
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocatalysis: landmark discoveries and applications in chemical synthesis
Adam
O’Connell†
 a,
Amber
Barry†
a,
Ashleigh J.
Burke
b,
Amy E.
Hutton
b,
Elizabeth L.
Bell
c,
Anthony P.
Green
*b and
Elaine
O’Reilly
*a
a,
Amber
Barry†
a,
Ashleigh J.
Burke
b,
Amy E.
Hutton
b,
Elizabeth L.
Bell
c,
Anthony P.
Green
*b and
Elaine
O’Reilly
*a
aSchool of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: elaine.oreilly@ucd.ie
bManchester Institute of Biotechnology, School of Chemistry, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: anthony.green@manchester.ac.uk
cRenewable Resources and Enabling Sciences Center, National Renewable Energy Laboratory, Golden, CO 80401, USA
First published on 26th February 2024
Abstract
Biocatalysis has become an important tool in chemical synthesis, allowing access to complex molecules with high levels of activity and selectivity and with low environmental impact. Key discoveries in protein engineering, bioinformatics, recombinant technology and DNA sequencing have contributed towards the rapid acceleration of the field. This tutorial review explores enzyme engineering strategies and high-throughput screening approaches that have been applied for the discovery and development of enzymes for synthetic application. Landmark developments in the field are discussed and have been carefully selected to highlight the diverse synthetic applications of enzymes within the pharmaceutical, agricultural, food and chemical industries. The design and development of artificial biocatalytic cascades is also examined. This tutorial review will give readers an insight into the landmark discoveries and milestones that have helped shape and grow this branch of catalysis since the discovery of the first enzyme.
 Adam O’Connell | Adam O’Connell obtained his bachelor's degree in Chemical Sciences with Medicinal Chemistry from the Technological University Dublin. In 2020, Adam began his PhD in University College Dublin under the supervision of Dr Elaine O’Reilly. His research aims to investigate new methodology that incorporates enzyme-triggered reactions for the synthesis of high-value natural products and their analogues. |
 Amber Barry | Amber Barry received her bachelor's degree in Medicinal Chemistry and Chemical Biology from University College Dublin. Amber is currently completing her PhD in Dr Elaine O’Reilly's research group. Her research focuses on the development of novel biocatalytic cascade methodology, for the synthesis of high-value chiral compounds and natural products. |
 Anthony P. Green | Following his PhD in synthetic organic chemistry under the supervision of Professor E. J. Thomas, Dr Anthony Green began postdoctoral research with Professor Nicholas Turner and Professor Sabine Flitsch based in the Manchester Institute of Biotechnology (MIB), working in the field of industrial biocatalysis. Professor Green worked as a postdoctoral research associate in the group of Professor Donald Hilvert at ETH (Zurich), before starting his independent research career in 2016 based in the MIB (Department of Chemistry) at the University of Manchester where he is a BBSRC David Phillips research fellow, a Professor in organic & biological chemistry and holder of an ERC starter grant. |
 Elaine O’Reilly | Elaine O’Reilly obtained her PhD in Chemistry from University College Dublin under the supervision of Prof. Francesca Paradisi. From 2010–2014 she carried out postdoctoral research at the University of Manchester before joining Manchester Metropolitan University as a Lecturer. Elaine was an Assistant Professor, then Associate Professor in Chemical Biology at the University of Nottingham between 2015 and 2018, before returning to University College Dublin as Associate Professor of Chemical Biology in 2019. Her research focuses on the development and application of biocatalysts in organic chemistry. |
Key learning points• Importance of biocatalysts in chemical synthesis.• Enzyme engineering methodologies for improved biocatalysts. • High-throughput screening methods for discovery of improved variants. • Milestone applications of enzymes in industry. • Incorporation of enzymes into cascades and the important design considerations when developing biocatalytic cascades. |
1. What are enzymes and how were they discovered?
Enzymes are biological catalysts that accelerate chemical reactions occurring within biological systems.1 Nature uses enzymes to perform many chemical reactions, including the synthesis of important building blocks, such as amino acids, and the assembly of complex natural products. The active site pocket associated with enzymes is responsible for the enormous rate accelerations and unparalleled levels of selectivity that can be achieved using these catalysts. The network of active site amino acid residues functions to precisely position the substrate in the binding pocket in the correct orientation for catalysis, by exploiting multiple weak interactions, for example, hydrogen-bonding, electrostatic, dipole–dipole and van der Waals interactions (Fig. 1). The finely tuned interactions can function; (i) to bring reactants into proximity; (ii) bind the substrate(s) in a favourable conformation to allow the reaction to occur; and (iii) stabilise and lower the energy of the transition state. The result of this substrate constraint and stabilisation is a significantly accelerated reaction displaying high levels of chemo-, regio- and stereoselectivity.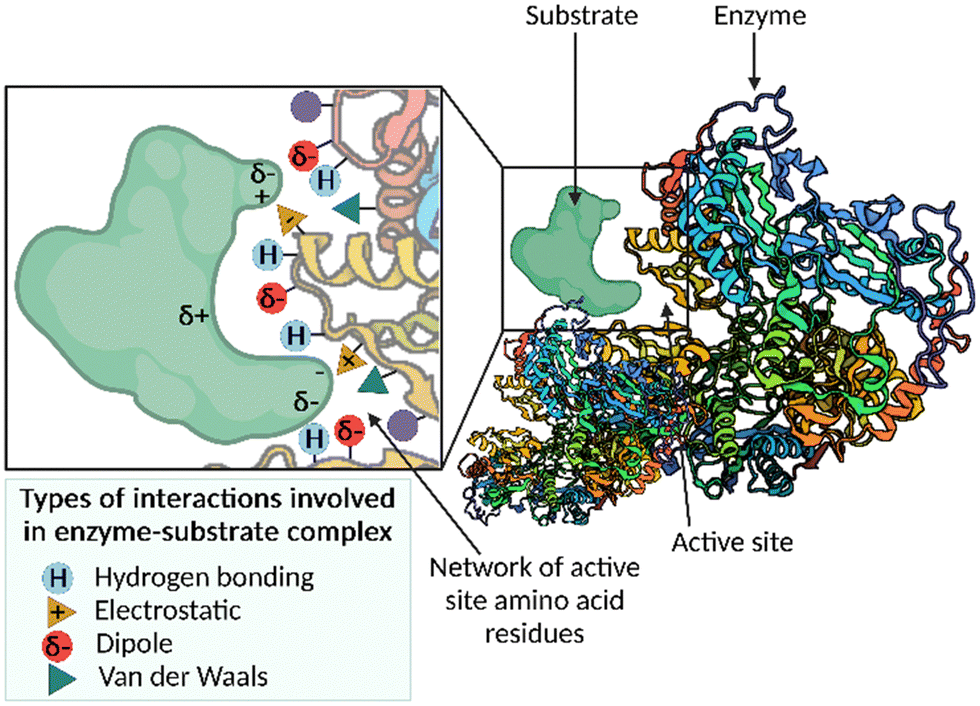 | ||
| Fig. 1 A cartoon depicting the network of active site amino acid residues, and the possible interactions with a given substrate to form the enzyme–substrate complex. | ||
Our understanding of enzymes developed incrementally, with key discoveries leading to landmark progressions in the field. Advances in molecular biology, bioinformatics, enzyme engineering and high throughput screening have developed our understanding of enzymes to a point where they now find widespread synthetic applications in the rapidly expanding field of biocatalysis and can be considered alongside other asymmetric homogeneous catalysts. The term ‘biocatalyst’ can refer to isolated (purified) enzymes, enzymes contained within a crude mixture, whole-cell systems containing the enzyme(s), as well as combinations of these.
This tutorial review will examine the landmark discoveries in the field of biocatalysis and highlight milestone examples of how enzymes have established themselves as an important part of any catalytic toolkit for chemical synthesis. Alternative applications of biocatalysis in fields such as diagnostics (biosensors) and biodegradation of polymers (plastic, cellulose, lignin, etc.) are not discussed in detail, however comprehensive reviews on these topics are referenced.2–4
The first known enzyme mixture, diastase, was discovered in 1833 by French chemists, Payen and Persoz,5 who found that the active catalyst converts starch to sugar and is found in germinating cereal.6 Fermentation chemistry, involving the conversion of sugar into alcohol by enzymes in yeast cells, had been used unknowingly for thousands of years in the brewing process, but explanations for how the process occurred were only answered as the field of enzymology developed. The details of fermentation chemistry were first published by Louis Pasteur in 1858, who attributed the phenomenon to “ferments” within living yeast cells carrying out a “vital force”.7 Nobel Prize winner, Buchner, later disproved the concept of vitalism in 1897, hypothesising instead that the yeast cells secrete proteins to facilitate fermentation and that living yeast cells are not required.8 It was later discovered these so called “ferments” were enzymes capable of catalysing the process that occurred inside the cells.
The 1946 Nobel Prize in Chemistry was awarded for the landmark work of Sumner, Northrop, and Stanley, who proved that enzymes are proteins. Sumner was the first to crystallise an enzyme (urease) in 1926, providing structural confirmation that the enzyme was a protein.9 Northrop and Stanley continued the work and crystallised several serine and aspartic proteases.10
Mechanistic and kinetic studies helped move the field of enzymology forward over the next number of years. The Lock and Key model, describing enzyme–substrate binding, was proposed by Fischer in 1894, following an observation that stereoisomeric hexoses interact differently with yeast (and its enzymes), therefore leading to the hypothesis that enzyme–substrate binding differs depending on a substrate's configuration, size and shape.11 This theory was later replaced by the Induced Fit Model,12 which took into account substrate positioning and 3D structural changes upon binding, and helped to deepen our understanding of enzyme-catalysed transformations. The precise binding between an enzyme and its substrate(s), due to an induced fit change, forms the enzyme–substrate complex (E–S-complex). The E–S-complex lowers the activation energy barrier of the given transformation by stabilising the transition state, allowing the reaction to proceed. This is followed by formation of the enzyme–product complex (E–P-complex) and release of the product from the active site, enabling subsequent catalytic transformations to take place.
Michaelis–Menten exploration on enzyme kinetics,13 alongside developments in X-ray crystallography, allowed elucidation of active site structures, cofactor requirements and mechanisms. Major advances in DNA technology, including the elucidation of DNA structure by Watson, Crick and Franklin, Sanger DNA sequencing, polymerase chain reaction (PCR) and recombinant expression (Fig. 2), accelerated advances in the field, allowing the modification of enzymes and the engineering of biocatalysts for desired applications.
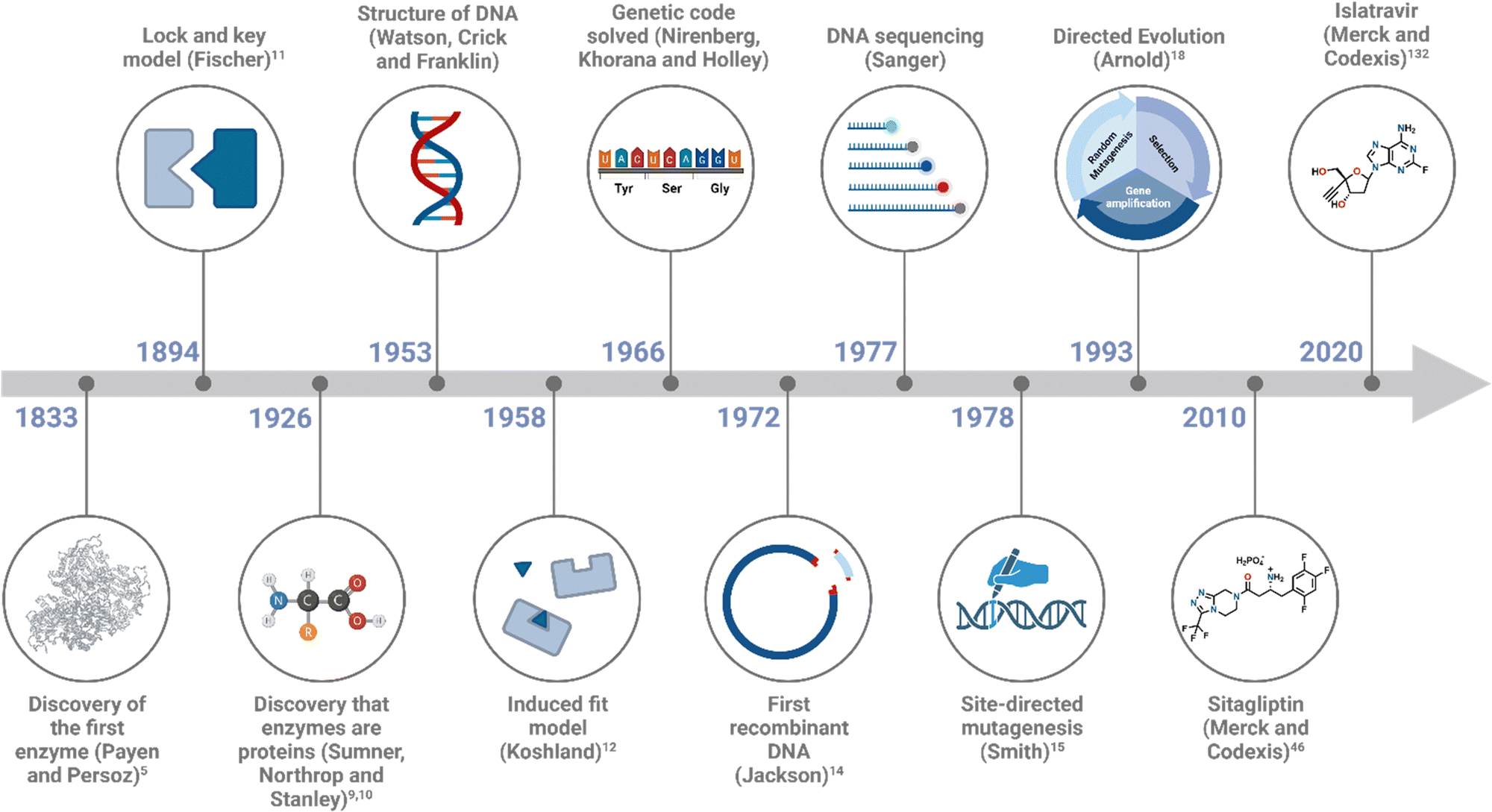 | ||
| Fig. 2 Timeline of some of the key milestones in the field of biocatalysis. | ||
2. Enzyme engineering
While the benefits of utilising an enzymatic step(s) in the asymmetric synthesis of high-value molecules is well established, the properties of many wild-type (WT) enzymes must be tuned to enable them to operate under the desired reaction conditions. Naturally occurring enzymes are typically evolved to carry out a very specific function under physiological conditions and are less effective at mediating reactions with unnatural substrates at varying temperatures, pH, substrate loadings and in the presence of a cosolvent. This section will discuss several enzyme engineering methodologies that have been developed to stabilise WT enzymes and enable them to operate in conditions distinct from their native environment or to increase the enzyme's substrate scope and selectivity.Since the discovery that an enzyme's structure is dictated by its amino acid sequence, which is in turn determined by the translated genetic code, modern advances in DNA technology (such as gene cloning and sequencing) have significantly accelerated the engineering process by allowing greater amounts of genetic material to be obtained and analysed on a nucleobase scale. The first example of DNA cloning was performed by Jackson et al.14 and since then, the utilisation of recombinant DNA has transformed the way enzymatic studies are carried out. The ability to insert a gene of interest into a vector that can be subsequentially over-expressed in a suitable host organism, allows access to larger quantities of enzymes than could ever be extracted previously from natural sources. DNA sequencing techniques, such as Sanger sequencing and the more recently developed massive parallel sequencing (also known as next-generation sequencing), have permitted correlations between function and essential residues within the enzyme. Preliminary mutagenesis attempts involved the use of radiation (e.g., ultraviolet light) to promote errors in the replication or repair cycles of DNA. However, this method was slow and unreliable due to the dependency on the organism being mutated, therefore a more robust strategy was required.
2.1. Rational design
In 1978, Smith and co-workers introduced the idea of rational design, in which precise changes in the amino acid sequence are integrated using site-directed mutagenesis.15 This technique involves the synthesis of an oligonucleotide that contains the desired mutation and the complementary base sequence to the template DNA, so that annealing can occur. The gene containing the mutation is extended using a DNA polymerase and can then be cloned and expressed in a suitable host organism. This approach was later used in a milestone study by the same group to demonstrate the systematic replacement of an amino acid residue in an enzyme (tyrosyl tRNA synthetase) for the first time.16 The method involved substituting a cystine residue, located in the enzyme's active site, to serine, and used several recombinant tools, such as oligonucleotide synthesis, DNA amplification and site-specific cut-and-paste. The result of this mutation was not beneficial to functionality, as it resulted in a reduction in the enzyme's ability to catalyse the aminoacylation of tRNA, but it gave an insight in to how the methodology could be used to investigate enzymatic mechanisms. The type of engineering strategy demonstrated by Smith and co-workers has been used to examine the effect of distinct point mutations on specific areas of the protein.Rational design forms the basis for de novo protein design and is an attractive strategy due to the smaller library sizes generated. It may require extensive knowledge of the protein structure, mechanism, and sequence to be able to produce a superior variant. It also typically explores a very small section of the genetic sequence from which the success depends entirely on the validity of the hypothesis, therefore a less target driven approach was developed in the form of directed evolution.
2.2. Directed evolution
Due to the vastness and immense complexity of the natural world, resulting from millions of years of evolution, the idea that humankind could potentially exploit this force for its own benefit was thought to be futile. That was until Darwin's On the Origin of Species in 1859 brought to light that humanity had been unknowingly harnessing this power for centuries in the form of selective breeding and domestication. During the second half of the twentieth century, evolution was brought into the laboratory, highlighted by the pioneering work by Mills et al. with their studies on bacteriophage Qβ RNA.17 While the following decades would find many more examples of adaptive evolution experiments, directed evolution in the contemporary sense would not manifest until the 1990's. The general approach when undertaking this evolutionary strategy is to first identify a gene of interest that encodes for the ‘parent’ enzyme to be engineered. The parent enzyme may only have minimal activity for the substrate of interest, yet still represent a good starting point, provided improvements in activity can be measured or ‘selected’ reliably (discussed further in Section 3). Gene diversification via random mutagenesis (e.g., error-prone PCR, mutator strains, chemical mutagens) and/or gene recombination (e.g., shuffling) generates a diverse library of gene products containing mutations at low frequencies throughout the sequence or at targeted regions of the gene. These are then screened for improved properties and the most optimal variant(s) are selected and amplified (Fig. 3). When variants with preferable phenotypes are identified, the corresponding gene must also be identified, therefore establishing a genotype–phenotype linkage is essential. The iterative directed evolution process continues until no further improvement is gained or the desired activity is obtained. A major advantage of this method is that it requires no previous structural or mechanistic knowledge of the gene/protein being mutated. The approach allows for random mutations to be incorporated at any position on the enzyme, and this may be beneficial, as properties such as activity, selectivity and substrate scope are not solely influenced by residues within the active site. Directed evolution can therefore produce highly valuable catalysts by substituting residues that may not have been considered in more rational engineering strategies. However, several rounds of mutagenesis are typically required to produce a variant with the desired properties and consequently, a robust and high-throughput screening assay must be developed for this type of engineering (discussed in Section 3).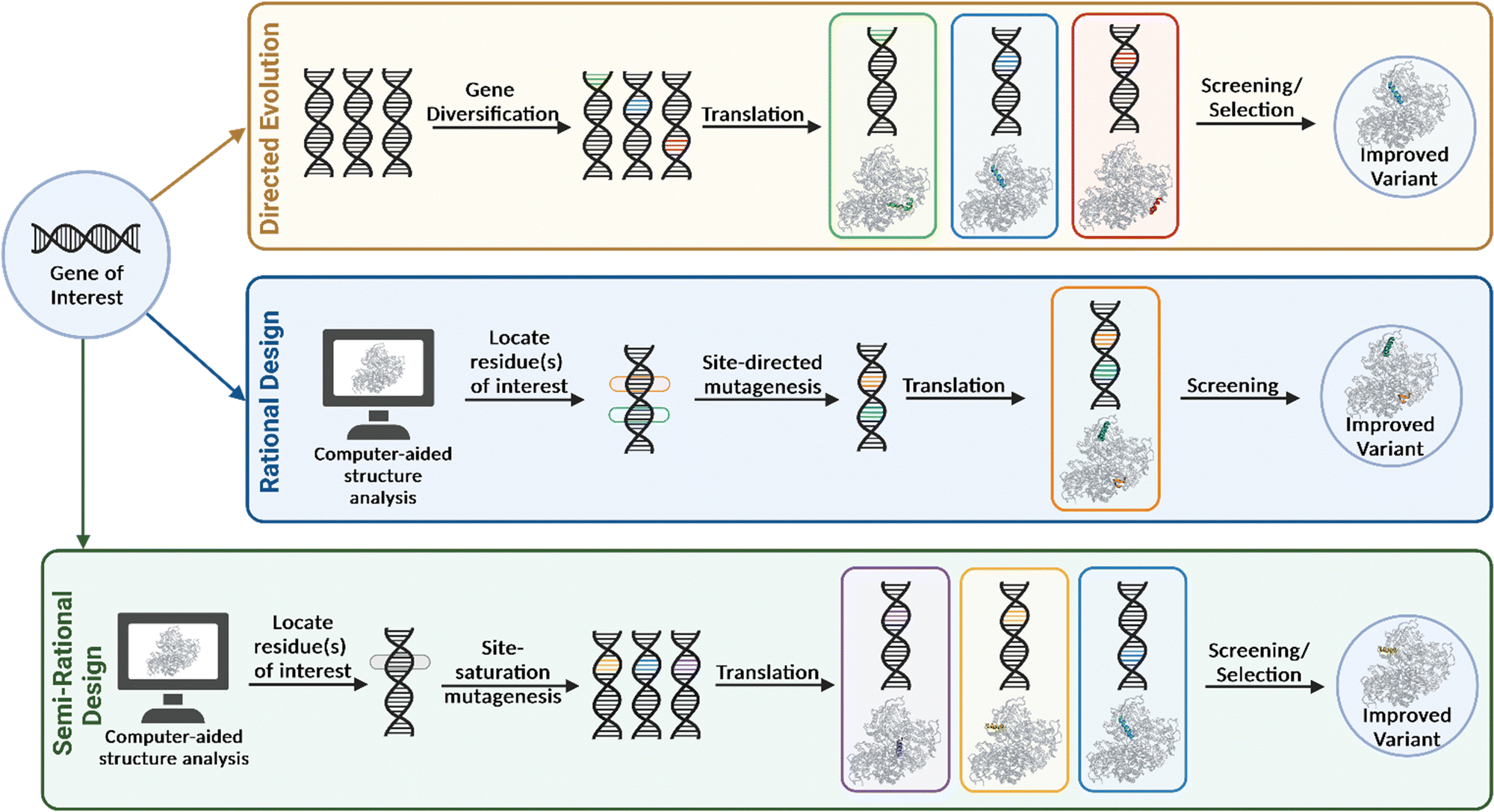 | ||
| Fig. 3 Pathways of protein engineering strategies for biocatalyst improvement. | ||
Directed evolution was brought to the spotlight by the pioneering work carried out by Arnold and co-workers when they demonstrated a landmark example of how this evolutionary approach could be employed when evolving the alkaline serine protease, subtilisin E.18 The objective of the work was to enable subtilisin E to carry out the hydrolysis of a peptide substrate (sAAPF-pna) in high concentrations of the polar organic solvent dimethylformamide (DMF). Starting from the gene of interest (a subtilisin E variant, containing four amino acid substitutions), three rounds of subsequent mutagenesis and screening was performed to yield a variant containing a total of ten mutations, which could tolerate high concentrations of DMF. The variant was over 500-fold more active in 60% DMF than the WT subtilisin E. Random mutations were incorporated using error prone PCR, which works on the basis that the fidelity of the Taq DNA polymerase used in the process is altered via the composition of the reaction buffer. These sub-optimal conditions introduce errors into the newly synthesised gene, which can then be transferred into a plasmid vector for over-expression and screening. In recent years, the implementation of directed evolution for the engineering of various enzymatic functions has become more commonplace and has had enormous impacts on our ability to tailor enzymes to specific functions. Prof. Frances Arnold shared the Nobel Prize in Chemistry in 2018 ‘for the directed evolution of enzymes’. Ever since this seminal methodology was performed, new gene diversification techniques have been developed and implemented into mutagenesis studies, such as DNA shuffling19 and mutator strains.20
2.3. Semi-rational design
Semi-rational protein design combines both the advantages of rational design and directed evolution in a technique that results in smaller, ‘smart’ libraries. This approach often utilises the new bioinformatic toolbox available to modern biochemists and involves mutating at “hot spots” on the enzyme that are known to play an important role in its function. The codon associated the chosen residue(s) for mutation is replaced with a degenerate codon. This degenerate codon has the ability to encode for all or a subset of all canonical amino acids. This process is termed site-saturation mutagenesis.Semi-rational design can be utilised as an alternative if residues vital to the mechanism of action are known or if high-throughput screening is not an option, as the library size produced is much smaller when compared to directed evolution. These ‘smarter’ libraries should in theory be more likely to contain variants with beneficial modifications, as they have been derived from previously obtained structural/mechanistic data. When addressing stereoselectivity and/or substrate scope, one of the most universally adopted approaches is combinatorial active site saturation test (CAST). CASTing involves the saturation of multiple residues surrounding the active site to induce favourable synergistic effects and produce a knowledge-driven systematisation of focused libraries.21 If an optimal variant is produced, the process can be repeated iteratively on the mutated gene of an improved mutant to screen for more complementary mutations, in a process known as iterative saturation mutagenesis (ISM). This strategy is often beneficial as it leads to reciprocal amino acids substitutions that may have been missed using single site-specific mutagenesis.
It is important to keep in mind that each type of engineering process is not mutually exclusive and with the combination of bioinformatic tools, a far more applicable protein design strategy can arise. An amalgamation of engineering methods was used to develop the multivariate optimisation technique known as ProSAR. Inspired by the concept of QSAR (quantitative structure–activity relationship), the optimisation strategy is based on protein sequence activity relationships (ProSAR). A landmark example of the effectiveness of this multivariate optimisation strategy was demonstrated by Codexis and co-workers, when a bacterial halohydrin dehalogenase was evolved to increase its cyanation process ∼4000-fold, to produce a starting material, (R)-4-cyano-3-hydroxybutyrate (HN), for the cholesterol-lowering drug atorvastatin (Lipitor).22
3. HT and uHT screening
The evolution of protein scaffolds provides a powerful solution to many of the challenges associated with WT enzymes, but such engineering strategies require an efficient and robust high-throughput screening (HTS) method for the successful selection of optimal variants. Due to the nature of the methodology, a high-throughput screen is especially important when carrying out random mutagenesis, as this approach often generates extremely large genetic libraries (ranging from ∼104–6 variants per evolution cycle) and an effective assay that links genotype to phenotype is the only way to identify variants with the desired properties. HT screening enables the identification and selection of genetic variants with improved properties, from potentially millions of genetic variants. Furthermore, as the gene is compartmentalised, for example within a cell, the improved variant has also been purified from a large library of genetic variants. There is no synthetic chemistry methodology that enables the generation of a library of thousands or millions of catalyst derivatives, in situ optimisation of the catalyst library, and concurrent selection and purification of the best catalyst; and therefore, this really is unique and powerful methodology.Medium-throughput assays can be designed to enable screening of purified enzyme, cell lysate or whole cells, with the latter often allowing higher-throughput. Effective whole cells assays rely on the screening components penetrating the cell wall and not being degraded by the host (temperature, pH, biocatalytic degradation). Additionally, to establish the necessary genotype–phenotype linkage, the assay product must remain within the cell. The application of screening approaches using cell lysate can be carried out in microtiter plates or other compartments, once endogenous enzymes do not interfere with the assay. While the use of assays relying on purified enzyme means issues with crossing cell walls or interference from endogenous enzymes and biomolecules are not present, such assays can be expensive and time-consuming.23
Creating an effective HTS can be challenging, as each assay is usually quite specific to the required task it was designed for, limiting its wider exploitation. An ideal HT screen should be general (can accommodate a range of enzymes/substrates/products), operationally simple, quantitative, carried out in its highest throughput form (e.g. whole cells, droplets, etc.) and sensitive. There are many screening platforms to choose from when developing a HT screening method depending on what suits the user, such as plate screening, surface display and the more recent development of virtual screening assisted HTS.24 When choosing the screening parameter (e.g., fluorescence, absorbance, changes in pH), the following characteristics are important: the assay should be (i) reproducible, so many iterations of the screen can be performed; (ii) low cost, so the screen is economically feasible; and (iii) highly sensitive and specific for identification of the target molecule(s) within a broad range of conversions. A range of measurable detection variables for high throughput screening efforts have been used successfully in protein engineering studies, some of which include spectrophotometric, colorimetric and fluorescence screening.
3.1. Spectrophotometric screening
Spectrophotometric assays rely on the reflectional or transmissional properties of a reaction reagent or product/coproduct as a function of wavelength. Homogenous absorbance assays are considered the most straightforward and classical detection method for monitoring an enzymatic reaction due to its simplicity and inexpensiveness. One screening methodology that absorbance measurement lends itself well to is coenzyme generation or consumption. Nicotinamide adenine dinucleotide (NAD+ or the reduced form NADH) and nicotinamide adenine dinucleotide phosphate (NADP+ or the reduced form NADPH) are common coenzymes used in redox reactions. Numerous studies have utilised simple absorption spectroscopy to detect a change in the coenzyme's concentration as a function of time, due to the reduced form (NADH and NADPH) strongly absorbing at 340 nm, while the oxidised forms (NAD+ and NADP+) do not absorb at this wavelength. This absorbance-concentration correlation has been particularly applicable in engineering studies that invert the cofactor specificity for enzymes that require the redox cofactor.25,26 NADH/NADPH detection is also well-suited to the subsequent reaction in a coupled enzyme assay, especially for monitoring reactions that are difficult to detect.27 However, there are also some notable limitations to nicotinamide-based absorbance assays. Typically, purified enzyme is required for these assays, due to the strong absorption of UV light by cells and cell lysates, making the screening approach more costly and low-throughput.An attractive alternative is to directly monitor the formation of products/co-products. One of the simplest and most widely used HT assays for screening amino acceptor specificity of ω-transaminase enzymes was developed by Schätzle et al. and involves using UV absorption spectrometry for the detection of acetophenone when α-methylbenzylamine is used as the amine donor (Scheme 1(a)).28 The elegance of this method stems from its simplicity, as it uses a cheap commercially available amine and UV detection for the screening of a relatively large library size. However, the limitation of only allowing analysis of low-absorbing amine acceptors and initial absorbance interference with the protein, hindered the wider usage of the methodology. Reetz et al. showed that a high-throughput UV/Vis-based method could be implemented for the enantioselectivity screening of a large library of lipase enzymes undergoing mutagenesis.29 The lipase-catalysed hydrolysis of a racemic ester (rac-p-nitrophenyl 2-methyldecanoate, rac-3) was chosen as the test reaction and four rounds of mutagenesis (error-prone PCR) were carried out to yield a mutant with 81% e.e. compared to the 2% e.e. in the starting wild-type enzyme.
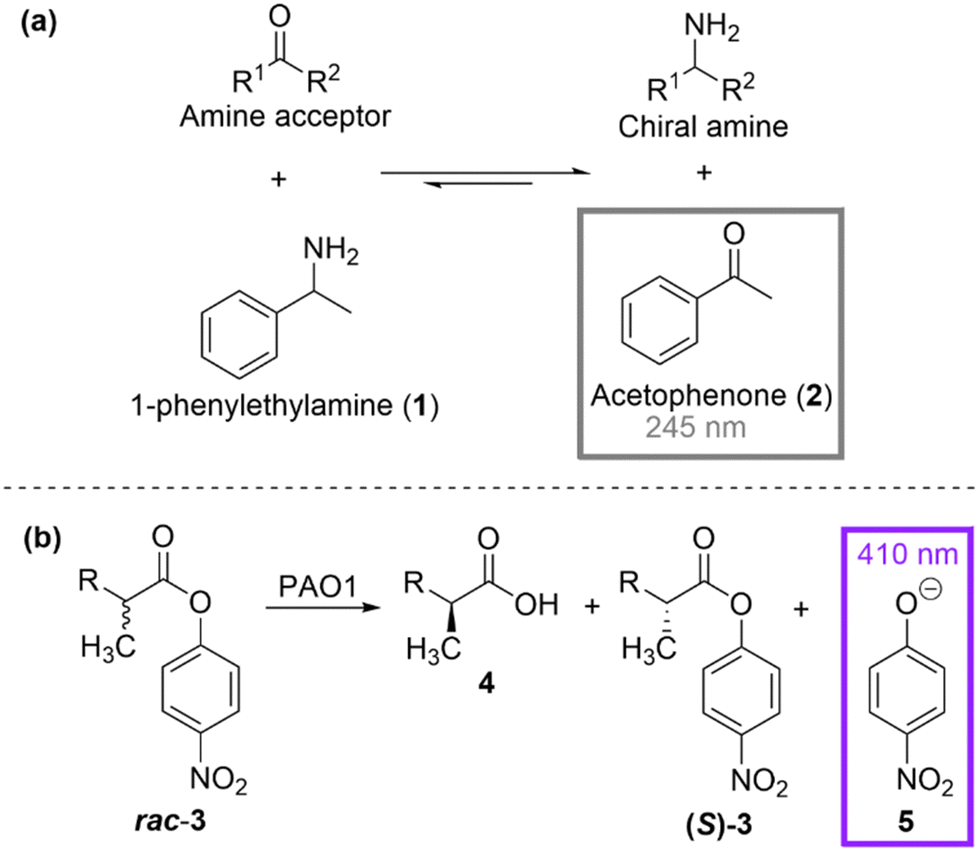 | ||
| Scheme 1 (a) Principle of the acetophenone assay for screening amino acceptor specificity28 (b) HT UV/Vis-based method for screening of enantioselective lipases.29 | ||
Measuring absorbance at 410 nm, the simple detection of the p-nitrophenolate anion by-product was used to monitor the hydrolysis as a function of time (Scheme 1(b)).
3.2. Colorimetric screening
Colorimetric screening is a sub-set of spectrophotometric screening that involves examining the analytes using the visible region of the electromagnetic spectrum (380–750 nm). The practicality and efficiency of the approach stem from the ability to visualise the ‘hits’ of a screening panel without the requirement of spectroscopic techniques and any specialised equipment. An elegant example of utilising a colorimetric approach in a mutagenesis study was demonstrated by Turner and co-workers when the substrate specificity of monoamine oxidase from Aspergillus niger (MAO-N) variants were screened for increased enantioselective preference for the oxidation of (S)-α-methylbenzylamine over the (R)-enantiomer. The colorimetric plate-based screening assay was based on the capture of the oxidation by-product, hydrogen peroxide, to monitor conversion. It did so by using a horseradish peroxidase alongside a colour-inducing substrate (3,3′-diaminobenzidine) to give rise to a pink, insoluble product that visually indicated the active colonies present on the nitrocellulose membranes. The same colorimetric screen was latter adapted by the Turner group to carry out further rounds of evolution on MAO-N to detect mutants that oxidised more challenging substrates such as secondary and tertiary amines as well as amines that have sterically demanding motifs.30–32A rapid and simple high-throughput screening method was also developed by the Turner group to identify and characterise a group of enzymes known as imine reductase (IREDs), which have been shown to synthesise a wide variety of primary, secondary and tertiary amine products.33 The screen, termed IREDy-to-go, takes advantage of the inherit reversibility associated with enzymatic transformations, as it monitors activity of the reverse oxidation of the target amine product. The NAD(P)H formed post-oxidation of the amine product was detected by a diaphorase-mediated reduction of a tetrazolium salt. Reduction of the salt led to the formation of a corresponding red/pink formazan dye that could be used to visually identify the most active IREDs. The authors screened metagenomic libraries to identify IREDs capable of performing reductive amination on several structurally demanding ketone and amine substrates for the synthesis of high-value chiral amines.
Our group previously reported the use of the amine donor o-xylylenediamine, which displays dual functionality as both an equilibrium mediator and as the colour-inducing reagent for colorimetric screening of ω-transaminases variants. Post-transamination, o-xylylenediamine spontaneously cyclises to the corresponding cyclic imine, which furthermore tautomerises to the unstable aromatic compound, isoindole. The isoindole produced rapidly polymerises resulting in intensely coloured by-products, which were used for qualitatively detecting ω-TA activity for both liquid- and colony-based assays. However, the wider application of the amine donor falls short as it exclusively allows for qualitative analysis of activity, not quantitative.34 To resolve this issue, our group developed a straightforward and comprehensive assay for ω-transaminases, which allowed for the identification of enzymes with activity towards a range of both amine donors and acceptors in liquid phase.35 The quantitative assay spectrophotometrically detects the formation of β-bis(indolyl)methane from the reaction of 2-aminoethylaniline and in situ produced or externally added indole. To increase the efficiency of the screen, it was adapted to a colony-based solid-phase format containing an engineered strain of E. coli that reduced background noise from endogenously produced indole.
Although these HTS methods are applicable to many engineering projects, the development of ultrahigh-throughput screening (uHTS) when working with extremely large amounts of variants is essential as it allows for the exploration of a larger pool of genetic diversity in a shorter amount of time.
3.3. uHTS (fluorescence screening)
The advancement of modern microfluidics has the potential to ramp up the enzymatic screening process by automatising the operation. Like microtiter plate and agar-based screens, uHTS requires a signal that corresponds to the activity of the enzyme, and fluorescence is often used to provide a method sensitive enough to enable single cell analysis. The basic requirements for fluorescent assays are that (i) the enzyme + probe produces a signal and (ii) a genotype–phenotype linkage can be established. The latter requires assay components to be compartmentalised. Using the cell itself as a natural compartment is attractive because Fluorescence Activated Cell Sorting (FACS)36,37 is capable of interrogating multiple fluorescent parameters of individual cells, and can both screen and sort during directed evolution. FAC sorters are instruments capable of interrogating multiple fluorescent parameters of individual cells. A challenge with the use of FACS is maintaining the genotype–phenotype linkage by ensuring the assay signal is contained within or tethered to the cell, but this approach has been successfully used to engineer many enzymes.38 When whole cells are not suitable fluorescence screening compartments, fluorescence activated droplet sorting (FADS) can be employed, which can sort single cells or cell components that have been encapsulated in emulsion droplets. The technique encapsulates the substrate and a single genetic variant (as a gene or whole cell containing the gene of interest) within a water-in-oil (w/o) emulsion. The emulsion can then be “incubated” for a certain period and temperature. This incubation period allows for either the in vivo expression of the enzyme and/or the conversion of the substrate to occur. Microfluidics commonly utilises fluorescence as the parameter corresponding to enzymatic activity and therefore requires the substrate and product to have differing fluorescence capabilities. The fluorescence levels of each droplet are typically detected as the emulsion passes through a laser being focused on the gap between two electrodes and are sorted via dielectrophoresis. A full comprehensive description on the specific applications of microdroplets in a chemistry and biology setting can be found in the review by Theberge et al.39Weitz and co-workers presented an elegant example of a uHTS platform for the identification of new optimised horseradish peroxidase (HRP) mutants utilising drop-based microfluidic technology. The procedure involved creating two generational libraries: the first using a mixed random mutagenesis approach via error-prone PCR (epPCR) and active site saturation mutagenesis, and the second using only epPCR with the 18 most active first-generation mutants. Yeast cells displayed the enzyme variants on their surface and the fluorogenic substrate Amplex Ultrared was used for the quantification of activity using fluorescence-activated droplet sorting (FADS). A total of ∼108 emulsion droplets within a time scale of 10 h were screened, identifying HRP mutants with more than ten times the catalytic turnover than its parent and some variants approaching diffusion-limited efficiency. This innovative screening technology proved to be 1000-fold faster with an estimated 10-million-fold reduction in cost, when compared to the state-of-the-art robotic screening at the time. Fluorescence-activated droplet sorting has evolved since, proving to be applicable to a range of different evolution studies.40 However, the procedure requires the use of a fluorogenic substrate to quantify and separate each droplet, which reduces its wider application to non-fluorescent substrates.
In a landmark study by Hilvert and co-workers, the required use of fluorogenic model substrates was circumvented by the implementation of a coupled enzyme assay within a chip-based microfluidic system.41 The assay screened for the directed evolution of a cyclohexylamine oxidase (CHAO), yielding improved catalytic efficiency for the deracemisation of 1-phenyl-1,2,3,4-tetrahydroisoquinoline to its (S)-enantiomer, which is a sterically demanding pharmaceutical intermediate for the production of the drug solifenacin. Although the system employed FADS as the sorting method, the fluorogenic species is produced by a reporter cascade that utilises the co-product, (FADH2) of the CHAO oxidation, which is then recycled and produces equimolar amounts of hydrogen peroxide (H2O2). This H2O2 is detected by a downstream oxidation of a fluorogenic substrate catalysed by an accompanying horseradish peroxidase (HRP) to produce the fluorophore. This study demonstrates how uHTS methods can radically outperform the conventional evolutionary optimisation methods such as typical microtiter plate assays by screening over 107 variants and sequencing the most enhanced protein genes all under a two-week timescale. Further advancements on the sorting process of ultrahigh-throughput droplet-based microfluidics has involved using labelled (fluorescence-, absorbance-activated) and label free (electrochemical, mass spectrometry, NMR, Raman) sorting.42
4. Applications in synthetic chemistry
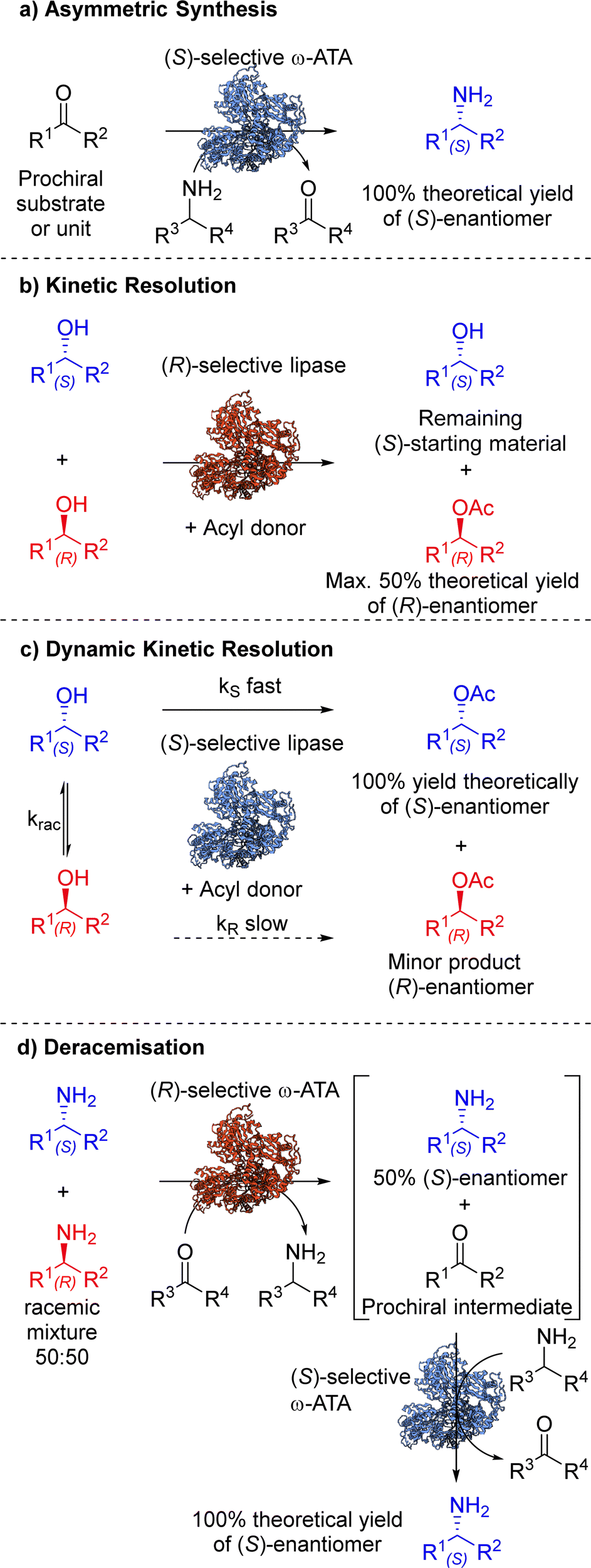
The inclusion of biocatalytic steps in synthetic strategies has become increasingly appealing to chemists in recent years. The ever-growing toolbox of enzymes and our ability to tailor their properties means that biocatalysts are now routinely considered alongside other asymmetric homogeneous catalysts when planning routes to important synthetic targets. There is growing pressure within the chemical industry to improve life cycle assessment (LCA) of processes, by adopting more environmentally friendly and less hazardous practices. Biocatalysis allows transformations to be performed under mild conditions and without the use of precious metals, which are so often associated with traditional chemical synthesis. The development of biocatalytic methodology that enables asymmetric synthesis, (dynamic) kinetic resolution and deracemisation remains a vibrant research area, and some illustrative examples are discussed here.4.1. Asymmetric synthesis
Asymmetric synthesis involves the introduction of one or more units of chirality (chiral centre, axis or plane) into a substrate, producing stereoisomeric products in unequal amounts, where one stereoisomer is favoured over another.43 The synthetic approach enables the stereoselective preparation of enantiopure products from substrates containing pro-chiral structural units (Scheme 2(a)). The field of enzyme engineering has played a pivotal role in developing efficient biocatalysts for asymmetric applications. This progress is marked by enhancements in stability, substrate scope and activity,44 using various protein engineering methodologies (Section 2). Enzymes can facilitate several types of asymmetric reactions including: (i) enantioselective replacement of an enantiotopic group located at a prochiral sp3 hybridised atom, for example in P450 monooxygenase catalysed C–H hydroxylation; (ii) enantioselective introduction of a nucleophile at a prochiral sp2 hybridised carbon atom, for example C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO and CN stereoselective reduction, by ene-reductases, alcohol dehydrogenases and imine reductases, respectively, or by C–heteroatom bond formation, for example transaminase catalysed chiral amine synthesis; (iii) enantioselective desymmetrisation by loss of one or more symmetry elements and; (iv) atroposelective reaction, where a chiral axis is formed in the molecule.45
C, CO and CN stereoselective reduction, by ene-reductases, alcohol dehydrogenases and imine reductases, respectively, or by C–heteroatom bond formation, for example transaminase catalysed chiral amine synthesis; (iii) enantioselective desymmetrisation by loss of one or more symmetry elements and; (iv) atroposelective reaction, where a chiral axis is formed in the molecule.45
 | ||
| Scheme 2 Biocatalytic induction of stereocontrol via a range of techniques including (a) asymmetric synthesis, (b) kinetic resolution, (c) dynamic kinetic resolution and (d) deracemisation. | ||
One group of enzymes that have been engineered to carry out this sought after transformation in a more efficient manner than their natural capability are cytochrome P450 monooxygenases (P450s). P450s are commonly employed enzymes in the asymmetric synthesis of desired pharmaceutical and chemical compounds, in the biosynthesis of natural products (e.g. paclitaxel (anti-cancer) and artemisinin (anti-malarial)), as well as in applications in gene therapy and bioremediation. P450s have been evolved to accept a broad substrate scope and enhanced properties through engineering of their nonpolar active sites.46 Engineering of wild-type P450s is important for their use as robust biocatalysts, as natural P450s often have poor solubility and stability, as well as low expression levels and turnover rates.47 P450s are NAD(P)H-dependent enzymes that use molecular oxygen to oxidatively modify an organic substrate, through the insertion of an oxygen atom, through hydroxylation, epoxidation, or dealkylation. P450s can catalyse C–H functionalisation at inactivated carbons centres under mild conditions with high regio- and stereocontrol. This type of reaction is extremely challenging to achieve using more traditional catalytic approaches, and as a result, the potential of P450s as synthetically useful biocatalysts to facilitate asymmetric synthesis has been heavily explored.48–50
Arnold and co-workers evolved an improved P450 biocatalyst from Pseudomonas putida to hydroxylate aromatic compounds without the use of cofactors, using hydrogen peroxide to instead facilitate a ‘peroxide shunt’ pathway.51 The concept is attractive as the biotransformation does not rely on an expensive cofactor (e.g., NADH) and its associated regeneration system. As a direct result, less by-products are produced, and the system's practical utility is greatly enhanced. Directed evolution was used to produce an improved horseradish peroxidase variant (HRP1A6), with higher activity and improved protein folding in the non-native E. coli environment. An efficient fluorescent digital image screen was developed to monitor the evolution of improved mono- and di-oxygenase variants. The screen relied on the co-expression of the variants with horseradish peroxidase, to convert the P450 catalysed products into fluorescent compounds.
A significant constraint associated with ω-transaminase enzymes lies in their restricted catalytic capability, limited to the synthesis of exclusively primary amines. Therefore, the discovery and development of a biocatalyst capable of synthesising chiral amines of higher saturation was of the utmost interest. This gap in synthetic utility was recently solved with the discovery of imine reductase (IRED) and reductive aminase (RedAm) enzymes. IREDs are NADP(H)-dependant oxidoreductase enzymes capable of catalysing the asymmetric reduction of prochiral imines to their corresponding chiral amine. This enzyme class has found application in the preparation of pharmaceutical targets and precursors, and its utility was expanded with the discovery of a subgroup (RedAms) capable of reductive amination starting from a prochiral ketone.54
Monoamine oxidases (MAOs) have been successfully employed for the asymmetric synthesis of optically pure amines, often using deracemisation strategies involving the selective oxidation of monoamines. Ghislieri et al. engineered enantioselective MAO variants, with tolerances for sterically demanding bulky aryl substituents, showcasing how enzymes can be evolved to asymmetrically synthesise structures with high molecular complexity.32 Rational structure-guided engineering approaches, saturation mutagenesis and HTS were used to develop the improved MAO biocatalyst variants from Aspergillus niger (MAO-N). Crystal structures of the enzymes and their active site helped to rationally select amino acid residues in which to mutate. The site-directed mutagenesis approach helped increase the volume of the active site pocket, allowing binding, positioning, and interaction with bulky sterically demanding aryl substituents. The MAO-N biocatalyst was applied in the asymmetric oxidative Pictet–Spengler reaction in the synthesis of (R)-harmicine, a chiral, bulky natural product. The MAO-N variant carried out the oxidative generation of a reactive iminium ion in a pyrrolidine substrate, containing a β-arylethylamine, which could undergo a non-stereoselective cyclisation to form racemic harmicine. The amine oxidase, in combination with a non-selective chemical reducing agent (BH3–NH3) was applied in repeated cycles of selective oxidation and reduction, to deracemise the racemic mixture into the desired (R)-harmicine (>99% e.e., 83% conversion).
4.2. Kinetic resolution (KR)
While asymmetric synthesis remains the most favoured approach for accessing stereo-enriched chiral products, it is often more practical to resolve racemic mixtures, prepared via non-asymmetric routes. This tactic may be preferred if the asymmetric approach is too challenging or if both enantiomers are desired. Chemists have used a range of well-established resolution methods when attempting to isolate a single enantiomer from a racemic mixture, such as crystallisation, chiral phase chromatography, and kinetic resolution.55–57 Kinetic resolution relies on the differential rates of reaction of each stereoisomer with a chiral entity, such as a biocatalyst or chemocatalyst (Scheme 2(b)).One of the most employed biocatalysts for kinetic resolution are hydrolases. These enzymes catalyse the cleavage of a covalent bond using water (hydrolysis) and represent one of the most attractive enzymes in synthesis, due to their broad substrate specificity, lack of cofactor/coenzyme, and high substrate loading tolerance. These enzymes can be further broken down into subclasses depending on the substrate they act upon.58 Lipases are one of the most popular subclasses of hydrolases and are deemed naturally promiscuous enzymes due to their ability to mediate reactions in both aqueous and non-aqueous media. This propensity to mediate reactions in organic solvents has led to the advent of non-aqueous enzymology and is particularly applicable to resolving processes.59
Optically active alcohols and carboxylic acids are among the most desirable reagents for organic chemistry due to their versatility and recurrent appearance in bioactive molecules, which has led to many resolution studies being conducted on compounds containing these moieties.60,61 With the intrinsic value of these compounds in mind, Kilbanov and co-workers were the first to introduce the concept when resolving a racemic mixture of secondary alcohols and carboxylic acids using two lipases (yeast lipases originating from Candida cylindracea and porcine pancreatic lipases).62 In this body of work, seven racemic acids and alcohols were optically resolved in high enantiomeric excess alongside their corresponding (R)-ester at a multigram scale. The process is of particular interest due to enantiopure 2-chloro- and 2-bromo-propionic acids being used as an intermediate in the total synthesis of phenoxypropionic herbicides and some pharmaceuticals. A non-aqueous enzymatic resolution approach has inspired hundreds of academic studies63 and has been scaled up to a multi-kilogramme scale by several companies world-wide.64 However, the drawback of maximum 50% conversion and the accumulation of large amounts of unwanted enantiomer has encouraged further studies to be conducted on developing a more efficient and higher yielding process.
4.3. Dynamic kinetic resolution (DKR)
DKR combines in situ racemisation of the starting material with an enantioselective transformation step, which allows the process to surpass the 50% conversion limit associated with kinetic resolution. While one stereoisomer (usually an enantiomer) is selectively converted to the corresponding stereospecific product, a chemical or enzymatic catalyst may act as the racemisation mediator to interconvert the opposite stereoisomer (Scheme 2(c)). In order to design a successful and efficient DKR system, four parameters, standardised by Verho and Bäckvall,65 must be taken into account; (i) the kinetic resolution reaction must have sufficient enantioselectivity (kfast/kslow ≥ 20), (ii) each catalyst (racemisation and KR) must be compatible in a one-pot system, (iii) rate of racemisation (krac) must be 10-fold faster than the rate of the KR reaction of the slow reacting enantiomer (kslow), and (iv) the racemisation catalyst must not react with the resolved product.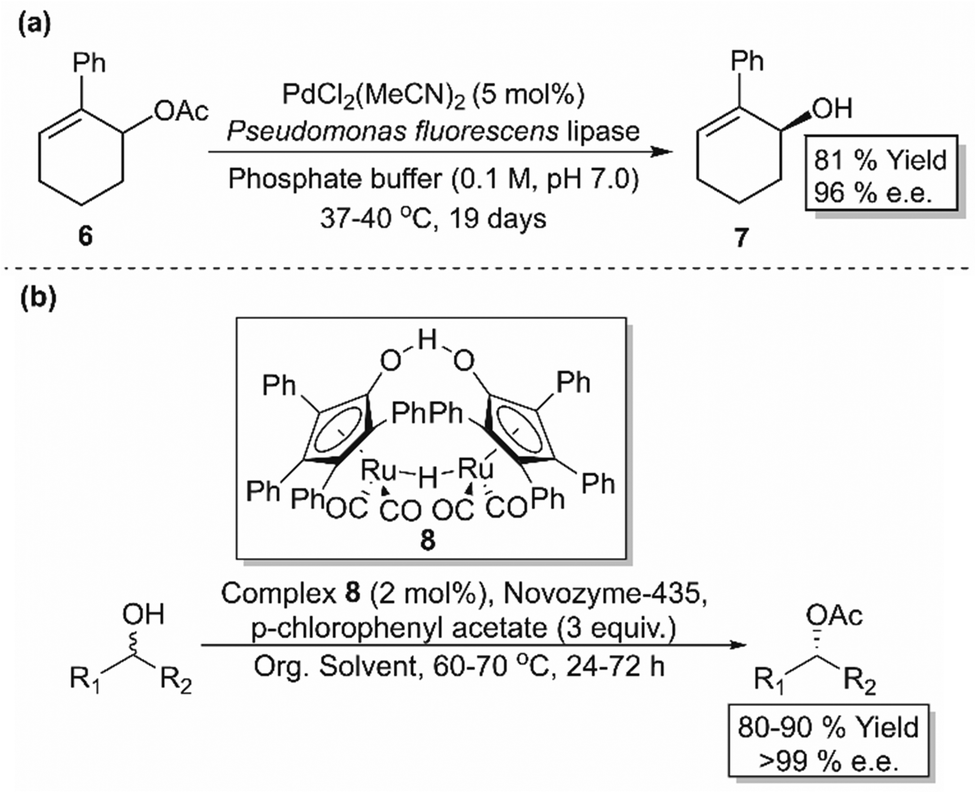 | ||
| Scheme 3 (a) DKR of 6-acetoxy-1-phenylcyclohexene (6) using PdCl2(MeCN)2 and a lipase enzyme.66 (b) DKR for the resolution of a range of secondary alcohols using ruthenium catalyst 8 and CALB.67,68 | ||
Being the most popular catalysts for alcohol racemisation, improved generations of Ru-based racemisation catalysts were developed in later years, including those that work on secondary alcohols at room temperature, those compatible with (S)-selective KR enzymes, and those that racemise various alcoholic functionalities including allylic alcohols, diols and chlorohydrins.65 An in-depth analysis of these racemisation catalysts is beyond the scope of this review, however there are comprehensive works that cover this topic.70–72 Alternative racemisation catalysts have also been developed using more cost-efficient and environmentally friendly metals such as aluminium and vanadium, with the latter being able to racemise tertiary alcohols, which are more difficult due to the lack of a hydrogen substituent (not compatible with transfer hydrogenation-type racemisation catalysts).70,73
 | ||
| Scheme 4 Dynamic kinetic resolution of methylbenzylamine (9) using CALB and Pd/C.74 | ||
Transaminase enzymes have also been shown to be a viable KR catalyst for dynamic kinetic resolutions of α-substituted aldehydes,79 α-substituted β-keto esters,80 and α- or β-substituted ketones.81,82 An elegant example of this was shown in the DKR of an α-fluoroketone by Molinaro and co-workers. A key building block, syn-1,2-amino-fluoropiperidine, in the synthesis of the calcitonin gene-related peptide receptor antagonist was synthesised using a dynamic asymmetric transamination route. The methodology utilised high pH conditions as the racemisation agent to promote epimerisation of the α-fluoroketone starting material. Optimisation of the reaction conditions yielded 96% conv. to the aminated product in 94% e.e.81
Enzymatic immobilisation can often lead to advantageous catalysts with enhanced stability, simplified recovery, and greater reusability. Immobilised enzymes can be obtained in different forms such as bound to a support carrier (inorganic solid, synthetic resin, polymers, etc.), entrapment (enzyme incorporated into polymer network) and CLEAs (cross-linking enzyme aggregates).89 The favourable properties obtained from using an immobilisation technique in a DKR system was demonstrated by Engström et al. when both CALB and a nanopalladium species were co-immobilised on mesoporous silica for the dynamic kinetic resolution of methylbenzylamine in high yield and enantioselectivity. Not only does the procedure take advantage of the benefits of an immobilised enzyme, but also the proximity of the two catalysts with relation to each other, which was proven to boost the efficiency of the dynamic kinetic resolution process.90
4.4. Deracemisation
Deracemisation is a protocol used to obtain an enantiopure product from a racemic mixture of starting material, proceeding through a prochiral intermediate (Scheme 2(d)), providing a theoretical 100% conversion to the desired enantiomer, and thereby overcoming the 50% conversion limitation experienced in kinetic resolutions. Deracemisation differs from DKR, in that it proceeds through a prochiral intermediate, rather than prior racemisation. An enzymatically catalysed deracemisation can proceed in two ways, (i) using two enantiocomplementary enzymes working in tandem, or (ii) by using one enantioselective redox enzyme and a non-selective chemical oxidising/reducing agent, which work together to achieve complete deracemisation after many redox cycles.A biocatalytic deracemisation procedure using two enantiocomplementary ω-transaminase (ω-TA) enzymes to access either enantiopure (R)- and (S)-mexiletine (11) from a racemic mixture was described by Koszelewski, Kroutil and co-workers.91 The deracemisation is achieved in a one-pot two-step cascade, involving a kinetic resolution followed by a stereoselective transamination, with the incorporation of by-product recycling. The design offers flexibility, allowing access to either enantiomer depending on the order the ω-TAs are added. The (S)-enantiomer can be accessed using a (R)-selective ω-TA for the resolution of a racemic amine mixture, leaving the (S)-amine untouched and transforming the (R)-amine into an intermediary ketone 14, which is subsequently transaminated into the desired (S)-amine by subsequent addition of a (S)-selective ω-TA (Scheme 5). The cascade incorporates recycling of pyruvate (12) as the amine acceptor and alanine (13) as the amine donor using auxiliary enzymes, amino acid oxidase and alanine dehydrogenase respectively. This biocatalytic approach allowed the sustainable deracemisation of the racemic amine mixture of mexiletine, resulting in >99% e.e. and 97% isolated yields of both enantiomers.
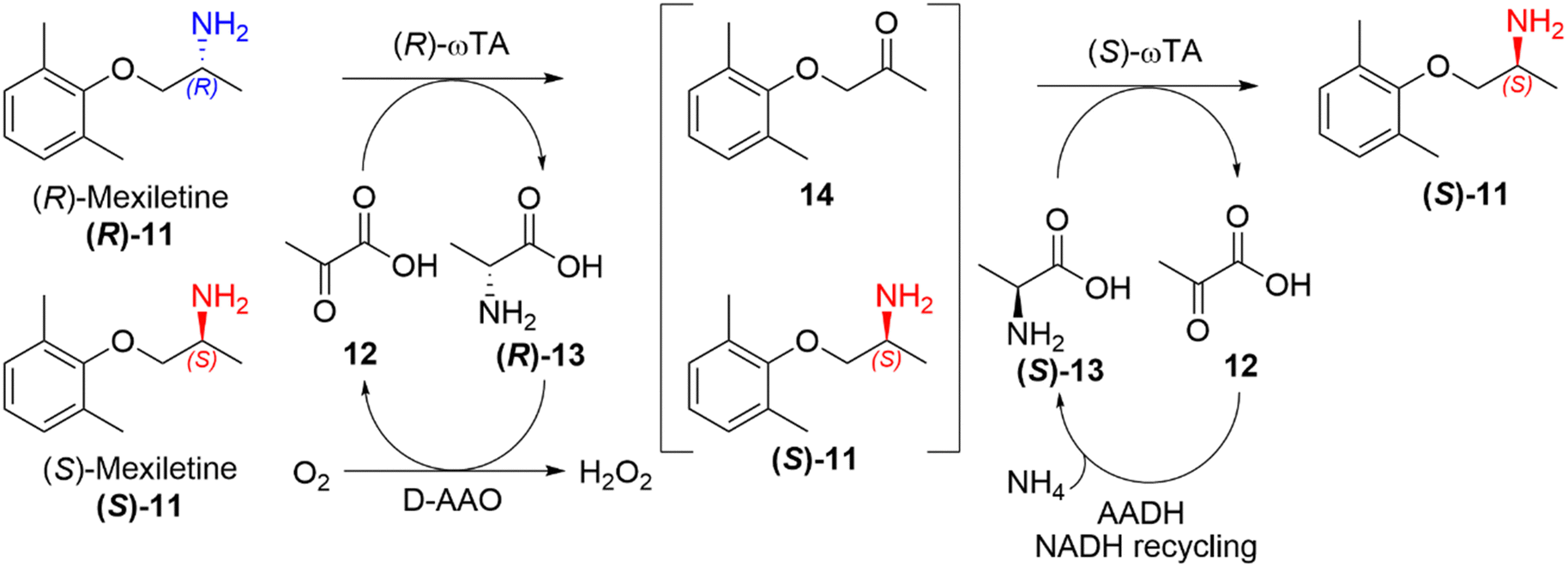 | ||
| Scheme 5 Deracemisation of racemic mexiletine (11) into enantiopure (S)-mexiletine using (R)-selective TA catalysed kinetic resolution and a subsequent stereoselective transamination using and (S)-selective TA.78 | ||
Amine oxidoreductase enzymes are widely used in the deracemisation of valuable amines. Their importance was first demonstrated using amino acid oxidases, structural family members of flavin amine oxidases,92 for the enantiomeric enrichment of racemic amino acid mixtures. L-Alanine and L-leucine were prepared from their corresponding D-enantiomers, using a D-amino acid oxidase and a non-selective chemical reducing agent.93 Although the process resulted in low yields, it was an important seminal example in this field. The chemistry was later improved and expanded enabling high yields and optical purities of both cyclic and acyclic D- and L-amino acids.94
Although amino acid oxidases are important in deracemisation and stereoinversion processes, they are limited in their scope to amino acid substrates only. Turner and co-workers have developed deracemisation processes, using engineered MAO enzymes to expand the scope of this chemistry, providing a new method to access chiral amines.
Extensive studies have been carried out to identify an enantioselective MAO enzyme, with the evolutionary starting point being a Type II MAO from Aspergillus niger (MAO-N). The enzyme showed high activity towards simple aliphatic amines, and low but measurable activity towards α-methylbenzylamine, oxidatively deaminating the substrate to the corresponding imine using molecular oxygen. A series of in vitro directed evolutionary experiments, employing multiple mutagenesis cycles, with a colorimetric screen was used to discover and engineer an amine oxidase with the ability to deracemise α-methylbenzylamine, in a chemoenzymatic route via a redox catalytic cycle.30
The substrate scope of amine oxidases was expanded to aliphatic, aromatic, and tetrahydro-β-carboline (THBC) fused ring systems, with further directed evolutionary approaches, whilst retaining high enantioselectivity.95,96 MAO-N variants were also successfully applied to the deracemisation of racemic mixtures of alkaloid natural products, e.g. (R)-coniine, (R)-eleagnine, and (R)-leptaflorine, and pharmaceutical building blocks, e.g. Solifenacin and Levocetirizine.32
Recent research has focused on developing a one-pot two-enzyme cascade, involving an amine oxidase and an imine reductase (IRED) for the deracemisation of racemic 2-substituted piperidines and pyrrolidines.97 The amine oxidase generates an imine from a racemic mixture of cyclic N-heterocycles, which is enantioselectively reduced by an (R)-IRED to give the (S)-amine, replacing the previous need of a nonselective chemical reducing agent and multiple cycles to give full deracemisation.
5. Enzymes in industry
Biocatalysts have been applied to a diverse range of industrial applications within the pharmaceutical, agricultural, chemical, cosmetic, and food industries (Fig. 4) including; the multi-ton production of the synthetic polymer polyacrylamide using nitrile hydratases,98 enzyme catalysed biofuel production,99 enzyme catalysed degradation of plastics,2,100,101 the industrial production of agrichemicals, such as the herbicide Frontier X2®,102 and in the biocatalysed production of the important drug intermediate, L-phenylacetylcarbinol (L-PAC).103 The food industry applies a range of biocatalysts for additive, catalytic and ingredient requirements in beverage fermentation, baking, meat and dairy processes.104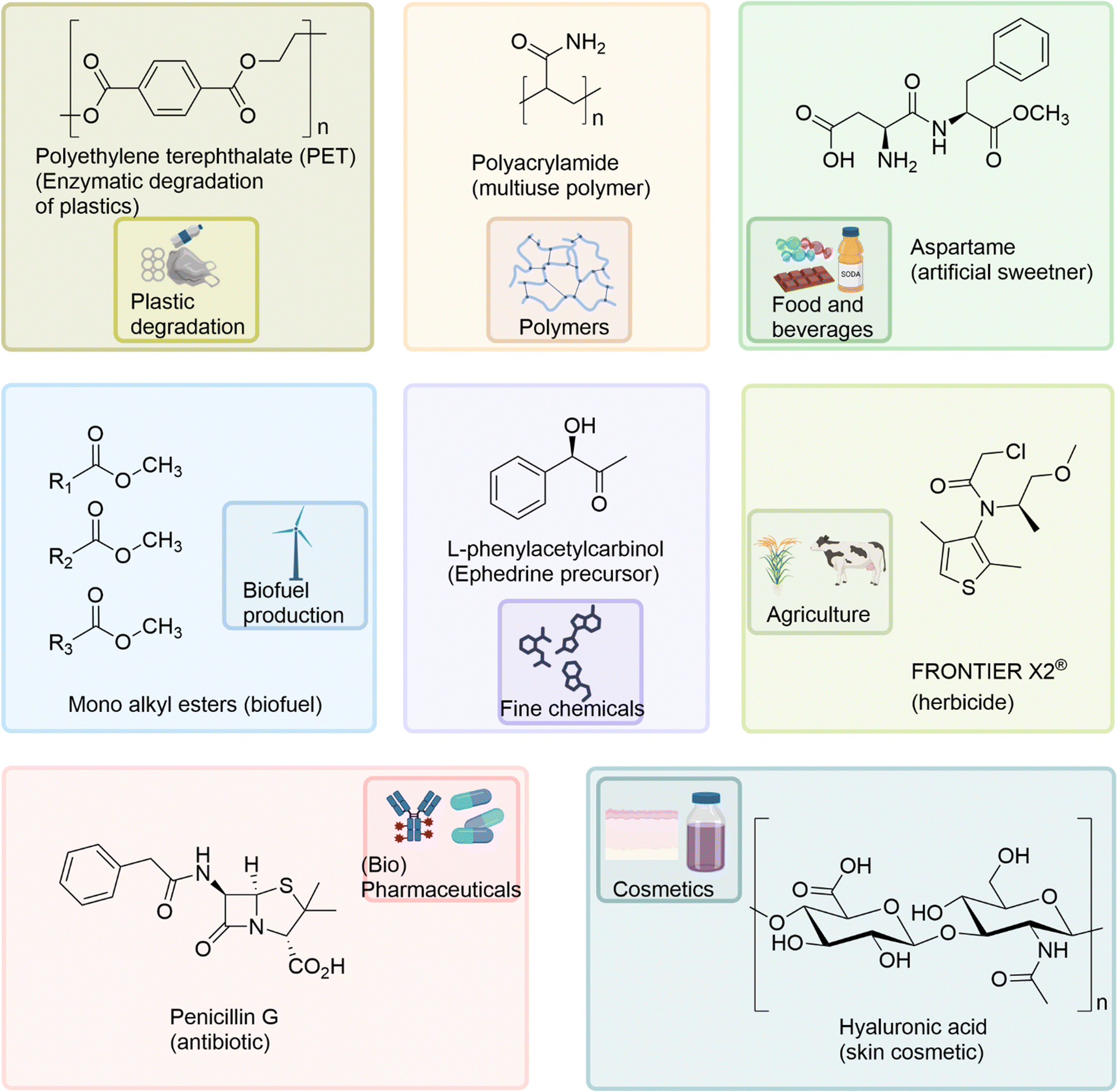 | ||
| Fig. 4 The diverse industrial applications of biocatalysts and their exemplar products, within the pharmaceutical, agricultural, chemical, cosmetic, and food industries. | ||
Enzyme's selectivity, tuneability and sustainable nature have made them an attractive alternative, to some more traditional catalysts, offering simplified synthetic routes to the assembly of complex targets. The economic and environmental attributes of enzymes, have furthered their incorporation into large-scale compound manufacture, as seen by many pharmaceutical companies, including Merck, Pfizer, Novartis, BASF, and Bayer.
Drawbacks surrounding enzyme stability and low turnover, long process development times and issues with scale-up to industrially relevant amounts, have been encountered and continued research is required to address these concerns. Nevertheless, biocatalysis continues to become an important consideration in target retrosynthesis, with the following examples chosen to provide a flavour of the diverse range of applications offered by biocatalysis.
5.1. Sitagliptin
The incorporation of a biocatalytic step in the large-scale pharmaceutical manufacture of the antidiabetic drug, sitagliptin ((R)-20), replacing the previously used transition-metal catalysed step, was a milestone development in the field of biocatalysis.46 An engineered transaminase enzyme was used for the successful installation of a chiral amine in the final step of the drug synthesis, replacing the original rhodium-catalysed asymmetric enamine hydrogenation and thereby negating the need of the high pressure (250 psi) hydrogenation, ligand optimisation and synthesis for the chiral Rh-catalyst, and additional purification steps for the removal of the precious and toxic rhodium catalyst (Scheme 6). The implementation of the key biocatalytic step was not only beneficial to the synthetic process, reducing cost and waste, it also enabled an enhancement of yields (increase of 10–13%), productivities (53% increase, kg L−1 per day) and high enantioselectivity. The incorporation of the biocatalytic step was also a landmark progression in the capabilities of enzyme engineering and called for an impressive engineering feat, involving a ‘substrate-walking’ approach alongside computer modelling, docking studies, and directed evolution. Substrate-walking, modelling and mutagenesis were first used to evolve the enzyme, (R)-ATA-117, limited to accepting only methyl or small cyclic ketones, to have activity for the bulky prochiral prositagliptin ketone, first using a surrogate-type truncated ketone substrate. The evolved enzyme with a broadened substrate scope and marginal activity to the prositagliptin ketone, was further engineered by directed evolution to improve its practical utility and ability to withstand industrial process conditions. Improved enzyme variants exhibited enhanced stability, withstanding organic solvents (50% DMSO, acetone and isopropyl amine), high substrate concentrations (prositagliptin ketone 200 g L−1) and elevated temperatures (>40 °C). The best and final enzyme variant, incorporating 27 mutations, exhibited four orders of magnitude increase in activity towards the prositagliptin substrate, while providing high enantioselectivity of >99.5% e.e. The achievement demonstrated how a combination of in silico design and directed evolution can evolve an enzyme into a robust biocatalyst, capable of working in industrial process conditions, possessing high productivity, activity and selectivity at an industrial scale. The landmark advancement served as an example of how biocatalysis is an attractive alternative to traditional catalysis.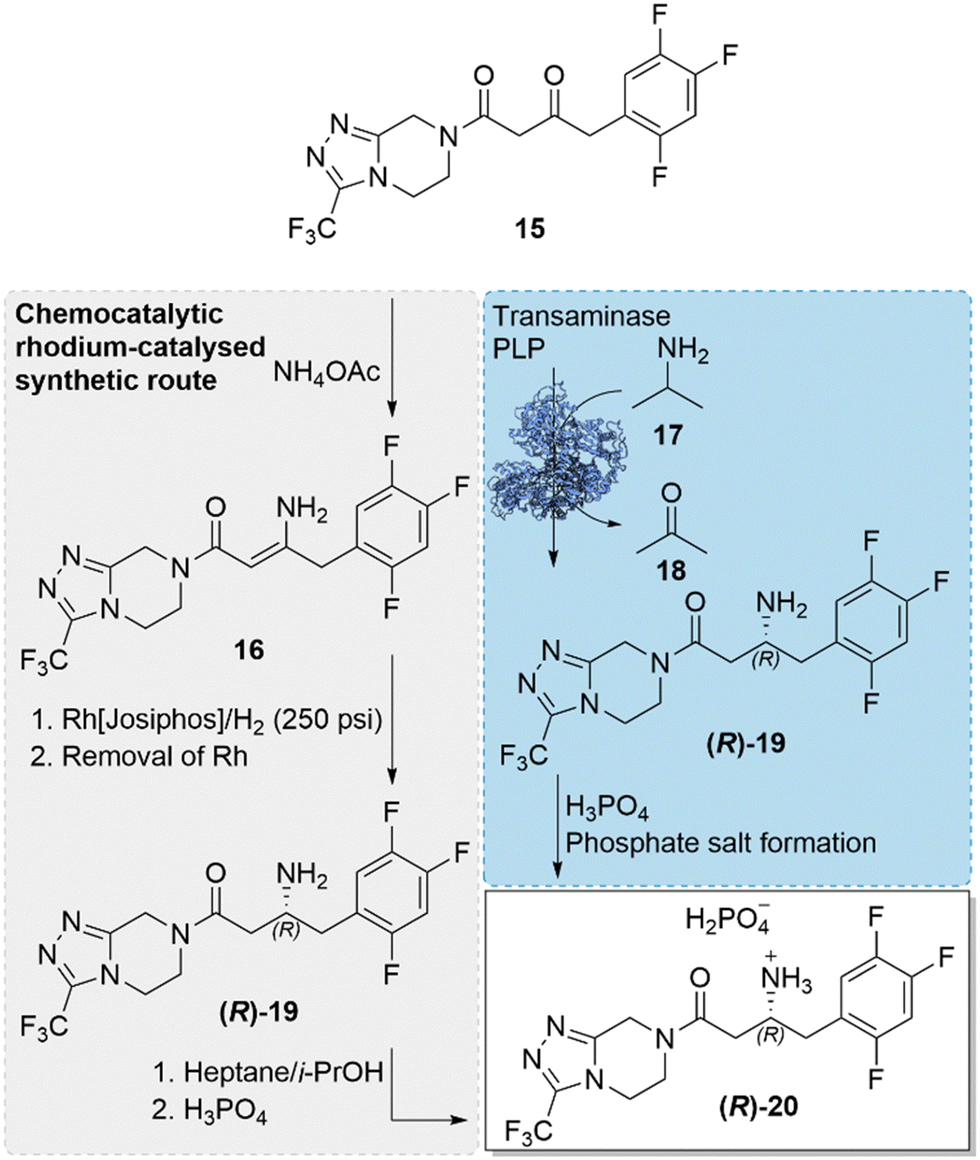 | ||
| Scheme 6 Industrial synthetic routes towards the synthesis of sitagliptin phosphate ((R)-20). The original rhodium-catalysed step was replaced with the transaminase catalysed approach, installing a chiral amine, in a key step of the antidiabetic drug.46 | ||
5.2. Hydrolase enzymes and the industrial relevance of CALB lipase
Hydrolases and the subclass, lipases, are an important enzymatic class in industry, used to carry out the kinetic resolution of racemic mixtures, allowing access to enantiopure products (Section 4.2). Their broad substrate specificity, lack of cofactor/coenzyme requirements and their ability to work in both aqueous and organic solvents, have enabled them to become a widely used enzyme in industry, with companies such as BASF incorporating such enzymes into routine industrial synthetic processes, a landmark development in the biocatalytic field.Candida antarctica lipase B (CALB) is a routinely used industrial biocatalyst lipase for the synthesis of amides and esters, as well as in the dynamic kinetic resolution of high value amine and alcohol products. CALB offers a low cost, easy to use and recyclable catalytic process for industrial manufacture of important optically active molecules, known as “ChiPros”, serving as intermediates, building blocks and auxiliaries. BASF uses their patented biocatalytic approach on an industrial scale to access optically active building blocks of alcohols and amines, using lipases to kinetically resolve racemic mixtures.105 The chemistry has been expanded to include nitrilase enzymes, which can convert a racemic mixture of mandelonitrile (rac-21), the cyanohydrin derivative of benzaldehyde, to the corresponding amino- and hydroxycarboxylic acid, namely (R)-mandelic acid (22), an important drug precursor. The chemistry has been achieved on a multi-ton scale, with high levels of enantioselectivity,106 a major development and proof of concept for the utility of biocatalysis at an industrially relevant scale (Scheme 7).
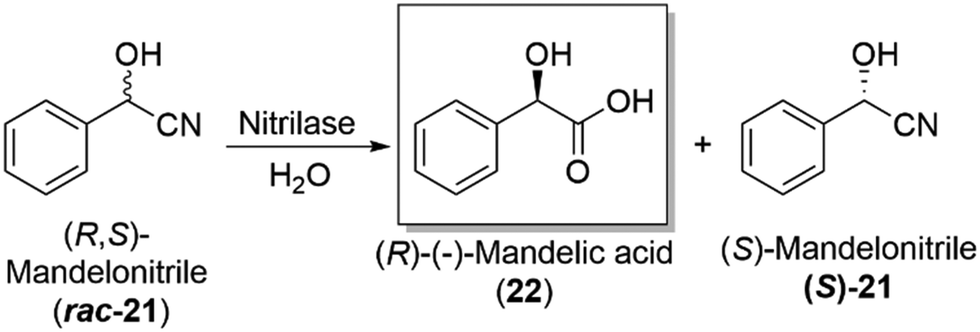 | ||
| Scheme 7 Industrial production of the important drug precursor, (R)-mandelic acid (22), employing a nitrilase enzyme as a key step in the overall production of (S)-mandelonitrile ((S)-21).106 | ||
The requirement of high biocatalytic loadings has hindered uptake of certain biocatalytic processes in industry,107 but it paves the way for further chemical discoveries. One such example, was the use of a salt-activated CALB lipase in a two-step enzymatic derivatisation strategy of paclitaxel (Taxol) derivatives, to improve the water-solubility and therefore the bioavailability, for use as potential anticancer compounds. A thermolysin enzyme was used for selective acylation of paclitaxel, and a CALB enzyme was used in the selective hydrolysis of the terminal vinyl ester in Taxol 2′-vinyladipate to give the final Taxol 2′-adipic acid derivative.108 The two step- synthetic strategy involving the CALB lipase enzyme provided water-soluble potential prodrugs, and the demonstrated the importance of the CALB enzyme in industry.
5.3. Penicillin – preparation of penicillin analogues with penicillin acylase
The group of enzymes known as penicillin acylases play an important role within the biotechnological industry, replacing traditional chemical methods, offering safer and more efficient enzymatic routes for the industrial production of semi-synthetic penicillin (amoxicillin and ampicillin) and cephalosporin (cefadroxil and cephalexin) antibiotic products. Immobilised penicillin acylases are used to catalyse the condensation of 6-aminopenicillanic acid with a D-(−)-amino acid derivative, in a kinetically controlled route to the β-lactam semi-synthetic products.109 The penicillin acylase enzymes are also employed to catalyse the hydrolysis of the fermentation product, penicillin G, into 6-aminopenicillanic acid, for use as the starting material in the overall process. The biocatalytic machinery exploited for this industrial process has enabled the production of natural products derivatives aimed at overcoming antibiotic resistance, and has done so in an enantiopure, economically feasible approach.5.4. Recent examples of enzymes used in industry
Novartis in collaboration with Codexis, recently evolved an amine transaminase enzyme, CDX-043, to asymmetrically install an amine into the bulky molecular architecture of the chiral precursor, sacubitril, a key component of the cardiovascular blockbuster drug, Entresto.110 The chemical synthetic route to sacubitril, although offers high yields, enantiomeric and diastereomeric purity, requires a lengthy multistep synthesis involving environmentally hazardous reagents, protecting groups and the generation of significant amounts of organic waste. An efficient and cost-effective biocatalytic transamination approach was sought, requiring extensive engineering to an enzyme capable of accepting the sacubitril precursor and operating at industrially relevant conditions. Following 11 rounds of directed evolution and mutagenesis, the starting enzyme, which suffered from trace activity and a preference for the undesired diastereomer, was evolved into the variant, CDX-043, capable of providing a 90% conversion to the desired compound (2R,4S)-5-([1,1′-biphenyl]-4-yl)-4-amino-2-methylpentanoic acid, and with high diastereoselective purity (>99.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1 d.r.). CDX-043 was also capable of operating in elevated temperatures up to 65 °C, in high substrate concentrations (2 molar amine donor, 75 g L−1 ketone substrate) and for 5 day reaction times.
:0.1 d.r.). CDX-043 was also capable of operating in elevated temperatures up to 65 °C, in high substrate concentrations (2 molar amine donor, 75 g L−1 ketone substrate) and for 5 day reaction times.
In work published in 2017, Pfizer opted to employ two enzymes, chosen using bioinformatics, to construct two key chiral intermediates in the synthesis of their novel anticancer, gamma secretase drug candidate.111 The chemoenzymatic route first used a transaminase (ATA-47 from cLEcta) to reductively aminate a fluorine substituted tetralone to the corresponding (S)-amine in a 95% yield and >99% e.e. The second desired intermediate, (R)-α-hydroxyester, was formed from an alcohol dehydrogenase catalysed reduction of an α-ketoester, which occurred on a multikilogram scale, with an 88% yield and 96.7% e.e. The enzymatically synthetised amine and alcohol products could then be coupled to form a key intermediate in the gamma secretase inhibitor. The work is an example of how biocatalysts are being considered as a viable technology and are finding increased use within industrial synthetic routes, operating at multikilogram scales, and displaying the necessary robustness to synthesise compounds of high purity and yield.
6. Biocatalytic cascades
Biocatalytic cascades, where several enzymes work in tandem, are prevalent in nature for the synthesis of many natural products. Enzyme activity in nature is tightly regulated, with the occurrence, location and extent of the transformations taking place being precisely controlled. In recent years, industrial and academic groups have constructed artificial biocatalytic cascades, inspired from natural biosynthetic routes, to develop strategies accessing synthetically challenging compounds.112 The design of artificial enzyme cascades is made possible due to advances in enzyme discovery and screening, alongside extensive protein engineering to create robust biocatalysts. The chemoselectivity of biocatalysts and the similar aqueous reaction conditions required, allows enzymes to operate in a one-pot approach, concurrently, sequentially or in a telescoped manner, while displacing reaction equilibria towards the ultimate product compound. Enzymes offer unparallel chemo-, regio-, and stereoselectivity, negating the need of protecting groups, isolation and purification of intermediates, and lengthy multistep synthesis. The use of expensive and unsustainable organic solvents which maybe possess various health, environmental and safety hazards, alongside energy intensive synthetic routes (high pressures, very low/high temperatures) and significant waste disposal encountered in traditional chemical strategies, makes biocatalysis an extremely attractive alternative.6.1. Biocatalytic cascade design considerations
This section will discuss the design considerations necessary to develop an efficient, sustainable, and practical biocatalytic cascade. Retrosynthetic analysis, the type of cascade, type and form of enzyme, cofactor requirements and optimisation requirements will be examined below. The landmark design and development of the nine-enzyme in vitro cascade, for the synthesis of islatravir, will be used as an exemplar model case study of how to build and optimise an efficient biocatalytic cascade.Computer-aided synthesis planning (CASP) has revolutionised retrosynthetic analyses, using computers to automate the process, learning from literature sources and databases of organic reactivity and experimental reaction, to create potential synthetic routes towards targets. RetroBioCat is a powerful CASP tool enabling retrosynthetic analysis for the design of biocatalytic reactions and cascades,116,117 an area other retrosynthetic databases does not sufficiently depict. RetroBioCat uses programmed reaction rules and encoded potential biocatalysts with their associated substrate specificity's, to automate the synthetic biotransformation design process. An enzyme toolbox, available on the database, displays the many enzyme types, their substrate scopes, and other important information regarding the biocatalyst, required for the construction of viable synthetic pathways. The database is an open access platform.
CASP technology is a rapidly growing area, with effective machine-learning enzymatic transformer models being developed, using multi-task transfer learning, to efficiently predict structure and stereochemistry of enzymatic transformations.118 Reymond and co-workers continued to expand the CASP technology to encapsulate and predict more information, with a recently developed open-source CASP resource to enable realistic and practical disconnections for short simple synthetic routes.119 The technology employing a triple transformer loop (TTL) involving necessary starting materials (T1), required reagents (T2) and resulting product (T3). The TTL works by tagging reactive atoms in the products, to propose efficient and sensible single-step retrosynthesis. The TTL in combination with a multistep tree-exploration algorithm (TTLA) can be employed for planning longer synthetic strategies to more complex molecules, by involving a route penalty score (RPScore) to predict practical syntheses.
The use of CASP for retrosynthetic analyses involving biocatalysis, offers a simplified, automatic approach to artificial biocatalytic cascade. The continued development of more accurate and efficient software to design increasingly complex synthetic routes involving enzymes, will be instrumental in the wider uptake and establishment of the importance of biocatalysis in synthesis.
Enzyme availability, cofactor recycling needs and operational process requirements may influence whether an in vitro or in vivo approach is adopted. In vitro cascades, employing crude cell free extracts, purified isolated enzymes, or freeze-dried whole cells, are advantageous as a controlled, reproducible amount of enzyme is added each time. However, such in vitro approaches often require the use of stoichiometric amounts of expensive cofactors and additional recycling systems, which can complicate the operation of the cascade. The design of in vivo cascades, where whole cells are used to continually express the desired enzyme and produce the necessary cofactors, can overcome some of these challenges. However, undesired side reactions, downstream processing and more difficult purifications are experienced with a whole-cell approach.
Biocatalytic cascades used in organic synthesis often work towards building molecular complexity,121 adding functionality and units of chirality to compounds, often transforming cheap achiral materials into high value chiral products. Multi-enzyme cascades can also be seen as an important design consideration to build more sustainable and atom-efficient processes, by incorporating auxiliary enzymes to facilitate co-factor regeneration and/or removal of (by-)products. Cascades remove the need for isolation of potentially hazardous intermediates, thereby offering a safer synthetic approach.
Wong and Whiteside developed an enzyme-catalysed regeneration system of the expensive reduced nicotinamide cofactors (NAD(P)H), allowing valuable asymmetric reactions, relying on such cofactors, feasible and applicable for incorporation into larger scale processes.124 The regeneration system involved the dehydrogenation of glucose-6-phosphate (G-6-P) by the commercially available, inexpensive, and stable enzyme, glucose-6-phosphate dehydrogenase, which could be immobilised further improving stability. The reaction is essentially irreversible, which aids to displace the equilibrium of the overall process towards product formation. The enzyme is importantly able to almost equally reduce both NADH and NADPH, making the system an operationally simple process for both types of cofactors. The cascade can be widely applied and simple preparation of the G-6-P starting material, is possible by several different methods. The regeneration system was showcased by incorporating it into the asymmetric synthesis of three important chiral building blocks; D-lactic acid, (S)-benzyl-α-d1 alcohol and threo-Ds(+)-isocitric acid. In all three cases, the dehydrogenase enzymes required, use a reduced nicotinamide cofactor, and therefore the cofactor regeneration system can be used in each process with the innocuous by-product 6-phospho gluconate.
In 2015, Mutti et al., showcased a two-enzyme in vitro hydrogen borrowing cascade incorporating in situ cofactor regeneration and reuse.126 The cascade involves the oxidation of an alcohol to the corresponding intermediary ketone using an alcohol dehydrogenase and NAD+ cofactor. The subsequent step employs an amine dehydrogenase to reduce the ketone to a chiral amine while simultaneously oxidising and recycling the NAD(H) cofactor. The research showcased here highlights the use of cascades for the recycling and supply of cofactors and by-products.
Chemical transformations catalysed by enzymes may experience thermodynamic limitations requiring consideration on driving the equilibrium towards the desired product formation. One such example is transaminase catalysed reactions, where careful choice of an amine donor, to facilitate the transfer of an amino group to the acceptor molecule, and a possible recycling system to remove the co-product, must be considered to drive equilibrium towards product formation. Celgene (now Celgro) pioneered the use of isopropylamine (IPA) as a cheap practical amine donor for transaminase enzymes for use at an industrial scale.127 The removal of acetone, as the resulting co-product, via evaporation, can drive equilibrium towards the amine product. Celgene showcased their improved transaminase technology for the synthesis of a range of stereocontrolled compounds, including (S)-Moipa, an intermediate in the synthesis of an herbicide. Despite the production of Moipa continuing with the optimised and well-established lipase enzyme technology, the research paved the way for IPA/acetone to become the amine donor/amine acceptor pair of choice in subsequent industrial applications using transaminases.
Smart amine donors can be used in transaminase catalysed transformations to push the unfavourable reaction equilibrium position towards chiral amine products, through spontaneous cyclisation or polymerisation of the resulting co-product. The commercially available diamine donor, ortho-xylylenediamine dihydrochloride can be used as an effective amine donor to shift equilibrium, by forming a cyclic imine by-product which tautomerises into the more stable aromatic isoindole form.34 The approach was an advantageous development, contributing towards thermodynamic and equilibrium control, but also as a substrate-independent colorimetric high-throughput screen due to spontaneous polymerisation giving a strongly coloured precipitate (see more in Section 3.2). The approach has been expanded to include further diamine donors, for example cadaverine, capable of displacing challenging reaction equilibria, through spontaneous cyclisation of the resulting by-products, shifting the thermodynamics in the transaminase catalysed transformations.128–131
The development of a biocatalytic cascade may require further process optimisation involving evolution of a suitable and efficient biocatalyst and its immobilisation. The key cascade design considerations discussed in this section, are highlighted below in the model cascade case study towards the synthesis of islatravir, contributing towards the design and the development of an efficient, selective and state-of-art synthetic approach.
6.2. Model biocatalytic cascade case study: islatravir
Merck's and Codexis’ sophisticated nine-enzyme in vitro cascade, for the synthesis of the structurally complex drug, islatravir (29), marked a landmark progression in the field of biocatalysis (Scheme 8).132 Five enzymes, namely purine nucleoside phosphorylase (PNPRd5BB), phosphopentomutase (PPMRd3BB), deoxyribose 5-phosphate aldolase (DERARd3BB), pantothenate kinase (PanKRd4BB) and galactose oxidase (GOaseRd13BB) were engineered by directed evolution for improved activity, stability, and ability to withstand high substrate concentrations needed for industrially relevant processes. Four auxiliary enzymes were incorporated into the final biocatalytic cascade for by-product removal, favourable reaction equilibria, cofactor regeneration and maintaining the oxidation state of copper-dependent enzymes. The biocatalytic cascade employed the bacterial purine nucleoside salvage pathway, which was engineered to allow it to occur in the reverse direction. The cascade was able to synthesise islatravir, the deoxyribonucleoside type compound, from simple achiral building blocks, using five engineered enzymes in a three-step biocatalytic cascades, with four of the enzymes working simultaneously to complete the final third step and to shift equilibrium towards the product. The finished cascade synthesis was a landmark progression in the field of biocatalysis exhibiting multiple enzyme engineering examples for a concurrent synthesis strategy with amplification of the stereochemistry at each successive step for the construction of a complex target.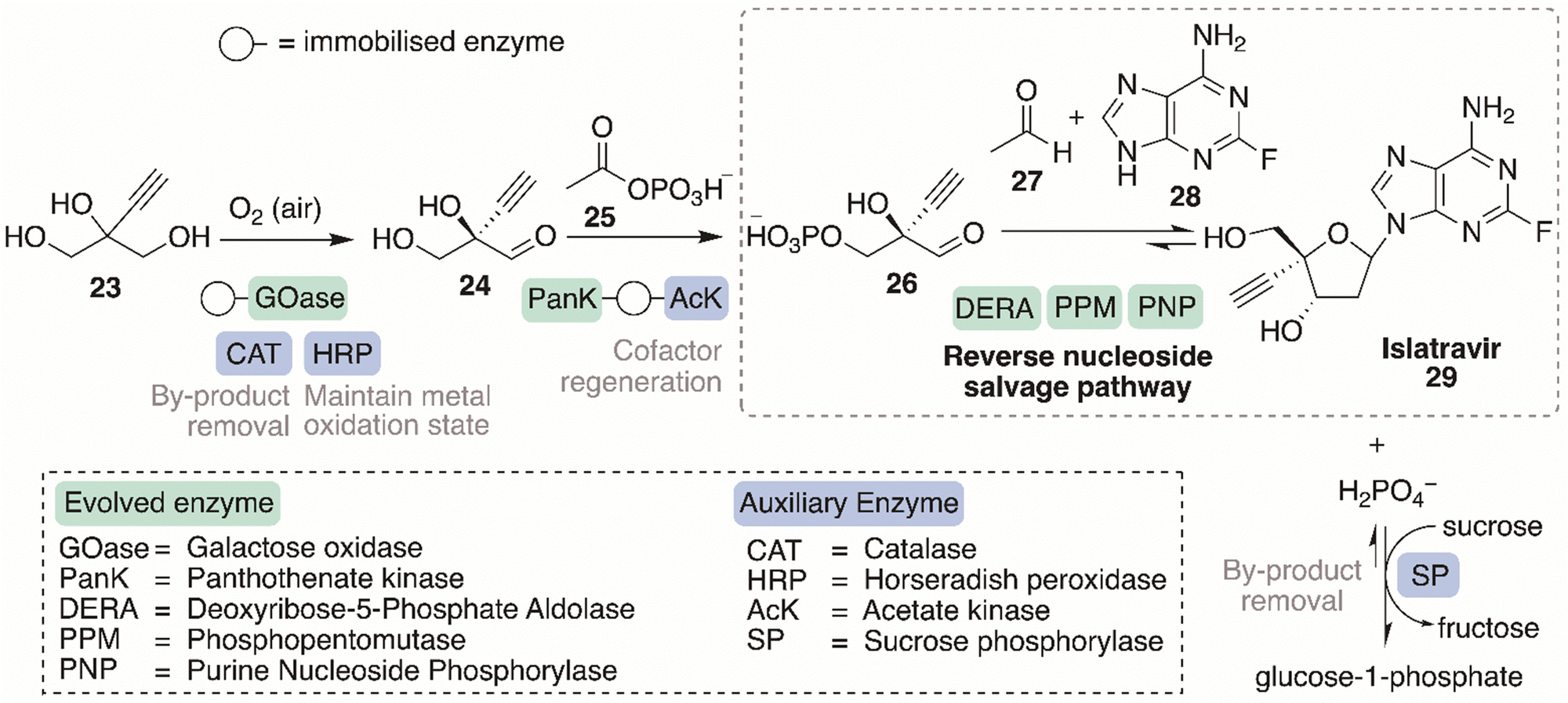 | ||
| Scheme 8 The engineered biocatalytic cascade, involving nine enzymes, for the production of the deoxyribonucleoside type drug islatravir. | ||
6.3. Hybrid bio-organocatalytic cascade approach
The combination of biocatalysis with chemocatalysis in a hybrid cascade approach has gained a lot of attention, and many systems have been designed to construct complex targets.68,133–135 As previously discussed in Section 4, Williams and Backvall developed metal-based chemocatalysts to carry out racemisations before using an enzyme-catalysed kinetic resolution approach to access the desired stereoisomeric product. Kajimoto, Wong and co-workers developed a chemo-enzymatic route for the asymmetric synthesis of various azasugars and their derivatives, including N-acetylglucosamine, N-acetylmannosamine, and deoxyhexoses.136 The synthetic route developed was important for showcasing the value of enzymes in asymmetric synthesis and for developing azasugars with the potential to be valuable glycosidase inhibitors. The asymmetric synthesis of the azasugars involved over 5 steps including an aldol condensation reaction, catalysed by the enzyme fructose 1,6-diphosphate aldolase (FDP-aldolase) and a palladium on carbon (Pd/C) hydrogenation for a catalytic reductive animation step. A phosphatase enzyme was used in cases where removal of a phosphate group was required. The starting materials for the aldol reaction, (R)- and (S)-3-azido-2-acetamidopropanal, were prepared from a lipase-resolved 2-hydroxy species. The synthetic route demonstrates the use of a number of different enzymes facilitating various functions including diverse chemical transformations, stereocontrol and resolutions, while being used alongside chemocatalysis for the preparation of biologically active compounds.Our group recently published work on a hybrid bio-organocatalytic cascade, inspired from the nature's biosynthetic route, to construct 2-substituted piperidines.137 The cascade involves the in situ generation of Δ1-piperideine in a transaminase-catalysed reaction, followed by a L-proline-catalysed Mannich-type reaction employing a panel of aliphatic ketones. The cascade employs the aliphatic ketones in a dual function as both the amine acceptor in the enzyme-catalysed step and as the nucleophile in the chemocatalysed Mannich step.
Turner and Marsden have developed an asymmetric route for the synthesis of 2-substituted tetrahydroquinolines, using both biocatalysis and chemocatalysis, to achieve this a C–C bond forming conjugate addition/condensation reaction between alkyl vinyl ketones and aminophenylboronic acids, catalysed by a rhodium(I) chemocatayst was used to yield a panel of racemic tetrahydroquinolines.138 An amine oxidase enzyme was then used to deracemise the racemic mixtures into enantioenriched tetrahydroquinoline natural products. The work showcased the benefits of employing both forms of catalysis alongside one another to construct complex molecules.
7. Conclusions
The field of biocatalysis has come a long way since the discovery of the first enzyme and confirmation that these catalysts are proteins. We are no longer restricted to the use of natural enzymes or to reaction conditions that directly mimic the biological cell. Advances in molecular biology and the development of enzyme engineering strategies means that enzymes can now be developed for bespoke applications and used to produce anything from milligram to tonne quantities of high-value molecules. The incredible specificity and selectivity of enzymes also allows for the design of complex biocatalytic cascades, meaning simple starting materials can be rapidly converted to complex molecules, without the need for traditional protecting group manipulations or purification steps. As the library of enzymes has grown over the past few decades, elegant enzymatic methodology has been developed and this has enabled the preparation of a wealth of complex chiral small molecules. Importantly, new enzymes with novel activity are still being discovered and existing enzymes being engineered towards completely new chemistries. Although powerful, top-down enzyme engineering has limitations. An ideal starting template for evolution should possess basal activity, which can hamper top-down engineering towards non-natural substrates. Additionally, enzymes are broadly restricted to catalysing biologically relevant reactions, and this restricts the chemistry that can be performed with enzymes. To ensure that biocatalysts can mediate a greater range of reactions, it is necessary to turn to computational enzyme design and approach biocatalyst development from the ‘bottom-up’. In the subsequent article in this issue, Bell et al. explore key developments in this area.139Author contributions
AO’C, AB and EOR conceived and wrote the article. APG contributed towards ideas for content and together with AJB, AEH and ELB, provided critical commentary during manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Science Foundation Ireland (SFI) Frontiers for the Future Project (19/FFP/6469) and from the A2P-Centre for Doctoral Training, which is supported by Science Foundation Ireland (SFI) and Engineering and Physical Sciences Research Council (EPSRC) (18/EPSRC-CDT/3582). AB is supported by BiOrbic under the SFI Research Centres Programme (16/RC/3889). For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.Notes and references
- Nature – Subjects – Biocatalysis, https://www.nature.com/subjects/enzymes, (accessed 24 October 2022).
- C. Jönsson, R. Wei, A. Biundo, J. Landberg, L. S. Bour, F. Pezzotti, A. Toca, L. M. Jacques, U. T. Bornscheuer and P. O. Syrén, ChemSusChem, 2021, 14, 4028–4040 CrossRef PubMed.
- M. M. M. Raja, A. Raja, M. M. Imran, A. M. I. Santha and K. Devasena, Biotechnology, 2011, 10, 51–59 CrossRef CAS.
- S. K. Metkar and K. Girigoswami, Biocatal. Agric. Biotechnol., 2019, 17, 271–283 CrossRef.
- A. Payen and J. Persoz, Ann. Chim. Phys., 1833, 53, 73–92 Search PubMed.
- D. E. F. Armstrong, Nature, 1933, 131, 535–537 CrossRef.
- L. Pasteur, C. R. Chim, 1858, 52, 404–418 Search PubMed.
- Eduard Büchner – Nobel Lecture: Cell-free fermentation, https://www.nobelprize.org/prizes/chemistry/1907/buchner/lecture/(accessed 24 October 2022).
- J. B. Sumner, in A Source Book in Chemistry, 1900–1950, ed. H. M. Leicester, The Isolation and Crystallization of the Enzyme Urease, Preliminary Paper, Harvard University Press, Cambridge, MA and London, England, 1968, pp. 322–327 Search PubMed.
- J. H. Northrop, Biol. Rev., 1935, 10, 263–282 CrossRef CAS.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1894, 27, 3479–3483 CrossRef CAS.
- D. E. Koshland Jr, Proc. Natl. Acad. Sci. U. S. A., 1958, 44, 98–104 CrossRef PubMed.
- L. Michaelis and M. L. Menten, Die Kinetik der Invertinwirkung, Biochem. Z., 1913, 49, 333–369 CAS.
- D. A. Jackson, R. H. Symons and P. Berg, Proc. Natl. Acad. Sci. U. S. A., 1972, 69(10), 2904–2909 CrossRef CAS PubMed.
- C. A. Hutchison, S. Phillips, M. H. Edgell, S. Gillam, P. Jahnke and M. Smith, J. Biol. Chem., 1978, 253(18), 6551–6560 CrossRef CAS PubMed.
- G. Winter, A. R. Fersht, A. J. Wilkinson, M. Zoller and M. Smith, Nature, 1982, 299, 756–758 CrossRef CAS PubMed.
- D. R. Mills, R. L. Peterson and S. Spiegelman, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 217–224 CrossRef CAS PubMed.
- K. Chen and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5618–5622 CrossRef CAS PubMed.
- W. P. C. Stemmer, Nature, 1994, 370, 389–391 CrossRef CAS PubMed.
- A. Greener, M. Callahan and B. Jerpseth, Mol. Biotechnol., 1997, 7, 189–195 CrossRef CAS PubMed.
- M. T. Reetz, M. Bocola, J. D. Carballeira, D. Zha and A. Vogel, Angew. Chem., Int. Ed., 2005, 44, 4192–4196 CrossRef CAS PubMed.
- R. J. Fox, S. C. Davis, E. C. Mundorff, L. M. Newman, V. Gavrilovic, S. K. Ma, L. M. Chung, C. Ching, S. Tam, S. Muley, J. Grate, J. Gruber, J. C. Whitman, R. A. Sheldon and G. W. Huisman, Nat. Biotechnol., 2007, 25(3), 338–344 CrossRef CAS PubMed.
- M. G. Acker and D. S. Auld, Perspect. Sci., 2014, 1, 56–73 CrossRef.
- L. Ye, C. Yang and H. Yu, Appl. Microbiol. Biotechnol., 2018, 102, 559–567 CrossRef CAS PubMed.
- J. K. B. Cahn, C. A. Werlang, A. Baumschlager, S. Brinkmann-Chen, S. L. Mayo and F. H. Arnold, ACS Synth. Biol., 2017, 6, 326–333 CrossRef CAS PubMed.
- S. Brinkmann-Chen, T. Flock, J. K. B. Cahn, C. D. Snow, E. M. Brustad, J. A. McIntosh, P. Meinhold, L. Zhang and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(27), 10946–10951 CrossRef CAS PubMed.
- H. Bisswanger, Perspect. Sci., 2014, 1, 41–55 CrossRef.
- S. Schätzle, M. Höhne, E. Redestad, K. Robins and U. T. Bornscheuer, Anal. Chem., 2009, 81, 8244–8248 CrossRef PubMed.
- M. T. Reetz, A. Zonta, K. Schimossek, K. Liebeton and K. Jaeger, Angew. Chem., Int. Ed. Engl., 1997, 36(24), 2830–2832 CrossRef CAS.
- M. Alexeeva, A. Enright, M. J. Dawson, M. Mahmoudian and N. J. Turner, Angew. Chem., Int. Ed., 2002, 41, 3177–3180 CrossRef CAS PubMed.
- R. Carr, M. Alexeeva, M. J. Dawson, V. Gotor-Fernández, C. E. Humphrey and N. J. Turner, ChemBioChem, 2005, 6, 637–639 CrossRef CAS PubMed.
- D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, J. Am. Chem. Soc., 2013, 135, 10863–10869 CrossRef CAS PubMed.
- J. R. Marshall, P. Yao, S. L. Montgomery, J. D. Finnigan, T. W. Thorpe, R. B. Palmer, J. Mangas-Sanchez, R. A. M. Duncan, R. S. Heath, K. M. Graham, D. J. Cook, S. J. Charnock and N. J. Turner, Nat. Chem., 2021, 13, 140–148 CrossRef CAS PubMed.
- A. P. Green, N. J. Turner and E. O’Reilly, Angew. Chem., Int. Ed., 2014, 53, 10714–10717 CrossRef CAS PubMed.
- R. Cairns, A. Gomm, C. Peel, M. Sharkey and E. O’Reilly, ChemCatChem, 2019, 11, 4738–4743 CrossRef CAS.
- G. Yang and S. G. Withers, ChemBioChem, 2009, 10, 2704–2715 CrossRef CAS PubMed.
- C. K. Longwell, L. Labanieh and J. R. Cochran, Curr. Opin. Biotechnol., 2017, 48, 196–202 CrossRef CAS PubMed.
- H. A. Bunzel, X. Garrabou, M. Pott and D. Hilvert, Curr. Opin. Struct. Biol., 2018, 48, 149–156 CrossRef CAS PubMed.
- A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. S. Huck, Angew. Chem., Int. Ed., 2010, 48, 5846–5868 CrossRef PubMed.
- U. Markel, K. D. Essani, V. Besirlioglu, J. Schiffels, W. R. Streit and U. Schwaneberg, Chem. Soc. Rev., 2020, 49, 233–262 RSC.
- A. Debon, M. Pott, R. Obexer, A. P. Green, L. Friedrich, A. D. Griffiths and D. Hilvert, Nat. Catal., 2019, 2, 740–747 CrossRef CAS.
- X. Fu, Y. Zhang, Q. Xu, X. Sun and F. Meng, Front. Chem., 2021, 9, 666867 CrossRef CAS PubMed.
- A. V. Karnik and M. Hasan, Stereochemistry, Elsevier, 2021, pp. 407–463 Search PubMed.
- M. Alexeeva, R. Carr and N. J. Turner, Org. Biomol. Chem., 2003, 1, 4133–4137 RSC.
- M. Hall, RSC Chem. Biol., 2021, 2, 958–989 RSC.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, J. Sci., 2010, 329, 305–309 CAS.
- S. T. Jung, R. Lauchli and F. H. Arnold, Curr. Opin. Biotechnol., 2011, 22, 809–817 CrossRef CAS PubMed.
- Y. Wei, E. L. Ang and H. Zhao, Curr. Opin. Chem. Biol., 2018, 43, 1–7 CrossRef CAS PubMed.
- E. O’Reilly, V. Köhler and S. L. Flitsch, Chem. Commun., 2011, 47, 2490–2501 RSC.
- R. Bernhardt, J. Biotechnol., 2006, 124, 128–145 CrossRef CAS PubMed.
- J. Ryan, M. Šiaučiulis, A. Gomm, B. Maciá, E. O’Reilly and V. Caprio, J. Am. Chem. Soc., 2016, 138, 15798–15800 CrossRef CAS PubMed.
- F. Taday, J. Ryan, S. P. Argent, V. Caprio, B. Maciá and E. O’Reilly, Chem. – Eur. J., 2020, 26, 3729–3732 CrossRef CAS PubMed.
- B. Yuan, D. Yang, G. Qu, N. J. Turner and Z. Sun, Chem. Soc. Rev., 2024, 53, 227–262 RSC.
- H. Joo, Z. Lin and F. H. Arnold, Nature, 1999, 399, 670–673 CrossRef CAS PubMed.
- H. Lorenz and A. Seidel-Morgenstern, Angew. Chem., Int. Ed., 2014, 53, 1218–1250 CrossRef CAS PubMed.
- G. Betzenbichler, L. Huber, S. Kräh, M. L. K. Morkos, A. F. Siegle and O. Trapp, Chirality, 2022, 34, 732–759 CrossRef CAS PubMed.
- M. Lämmerhofer, J. Chromatogr. A, 2010, 1217, 813–856 CrossRef PubMed.
- E. Busto, V. Gotor-Fernandez and V. Gotor, Chem. Soc. Rev., 2010, 39, 4504–4523 RSC.
- A. S. de Miranda, L. S. M. Miranda and R. O. M. A. de Souza, Biotechnol. Adv., 2015, 33, 372–393 CrossRef CAS PubMed.
- N. J. Turner, Curr. Opin. Chem. Biol., 2004, 8, 114–119 CrossRef CAS PubMed.
- M. Rachwalski, N. Vermue and F. P. J. T. Rutjes, Chem. Soc. Rev., 2013, 42, 9268–9282 RSC.
- G. Kirchner, M. P. Scollar and A. M. Kilbanov, J. Am. Chem. Soc., 1985, 107, 7072–7076 CrossRef CAS.
- G. Carrea and S. Riva, Angew. Chem., Int. Ed., 2000, 39, 2226–2254 CrossRef CAS PubMed.
- A. Zaks and D. R. Dodds, Drug Discovery Today, 1997, 2(12), 513–531 CrossRef CAS.
- O. Verho and J.-E. Bäckwall, J. Am. Chem. Soc., 2015, 137(12), 3996–4009 CrossRef CAS PubMed.
- J. V. Allen and J. M. J. Williams, Tetrahedron Lett., 1996, 37(11), 1859–1862 CrossRef CAS.
- A. L. E. Larsson, B. A. Persson and J.-E. Bäckvall, Angew. Chem., Int. Ed. Engl., 1997, 36(11), 1211–1212 CrossRef CAS.
- B. A. Persson, A. L. E. Larsson, L. Ray and J.-E. Bäckvall, J. Am. Chem. Soc., 1999, 121(8), 1645–1650 CrossRef CAS.
- R. J. Kazlauskas, A. N. E. Weissfloch, A. T. Rappaport and L. A. Cuccia, J. Org. Chem., 1991, 56, 2656–2665 CrossRef CAS.
- O. Verho and J. E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 3996–4009 CrossRef CAS PubMed.
- Y. Ahn, S. B. Ko, M. J. Kim and J. Park, Coord. Chem. Rev., 2008, 252, 647–658 CrossRef CAS.
- O. Langvik, T. Saloranta, D. Yu. Murzin and R. Leino, ChemCatChem, 2015, 7, 4004–4015 CrossRef CAS.
- R. Kourist, P. D. de Maria and U. T. Bornschuer, ChemBioChem, 2008, 9, 491–498 CrossRef CAS PubMed.
- M. T. Reetz and K. Schimossek, Chimia, 1996, 50, 668–669 CrossRef CAS.
- M. J. Kim, W. H. Kim, K. Han, K. C. Yoon and J. Park, Org. Lett., 2007, 9, 1157–1159 CrossRef CAS PubMed.
- A. Parvulescu, D. De Vos and P. Jacobs, Chem. Commun., 2005, 5307–5309 RSC.
- X. Li, Y. Cao, K. Luo, Y. Sun, J. Xiong, L. Wang, Z. Liu, J. Li, J. Ma, J. Ge, H. Xiao and R. N. Zare, Nat. Catal., 2019, 2, 718–725 CrossRef CAS.
- C. K. Chung, P. G. Bulger, B. Kosjek, K. M. Belyk, N. Rivera, M. E. Scott, G. R. Humphrey, J. Limanto, D. C. Bachert and K. M. Emerson, Org. Process Res. Dev., 2014, 18, 215–227 CrossRef CAS.
- A. Cuetos, I. Lavandera and V. Gotor, Chem. Commun., 2013, 49, 10688–10690 RSC.
- C. Molinaro, E. M. Phillips, B. Xiang, E. Milczek, M. Shevlin, J. Balsells, S. Ceglia, J. Chen, L. Chen, Q. Chen, Z. Fei, S. Hoerrner, J. Qi, M. L. Ruiz, L. Tan, B. Wan and J. Yin, J. Org. Chem., 2019, 84, 8006–8018 CrossRef CAS PubMed.
- Z. Peng, J. W. Wong, E. C. Hansen, A. L. A. Puchlopek-Dermenci and H. J. Clarke, Org. Lett., 2014, 16, 860–863 CrossRef CAS PubMed.
- K. Engström, E. v Johnston, O. Verho, K. P. J. Gustafson, M. Shakeri, C. W. Tai and J. E. Bäckvall, Angew. Chem., Int. Ed., 2013, 52, 14006–14010 CrossRef PubMed.
- M. M. Musa, Chirality, 2020, 32, 147–157 CrossRef CAS PubMed.
- J. S. Dehovitz, Y. Y. Loh, J. A. Kautzky, K. Nagao, A. J. Meichan, M. Yamauchi, D. W. C. Macmillan and T. K. Hyster, Science, 2020, 369, 1113–1118 CrossRef CAS PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- S. H. Albayati, M. Masomian, S. N. H. Ishak, M. S. B. M. Ali, A. L. Thean, F. B. M. Shariff, N. D. B. M. Noor and R. N. Z. R. A. Rahman, Catalysts, 2020, 10(7), 747 CrossRef CAS.
- M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed.
- M. R. Maldonado, R. C. Alnoch, J. M. de Almeida, L. A. dos Santos, A. T. Andretta, R. del, P. C. Ropaín, E. M. de Souza, D. A. Mitchell and N. Krieger, Biochem. Eng. J., 2021, 172, 10847 CrossRef.
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290 RSC.
- K. Engström, E. V. Johnston, O. Verho, K. P. J. Gustafson, M. Shakeri, C. W. Tai and J. E. Bäckvall, Angew. Chem., Int. Ed., 2013, 52, 14006–14010 CrossRef PubMed.
- D. Koszelewski, D. Pressnitz, D. Clay and W. Kroutil, Org. Lett., 2009, 11, 4810–4812 CrossRef CAS PubMed.
- H. Gaweska and P. F. Fitzpatrick, Biomol. Concepts, 2011, 2, 365–377 CAS.
- E. W. Hafner and D. Wellner, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 987–991 CrossRef CAS PubMed.
- T. M. Beard and N. J. Turner, Chem. Commun., 2002, 246–247 RSC.
- R. Carr, M. Alexeeva, A. Enright, T. S. C. Eve, M. J. Dawson and N. J. Turner, Angew. Chem., 2003, 115, 4955–4958 CrossRef.
- D. Ghislieri, D. Houghton, A. P. Green, S. C. Willies and N. J. Turner, ACS Catal., 2013, 3, 2869–2872 CrossRef CAS.
- R. S. Heath, M. Pontini, S. Hussain and N. J. Turner, ChemCatChem, 2016, 8, 117–120 CrossRef CAS.
- H. Yamada and M. Kobatashi, Biosci., Biotechnol., Biochem., 1996, 60, 1391–1400 CrossRef CAS PubMed.
- N. Mohanan, Z. Montazer, P. K. Sharma and D. B. Levin, Front. Microbiol., 2020, 11, 580709 CrossRef PubMed.
- B. Zhu, Q. Ye, Y. Seo and N. Wei, Environ. Sci. Technol. Lett., 2022, 9, 650–657 CrossRef CAS.
- A. Robles-Medina, P. A. González-Moreno, L. Esteban-Cerdán and E. Molina-Grima, Biotechnol. Adv., 2009, 27, 398–408 CrossRef CAS PubMed.
- B. Schulze and M. G. Wubbolts, Curr. Opin. Biotechnol., 1999, 10, 609–615 CrossRef CAS PubMed.
- G. Hildebrandt and W. Klavehn, US Pat., US1956950, 1934 Search PubMed.
- H. Kaur and P. K. Gill, Engineering Tools in the Beverage Industry, The Science of Beverages, Elsevier, 2019, vol. 3, pp. 255–282 Search PubMed.
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- M. Ress-Löschke, T. Friendrich, B. Hauer and R. Mattes, De Pat., DE19848129A1, 1998.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- Y. L. Khmelnitsky, C. Budde, J. M. Arnold, A. Usyatinsky, D. S. Clark and J. S. Dordick, J. Am. Chem. Soc., 1997, 119, 11554–11555 CrossRef CAS.
- M. A. Wegman, M. H. A. Janssen, F. van Rantwijk and R. A. Sheldon, Adv. Synth. Catal., 2001, 343, 559–576 CrossRef CAS.
- S. J. Novick, N. Dellas, R. Garcia, C. Ching, A. Bautista, D. Homan, O. Alvizo, D. Entwistle, F. Kleinbeck, T. Schlama and T. Ruch, ACS Catal., 2021, 11, 3762–3770 CrossRef CAS.
- M. Burns, C. A. Martinez, B. Vanderplas, R. Wisdom, S. Yu and R. A. Singer, Org. Process Res. Dev., 2017, 21, 871–877 CrossRef CAS.
- J. Kuska and E. O’Reilly, Curr. Opin. Chem. Biol., 2020, 58, 146–154 CrossRef CAS PubMed.
- E. J. Corey and X. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons, 1991 Search PubMed.
- E. J. Corey and W. T. Wipke, Science, 1969, 166, 178–192 CrossRef CAS PubMed.
- N. J. Turner and E. O’Reilly, Nat. Chem. Biol., 2013, 9, 285–288 CrossRef CAS PubMed.
- W. Finnigan, L. J. Hepworth, S. L. Flitsch and N. J. Turner, Nat. Catal., 2021, 4, 98–104 CrossRef CAS PubMed.
- W. Finnigan, M. Lubberink, L. J. Hepworth, J. Citoler, A. P. Mattey, G. J. Ford, J. Sangster, S. C. Cosgrove, B. Zucoloto da Costa., R. S. Heath, T. W. Thorpe, Y. Yu, S. L. Flitsch and N. J. Turner, ACS Catal., 2023, 13, 11771–11780 CrossRef CAS PubMed.
- D. Kreutter, P. Schwaller and J. Reymond, Chem. Sci., 2021, 12, 8648–8659 RSC.
- D. Kreutter and J. Reymond, Chem. Sci., 2023, 14, 9959–9969 RSC.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- S. A. Ferrinho, S. A. Molyneux, R. J. M. Goss, E. L. Bell, A. Crossley, A. P. Green, K. Yeow and E. O’Reilly, in Modern Developments in Catalysis, G. Hutchings, M. Davidson, R. Catlow, C. Hardacre, N. Turner, C. Williams, A. Mulholland, J. Goodall and C. Mitchell, World Scientific, Europe, 2023, ch. 14, pp. 535–594 Search PubMed.
- C. K. Winkler, J. H. Schrittwieser and W. Kroutil, ACS Cent. Sci., 2021, 7, 55–71 CrossRef CAS PubMed.
- V. Uppada, S. Bhaduri and S. B. Noronha, Curr. Sci., 2014, 106, 946–957 CAS.
- C.-H. Wong and G. M. Whiteside, J. Am. Chem. Soc., 1981, 103, 4890–4899 CrossRef CAS.
- J. S. Shin and B. G. Kim, Biotechnol. Bioeng., 1999, 65, 206–211 CrossRef CAS PubMed.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, J. Sci., 2015, 349, 1525–1529 CAS.
- W. Wu, M. B. Bhatia, C.M. Lewis, W. Lang, A. Wang and G. W. Matcham, World Intellectual Property Organization, WO99/46398, 1999 Search PubMed.
- A. Gomm, W. Lewis, A. P. Green and E. O’Reilly, Eur. J. Chem., 2016, 22, 12692–12695 CrossRef CAS PubMed.
- J. L. Galman, I. Slabu, N. J. Weise, C. Iglesias, F. Parmeggiani, R. C. Lloyd and N. J. Turner, Curr. Green Chem., 2017, 19, 361–366 RSC.
- A. Gomm and E. O’Reilly, Curr. Opin. Chem. Biol., 2018, 43, 106–112 CrossRef CAS PubMed.
- A. Gomm, S. Grigoriou, C. Peel, J. Ryan, N. Mujtaba, T. Clarke, E. Kulcinskaja and E. O’Reilly, Eur. J. Org. Chem., 2018, 5282–5284 CrossRef CAS.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, J. Sci., 2019, 366, 1255–1259 CAS.
- J. V. Allen and J. M. J. Williams, Tetrahedron Lett., 1996, 37, 1859–1862 CrossRef CAS.
- H. Gröger and W. Hummel, Curr. Opin. Chem. Biol., 2014, 19, 171–179 CrossRef PubMed.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef.
- T. Kajimoto, K. K.-C. Liu, R. L. Pederson, Z. Zhong, Y. Ichikawa, J. A. Porco and C. H. Wong, J. Am. Chem. Soc., 1991, 113, 6187–6196 CrossRef CAS.
- F. Taday, R. Cairns, A. O’Connell and E. O’Reilly, Chem. Commun., 2022, 58, 1697–1700 RSC.
- S. C. Cosgrove, S. Hussain, N. J. Turner and S. P. Marsden, ACS Catal., 2018, 8, 5570–5573 CrossRef CAS.
- E. L. Bell, A. J. Burke, A. E. Hutton, A. O’Connell, A. Barry, E. O’Reilly and A. P. Green, Chem. Soc. Rev., 2024 10.1039/D3CS00972F.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |