe how they enable the self-assembly of anions into discrete adducts or 1D, 2D, and 3D supramolecular architectures.15The ability of the hydrogen bond (HB) to overcome the Coulombic repulsion between protic hydroxyanions and to drive their self-assembly into stable anion⋯anion adducts has been recognized since long ago. Anion⋯anion adducts formed by H2PO4− are of paramount importance. The dimerization of this anion in water solution and the value of the association constant were reported as early as 1969.16 The selectivity of phosphate binding proteins for phosphate over sulphate is ensured by the ability of hydroxy group(s) of phosphate anion to bind a carboxylate residue present, at physiological pH, in the protein active site.17 ADP and ATP synthesis and hydrolysis are ubiquitous processes and of immense relevance in living organisms; the involvement of their phosphoric residues in the formation of anion⋯anion short contacts under the control of HB plays a key role in these processes.18–20 The HB driven self-assembly of monoanions of di- and poly-carboxylic acids of biopharmacological relevance is a widespread phenomenon.4,5 The versatility and effectiveness of the HB in enabling anion⋯anion adducts formation is confirmed by the formation of not only homomeric adducts (e.g., starting from HCO3−, H2PO4−, H2AsO4−, HSO4−),21,22 but also heteromeric trimers (HSO4−⋯H2PO4−⋯HSO4−)23 and dianions dimers (HPO42−⋯HPO42− and HAsO42−⋯HAsO42−).22
σ- and π-hole interactions complement the potential of HB in directing anion⋯anion self-assembly by extending this process to anions wherein no hydroxy group is present. The versatility and effectiveness of σ/π-hole interactions in forming anion⋯anion adducts are probably as impactful as those of HBs. For instance, discrete adducts or infinite supramolecular architectures have been obtained through the self-assembly of mono- and dianions as different as IO3−,24 MnO4−,25 AuCl4−,26 XeCl5−,27 PdCl42−,28 and Se2O52−.29
The halogen bond (HaB), an attractive and directional bonding where group 17 elements behave as electrophiles,30 is the first identified and best studied σ-hole interaction. The history of HaB dates back two hundred years31 and its present understanding is the result of many studies wherein different rationalization models were proposed.32 The struggle to understand the phenomenon is emblematically declared by the twenty descriptive phrases listed in 1968 by H. A. Bent33 to indicate what is now named HaB.34–40 The wording “bumps-in-hollows” and “pair-pocket” are two of the descriptors mentioned by Bent, which closely resemble the hole model adopted here. Importantly, in their seminal paper published on the HaB in 1996, F. H. Allen et al. stated that “The attractive nature of the interaction is mainly due to electrostatic effects, but polarization, charge-transfer, and dispersion contributions all play an important role in causing interpenetration of van der Waals volumes”.41
The chalcogen bond (ChB), a bonding interaction wherein the electrophile is a group 16 element,42 is the second most impacting σ/π-hole interaction.43,44 Similar to the HaB, the present understanding of this interaction has developed through a rather patchy course. The contributions focussing on the charge-transfer33,35,36 and geometric37,45 viewpoints were important in constructing the present-day ChB concept, but the topic boomed and was clearly defined42 only after modelling based on the electrostatic standpoint was proposed.46 It is commonly accepted that Allen's generalization on the HaB equally holds for the ChB and other σ/π-hole interaction.47–49
In order to further present the basics of σ/π-hole interactions with a focus on those affording anion⋯anion adducts, we will not delve into the details of all theoretical models that have been proposed to rationalize these interactions and which contemplates the presence of covalent50 and dative51 components. Different computational methods give different relative weights to the different components of σ/π-hole interactions and providing a critical assessment of related problems is out of the scope of this review.52 We will concentrate only on the electrostatic and the charge-transfer models as experimental evidence supports their relevance in all σ/π-hole bonds. Emphasis will be given to the electrostatic viewpoint53 as it is the most frequently employed one and the pictures representing the molecular electrostatic potential (MEP) are very intuitive and have a strong communicative effectiveness.
The basis of the electrostatic model of σ/π-hole interactions is the anisotropic distribution of the electron density in bonded atoms. This anisotropy determines the presence of regions of depleted electron density that are localized along the elongation of the σ covalent bonds formed by atoms (σ-holes) or above and below a planar region of a molecular entity (π-holes). According to this perspective, the σ-11,12 and π-hole bonds13,14 are the short and attractive contacts between these regions, acting as electrophilic sites (Lewis acidic sites), and regions of excess electron density (e.g., lone pair orbitals or π covalent bonds), acting as nucleophilic sites (Lewis basic sites). The linear and orthogonal geometries of σ- and π-hole bonds are a consequence of the location of σ- and π-holes in the outer regions of atoms and molecules.
When a σ covalent bond is formed, the accumulation of the electron density in the bond region occurs at the cost of a depletion outside the bond, this depletion aligns collinearly with the bond and generates the σ-hole (electrostatic viewpoint). But upon the formation of a σ covalent bond, an antibonding (higher energy) and non-occupied orbital (σ*) is also formed. An attractive interaction occurs when this empty orbital (electrophilic site) overlaps with a lone-pair or a π-bond orbital (nucleophilic sites) and a transfer of electron density ensues (n/π → σ* charge-transfer interaction).
Similarly, an attractive interaction results from the overlap and transfer of electron density between a lone-pair or a π-bond orbital and an empty π* orbital. Understanding the distribution of electron density in molecules similar to the approach used by electronic engineers when talking about electron holes, the empty σ*/π*orbitals that can accept electron density from an occupied orbital (and indeed do so) can be considered σ/π-holes and the resulting interactions can be named σ/π-hole bonds. According to the charge-transfer rationalization, the linear and perpendicular geometries of σ- and π-hole bonds are a consequence of the localization and orientation of the antibonding orbitals accepting electron density from the nucleophile.
Finally, it may be useful to observe that the anion⋯anion self-assembly driven by HBs and σ/π-hole bonds are not mutually exclusive processes but can occur concomitantly and cooperate in constructing anionic supramolecular architectures. This is the case, for instance, with hydroselenite anions (HSeO3−)29 in hybrid organic–inorganic salts which form structurally different infinite chains where anions are doubly pinned to each other by a synthon composed of one HB and one ChB (Scheme 1, top) or where dimers bonded via two H⋯O HBs are connected via two Se⋯O ChBs (Scheme 1, bottom).
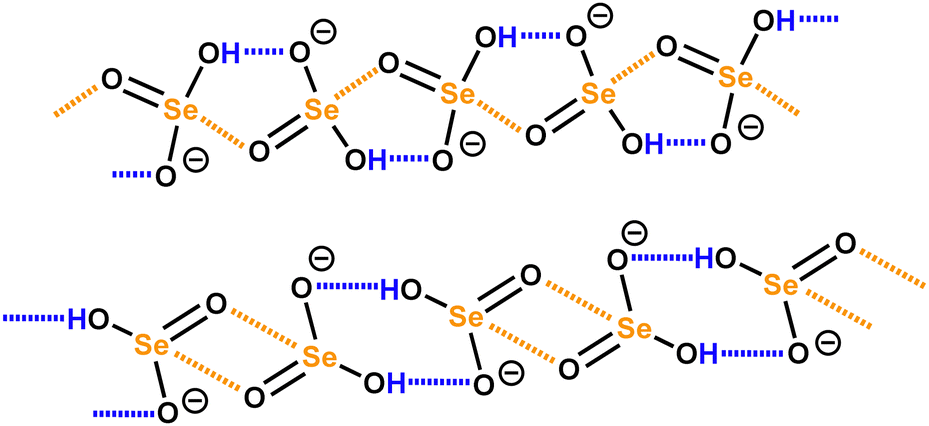 | ||
| Scheme 1 Infinite chains formed by HSeO3− anions under synergistic action of ChBs and HBs.29 | ||
2. Generalities on anion⋯anion adducts in the solid, liquid, and gas phases
In isolated anions, σ- and π-holes are found at the expected positions and the MEP value is also negative in these regions, while less negative than elsewhere. This may lead to the expectation that the approach of an anion to these holes is electrostatically disfavoured, but, as exemplified onwards, this may not always be the case. In general, attractive interactions between like-charges54 can be treated as Coulombic if electrostatics and polarization are both considered.55 In this analysis, dispersion is included as part of polarization.56 The electrostatic potential of any region in a molecular entity creates an electric field which affects the charge distribution in any other region of the same and nearby molecular entities (polarization). The electrostatic potential of two, or more, units in an adduct is thus different from that of the same units when isolated and in their ground state. If the ground state potentials of two isolated units have the same sign and one of them is weak, this polarizing force is enough to overcome the ground state electrostatic repulsion of the isolated units and to enable attractive interactions. In anion⋯anion adducts, the interacting region with the locally more negatively charged potential functions as the donor of electron density (nucleophile) while the interacting region with the locally less negatively charged potential is still capable of functioning as the acceptor of electron density (nucleophile).57In the solid state and in solution, anions are not present as isolated species as they are in close proximity to surrounding cations and solvent molecules. The MEP of anion–cation pairs (computed either in the crystallographic or optimized geometry) show the presence of σ/π-holes on the anionic units where the electrostatic potential is less negative than in isolated anions or it is even positive, making them suitable to interact attractively with atoms having a negative potential (Fig. 1 and 2).25,26
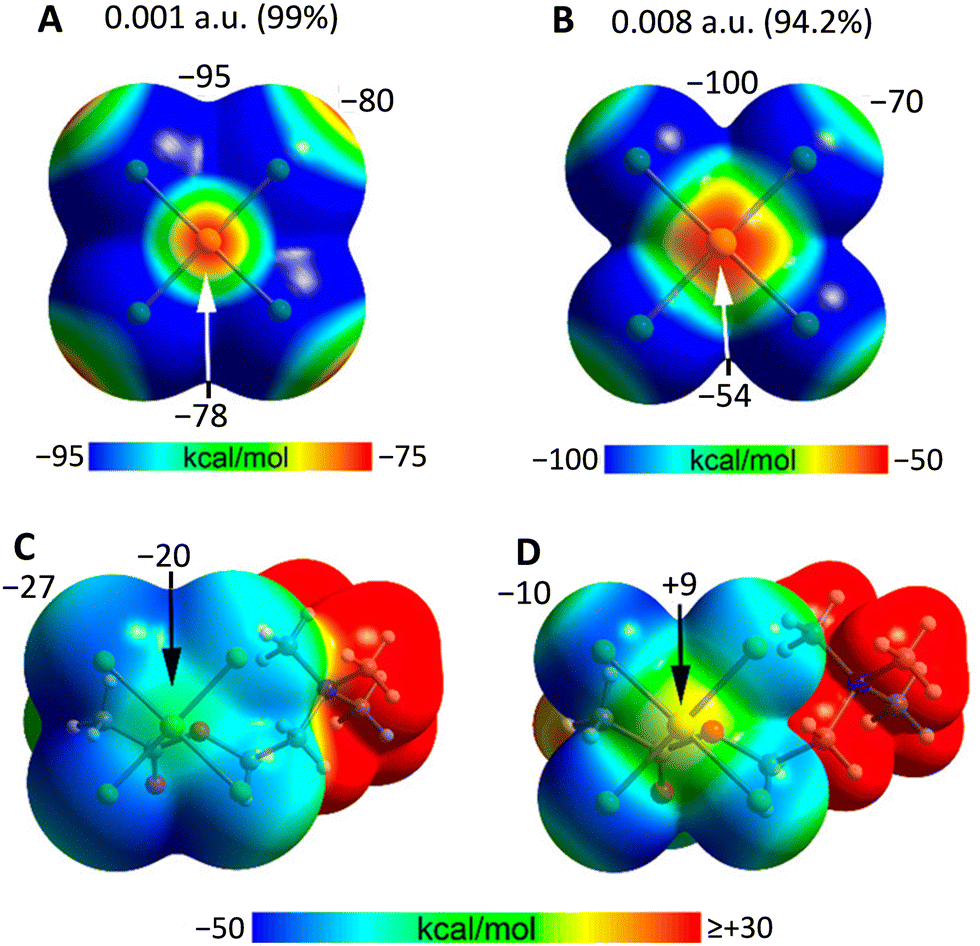 | ||
| Fig. 1 MEP surfaces of the isolated AuCl4− anion and its acetylcholine ion pair26 using 0.001 a.u. (A) and (C) and 0.008 a.u. (B) and (D) isovalues at the PBE0-D3/def2-TZVP level of theory. Some values of the surface electrostatic potential are given in kcal mol−1 at the given points. | ||
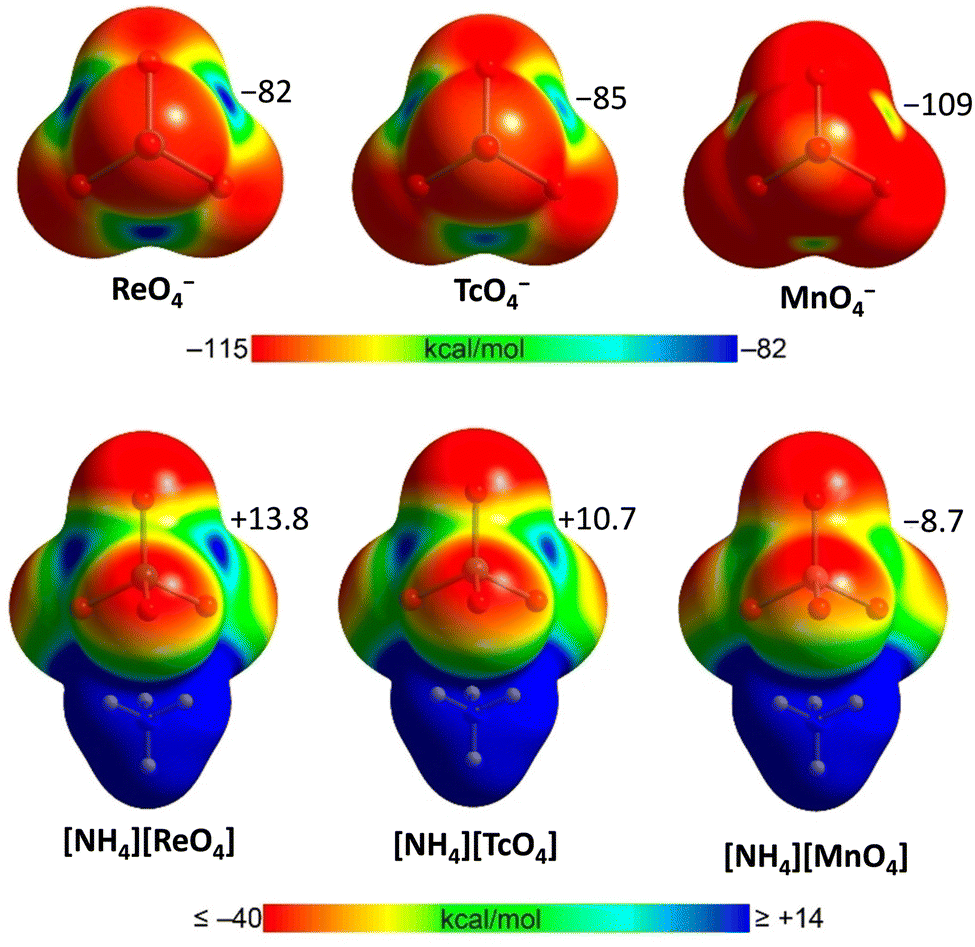 | ||
| Fig. 2 MEP surfaces of MaO4− (Ma = Mn, Tc and Re) anions and their ammonium salts25 at the PBE0-D3/def2-TZVP level of theory. The values at σ-holes are given in kcal mol−1. | ||
It is also important to note that, following Bader's recommendation58 MEPs are routinely calculated using the 0.001 a.u. isovalue as it embraces ca. 99% of the electron density and provides a quite good estimate of the van der Waals surface. But the anion⋯anion interactions considered in this review occur at distances shorter than the van der Waals radii and the electrostatic component of their nature may be better estimated by using surfaces smaller than the van der Waals one, i.e., by considering isovalues greater than 0.001 a.u. The MEP values at σ- and π-hole sites progressively increase with the isovalue,26 particularly when they approach the nuclei of atoms. Indeed, at distances comparable to those of the observed anion⋯anion interactions they may become positive when they are negative if considered on the 0.001 a.u. isovalue surface (Fig. 1).
According to the charge-transfer viewpoint, in anion⋯anion adducts, the superposition between an occupied orbital of the anion acting as the donor of electron density and an empty antibonding orbital in the hole region of the other anion results in a charge-transfer stabilization which, depending on the adduct and the adopted modelling tool, can be a major attractive component in the interactions.50,59 Importantly, this stabilizing force acts independent of the like-charge of the two anions and of the electrostatic potential in the regions involved in the interaction.
The ability of anions to form anion⋯anion adducts in the condensed phases is also affected by the overall interactional landscape in which they are involved. Specifically, interactions formed by anions with electrophiles (e.g., the HBs or HaBs formed by oxygen atoms of oxoacid anions with positively charged hydrogen atoms of cationic units or with electrophilic halogen atoms) dissipate the net negative charge of the anions; this increases their surface electrostatic potential and helps in the possible formation of close and attractive contacts with other anions.60,61 The stronger and more numerous these anion–electrophile interactions are, the more extended and less negative, or more positive, the anion σ/π-hole(s) are, and the more favoured the formation of stable anion⋯anion adducts is.
σ/π-Hole interactions frequently allow anion⋯anion adducts to be metastable species in the gas phase, namely, the diagram of the binding energies of the two anions vs. the anions separation show a local minimum which is positive. Calculations on the 32 dimers formed by the four halide anions with the eight anionic HaB donors depicted in Table 1 reveal that in the gas phase 18 of them are thermodynamically unstable but kinetically stable with respect to the isolated species and the others dissociate spontaneously (Table 1 and Fig. 3).62 The relatively large set of data allow making some general observations. The local minimum of the dimer binding energy can be fairly deep. The largest dissociation barriers are found for the most stable minima, while the smallest ones are obtained for the least stable minima. Iodocarbons form more stable dimers than structurally analogous bromocarbons and as far as the donor of electron density is concerned the binding energies follow the following order: F− < Cl− < Br− < I−.63
| Halide anion |
|
|
|
|
||||
|---|---|---|---|---|---|---|---|---|
| X = Br | I | Br | I | Br | I | Br | I | |
| Gas phase | ||||||||
| F− | 133.2 (259.7) | 89.8 (241.9) | 147.4 (268.2) | 109.9 (247.9) | 155.9 (272.3) | 118.2 (248.9) | 115.9 (233.3) | 58.5 (227.8) |
| Cl− | — | 125.6 (335.9) | — | 137.8 (349.9) | — | 143.3 (356.9) | 140.0 (328.5) | 111.9 (310.8) |
| Br− | — | 127.9 (362.6) | — | 138.6 (385.4) | — | — | 140.4 (366.9) | 116.8 (335.6) |
| I− | — | — | — | — | — | — | — | 120.2 (366.2) |
| Water solution | ||||||||
| F− | −2.5 (286.5) | −16.28 (262.6) | 2.0 (309.9) | −5.8 (279.2) | 0.9 (413.7) | −2.4 (288.1) | −10.0 (263.9) | −34.0 (241.4) |
| Cl− | — | −7.9 (342.5) | — | −3.3 (357.6) | — | −2.2 (366.7) | −6.5 (330.5) | 16.2 (321.1) |
| Br− | — | −8.1 (358.0) | — | −3.3 (372.2) | — | — | −7.2 (345.5) | −15.9 (339.2) |
| I− | — | — | — | — | — | — | — | −15.4 (361.7) |
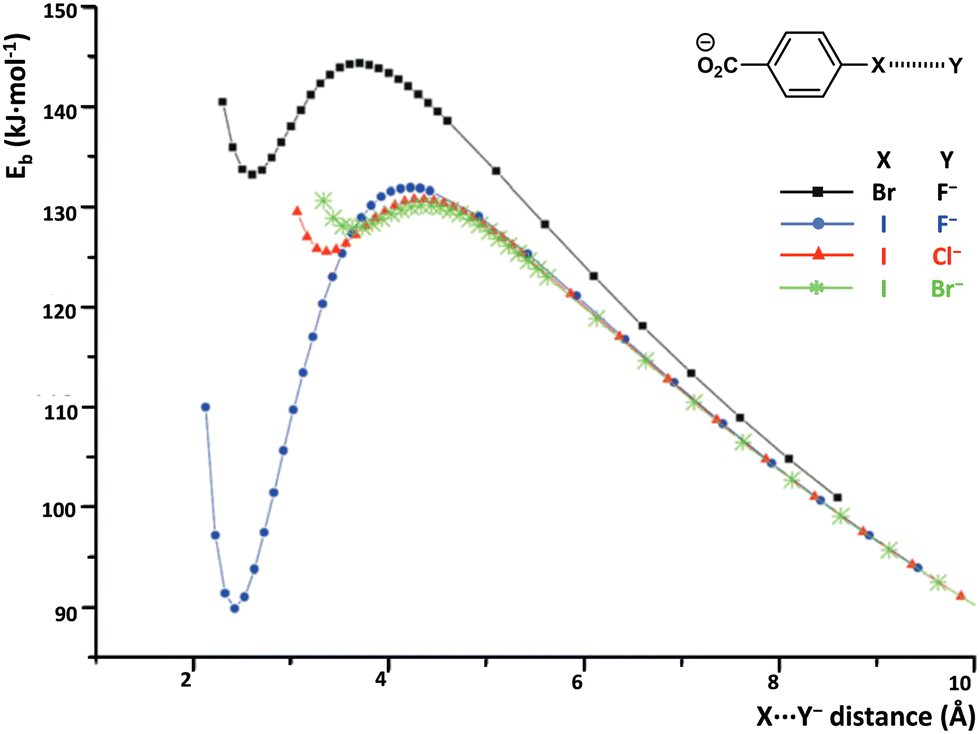 | ||
| Fig. 3 Binding energy (Eb, kJ mol−1) vs. Br/I⋯halide anion distance of 4-bromo- and 4-iodo-benzoate complexes in the gas phase.62 | ||
In solution, the dielectric environment of polar solvents effectively lowers the electrostatic repulsion between like-charges, stabilizing the anion⋯anion adducts, and the local minimum in the diagram of the binding energies of the two anions vs. their distance decreases in all cases and frequently becomes negative (Table 1), indicating that the interactions become thermodynamically stable. Computational analyses24,25,64–66 and experimental results67 on various systems consistently show that the stabilizing effect increases with the dielectric constant of the solvent. UV-vis analyses of solutions containing an anionic and iodinated cyclopropenylium-based donor of HaB and chloride, bromide, or iodide anions gave experimental proof of the presence of complexes between the organic anion and all three inorganic anions. Stabilities of these adducts are comparable to those formed when the same halides bind to common neutral HaB donors, e.g., bromo- and iodo-alkanes and -arenes (Scheme 2).67 These findings confirm that the HaBs between anionic HaB donors and halide anions are strong enough to overcome the Coulombic repulsion between like-charges, even in solution.68 Moreover, they experimentally support the numerous computational studies which suggest that a weak covalent component contributes to the formation of halogen bonded complexes between two anions.
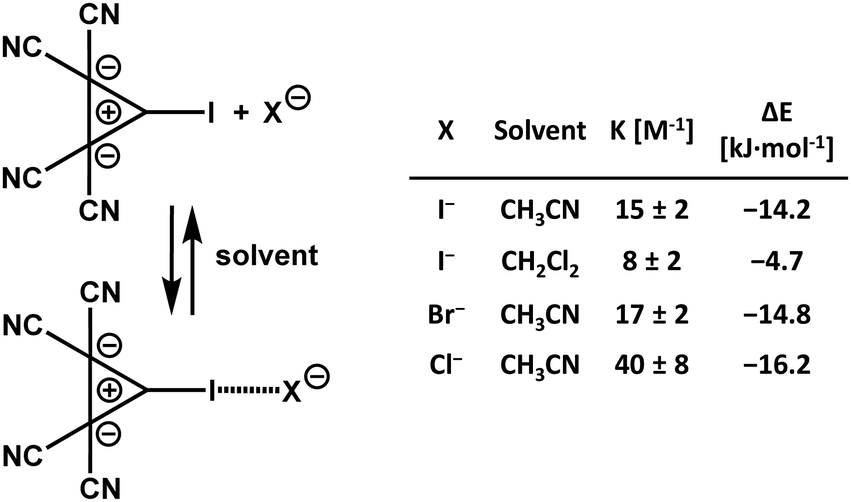 | ||
| Scheme 2 Formation of halogen bonded adducts between an anionic and iodinated bis(dicyanomethylene)cyclopropanide derivative and halide anions.67 | ||
The dielectric constant of any crystalline solid is unknown, but it can be expected that the ionic environment in a crystalline salt is even more effective than a polar solvent in lowering the Coulombic repulsion between anions and in stabilizing anion⋯anion adducts. This may explain why numerous structures in the Cambridge structural database (CSD)69 show numerous short anion⋯anion contacts which can be rationalized as σ/π-hole attractive interactions. It also supports the general ability of σ/π-hole interactions between anions to function as reliable tools in crystal engineering.
Finally, in order to evince how the overall molecular structure of a polyatomic anion affects its ability to function as the donor of σ-hole interactions and determines the stability of the adducts formed with other anions, a computational study was performed on anion⋯anion dimers formed by ω-iodo-carboxylates wherein the iodine atom and the carboxylate group are separated by polyyne, polyene, or polymethylene chains of different lengths. As the distance between the iodine (the HaB donor atom, i.e., the electrophilic site) and the carboxylate group (the HaB acceptor site, i.e., the nucleophilic site) increases, the energy of the dimer tends to decrease with respect to the two separated anions as reported in Scheme 3 for dimers between iodocarbon anions and a chloride anion.70 While shorter spacing chains produce anion⋯anion dimers which are metastable in the gas phase (positive binding energies), the longer ones can yield stable dimers (negative binding energies). Polymethylene chains are more effective in stabilizing the anion dimers than polyene chains with the same number of carbon atoms.
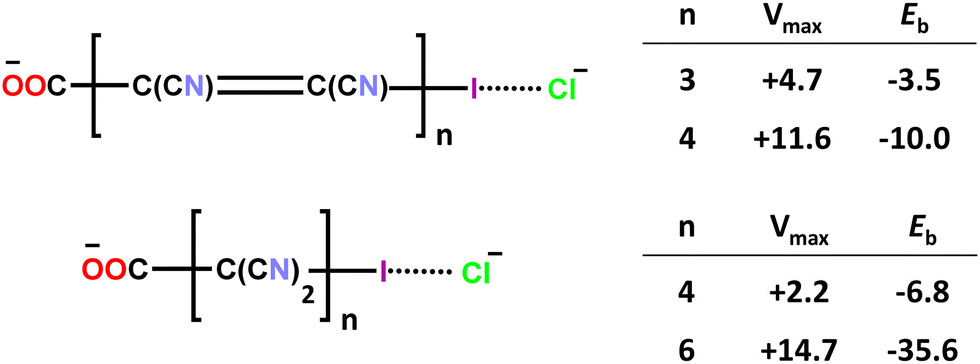 | ||
| Scheme 3 Electrostatic potential at the σ-hole of iodine (Vmax) in some ω-iodocarboxylates and binding energy (Eb) of these anions with chloride anions; both values are in kcal mol−1.70 | ||
3. Adducts assembled by σ–hole interactions
Donors of electron density can engage in attractive interactions when they approach σ/πholes centred on a wide diversity of elements. This also applies to the subset of interactions wherein the donor and the acceptor of the electron density are both anions. This section and the following one record the anion⋯anion adducts formed under the influence of σ- and π-hole interactions by dividing them in subsections devoted to systems wherein the element hosting the hole belongs to the p or d blocks of the periodic table.3.1. σ-Hole interactions at p block elements
Boron, aluminium, gallium, indium, and thallium, the five common elements of group 13, are collectively named triels (Trs) and the σ/π-hole interactions involving them as electrophilic atoms are usually designated as triel bonds (TrBs).71 In the solid state, their tetrahalide anions (TrX4−) adopt a tetrahedral geometry and structures are present in the CSD wherein these anions are assembled into X4Tr−⋯TrX4− adducts (Tr = B,72–75 Al,76 In,77,78 and Tl;79 X = F,72–75 Cl,76,77,79 and Br78) via one or more Tr⋯X short contacts between different anions. A systematic study of several triel tetrahalides confirmed computationally the attractive nature of these TrBs.80 This study showed that the Coulombic repulsion is counteracted by large polarization energies and the formed dimers are typically metastable in the gas phase and stable in solution. Bond paths and bond critical points (CPs) were frequently observed for the Tr⋯X TrBs. Similar to the σ/π-hole interactions involving neutral units, the strength of interanion TrBs increases with the atomic weight of the Tr atom.Computational analyses at different levels indicate that heteromeric anion adducts can be formed on interaction with CN− anions. Specifically, for the tetrachlorides of the four heaviest triels (Al, Ga, In, and Tl), the more stable adducts are two structurally different Cl4Tr−⋯CN− complexes (encoded as E and A isomers, respectively, Table 2), both of which have carbon of the cyano group positioned close to the triel and both adopt a distorted trigonal bipyramidal geometry with the cyano group in the equatorial or axial position.81 Two other complexes with the nitrogen of the cyano group close to the triel are less stable. When the Cl4Tr− and CN− anions come close to each other, the tetrahedral geometry of the isolated TrCl4− anion restructures and forms either the E or A isomers, locating the four chlorine atoms in a distorted see-saw geometry or in a pseudo trigonal pyramidal geometry. The energetic cost of these rearrangements, while smaller for the larger triels, is non-minor in all cases. These restructurings nevertheless occur as they help complex formation by inducing a much more positive, or less negative, electrostatic potential at the positions that are to be occupied by the cyano group in the complexes. The Cl4Tr−⋯CN− dimers are formed as the resulting σ-hole TrBs between the TrCl4− and CN− anion are strong enough to overcome the long-range Coulombic repulsion between their like charges. In the gas phase, these complexes are characterized by positive binding energies, but they are metastable, namely, an energy barrier opposes their dissociation. In water solution they become stable and the binding energies are more negative than those of some complexes wherein anions interact with neutral donors of electron density (e.g., Cl4Tr−⋯NH3), consistent with the general tendency of anionic donors of electron density to form stronger σ-hole interactions than neutral donors.
E
b(gas![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) phase) phase)
|
E
b(watersol.)
|
|||
|---|---|---|---|---|
| MP2 | CCSD(T) | MP2 | CCSD(T) | |
| E isomer | ||||
| Cl4Al−⋯CN− | 48.78 | 50.99 | −9.11 | −6.63 |
| Cl4Ga−⋯CN− | 49.10 | 51.53 | −8.54 | −5.80 |
| Cl4In−⋯CN− | 36.98 | 39.13 | −17.06 | −14.59 |
| Cl4Tl−⋯CN− | 37.78 | 40.23 | −15.61 | −12.72 |
| A isomer | ||||
| Cl4Al−⋯CN− | 48.85 | 50.98 | −8.81 | −6.47 |
| Cl4Ga−⋯CN− | 50.59 | 52.89 | −6.45 | −3.92 |
| Cl4In−⋯CN− | 37.28 | 39.31 | −16.01 | −13.64 |
| Cl4Tl−⋯CN− | 38.13 | 40.44 | −13.85 | −11.22 |
| Cl4Tl−⋯NH3 | −22.07 | −19.95 | −6.01 | −4.43 |
A computational study rationalizes82 the Cl4Pn−⋯CN− complexes (Pn = P, As, and Sb) as systems formed under the control of the pnictogen bond (PnB), the σ/π-hole interaction wherein the electrophilic atom is the group 15 element.83 On approach of the CN− anion, PnCl4− rearranges from its see-saw geometry, the preferred conformation in the solid and gas phases, to the square geometry and a minimum on the potential energy surface is found for complexes with the CN− ion in apical position with the carbon atom pointing to the pnictogen atom. These complexes are metastable in the gas phase, meaning their dissociation requires the two monoanions to pass an energy barrier, but in water solution they are more stable than the isolated monoanions, as it happens in several other systems. These polyanion complexes can be rationalized not only as systems formed under the control of σ-hole PnBs involving the PnCl4− anion in the see-saw conformation, as described above, but also as systems formed under the control of π-hole PnBs involving the PnCl4− anion in the rearranged square conformation. An analysis of the CSD confirms the tendency of the pnictogen atom in PnX4− monoanions (Pn = As, Sb, Bi; X = F, Cl, and occasionally Br, I) and in related species (e.g. Pn2X7−) to form short contacts with other anions (e.g., with X− or with another PnX4−) and to form supramolecular anionic adducts having quite different structures and charges.84–87
Experimental and theoretical results show that the self-assembly of anion⋯anion adducts can be driven by σ-hole ChBs involving S, Se, and Te atoms of both organic and inorganic anions. Dithieno[3,2-b:2′,3′-d]thiophene and its analogues can bind anions via convergent bidentate ChBs.88 The ChB driven binding of a chloride anion by some dithieno[3,2-b:2′,3′-d]pyrroles bearing a deprotonated dicyanomethylene pendant on the bridging nitrogen, and by the diselenopheno and ditelluropheno analogues, has been studied computationally.89 The stabilizing charge-transfer, polarization, and dispersion components of the binding energy become monotonically more and more negative as the anions get together. As expected from classic electrostatics, the electrostatic component experiences a Coulombic repulsion that increases with the shortening of the Ch⋯X− distance. This happens before reaching a given threshold (the barrier separation), and after passing this threshold it decreases as the Ch⋯X− distance shortens and at quite close separations it becomes even more favourable than some other energy components. As a result, characteristic energy minima are present in the diagrams of the binding energy vs. the Ch⋯X− distance for all the examined systems. The local minimum of the dimer binding energy is positive in most dimers (kinetically stable adducts), it decreases when moving from S, to Se, to Te, and it is negative for some of the studied ditelluropheno dimers (thermodynamically stable adducts). An analogous dependence of the stability of anion⋯anion adducts assembled by σ/π–hole interactions on the polarizability and electronegativity of the atom hosting the hole has been described above for triel bonded systems and will be discussed further for systems involving elements of groups 7 and 17. Clearly, the general tendency of σ/π-hole interactions involving neutral units to become more stabilizing with the increase in the polarizability and the decrease in the electronegativity of the atom hosting the hole, is equally applicable in σ/π-hole interactions involving anionic units.
Approximately one fourth of the CSD structures that contain the HSeO3− unit shows the presence of anion⋯anion adducts assembled via Se⋯O ChBs between two anions.90–92 The effectiveness of this interaction and its reliability in crystal engineering has been proven by the design of chalcogen bonded anion⋯anion dimers and infinite chains (Scheme 1).29 Interestingly, the same paper describes that selenium is a quite strong ChB donor also in the diselenite dianion (Se2O52−) as it forms two-dimensional (2D) anion⋯anion architectures via Se⋯O ChBs which can be 302.9 pm long, characterized by a normalized contact93 as small as 0.88 (Fig. 4). A combined quantum theory of atoms-in-molecules (QTAIM) and noncovalent interaction plot (NCIPlot) analysis identifies a bond path and a bond CP for the short Se⋯O contacts observed in the crystals. A green and bluish reduced density gradient (RDG) isosurface characterizes these interactions so that their attractive nature is confirmed, in spite of the overall Coulombic repulsion between the two anions.
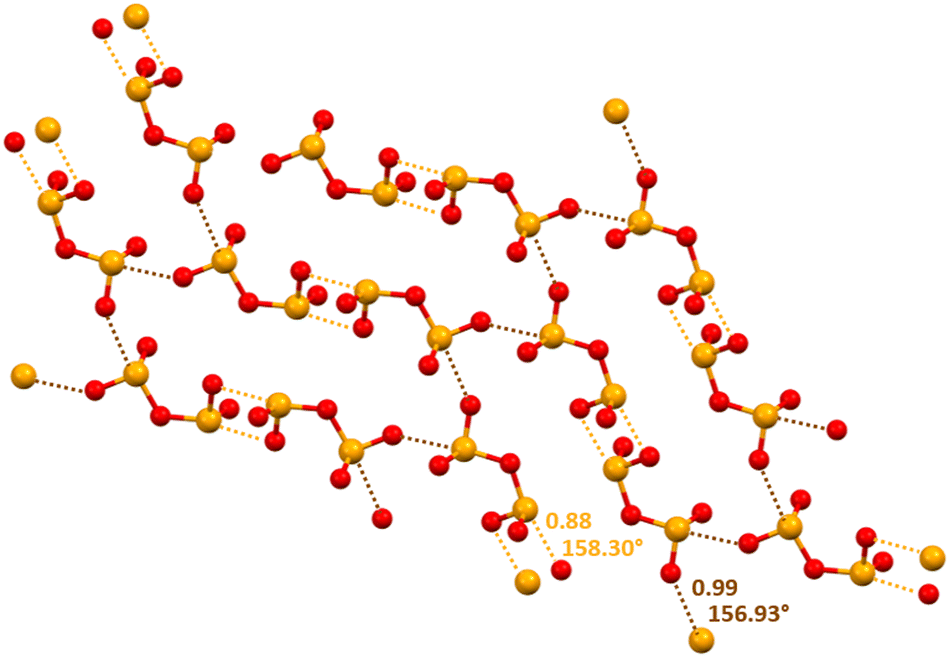 | ||
| Fig. 4 Partial representation (ball and stick, Mercury 2022.1.0) of the 2D net assembled via ChBs in a salt between diprotonated diazabicylooctane and diselenite dianion (Refcode BOGWEG).29 Short and long ChBs are ocher and brown dotted lines; ChB angles and Nc values are reported close to the interactions. Colour code: red, oxygen and orange, selenium. | ||
Structures in the CSD94,95 and computational studies96 indicate that the aryl-tetrahalotellurate anion and the alkyl-tetrahalo analogues as well as the corresponding selenium and sulfur species (aryl/alkyl-ChX4−, Ch = S, Se, and Te; X = Cl, Br, and I) form dimers through the antiparallel pairing of the Ch–X moiety of different anions. The main attractive components of these ChBs are polarization and charge-transfer (from the halogen lone pairs to the chalcogen), and the formed dimers are metastable in the gas phase and stable in water solution.
The casuistry of anion⋯anion adducts assembled via HaBs is wider and more diversified than that of adducts assembled via other σ-hole interactions,15 consistent with HaBs being the most extensively studied σ-hole bond.
Polyiodides are well-known systems which have received great attention thanks to their useful applications and highly diversified structures. As stated by L. Kloo in his review “Examples of everything from very simple discrete units to one-dimensional chains and complicated two and three-dimensional networks all occur.”97 To a lesser extent, this is also true for other homo- and heteromeric polyhalides.98 Even the formation of as extreme systems as stable Cl−⋯Cl− dimers has been reported.99 Some polyhalide anions result from the interaction of an halide anion with one or more dihalogen molecules; some others are formed via the interaction of two or more (poly)halides and here we will discuss these latter systems only. The bonding nature in polyiodides has been the subject of much theoretical speculation and traditionally their formation has been rationalized via the donor–acceptor and multicenter bonding modellings. Often their geometric features are consistent with those expected for σ-hole adducts and recent theoretical studies have confirmed this rationalization for an ever-increasing number of such systems.98
The electrostatic potential of I3− is negative on its entire surface but more negative in regions orthogonal to the main axis of the ion and least negative at two caps located on the external iodine atoms and along the main axis (Fig. 5A). This implies that the electrostatic repulsion in I3−⋯I3− adducts is minimized when the region orthogonal to the ion main axis of one I3− anion approaches the cap of the external iodine of the other I3− anion. Indeed, in several structures of the CSD, nearby triiodide units adopt an orthogonal orientation and dimers100 or infinite chains101 can be formed (Fig. 5B and D).
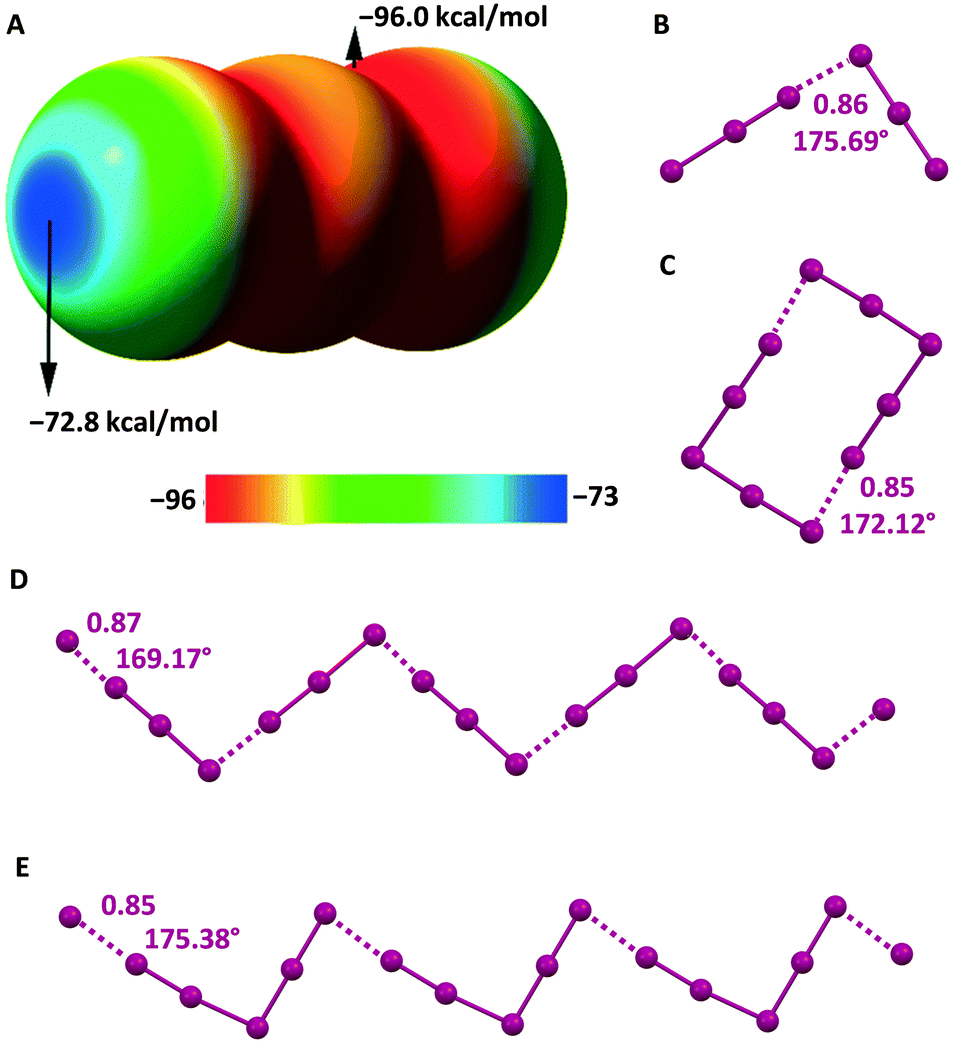 | ||
| Fig. 5 MEP surface of I3− using the 0.001 au isosurface (calculated at the M06-2X/def2-TXVP level of theory) (A).102 Ball and stick representation of: I3−⋯I3− dimers in an L-tyrosinium salt (Refcode ILUSUF)100 (B) and the I3−⋯I3− infinite chain in the salt of a (tetrathiacyclotetradecane)–palladium(II) derivative (Refcode UHANUM)101 (D); I5−⋯I5− dimers in the salt of the radical cation of a tetrathiafulvalene derivative (Refcode CUTXUN)103 (C) and the I5−⋯I5− infinite chain in 4-(pyrimidin-2(1H)-ylidenesulfamoyl)anilinium pentaiodide (Refcode JUPDEF)107 (E). Cations have been omitted for the sake of clarity; HaBs are violet dotted lines; HaB angles and Nc values are reported close to the interactions. | ||
Interestingly, NCIPlot analysis reveals that a green isosurface is found between the interacting iodine atoms, namely, the HaB observed in the crystal is attractive, and this is also the case when the two anions adopt a binding geometry quite different from the preferred orthogonal arrangement described above.102
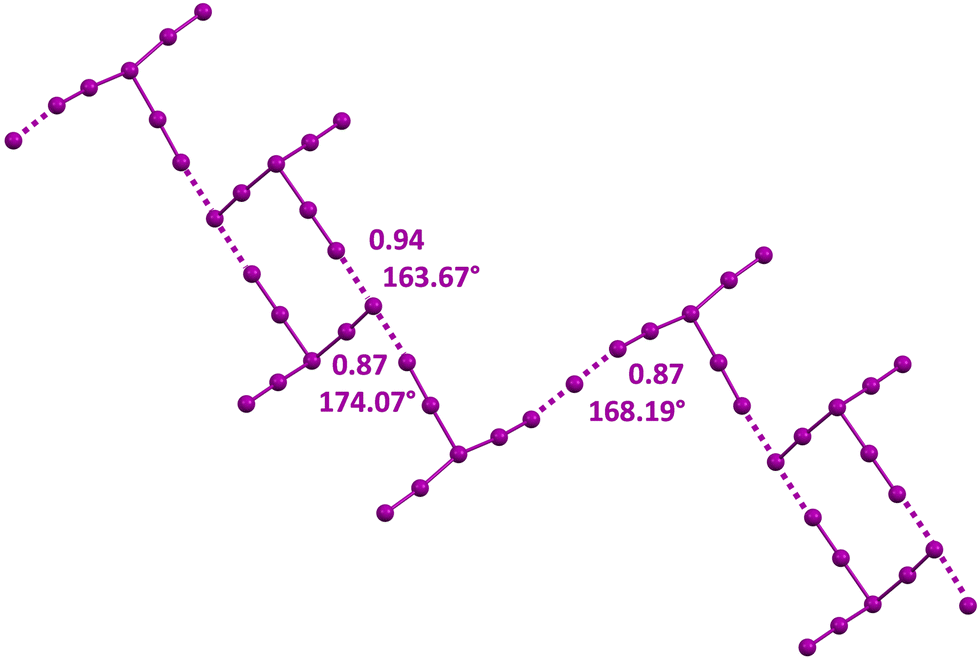
An anisotropic distribution of electron density is also present in polyiodides wherein two or three iodine molecules are bonded to a central iodide, namely, in the V shaped I5− anion and in the I7− anion adopting a trigonal pyramidal geometry. In these anions, a higher and lower electron density is present orthogonal to the I–I covalent bonds and at the cap of the external iodine atoms opposite to these bonds. Consequently, I5− and I7− anions self-assemble into adducts preferentially adopting an orthogonal binding geometry similar to that described above for the I3− anion. Discrete supramolecular polyanions,103,104 infinite chains,105–107 (Fig. 5C and E) or even more complex topologies108 (Fig. 6) are formed. The convenient design of the cation enables the templating of the structure of the obtained polyhalide through a tailored combination of geometric and electronic features. This is the case, for instance, for the dianions X−⋯I2⋯X−, (X = I,106,109–113 Br,106,111 and Cl59,106,111) and for the trianion114 I−⋯I2⋯I−⋯I2⋯I− (Fig. 7).
 | ||
| Fig. 6 Ball and stick representation of the infinite chain with pendants assembled via HaBs involving I− and I7− anions in a salt of the tetrakis(10H)-phenothiazin-10-ium radical cation (Refcode EWEKUP).108 Cations have been omitted for the sake of clarity; HaBs are violet dotted lines; HaB angles and Nc values are reported close to the interactions. | ||
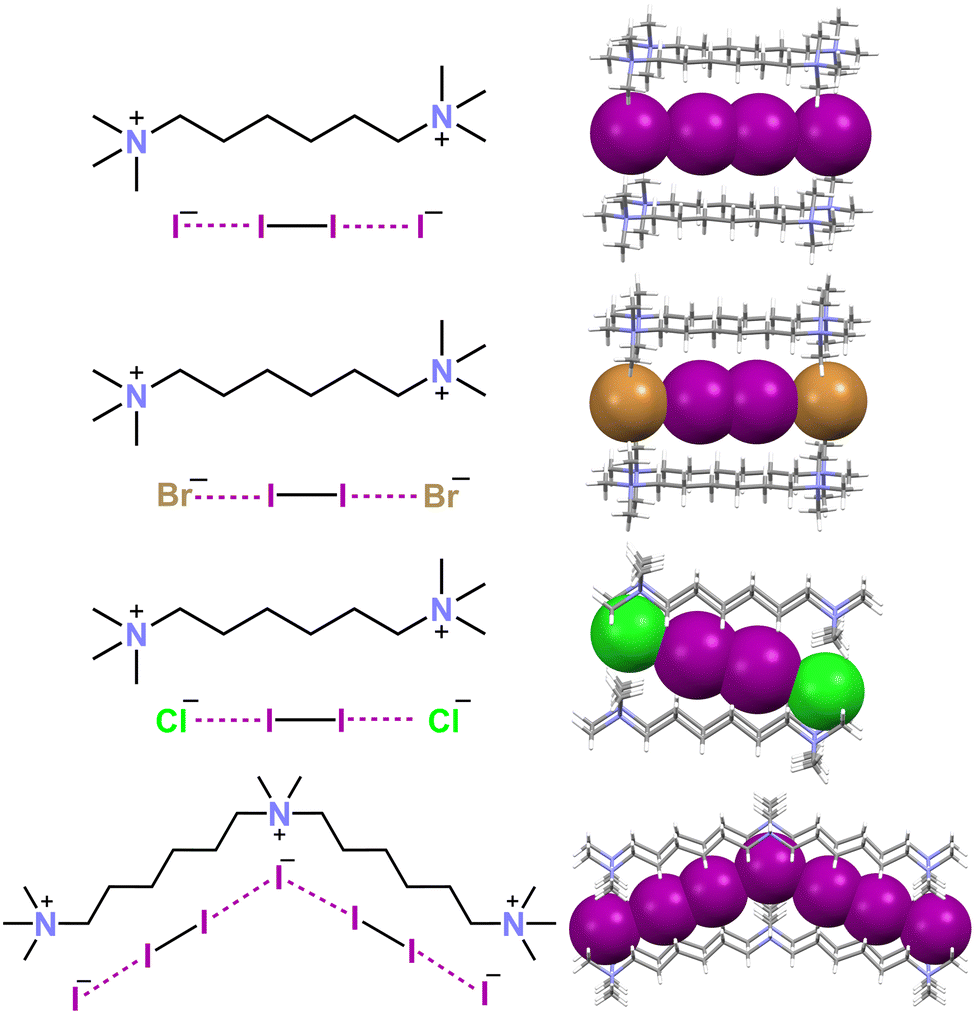 | ||
| Fig. 7 Structural formulas and partial representation of the crystal packing of the dianions X−⋯I2⋯X− (X = I, Br, and Cl) in respective decamethonium salts (Refcodes NUTSOL, DIQBOW, and DIQBUC),109,111 and the trianion I−⋯I2⋯I−⋯I2⋯I− in the N,N,N,N',N'-pentamethyl-N'-(6-(trimethylammonio)hexyl)hexane-1,6-diaminium salt (Refcode PEKKEX).114 Cationic units are depicted as sticks and the anionic ones are shown in space filling representation. Colour code: grey, carbon; indigo, nitrogen; violet, iodine; green, chlorine; and ocher, bromine. | ||
The tendency of different halogen atoms to form HaBs decreases in the order I > Br > Cl > F and the number of CSD structures containing polyhalide anions decreases in the same order but the preferred arrangement of bonded units remains as described above.115 The Br3− anion shows an anisotropic distribution of the electron density similar to that of the I3− anion and its HaB driven assembly into polyanionic supramolecular adducts occurs adopting analogous orthogonal arrangements of interacting units (Fig. 8).116
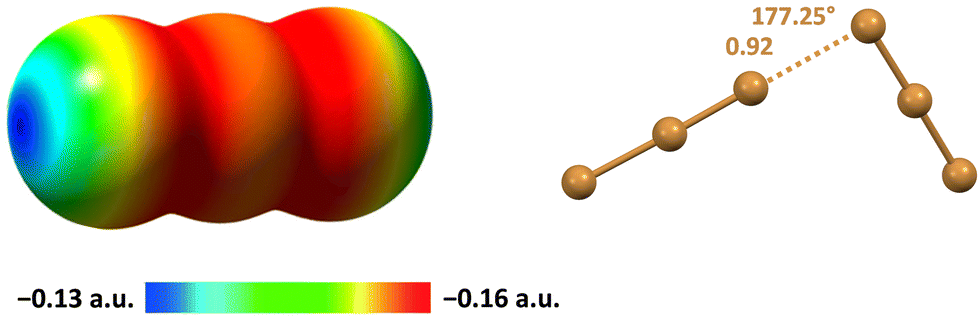 | ||
| Fig. 8 MEP of Br3− using the 0.001 a.u. isosurface (left).115 Ball and stick representation of the Br3−⋯Br3− adduct in the crystal of the tribromide of a tetrahydropyrrolo[2′,3′:3,4]pyrrolo[1,2-a](benzimidazole-1,10-diium) derivative (Refcode IYEVAL) (right).116 Cations have been omitted for the sake of clarity; HaBs are ocher dotted lines; HaB angles and Nc values are reported close to the interaction. | ||
In the CSD, crystalline solids can be found wherein the O4I−⋯IO4− supramolecular synthon drives the formation of infinite chains117 or discrete adducts118,119 (Fig. 9). In these structures oxygen atoms of periodate anions typically act as donors of electron density in quite short HBs with hydrogen atoms of the cation and this may contribute to unmasking the HaB donor potential of iodine atoms by decreasing the electron density on the anion. Moving from this hypothesis some pyridinium periodates (wherein the cation could act as an effective HB donor) were prepared. The single crystal X-ray analyses of these salts confirmed the presence of short HBs and of O4I−⋯IO4− adducts assembled via different patterns of HaBs.119 For instance, in two crystals, dimers were present where the two anions were coupled through the antiparallel pairing of the I–O moieties of different IO4− units (Fig. 9, bottom).
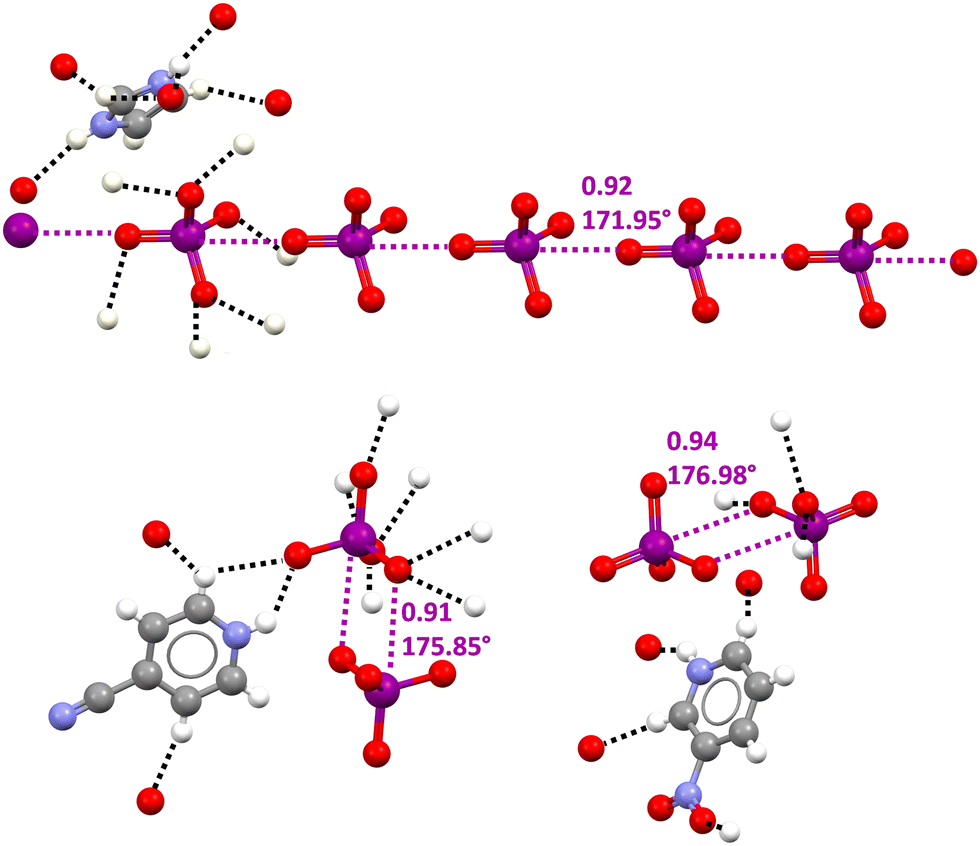 | ||
| Fig. 9 Ball and stick representation of an anionic supramolecular chain assembled via O4I−⋯IO4− HaBs in imidazolium periodate (Refcode JOJYOY)117 (top) and O4I−⋯IO4− dimers assembled via the antiparallel pairing of two O–I bonds in 4-cyanopyridinium and 3-nitropyridinium periodates (Refcode BEKNIS and BEKNOY)119 (bottom). One cationic unit is reported for each structure and the HBs involving this cation and one anionic unit are given (black dotted lines); HaBs are shown in violet dotted lines; HaB angles and Nc values are reported close to the interactions. Colour code: whitish, hydrogen; grey, carbon; red, oxygen; indigo, nitrogen; and violet, iodine. | ||
The MEP of the naked periodate anion is negative on the whole surface but the anisotropic distribution of the electron density generates four symmetric and negative σ-holes on the extension of the four O–I bonds. Differently, the MEP of a pyridinium periodate ion pair in the geometry adopted in the corresponding crystal shows that the electrostatic potential at the σ-holes becomes positive so that also the electrostatic component of the O–I⋯O HaBs observes in the crystals may act as a stabilizing force. The attractive nature of these short O–I⋯O contacts was confirmed by the bond path, bond CPs, and bluish RDG isosurfaces identified by QTAIM/NCIPlot analyses.
CSD analyses reveal that the iodine atom in iodate anions can function as a remarkably effective HaB donor as it can act as a mono-, bi-, and tridentate HaB donor and assemble one,120 two24 and three121 dimensional supramolecular polyanions (Fig. 10, top). Also, bromine in bromate anions can function as a HaB donor24,122 and not surprisingly it seems to be a weaker donor than iodine in iodate anions. In these systems, QTAIM/NCIPlot analyses identified the presence of bond paths, bond CPs, and blue RDG isosurfaces in each O–I/Br⋯O contact. The interaction energies for the dimers in X-ray geometry are positive (repulsive) in the gas phase, but they become negative (attractive) in water solution. The natural bond orbital (NBO) methodology confirms the σ-hole nature of the O–I⋯O contacts identifying the orbital donor–acceptor component (LP(O) → σ*(I–O)) (Fig. 10, bottom).
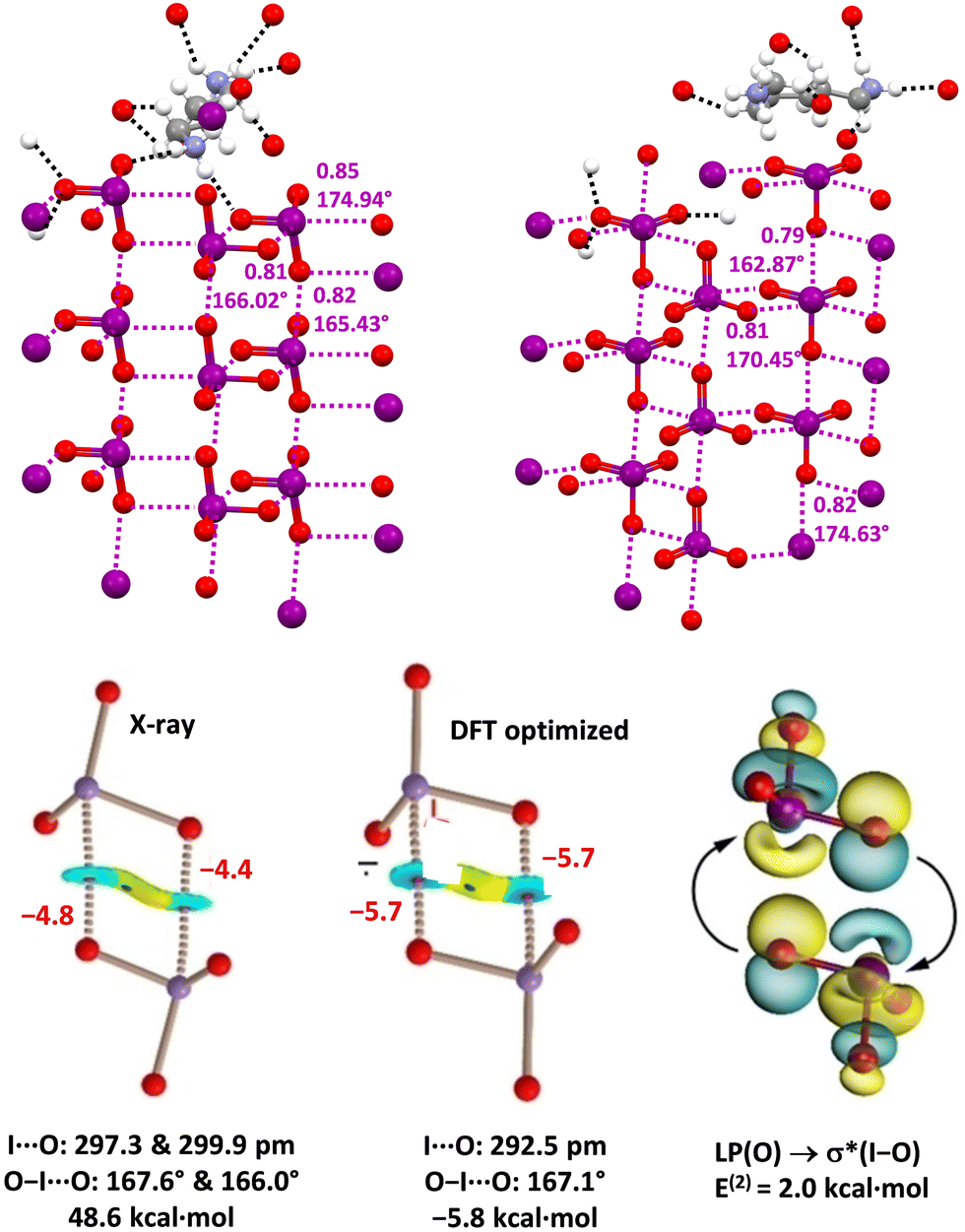 | ||
| Fig. 10 Crystal packing (ball and stick representation) of iodates of 1,4-diammonium-butane (Refcode VITFEN)24 (top, left) and 1,6-diammonium-hexane (Refcode VITFIR)24 (top, right); one cationic unit for each salt and the HBs (black dotted lines) involving this cation and one anionic unit are reported; HaBs are shown in violet dotted lines; HaB angles and Nc values are given close to the interactions. Colour code: whitish, hydrogen; grey, carbon; red, oxygen; indigo, nitrogen; and violet, iodine. QTAIM/NCIPlot analyses of the anion⋯anion dimer in 1,4-diammonium butane iodate with X-ray geometries and in the gas phase (bottom, left) and with optimized geometries with a continuum solvation model in water (bottom, mid); geometric features and interaction energies are shown in black; HaBs energies (kcal mol−1) are shown in red. NBO characterization of LP(O) → σ*(I–O) interactions via analysis for the anion⋯anion optimized dimer (bottom. right); E(2) energy is shown in black. | ||
The HaB driven self-assembly of halocarbon anions into anion⋯anion adducts15 has been studied and identified in the solid state via single crystal X-ray analyses,64,123 in solution via UV-vis67,123 and modelling,57,63 and in the gas phase via modelling.57,62,63 The molecular structures of the used iodocarbon anions are quite different including halo-alkanes, -alkenes, -alkynes, and -arenes; some structures are reported in Table 1 and Scheme 2 while some others are shown in Scheme 4. As expected, the stability of the adducts decreases in the order C–I > C–Br > C–Cl.57,62 Some of the observed HaBs are quite short (e.g., Nc = 0.80)64 and strong (e.g. ΔEint.,water = −5.6 kcal mol−1),63 probably due to the non-minor geometric and electronic separation of the halogen and the atom(s) where the anion negative charge is mainly localized.
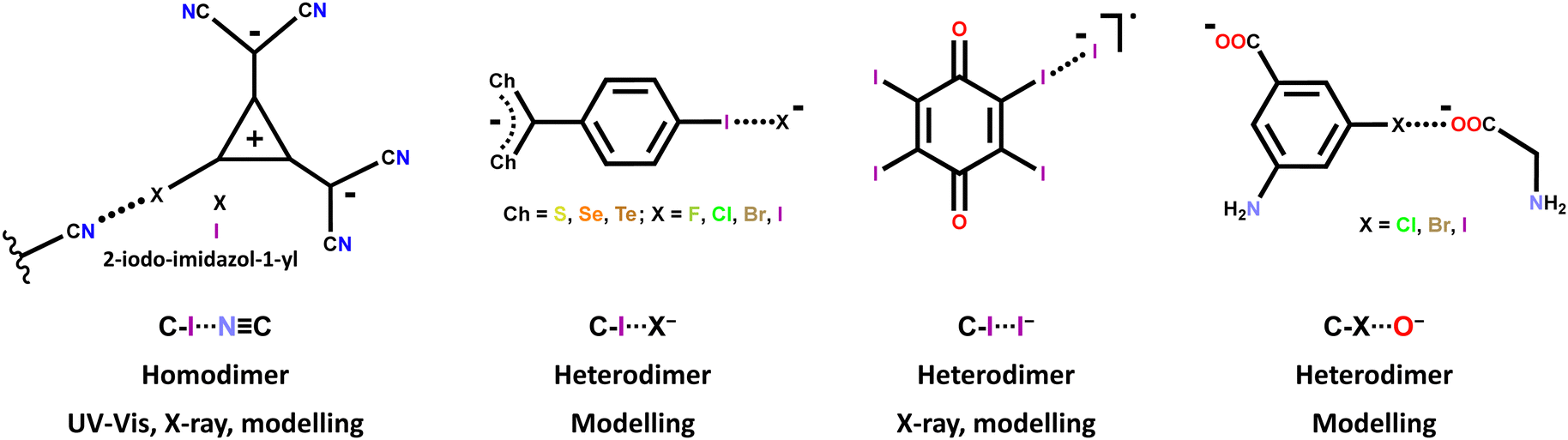 | ||
| Scheme 4 Structural formulas of some halocarbons forming halogen bonded anion⋯anion adducts. The interaction and adduct features and the used analytical techniques are also given. | ||
3.2. σ–Hole interactions at d block elements
Initial studies on σ–hole interactions focused on systems wherein the electrophilic atom was a p block element. In 2014, it was first anticipated that d block elements could also act as the electrophile in this type of interactions.124 The first papers confirming this possibility appeared in 2017125 when it was described that the catalytic abilities of gold nanoparticles is related to the presence of σ-holes at their surface and that these holes serve as binding sites for Lewis bases.CSD structures wherein d block elements form anion⋯anion adducts by acting as electrophiles and forming short contacts with geometric and electronic features typical for σ–hole interactions were reported as early as the nineteen seventies126 but, to the best of our knowledge, the rationalization of these interactions within the framework of σ–hole bonds was first published in 202125 when O4Ma−⋯ O4Ma− adducts (Ma = Re, Tc, and Mn) were obtained by design and their characteristics were thoroughly studied. The term matere bond (MaB) has been proposed to name these σ–hole interactions wherein the electrophile is an element of group 7. Similar to what is observed for the elements of other groups of the periodic table, the tendency of group 7 elements to act as electrophiles in MaO4− anions and to form anion⋯anion complexes, increases with their atomic weight.25 In crystals, two adjacent MaO4− anions can interact with each other through a singly pinning supramolecular synthon (i.e., through the approach of the O of one anion to the metal of the nearby anion) and supramolecular anionic dimers25,127,128 or infinite chains25,129–131 are formed (Fig. 11A and B). Alternatively, two adjacent MaO4− anions can be bonded to each other through a doubly pinning supramolecular synthon (i.e., though the antiparallel pairing of the Ma–O moieties of one anion with the Ma–O moieties of a nearby anion). Most frequently, the group 7 elements function as monodentate donors of MaB, and in such cases, the doubly pinning synthons affords dimers25,60,129,132,133 (Fig. 11D). But occasionally rhenium can also act as a bidentate donor of MaB and in this case discrete adducts134 and 1D or 2D60,134–136 networks are formed wherein the singly pinning synthon can also be present (Fig. 11C, E and F). Interestingly, two rhenium atoms in the polyoxorhenate(VII) dianion Re4O152− can act as MaB donors and form a ribbon with a nice structure (Fig. 11G).130
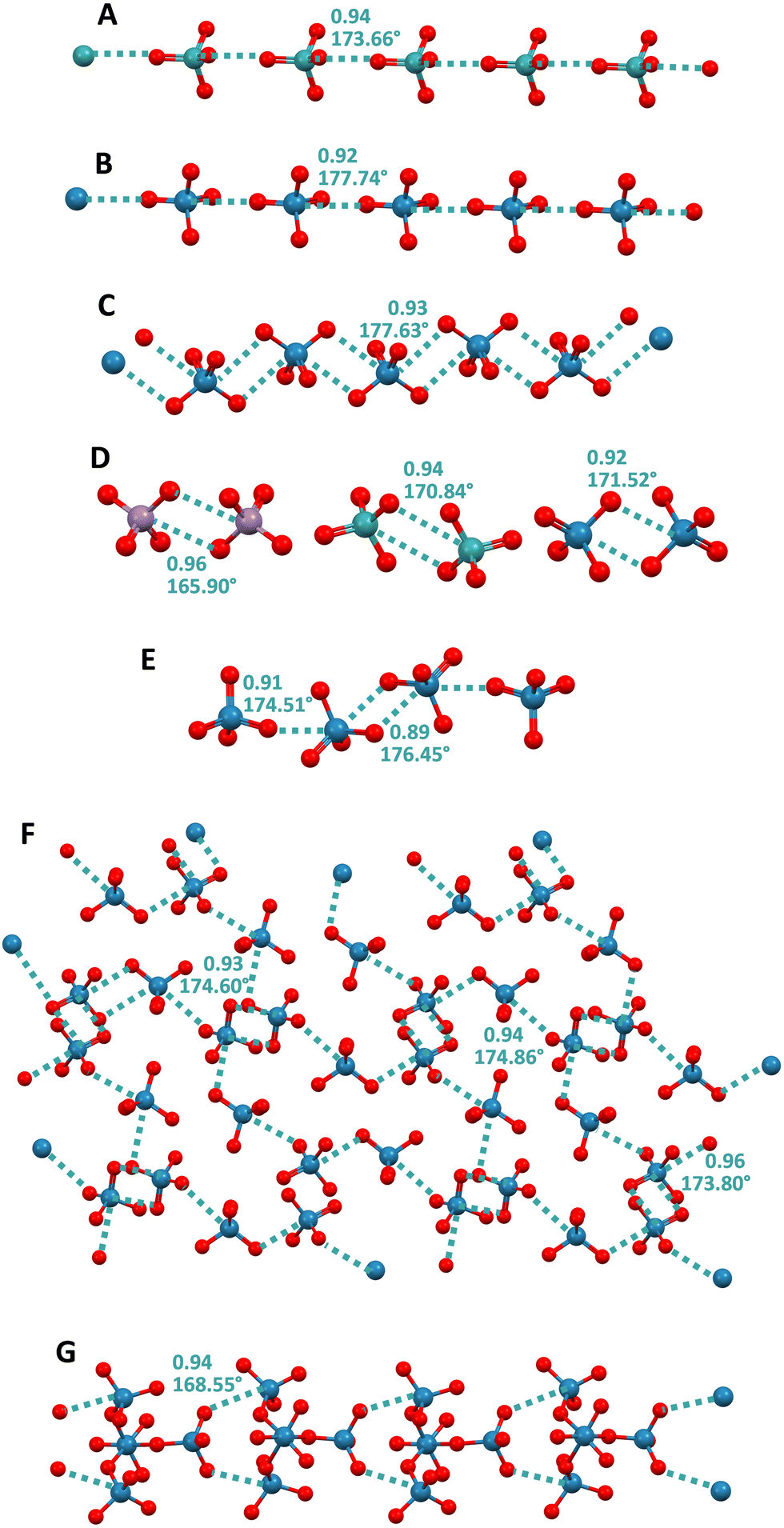 | ||
| Fig. 11 Ball and stick representation of infinite chains formed by TcO4− in its pyrazolium salt (Refcode VISXII)130 (A) and ReO4− in its melaminium salt (Refcode NAPVUZ)25 (B) and in a Rh2+/3+ salt (Refcode DELZUQ)135 (C); dimers of MnO4− in melaminium salt (Refcode NAPWAG)25 (D, left) and of TcO4− and ReO4− in guanidinium salts (Refcodes GEQHUJ and GEQJAR)60 (C, mid and right); tetramer of ReO4− in its salt with a pyridinium derivative (Refcode ZUYKAH)134 (E); 2D net of ReO4− in a salt with protonated N-methyl-DABCO (Refcode FAWDEQ)136 (F); ribbon of Re4O152− in a pyrazolium salt (Refcode VISXAA)130 (G). Cations have been omitted for clarity; MaBs are teal dotted lines. Colour code: red, oxygen; lavender indigo, manganese; teal, technetium; and imperial blue, rhenium. | ||
Experimental techniques (single crystal X-ray analysis, 135/137 NMR and NQR spectroscopy)25,60,129–132,137 and computation (MEP surface, QTAIM/NCIPlot, and Hirshfeld surface analyses)25,60,129,130,133 consistently prove the σ-hole character and the attractive nature of the short contacts observed in the crystals. In isolated MaO4− anions there are four identical σ-holes where the electrostatic potential is negative but less negative than in other regions. In anion–cation pairs, the holes are no more identical and, more importantly, their potential increases and becomes positive in TcO4− and ReO4− (Fig. 2). Bond paths and bond CPs have been found for these interactions and NCIPlot analyses revealed extended green/blue surfaces.25,129
Tetrachloro-nitrido-technetate anions adopt a square pyramidal geometry and afford matere bonded infinite chains129,138via the approach of the nitrogen to the σ-hole at technetium opposite to the Tc![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond (Fig. 12).
N bond (Fig. 12).
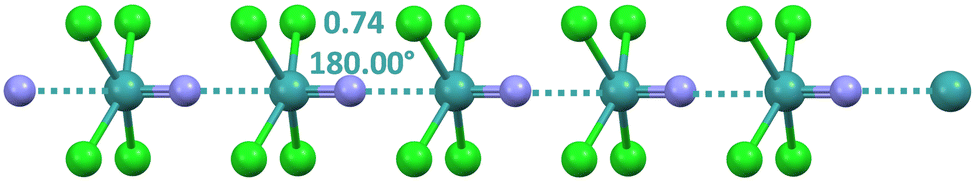 | ||
| Fig. 12 Ball and stick representation of the infinite chain formed by the tetrachloro-nitrido-technetate anion (Refcode SODWIR10).138 Cations have been omitted; MaBs are shown in teal dotted lines. MaB angles and Nc values are reported close to the interaction. Colour code: green, chlorine; indigo, nitrogen; and teal, technetium. | ||
Some CSD structures containing chromate, bichromate, and trichromate dianions (CrO42−, Cr2O72−, and Cr3O102−, respectively) form discrete adducts, chains,139 ribbons,140 and 2D networks126via short and almost linear O–Cr⋯O contacts between adjacent anions. These anion⋯anion interactions have the geometric and electronic features141 for being rationalized as wolfium bonds (WfBs),142 which are σ-hole bonds wherein an element of group 6 of the periodic table interacts with an electron donor.
Similarly, some orthovanadate anions, specifically the monoanions of orthovanadate diesters (RO)2VO2−, form dimers143 and infinite chains144via short and almost linear O–V⋯O contacts between nearby anions. These anion⋯anion interactions can be considered erythronium bonds (EyB),145 the σ-hole interaction wherein an element of group 5 is the electrophile.
4. Adducts assembled by π-hole interactions
4.1. π-Hole interactions at s- and p-block elements
Computation shows that in the dihalides of alkali metals (AX2−, A = Li and Na; X = F and Cl) adopting a linear geometry, the less negative (most positive) region is a belt around the alkali metal atom and orthogonal to the main axis of the anion. The approach of a halide anion to this region results in the formation of X2A−⋯X− dianion systems whose optimized structure exhibits minima with trigonal geometry (D3h symmetry).146 The formation of X2A−⋯X− dianions through binding of the X− and AX2− monoanions is endothermic for all examined systems, but these dianions are kinetically stable as their spontaneous dissociation is prevented by a maximum that is present along the dissociation profile. QTAIM analyses reveal that a bond CP is present in the three metal–halogen bonds of all dianion adducts.CSD analyses show that in the solid state, the dihalides of some alkaline earth metals can form tetrahalide dianions that typically adopt a tetrahedral geometry (e.g., AeX42−, Ae = Be and Mg; X = F, Cl, and Br).147–149 Computational studies on X3Ae−⋯X− and X3Ae−⋯CN− systems (Ae = Be, Mg, Ca, Sr, and Ba; X = F and Cl)65,146 have been performed, yielding analogous results for all systems, and these studies support the rationalization of these dianions as stable π-hole species in the solid state. The following discussion concentrates on AeX3(CN)2− complexes. The AeCl3− monoanions all adopt a planar trigonal geometry with D3h symmetry in the gas phase. In aqueous solution, this is the case for the two lighter alkaline earth metal anions while the three heavier ones exhibit a distorted trigonal pyramidal geometry. For BeCl3− and MgCl3−, the electrostatic potential is negative on the whole surface, but less negative at the π-holes above and below the metal atom. In CaCl3−, SrCl3−, and BaCl3−, the potential distribution is similar in shape to that of the lighter anions, but the numerical value at the π-hole is positive. On approaching one of these holes, the CN− anion promotes the pyramidalization around the metal centre. Importantly, in pyramidal AeCl3− the potential in the hole region becomes positive for all the five anions; in this geometry monoanions are ready to form an attractive electrostatic interaction with the incoming CN− anion. The X3Ae−⋯CN− dianions are metastable species in the gas phase, requiring an energy barrier of approximately 20 kcal mol−1 to overcome in order to transform into the more stable separated monoanions (Table 3). In water these barriers disappear and the dianions become thermodynamically more stable than the two separated monoanions.
| Complex |
E
b(gasphase)
|
E
b(watersol.)
|
||
|---|---|---|---|---|
| MP | CCSD | MP | CCSD | |
| Cl3Be−⋯NC− | 42.76 | 44.49 | −19.55 | −17.56 |
| Cl3Mg−⋯CN− | 27.70 | 27.85 | −15.75 | −15.61 |
| Cl3Ca−⋯CN− | 20.31 | 20.20 | −7.80 | −7.83 |
| Cl3Sr−⋯CN− | 19.30 | 18.97 | −5.47 | −5.44 |
| Cl3Ba−⋯CN− | 15.76 | 15.23 | −1.24 | −1.20 |
The case of nitrate anions (NO3−) is particularly important due to the common presence of this anion in both synthetic and natural systems. Already in 2006, the formation of nitrate⋯nitrate dimers was recognized as a structure determining factor in a crystalline aminoguanidine salt.150 One year later, a CSD analysis proved that O3N−⋯O3N− interactions are relatively common in crystalline solids and can play a non-minor role in determining the structure of the corresponding salts.151 Adjacent anions can interact with each other either via a singly pinning supramolecular synthon (wherein the O of one NO3− unit approaches the N atom of a nearby NO3− unit forming O–N⋯O angles close to 90°) or via a doubly pinning supramolecular synthon (wherein there is the antiparallel pairing of the N–O moieties of two nitrate anions, once again the O–N⋯O angles being close to 90°). Anionic adducts with different topologies (e.g., either discrete systems such as dimers,150,151 trimers,152 and tetramers153,154 or infinite chains155,156) are formed thanks to the presence of one or both supramolecular synthons (Fig. 13).
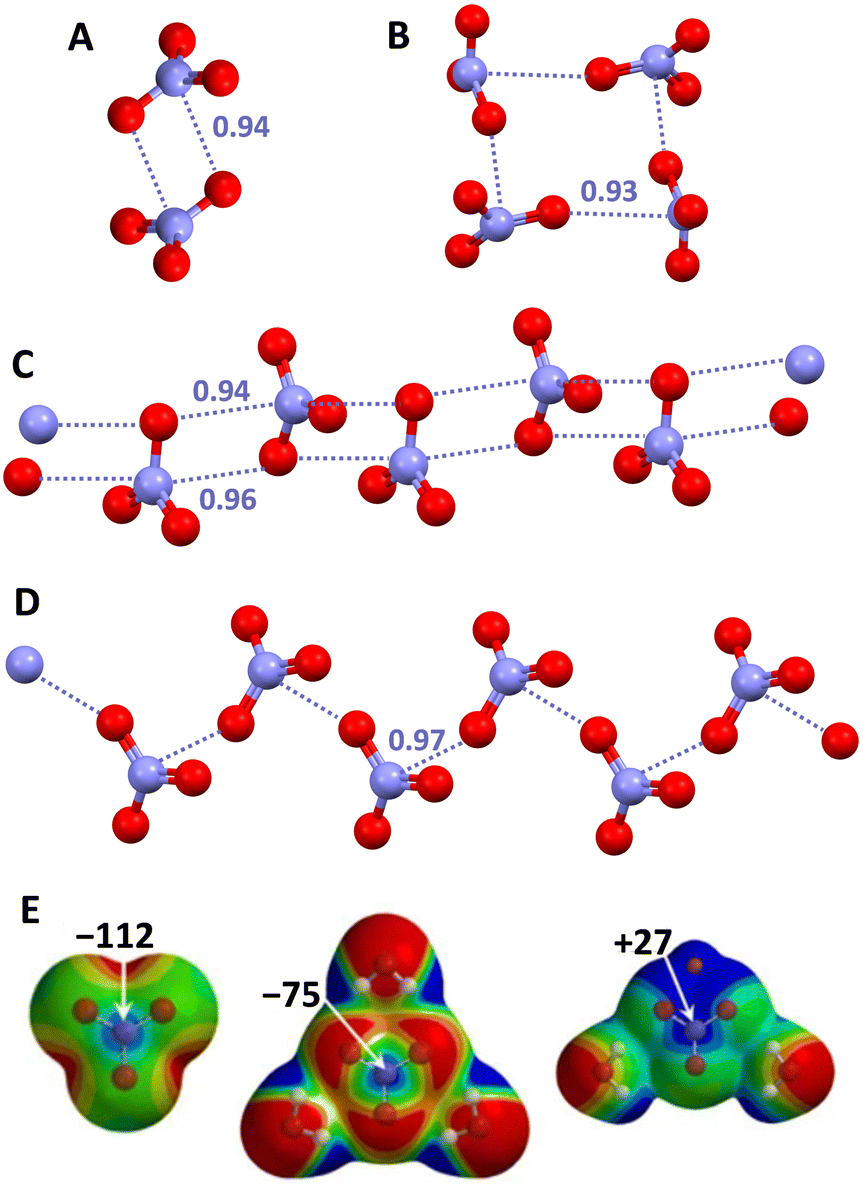 | ||
| Fig. 13 Ball and stick representation of supramolecular anionic adducts formed by NO3−: dimer in an aminoguanidine salt (Refcode GEMMUH)150 (A); cyclic tetramer in a Pt(II) organometallic complex (Refcode EWIDIY)153 (B); infinite chains assembled via singly (Refcode JUMSOR)156 (C) and doubly (Refcode EDOHOU)155 (D) pinning supramolecular synthons in a copper(II) coordination complex and in the triethylenetetra-ammonium salt, respectively; cations have been omitted for clarity; PnBs are shown with indigo dotted lines. Nc values of PnBs are reported close to the interactions. Colour code: red, oxygen and indigo, nitrogen. MEP of naked (E, left) or trihydrated (E, mid) NO3−, and of LiNO3·2H2O (E, right) obtained at the MP2/6-311pG** level (energy values in kcal mol−1); colour scale ranges from more negative (red) to more positive (blue) potentials in between −155 and −122 (E, left), −102 and −75 (E, mid), and −48 and −27 (E, right).157 | ||
This behaviour is consistent with the anisotropic distribution of charge in NO3−. Above the nitrogen atom there is a π-hole which is remarkably negative in the naked anion but is substantially dampened if water molecules are close to the oxygen atoms and becomes positive in the neutral species LiNO3·2H2O (Fig. 13, bottom).157 Interestingly, the single crystal study of urea nitrate, a high explosive, showed the presence of nitrate dimers assembled via doubly pinning supramolecular synthons. The topological analysis of the distribution of electron density in this salt, revealed that the observed N⋯O PnBs are not solely driven by the tight network of cation⋯anion interactions but also result from the electrostatic nature of the interaction between the like-charged moieties.151
After discussing anion⋯anion adducts assembled through π-hole interactions wherein the electrophilic atom is an element of groups 1, 2, or 15, we consider adducts formed by the noble gas bond (NgB), the interaction wherein the element hosting the hole belongs to group 18.27 Calculations predict that NgX5− (Ng = Kr and Xe; X = F and Cl) are planar systems with approximate D5h symmetry. Above and below the noble gas atom, the surface electrostatic potential presents regions (π-hole) where the electrostatic potential is the least negative (Fig. 14, left) and these are the regions of preferential approach of anions such as F−, Cl−, and CN−. The formed supramolecular dianions are stable in polar media (e.g., aqueous solution) and metastable in less polar environments (e.g., THF and DMF solvents). The anionic infinite chain present in crystalline NMe4+XeF5− (Fig. 14, right) confirms the theoretical results.158 The ability of π-hole interactions to drive the self-assembly of as exotic systems as XeF5− forcefully confirms how general and reliable the possibility to direct anion⋯anion assembly via σ/π-hole interactions is.
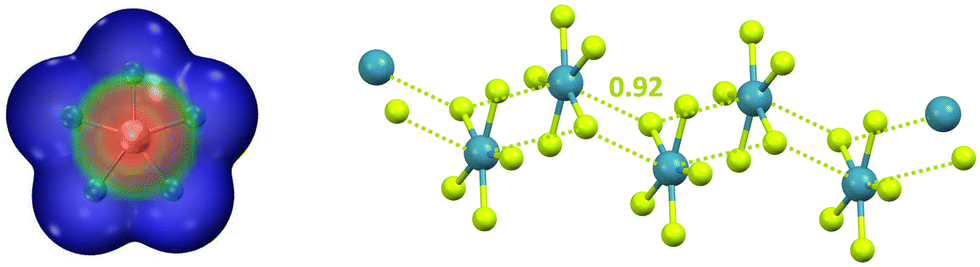 | ||
| Fig. 14 Left: MEP of naked XeF5− (obtained at the MP2/aug-cc-pVDZ level of theory); colour scale ranges from −0.13 (blue) to −0.11 (red) a.u.27 Right: ball and stick representation of the infinite chains formed by XeF5− in its tetramethylammonium salt (Refcode SOBWAH);158 cations have been omitted for clarity; NgBs are represented by acid yellow dotted lines; Nc value of NgB is reported close to the interaction; colour code: light teal xenon and acid yellow, fluorine. | ||
4.2. π-Hole interactions at d-block elements
The tetrachloridopalladate and tetrabromidopalladate anions (PdX42−, X = Cl and Br) adopt in the solid a square planar geometry. Salts wherein these anions are paired with cations that can form a tight network of H⋯X HBs (e.g., α,ω-diammoniumalkanes) can occasionally show the presence, in the solid, of anions dimers159 and trimers28,160 which are assembled via Pd⋯X short contacts wherein the halogen of one unit approaches the palladium of another unit orthogonal to the plane of this second unit. A computational study28 rationalizes these contacts as π-hole interactions. In fact the MEP of the anion reveals two regions of less negative electrostatic potential above and below the metal and QTAIM/NCIPlot analyses identify a bond path connecting palladium and halogen atoms as well as a bond CP and a green disk along the path. The NBO analyses detect a halogen → metal charge transfer associated with the overlap of a halogen lone pair with a vacant Rydberg orbital on palladium.A similar ability of AuCl4− anions to self-assemble into supramolecular anionic chains in the solid via short Au⋯Cl contacts was first reported nearly fifty years ago.161 Various other studies have subsequently acknowledged the propensity of AuX4− anions (X = Cl and Br) to self-assemble into discrete adducts or infinite networks,162,163 either through a singly pinning supramolecular synthon (in which the halogen of one unit gets close to the gold atom of another unit and the X–Au⋯X angle is close to 90°), or through a doubly pinning supramolecular synthon164 (in which the Au–X moiety of one anion couples with the Au–X moiety of a nearby anion after an antiparallel orientation and a nearly orthogonal geometry) (Fig. 15A–C). The tetracyanido anion Au(CN)4− shows a similar interactional landscape forming discrete adducts and 1D165 or 2D networks166via orthogonal C–Au⋯N short contacts (Fig. 15D). Recently, these interactions have been rationalized as coinage bonds (CiBs), namely the interactions wherein an element of the group 11 acts as an electrophile and attractively interacts with a donor of electron density (nucleophile). The MEP analyses of AuCl4− shows the presence of two regions above and below the gold atom where the electrostatic potential is maximum (π-holes) in both the isolated anion and in cation–anion pairs.26 The value of the potential is negative using the 0.001 a.u. isovalue but becomes positive for the ion pair when the 0.008 a.u. isovalue is used (Fig. 1). The QTAIM/NCIPlot analyses of Cl–Au⋯Cl contacts in the geometries observed in the crystals reveal the presence of a bond CP and a bond path connecting the gold and chlorine atoms, indicating that the anion⋯anion interactions between AuCl4− units serve as robust and reliable supramolecular synthons for the control of the overall packing in crystalline structures. Similar computational results were obtained for the anion⋯anion interactions between Au(CN)4− and several different mono- and polyatomic anions.167 For instance, the dimers with fluoride, chloride, cyanide, and hypochlorite anions are metastable species characterized by potential wells. The electron withdrawing ability of the CN− residues in Au(CN)4− units and the small size of the anion approaching the gold atom seem to be two critical aspects for the existence of these metastable dianions. QTAIM analysis of the interaction between gold and the incoming anion shows a bond CP with appropriate electron density and positive Laplace values, as typical for close–shell interactions.
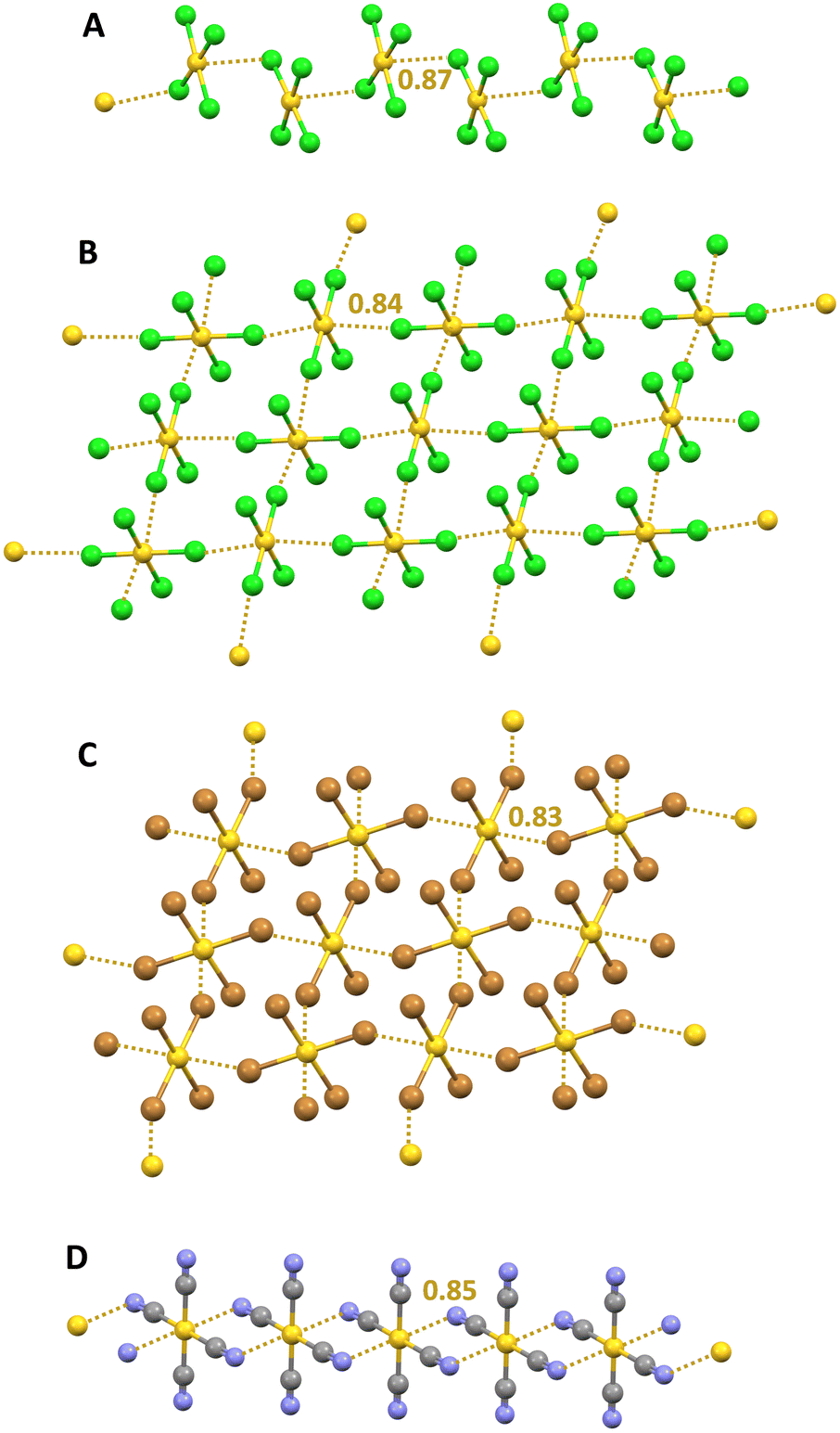 | ||
| Fig. 15 Ball and stick representation of supramolecular anionic: 1D networks formed by AuCl4− in its S-benzyl-isothiouronium salt (Refcode BTURAN)161 (A) and by AuCN4− in an (ethylenedithio)(diselenadithiafulvalene) salt (Refcode XIZCOY);166 2D networks formed by AuCl4− in its pyrazinium salt (Refcode BIHXIC)162 (B) and by AuBr4− in its methylammonium salt (Refcode LONBUO)163 (C); cations have been omitted for clarity; CiBs are shown in ocher dotted lines. Nc values of CiBs are reported close to the interactions. Colour code: yellow, gold; green, chlorine; ocher, bromine; grey, carbon; and indigo, nitrogen. | ||
In the CSD, the trihalides of the group 12 elements SpX3− (Sp = Zn, Cd, and Hg; X = Cl, Br, and I) can exist as isolated units adopting a trigonal and nearly planar geometry or as dimeric, oligomeric, and polymeric adducts wherein one or two halogen atoms bridge adjacent metal centres. The structural diversity of the anion complexes present in the CSD is attributed to the different geometries around the metal and the diverse nature of the metal–halogen bonds. In many crystalline systems, anion adducts show a deformed tetrahedral arrangement around the metal which forms four short and nearly equivalent Sp–X bonds characterized by a strong covalent nature.168 Calculations indicate that these systems can be rationalized as covalently bonded adducts which are stable in the vacuum, ethanol, and water environment. In a smaller number of adducts, the SpX3− units largely retain their native planar D3h geometry and are held together by noncovalent Sp⋯X interactions which can be rationalized as π-hole spodium bonds (SpBs), the interactions wherein the group 12 element acts as the electrophilic site.169 The Sp⋯X interactions are much longer than the other three Sp–X bonds forming the SpX3− units. Also these discrete or infinite supramolecular systems170,171 (Fig. 16) wherein SpX3− units are connected by longer and non-covalent bonds, can be assembled via singly or doubly pinning supramolecular synthons, analogous to those described above for AuX4−. As expected, calculations show that the spodium bonded adducts are not a stable minimum in the gas phase, but they do exist in ethanol and water solution and for systems involving Hg, the heaviest element of the group, they are more stable than the corresponding covalent anion complexes. Analysis of the electron density topology in Hg⋯Cl interactions of HgCl3− homodimers revealed a bond path between the mercury and chlorine atoms and a density at the critical point having the characteristic value of noncovalent interactions of reasonable strength.172 A green RDG isosurface characterizes these interactions so that their attractive nature is confirmed, notwithstanding the Coulombic repulsion between the two anions. Similarly, CN− and SpCl3− form heteromeric dimers that are less stable than the pairs of separated ions in gas phase, but in aqueous solution, the dimerization process is exothermic.173
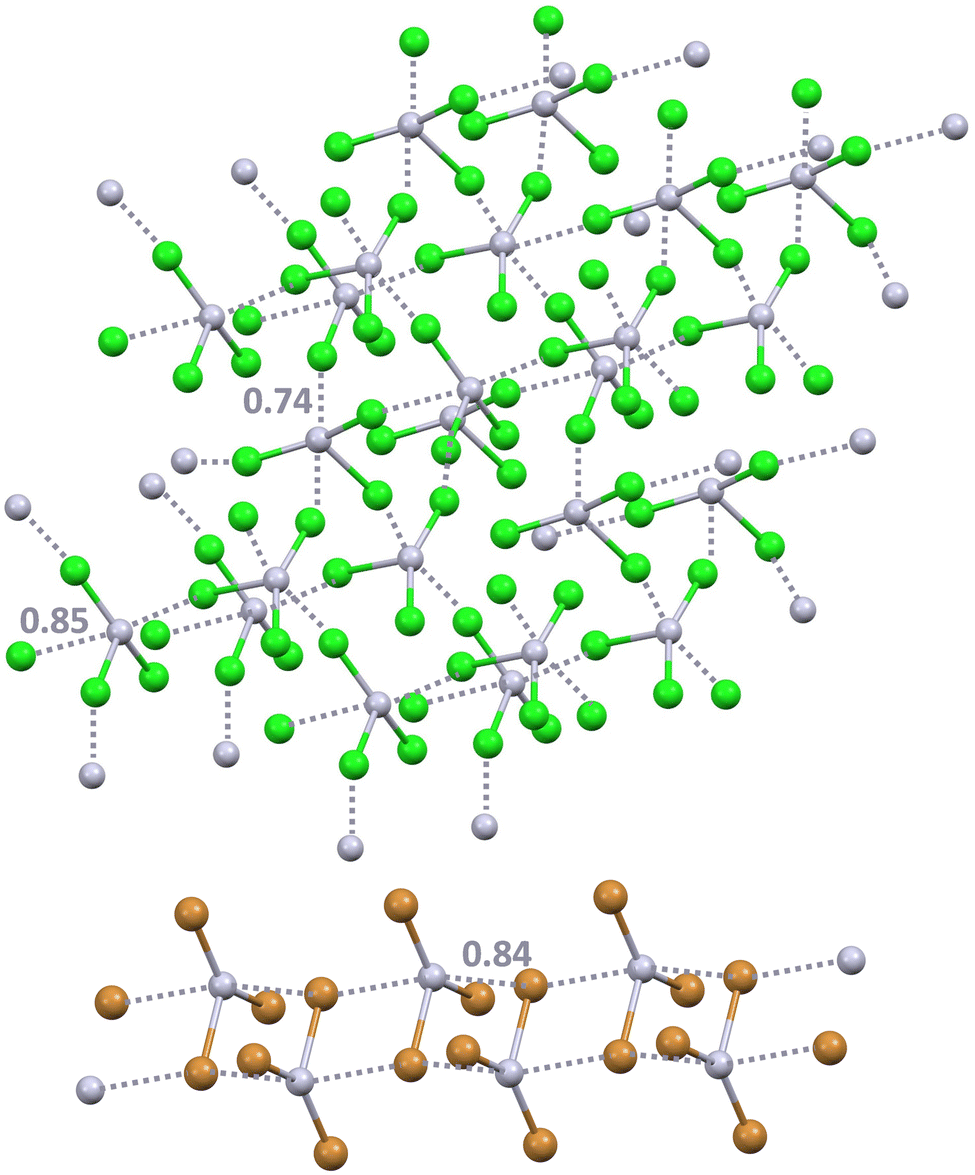 | ||
| Fig. 16 Ball and stick representation of the supramolecular anionic 3D network formed by HgCl3−via SpBs in the methylammonium salts (Refcode QQQBVJ04)170 (top) and the 1D network formed by HgBr3− in the salt with a benzymidazoliun derivative (Refcode WURPOP)171 (bottom). Cations have been omitted for clarity; SpBs are shown in grey dotted lines. Nc values of SpBs are reported close to the interactions. Colour code: light grey, mercury; green, chlorine; and ocher, bromine. | ||
5. Applicative impacts
5.1. Biorelevant systems
PDB analyses show that anion⋯anion interactions are not exceptional in the protein environment. For instance, 24% of the halogenated ligands bonded to a protein and showing structural features consistent with the presence of a HaB are expected to exist as negatively charged species at physiological pH. Quantum mechanics/molecular mechanics studies reveal that the HaBs observed in these crystal structures are stabilizing interactions in the protein environment, while they are unstable or metastable in the vacuum.66 These interactions may increase the biopharmacological activity of halogenated compounds by increasing their affinity for the target protein pocket.57 The HaB acceptor is often the oxygen of a backbone amide moiety, although it can also be the oxygen of the carboxylate residue of an aspartic or glutamic acid.174 For instance, this is the case in the complex between bovine and equine serum albumin and 3,5-diiodosalicilate175 as well as in the complex between human serum albumin and iodipamide, a well-known contrast agent176 (Fig. 17).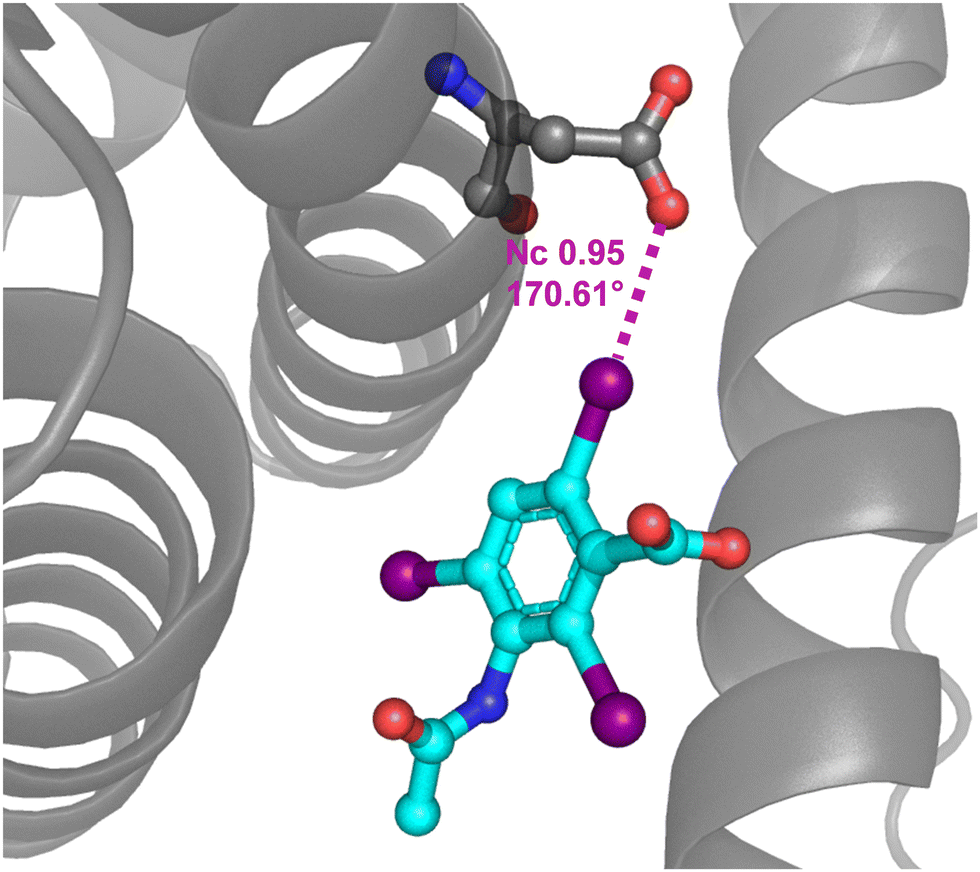 | ||
| Fig. 17 Partial view of the complex between a iodipamide fragment and human serum albumin (PDB code 2BXN)176 evidencing the C–I⋯O HaB between the carboxylate of Asp 451 and the iodocarbon moiety. | ||
At physiological pH, ATP and ADP exist as anionic species. According to the associative ATP hydrolysis mechanism, the trigonal–bipyramidal transition state producing ADP and a phosphoric unit involves the attack of an OH− to the phosphorous of the terminal phosphoric unit of ATP.18 It may be expected that an anion⋯anion O–P⋯O PnB wherein the OH− unit is the PnB acceptor and the terminal phosphorous atom of ATP is the PnB donor is en route to the transition state. Analogously, according to the dissociative ATP synthesis mechanism, it can be expected that an anion⋯anion O–P⋯O PnB wherein an oxygen atom of ADP is the PnB acceptor and a metaphosphate anion is the PnB donor is en route to the formation of ATP from ADP.20
At physiological pH, the phosphoric unit also exists in its anionic form and its binding to proteins can be mediated by an anion⋯anion interaction, e.g., when the selectivity versus sulphate anion binding is secured by an HB between a carboxylate residue at the protein active site and the phosphate –OH group(s).17 In other cases the anion⋯anion interaction locking the phosphate within the cavity, can be a O–P⋯O PnB wherein the PnB donor is phosphate phosphorous and the acceptor is a peptide carboxylate group. For instance, this is the case in the hydrolase enzyme from a thermophilic organism177 (Fig. 18).
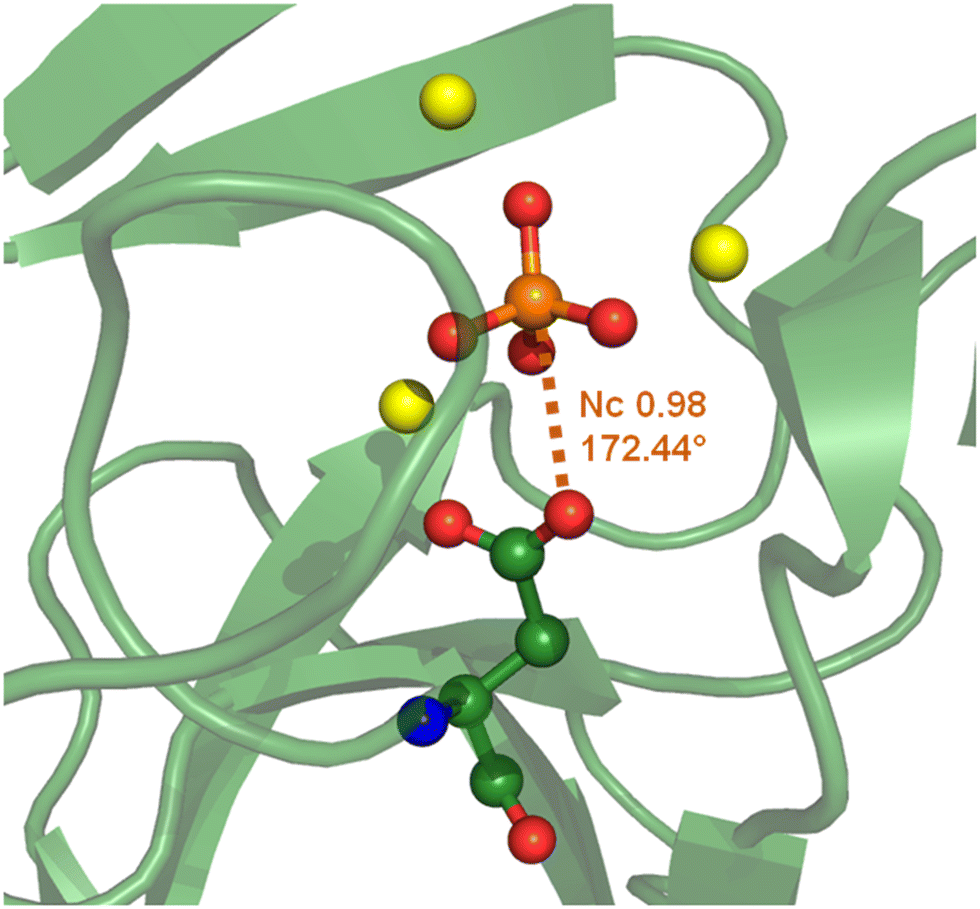 | ||
| Fig. 18 Partial view of the complex between a phosphoric unit and a hydrolase from Thermococcus thioreducens (PDB code 3Q46)177 evidencing the O–P⋯O PnB between the carboxylate of Asp 71 and the phosphate anion. | ||
Interestingly, it is known that MgF42− dianion acts as a phosphate anion(s) analogue and binds to proteins. At the active site, MgF42− can form a short F–Mg⋯O contact with a carboxylate group of the protein. This anion⋯anion interaction can be quite close to linearity and can be rationalized as a σ-hole bond. For instance, such an interaction is found in MgF42− bonded to a hydrolase transport protein (sodium/potassium pump)178 wherein the Mg⋯O separation corresponds to an Nc value of 0.67 and the F–Mg⋯O angle is 176.68° (Fig. 19, top). When MgF42− binds to a human ATPase, both oxygen atoms of the carboxylate residue of Asp 381 get close to the magnesium, namely, the carboxylate group acts as a bifurcated σ-hole bond acceptor (Fig. 19, bottom).179 This bifurcated binding mode is well-documented in the literature for σ-hole interactions whenever the acceptor of electron density approaches a moiety having two geminal oxygen atoms.40
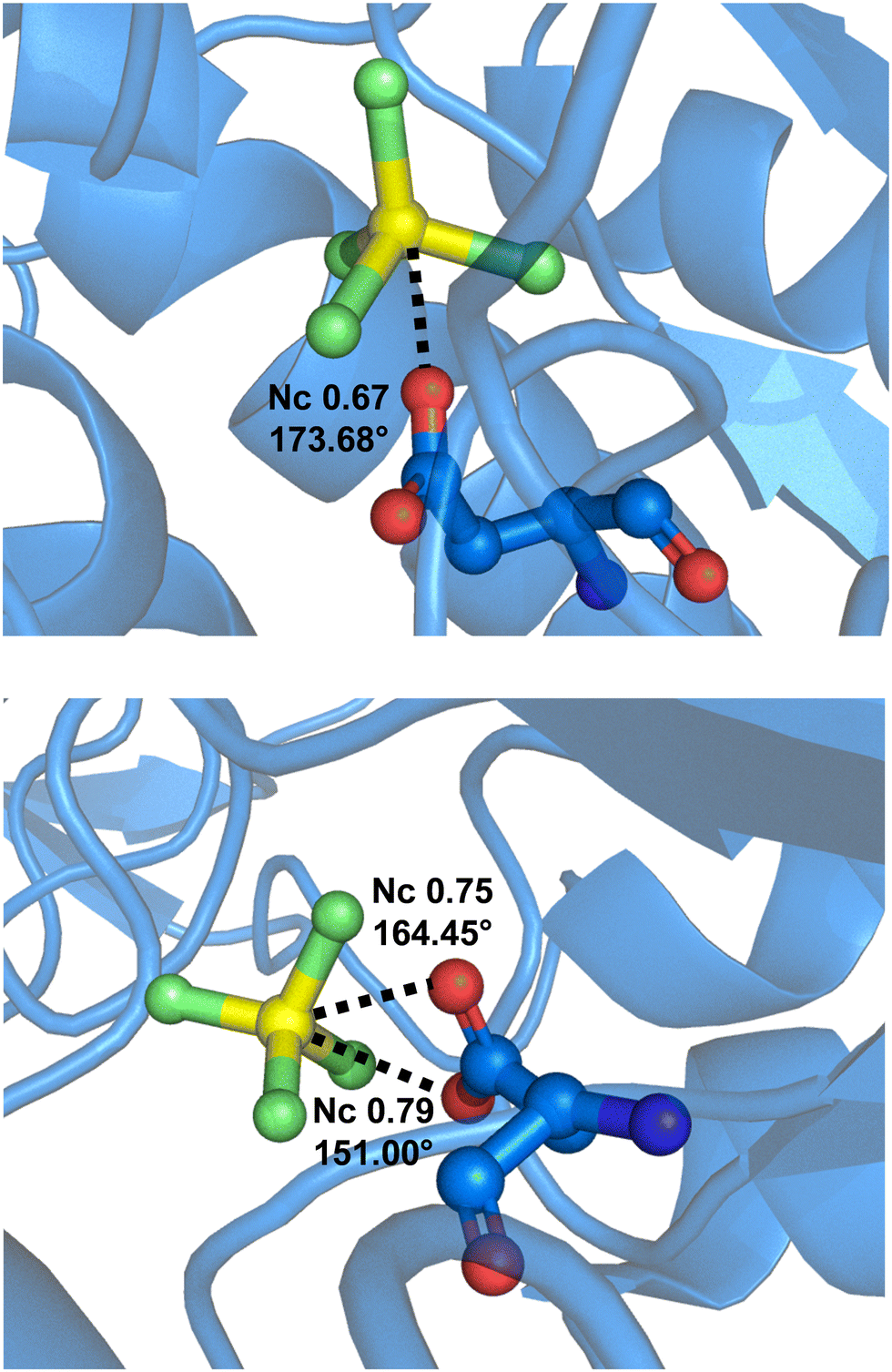 | ||
| Fig. 19 Partial view of the complex between MgF42− and a hydrolase transport protein (PDB code 2ZXE)178 (top) and a human Na+/K+-ATPase (PDB code 8D3X)179 (bottom) wherein the carboxylate residue of Asp 381 forms an anion⋯anion σ-hole bond with MgF42− acting as monofurcated and bifurcated donors of electron density. | ||
5.2. Materials
Hybrid organic–inorganic salts are of interest (e.g., when pursuing a variety of electric and photophysical properties) as they enable the exploitation of the functional performances of both organic and inorganic moieties. The overall crystal packing of these systems can be characterized by the segregation of anions into 1D columnar arrangements or 2D layers and this segregation is more likely when anions self-assemble into extended supramolecular adducts.121 Indeed, hybrid organic–inorganic salts have been reported wherein the structure of the anionic domains is apparently affected or determined by σ/π-hole bonds connecting two or more anions. The strength of these interactions is typically promoted by a network of HBs connecting the organic cations (the HB donors) and the inorganic anions (the HB acceptors) and dissipating the net negative charge of the anions.Regarding π-hole bonds, gold(III) anions provide an example proving how anion⋯anion supramolecular architectures assembled by these interactions are not uncommon in useful functional materials. The methylammonium tetrabromidoaurate (CH3NH3+AuBr4−) and its tetrachlorido analogue form anionic 2D networks assembled via Au⋯X (X = Cl and Br) CiBs. These Au-based halides show relatively small band gap energies (lower than 2.5 eV) and can be considered as alternatives for Pb-based halides in optoelectronic applications.163 Anionic 2D nets assembled by Au⋯Cl π-hole CiBs are also present in pyrazinium tetrachloridoaurate (C4H5N2+AuCl4−) and these crystals show a giant 2-dimensional dielectric response.162 Au⋯I π-hole CiBs between AuI4− and AuI2− anions in the mixed-valent gold(I)/gold(III) iodides of α,ω-diammonium alkanes ([NH3(CH2)nNH3]22+[(AuII2)−(AuIIII4)−(I3−)2] (n = 7, 8)) form 2D networks. These organic-based layered perovskites present an optical band-gap smaller than that of fully inorganic analogous salts.180 In the tetracyanidoaurate of a diselenadithiafulvalene, fairly short Au⋯NC π-hole CiBs assemble the Au(CN)4− anions into columnar arrays and the crystals show superconductivity under uniaxial strain.166
The anionic 1D and 2D arrays present in crystals endowed with useful functional properties can also be structured under the control of σ-hole bonds. Imidazolium periodate (C3H5N2+IO4−) is an improper molecular ferroelectric crystal showing switchable dielectric, piezoelectric, and second-harmonic generation bistability. In its crystals, O–I⋯O σ-hole HaBs assemble IO4− anions into supramolecular chains which segregate into layers.117
Analogously, O–Re⋯O σ-hole MaBs assemble the ReO4− anions of some N-ethyl-Dabco perrhenate derivatives (e.g., [N-2-chloroethyl-Dabco2+(ReO4)22−]) into supramolecular anionic networks. These hybrid organic–inorganic salts show tunable and switchable dielectric properties.136 In crystalline 4-aminopyridinium tetrachloroantimony(III) (4-NH2-C5H5N+SbCl4−), adjacent SbCl4− anions are assembled into supramolecular columnar arrangements by Cl–Sb⋯Cl σ-hole PnBs formed through the antiparallel pairing of the Cl–Sb moiety of nearby anions. Below 270 K, these crystals show ferroelectric properties.181 1,4-Dimethylpiperazine-1,4-diium tetrachloroantimony(III) chloride [(C6H16N2)]2+[(SbIIICl4)−Cl−] shows enhanced photoluminescent efficiency87 and Cl–Sb⋯Cl PnBs are present in its crystals. Specifically, Cl− anions bridge SbIIICl4− anions and supramolecular anion chains are formed.
6. Summary and outlook
The anion⋯anion self-assembly is a relatively new entry in the palette of opportunities for controlling the organization of molecular entities in condensed phases. It has become apparent that attractive forces between anions can occur and some of them can be robust enough to overcome the electrostatic repulsion between the net like charge of two anions and to drive their assembly into supramolecular anion⋯anion adducts.The σ- and π-hole bonds are attractive noncovalent interactions that occur between nucleophiles and the regions of depleted electron density, which most molecular entities have opposite to their single covalent bonds (σ-holes)11,12 and perpendicular to their planar regions (π-holes).13,14 In this review we show that σ- and π-hole bonds wherein anions act as both the nucleophile (via one or more of their regions of high electron density) and the electrophile (via one or more of their σ- and π-holes) can effectively overcome the electrostatic repulsion between the anions and drive their assembly into a variety of structurally different adducts (e.g., discrete dimers or oligomers as well as 1D, 2D, or 3D networks). Typically, these adducts are stable in the solid and in polar solutions and are metastable or unstable in the gas phase.
Regarding the ability of some specific attractive forces between like charges to overcome electrostatic repulsion, it may be useful to summarize that isolated polyatomic anions in their ground state geometry show the presence of σ/π-holes where the electrostatic potential is, in most cases, negative on a surface that is a quite good approximation of the van der Waals surface, namely if the 0.001 a.u. isovalue is used, but occasionally the potential on this surface can also be positive.65 An incoming and negatively charged nucleophile experiences, at this surface, an electrostatic repulsion in the former cases and an attraction in the latter ones. But the separations in anion⋯anion interactions considered in this review are shorter than the sum of van der Waals radii of involved atoms and the contribution of the electrostatic forces to the interaction energy may be better understood by considering surfaces smaller than the van der Waals one, namely, by considering isovalues greater than 0.001 a.u. The electrostatic potential values progressively increase with the isovalue, namely, when getting closer to atoms nuclei, and at interaction distances similar to those observed in anion⋯anion adducts these values may become positive when they are negative on the 0.001 a.u. isovalue surface.26 In most cases, the electrostatic force is thus repulsive at long distances and initially increases as anions approach each other, in accordance with the classical Coulomb's law. But, after reaching the Coulombic repulsion barrier, it decreases drastically; at short ranges, the inter-anion electrostatic repulsion among electrons and among nuclei can be overwhelmed by the inter-anion nucleus–electron attractions.53,89,167
Anions change their geometry when assembling into adducts and changes tend to be greater when interactions are shorter. These structural rearrangements are invariably endothermic (deformation energies are always positive), but may be favoured by the fact that the electrostatic potential at σ/π-holes of anions adopting the geometry of the adducts is usually more positive (less negative) than when adopting the ground state geometry.65
The Pauli exchange repulsion component of the interaction energy is positive (destabilizing) at any interaction distance and becomes greater when the distance decreases. Also the absolute values of charge transfer, dispersion, and polarization energies become greater when the interaction distance decreases, but, differently, they are negative (stabilizing) at any length. The dependence on the interaction distance of all the component energies mentioned above varies from one anion⋯anion adduct to another, and their combination determines the separation and energy of the interaction.
Cases are described wherein the electrophilic atom of the anion belongs to the 1, 2, 5, 7, and 11–18 groups of the periodic table. Clearly, the electrophilic atom can vary across many groups of the s-, p- or d-blocks, namely the σ/π-hole bonds open the possibility for the self-assembly of a particularly wide diversity of anions. By doing so, σ/π-hole bonds effectively complement the opportunities offered by the HB, probably the most frequently used interaction when the anion⋯anion self-assembly is pursued.21,22
While the field of anion⋯anion adduct formation under the control of σ/π-hole bonds is possibly leaving its infancy, it may be expected that some anions that can be assembled via these interactions remain to be identified. Other aspects where major advances may occur in the close future are the understanding of the molecular and supramolecular features favouring the anion⋯anion self-assembly under the control of σ/π-hole bonds (e.g., the most convenient molecular structures of the cations and the overall set of interactions involving the anions). The specific role that σ/π-hole bonds controlling anion⋯anion self-assembly have in known processes, both chemical and biological, is largely unexplored and expectedly the rational design of anion⋯anion adducts will offer new and useful opportunities in various fields such as biopharmacology, catalysis, and molecular or polymeric materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge PRIN-2020, project NICE, no. 2020Y2CZJ2; PRIN-2022, project FLIPPER, no. 202224KAX8; and PRIN-2022, project GEMSTONE, no. 2022SK7JPA.Notes and references
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- Y. V. Nelyubina, M. Y. Antipin and K. A. Lyssenko, Russ. Chem. Rev., 2010, 79, 167–187 CrossRef CAS.
- V. Y. Kotov and S. L. Gorei'sky, Russ. Chem. Bull., 1999, 48, 823–830 CrossRef CAS.
- S. R. Kass, J. Am. Chem. Soc., 2005, 127, 13098–13099 CrossRef CAS PubMed.
- R. Barbas, R. Prohens, A. Bauza, A. Franconetti and A. Frontera, Chem. Commun., 2019, 55, 115–118 RSC.
- M. O. Miranda, D. J. R. Duarte and I. Alkorta, ChemPhysChem, 2020, 21, 1052–1059 CrossRef CAS PubMed.
- The wording short contact, or close contact, is used for any bonding when the separation between the involved atoms is longer than sum of respective covalent radii but shorter than the sum of van der Waals radii ( S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS ).
- R. Billing and D. E. Khoshtariya, Inorg. Chem., 1994, 33, 4038–4040 CrossRef CAS.
- Y. V. Nelyubina, K. A. Lyssenko, V. Yu. Kotov and M. Y. Antipin, J. Phys. Chem. A, 2008, 112, 8790–8796 CrossRef CAS PubMed.
- Q. He, P. Tu and J. L. Sessler, Chem, 2018, 4, 46–93 CAS.
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC.
- H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed.
- S. Scheiner, J. Phys. Chem. A, 2021, 125, 6514–6528 CrossRef CAS PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496–2517 CrossRef PubMed.
- J. M. Holthoff, R. Weiss, S. V. Rosokha and S. M. Huber, Chem. – Eur. J., 2021, 27, 16530–16542 CrossRef CAS PubMed.
- W. Childs, J. Phys. Chem., 1969, 73, 2956–2960 CrossRef PubMed.
- H. Luecke and F. A. Quiocho, Nature, 1990, 347, 402–406 CrossRef CAS PubMed.
- A. A. Malär, N. Wili, L. A. Völker, M. I. Kozlova, R. Cadalbert, A. Däpp, M. E. Weber, J. Zehnder, G. Jeschke, H. Eckert, A. Böckmann, D. Klose, A. Y. Mulkidjanian, B. H. Meier and T. Wiegand, Nat. Commun., 2021, 12, 5293 CrossRef PubMed.
- F. A. Kiani and S. Fischer, J. Biol. Chem., 2013, 288, 35569–35580 CrossRef CAS PubMed.
- T. Beke-Somfai, P. Lincoln and B. Nordéna, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4828–4833 CrossRef CAS PubMed.
- W. Zhao, A. H. Flood and N. G. White, Chem. Soc. Rev., 2020, 49, 7893–7906 RSC.
- N. G. White, CrystEngComm, 2019, 21, 4855–4858 RSC.
- M. E. Light and P. A. Gale, CSD Commun., 2016, Deposition No. 1491437, Refcode: IQOKOQ Search PubMed.
- M. Calabrese, A. Pizzi, R. Beccaria, A. Frontera and G. Resnati, ChemPhysChem, 2023, 24, e202300298 CrossRef CAS PubMed.
- A. Daolio, A. Pizzi, G. Terraneo, A. Frontera and G. Resnati, ChemPhysChem, 2021, 22, 2281–2285 CrossRef CAS PubMed.
- A. Daolio, A. Pizzi, G. Terraneo, M. Ursini, A. Frontera and G. Resnati, Angew. Chem., Int. Ed., 2021, 60, 14385–14389 CrossRef CAS PubMed.
- A. Grabarz, M. Michalczyk, W. Zierkiewicz and S. Scheiner, Molecules, 2021, 26, 2116 CrossRef CAS PubMed.
- W. Zierkiewicz, M. Michalczyk, T. Maris, R. Wysokiński and S. Scheiner, Chem. Commun., 2021, 57, 13305–13308 RSC.
- R. Beccaria, A. Dhaka, M. Calabrese, A. Pizzi, A. Frontera and G. Resnati, Chem. – Eur. J., 2024, 30, e202303641 CrossRef CAS PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- The I2⋯NH3 adduct, probably the first synthesized σ-hole bonded system, was prepared by J. J. Colin early nineteen century: J. J. Colin, Ann. Chim., 1814, 91, 252–272 Search PubMed.
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Halogen Bond: A Long Overlooked Interaction, in Topics in Current Chemistry, ed. P. Metrangolo and G. Resnati, Springer, 2015, vol. 358, pp. 1–17 Search PubMed.
- A. Bent, Chem. Rev., 1968, 68, 587–648 CrossRef.
- The charge-transfer mindset introduced by R. S. Mulliken (ref. 35, 36), the identification of the preferred geometry of adducts involving halogens and other p block elements by N. W. Alcock (ref. 37) and R. Parthasarathy (ref. 38), the computational investigations based on electrostatics by P. Politzer (ref. 39), and the experimental findings of G. Resnati in the solid (ref. 40) gave particularly important contributions to the development of the today concept and practice of the HaB.
- R. S. Mulliken, J. Am. Chem. Soc., 1950, 72, 600–608 CrossRef CAS.
- R. S. Mulliken, J. Phys. Chem., 1952, 56, 801–822 CrossRef CAS.
- N. W. Alcock, Adv. Inorg. Chem. Radiochem., 1972, 15, 1–58 CrossRef CAS.
- N. Ramasubbu, R. Parthasarathy and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308–4314 CrossRef CAS.
- T. Brinck, J. S. Murray and P. Politzer, Int. J. Quantum Chem., 1992, 44, 57–64 CrossRef.
- P. Metrangolo and G. Resnati, Chem. – Eur. J., 2001, 7, 2511–2519 CrossRef CAS PubMed.
- J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS.
- C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef CAS.
- P. Scilabra, G. Terraneo and G. Resnati, Acc. Chem. Res., 2019, 52, 1313–1324 CrossRef CAS PubMed.
- A. Dhaka, I.-R. Jeon and M. Fourmigué, Acc. Chem. Res., 2024, 57, 362–374 CrossRef CAS PubMed.
- R. E. Rosenfield, R. Parthasarathy and J. D. Dunitz, J. Am. Chem. Soc., 1977, 99, 4860–4862 CrossRef CAS.
- J. S. Murray, P. Lane, T. Clark and P. Politzer, J. Mol. Model., 2007, 13, 1033–1038 CrossRef CAS PubMed.
- M. H. Kolář and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541–548 CrossRef CAS PubMed.
- F. Weinhold, Molecules, 2022, 27, 377 CrossRef CAS PubMed.
- A. K. Guha, ChemPhysChem, 2023, 24, e202300403 CrossRef CAS PubMed.
- N. Tarannam, R. Shukla and S. Kozuch, Phys. Chem. Chem. Phys., 2021, 23, 19948–19963 RSC.
- L. Chen, Q. Feng, C. Wang, S. Yin and Y. Mo, J. Phys. Chem. A, 2021, 125, 10428–10438 CrossRef CAS PubMed.
- S. Scheiner, Phys. Chem. Chem. Phys., 2023, 25, 7184–7194 RSC.
- J. S. Murray and P. Politzer, ChemPhysChem, 2021, 22, 1201–1207 CrossRef CAS PubMed.
- K. L. C. Hunt, J. Chem. Phys., 1990, 92, 1180–1187 CrossRef CAS.
- Z. Zhu, G. Wang, Z. Xu, Z. Chen, J. Wang, J. Shia and W. Zhu, Phys. Chem. Chem. Phys., 2019, 21, 15106–15119 RSC.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- C. Wang, Y. Fu, L. Zhang, D. Danovich, S. Shaik and Y. Mo, J. Comput. Chem., 2018, 39, 481–487 CrossRef CAS PubMed.
- A. P. Novikov, K. E. German, A. V. Safonov and M. S. Grigoriev, ChemistrySelect, 2022, 7, e202202814 CrossRef CAS.
- R. Wysokiński, W. Zierkiewicz, M. Michalczyk, T. Maris and S. Scheiner, Molecules, 2022, 27, 2144 CrossRef PubMed.
- D. Quiñonero, I. Alkorta and J. Elguero, Phys. Chem. Chem. Phys., 2016, 18, 27939–27950 RSC.
- Y. Li, L. Meng and Y. Zeng, ChemPlusChem, 2021, 86, 232–240 CrossRef CAS PubMed.
- J. M. Holthoff, E. Engelage, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 11150–11157 CrossRef CAS PubMed.
- W. Zierkiewicz, R. Wysokiński, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 870–877 CrossRef CAS PubMed.
- Z. Yang, Z. Xu, Y. Liu, J. Wang, J. Shi, K. Chen and W. Zhu, J. Phys. Chem. B, 2014, 118, 14223–14233 CrossRef CAS PubMed.
- C. Loy, J. M. Holthoff, R. Weiss, S. M. Huber and S. V. Rosokha, Chem. Sci., 2021, 12, 8246–8251 RSC.
- F. Groenewald, C. Esterhuysen and J. Dillen, Theor. Chem. Acc., 2012, 131, 1281 Search PubMed.
- C. R. Groom and F. H. Allen, Angew. Chem., Int. Ed., 2014, 53, 662–671 CrossRef CAS PubMed.
- S. Scheiner, Phys. Chem. Chem. Phys., 2022, 24, 6964–6972 RSC.
- S. J. Grabowski, ChemPhysChem, 2014, 15, 2985–2993 CrossRef CAS PubMed.
- A. Ferguson, M. A. Squire, D. Siretanu, D. Mitcov, C. Mathoniere, R. Clerac and P. E. Kruger, Chem. Commun., 2013, 49, 1597–1599 RSC.
- D. Leitz, M. C. Bayer, Y. Morgenstern, F. Zischka and A. J. Kornath, Chem. – Eur. J., 2018, 24, 15825–15830 CrossRef CAS PubMed.
- Q. Michaudel, D. Thevenet and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 2547–2550 CrossRef CAS PubMed.
- P. P. Nievergelt, M. Babor, J. Cejka and B. Spingler, Chem. Sci., 2018, 9, 3716–3722 RSC.
- S.-S. Liu, J. W. Ziller, Y.-Q. Zhang, B.-W. Wang, W. J. Evans and S. Gao, Chem. Commun., 2014, 50, 11418–11420 RSC.
- K. Chainok, S. M. Neville, B. Moubaraki, S. R. Batten, K. S. Murray, C. M. Forsyth and J. D. Cashion, Dalton Trans., 2010, 39, 10900–10909 RSC.
- T. Iawakiri, H. Terao, E. Lork, T. M. Gesing and H. Ishihara, Z. Naturforsch. B, 2017, 72, 141–151 CrossRef.
- R. Castillo, J. Cisterna, I. Brito, S. Conejeros and J. Llanos, Inorg. Chem., 2020, 59, 9471–9475 CrossRef CAS PubMed.
- M. Michalczyk, W. Zierkiewicz, R. Wysokiński and S. Scheiner, Phys. Chem. Chem. Phys., 2021, 23, 25097–25106 RSC.
- R. Wysokiński, M. Michalczyk, W. Zierkiewicz and S. Scheiner, Phys. Chem. Chem. Phys., 2021, 23, 4818–4828 RSC.
- S. Scheiner, R. Wysokiński, M. Michalczyk and W. Zierkiewicz, J. Phys. Chem. A, 2020, 124, 4998–5006 CrossRef CAS PubMed.
- G. Resnati, D. L. Bryce, G. R. Desiraju, A. Frontera, I. Krossing, A. C. Legon, P. Metrangolo, F. Nicotra, K. Rissanen, S. Scheiner and G. Terraneo, Pure Appl. Chem., 2024, 96, 135–145 CrossRef CAS.
- K. Kamada, S.-I. Fuku-en, S. Minamide, K. Ohta, R. Kishi, M. Nakano, H. Matsuzaki, H. Okamoto, H. Higashikawa, K. Inoue, S. Kojima and Y. Yamamoto, J. Am. Chem. Soc., 2013, 135, 232–241 CrossRef CAS PubMed.
- V. Y. Lee, K. Ota, Y. Ito, O. A. Gapurenko, A. Sekiguchi, R. M. Minyaev, V. I. Minkin and H. Gornitzka, J. Am. Chem. Soc., 2017, 139, 13897–13902 CrossRef CAS PubMed.
- E. I. Davydova, A. Virovets, E. Peresypkina, A. V. Pomogaeva, A. S. Lisovenko and A. Y. Timoshkin, Dalton Trans., 2021, 50, 13357–13367 RSC.
- J.-Q. Zhao, M.-F. Han, X.-J. Zhao, Y.-Y. Ma, C.-Q. Jing, H.-M. Pan, D.-Y. Li, C.-Y. Yue and X.-W. Lei, Adv. Opt. Mater., 2021, 9, 2100556 CrossRef CAS.
- K. Strakova, L. Assies, A. Goujon, F. Piazzolla, H. V. Humeniuk and S. Matile, Chem. Rev., 2019, 119, 10977–11005 CrossRef CAS PubMed.
- D. Fan, L. Chen, C. Wang, S. Yin and Y. Mo, J. Chem. Phys., 2021, 155, 234302 CrossRef CAS PubMed.
- Q. Ai, D. M. Williams, M. Danielson, L. G. Spooner, J. A. Engler, Z. Ding, M. Zeller, A. J. Norquist and J. Schrier, J. Chem. Phys., 2021, 154, 184708 CrossRef CAS PubMed.
- R. Takouachet, R. Benali-Cherif, E.-E. Bendeif, N. Benali-Cherif, S. Pillet and D. Schaniel, Inorg. Chim. Acta, 2016, 446, 6–12 CrossRef CAS.
- N. Kamali, C. OMalley, M. F. Mahon, A. Erxleben and P. McArdle, Cryst. Growth Des., 2018, 18, 3510–3516 CrossRef CAS.
- The “normalized contact” Nc for an interaction between atoms i and j is the ratio Dij/(rvdWi + rvdWj), wherein Dij is the experimental separation between atoms i and j and rvdWi and rvdWj are the van der Waals radii (ref. 7) of atoms i and j. If the electron donor j is a monoatomic anion, rvdWj is substituted by the anionic radius of j (D. C. Ghosh and R. Biswas, Int. J. Mol. Sci., 2003, 4, 379–407). Nc is a particularly useful way to measure the interaction separation as it allows for a more meaningful comparison of separations between different interacting atoms than the absolute values of interaction distances.
- S. Santos dos Santos, E. Schulz Lang and G. Manzoni de Oliveira, J. Organomet. Chem., 2007, 692, 3081–3088 CrossRef CAS.
- G. A. Casagrande, E. Schulz Lang, G. Manzoni de Oliveira, S. S. Lemos and V. A. S. Falcomer, J. Organomet. Chem., 2006, 691, 4006–4011 CrossRef CAS.
- R. Wysokiński, Phys. Chem. Chem. Phys., 2022, 24, 12860–12869 RSC.
- P. H. Svensson and L. Kloo, Chem. Rev., 2003, 103, 1649–1684 CrossRef CAS PubMed.
- K. Sonnenberg, L. Mann, F. A. Redeker, B. Schmidt and S. Riedel, Angew. Chem., Int. Ed., 2020, 59, 5464–5493 CrossRef CAS PubMed.
- Y. V. Nelyubina, M. Y. Antipin and K. A. Lyssenko, J. Phys. Chem. A, 2007, 111, 1091–1095 CrossRef CAS PubMed.
- K. Lamberts, P. Handels, U. Englert, E. L. Aubert and E. Espinosa, CrystEngComm, 2016, 18, 3832–3841 RSC.
- A. J. Blake, R. O. Gould, W.-S. Li, V. Lippolis, S. Parsons and M. Schroder, Cryst. Eng., 1999, 2, 153–170 CrossRef CAS.
- K. Ghosh, A. Frontera and S. Chattopadhyay, CrystEngComm, 2021, 23, 1429–1438 RSC.
- J. Short, T. J. Blundell, S. Yang, O. Sahin, Y. Shakespeare, E. L. Smith, J. D. Wallis and L. Martin, CrystEngComm, 2020, 22, 6632–6644 RSC.
- Z. Wang, Y. Cheng, C. Liao and C. Yan, CrystEngComm, 2001, 3, 237–242 RSC.
- K.-F. Tebbe and R. Buchem, Angew. Chem., Int. Ed. Engl., 1997, 36, 1345–1346 CrossRef CAS.
- J. Blake, R. O. Gould, W.-S. Li, V. Lippolis, S. Parsons, C. Radek and M. Schroder, Angew. Chem., Int. Ed., 1998, 37, 293–296 CrossRef PubMed.
- F. Pan, R. Puttreddy, K. Rissanen and U. Englert, CrystEngComm, 2015, 17, 6641–6645 RSC.
- A. J. Peloquin, C. D. McMillen, S. T. Iacono and W. T. Pennington, Chem. – Eur. J., 2021, 32, 8398–8405 CrossRef PubMed.
- A. Abate, M. Brischetto, G. Cavallo, M. Lahtinen, P. Metrangolo, T. Pilati, S. Radice, G. Resnati, K. Rissanen and G. Terraneo, Chem. Commun., 2010, 46, 2724–2726 RSC.
- M. D. García, J. Martí-Rujas, P. Metrangolo, C. Peinador, T. Pilati, G. Resnati, G. Terraneo and M. Ursini, CrystEngComm, 2011, 13, 4411–4416 RSC.
- J. Martí-Rujas, L. Meazza, G. K. Lim, G. Terraneo, T. Pilati, K. D. M. Harris, P. Metrangolo and G. Resnati, Angew. Chem., Int. Ed., 2013, 52, 13444–13448 CrossRef PubMed.
- K. F. Konidaris, T. Pilati, G. Terraneo, P. Politzer, J. S. Murray, P. Scilabra and G. Resnati, New J. Chem., 2018, 42, 10463–10466 RSC.
- S. P. Kelley, H. Pei, V. Smetana, A.-V. Mudring and R. D. Rogers, Cryst. Growth Des., 2020, 20, 498–505 CrossRef CAS.
- J. Lin, J. Martí-Rujas, P. Metrangolo, T. Pilati, S. Radice, G. Resnati and G. Terraneo, Cryst. Growth Des., 2012, 12, 5757–5762 CrossRef CAS.
- K. Sonnenberg, P. Pröhm, C. Meller, H. Beckers, S. Steinhauer, D. Lentz and S. Riedel, Chem. – Eur. J., 2018, 24, 1072–1075 CrossRef CAS PubMed.
- D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886–12890 CrossRef CAS PubMed.
- Y. Zhang, H.-Y. Ye, H.-L. Cai, D.-W. Fu, Q. Ye, W. Zhang, Q. Zhou, J. Wang, G.-L. Yuan and R.-G. Xiong, Adv. Mater., 2014, 26, 4515–4520 CrossRef CAS PubMed.
- X.-H. Ding, S. Wang, Y.-H. Li and W. Huang, J. Mol. Struct., 2015, 1079, 266–273 CrossRef CAS.
- M. Calabrese, A. Pizzi, A. Daolio, A. Frontera and G. Resnati, Chem. Commun., 2022, 58, 9274–9277 RSC.
- T. T. da Cunha, W. X. C. Oliveira, C. B. Pinheiro, E. F. Pedroso, W. C. Nunes and C. L. M. Pereira, Cryst. Growth Des., 2016, 16, 900–907 CrossRef CAS.
- M. A. B. Abdallah, A. Bacchi, A. Parisini, S. Canossa, P. P. Mazzeo, L. Bergamonti and S. Kamoun, Struct. Chem., 2019, 30, 1911–1928 CrossRef.
- M. H. H. Wurzenberger, N. Szimhardt and J. Stierstorfer, Inorg. Chem., 2018, 57, 7940–7949 CrossRef CAS PubMed.
- T. Maxson, A. S. Jalilov, M. Zeller and S. V. Rosokha, Angew. Chem., Int. Ed., 2020, 59, 17197–17201 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Cryst. Growth Des., 2014, 14, 2697–2702 CrossRef CAS.
- J. H. Stenlid and T. Brinck, J. Am. Chem. Soc., 2017, 139, 11012–11015 CrossRef CAS PubMed.
- A. Stepien and M. J. Grabowski, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 2924–2927 CrossRef.
- A. Daolio, A. Pizzi, M. Calabrese, N. Demitri, J. S. Murray, P. Politzer and G. Resnati, Cryst. Growth Des., 2023, 23, 574–579 CrossRef CAS.
- M. A. Volkov, A. V. Borisov, V. G. Nenajdenko, E. A. Dukhnovsky, A. E. Bely, M. M. Grishina, A. S. Kritchenkov, R. M. Gomila, A. Frontera and A. G. Tskhovrebov, Inorg. Chim. Acta, 2024, 563, 121929 CrossRef.
- S. Burguera, R. M. Gomila, A. Bauzá and A. Frontera, Crystals, 2023, 13, 187 CrossRef CAS.
- M. A. Volkov, A. P. Novikov, N. E. Borisova, M. S. Grigoriev and K. E. German, Inorg. Chem., 2023, 62, 13485–13494 CrossRef CAS PubMed.
- A. P. Novikov, A. V. Safonov, K. E. German and M. S. Grigoriev, CrystEngComm, 2024, 26, 61–69 RSC.
- R. Alberto, G. Bergamaschi, H. Braband, T. Fox and V. Amendola, Angew. Chem., Int. Ed., 2012, 51, 9772–9776 CrossRef CAS PubMed.
- Y.-Y. Tang, Y. Xie, Y. Ai, W.-Q. Liao, P.-F. Li, T. Nakamura and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 21932–21937 CrossRef CAS PubMed.
- A. M. S. Riel, D. A. Decato and O. B. Berryman, Cryst. Growth Des., 2016, 16, 974–980 CrossRef CAS PubMed.
- Y. Fuma and M. Ebihara, Acta Crystallogr., 2006, E62, m1898–m1900 Search PubMed.
- M. Wan, Y.-N. Wang, J.-Y. Liu, L. Tong, S.-Y. Ye, J.-Y. Li and L.-Z. Chen, CrystEngComm, 2022, 24, 782–787 RSC.
- Y. Xu, M. Calabrese, N. Demitri, A. Pizzi, T. Nag, I. Hung, Z. Gan, G. Resnati and D. L. Bryce, Chem. Commun., 2023, 59, 12609–12612 RSC.
- J. Baldas, J. F. Boas, S. F. Colmanet, A. D. Rae and G. A. Williams, Proc. R. Soc. London, Ser. A, 1993, 442, 437–461 CrossRef CAS.
- M. R. Pressprich, R. D. Willett, R. D. Poshusta, S. C. Saunders, H. B. Davis and G. L. Gardlc, Inorg. Chem., 1988, 27, 260–264 CrossRef CAS.
- C.-R. Ding, Z.-M. Jin, H.-B. Wang, M.-L. Huc and H. Lind, Acta Crystallogr., 2004, C60, m203–m204 CrossRef CAS PubMed.
- R. Beccaria, M. Calabrese, A. Pizzi, A. Frontera and G. Resnati, submitted.
- A. Bauzá and A. Frontera, Chem. – Eur. J., 2022, 28, e202201660 CrossRef PubMed.
- C. Redshaw, M. J. Walton, D. S. Lee, C. Jiang, M. R. J. Elsegood and K. Michiue, Chem. – Eur. J., 2015, 21, 5199–5210 CrossRef CAS PubMed.
- T. Koleša-Dobravc, A. Meden and F. Perdih, New J. Chem., 2015, 39, 4265–4277 RSC.
- M. Calabrese, R. M. Gomila, A. Pizzi, A. Frontera and G. Resnati, Chem. – Eur. J., 2023, 29, e202302176 CrossRef CAS PubMed.
- D. Quiñonero, I. Alkorta and J. Elguero, ChemPhysChem, 2020, 21, 1597–1607 CrossRef PubMed.
- D. Rottschafer, B. Neumann, H.-G. Stammler, T. Sergeieva, D. M. Andrada and R. S. Ghadwal, Chem. – Eur. J., 2021, 27, 3055–3064 CrossRef PubMed.
- M. R. Buchner, M. Muller and N. Spang, Dalton Trans., 2020, 49, 7708–7712 RSC.
- J. Pena, G. Talavera, B. Waldecker and M. Alcarazo, Chem. – Eur. J., 2017, 23, 75–78 CrossRef CAS PubMed.
- Y. V. Nelyubina, K. A. Lyssenko, A. S. Sigachev, M. Y. Antipin and A. N. Kravchenko, Russ. Chem. Bull. Int. Ed., 2006, 55, 399–407 CrossRef CAS.
- Y. V. Nelyubina, K. A. Lyssenko, D. G. Golovanov and M. Y. Antipin, CrystEngComm, 2007, 9, 991–996 RSC.
- V. Blanco, M. Chas, D. Abella, E. Pia, C. Platas-Iglesias, C. Peinador and J. M. Quintela, Org. Lett., 2008, 10, 409–412 CrossRef CAS PubMed.
- K. Otsubo, Y. Wakabayshi, J. Ohara, S. Yamamoto, H. Matsuzaki, H. Okamoto, K. Nitta, T. Uruga and H. Kitagawa, Nat. Mater., 2011, 10, 291–295 CrossRef CAS PubMed.
- D. B. Hassan, W. Rekik, A. O. Roza, F. B. Mefteh, T. Roisnel, T. Bataille and H. Naili, Polyhedron, 2016, 119, 238–247 CrossRef.
- C. A. Ilioudis, D. G. Georganopoulou and J. W. Steed, CrystEngComm, 2002, 4, 26–36 RSC.
- M. Murphy, A. A. E. Gaertner, A. M. Owen, S. Struder, C. D. McMillen, M. Wetzler and J. L. Brumaghim, Inorg. Chim. Acta, 2020, 507, 119568 CrossRef.
- A. Bauza, A. Frontera and T. J. Mooibroek, Nat. Commun., 2017, 8, 14522 CrossRef CAS PubMed.
- K. O. Christe, E. C. Curtis, D. A. Dixon, H. P. Mercier, J. C. P. Sanders and G. J. Schrobilgen, J. Am. Chem. Soc., 1991, 113, 3351–3361 CrossRef CAS.
- R. Zouari, J. M. Leger, T. Maris and N. B. Chanh, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 1253–1255 CrossRef.
- I. A. Efimenko, A. V. Churakov, N. A. Ivanova, O. S. Erofeeva and L. I. Demina, Russ. J. Inorg. Chem., 2017, 62, 1469–1478 CrossRef CAS.
- L. E. Pope and J. C. A. Boeyens, J. Cryst. Mol. Struct., 1975, 5, 47–58 CrossRef CAS.
- D. Paliwoda, M. Szafrański, M. Hanfland and A. Katrusiak, J. Mater. Chem. C, 2018, 6, 7689–7699 RSC.
- C. Worley, A. Yangui, R. Roccanova, M.-H. Du and B. Saparov, Chem. – Eur. J., 2019, 25, 9875–9884 CrossRef CAS PubMed.
- A. Shaikjee, D. C. Levendis, H. M. Marques and R. Mampa, Inorg. Chem. Commun., 2011, 14, 534–538 CrossRef CAS.
- A. R. Geisheimer, J. E. C. Wren, V. K. Michaelis, M. Kobayashi, K. Sakai, S. Kroeker and D. B. Leznoff, Inorg. Chem., 2011, 50, 1265–1274 CrossRef CAS PubMed.
- T. Imakubo, N. Tajima, M. Tamura, R. Kato, Y. Nishio and K. Kajita, J. Mater. Chem., 2002, 12, 159–161 RSC.
- J. Li, Q. Feng, C. Wang and Y. Mo, Phys. Chem. Chem. Phys., 2023, 25, 15371–15381 RSC.
- R. Wysokiński, W. Zierkiewicz, M. Michalczyk and S. Scheiner, Phys. Chem. Chem. Phys., 2021, 23, 13853–13861 RSC.
- A. Bauzá, I. Alkorta, J. Elguero, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2020, 59, 17482–17487 CrossRef PubMed.
- M. Körfer and H. Fuess, Zeit. Kristall., 1988, 183, 27–41 Search PubMed.
- R. P. Sharma, A. Singh, P. Venugopalan, H. Ishihara, M. Nakashima, H. Terao and K. Horiuchi, J. Mol. Struct., 2010, 973, 27–35 CrossRef CAS.
- R. Wysokiński, W. Zierkiewicz, M. Michalczyk and S. Scheiner, ChemPhysChem, 2021, 22, 818–821 CrossRef PubMed.
- R. Wysokiński, W. Zierkiewicz, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 1119–1125 CrossRef PubMed.
- G. Wang, Z. Chen, Z. Xu, J. Wang, Y. Yang, T. Cai, J. Shi and W. Zhu, J. Phys. Chem. B, 2016, 120, 610–620 CrossRef CAS PubMed.
- B. Sekula, A. Bujacz, K. Zielinski and G. Bujacz, Int. J. Biol. Macromol., 2013, 60, 316–324 CrossRef CAS PubMed.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed.
- R. C. Hughes, L. Coates, M. P. Blakeley, S. J. Tomanicek, P. Langan, A. Y. Kovalevsky, J. M. Garcia-Ruiz and J. D. Ng, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2012, 68, 1482–1487 CrossRef CAS PubMed.
- T. Shinoda, H. Ogawa, F. Cornelius and C. Toyoshima, Nature, 2009, 459, 446–450 CrossRef CAS PubMed.
- P. T. Nguyen, C. Deisl, M. Fine, T. S. Tippetts, E. Uchikawa, X.-C. Bai and B. Levine, Nat. Commun., 2022, 13, 5293 CrossRef CAS PubMed.
- L. M. Castro-Castro and A. M. Guloy, Angew. Chem., Int. Ed., 2003, 42, 2771–2774 CrossRef CAS PubMed.
- R. Jakubas, Z. Ciunik and G. Bator, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 024103 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |