
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From sugars to aliphatic amines: as sweet as it sounds? Production and applications of bio-based aliphatic amines†
Benjamin
Vermeeren
 a,
Sofie
Van Praet
a,
Wouter
Arts
a,
Thomas
Narmon
a,
Yingtuan
Zhang
a,
Cheng
Zhou
a,
Hans P.
Steenackers
b and
Bert F.
Sels
*a
a,
Sofie
Van Praet
a,
Wouter
Arts
a,
Thomas
Narmon
a,
Yingtuan
Zhang
a,
Cheng
Zhou
a,
Hans P.
Steenackers
b and
Bert F.
Sels
*a
aCenter for Sustainable Catalysis and Engineering (CSCE), KU Leuven, Belgium. E-mail: bert.sels@kuleuven.be
bCentre of Microbial and Plant Genetics (CMPG), KU Leuven, Belgium
First published on 4th October 2024
Abstract
Aliphatic amines encompass a diverse group of amines that include alkylamines, alkyl polyamines, alkanolamines and aliphatic heterocyclic amines. Their structural diversity and distinctive characteristics position them as indispensable components across multiple industrial domains, ranging from chemistry and technology to agriculture and medicine. Currently, the industrial production of aliphatic amines is facing pressing sustainability, health and safety issues which all arise due to the strong dependency on fossil feedstock. Interestingly, these issues can be fundamentally resolved by shifting toward biomass as the feedstock. In this regard, cellulose and hemicellulose, the carbohydrate fraction of lignocellulose, emerge as promising feedstock for the production of aliphatic amines as they are available in abundance, safe to use and their aliphatic backbone is susceptible to chemical transformations. Consequently, the academic interest in bio-based aliphatic amines via the catalytic reductive amination of (hemi)cellulose-derived substrates has systematically increased over the past years. From an industrial perspective, however, the production of bio-based aliphatic amines will only be the middle part of a larger, ideally circular, value chain. This value chain additionally includes, as the first part, the refinery of the biomass feedstock to suitable substrates and, as the final part, the implementation of these aliphatic amines in various applications. Each part of the bio-based aliphatic amine value chain will be covered in this Review. Applying a holistic perspective enables one to acknowledge the requirements and limitations of each part and to efficiently spot and potentially bridge knowledge gaps between the different parts.
 Benjamin Vermeeren | Benjamin Vermeeren earned his PhD in 2024 from KU Leuven, where he conducted his research under the supervision of Professor Bert F. Sels within the CSCE research group. His doctoral work focused on the selective production of bio-based aliphatic amines, specifically through the catalytic reductive amination of α-hydroxy carbonyl oxygenates. Before pursuing his PhD, Benjamin completed his MSc in Bioscience Engineering, specializing in Catalytic Technology, at KU Leuven in 2019. |
 Sofie Van Praet | Sofie Van Praet completed her PhD in 2023 at KU Leuven, working under the supervision of Professor Bert F. Sels in the CSCE research group. Her research centered on developing sustainable catalytic pathways for the production of amines from carbohydrates, conducted in close collaboration with Eastman Chemical Company. Prior to her doctoral studies, Sofie earned her MSc in Bioscience Engineering, with a specialization in Catalytic Technology, from KU Leuven in 2019. |
 Wouter Arts | Wouter Arts is a postdoctoral researcher in the CSCE research group at KU Leuven, where he also earned his PhD in 2024 under the supervision of Professor Bert F. Sels. His research focuses on biorefinery processes, combining process modeling with laboratory experiments. Before pursuing his PhD, Wouter completed an MSc in Bioscience Engineering, specializing in Catalytic Technologies, at KU Leuven in 2018, followed by a postgraduate degree in Chemical Engineering from Imperial College London in 2019. |
 Thomas Narmon, Yingtuan Zhang, and Cheng Zhou | Thomas Narmon, Yingtuan Zhang, and Cheng Zhou are currently conducting their PhD research under the supervision of Professor Bert F. Sels in the CSCE research group at KU Leuven. Thomas Narmon's research focuses on the synthesis of lignin-derived amines for applications in functionalized polymers. He obtained his MSc in Chemistry from KU Leuven in 2019 and subsequently worked for two years as a Research and Teaching Assistant in Pharmaceutical (Neuro)sciences at the University of Brussels. Yingtuan Zhang is focused on the valorization of lignin oligomers and the synthesis of novel bio-based polymers. He earned his MSc in Polymer Chemistry and Physics from the Guangzhou Institute of Chemistry in 2019. Cheng Zhou's research is centered on CO2 capture and utilization. He received his MSc in Chemistry from Xiamen University in 2019. |
 Hans P. Steenackers | Hans P. Steenackers received his PhD from KU Leuven in 2011, where his research focused on the structure–activity relationship and mode of action of small inhibitors of biofilm formation. He is currently an associate professor at KU Leuven and leads the MICA laboratory, which is part of the Center of Microbial and Plant Genetics. His research group investigates microbial community behavior and develops antimicrobial strategies targeting these communities, with the aim of advancing the understanding of social interactions within microbial communities, resistance development, and in situ monitoring methodologies. He has published over 50 peer-reviewed papers in this field. |
 Bert F. Sels | Bert F. Sels, currently a full professor at KU Leuven and head of the CSCE research group, earned his PhD in 2000 in the field of heterogeneous oxidation catalysis. His research focuses on heterogeneous catalysis, addressing future challenges in industrial organic and environmental catalysis. A key area of his work is advancing carbon circularity through the use of renewable carbon sources such as biomass, carbon dioxide, and plastic waste. His group has published over 400 peer-reviewed articles and holds more than 40 patents. Professor Sels is the co-chair of the Catalysis Commission of the International Zeolite Association (IZA), a co-founder of the European Research Institute of Catalysis (ERIC), and a member of the European Academy of Sciences and Arts. |
1. Introduction
Amines are defined as compounds in which at least one of the hydrogen atoms of ammonia (NH3) is replaced by an organic substituent, making them a versatile class of chemicals. In aliphatic amines, these substituents comprise (functionalized) alkyl groups. Alkylamines, alkyl polyamines, aliphatic heterocyclic amines, and alkanolamines are all different types of aliphatic amines. Alkylamines contain up to three alkyl substituents while in alkyl polyamines two or more amino groups are present, and in aliphatic heterocyclic amines at least one amino group is part of a saturated cyclic structure. As bifunctional aliphatic amines, alkanolamines contain at least both one amino and one hydroxyl group. This structural diversity makes aliphatic amines of great industrial importance as they are used in a wide range of applications in almost every field of chemistry, technology, agriculture, and medicine.1,2 As an introduction, all elementary steps in today's aliphatic amine industry will be concisely discussed (Fig. 1).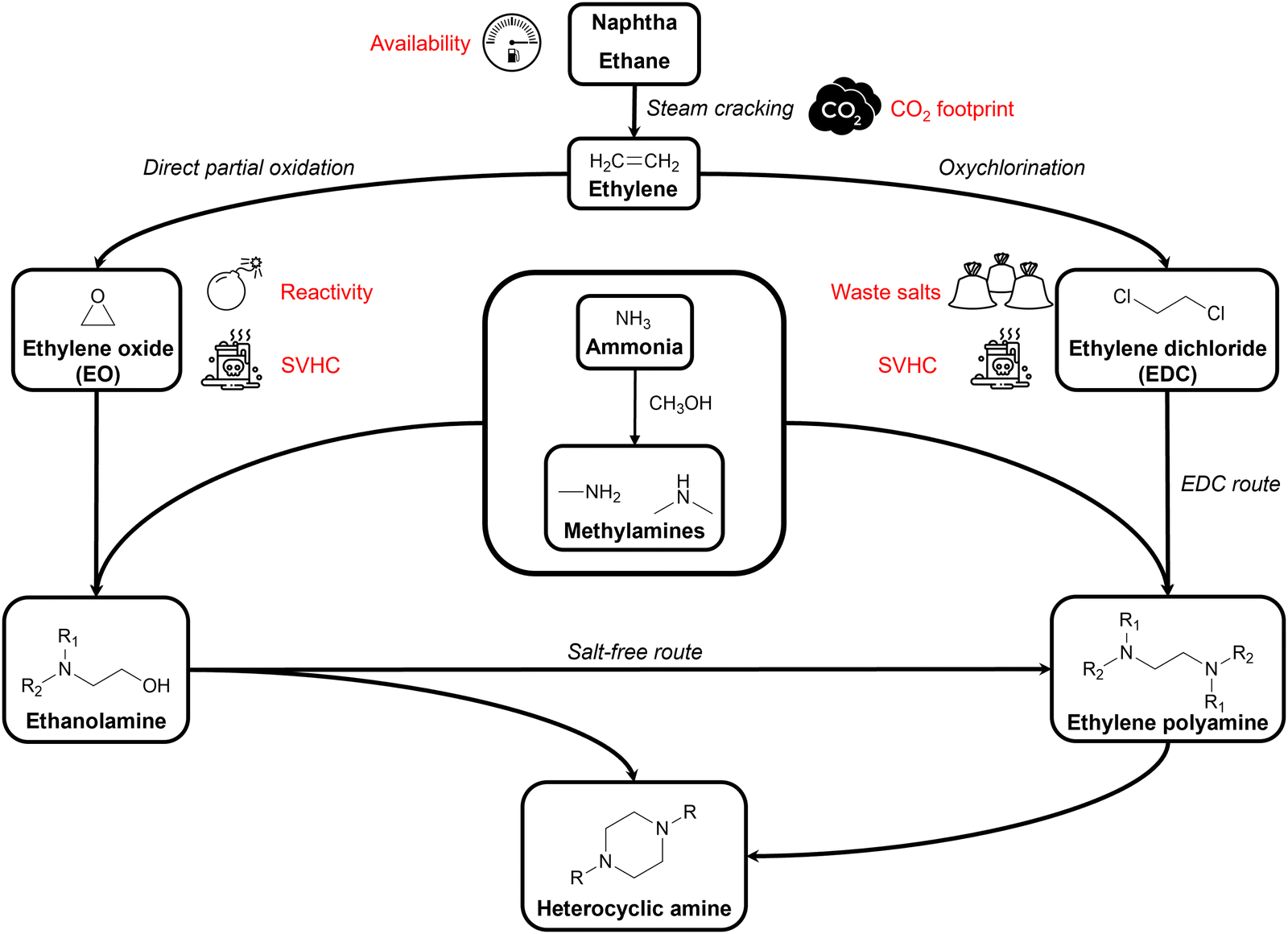 | ||
| Fig. 1 Schematic overview of the traditional aliphatic amine industry and its shortcomings. | ||
1.1. Overview of the traditional aliphatic amine industry
Nearly all nitrogen-containing chemicals, including amines, originate from ammonia (NH3), the second most produced chemical worldwide.3,4 Since natural ammonia sources are scarce, NH3 is predominantly produced via the Haber–Bosch process where hydrogen (H2) and nitrogen (N2) are converted into NH3 using an iron catalyst at an elevated temperature (400–500 °C) and pressure (>10 MPa). N2 and H2 are typically sourced from air and syngas, respectively.5,6 Methylamines, the simplest and industrially most relevant alkylamines, are produced through an exothermic reaction of NH3 with methanol (MeOH) over amorphous silica-alumina catalysts (i.e., solid acid catalysts or shape-selective zeolites) at 350–450 °C. This process yields a mixture of monomethylamine (MMA), dimethylamine (DMA) and trimethylamine (TMA), with product selectivity controlled by reaction conditions and catalyst choice.7–10 NH3, MMA and DMA are the indispensable nitrogen sources for all larger aliphatic amines.The primary carbon sources in today's aliphatic amine industry are derived from fossil feedstock. For instance, industrially important aliphatic C2 amines (i.e., ethanolamines, ethylene polyamines and piperazine derivatives) originate from ethylene, a key petrochemical produced by steam cracking petroleum hydrocarbons.11–13 From ethylene, two substrates of our interest can be derived, namely ethylene oxide (EO) and ethylene dichloride (EDC), which after amination lead to ethanolamines and ethylene polyamines, respectively. The former, EO, is produced via partial oxidation of ethylene with oxygen in the presence of a silver catalyst at 200–300 °C. Ethylene conversion rates are intentionally kept low (<10%) to minimize complete combustion of CO2.14,15 The latter, EDC, is produced by gas- or liquid-phase oxychlorination of ethylene. In the gas-phase process, ethylene and HCl are reacted with oxygen in the presence of a supported CuCl2 catalyst at 220–250 °C, while in the liquid-phase an aqueous CuCl2 solution is used at 170–185 °C.16
By reacting these nitrogen and carbon sources, aliphatic amine products are formed. Ethanolamines are formed by reacting EO with NH3, MMA or DMA at an elevated temperature (50–200 °C) and pressure (up to 16 MPa) producing monoethanolamine (MMA), diethanolamine (DEA), and triethanolamine (TriEA). Product distribution primarily depends on the amine-to-EO molar ratio.17,18 Propanolamines are similarly formed by reacting NH3 or an alkylamine with propylene oxide (PO).19 Ethylene polyamines are currently manufactured via two production methods: the EDC process and the salt-free process.1,2 The EDC process involves reacting EDC with an amine reactant at ∼100 °C, producing ethylene polyamines and HCl. This process is unselective, yielding various polyamines such as ethylenediamine (EDA) and higher analogs including diethylenetriamine (DETriA), and triethylenetetramine (TriETA). The salt-free process avoids the use of EDC by reacting an ethanolamine with an amine reactant under catalytic hydrogenation conditions (150–250 °C, up to 20 MPa H2 pressure) using modified Ni, Co, or Ru catalysts. Propylene amines, such as 1,2-propylenediamine (1,2PDA), are exclusively formed via this salt-free route.2,20 Additionally, piperazine (PZ) and other heterocyclic derivatives are produced as by-products in both the EDC and salt-free processes by optimizing the reaction conditions to favor their formation.2,21
1.2. The aliphatic amine industry in numbers
From an economic perspective, the aliphatic amine industry represents a multibillion-dollar market that steadily increases with a compound annual growth rate (CAGR) of 5–6% per year.22 In today's industry, the demand for alkanolamines is significantly higher than that for alkyl polyamines. In 2022, the global demand for alkanolamines and alkyl polyamines approximately amounted to 2 and 0.5 million tons, respectively.23,24 Notable alkanolamines include ethanolamines such as MEA, DEA and TriEA, as well as N-substituted ethanolamines such as N,N-dimethylethanolamine (DMEA), and propanolamines such as 1-amino-2-propanol (1A2P). These alkanolamines play an essential role as precursors or final products in surfactants, cosmetics, pharmaceuticals, polymers and coatings, agrochemicals, water treatment applications, gas purification, etc.17,19 The most prominent alkyl polyamines encompass the ethylene polyamines EDA, DETriA, and TriETA, and the propylene polyamines 1,2PDA and 1,3-propylenediamine (1,3PDA). These alkyl polyamines are crucial components in polymer synthesis, surfactants, agrochemicals, gas purification applications, corrosion inhibitors, lubricant oils, etc.2,201.3. Shortcomings of the current aliphatic amine industry
Not only the amine industry, but the entire chemical industry currently is a petrochemical industry, as it heavily relies on fossil fuels such as crude oil and natural gas. According to the organization of petroleum exporting countries (OPEC), the chemical industry accounted for 14% of the total oil demand in 2022.25 Meanwhile, the international energy agency (IEA) claimed in their most recent report that: “petrochemicals are rapidly becoming the largest driver of global oil consumption. They are set to account for more than a third of the growth in oil demand to 2030, and nearly half to 2050.”26 This is in contrast with other sectors, including transportation and residential use, as their projected growth in oil demand will stagnate or even decline in the future. This discrepancy is linked to the chemical industry's unique dependency on fossil fuels. More than half of the fossil fuel demand in the chemical industry is used as feedstock for value-added components rather than merely as an energy resource, setting it apart from all other sectors. This strong dependency on a finite, geographically unequally distributed, non-renewable feedstock poses a significant challenge for the chemical industry.27 Another drawback associated with the consumption of fossil fuels is their greenhouse gas (GHG) emission, which can be illustrated by the ethylene production process. As an energy- and carbon-intensive process, steam cracking generates a carbon footprint that varies from 1.0 to 1.7 tons of CO2 released per ton of ethylene produced, depending on the hydrocarbon feedstock. In total, this emission accounts for approximately 20% of the annual petrochemical CO2 emission and 5% of the annual total industrial CO2 emission.26,28,29Furthermore, apart from these drawbacks related to fossil fuels, today's aliphatic amine industry faces significant safety and health concerns. Its highly strained epoxide ring makes EO thermally unstable, highly flammable and very reactive to other substances, causing a permanent explosion risk and complicating its use and transport. Moreover, EO is classified as carcinogenic, mutagenic and reprotoxic according to the REACH regulations.30,31 Similarly, REACH classifies EDC as a hazardous compound due to its high flammability, toxicity and carcinogenicity.32 Additionally, the EDC process leads to stoichiometric amounts of HCl which, in order to prevent corrosion, are neutralized using caustic soda (NaOH) or lime, resulting in significant amounts of waste salts. According to REACH regulations, both substances are listed as substances of very high concern (SVHCs) which should be progressively replaced by less dangerous substances or technologies.31,32
1.4. Scope of the Review
All mentioned shortcomings (in)directly arise from the use of fossil-derived resources, highlighting the need for safer and more sustainable resources and production routes. A promising solution can be found in the use of biomass as a renewable feedstock for the chemocatalytic production of aliphatic amines. The increasing academic and industrial interest in bio-based aliphatic amines can be exemplified by the number of high-quality reviews dedicated to this topic that have been published in recent years.33–37 To date, however, all these reviews primarily focus on the state-of-the-art amination methods, thereby reporting only one, yet essential, aspect of the future bio-based aliphatic amine industry. This Review aims to cover the bio-based aliphatic amine industry from a holistic perspective by relating their production methods to biomass valorization and addressing potential sustainable applications of these amines. In this way, this Review consists of three sections: (i) from biomass as a feedstock to renewable substrates, (ii) from renewable substrates to bio-based aliphatic amines, and (iii) from bio-based aliphatic amines to sustainable applications. The first section, from biomass feedstock to renewable substrates, summarizes the key chemocatalytic valorization reactions of cellulose and hemicellulose. In these reactions, the feedstock is converted into a range of oxygenate substrates characterized by the presence of hydroxyl and carbonyl functional groups. The second section, from renewable substrates to bio-based aliphatic amines, reports the most prominent catalytic amination methods that convert these oxygenate substrates into aliphatic amines. Here, particular emphasis is placed on the range of oxygenate substrates used and the product selectivity. Notably, the use of biomass enables the formation of both drop-in and novel aliphatic amine products. The final section, from bio-based aliphatic amines to sustainable applications, highlights three sustainable domains and related applications, namely CO2-reactive applications, polymerization and quaternary ammonium compounds (QACs), where different types of aliphatic amines, drop-in or novel, are indispensable components or improve the status quo of the current state-of-the-art technologies.The other, complementary approach toward sustainable bio-based aliphatic amines, i.e., handling the sustainability concerns related to the synthesis of ammonia and other nitrogen sources, is out of the scope of this Review. Nevertheless, the amount of research devoted to green ammonia production proves its utmost importance.38–41
2. From biomass as a feedstock to renewable substrates
2.1. Biomass: setting the scene
Biomass is an umbrella term that encompasses all organic (macro)molecules produced by living organisms, covering a wide range of different chemical structures. Contrary to fossil resources, it is defined as a renewable carbon source since it is replenished within a reasonable period. In general, biomass is considered a promising alternative resource for the production of fuels and chemicals if it is abundantly available, has little initial value, and is susceptible to chemical transformations.42–45 A type of biomass that fulfills all these requirements is lignocellulose. As the major structural building block of plant cell walls, lignocellulose is universally recognized as the largest source of organic carbon on Earth and the largest fraction of biomass. Furthermore, it is readily available from various currently underused, low-value, non-edible waste streams such as wood residues from the construction or paper industry and agricultural waste (e.g., corn stover, rice and wheat straw, sugarcane bagasse), bypassing land-use competition with food crops. Lignocellulose is a composite material primarily consisting of cellulose, hemicellulose and lignin. Cellulose is a linear homopolysaccharide containing up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 glucose units linked via β-1,4-glycosidic bonds, whereas hemicellulose is a branched heteropolysaccharide containing both pentose (e.g., xylose, arabinose) and hexose (e.g., galactose, glucose, mannose) monomers with a degree of polymerization from 50 up to 300 units. Lignin, on the other hand, is an irregularly crosslinked aromatic polymer formed by radical polymerization of three monolignol monomers, namely p-coumaryl alcohol, coniferyl alcohol and sinapyl alcohol.46–48 In the context of bio-based aliphatic amines, the carbohydrate fraction of lignocellulose (i.e., cellulose and hemicellulose) emerges as a promising feedstock. Carbohydrates are safe in use and consist of a functionalized and tunable aliphatic carbon backbone. In addition to lignocellulose, natural oils and lipids, encompassing fatty acid constituents, can serve as a potentially interesting feedstock in the production of long-chain aliphatic amines.34,35
000 glucose units linked via β-1,4-glycosidic bonds, whereas hemicellulose is a branched heteropolysaccharide containing both pentose (e.g., xylose, arabinose) and hexose (e.g., galactose, glucose, mannose) monomers with a degree of polymerization from 50 up to 300 units. Lignin, on the other hand, is an irregularly crosslinked aromatic polymer formed by radical polymerization of three monolignol monomers, namely p-coumaryl alcohol, coniferyl alcohol and sinapyl alcohol.46–48 In the context of bio-based aliphatic amines, the carbohydrate fraction of lignocellulose (i.e., cellulose and hemicellulose) emerges as a promising feedstock. Carbohydrates are safe in use and consist of a functionalized and tunable aliphatic carbon backbone. In addition to lignocellulose, natural oils and lipids, encompassing fatty acid constituents, can serve as a potentially interesting feedstock in the production of long-chain aliphatic amines.34,35
Although out of the Review's scope, it is worth mentioning that the polysaccharide chitin is another important biomass resource. Chitin is present in the cell wall of certain fungi and in the exoskeletons of arthropods (i.e., crustaceans and insects) and cephalopods (i.e., octopuses and squids). It is generated in significant amounts as a waste product in the food industry (e.g. seafood industry, insect farms). Remarkably, being a linear amino polymer composed of N-acetol-D-glucosamine monomers, chitin is one of the few forms of biomass, next to proteins, containing biogenic nitrogen. Chitin valorization, also referred to as “shell biorefinery”, presents a unique opportunity for the formation of fully bio-based amines. Current research efforts are mainly focused on chitin recovery and its conversion into value-added components.49–53
2.2. Lignocellulose valorization: general principles
In the biorefinery, lignocellulose valorization comprises three consecutive steps, namely fractionation, depolymerization and upgrading, comprehensively reviewed by Schutyser et al.54Fractionation, the first step in the lignocellulose biorefinery consists of lignocellulose pretreatment into its three main constituents. A variety of physical, chemical, thermochemical and biological pretreatment methods exist, targeting the different lignocellulose fractions.55–57 So does, for example, Kraft pulping, a well-established process in the paper and pulp industry, result in high-quality cellulose and hemicellulose fractions,58 whereas various delignification methods, such as lignin-first techniques (e.g., reductive catalytic fractionation (RCF)),59,60 focus on lignin recovery.61–63
In the next two steps, the isolated cellulose and/or hemicellulose fractions are first depolymerized into their respective monomers which subsequently can be further upgraded into value-added compounds. Depolymerization and upgrading are often, but not necessarily, combined in one process. Three distinctive strategies are generally applied: (i) thermochemical (e.g., gasification,64,65 pyrolysis,64,66 thermal cracking67), (ii) biotechnological (e.g., enzymatic hydrolysis, fermentation)68,69 and (iii) chemocatalytic transformation. The latter will be discussed in more detail (Fig. 2).
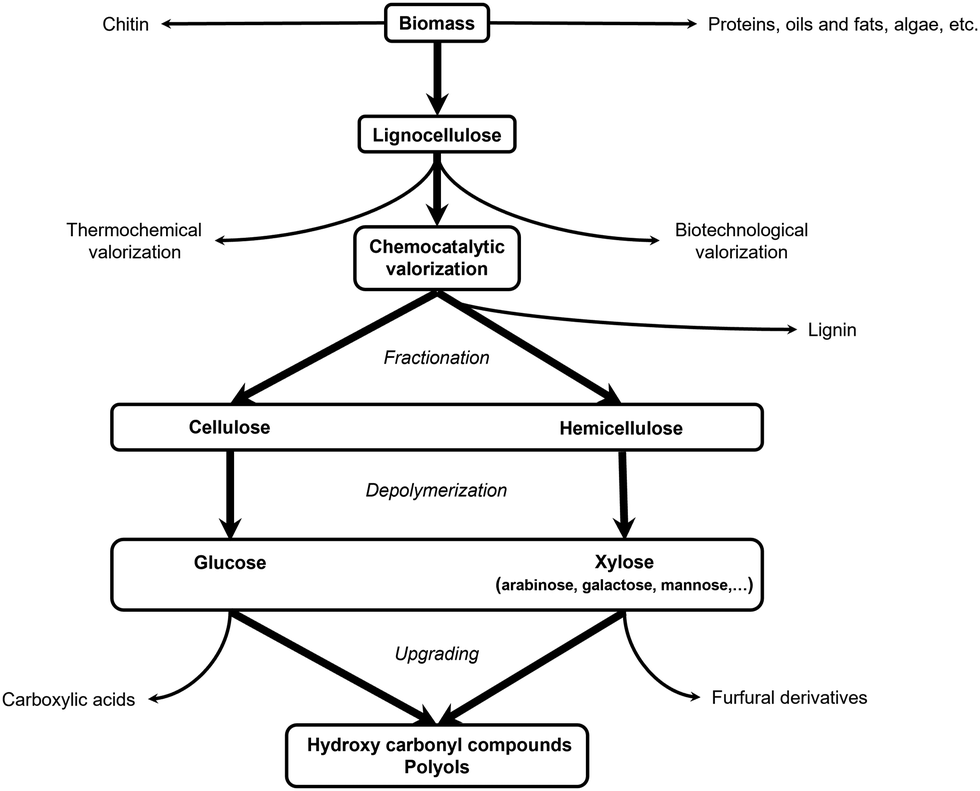 | ||
| Fig. 2 Schematic overview of the biomass valorization pathway covered in this Review. | ||
2.2.1.1. Isomerization. Carbohydrate isomerization, comprehensively reviewed by Delidovich et al.72,73 and Li et al.,74 is the interconversion reaction between an aldose and ketose isomer. In general, carbohydrate isomerization reactions are carried out in an aqueous solution and are restricted by both their thermodynamic equilibrium and thermal stability.75 Isomerization can be catalyzed by both homogeneous and heterogeneous basic or acidic catalysts.
Base-catalyzed isomerization, or Lobry de Bruyn-Alberda van Ekkenstein isomerization, follows an enediol mechanism by proton abstraction at the C2 position of the aldose. Initially, soluble alkali catalysts, such as NaOH and KOH, were employed but they suffer from a low isomerization rate together with the formation of various acidic by-products through alkaline degradation.76 However, the isomerization efficiency of these soluble base catalysts can be significantly improved by adding a complexing agent such as borate (Na2B4O7). This complexing agent preferentially forms a more stable complex with ketoses than with aldoses, resulting in an equilibrium shift in favor of the ketose isomer.72 Interestingly, organic amines (e.g., triethylamine, ethylenediamine, piperidine, morpholine, etc.) can be used as effective isomerization catalysts as they display desirable isomerization yields combined with reduced saccharide degradation.77 Furthermore, various suitable heterogeneous basic isomerization catalysts have been reported such as Mg–Al hydrotalcites, metal oxides (e.g., ZrO2, MgO) and cation-exchanged zeolites (e.g., type A zeolite).72,74,78
The acid-catalyzed isomerization, on the other hand, follows a 1,2-hydride transfer mechanism in which an intramolecular hydride shift takes place between the C2 and C1 positions of the aldose. Lewis acids, especially in heterogeneous forms such as Sn-BEA zeolites, are preferred over Brønsted acids as they minimize the formation of dehydrated by-products. Sn-BEA zeolites, in contrast to homogeneous Lewis acids, maintain their catalytic activity as their catalytic sites are protected from hydration and deactivation by the hydrophobic zeolite matrix.75,79–81
Both bases and Lewis acids also catalyze the epimerization reaction.72,74 While most catalytic systems favor the isomerization reaction, some are more selective toward epimerization. Most notable are the molybdenum catalysts developed by Bílik.82 This reaction, also referred to as the Bílik reaction, follows a 1,2-intramolecular carbon shift. Bílik initially studied molybdic acid but the catalyst scope has expanded over time to other highly active Mo(VI)-catalysts such as Mo-based heteropolyacids (e.g. H3PMo12O40, Ag3PMo12O40, etc.).83
2.2.1.2. Retro-aldol condensation. In the retro-aldol condensation of carbohydrates, the C–C bond of monosaccharide substrates is cleaved at the β-position relative to their carbonyl group to form shorter oxygenates.84 The non-catalytic retro-aldol condensation of carbohydrates in the presence of (supercritical) water, referred to as hydrothermal cracking, proceeds at elevated temperatures (400–600 °C).67,85 The use of a catalyst can significantly reduce this temperature requirement to around 200 °C.86
As reviewed by Zheng et al.,84 both basic and transition metal catalytic systems can be used in the retro-aldol reaction of carbohydrates. In general, all metal oxides and hydroxides containing alkali (e.g., NaOH, KOH) and alkaline earth elements (e.g., Ca(OH)2) are active retro-aldol catalysts via their basic sites. However, their selectivity toward retro-aldol is limited due to competing reactions such as the abovementioned isomerization.84 Similarly, in addition to their isomerization activity, tin-based catalysts such as Sn-BEA also demonstrate retro-aldol activity.87,88 Other transition metal catalysts, in specific tungsten and related metals (i.e., Mo and Cr), are regarded as the most efficient and selective retro-aldol catalysts.84,89
Recently, Liu et al. proposed an alternative mechanism for the tungsten-catalyzed retro-aldol reaction, deviating from the conventional base-catalyzed mechanism where only the β-hydroxyl group is required.90,91 They studied the WO3-catalyzed retro-aldol reaction of various carbohydrate substrates and found that both an α- and β-hydroxyl group are essential for the selective C–C bond cleavage. Once absorbed on the catalytic surface, the substrate forms a tridentate complex by coordinating its carbonyl group and α- and β-hydroxyl groups with two adjacent tungsten atoms, identifying the W–O–W structure as the catalytic active site. According to Liu and co-workers, all literature-reported active tungsten catalysts, including tungstic acid (H2WO4), hydrogen tungsten bronze (HxWO3), meta- and paratungstate salts, heteropolyacids (e.g., H3PW12O40), and even tungsten metal and tungsten carbide (W2C), derive their catalytic activity via intrinsic or in situ generated W–O–W sites despite their diverse compositions and structures.91
2.2.1.3. Dehydration. Dehydration, another key cleavage reaction, involves the breaking of C–O carbohydrate bonds via acid-catalyzed H2O elimination. A wide variety of acidic catalysts has been used for these dehydration reactions: mineral (e.g., HCl, H2SO4) and organic acids (e.g., oxalic acid), soluble Lewis acids (e.g., CrCl3, ZnCl2), acid resins (e.g., Amberlyst-15), zeolites (e.g., MFI, BEA), metal oxides (e.g., Al2O3, ZrO2, TiO2), etc. Besides catalyst type, the effect of other reaction conditions such as reaction temperature (100–200 °C) and solvent (H2O, DMSO, ionic liquids, etc.) have been studied extensively.92–97
2.2.1.4. (De)hydrogenation. Carbohydrates, containing a carbonyl functional group, are reduced to polyols or sugar alcohols by catalytic hydrogenation. In general, hydrogenation occurs as the final step in a carbohydrate upgrading process as polyol products are more stable and less susceptible to side reactions than their hydroxy carbonyl equivalents.42 In the presence of molecular hydrogen, carbohydrates can be hydrogenated by both homogeneous and heterogeneous metal catalysts. The latter, however, are preferred on an industrial scale due to their ease of recovery and reusability in combination with high product yields and selectivity.98 The catalytic efficiency of heterogeneous hydrogenation catalysts depends on both the active metal species and the catalytic support. Metals such as Ru, Pt and Ni typically demonstrate the highest hydrogenation activity of carbonyl functional groups. Contrary, Pd, another well-known hydrogenation metal, displays limited activity in reducing carbonyl bonds.99 The use of support materials beneficially leads to improved structural stability, surface area, and spatial distribution and can additionally induce metal–surface interactions that affect the catalyst's performance. Frequently used support materials include, but are not limited to, activated carbon, alumina, silica and titania.100
In dehydrogenation, the reverse reaction, a polyol or sugar alcohol is oxidized by catalytical removal of hydrogen yielding a carbonyl-containing product (e.g. sorbitol to glucose or fructose, and xylitol to xylose). While hydrogenation is an exothermic reaction, dehydrogenation is an endothermic reaction and requires a higher reaction temperature. Dehydrogenation is usually the first step in the catalytic upgrading of sugar alcohols as more reactive hydroxy carbonyl substrates are obtained.101 The same heterogeneous metal catalysts used for hydrogenation can be applied in dehydrogenation reactions. Similar to hydrogenation, the dehydrogenation capacity of a heterogeneous metal catalyst depends on both the metal active species and support material.101–103
2.3. (Hemi)cellulose-derived oxygenates
In (hemi)cellulose valorization, hydrolysis and the four upgrading reactions can be combined to unravel a plethora of oxygenate molecules. The product scope ranges from large monosaccharides containing five or six oxygen atoms (O5 and O6 oxygenates) to small diols (O2 oxygenates). In this regard, selectivity control, i.e., favoring desired reactions and inhibiting undesired reactions, is of critical importance. Zheng et al. describe this selectivity challenge as “dancing on eggs”, in which the dancer's weight needs to be distributed subtly on each egg to achieve a desirable result.84 As a heavily studied topic in the literature, this overall conversion of (hemi)cellulose is referred to as hydrogenolysis.104,105 In general, the complexity of these hydrogenolysis reactions increases when targeting smaller oxygenates, as more consecutive reaction steps are required. Hence, hydrogenolysis of (hemi)cellulose targeting their corresponding monomers (O6-5 oxygenates) is first evaluated, after which the complexity gradually increased by assessing O4 and O3 oxygenates (containing four and three oxygen atoms) ultimately followed by the discussing O2 oxygenates.2.3.1.1. Monosaccharides (glucose and xylose). Depolymerization of cellulose into glucose monomers is challenging due to its rigid, crystalline structure. This characteristic originates from the excessive network of both inter- and intramolecular hydrogen bonds as well as the stable β-1,4-glycosidic bonds.48 Chemocatalytic depolymerization of cellulose is an acid-catalyzed hydrolysis reaction in which traditionally aqueous soluble mineral acids, such as H2SO4, are used as they enable good accessibility to insoluble, solid cellulose chains. This process can either be performed using diluted or concentrated mineral acids.70,71 In the former, hydrolysis is typically conducted with 0.5–1% H2SO4 at elevated temperature (>200 °C) and pressure (>2 MPa). Under these conditions, glucose is significantly degraded, limiting its yield to 60%. In the latter process, glucose yields above 80% can be obtained by operating with a concentrated H2SO4 solution (60–90%) due to decrystallization of the cellulose feed.106,107 However, mineral acids are highly corrosive to the equipment making their industrial use not viable. Alternatively, organic acids (e.g., carboxylic acids) or heteropolyacids (e.g., H3PW12O40, H4SiW12O40) can be used. Nevertheless, all these acid catalysts suffer from difficult recyclability and product separation.70,71 As a result, solid acids have gained increasing research interest over the past years. The explored heterogeneous catalysts range from metal oxides (both single and complex metal oxides) to H+-form zeolites, acidic resins, sulfonated carbon catalysts, etc.108–110 To prevent mass transfer limitations between the solid catalyst and solid cellulose feed catalyst design primarily focuses on creating a catalyst with strong binding sites for adsorption, strong acid sites for hydrolysis, and a high specific area to enhance the catalytic activity. Moreover, various feedstock pretreatment methods exist to increase its accessibility.108 Optimized heterogeneous catalytic systems with glucose yields exceeding 90% have been reported in the literature.111
Hemicellulose, in contrast to cellulose, contains shorter and branched chains, which inhibit crystallization and enhance its solubility, thereby facilitating hemicellulose depolymerization.44,112 This difference in ease of hydrolysis is illustrated by the work of Kobayashi et al.113 In their work, a comparison between the acid-catalyzed hydrolysis of cellulose and hemicellulose (xylan type) was made using a heterogeneous carbon-based catalyst. Performing the hydrolysis reaction in an aqueous solution containing traces of HCl for 17 minutes at 215 °C resulted in glucose and xylose yields of 78% and 94%, respectively.113
2.3.1.2. Sugar alcohols (sorbitol and xylitol). Early research addressed the conversion of (hemi)cellulose into sugar alcohols, such as sorbitol and xylitol, using a binary catalytic system.114 This system consists of (i) a soluble mineral acid to facilitate hydrolysis and (ii) a transition metal catalyst, typically Ru, to subsequently perform the hydrogenation of the monosaccharide into the sugar alcohol.115,116 In 2006, Fukuoka and co-workers were the first to report a different approach, using an integrated multifunctional catalyst, bypassing the drawbacks of mineral acids.117 In their work, cellulose was converted to sorbitol (25% yield) and mannitol (6% yield) using a Pt/γ-Al2O3 catalyst in the presence of 5 MPa H2 pressure, at 190 °C for 24 h. The solid catalyst, consisting of a transition metal and an acidic support, both catalyzed the hydrolysis of cellulose and hydrogenation of glucose.117 This finding paved the way for much follow-up research exploring various transition metals (e.g., Ni, Ru, Pt, Rh) and carbon-based supports (e.g., activated carbon, zeolites, carbon nanotubes). Various of these studies have reported sugar alcohol selectivities exceeding 70%.118–121 Importantly, selective multifunctional catalytic systems balance the activity of their acid sites and the activity of their hydrogenation sites. A catalyst with dominant acid sites results in fast hydrolysis followed by glucose degradation due to the high reaction temperature. Contrary, a catalyst with dominant hydrogenation sites additionally favors other metal-catalyzed reactions (e.g., retro-aldol condensation, dehydration) resulting in smaller oxygenate by-products.122,123
2.3.2.1. O4 oxygenates. In theory, the O4 oxygenate erythrose can be obtained via retro-aldol condensation of glucose. Erythrose, in its turn, can subsequently undergo keto–enol isomerization to form erythrulose or hydrogenation to yield erythritol.124 Since currently no chemocatalytic processes exist, biotechnological routes dominate industrial production of these O4 oxygenates.125,126 Notably, the research team at Haldor Topsøe has proposed a chemocatalytic bottom-up pathway to produce these O4 oxygenates.127 In their study, they achieved the formation of these O4 oxygenates through the aldol condensation of two glycolaldehyde (GA) molecules using shape-selective zeolites. For example, using a Sn-MFI catalyst at 80 °C in water for 30 minutes, they reported a total yield and selectivity for O4 oxygenates of 74% and 97%, respectively.127
2.3.2.2. O3 oxygenates. Within the scope of (hemi)cellulose valorization, three main O3 oxygenates can be formed: 1,3-dihydroxyacetone (DHA), glyceraldehyde, and glycerol. In theory, the isomers DHA and glyceraldehyde can be produced via retro-aldol condensation of fructose, the ketose isomer of glucose. Subsequent hydrogenation of these O3 isomers results in the formation of glycerol. In practice, glycerol is abundantly available being a major by-product in the biodiesel process. Approximately 100 g of glycerol is generated for every kg of biodiesel produced via transesterification of triglycerides.128,129 Consequently, glycerol is the industrial feedstock to produce DHA and glyceraldehyde via fermentative or catalytic oxidation.130,131
2.3.3.1. Ethylene glycol (EG) and glycolaldehyde (GA). The chemocatalytic conversion of glucose to EG passes through three consecutive steps and two different key upgrading reactions: (i) retro-aldol condensation of glucose in erythrose and GA, (ii) consecutive retro-aldol condensation of erythrose in two additional GA molecules, and (iii) hydrogenation of GA into EG. Dehydration, isomerization, and preliminary hydrogenation of glucose and erythrose should all be suppressed or be less competitive than retro-aldol condensation to selectively form EG. A successful strategy consists in designing a catalytic system that contains both retro-aldol and hydrogenation activity. On the one hand, the catalyst can be a binary system comprising both a metal-based hydrogenation catalyst and a tungsten-based retro-aldol catalyst. For example, in Avantium's patented RAY process, a 72% EG yield could be obtained from an aqueous glucose solution by using a binary 5 wt% Ru/C–H2WO4 catalytic system (W-to-Ru molar ratio of 16
:1) in a batch process at 180 °C and 5 MPa H2.135 Zhao et al. studied the conversion of an aqueous glucose solution with a binary 4 wt% Ru/C–ammonium metatungstate (AMT) catalytic system. They achieved a 76% EG yield by performing the reaction in a fed-batch set-up at 220 °C and 5 MPa H2.136 On the other hand, both hydrogenation and retro-aldol activity can be incorporated into one bimetallic catalyst. For instance, Ooms et al. developed a heterogeneous nickel–tungsten carbide catalyst (2 wt% Ni–30 wt% W2C/AC) for the conversion of an aqueous glucose solution in a fed-batch process. Operating the reaction at 260 °C and 6 MPa H2 yielded 66% EG.137 It is noticeable that all reactions targeting EG are conducted at temperatures around or above 200 °C. Under applied reaction conditions, preliminary hydrogenation of glucose into sorbitol is the dominant side reaction. Although system-dependent, it is generally acknowledged that the activation energy of the retro-aldol condensation is significantly higher than that of the hydrogenation. For example, in the abovementioned Ru/C–AMT catalytic system, the activation energy for the retro-aldol condensation and hydrogenation of glucose are around 160 kJ mol−1 and 65 kJ mol−1, respectively.136 Therefore, conducting the reaction at an elevated temperature should beneficially affect the retro-aldol condensation at the expense of the preliminary hydrogenation.137
The chemocatalytic conversion of sorbitol to EG follows the same pathway as the hydrogenolysis of glucose, with the distinction that this substrate requires an initial dehydrogenation step. The dehydrogenation–hydrogenation activity of the catalytic system is a crucial property of this hydrogenolysis reaction as substrate conversion generally is the rate-determining step. Liang et al. studied the dehydrogenation of sorbitol and observed the importance of the synergetic effect of ample acid–base pair sites. Comparing the catalytic activity of Ni/Al2O3 and Ni/ZSM-5, the first catalyst had both strong acidity and basicity and resulted in a higher dehydrogenation activity than the latter catalyst, which contained sufficient acidic but less basic sites.103 Annuar et al. recently reviewed numerous heterogeneous catalysts used in sorbitol hydrogenolysis.101
In hydrolytic hydrogenolysis, acid-catalyzed hydrolysis of (hemi)cellulose is coupled with monosaccharide hydrogenolysis. Most hydrogenolysis catalytic systems are also effective hydrolytic hydrogenolysis catalysts since these catalysts already contain acidic properties to perform the retro-aldol condensation, isomerization and/or dehydration steps.138,139 For example, the nickel–tungsten catalysts active in the hydrogenolysis of glucose to EG are also active catalytic systems in the conversion of cellulose to EG.84
2.3.3.2. 1,2-Propylene glycol (1,2PG) and acetol. The chemocatalytic conversion of glucose to 1,2PG involves a series of five sequential steps encompassing all four key reactions: (i) glucose isomerization to fructose, (ii) retro-aldol condensation of fructose leading to the isomers glyceraldehyde and DHA, (iii) dehydration of glyceraldehyde to pyruvaldehyde, (iv) hydrogenation of pyruvaldehyde to acetol, and finally (v) hydrogenation of acetol to yield 1,2PG. The primary challenge in achieving a desired 1,2PG selectivity via this hydrogenolysis process lies in the initial step, namely the isomerization reaction. To selectively obtain 1,2PG, it is important to favor glucose isomerization while minimizing preliminary glucose hydrogenation to sorbitol and retro-aldol condensation to erythrose and GA. Another selectivity challenge lies in the dehydration of glyceraldehyde as two other side reactions can occur, namely the retro-aldol condensation to GA and formaldehyde, and hydrogenation to glycerol. Consequently, a 1,2PG selective catalytic system should contain sufficient isomerization capacity alongside retro-aldol, dehydration and hydrogenation capacity. Analogous to selective EG formation, the two most studied strategies in selective 1,2PG formation are a binary catalytic system and a modified hydrogenation catalyst. For both strategies, numerous catalytic systems have been studied and were recently reviewed.140,141 Here, both strategies are illustrated with a couple of suitable examples. Hirano et al. studied a binary Ru/C–ZnO catalytic system.142 When only Ru/C was present, the direct hydrogenation of glucose into sorbitol was the dominant reaction pathway while the addition of ZnO steered the selectivity toward 1,2PG. They proposed that ZnO, acting as an amphoteric metal oxide, positively affected the isomerization, retro-aldol condensation and dehydration steps. The 1,2PG yield increased from 9% to 38% by adding ZnO to the 5 wt% Ru/C catalyst after 20 h reaction time at 180 °C and a stoichiometric hydrogen pressure (0.4 MPa H2) to limit preliminary hydrogenation. Other tested metal oxides, including Fe3O4, La2O3 and CeO2, were less effective but still selective toward 1,2PG.142 Another selective 1,2PG binary system was proposed by Xiao et al.143 They explored a binary system with on the one hand a Cu–Cr catalyst containing both hydrogenation and dehydration properties and on the other hand the base Ca(OH)2 as an isomerization and retro-aldol condensation catalyst. With this catalytic system, they achieved a 1,2PG yield of 53% by conducting the reaction at 6 MPa H2, first for 2 hours at 140 °C followed by 5 hours at 220 °C. In the second strategy, i.e., developing a modified hydrogenation catalyst, the choice of support is crucial as it should introduce acid–base properties in the system. Liu et al. studied the Cu/Al2O3 catalytic system for the selective formation of 1,2PG.144 Similar to ZnO used by Hirano et al.,142 the amphoteric properties of the Al2O3 support significantly contributed to the isomerization, retro-aldol condensation and dehydration steps. At 180 °C and 4 MPa H2, a 43% 1,2PG yield was obtained. Additionally, the incorporation of WOx in the catalytic system (Cu–WOx(0.8)/Al2O3) further increased the 1,2PG yield to 55% by beneficially influencing the retro-aldol condensation and glyceraldehyde dehydration.144 In a successive study, the same researchers investigated the synergetic effect between Cu and Al2O3 in more detail. In addition to the intrinsic amphoteric character of the support, they found that Al2O3 could also induce the formation of oxidized Cuδ+ species which in their turn could act as Lewis acid sites promoting the isomerization, retro-aldol condensation and dehydration steps.145 In line with these findings, Kirali et al. developed a Ce-promoted Cu/Al2O3 catalyst with optimized acid–base properties for the selective conversion of glucose into 1,2PG. After 6 h reaction time at 200 °C and 4 MPa H2, the reaction catalyzed by 8 wt% Cu/Al2O3 yielded 31% 1,2PG. The 1,2PG yield was improved to 61% by performing the reaction with the Ce–Cu/Al2O3 catalyst.146 While most studies focus on the production of 1,2PG, Deng et al. reported a catalytic system (Ni–SnOx/Al2O3) with limited hydrogenation activity predominantly leading to acetol formation. The reaction yielded 3% 1,2PG and 53% acetol after 0.5 h at 200 °C and 6 MPa H2.147
Glycerol hydrogenolysis mainly results in three diols, namely 1,2PG and 1,3PG and to a lesser extent in EG. 1,2PG and 1,3PG are produced via two consecutive steps: (i) acid-catalyzed dehydration of glycerol followed by (ii) hydrogenation of the hydroxy carbonyl intermediates. The dehydration of glycerol's two primary hydroxyl groups results in acetol which leads to 1,2PG after hydrogenation. The dehydration of the secondary hydroxyl group yields 3-hydroxypropanal and 1,3PG after hydrogenation.134,148 From a kinetic point of view, the dehydration of the two primary hydroxyl groups and the subsequent formation of 1,2PG is favored. Contrary, from a thermodynamic point of view, the formation of 1,3PG via its more stable secondary carbocation is favored.149 In practice, however, most investigated catalytic systems mainly yield the kinetic product 1,2PG. Moreover, 1,2PG can also be formed via a second pathway: (i) glycerol dehydrogenation into glyceraldehyde, (ii) glyceraldehyde dehydration into pyruvaldehyde, (iii) subsequent hydrogenation of pyruvaldehyde into 1,2PG. EG can be formed as a minor by-product via this pathway if retro-aldol condensation of glyceraldehyde is competitive with its dehydration. Similar to other mentioned hydrogenolysis reactions, the catalytic system for glycerol hydrogenolysis consists of a transition hydrogenation metal (Cu, Ni, Ru, Pt, etc.), potentially accompanied by a promoter, and a support material. Consequently, product selectivity is influenced by the dehydrogenation–hydrogenation activity and, especially, by the acid–base properties of the overall catalytic system.104 An overview of tested catalytic systems can be found in the reviews by Wang et al.134 and Basu et al.148
3. From renewable substrates to bio-based aliphatic amines
3.1. Reductive amination: principles and mechanism
The catalytic transformation of (hemi)cellulose-derived oxygenates into aliphatic amines essentially involves the transformation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or C–OH bond into a C–N bond via a reductive amination reaction. Over the years, multiple reductive amination approaches have been studied, ranging from biocatalytic techniques using transaminase150,151 and amine dehydrogenase enzymes152,153 to electrocatalytic154,155 and chemocatalytic techniques. The latter technique will be reviewed in detail, in correspondence with the abovementioned chemocatalytic valorization of (hemi)cellulose.
O or C–OH bond into a C–N bond via a reductive amination reaction. Over the years, multiple reductive amination approaches have been studied, ranging from biocatalytic techniques using transaminase150,151 and amine dehydrogenase enzymes152,153 to electrocatalytic154,155 and chemocatalytic techniques. The latter technique will be reviewed in detail, in correspondence with the abovementioned chemocatalytic valorization of (hemi)cellulose.
From a mechanistic perspective (Scheme 2), the reductive amination of a carbonyl-containing substrate (i.e., aldehyde or ketone) is a multi-step reaction that starts with the nucleophilic addition of an amine reactant onto the carbonyl group of the substrate resulting in a hemi-aminal intermediate. Subsequent dehydration of this intermediate yields an imine or enamine if the reactant is NH3/primary amine or a secondary amine, respectively. This imine/enamine is ultimately reduced to yield an amine product.2,156,157 In the chemocatalytic reductive amination process, various reducing agents can be used.158 The most widespread reducing agents include metal hydrides (e.g., borohydrides such as NaBH4 and NaBH3CN),159 formic acid/formate,160–162 CO,163,164 and molecular H2 in combination with a homogeneous or heterogeneous transition metal hydrogenation catalyst.165,166 The latter heterogeneous reducing system is predominantly used in research and industry because it uses non-toxic, relatively inexpensive and potentially green H2 gas, only leads to H2O as a by-product, and the metal catalyst can be recycled.158,167 The amination of a hydroxyl-containing substrate (i.e., alcohol) starts with its activation into a more reactive aldehyde/ketone via oxidation. Afterward, the process proceeds via the same elementary steps as the reductive amination reaction. In the literature, this process is referred to as hydrogen borrowing amination since overall no net H2 is consumed.168 The hydrogen atoms necessary for imine/enamine reduction are initially obtained via alcohol activation. In practice, however, most hydrogen borrowing amination reactions are conducted under (limited) hydrogen pressure to suppress catalyst deactivation and detrimental amine disproportionation (vide infra).169 In hydrogen borrowing amination, substrate activation by oxidation is generally recognized as the rate-determining step. Consequently, these reactions typically require higher reaction temperatures than reductive amination (150–250 °C) and the presence of a transition metal catalyst that contains both dehydrogenation and hydrogenation activity.170–172
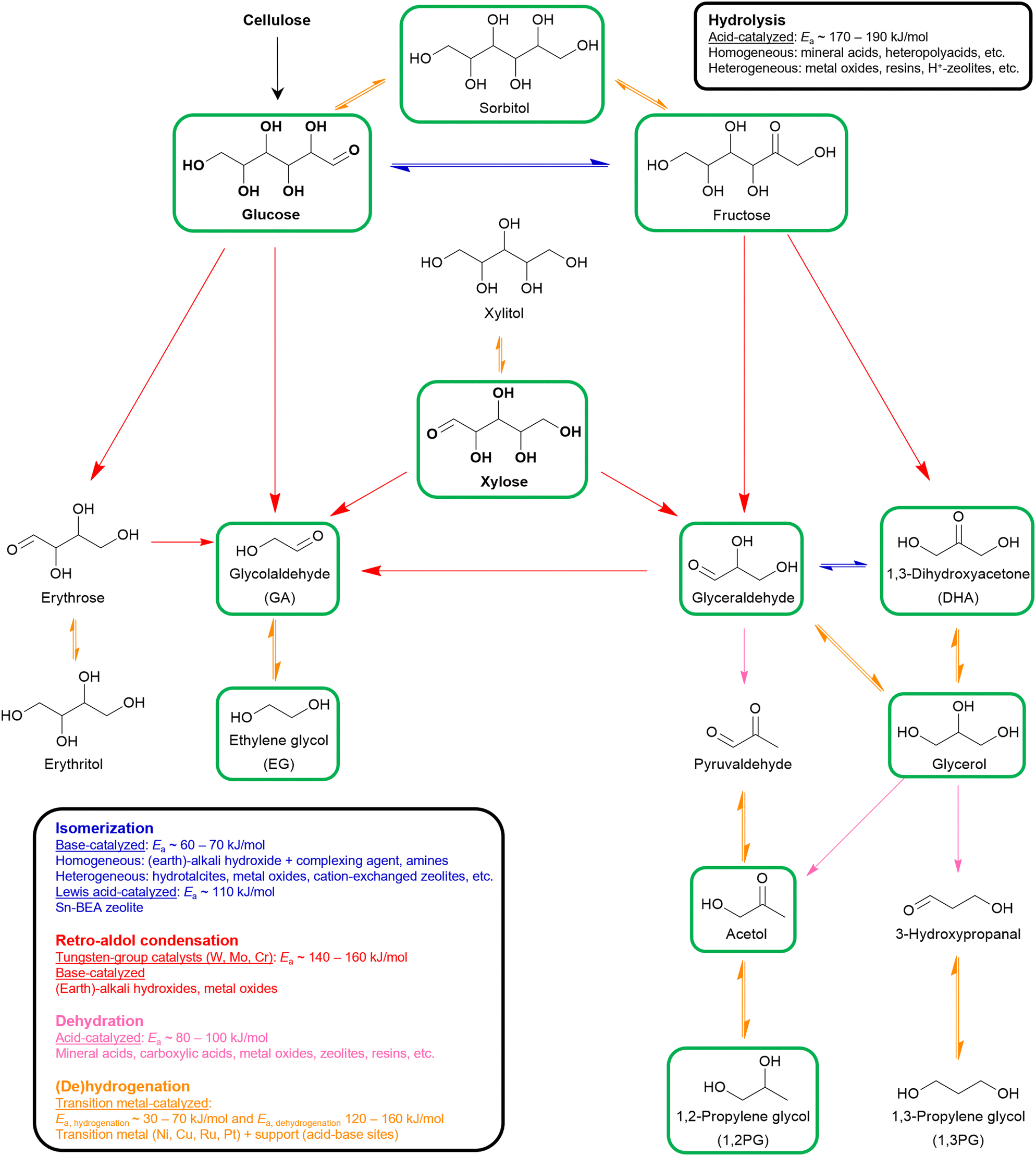 | ||
| Scheme 1 Overview of the chemocatalytic valorization of (hemi)cellulose-derived monomers into hydroxy carbonyl and polyol oxygenates through hydrolysis and four upgrading key reactions. Oxygenates framed in green have already been assessed as viable substrates in the production of bio-based aliphatic amines. Reported activation energies (Ea) should be interpreted as an indication of the order of magnitude as they strongly depend on the studied catalytic system. References for hydrolysis,70,71,108,173 isomerization,72–74,174–176 retro-aldol condensation,84,86,88,89,136 dehydration,134,148,149,177 and (de)hydrogenation.98,99,101,103 | ||
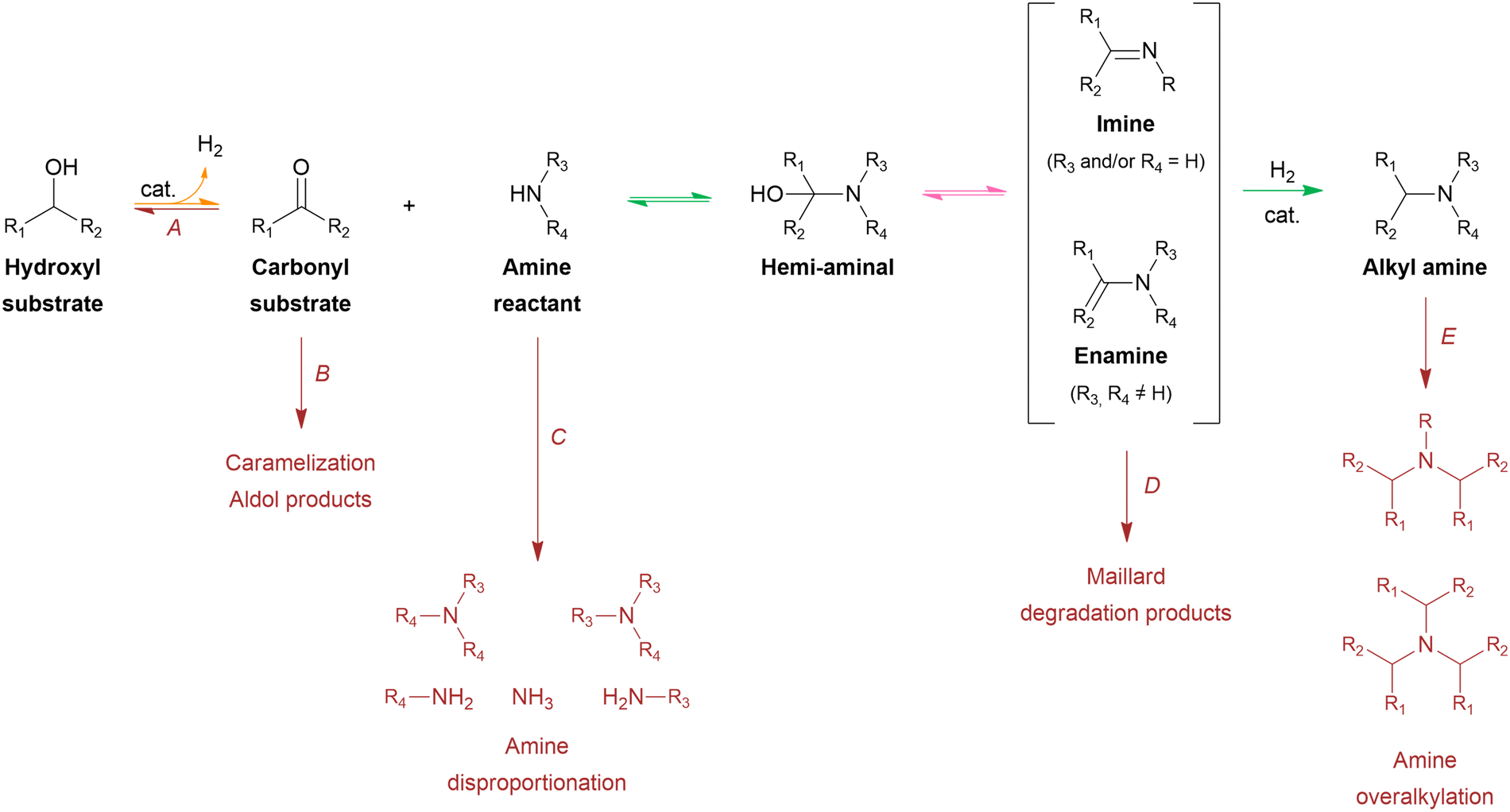 | ||
| Scheme 2 General mechanism of the reductive amination of an oxygenate into an alkyl amine. Each elementary reaction is represented by a color; amination (green), dehydration (pink) and (de)hydrogenation (orange). Reactions in dark red represent the most important side reactions. | ||
Both the reductive amination and hydrogen borrowing amination reactions encompass a wide scope of potential side reactions, influenced by the activity of the catalyst and the reactivity of the various components involved.156,157 First, the choice of catalyst is crucial as it significantly contributes to the overall efficiency and selectivity of the process. In reductive amination, uncontrolled, fast hydrogenation of the unreacted aldehyde/ketone substrate into the corresponding alcohol inevitably depletes the substrate (Scheme 2, side reaction A).166 In hydrogen borrowing amination, substrate activation, as the rate-determining step, emphasizing the need for a catalytic system with excellent dehydrogenation properties.172 Second, all components throughout the reaction pathway (i.e., substrate, reactant, intermediates and product) can undergo various side reactions, thereby decreasing the selectivity of the desired product. In general, aldehydes are considered to be more reactive than ketones due to electronic and steric effects. Consequently, aldehyde-containing substrates are not only more susceptible to the amination reaction but also more reactive to side reactions such as aldol condensation, caramelization and other degradation reactions, leading to depletion of the substrate (Scheme 2, side reaction B).178,179 Moreover, both the amine reactant and product can undergo amine disproportionation, as it is catalyzed by a metal catalyst at elevated temperatures (i.e., typical for hydrogen borrowing amination), resulting in the interconversion of primary, secondary and tertiary amines (Scheme 2, side reaction C).169 In addition, the amine product can suffer from overalkylation, since the alkylated amine product, in general, exhibits greater reactivity compared to the amine reactant due to its increased nucleophilicity.180 Similar to amine disproportionation, overalkylation results in a mixture of alkylated amine products (Scheme 2, side reaction E). Furthermore, all amine intermediates, particularly the imine/enamine intermediates, are labile compounds that are prone to Maillard-type degradation reactions, which adversely impact product selectivity (Scheme 2, side reaction D).178,179
3.2. Oxygenate-derived amines
Over the past decades, various lignocellulose-derived compounds have been explored as potential substrates in the catalytic reductive amination yielding a plethora of amines.181 Amination of lignin and its monomers results in phenolic and cycloalkyl amine products such as aniline, cyclohexylamine and their derivatives.182–186 (Hemi)cellulose-derived carboxylic acids, for example levulinic acid or lactic acid, can be converted into heterocyclic pyrrolidines and pyrrolidones,187,188 amino acids189 or even alkyl amines,190 whereas (hemi)cellulose-derived furfurals predominantly yield furfuryl amines.191–193In the scope of the Review, aliphatic amines are obtained from aliphatic (hemi)cellulose-derived oxygenates. Moreover, all three types of aliphatic amines, namely alkanolamines, alkyl polyamines and heterocyclic amines, are formed via the same reaction pathway due to the multi-oxygen nature of these oxygenate substrates. Hence, steering the selectivity toward one desired product while reducing the formation of other products and undesired side reactions is the key challenge in governing the catalytic reductive amination of carbohydrate substrates. Strikingly, product selectivity is affected by the same four elementary key reactions that influence the chemocatalytic valorization of (hemi)cellulose, in addition to the nucleophilic amination step. While, two key reactions are inherently present in the reductive amination mechanism (i.e., (de)hydrogenation and dehydration, Scheme 2), all four key reactions can occur as pre-amination reactions preceding reductive amination and further complicating product selectivity. In general, the complexity of the overall amination reaction, associated with the occurrence of these pre-amination reactions, increases with an increasing number of oxygen atoms in the oxygenate substrate (Scheme 3). In this regard, the reductive amination of O2 substrates is first evaluated, after which the complexity gradually increases by assessing O3 substrates followed by O5 and O6 substrates.
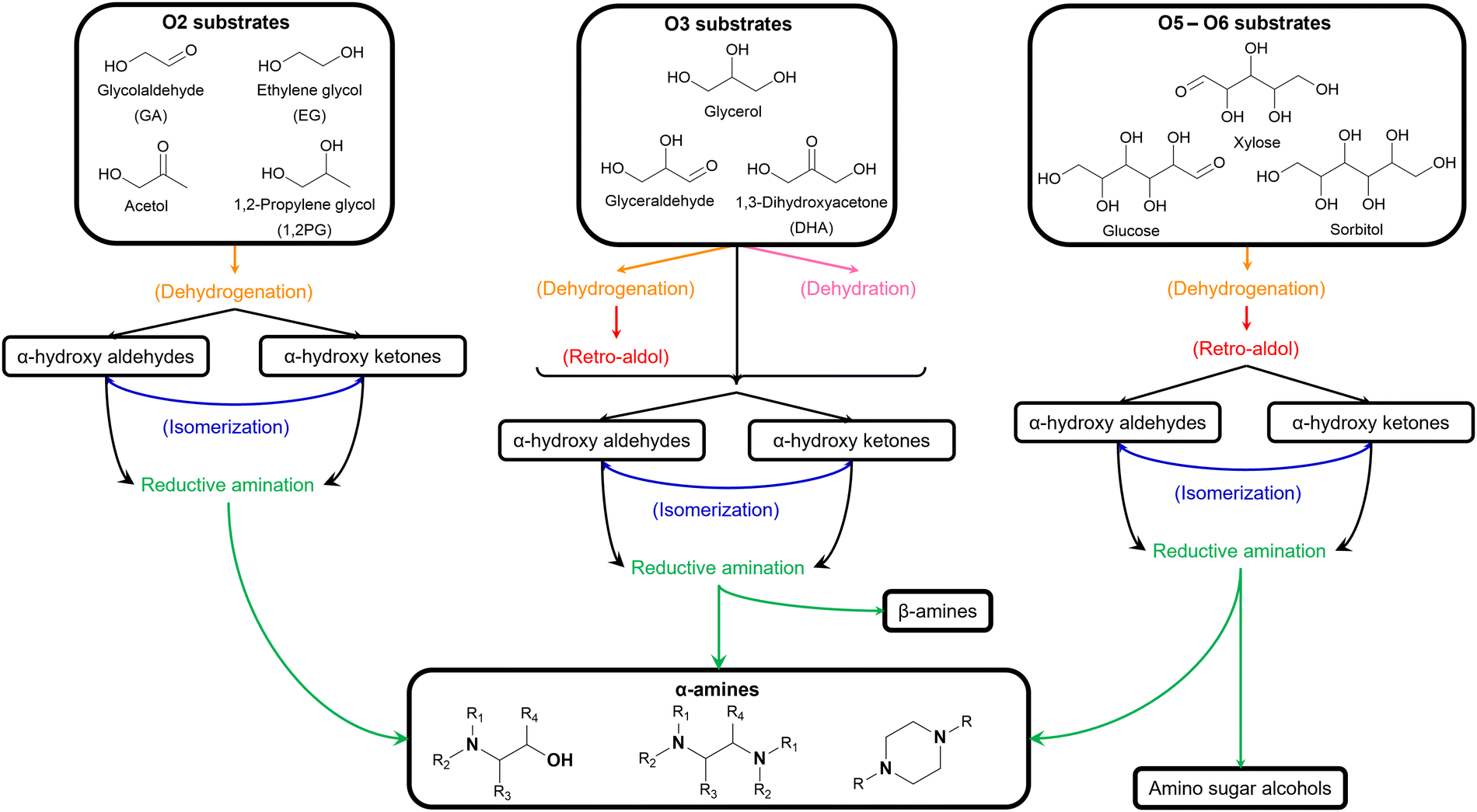 | ||
| Scheme 3 Overview of the possible reductive amination pathways of different (hemi)cellulose-derived oxygenate substrates. | ||
The general pathway for the catalytic reductive amination of O2 substrates is given in Scheme 4. Prior to the reductive amination reaction steps, two additional pre-amination reactions can or need to take place. First, when using a diol (Scheme 4, S1), an initial activation step is required. This activation involves the dehydrogenation of the diol, resulting in the formation of an α-hydroxy carbonyl component (S2 or S3). Second, when using an asymmetric α-hydroxy carbonyl (i.e., if R1 ≠ R2), an isomerization reaction can occur. If one of the substituents is a proton (i.e., if R1 or R2 = H), the isomerization involves the interconversion between an α-hydroxy ketone and an α-hydroxy aldehyde, resembling the aldose – ketose isomerization in carbohydrate upgrading. After these pre-amination reactions, the reductive amination of the α-hydroxy carbonyl substrates initially proceeds via the general reductive amination mechanism. First, the nucleophilic addition of an amine reactant onto the O2 substrate results in a hemi-aminal intermediate (I1). Secondly, dehydration of I1 leads to an imine (I2) or enamine (I3) intermediate. In their turn, I2 and I3 can interconvert via an imine-enamine tautomerization.194,195 Due to the unique structure of α-hydroxy carbonyl components, the formed enamine (I3) can further undergo a keto–enol tautomerization reaction, yielding an α-amino carbonyl intermediate (I4). The overall isomerization of I2 to I4, passing through two successive tautomerization reactions, is commonly referred to as the acid-catalyzed Amadori or Heyns rearrangement, depending on whether the substrate is an α-hydroxy aldehyde or α-hydroxy ketone, respectively.196,197 In the presence of molecular hydrogen and a heterogeneous metal catalyst, I2, I3 and I4 can all be hydrogenated into a (substituted) alkanolamine product (P1). Alternatively, I4 can undergo a second nucleophilic addition reaction on its carbonyl functional group, resulting in a second hemi-aminal intermediate (I5). Dehydration of I5 results in an imine (I6) or enamine (I7) intermediate, which both yield the (substituted) alkyl polyamine product (P2) after hydrogenation. Under dehydrogenation conditions, the alkanolamine product (P1) could potentially be reactivated into I4, eventually resulting in the alkyl polyamine product (P2). Aliphatic heterocyclic piperazines (P3) can be formed from both amine products.21 In Scheme 4 the amination reaction is depicted using the S2 hydroxy carbonyl substrate. In the case of asymmetry, the amination of the isomer S3 would result in the same polyamine (P2) and an isomeric ethanolamine (P1) compared to the S2-related amine products.
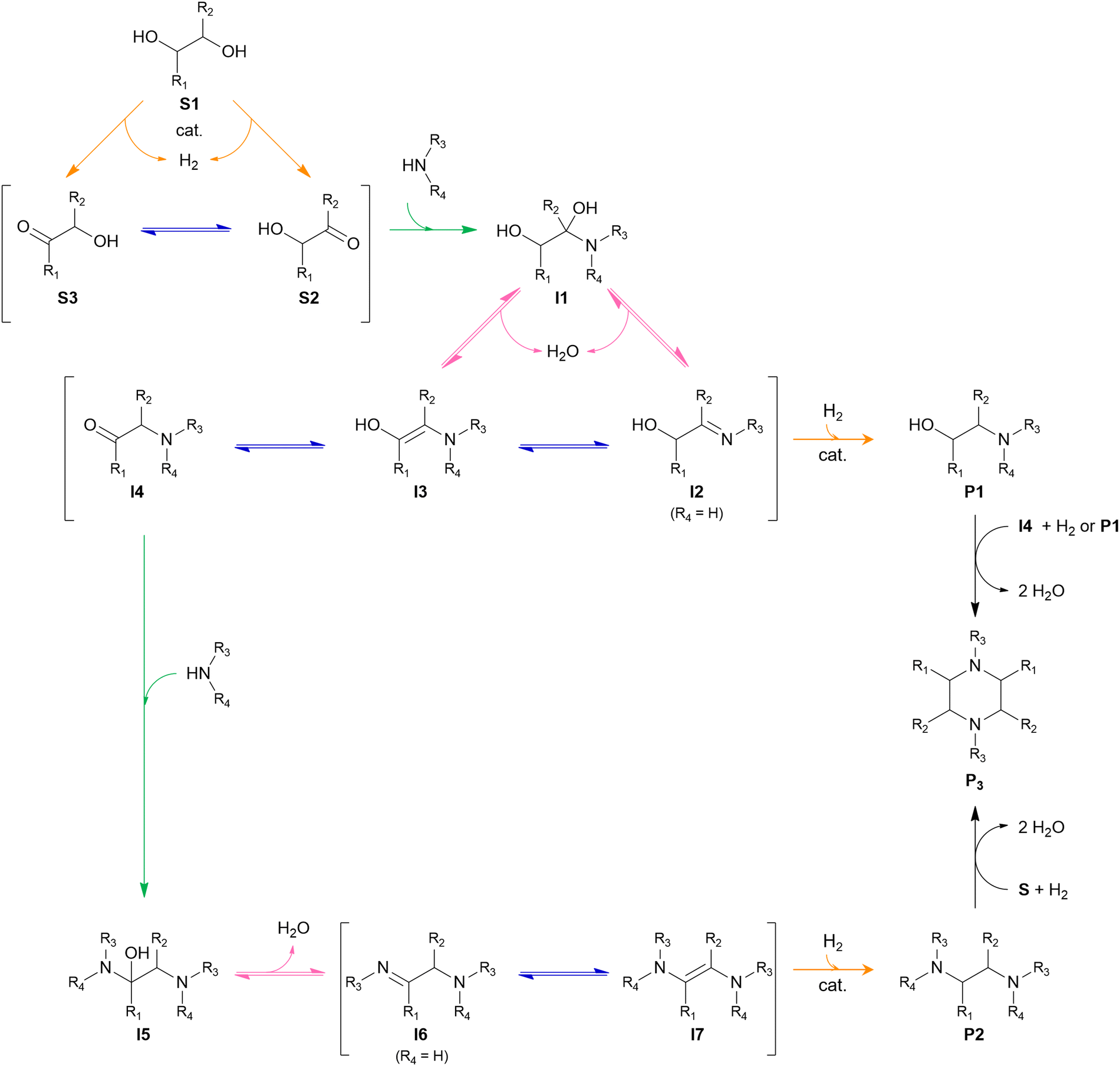 | ||
| Scheme 4 General reaction mechanism of the reductive amination of O2 substrates. Each elementary key reaction is represented by a color: amination (green), isomerization (blue), dehydration (pink), and (de)hydrogenation (orange). | ||
Four different O2 substrates have been predominantly studied in literature and will be here reviewed systematically: (i) the symmetric α-hydroxy aldehyde glycolaldehyde (GA), (ii) the symmetric α-diol ethylene glycol (EG), (iii) the asymmetric α-hydroxy ketone acetol, and (iv) the asymmetric α-diol 1,2-propylene glycol (1,2PG).
3.2.1.1. Glycolaldehyde (GA). As the smallest reducing sugar, GA (Scheme 4, S2: R1 = R2 = H) is the most suitable model component to study the selectivity control challenge in catalytic reductive amination.30
Multiple researchers have used GA to qualitatively study the reductive amination reaction in order to develop handles that steer the product selectivity toward the formation of either ethanolamines or ethylene polyamines. In their work, Faveere et al. developed a set of guidelines for highly selective ethanolamine formation from GA.198,199 They achieved a quantitative N,N-dimethylethanolamine (DMEA, Table 1 entry 3) yield (97%) in the one-step reductive amination of GA with DMA after 1 h at 100 °C. Three main aspects contribute to this high yield. First, they performed a fast and selective hydrogenation reaction of I3 (Scheme 4) by conducting the reaction under high H2 pressure (7 MPa H2) in the presence of an effective CC hydrogenation catalyst (e.g., 5 wt% Pd/C). Second, they conducted the reaction in MeOH, a protic solvent that assists the different proton-transfer reactions throughout the amination pathway. Performing the same reaction in H2O or THF decreased the DMEA yield to 67% and 57%, respectively. Although a protic solvent, performing the reaction in the presence of H2O negatively affects the dehydration step. Third, they applied a low amine-to-substrate (ATS) molar ratio to stoichiometrically prevent the formation of the polyamine product. Similar to the fossil-based processes, the ATS molar ratio is an efficient tool to influence product selectivity. For example, in the reductive amination of GA with MMA in MeOH for 1 h at 100 °C and 7 MPa H2 with a 5 wt% Pd/C hydrogenation catalyst, increasing the ATS molar ratio drastically shifted the product selectivity. At a molar ratio of 0.5:1, the yield of the overalkylated diethanolamine N-methyldiethanolamine (MDEA, Table 1 entry 5) amounted to 91%, while its yield strongly decreased to 18% at a stoichiometric molar ratio of 1:1 in favor of the ethanolamine N-monomethylethanolamine (MMEA, Table 1 entry 2), yielding 64%. A maximum MMEA yield of 91% was achieved at a molar ratio of 3:1.198,199 These three control handles were further validated by expanding the amine reactant scope.200 In agreement with the insights of Faveere et al.,199 the reductive amination of GA with the reactant N-monoethylamine yielded 94% of the ethanolamine N-monoethylethanolamine (MEEA, Table 1 entry 4) under optimized conditions (MeOH as solvent, 1 h, 120 °C, 8 MPa H2, 5 wt% Pd/C hydrogenation catalyst, ATS molar ratio of 2:1).200 The ethanolamine guidelines are also applied in a recent patent application by Solvay.201 A 94% DMEA yield was achieved by performing the reductive amination of GA with DMA for 3 h at 25 °C under selective hydrogenation conditions (4 MPa H2 with 5 wt% Pd/C), in EtOH as the protic solvent, and a low ATS molar ratio of 2.4:1.201 Recently, Van Praet et al. strengthened the viability of the ethanolamine guidelines in a scale-up study.202 Whereas Faveere et al. typically performed their reactions at a 5 wt% GA concentration, Van Praet et al.199 conducted the reductive amination of GA using a significantly more concentrated GA solution (40 wt%) to align with industrial productivity standards.202 Although all control handles were validated, performing the reductive amination using this highly concentrated GA solution negatively impacted the DMAE selectivity compared to the conventional low GA concentration reactions. These results were ascribed to H2 transfer limitations induced by the increased GA concentration. To overcome these transfer limitations, Van Praet et al. proposed to enhance the gas-phase mass transfer of the system. This could be achieved by increasing the stirring rate or implementing baffles adjacent to the stirrer to facilitate mixing while minimizing excessive stirring. For example, performing the reductive amination of a 40 wt% GA MeOH solution for 1 h at 100 °C and 2 MPa H2, with a 10 wt% Pd/C hydrogenation catalyst, an ATS molar ratio of 2.5:1, and a stirring rate of 800 rpm resulted in unselective DMEA and TMEDA formation with yields of 44% and 45%, respectively. However, conducting the reaction under the same conditions in the presence of baffles shifted product selectivity if favor of the ethanolamine, with DMEA and TMEDA yields amounting to 88% and 2%, respectively.202
| # | Name | Structure | Oxygenate substrate | Reaction conditions [cat., ATS molar ratio, T, solvent] | X–S [%] | Ref. | Applications |
|---|---|---|---|---|---|---|---|
| Alkanolamines | |||||||
| Ethanolamines | |||||||
| 1 | MEA |
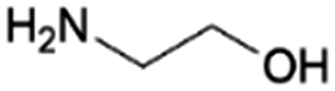
|
GA | Ru/ZrO2, 10:1, 75 °C, H2O |
100–93 | 203 | CCUS |
| Co/MnO, 35:1, 100 °C, THF |
100–83 | 204 | |||||
| EG | (1) Ni–Cu–Mo/ZrO2, 15:1, 150 °C, — |
43–29 | 205 | ||||
| (2) Ru–Co/Al2O3, 15:1, 170 °C, — |
|||||||
| Re–Ru–Co/ZrO2, 20:1, 170 °C, — |
35–32 | 206 and 207 | |||||
| Co—Cu/Al2O3, 5:1, 190 °C, H2O |
42–23 | 208 and 209 | |||||
| Cellulose | (1) H2WO4, —, 290 °C, H2O | 100–12 | 203 | ||||
| (2) Ru/ZrO2, 3.5:1, 75 °C, H2O |
|||||||
| 2 | MMEA |
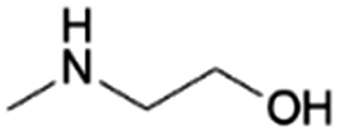
|
GA | Pd/C, 3:1, 100 °C, MeOH |
100–91 | 199 | CCUS |
| 3 | DMEA |
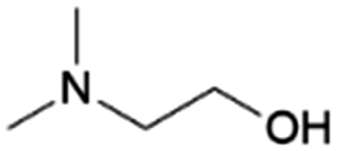
|
GA | Pd/C, 1:1, 100 °C, MeOH |
100–97 | 199 | Epoxy |
| Pd/C, 2.4:1, 25 °C, EtOH |
100–94 | 201 | |||||
| Pd/C, 2.5:1, 100 °C, MeOH |
100–88 | 202 | |||||
| Cu/Al2O3, 12:1, 165 °C, — |
100 - 74 | 210 | |||||
| Ni/SiO2, 2.8:1, 130 °C, THF |
100–76 | 211 | |||||
| EG | Cu/Al2O3, 1:1, 230 °C, — |
91–68 | 212 | ||||
| Glyceraldehyde | Ni oxide, 8:1, 130 °C, MeOH |
100–1 | 213 and 214 | ||||
| Xylose | 100–2 | ||||||
| Fructose | 100–2 | ||||||
| Glucose | 100–4 | ||||||
| 4 | MEEA |
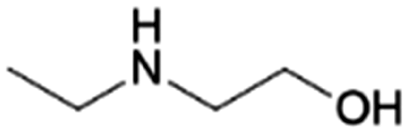
|
GA | Pd/C, 2:1, 120 °C, MeOH |
100–94 | 200 | CCUS |
| 5 | MDEA |
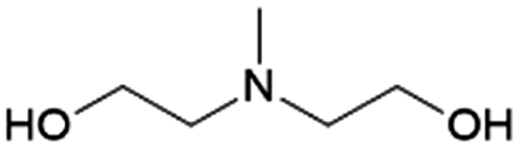
|
GA | Pd/C, 0.5:1, 100 °C, MeOH |
100–91 | 199 | CCUS |
| 6 | TriMAEEA |
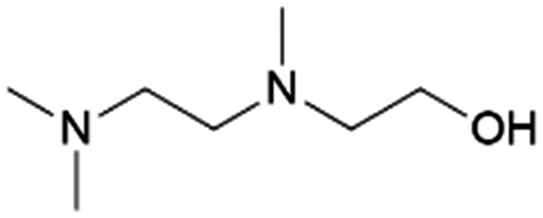
|
GA | Pd/C, 1:1, 100 °C, MeOH |
100–95 | 215 | PUR |
| Propanolamines | |||||||
| 7 | 1A2P |
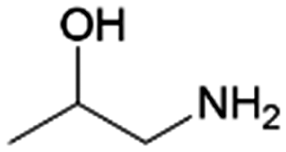
|
Acetol | Cu/Cr2O3, 50:1, 210 °C, — |
100–2 | 216 | |
| 1,2PG | Rh–In/C, 10:1, 180 °C, H2O |
38–42 | 217 | ||||
| Co/Nb2O5, 9:1, 160 °C, H2O |
37–36 | 218 | |||||
| (1) Cu–PdO–Bi2O3–In2O3/Al2O3, —, 180 °C, — | 100–99 | 219 | |||||
| (2) Ni–V2O5–Y2O3/Al2O3, 3:1, 200 °C, — |
|||||||
| 8 | 1DMA2P |
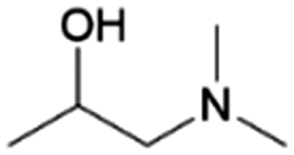
|
Acetol | (1) —, 2:1, 100 °C, EG |
100–90 | 220 | CCUS |
| (2) Ru/C, 2:1, 50 °C, EG |
|||||||
| Glyceraldehyde | Ni oxide, 8:1, 130 °C, MeOH |
100–14 | 213 and 214 | ||||
| Xylose | 100–13 | ||||||
| Fructose | 100–11 | ||||||
| Glucose | 100–5 | ||||||
| 9 | 1TriMEDA2P |
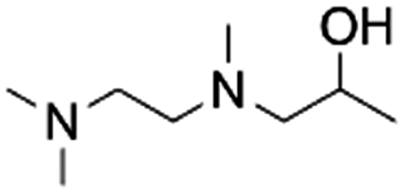
|
Acetol | (1) —, 2:1, 100 °C, EG |
100–89 | 220 | |
| (2) Ru/C, 2:1, 50 °C, EG |
|||||||
| 10 | 2A1P |
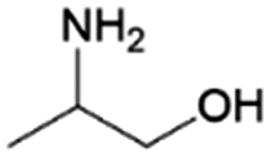
|
Acetol | Ru/ZrO2, 10:1, 65 °C, H2O |
100–26 | 203 | |
| Ni–Cu/Cr2O3, 4:1, 150 °C, H2O |
100–51 | 221 | |||||
| Ru–Ni/C, 11:1, 65 °C, H2O |
100–52 | 222 | |||||
| (1) —, 3:1, RT, H2O |
100–94 | 223 | |||||
| (2) Ni oxide, 3:1, 85 °C, H2O |
|||||||
| Cu/Cr2O3, 50:1, 210 °C, — |
100–47 | 216 | |||||
| 1,2 PG | Rh–In/C, 10:1, 180 °C, H2O |
38–26 | 217 | ||||
| Co/La3O4, 9:1, 160 °C, H2O |
69–89 | 218 | |||||
| Fructose | Ru–W2C/C, 80:1, 180 °C, H2O |
100–1 | 222 | ||||
| Cellulose | (1) W2C, —, 235 °C, H2O | 100–2 | 222 | ||||
| (2) Ru–W2C, 20:1, 65 °C, H2O |
|||||||
| 11 | 2iPA1P |
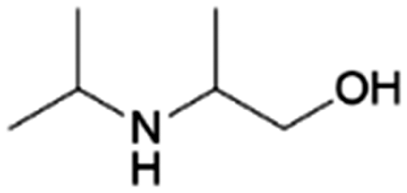
|
Acetol | Ni–Cu/Cr2O3, 4:1, 100 °C, EtOH |
100–64 | 221 | |
| 12 | 2TriMEDA1P |
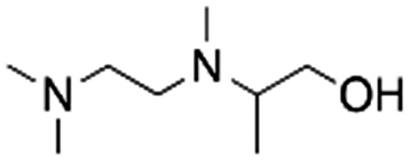
|
Acetol | Pd/C, 1:1, 50 °C, MeOH |
77–75 | 220 | |
| 13 | 2A1,3PG |
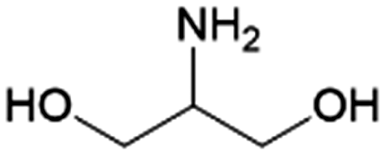
|
DHA | (1) —, 2.8:1, 20 °C, H2O–MeOH |
100–99 | 224 | |
| (2) RANEY® Ni, 2.8:1, 70 °C, H2O–MeOH |
|||||||
| RANEY® Ni, 10:1, 65 °C, H2O–MeOH |
100–91 | 225 | |||||
| 14 | 3A1,2PG |
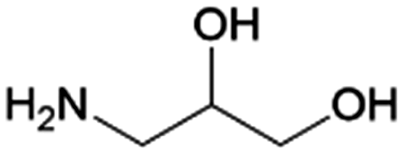
|
Glyceraldehyde | Ru/ZrO2, 10:1, 55 °C, H2O |
100–82 | 203 | |
| (1) —, 2.8:1, 20 °C, H2O |
100–95 | 224 | |||||
| (2) Pd/C, 2.8:1, 50 °C, H2O |
|||||||
| 15 | 1,2DA3P |
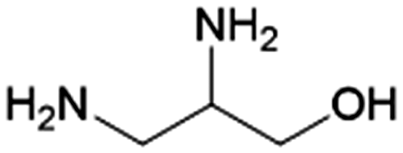
|
Glycerol | RANEY® Ni, 32:1, 200 °C, H2O |
91–21 | 226 | |
| Sugar alcohols | |||||||
| 16 | Glucamine |
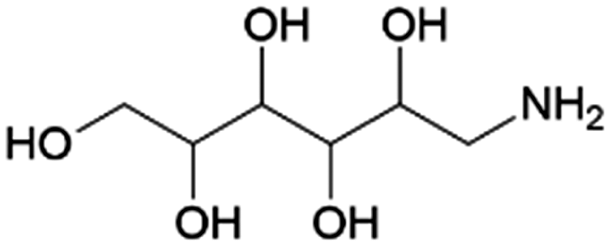
|
Glucose | RANEY® Ni, 5:1, 100 °C, MeOH |
100–26 | 227 | |
| 17 | DEGA |
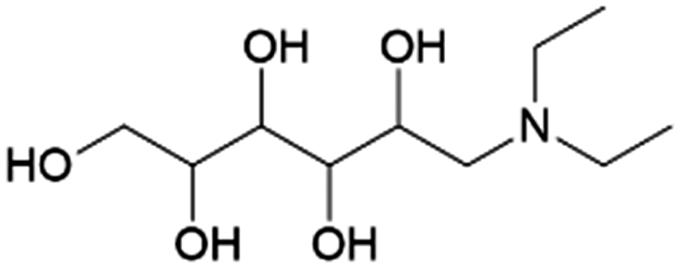
|
Glucose | Ru/C, 3:1, 45 °C, MeOH |
100–95 | 228 | |
| Alkyl polyamines | |||||||
| Ethylene polyamines | |||||||
| 18 | EDA |
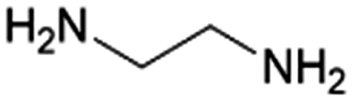
|
GA | Ru/ZrO2, 10:1, 75 °C, H2O |
100–2 | 203 | CCUS Epoxy |
| Co/MnO, 35:1, 100 °C, THF |
100–6 | 204 | |||||
| EG | (1) Ni–Cu–Mo/ZrO2, 15:1, 150 °C, — |
43–50 | 205 | ||||
| (2) Ru–Co/Al2O3, 15:1, 170 °C, — |
|||||||
| Re–Ru–Co/ZrO2, 20:1, 170 °C, — |
35–52 | 206 and 207 | |||||
| Co–Cu/Al2O3, 5:1, 190 °C, H2O |
42–46 | 208 and 209 | |||||
| Glycerol | RANEY® Ni, 32:1, 200 °C, H2O |
91–8 | 226 | ||||
| Glucose | RANEY® Ni, 32:1, 200 °C, H2O |
100–15 | 229 | ||||
| Sorbitol | 100–8 | ||||||
| 19 | TMEDA |
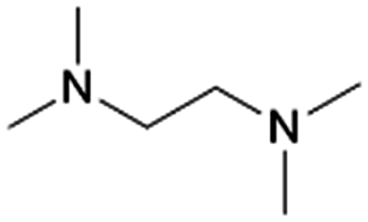
|
GA | (1) —, 12:1, 25 °C, EG |
100–91 | 199 | multiQAC |
| (2) Pd/C, 12:1, 130 °C, EG |
|||||||
| Ni/SiO2, lactic acid, 2.8:1, 130 °C, THF |
100–63 | 211 | |||||
| EG | Cu/Al2O3, 1:1, 230 °C, — |
91–21 | 212 | ||||
| Glyceraldehyde | Ni oxide, 8:1, 130 °C, MeOH |
100–4 | 213 and 214 | ||||
| Xylose | 100–34 | ||||||
| Fructose | 100–24 | ||||||
| Glucose | 100–66 | ||||||
| 20 | BHEDMEDA |
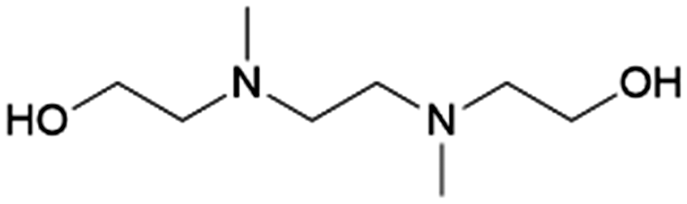
|
Glucose | (1) —, 70:1, 130 °C, MMEA |
100–92 | 213 and 214 | |
| (2) Ni oxide, 70:1, 130 °C, MMEA |
|||||||
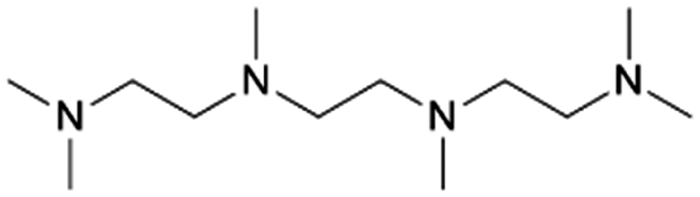
| 21 | HMTriETA |
|
GA | (1) —, oxalic acid, 4:1, 30 °C, EG |
100–82 | 215 | ATRP |
| (2) Pd/C, oxalic acid, 4:1, 100 °C, EG |
multiQAC | ||||||
| Propylene polyamines | |||||||
| 22 | 1,2PDA |
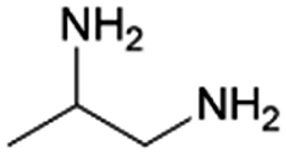
|
Acetol | Ru/ZrO2, 10:1, 65 °C, H2O |
100–10 | 203 | |
| Ru/C, 11:1, 65 °C, H2O |
100–5 | 222 | |||||
| 1,2PG | Co/Fe3O4, 9:1, 160 °C, H2O |
25–15 | 218 | ||||
| Glycerol | RANEY® Ni, 32:1, 200 °C, H2O |
91–22 | 226 | ||||
| Glucose | RANEY® Ni, 32:1, 200 °C, H2O |
100–13 | 229 | ||||
| Sorbitol | 100–12 | ||||||
| 23 | 1,2TMPDA |
|
Acetol | (1) —, 2:1, 100 °C, MeOH |
100–87 | 220 | |
| (2) Pd/C, 4:1, 100 °C, MeOH |
|||||||
| Glyceraldehyde | Ni oxide, 8:1, 130 °C, MeOH |
100–6 | 213 and 214 | ||||
| Xylose | 100–7 | ||||||
| Fructose | 100–6 | ||||||
| Glucose | 100–7 | ||||||
| 24 | 1,2HMPBEDA |
|
Acetol | (1) —, 2:1, 100 °C, MeOH |
100–90 | 220 | |
| (2) Pd/C, 4:1, 100 °C, MeOH |
|||||||
| 25 | PTriA |
|
Glycerol | Ni–Cu–Co/ZrO2, 32:1, 200 °C, H2O |
77–17 | 226 | |
| Aliphatic heterocyclic amines | |||||||
| 26 | PZ |
|
EG | Ni–Cu–Mo/ZrO2, 15:1, 150 °C, — |
43–7 | 205 | CCUS |
| Ru–Co/Al2O3, 15:1, 170 °C, — |
|||||||
| Re–Ru–Co/ZrO2, 20:1, 170 °C, — |
35–10 | 206 and 207 | |||||
| Co–Cu/Al2O3, 5:1, 190 °C, H2O |
42–18 | 208 and 209 | |||||
| 27 | DMPZ |
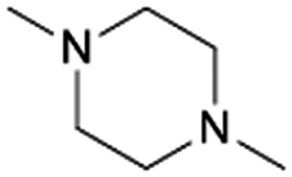
|
1,2PG | Rh–In/C, 10:1, 180 °C, H2O |
38–26 | 217 | |
| Amino ketones | |||||||
| 28 | 1-DMA-2-propanone |
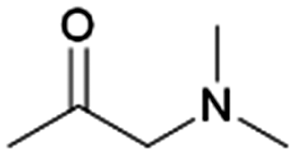
|
Acetol | —, 2:1, 100 °C, EG |
100–93 | 220 | |
| Glycerol | Cs2.5H0.5PMo12O40, 1.5:1, 250 °C, — |
47–70 | 230 | ||||
| H3PW12O40, 2.5:1, 300 °C, — |
100 - 33 | 231 | |||||
| Glyceraldehyde | Ni oxide, 8:1, 130 °C, MeOH |
100–8 | 213 and 214 | ||||
| Xylose | 100–4 | ||||||
| Fructose | 100–6 | ||||||
| Glucose | 100–1 | ||||||
| 29 | 1-TriMEDA-2-propanone |
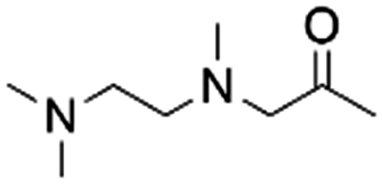
|
Acetol | —, 2:1, 100 °C, EG |
100–93 | 220 | |
| 30 | 1-DMA-3-propanone |
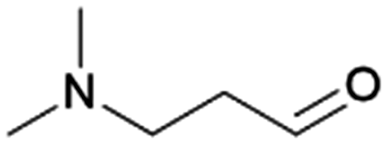
|
Glycerol | H6P2W18O62, 2.5:1, 300 °C, — |
100–81 | 231 | |
Other researchers have primarily focused on catalyst development to obtain high ethanolamine yields under conditions which diverge from the benchmark approach. BASF patented a gas-phase reductive amination reaction to overcome the limitations of working with aqueous solutions.210 In this gas-phase reaction, an aqueous GA solution was evaporated and reacted with gaseous DMA in the presence of a 56 wt% Cu/Al2O3 catalyst. Under investigated conditions (165 °C, ATS molar ratio of 12:1, H2-to-GA molar ratio of 56:1 and a catalyst loading of 0.17 kg L h−1), the DMEA, TMEDA and EG yields amounted to 74%, 4% and 1%, respectively.210 In their work, Liang et al. conducted the reductive amination of GA with aqueous NH3, for 12 h at 75 °C, 3 MPa H2, and an ATS molar ratio of 10:1. They achieved a 93% MEA yield by using a bifunctional 5 wt% Ru/ZrO2 hydrogenation catalyst. The remarkable activity of the Ru/ZrO2 catalyst stood out as Ru on other supports (e.g., activated carbon, Al2O3, SiO2) or other transition metals (e.g., Pd, Pt, Ir) on ZrO2 did not result in a comparably high yield. They reasoned that RuO2 species act as Lewis acid sites, facilitating imine formation by activating the carbonyl group of GA, whereas the metallic Ru0 species function as active hydrogenation sites to subsequently yield MEA.203,232 In another patent by BASF, researchers developed a Co/MnO catalyst for the reductive amination of GA with NH3 in THF. An 82% MEA yield was obtained by conducting the reaction for 8 h, at 100 °C, 8 MPa H2, and an excessive ATS molar ratio of 35:1.204 Both the work by Liang and the latter patent by BASF conducted the reductive amination with an excess of NH3 reactant to limit overalkylation of the amine product.203,204,232 Contrary, overalkylation does not occur when performing the reaction with secondary amines, which allows for the use of stoichiometric amounts of amine reactant.
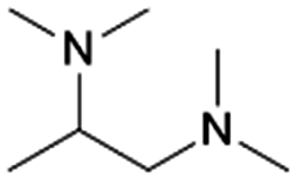
Vermeeren et al. recently formulated a set of guidelines for the selective formation of ethylene polyamines from GA by investigating the reductive amination with various diamines such as N,N,N′-trimethylethylenediamine (TriMEDA).215 In their work, the selectivity was shifted from the ethanolamine, obtainable in quantitative yields when employing the ethanolamine guidelines (vide supra), toward the ethylene polyamine product by three rational-design handles based on a profound understanding of the reaction network.233 These handles were developed by demystifying the reaction network via intermediate analysis. The first hemi-aminal (Scheme 4, I1) and the unsaturated polyamine (Scheme 4, I7) were identified as the predominant intermediates in the studied reaction system. Each selectivity control strategy successfully contributed to the fast and selective reaction of I1 to I7. The first handle consisted of kinetically and thermodynamically enhancing the dehydration reaction by smart solvent choice. Out of a comprehensive polar solvent screening, EG stood out as the solvent with the highest dehydration capacity. In the second handle, I7 formation was favored over hydrogenation of I3 and I4 by physically separating in time the amination reactions and hydrogenation, employing a one-pot-two-step method. The third handle involved the use of trace amounts of a carboxylic acid catalyst, already present as natural impurities in crude GA, to increase both the rate of dehydration and keto–enol tautomerization. Integrating these three handle in one general selectivity control strategy yielded ethylene polyamine products exceeding a yield of 80%. For example, carrying out the reductive amination of GA with the diamine TriMEDA in a one-pot-two-step approach in EG, with an ATS molar ratio of 4:1, 10 mol% oxalic acid, and a 5 wt% Pd/C hydrogenation catalyst, yielded 82% of the ethylene polyamine N,N,N′,N′,N′′,N′′,N′′′,N′′′-hexamethyltriethylenetetramine (HMTriETA, Table 1 entry 21). The first, intermediate, step was performed for 1 h at 30 °C under an inert atmosphere, whereas the second, hydrogenation, step was performed for 1 h at 100 °C and 3 MPa H2.215 The work by Faveere et al.199 and a patent by BASF211 both support the different control handles of this guideline for selective ethylene polyamine formation. Faveere et al. obtained a 91% yield of the polyamine N,N,N′,N′-tetramethylethylenediamine (TMEDA, Table 1 entry 19) by performing the reductive amination of GA with DMA in a one-pot-two-step reaction in EG. Contrary to Vermeeren et al., they performed the reaction with an ATS molar ratio of 12:1, for 5 h under an inert atmosphere during the intermediate step, and at an elevated temperature of 130 °C during the hydrogenation step.199 In the BASF patent, carboxylic acids were used to shift the selectivity from the ethanolamine DMEA to the ethylene polyamine TMEDA. The reductive amination of GA with DMA was performed in THF, for 1 h, at 130 °C, 17.5 MPa, with a 64 wt% Ni/SiO2 catalyst, and an ATS molar ratio of 2.8:1. Conducting the reaction in absence of lactic acid yielded 26% TMEDA and 76% DMEA, while the presence of 20 mol% lactic acid shifted the yields to 63% TMEDA and 23% DMEA.211
3.2.1.2. Ethylene glycol (EG). As the simplest symmetric diol, EG (Scheme 4, S1: R1 = R2 = H) is the ideal model component to study the hydrogen borrowing amination, i.e., dehydrogenation followed by reductive amination. Notably, most of the research up to now has essentially been focused on the dehydrogenation reaction, which is regarded as the rate-determining step, and centered around the search for an optimal catalytic dehydrogenation–hydrogenation system.234
Already since the 1960s, multiple articles and patents have been published on the gas- and liquid-phase amination of EG.235,236 For example in 1964, Moss et al. patented a Ni–Cu/Cr2O3 dehydrogenation–hydrogenation catalyst for the hydrogen borrowing amination of EG with aqueous NH3 in a fixed bed reactor.235 More recently, van Cauwenberge and co-workers patented a continuous two-step fixed bed process.205 The first fixed bed contained a Ni–Cu–Mo/ZrO2 dehydrogenation catalyst while the second bed was equipped with a Ru–Co/Al2O3 hydrogenation catalyst. The amination of EG with NH3 was performed at 150 °C (first reactor) and 170 °C (second reactor), with a total H2 pressure of 20 MPa, and an ATS molar ratio of 15:1. At an EG conversion of 43%, the product selectivity of EDA, MEA and PZ amounted to 50%, 29% and 7%, respectively.205 In two related patents, Heidemann and Becker developed several dehydrogenation–hydrogenation catalysts containing at least one or a combination of the following metals: Co, Ru, Ni, Cu or Sn dispersed on a ZrO2 or Al2O3 support.206,207 The hydrogen borrowing amination of EG with NH3 was tested in a fixed bed reactor at 150 °C, 17 MPa H2, and an ATS molar ratio of 10:1. At an EG conversion of 35%, the selectivities toward EDA, MEA and PZ on average amounted to 50%, 30% and 10%, respectively.206,207 Recently, An and co-workers quantitatively and qualitatively investigated the performance of various Co-based dehydrogenation–hydrogenation catalysts for the hydrogen borrowing amination of EG with aqueous NH3.208 They were able to ascribe the catalytic activity of these Co-based catalysts to the amount of acid and base sites of the different metal oxide catalyst supports as determined by NH3- and CO2-TPD. The catalytic activity (i.e., EG conversion) significantly increased when the metal oxide support possessed sufficient acid–base amphoteric sites (e.g., Al2O3, ZrO2, MgO) in comparison with metal oxide supports with a reduced number of base sites (e.g., SiO2, TiO2, Nb2O5). DFT calculations indicated that the base sites promote O–H bond cleavage in the substrate while the acid sites promote C–H bond cleavage, underlining the importance of this synergetic effect between acid–base sites. Co/Al2O3, as the most active catalyst, resulted in an EG conversion of 57% alongside an unselective formation of EDA (29% selectivity), MEA (25%) and PZ (23%). This optimized reaction was carried out with aqueous NH3, for 12 h, at 175 °C, 3 MPa H2, and an ATS molar ratio of 12:1.208 In a supplementary study, An et al. evaluated different Co-based bimetallic dehydrogenation–hydrogenation catalytic systems.209 Compared to Co/Al2O3, the presence of a second metal (e.g., Cu, Ni, Ru or Pt) improved the activity and selectivity of the ethylene polyamine product. It was hypothesized that Co and the second metal could separately facilitate the dehydrogenation of EG and the subsequent reductive amination. In addition to the choice of the second metal, other preparation conditions, such as metal loading and calcination and reduction temperature, strongly affected the catalytic performance. The Co–Cu/Al2O3 catalytic system demonstrated the highest activity and selectivity as it led to an EG conversion of 42% and an EDA, MEA and PZ selectivity of 46%, 23% and 18%, respectively. The optimized reaction was conducted with aqueous NH3, for 12 h, at 190 °C, 4 MPa H2, and an ATS molar ratio of 5:1.209
To the best of our knowledge, only one research article qualitatively addresses the product selectivity challenge in the hydrogen borrowing amination of EG.212 Runeberg et al. systematically screened four reaction conditions (i.e., temperature, ATS molar ratio, H2 pressure and addition of H2O) and assessed their effect on the selectivity by monitoring both the ethanolamine and ethylene polyamine product as well as the unsaturated polyamine intermediate (Scheme 4, I7). Moreover, they related these conditions and corresponding results to the proposed reaction mechanism. Of all screened conditions, only the temperature had a significant, positive effect on EG conversion. The ATS molar ratio, H2 pressure and presence of water all had no significant effect on EG conversion, suggesting that neither the amine nor H2 nor H2O are involved in the rate-determining step. This is in agreement with the general assumption that the dehydrogenation of EG is the rate-determining step. In contrast, these three reaction conditions did have a significant effect on product selectivity. Increasing the ATS molar ratio negatively affected the ethanolamine yield in favor of both the polyamine and I7. In the absence of H2 pressure, the reaction favored I7 formation. Increasing the H2 pressure positively affected the yields of both the ethanolamine and polyamine at the expense of I7. Finally, the addition of H2O to the reaction system steered the product selectivity in favor of the ethanolamine product.212 All these findings unambiguously support the selectivity control handles that were applied in the guidelines for GA amination (vide supra).
3.2.1.3. Acetol. Acetol (Scheme 4, S2: R1 = H, R2 = CH3) can be regarded as a potential model substrate for asymmetric α-hydroxy ketone substrates. As an asymmetric oxygenate, acetol possesses the ability to undergo an aldose–ketose isomerization reaction forming the α-hydroxy aldehyde 2-hydroxypropanal (lactaldehyde).237,238 Interestingly, the amination of acetol or lactaldehyde would lead to the same α-propylene polyamine but result in two different isomeric α-propanolamine products. To date, the reductive amination of acetol has gained limited research interest and has mainly been focused on the formation of the acetol-derived α-propanolamines.
In their patent, Cavitt and co-workers studied the formation of the α-propanolamine 2-amino-1-propanol (2A1P, Table 1 entry 10) by the reductive amination with aqueous NH3 and a Ni–Cu/Cr2O3 hydrogenation catalyst.221 They obtained a product yield of 51% by carrying out the reaction for 1 h, at 150 °C, 3.4 MPa H2, and an ATS molar ratio of 4:1. Furthermore, they performed the reductive amination of acetol and isopropylamine both in H2O and EtOH as the solvent. The reaction in EtOH significantly outperformed the reaction in H2O as the corresponding α-propanolamine (i.e., 2-isopropylamino-1-propanol, Table 1 entry 11) yield in EtOH and H2O amounted to 64% and 33%, respectively. Both reactions were performed for 1 h, at 100 °C, 20.6 MPa H2, and an ATS molar ratio of 4:1. Throughout the patent, no α-propylene polyamine or other amine products were reported.221 Liang et al. reported a low 2A1P yield in the reductive amination of acetol with aqueous NH3, although their RuZrO2 catalyst was very selective toward the formation of MEA in the reductive amination of GA. Under optimized conditions (6 h, 65 °C, 3 MPa H2, ATS molar ratio of 10:1), they achieved a 2A1P and 1,2-propylenediamine (1,2PDA, Table 1 entry 22) yield of 26% and 10%, respectively.203 Recently, Boulos et al. studied the use of bimetallic Ru–Ni/C catalysts in the reductive amination of acetol with aqueous NH3.222 Compared to Ru/C (5 wt%), using a Ru–Ni/C (4.5 wt% Ru and Ni) enhanced the 2A1P yield from 37% to 52% at full acetol conversion under optimized reaction conditions (3 h, 65 °C, 6 MPa H2, ATS molar ratio of 11:1). Next to 2A1P, this optimized reaction resulted in a 1,2PG yield of 28%.222 In their granted patent, P&G reported a highly selective two-step reductive amination process for the production of 2A1P.223 In the first step, aqueous NH3 was dropwise added to a reactor containing acetol while stirring at room temperature for 90 minutes until an ATS molar ratio of 3:1 was obtained. In the second step, the reactor was loaded with a Ni oxide on kieselguhr hydrogenation catalyst, pressurized with H2 to 15 MPa and heated to 85 °C. In this way, a 94% 2A1P yield was obtained after the reaction.223 Trégner et al. studied the reductive amination of acetol to 2A1P in a gas-phase continuous fixed bed reactor with a Cu/Cr2O3 catalyst.216 In the optimized reaction (WHSV = 0.078 h−1, 210 °C, molar ratio of acetol:H2:NH3 of 1:50:50) a 2A1P yield of 47% was obtained at full acetol conversion. In addition, they identified numerous by-products via GC-MS and proposed their corresponding reaction mechanism. Under these conditions, the major by-products were heterocyclic PZ-related and aromatic amines. Interestingly, they also reported trace amounts (∼2% yield) of 1-amino-2-propanol (1A2P, Table 1 entry 7), the α-propanolamine product that originates from lactaldehyde, the isomer of acetol.216 In line with their work on GA,215 Vermeeren et al. recently used the same bottom-up methodology to qualitatively study the selectivity control challenge in the catalytic reductive amination of acetol with TriMEDA as the aminating agent.220 This methodology, consisting of intermediate identification and control handle evaluation, ultimately resulted in three distinct control strategies targeting the two isomeric alkanolamines: N,N,N′-trimethyl-2-ethylenediamino-1-propanol (2TriMEDA1P, Table 1 entry 12) and N,N,N′-trimethyl-1-ethylenediamino-2-propanol (1TriMEDA2P, Table 1 entry 9), analogues to 2A1P and 1A2P, respectively, and the propylene polyamine product N,N,N′,N′′,N′′′,N′′′-hexamethyl-1,2-propylene-bis(ethylenediamine) (1,2HMPBEDA, Table 1 entry 24). The first control strategy, targeting 2TriMEDA1P, encompassed catalyst selection, solvent choice and reaction temperature as the most influential control handles. After fine-tuning, this strategy obtained a product selectivity up to 75%. Notably, the two other strategies, targeting 1TriMEDA2P and 1,2HMPBEDA, harnessed the formation of a highly stable α-amino ketone intermediate, N,N,N′-trimethyl-1-diamino-2-propanone (1-TriMEDA-2-propanone, Table 1 entry 29) in a one-pot-two-step reaction configuration. In both strategies, the reaction temperature proved to be the crucial control handle in the first process step to achieve intermediate selectivities exceeding 90%. Subsequently in the second process step, product selectivity could be consciously steered toward 1TriMEDA2P or 1,2HMPBEDA by judicious hydrogenation catalyst selection. In this way, these two strategies resulted in excellent 1TriMEDA2P and 1,2HMPBEDA selectivity, amounting to 95% and 90%, respectively. These two one-pot-two-step strategies were successfully validated by expanding the reactant and substrate scope. As a proof of concept, the one-pot-two-step polyamine strategy could be modified to accommodate the formation of high-value asymmetric polyamines consisting of different vicinal amino groups. This proof of concept elucidated the importance of both the relative reactivity of the two amine reactants and the stability of the formed α-ketone intermediate.220
3.2.1.4. 1,2-Propylene glycol (1,2PG). The asymmetric α-diol 1,2PG (Scheme 4, S1: R1 = CH3, R2 = H) has received limited research interest, despite its potential significance. In theory, the dehydrogenation of 1,2PG can yield acetol (α-hydroxy ketone) or lactaldehyde (α-hydroxy aldehyde). In practice, most research has still targeted the formation of the acetol-derived α-propanolamine. However, it can be noticed that in general the selectivity toward the lactaldehyde-derived α-propanolamine increases by using 1,2PG instead of acetol. Analogous to EG amination research, all 1,2PG hydrogen borrowing amination studies essentially focus on the development of a suitable dehydrogenation–hydrogenation catalytic system.
Takanashi et al. studied the hydrogen borrowing amination of 1,2PG with aqueous NH3 in the presence of a 5 wt% Rh–In/C catalyst.217 With this catalytic system under applied conditions (24 h, 180 °C, 5 MPa H2, ATS molar ratio of 10:1), the two isomeric α-propanolamines, namely 2A1P (42% selectivity) and 1A2P (47%), were obtained as major products and dimethylpiperazine (DMPZ, Table 1 entry 27) (10%) as the by-product at a 1,2PG conversion of only 11%. The Rh/C catalyst, in the absence of In, was unable to convert 1,2PG while the combination of the individually supported metals, Rh/C and In/C, resulted in a reduced 1,2PG conversion of 6% with similar product distribution. They reasoned that the presence of In enhanced the resistance to catalyst deactivation during the dehydrogenation step. By extending the reaction time from 24 h to 160 h with the Rh–In/C catalyst, the 1,2PG conversion increased to 38%. In addition, extending the reaction time had a significant effect on product selectivity. While the selectivity of 1A2P remained unchanged (42%), the selectivity of 2A1P decreased to 26% in favor of DMPZ (26%).217 Niemeier et al. investigated Ru/C as an effective catalytic system in hydrogen borrowing amination of various alcohols. Although the catalyst was active for some alcohol substrates, only a 1,2PG conversion of 10% was obtained by performing the reaction with aqueous NH3 for 6 h, at 170 °C, 1 MPa H2, and an ATS molar ratio of 10:1. No distinctive product analysis was performed.239 Yue et al. evaluated the activity and selectivity of various Co-based dehydrogenation–hydrogenation catalysts in the hydrogen borrowing amination of 1,2PG with aqueous NH3.218 Next to the formation of the acetol-derived α-propanolamine 2A1P, they also monitored the formation of the lactaldehyde-derived α-propanolamine 1A2P and the diamine 1,2PDA. During the initial catalyst screening (6 h, 160 °C, N2 atmosphere, ATS molar ratio of 9:1), three catalysts displayed promising activity. Co/Al2O3, Co/La3O4 and Co/Nb2O5 achieved 1,2PG conversions of 50%, 40% and 38%, respectively. Although Co/Al2O3 exhibited the highest activity, its selectivity toward 2A1P (54%) was moderate, while 1A2P (21%) and 1,2PDA (13%) were also formed at lower selectivities. In contrast, Co/Nb2O5 demonstrated comparable selectivities toward both α-propanolamine isomers 2A1P (40%) and 1A2P (36%), with a minor formation of 1,2PDA (14%). Notably, Co/La3O4 exhibited a high selectivity toward 2A1P (75%), with significantly lower selectivities toward 1A2P (18%) and 1,2PDA (3%). After finetuning the Co/La molar ratio, the 2A1P selectivity reached an optimum at 89%, together with a 1A2P selectivity of 9%, at a 1,2PG conversion of 69%. No further efforts were made to elucidate the role of the support in these selectivity differences. Without additional experimental insights, it is impossible to pinpoint if this support-induced selectivity was established during the initial dehydrogenation or later in the isomerization step.218 A highly selective and quantitative two-step 1,2PG hydrogen borrowing amination process was patented by Shujie and co-workers.219 In contrast to other research, this process targeted the formation of the lactaldehyde-derived α-propanolamine 1A2P. In the first step, a Cu–Pd–Bi2O3–In2O3/Al2O3 catalyst was used to selectively dehydrogenate 1,2PG into lactaldehyde. In the second step, the formed lactaldehyde subsequently underwent reductive amination into the corresponding 1A2P product in the presence of a Ni–V2O5–Y2O3/Al2O3 hydrogenation catalyst. Under optimized first-step reaction conditions (SV = 2 L h−1 Lcat−1, 180 °C), 1,2PG was fully converted into lactaldehyde. Under optimized second-step reaction conditions (SV = 6 L h−1 Lcat−1, 200 °C, H2:lactaldehyde molar ratio of 2:1, ATS molar ratio of 3:1), lactaldehyde, in its turn, was fully converted into 1A2P. Furthermore, the role of each metal oxide present in both catalysts was clarified through a series of comparative experiments. In the dehydrogenation catalyst, Cu was identified as the primary active site responsible for the dehydrogenation capacity, whereas Pd enhanced the catalyst activity. Additionally, the presence of Bi2O3 and In2O3 positively affected the selectivity toward lactaldehyde. In the hydrogenation catalyst, Ni contained the hydrogenation capacity while the presence of the metal oxides V2O5 and Y2O3 beneficially increased the selectivity toward 1A2P.219
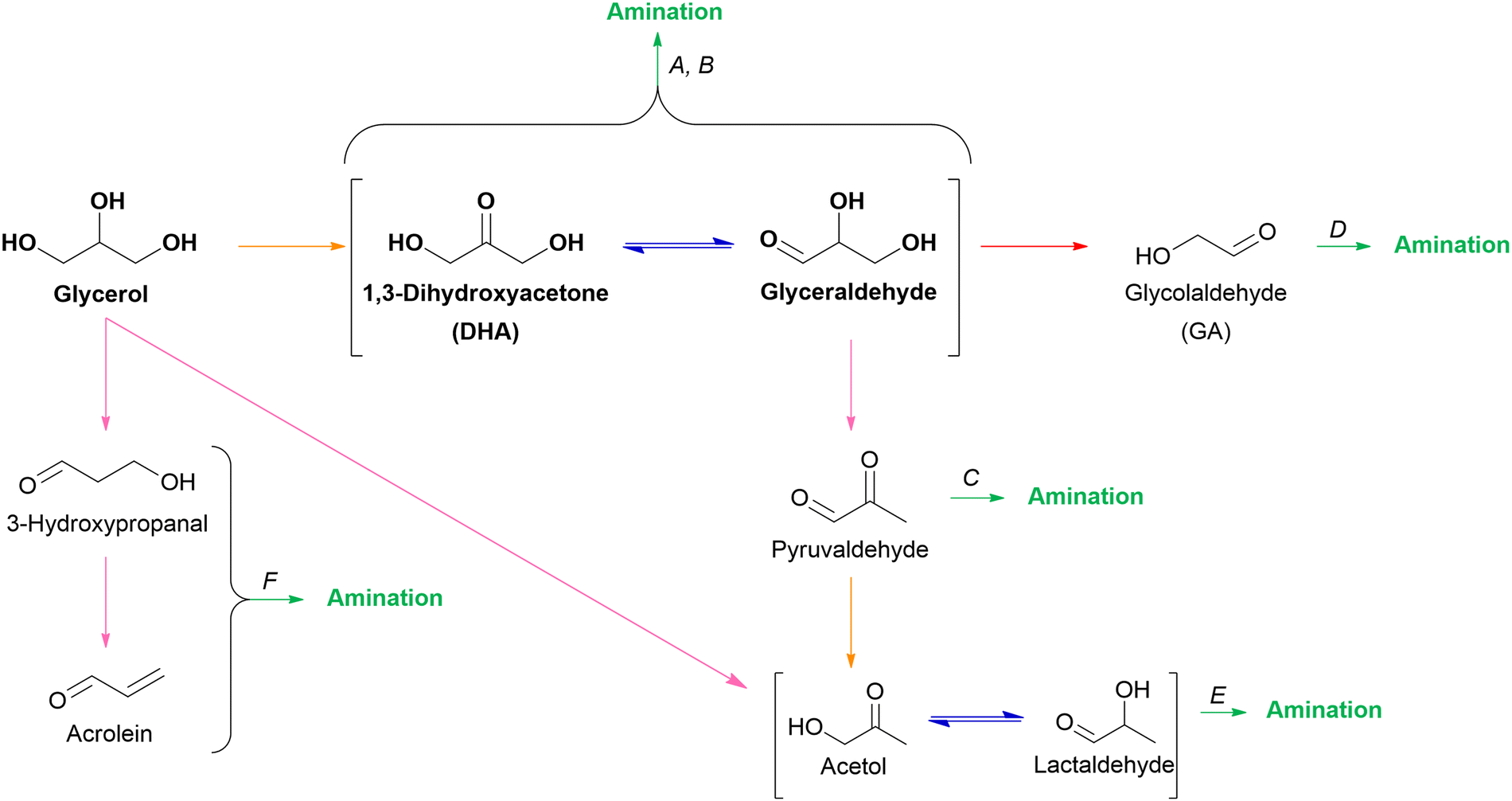 | ||
| Scheme 5 Overview of different pre-amination pathways in O3 oxygenate reductive amination. Each elementary step is represented by a color: amination (green), isomerization (blue), retro-aldol condensation (red), dehydration (pink) and (de)hydrogenation (orange). | ||
3.2.2.1. Glyceraldehyde. In theory, four different amination routes are possible from the α-hydroxy aldehyde glyceraldehyde (Scheme 5). The first route involves the direct reductive amination of glyceraldehyde's available carbonyl group, leading to a product containing one up to three amino groups (Scheme 5, route A). In the second route, glyceraldehyde isomerizes to DHA which can subsequently undergo reductive amination (Scheme 5, route B). In the third and fourth routes, amination is preceded by a cleavage reaction; dehydration (Scheme 5, route C) and retro-aldol condensation (Scheme 5, route D), respectively. Dehydration of glyceraldehyde yields pyruvaldehyde and acetol, whereas retro-aldol condensation leads to GA, which are all suitable amination substrates (vide supra). To date, research efforts concerning the reductive amination of glyceraldehyde have been limited.
Two authors have targeted the first route (Scheme 5, route A), namely the direct reductive amination of glyceraldehyde. In addition to GA and acetol, Liang et al. also explored glyceraldehyde as a substrate to assess the effectiveness of their Ru/ZrO2 reductive amination catalyst. In an optimized one-step reaction with aqueous NH3 (6 h, 55 °C, 2 MPa H2, ATS molar ratio of 10:1), they reported a yield of 82% for the diolamine 3-amino-1,2-propylene glycol (3A1,2PG, Table 1 entry 14).203 A patent by Merck outlined a two-step reductive amination process for the selective formation of 3A1,2PG.224 In the first step, aqueous NH3 is gradually added to an aqueous glyceraldehyde solution at a reduced temperature (20 °C) under continuous stirring for 1 h until an ATS molar ratio of 2.8:1 is obtained. In the second step, hydrogenation conditions are introduced (6.5 MPa H2, 10 wt% Pd/C hydrogenation catalyst) and the reaction is further conducted at 50 °C for an additional 40 minutes, resulting in a notable 95% yield of 3A1,2PG.224
In the context of their glucose amination research (vide infra), Pelckmans et al. extended the substrate scope in one experiment to glyceraldehyde.213 Noteworthy, this experiment solely focused on the amine products obtained via both cleavage routes (Scheme 5, routes C–D). They performed the reductive amination of glyceraldehyde with DMA and a commercial Ni oxide catalyst (Ni-6458P, Engelhard) in MeOH, for 1 h, at an elevated temperature (130 °C), 8.5 MPa H2, and an ATS molar ratio of 8:1. Under these conditions, product yields indicated that dehydration (Scheme 5, route C) was more prominent than cleavage via retro-aldol condensation (Scheme 5, route D). The dehydration-derived amination products, including the diamine N,N,N′,N′-tetramethyl-1,2-propylenediamine (1,2TMPDA, Table 1 entry 23), the α-propanolamine N,N-dimethyl-1-amino-2-propanol (1DMA2P, Table 1 entry 8), and the amino ketone intermediate (Scheme 4, I4) N,N-dimethyl-1-amino-2-propanone (1DMA-2-propanone, Table 1 entry 28), were formed in yields of 6%, 14%, and 8%, respectively. The formation of 1DMA2P and 1DMA-2-propanone illustrate the selectivity preference of the amine reactant to perform the nucleophilic addition at the aldehyde carbon of pyruvaldehyde rather than at its ketone carbon. The retro-aldol-derived products, namely the diamine TMEDA and the ethanolamine DMEA, were formed in trace amounts, with yields of 5% and 1%, respectively.213
3.2.2.2. 1,3-Dihydroxyacetone (DHA). The O3 α-hydroxy ketone DHA can potentially undergo two amination routes. DHA can undergo direct reductive amination at its carbonyl group (Scheme 5, route A), or it can first isomerize to glyceraldehyde before proceeding with its corresponding amination routes (Scheme 5, route B). The first route has been targeted in the scarce literature available.
Complementary to the two-step process for glyceraldehyde reductive amination, the patent by Merck likewise reported a two-step process for DHA reductive amination under similar conditions.224 In the first step, aqueous NH3 was gradually added to a MeOH solution containing DHA at 20 °C until an ATS molar ratio of 2.8:1 was obtained. Hydrogenation conditions (10 MPa H2, RANEY® Ni hydrogenation catalyst) were introduced in the second step and the reaction was performed at 70 °C for an additional 40 minutes. The diethanolamine 2-amino-1,3-propylene glycol/serinol (2A1,3PG, Table 1 entry 13) was formed in a quantitative yield of 99%.224 In another patent, Hegde and co-workers performed the same reaction using a one-step process.225 During a period of 3 h, a DHA–MeOH mixture was gradually added to the reaction mixture containing aqueous NH3 and RANEY® Ni, operating at 65 °C and 1.7 MPa H2, until an ATS molar ratio of 10:1 was achieved. The reaction was then prolonged for an additional 3 h. This one-step approach resulted in an excellent 2A1,3PG yield of 91% with the overalkylated bis adduct as a minor by-product (7%).225
3.2.2.3. Glycerol. As an α-triol substrate, glycerol requires an activation step before amination can occur. In this regard, two distinctive activation routes can be applied. In the first route, glycerol is dehydrogenated to form its corresponding aldehyde (i.e., glyceraldehyde) or ketone (i.e., DHA) substrate, similar to O2 diol activation. In the second route, glycerol is activated via dehydration, leading to the formation of acetol (Scheme 5, route E) or 3-hydroxypropanal (Scheme 5, route F). Both routes have been explored as viable amination strategies in the literature.
In the first activation strategy, glycerol is dehydrogenated into glyceraldehyde/DHA in the presence of a dehydrogenation catalyst. To promote the initial dehydrogenation step, this strategy is typically carried out at elevated temperatures which additionally also favor cleavage reactions such as retro-aldol condensation and dehydration. Therefore, although glyceraldehyde and DHA are susceptible to undergo reductive amination (vide supra), most of the formed amine products via this strategy are obtained by the amination of smaller oxygenates such as GA and pyruvaldehyde/acetol. In a patent by BASF, Ernst et al. performed the hydrogen borrowing amination of glycerol with aqueous NH3 in the presence of a number of dehydrogenation–hydrogenation catalysts (e.g., RANEY® Ni, RANEY® Co, Cu–Ni–Co/ZrO2).226 At standardized conditions (200 °C, 2 MPa H2 while heating and 20 MPa H2 when attaining the desired reaction temperature, and an ATS molar ratio of 32:1) resulting in full glycerol conversion, the amination reactions led to a plethora of amine products. On the one hand, they monitored O3-derived amines such as the propanoldiamine 1,2-diamino-3-propanol (1,2DA3P, Table 1 entry 15) and the triamine 1,2,3-propylenetriamine (PTriA, Table 1 entry 25). On the other hand, they also observed O2-derived amines such as EDA, 1,2PDA and three PZ derivatives. In general, all tested catalytic systems favored the formation of PZ and its derivatives at the expense of the other amine products. For example, the reaction in the presence of a commercial RANEY® Ni catalyst resulted in three major products after 36 h: 1,2DA3P at a 21% yield, 1,2PDA at 22%, and the PZ derivatives at 26%. By extending the reaction time to 48 h, the yield of the PZ derivatives increased to 59% at the expense of 1,2DA3P and 1,2PDA.226 In a similar concept, Du et al. studied the reductive amination of glycerol with aqueous NH3 in the presence of 5 wt% Ru/C as the dehydrogenation–hydrogenation catalyst.240 However, the main objective of this research was not to form O3- and O2-derived amines, but rather simple alkylamines such as MMA, N-monoethylamine and N-monopropylamine. In their proposed reaction mechanism, these alkylamines result from excessive hydrogenation and dehydration of initially formed O3 and O2 substrates and amines. As a consequence, these harsh reaction conditions additionally led to undesirable glycerol hydrogenolysis products such as 1,2PG, EG and MeOH. Under optimized conditions (48 h, 200 °C, 10 MPa H2, and an ATS molar ratio of 6:1), the selectivity toward these alkylamines, PZ derivates and hydrogenolysis products were 51%, 8% and 19%, respectively, at full glycerol conversion.240
In the second activation strategy, glycerol is dehydrated into either acetol or 3-hydroxypropanal, which can subsequently undergo amination. In a study conducted by Safariamin et al., the dehydrative amination of glycerol was carried out using DMA as the reactant in a fixed bed reactor equipped with a silica-supported heteropolyacid catalyst (Cs2.5H0.5PMo12O40).230 This research primarily targeted the amino ketone components (Scheme 4, I4) derived from acetol and 3-hydroxypropanal, as the catalyst lacked any inherent hydrogenation capacity. The experimental results, however, demonstrated only the occurrence of 1DMA-2-propanone, the α-amino ketone derived from the acetol-isomer lactaldehyde. These findings suggest that the catalyst, under applied conditions, facilitated both the selective dehydration of the primary hydroxyl groups of glycerol and the isomerization of acetol to lactaldehyde. Under optimized conditions (reactant flow of 10 L h−1, 250 °C, and an ATS molar ratio of 1.5:1) a 1DMA-2-propanone selectivity of 70% was achieved at a glycerol conversion of 47%.230 Ding et al. further optimized this strategy and elaborated on the mechanism as they explored the same reaction with two different Zr-MCM-41-supported heteropolyacid catalysts (i.e., H3PW12O40 and H6P2W18O62).231 Next to 1DMA-2-propanone, the reaction also produced N,N-dimethyl-1-amino-3-propanone (1DMA-3-propanone, Table 1 entry 30), the β-amino ketone derived from 3-hydroxypropanal. Based on the presence of trace amounts of acrolein, the researchers proposed an alternative mechanism for the formation of 1DMA-3-propanone. According to this pathway, glycerol undergoes two consecutive dehydration reactions resulting in acrolein. Subsequently, acrolein preferentially undergoes hydroamination at its unsaturated carbon site rather than amine addition at its carbonyl group, yielding 1DMA-3-propanone. Under optimized reaction conditions (GHSV = 3 h−1, 300 °C, ATS molar ratio of 2.5:1, full glycerol conversion), both heteropolyacid catalysts favored the formation of 1DMA-3-propanone over 1DMA-2-propanone. In the dehydrative amination catalyzed by the heteropolyacid H3PW12O40, the selectivities of 1DMA-3-propanone and 1DMA-2-propanone were 62% and 33%, respectively. The use of the heteropolyacid H6P2W18O62 additionally enhanced the product distribution in favor of 1DMA-3-propanone. In this reaction, the selectivities of 1DMA-3-propanone and 1DMA-2-propanone reached 81% and 11%, respectively. Ding et al. related this difference in selectivity to the relative amount of Brønsted acid sites in both catalysts. Glycerol, so they postulated, would preferentially be dehydrated into 3-hydroxypropanal on Brønsted acid sites, whereas glycerol dehydration would favorably produce acetol on Lewis acid sites. In accordance with this hypothesis, the heteropolyacid with relatively more Brønsted acid sites resulted in a higher selectivity toward 1DMA-3-propanone.231 Dai et al. reported a glycerol amination process that seems to balance between the two activation strategies.241 In this process, glycerol is reacted with morpholine into the α-amino ketone component 1-morpholine-2-propanone (Scheme 4, I4) using a Cu–Ni/AlOx catalyst in the presence of K2CO3. Although the authors provided no mechanistic insights, the formation of the amino ketone product indicates that glycerol has undergone dehydrative C–C cleavage during the reaction, directly or after dehydrogenation into glyceraldehyde. The presence of both the heterogeneous Cu–Ni/AlOx catalyst and the inorganic base K2CO3 were required to obtain excellent activity and selectivity. At a morpholine conversion of 91%, a quantitative selectivity of 99% was achieved by conducting the reaction in 1,4-dioxane for 12 h, at 150 °C, under an Ar atmosphere, and a sub-stoichiometric ATS ratio of 0.2:1. Performing the same reaction in absence of K2CO3 reduced both the conversion and selectivity to 80% and 41%, respectively.241
The direct reductive amination of glucose, resulting in glucamine-based products, has been studied throughout time.242,243 In 1940, Wayne and Adkins reported the straightforward formation of glucamine (Table 1 entry 16) from glucose with a NH3–MeOH solution in the presence of a RANEY® Ni hydrogenation catalyst.227 An isolated glucamine yield of 26% was obtained when conducting the reaction for 1 h, at 100 °C, 15 MPa H2, and an ATS molar ratio of 5:1. They attributed this poor yield to the purification step as they commented that the difficulty in obtaining glucamine was not in the amination reaction but solely in the isolation of the product.227 Recently, Seddig et al. obtained a N,N-diethylglucamine (DEGA, Table 1 entry 17) yield of 95% in the reductive amination of glucose with N,N-diethylamine.228 This excellent yield was obtained by performing the reaction in MeOH for 18 h, at 45 °C, 9 MPa H2, an ATS molar ratio of 3:1, and a 5 wt% Ru/C hydrogenation catalyst. Carrying out the reaction in H2O instead of MeOH drastically slowed down the reaction and additionally led to a significantly diminished DEGA yield of 35%.228
The other approach, referred to as reductive aminolysis in the literature, involves the formation of shorter aliphatic amine products. BASF has patented an aminolysis process that applies to both sorbitol and glucose.229 By employing a dehydrogenation–hydrogenation catalyst (e.g., RANEY® Ni, Cu–Ni–Co/ZrO2) and aqueous NH3, the substrates were converted into the diamines EDA and 1,2PDA, as well as various heterocyclic PZ-related amines. Under tested conditions (32 h, 200 °C, 20 MPa, and an ATS molar ratio of 32:1) and full glucose conversion, the amination of glucose catalyzed by RANEY® Ni resulted in equivalent yields of EDA and 1,2PDA, 15% and 13%, respectively, alongside a cumulative yield of 23% for the PZ-derivatives. The same reaction conducted with sorbitol yielded a similar product distribution, with yields of EDA, 1,2PDA and PZ-derivatives amounting to 8%, 12% and 20%, respectively.229 In their work, Boulos et al. expanded the substrate scope from acetol to fructose.222 They explored the reductive aminolysis of fructose with aqueous NH3 using a bimetallic Ru–W2C/C (7.5 wt% Ru and 36 wt% W2C). Under tested reaction conditions (3 h, 180 °C, 7.5 MPa H2, an ATS molar ratio of 80:1) full fructose conversion yielded a wide range of amine and oxygenate products in trace amounts: MEA (5%), EDA (1%), 2A1P (1%), 1,2PDA (1%), EG (1%), and 1,2PG (1%).222 The fundamental mechanistic insights of this aminolysis reaction were clarified by Pelckmans and co-workers (Scheme 6).213,214,244,245 They reasoned that the retro-aldol C–C scissions in the substrate are induced by the amine reactant itself, mimicking the mechanism of (retro-)aldolase enzymes. To start, the amine reactant performs a nucleophilic attack on the carbonyl of the substrate, forming a zwitterionic iminium intermediate through dehydration. Subsequently, two distinct reaction pathways can occur. On the one hand, this iminium intermediate can undergo intramolecular proton transfer, yielding an amino sugar alcohol product upon hydrogenation of the imine/enamine intermediate. On the other hand, this iminium intermediate can undergo amine-facilitated retro-aldol condensation, forming a C2-enamine intermediate and a smaller α-hydroxy carbonyl. The latter component can re-enter the reaction as a substrate, whereas the C2-enamine proceeds via the established O2 amination reaction pathway (Scheme 4), resulting in an ethanolamine or ethylene polyamine product. The proposed pathway was verified via theoretical DFT calculations and supported by experimental data. For example, carbohydrate hydrogenolysis was experimentally excluded as a cause of C–C scission as sorbitol was not converted under applied reaction conditions.213,244 Advantageously, the amine-facilitated retro-aldol condensation can be carried out at temperatures significantly lower than the typical retro-aldol temperatures of around 200 °C. As a result, this approach mitigates numerous temperature-induced side reactions. Notably, besides the formation of these retro-aldol-derived C2 amines, so-called C3 amines, such as the diamine 1,2TMPDA and the α-propanolamine 1DMA2P, were also observed. Based on their structure, these C3 amines essentially originate from the amination of pyruvaldehyde or acetol species that can be formed in situ via the retro-aldol condensation of fructose or xylose (as depicted in Scheme 1). Although the reaction mechanism remains unclear, the authors still provided some preliminary findings. In theory, glucose–fructose isomerization prior to amination could be a major pathway. However, the reductive aminolysis of glucose and fructose led to the same product distribution of C2 and C3 amines, indicating that fructose is not the main precursor of the C3 amines. Approximately 75% of formed aliphatic amines were C2 amines when using both substrates under reductive aminolysis conditions (1 h, 130 °C, 7.5 MPa H2, Ru/C hydrogenation catalyst, aqueous DMA, ATS molar ratio of 12:1). Alternatively, the reductive aminolysis of xylose did result in a 50–50 distribution of C2 and C3 amines, which strongly relates with its retro-aldol product distribution. Noteworthy, diamines were preferentially formed over alkanolamines in both C2 and C3 amines for each substrate. This phenomenon is most likely attributed to the retro-aldol requirements, such as an elevated reaction temperature and a high ATS molar ratio.214 Furthermore, the choice of solvent significantly affected the product yields and selectivity. Using MeOH instead of H2O as the solvent enhanced the total yield of all amine products and steered the selectivity more in favor of C2 amines relative to C3 amines. All these insights were combined in an optimization experiment (1 h, 130 °C, 8.5 MPa H2, commercial Ni oxide hydrogenation catalyst, 2 M DMA MeOH solution, ATS molar ratio of 8:1), yielding 66% of the C2 diamine TMEDA and minor amounts of the C3 diamine 1,2TMPDA (7%), the C2 ethanolamine DMEA (4%) and the C3 α-propanolamine 1DMA2P (5%). Additionally, the solvent-free reductive amination of glucose with MMEA, serving both as amine reactant and solvent, (2 h, 130 °C, 8.5 MPa H2, commercial Ni oxide hydrogenation catalyst) yielded a remarkable 84% product yield of the C2 polyamine N,N′-bis(2-hydroxyethyl)-N,N′-dimethylethylenediamine (BHEDMEDA, Table 1 entry 20). This notably high yield was attributed to the formation of a stable, cyclic C2-enamine adduct, namely a heterocyclic 5-membered oxazolidinic compound, favoring the subsequent formation of BHEDMEDA through hydrogenation.213
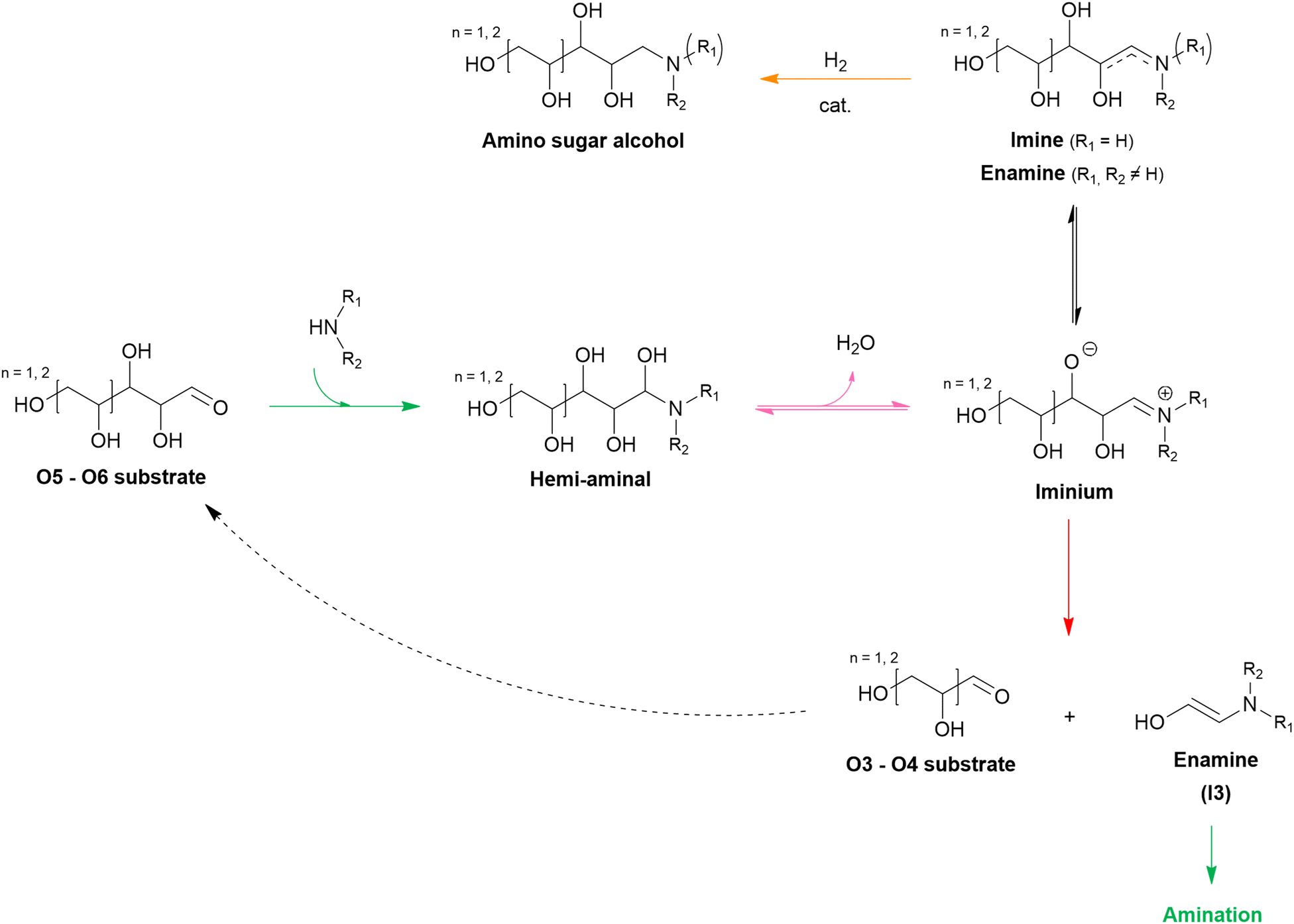 | ||
| Scheme 6 General reaction mechanism of the reductive amination of O5 and O6 substrates. Each elementary key reaction is represented by a color: amination (green), retro-aldol condensation (red), dehydration (pink) and (de)hydrogenation (orange). | ||
:1).203 The overall yield, from cellulose to MEA, amounted to 10%. Boulos et al. performed cellulose valorization with 30 wt% W2C/C in hot water (235 °C) for 30 minutes and 4 MPa H2, targeting acetol formation. An acetol yield of 12% was obtained after purification. Reductive amination of this recovered acetol with aqueous NH3 using a Ru–W2C/C (7.5 wt% Ru and 36 wt% W2C) catalyst yielded 18% 2A1P, resulting in an overall yield, from cellulose to 2A1P, of 2%.222
4. From bio-based aliphatic amines to sustainable applications
Aliphatic amines are omnipresent in a plethora of applications due to their reactivity and structural diversity. This is illustrated by considering three diverse domains in which aliphatic amines play a pivotal role as a catalyst or an active component. These three domains cover all different types of aliphatic amines as they each require distinct functional features of the amine in question. The three domains include (i) CO2-reactive applications, (ii) polymerization, and (iii) quaternary ammonium compounds.4.1. CO2-reactive applications
4.1.1.1. Introduction. The ever-rising importance of fossil fuels as an energy and chemical resource has led to a substantial increase in atmospheric CO2 concentration from below 300 ppm to above 400 ppm since the industrial revolution. The increase in atmospheric CO2 has been causing global greenhouse effects, which destabilize the climate and contribute to climate change and global warming.246 In the context of long-term climate change mitigation, a shift towards non-fossil alternatives, such as biomass, solar and wind energy, presents a promising strategy to reduce CO2 emissions. However, during this transition, the primary focus remains on curbing anthropogenic CO2 emissions in the existing fossil-based industries. In this regard, the concept of carbon capture, utilization and/or storage (CCUS) has gained extensive research interest.247,248 A wide range of technologies has been studied to perform CO2 capture, i.e., the initial step in CCUS, to remove CO2 from a gaseous stream such as flue gas. Among these, the post-combustion CO2 capture process utilizing aqueous (alkanol)amine solvents as CO2 absorbents has emerged as a prominent and well-established industrial-scale technology.249,250
This chemical absorption process involves the reversible, temperature-dependent reaction of CO2 with an aqueous amine solution. In a typical set-up, a CO2-rich gas stream is pumped through an absorption column where CO2 reacts with an aqueous amine solution at temperatures around 30–60 °C and atmospheric pressure, generating a CO2-lean gas stream and a CO2-rich amine solution. Subsequently, the CO2-rich amine solution is sent to a desorption column where CO2 is released from the amine absorbent upon heating, typically by steam. This regeneration step is usually carried out around 120–140 °C and results in a concentrated CO2 stream and a lean amine solution that can be recycled back to the absorption unit for a new absorption–desorption cycle.250 The mechanism of CO2 absorption is influenced by the type of amine. Primary and secondary amines following a zwitterion mechanism, forming a carbamate and a protonated amine upon reacting with CO2. Stoichiometrically, one CO2 molecule reacts with two amine molecules, yielding a maximum CO2 loading of 0.5 mol CO2/mol amine:
| CO2 + 2R1R2NH ⇌ R1R2NCOO− + R1R2NH2+ |
On the other hand, tertiary amines, which lack a nucleophilic proton, function as base catalysts, resulting in the formation of bicarbonate through CO2 hydration. Consequently, the theoretical maximum CO2 loading of tertiary amines is 1 mol CO2/mol amine:
| CO2 + H2O + R1R2R3N ⇌ HCO3− + R1R2R3NH+ |
From a kinetic point of view, primary and secondary amines generally demonstrate significantly higher CO2 absorption rates (i.e., one or two orders of magnitude higher) compared to tertiary amines. Thermodynamically, carbamates are more stable compounds than bicarbonate, leading to higher heat of reaction values for primary and secondary amines than tertiary amines. Therefore, tertiary amines are more readily regenerated than primary and secondary amines. Another important parameter to evaluate the performance of an amine absorbent is the cyclic capacity. It is defined as the difference between the CO2 concentration in the rich and lean amine solution and thus combines the CO2 loading and heat of reaction of the amine absorbent. The cyclic capacity is characteristic for a specific amine absorbent but also strongly depends on reaction conditions such as gas flow. Additionally, an ideal amine absorbent should also be resistant to degradation, non-volatile, non-toxic, non-corrosive, cheap, sustainable, and maintain low viscosity; properties which are not always taken into account.249,251
Aqueous solutions containing 20–30 wt% MEA and MDEA have been established as benchmark absorbents for primary/secondary and tertiary amines, respectively. These two reference solutions illustrate the tradeoff between fast absorption kinetics and ease of regeneration. The primary amine MEA readily absorbs CO2, however, CO2 desorption requires a substantial amount of energy. On the other hand, CO2 absorption proceeds slower with the tertiary amine MDEA, but less energy is required to desorb CO2.250,252 In the literature, various strategies have been investigated to address this tradeoff, aiming to reduce the energy required for CO2 desorption while maintaining favorable absorption kinetics. These strategies focus either on the reactor configuration,253 the use of a catalyst254,255 or the amine absorbent. In general, the latter strategy is approached by developing either new effective aqueous amine absorbents or so-called water-lean amine solutions. Both approaches will be discussed in more detail in following subsections.
4.1.1.2. Novel aqueous amine absorbents. The search for new aqueous amine absorbents should target the specific shortcoming of each type of amine without compromising its beneficial properties (Fig. 3). The high energy requirements in CO2 desorption, the bottleneck for primary and secondary amine, can be addressed by two approaches. First, the energy requirements can be reduced by using amines with a lower heat of reaction than MEA. When reacting with CO2, sterically hindered primary/secondary amines will form less stable carbamate species which correspond to a lower heat of reaction. The sterically hindered primary amine, 2-amino-2-methyl-1-propanol (AMP), has been studied by multiple researchers as a suitable alternative for MEA.251,256 The heat of reaction of AMP (ΔH = −80 kJ mol−1 CO2) was experimentally verified to be lower than that of MEA (ΔH = −85 kJ mol−1 CO2).257,258 Furthermore, stability studies indicated that AMP is more resistant to thermal and oxidative degradation than MEA.259,260 In addition to AMP, other sterically hindered primary/secondary amines have been investigated as potential CO2 absorbents based on their heat of reaction. In a study conducted by Chowdhury and co-workers, N-monoisopropylethanolamine (MiPEA) emerged as a favorable absorbent compared to MEA and other hindered amines due to its reduced heat of reaction (−64 kJ mol−1 CO2) combined with a comparable absorption rate to that of MEA.257 Other promising sterically hindered amines were observed in the research by El Hadri et al. The amines of interest, namely N-monomethylethanolamine (MMEA) and N-monoethylethanolamine (MEEA), exhibited similar kinetics as MEA in addition to a reduced heat of reaction of −74 kJ mol−1 CO2 and −69 kJ mol−1 CO2, respectively.258 Second, the energy requirements of the desorption step can be reduced by enhancing the cyclic capacity of the amine absorbent as less solvent is required to react with a given amount of CO2. In this regard, polyamines are a potentially interesting group of amines as, in theory, each nitrogen atom in the absorbent can react with CO2. Singh et al. systematically assessed the effect of the number of functional groups in the absorbent on the CO2 loading and the cyclic capacity. The tested polyamines (i.e., EDA, DETriA, TriETA and TEPA) all exhibited a higher CO2 loading and cyclic capacity than MEA. Moreover, both the CO2 loading and cyclic capacity gradually improved with an increasing number of nitrogen atoms present in the absorbent.261,262 Interestingly, both energy reduction approaches can be combined by using heterocyclic polyamine absorbents such as piperazine (PZ). Aqueous PZ solutions display a superior overall performance in CO2 capture compared to MEA since PZ demonstrates fast kinetics, a high cyclic capacity, and relatively low heat of reaction.263
 | ||
| Fig. 3 Overview of novel aqueous amine absorbents reported in the literature. Amine absorbents that have been produced from (hemi)cellulose-derived oxygenates are framed in green, whereas amines that could potentially be produced from these oxygenates are framed in dashed green. | ||
Tertiary amines, on the other hand, generally suffer from slow absorption kinetics. Multiple researchers have conducted screening experiments in the search for tertiary amines with faster absorption rates compared to the benchmark MDEA. In their study, Chowdhury and co-workers identified four better-performing aliphatic alkanolamines than MDEA, namely N,N-diethylethanolamine (DEEA), N,N-diethyl-3-amino-1,2-propylene glycol (3DEA1,2PG), N,N-diethyl-3-amino-1-propanol (3DEA1P), and N,N-dimethyl-1-amino-2-propanol (1DMA2P).264 In addition to 1DMA2P, El Hadri et al. also found that the β-propanolamine N,N-dimethyl-3-amino-1-propanol (3DMA1P) and the diamine N,N,N′,N′-tetramethyl-1,3-propylenediamine (1,3TMPDA) outperformed MDEA in terms of absorption rate.258 Xiao et al. drew similar conclusions as Chowdhury et al. and El Hadri et al. as they reported 3DMA1P and DEEA as interesting tertiary amine absorbents.265 Consistent with the primary and secondary amine absorbents, also various heterocyclic piperidine- and pyrrolidine-derived tertiary amines outperformed the MDEA in terms of absorption rate.264,266,267 From a mechanistic point of view, one could intuitively reason that the absorption rate predominantly correlates with the basicity of the amine, i.e., the corresponding pKa value. In practice, however, this straightforward relationship is rather limited and not entirely compelling.267,268 Recently, Rozanska et al. developed an empirical and predictive model to quantitatively assess the absorption rate of tertiary amines.269 In their model, the key determinants for the reaction rate were the concentrations of CO2 and OH− and the Gibbs free energy of activation of the reaction between these two species. Hence, the pKa value of the amine absorbent indirectly influences the absorption rate by affecting the concentration of the OH− species via acid–base chemistry.269 In a follow-up study, Orlov et al. successfully employed this model by screening 100 structurally diverse tertiary amines. This screening verified existing experimental reaction rates and additionally highlighted pyrrolidinol (e.g., 1-ethyl-3-pyrrolidinol (EPOL)) as a promising yet unexplored class of tertiary amines.270
Despite all research interests, it is still challenging to obtain an amine that combines the fast absorption kinetics of primary/secondary amines and the ease of regeneration of tertiary amines. Consequently, amine blends, which contain two or more amines with complementary properties, have gained attention as a suitable solution. Currently, the blend of PZ and AMP is the best-known absorbent formulation. The blend of these two absorbents exceeds the individual amines in terms of absorption rate, ease of desorption and cyclic capacity.249,271 In addition to the PZ/AMP formulation, PZ has been employed as a rate-enhancing additive for various aqueous tertiary amines including MDEA.270,272 The effectiveness of these amine blends is demonstrated by the fact that almost all industrial amine-based absorption technologies utilize commercial amine blends such as KS-1 (MHI), Cansolv (Shell) and OASE (BASF).250,252,273
4.1.1.3. Water-lean absorbents. The second strategy to decrease energy consumption involves the elimination of water from the solvent system. Amine absorbents typically contain water to maintain their solubility and minimize the viscosity of the CO2-rich solution. However, the relatively high specific heat capacity and vaporization heat of water have a large parasitic effect on the overall cost and energy requirements of heating and recondensing the absorbent solution throughout the absorption–desorption cycles.274,275 Different so-called water-lean approaches have been studied in the literature.276 The most straightforward water-lean approach consists of replacing water with an organic solvent that has a lower specific heat capacity and higher CO2 solubility such as MeOH, EtOH or EG. In practice, however, water is not completely eliminated from the capture system as most water-lean approaches still rely on steam to regenerate the CO2-rich solvent. This approach primarily focuses on solvent design while utilizing the same group of amines used in traditional aqueous amine absorbents.274
In the context of this Review, a noteworthy water-lean approach involves the substitution of aqueous amine solutions with a tailor-made single amine absorbent system (Fig. 4). Since no additional solvent is present, the amine absorbent should be liquid both in the absence and presence of CO2 while preventing a substantial increase in viscosity. Furthermore, the amine absorbent should at least contain one primary/secondary amino group to form carbamates upon CO2 absorption as no bicarbonate can be formed in the absence of water.276 Barzagli et al. investigated a number of N-alkylated ethanolamines as potential single-component absorbents. Among these, N-monobutylethanolamine (MBEA) stood out as the most promising absorbent due to its high absorption efficiency, high boiling point (i.e., low volatility), thermal stability and moisture tolerance.277 Recently, Heldebrant and co-workers have explored various single-component systems in a series of publications.278–280 In their initial work, they presented six promising single-amine absorbents based on computational screening via molecular simulations.278 Subsequently, they experimentally evaluated two of these amines, namely N-(2-ethoxyethyl)-3-morpholinopropyleneamine (2EEMPA)279 and N-(2-ethoxyethyl)-N′,N′-diisopropylethylenediamine (2EEDiPEDA).280 Comparing the two, 2EEDiPEDA exhibited a lower viscosity and stronger affinity for CO2, relative to 2EEMPA.279,280 Through a comprehensive techno-economic analysis, they estimated that employing the 2EEDiPEDA absorbent could potentially lead to a 20% reduction in the overall capture cost when compared to the commercially available Cansolv absorbent.280
 | ||
| Fig. 4 Examples of novel water-lean absorbents reported in the literature of which some could potentially be produced from (hemi)cellulose-derived oxygenates (green dashed frame). | ||
4.1.2.1. Introduction. Apart from CO2 capture, the acid–base interaction between CO2 and an amine can also be harnessed in so-called switchable materials such as switchable solvents. This group of designer solvents employs CO2 as a smart chemical switch to alter specific solvent properties. These property changes are reversible as CO2 can be removed from the solvent via steam stripping or sparging with non-acid gases such as N2 or air, with or without heating.281 Since 2005, Jessop and collaborators have been pioneers in the development of such CO2-switchable solvents.282,283 Two types of CO2-switchable solvents that require aliphatic amines, namely switchable water (SW) and switchable hydrophilicity solvents (SHS), will be discussed in more detail (Fig. 5).
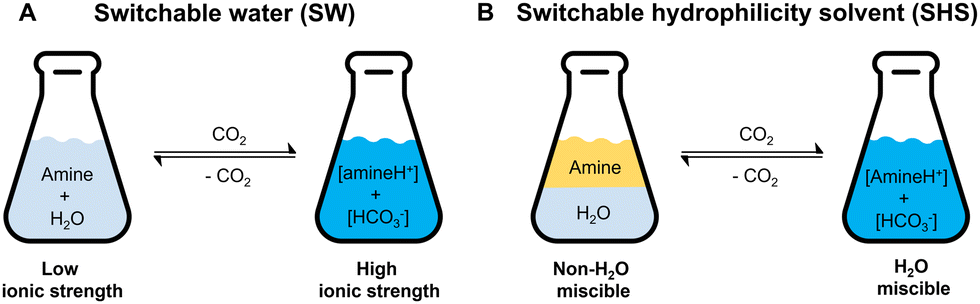 | ||
| Fig. 5 General principle of (A) switchable water and (B) switchable hydrophilicity solvents. | ||
4.1.2.2. CO2-switchable water (SW). Switchable water (SW) is a single-phase aqueous solution, both in the absence and presence of CO2, that contains a water-miscible organic base (referred to as the ionogen), generally an aliphatic amine. Upon introduction of CO2, the amine reacts with CO2 to yield a bicarbonate salt which triggers a significant increase in the ionic strength of the aqueous solution (Fig. 5A). Additionally, also the conductivity, viscosity and osmotic pressure will increase.284
The altering ionic strength can be exploited to efficiently separate or purify water-soluble organic compounds from an aqueous environment. In the CO2-lean solvent, the organic solute is highly soluble due to the ionogen acting as a hydrotrope. In the CO2-rich solvent, however, the ionic strength increases and the organic solute is salted out because the ionogen–solute interaction changes from attractive to repulsive.285 Mercer et al. have experimentally illustrated this salting-out effect by separating THF from an aqueous solution.286,287 Interestingly, these studies have postulated a number of structural requirements for the amine ionogen as it strongly affects the performance of the SW. First, tertiary and bulky secondary amines are preferred as they exclusively form bicarbonate salts instead of carbamate salts. The formation of carbamate salts is detrimental as it complicates the reversibility of the reaction and leads to the formation of fewer ions, thus, decreasing the ionic strength. Furthermore, using a polyamine ionogen will lead to a higher ionic strength than a monoamine ionogen as each amino group can enhance the ionic strength upon protonation.286 Second, incomplete protonation of the amino group(s) of an ionogen has an adverse effect on the performance of the SW. Consequently, the basicity of the amine (i.e., pKa) plays a vital role. If the basicity of the amine is too low, it will not be fully protonated in the presence of CO2. Conversely, if the basicity is too high, the amine will already be partially protonated in the CO2-lean aqueous solution. These lower and upper pKa limits are dependent on the amine concentration, temperature and CO2 pressure.288 In this regard, polyamines with a two-carbon linkage between the different amino groups tend to perform poorly. The protonation of one amino group diminishes the basicity of the adjacent amino group(s), rendering it unprotonated in the carbonated solution. On the other hand, polyamines with a three- or four-carbon linkage display an improved performance because the longer linkage ensures that each amino group can undergo protonation.287 In line with these insights, the best-performing ionogens in the studies of Mercer et al. were propylene polyamines, namely the diamine 1,3TMPDA and the tetramine N,N,N′,N′′,N′′′,N′′′-hexamethyltripropylenetetramine (HMTriPTA). Under reaction conditions (THF-to-H2O wt ratio of 1:1, 0.8 molal amine loading, 30 min, 25 °C), a THF separation of 82% and 85% was achieved by employing 1,3TMPDA and HMTriPTA, respectively, while both amines retained for more than 99% in the aqueous phase.286,287 By elaborating on this salting-out effect, SW can also be used as a reaction medium in a chemical process, facilitating the separation of the organic product after the reaction (Fig. 6A). For example, Püschel et al. performed an auto-tandem reductive hydroformylation reaction in a SW medium.289 Noteworthy, they employed the ethanolamine DEEA both as a catalytic ligand and as an ionogen in the SW. After completion of the reaction, CO2 was added to the reaction system in order to separate the alcoholic products from the aqueous phase containing DEEA and the catalyst. In this way, an alcohol yield of 99% and a turnover frequency of 764 h−1 were successfully achieved.289
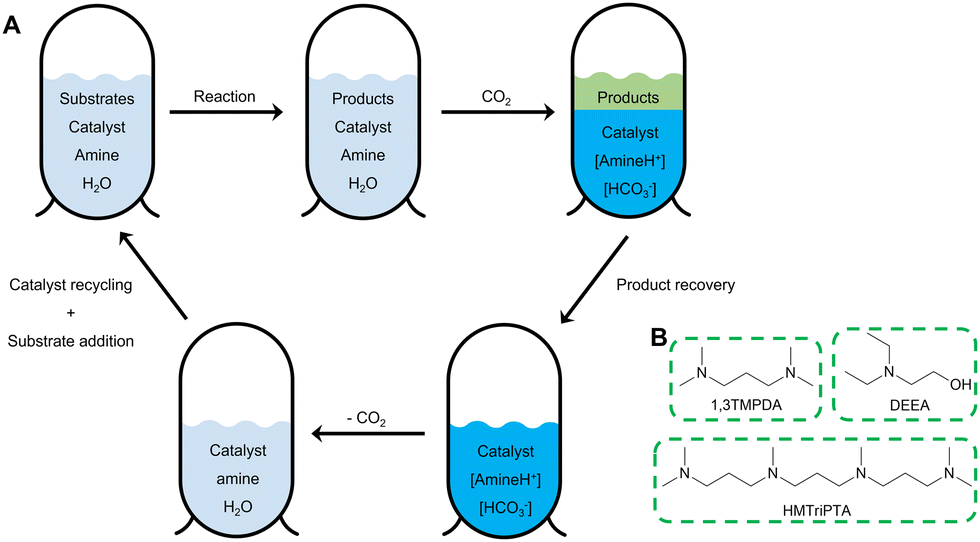 | ||
| Fig. 6 (A) Illustration of a reaction performed in switchable water. (B) Examples of switchable water ionogens which could potentially be produced from (hemi)cellulose-derived oxygenates (green dashed frame). | ||
4.1.2.3. CO2-switchable hydrophilicity solvents (SHSs). A switchable hydrophilicity solvent (SHS) represents a basic, hydrophobic liquid that, when combined with water, results in a biphasic system. Upon introducing CO2, however, the SHS becomes hydrophilic and readily miscible with water (Fig. 5B).281,290 The hydrophobic form of SHSs typically comprises a tertiary or bulky secondary amine, while its hydrophilic counterpart corresponds to its respective bicarbonate salt.291 The altering miscibility of SHSs can be exploited to efficiently dissolve or extract hydrophobic organic compounds without the need for a distillation step (Fig. 7A). Initially, these organic compounds are dissolved in the hydrophobic SHS. Afterward, the desired organic compound can be purified by removing the SHS with carbonated water. In this way, the hydrophobic phase solely contains the organic compound and is separated from the aqueous phase that comprises the amine as a bicarbonate salt.283,284
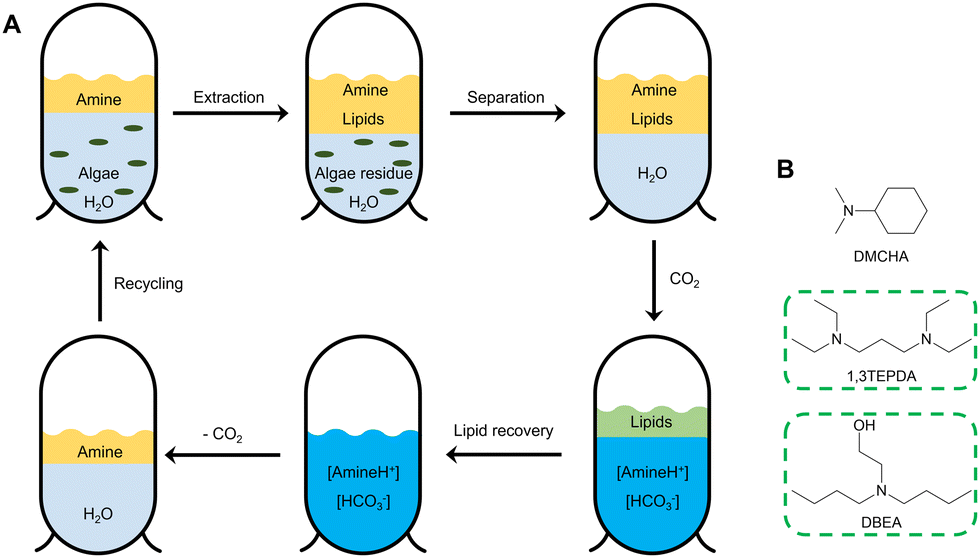 | ||
| Fig. 7 (A) Illustration of lipid extraction from algae using a switchable hydrophilicity solvent. (B) Examples of aliphatic amines as switchable hydrophilicity solvents which could potentially be produced from (hemi)cellulose-derived oxygenates (green dashed frame). | ||
An effective SHS must fulfill two primary requirements. First, the SHS should be able to fully dissolve the targeted hydrophobic compound in the absence of CO2. Second, the presence of CO2 should trigger the SHS to move completely toward the aqueous phase. These two requirements can be quantified by two intrinsic parameters, namely the octanol–water partition coefficient (logKow) and the basicity (pKa). In general, all tertiary and bulky secondary monoamines with logKow values between 1.2 and 2.5 and pKa values between 9.5 and 11 are applicable as SHSs.292 Amines with logKow values lower than 1.2 are miscible with water and do not result in a biphasic system, while amines with logKow values higher than 2.5 remain non-water-miscible regardless of the applied CO2 pressure. On the other hand, amines with pKa values below 9.5 are not easily protonated by the carbonated water, whereas those with pKa values above 11 lack reversible behavior under mild conditions.292 The most commonly studied monoamine as an SHS is N,N-dimethylcyclohexylamine (DMCHA). For example, DMCHA has been employed as an SHS to extract lipids from algae,293,294 phospholipids from dairy295 and the carotenoid astaxanthin from bacteria.296 Furthermore, Samori et al. employed DMCHA as an SHS to successfully recycle polyethylene and aluminum from multilayer packaging materials.297
In addition to monoamines, polyamines have also been explored as suitable SHSs. However, polyamines exhibit some differences in properties compared to monoamines. For example, polyamine SHSs generally require larger logKow values than monoamines, as they partition more favorably into the aqueous phase. Consequently, the use of polyamine SHSs can lead to an enhanced separation as the residual polyamine amount in the hydrophobic phase is reduced compared to monoamines. One drawback associated with polyamine SHSs is their relatively longer transition time when switching between the hydrophobic and hydrophilic states, in comparison to monoamine SHSs.298 The diamine N,N,N′,N′-tetraethyl-1,3-propylenediamine (1,3TEPDA) is an example of a frequently studied SHS. 1,3TEPDA has been used in multiple extraction studies, for example in the extraction of lipids from algae299 or extraction of heavy oil in oil-solid separation.300 Besides their use in extraction processes, SHSs can also be used in chemical reactions. A case in point is the work of Viner et al., who employed the ethanolamine N,N-dibutylethanolamine (DBEA) both as a solvent and recoverable base catalyst in the transesterification of soybean oil to long-chain fatty acid methyl esters (FAMEs). They achieved a very good FAME yield of 80–85% along with an excellent DBEA recovery rate of approximately 92%.301
4.2. Polymerization
Aliphatic amines are widespread as (reactive) catalysts in numerous polymerization reactions. This will be illustrated by discussing the role of aliphatic amines in three distinct polymers and their polymerization process, namely (i) epoxy polymers, (ii) polyurethane (PUR), and (iii) atom transfer radical polymerization (ATRP).4.2.1.1. Introduction. Epoxy polymers or epoxies are an important and versatile class of thermosetting polymers. They are known for their outstanding mechanical strength and toughness, excellent chemical, moisture and corrosion resistance, as well as remarkable thermal, adhesive and electrical properties. Consequently, epoxies find extensive applications in various fields, ranging from coatings and adhesives to electronic insulation, composites and construction.302 Epoxy polymers are produced by the polymerization and crosslinking of mono- and oligomeric epoxy resins or epoxy prepolymers. These epoxy resins are aliphatic, cycloaliphatic, aromatic or nonhydrocarbon structures containing at least one epoxide functional group. To date, diglycidyl ether of bisphenol A (DGEBA) is the most commonly used epoxy resin monomer.303–305 In recent years, however, the production of epoxy resins derived from various biomass sources such as lignin, carbohydrates, vegetable oils and plant extracts (e.g., tannins and terpenes) has gained increasing research interest and poses numerous sustainable alternatives.306–311
The polymerization and crosslinking of these resins occur in the presence of so-called curing agents or hardeners as they irreversibly transform the epoxy resins into a solid, infusible and insoluble three-dimensional thermoset network. The curing agent plays a pivotal role in determining the type of chemical bonds formed and the degree of crosslinking. Furthermore, the curing process can be either catalytic or co-reactive. In a catalytic curing process, the curing agent functions as a catalyst, activating another curing agent or the homopolymerization of the epoxy resin. In a co-reactive curing process, on the other hand, the curing agent acts as a reactive co-monomer and is incorporated into the polymeric network via polyaddition. Consequently, the choice of curing agent has a direct impact on the properties of the final epoxy polymer product.305 A wide scope of basic and acidic curing agents has been extensively studied and industrially applied.302–305 Notably, aliphatic amines stand out as a prominent group within this diverse range of curing agents.
4.2.1.2. Aliphatic amine curing agents. Primary and secondary aliphatic amines are widely used as co-reactive curing agents. They have a low viscosity and are highly reactive, which enables them to initiate curing at ambient temperatures. Adversely, their high reactivity makes primary and secondary aliphatic amines sensitive to amine blush, which can compromise both the performance and aesthetic properties of the cured epoxy material.312 Amine blush refers to the reaction between a primary or secondary amine hardener and moisture and CO2 from the air. Similar to CO2-related applications, this carbonation reaction will yield unreactive carbamate species, which appear as an oily, sticky film on the surface of the material.313
In the co-reactive polyaddition mechanism (Fig. 8A), the curing agent performs a nucleophilic addition onto the epoxide functionality of the epoxy resin, resulting in the formation of a hydroxyl group. This polyaddition process continues until all nucleophilic amine sites have either reacted or become unreactive (e.g., due to steric hindrance). In order to perform effective crosslinking, the curing agent should be a polyamine containing at least two amino groups.302–305 Recently, Mora et al. evaluated the reactivity of different types of primary and secondary polyamine curing agents.314 The reactivity of the curing agent depends on both its nucleophilicity and steric hindrance. In general, primary polyamines are presumed to be more reactive than secondary polyamines due to steric hindrance. Additionally, primary aliphatic polyamines are considered to be more reactive than their cycloaliphatic and aromatic counterparts. Among the primary aliphatic polyamines, DETriA and TriETA are regarded as the most reactive aliphatic amines due to the presence of two chain-end primary amino groups and one or two internal secondary amino groups, respectively.314 In another study by the same research group, primary ethanolpolyamines emerged as a potentially valuable, yet insufficiently explored type of highly reactive aliphatic polyamines. The researchers related the enhanced reactivity of these primary ethanolpolyamines to their ability to form additional hydrogen bonds with the epoxy reactant through their hydroxyl group.315 Secondary aliphatic amines are another type of underexplored curing agent. For example, little is known about the relative reactivity of N-methylated secondary aliphatic polyamines compared to primary aliphatic polyamines. Although more sterically hindered than primary aliphatic polyamines, N-methylation increases the nucleophilicity of these secondary aliphatic polyamines. In this regard, ethanolpolyamines, such as the DETriA adduct N-(2-monohydroxyethyl)-diethylenetriamine (MHEDETriA), are an interesting type of polyamines as they possess a primary amino group and a reactivity enhancing hydroxyl group.302,314 Noteworthy, preliminary research also suggests that this type of curing agent demonstrates reduced skin-sensitizing effects compared to the more common primary aliphatic polyamines.316
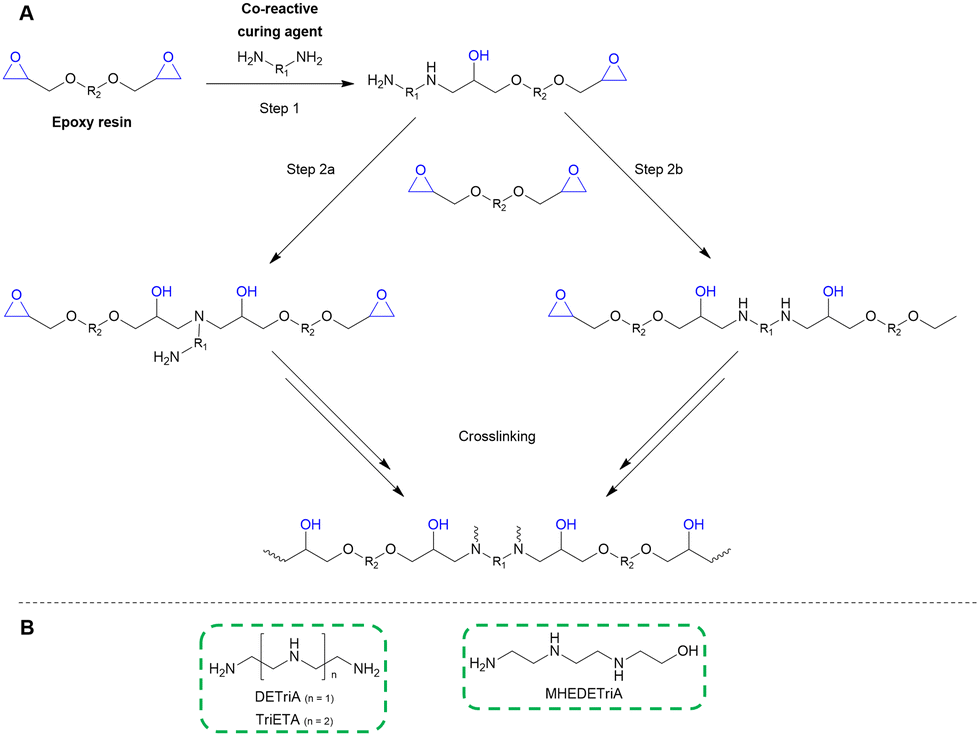 | ||
| Fig. 8 (A) Mechanism of co-reactive polymerization of epoxy resins. (B) Examples of aliphatic amines as co-reactive curing agents which could potentially be produced from (hemi)cellulose-derived oxygenates (green dashed frame). | ||
Tertiary amines, unable to perform polyaddition, catalyze the anionic homopolymerization of the epoxy resins (Fig. 9A). Through their Lewis basicity, tertiary amines activate either the epoxy monomers itself or another curing co-catalyst.314 In addition to commonly used aliphatic and aromatic monoamines (e.g., TriEA and benzyldimethylamine), various alkanolamines, including DMEA, DEEA and N,N-dimethyl-2-amino-2-methyl-1-propanol (DMAMP), are employed as catalytic curing agents.303–305,312
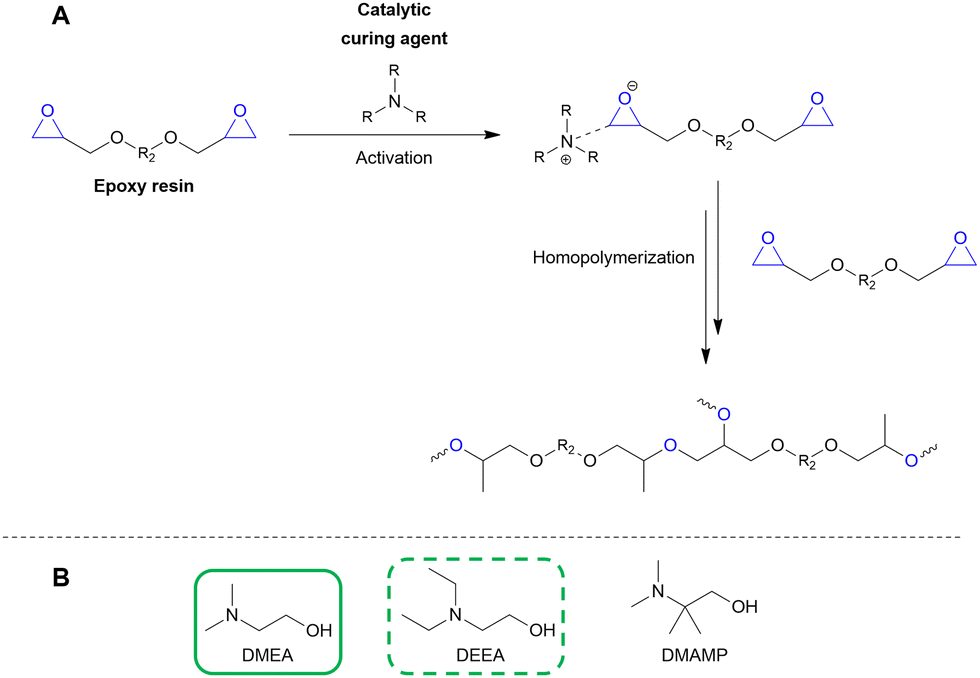 | ||
| Fig. 9 (A) Mechanism of catalytic homopolymerization of epoxy resins. (B) Examples of aliphatic amines as catalytic curing agents which have been (green fame) or could potentially be (green dashed frame) produced from (hemi)cellulose-derived oxygenates. | ||
4.2.2.1. Introduction. Polyurethane (PUR) is a highly versatile and commercially important group of specialty polymers. It encompasses all polymers formed by the polyaddition reaction that involves two types of monomers, namely polyfunctional isocyanates and polyols.317 Regarding these monomers, industrial PUR production is dominated by aromatic polyisocyanates, particularly methylene diphenyl diisocyanate (MDI) and toluene diisocyanate (TDI). More than 90% of PURs are industrially produced from these aromatic polyisocyanates while a handful of (cyclo)aliphatic polyisocyanates, e.g., hexamethylene diisocyanate (HDI) and isophorone diisocyanate (IPDI), are used in niche applications (e.g., applications requiring stability to UV light). In contrast, a diverse set of polyols is used as a reaction partner for these isocyanates. The two most common types of polyols are polyether polyols and polyester polyols. Polyether and polyester polyols result from the polycondensation reaction of an initiator with a cyclic ether (i.e., EO, PO) or a dicarboxylic acid (e.g., adipic acid, terephthalic acid), respectively. These initiators typically include glycols (e.g., EG, diethylene glycol, 1,2PG, 1,3PG), glycerol, 1,6-hexanediol, trimethylolpropane, bisphenol A, etc.318 Some of these initiators can be derived from biomass. Moreover, partially or fully bio-based PUR polymers have experienced increased research activity in recent years. All different fractions of lignocellulose, as well as various vegetable oils, have been reported as suitable resources for the formation of bio-based drop-in polyols and isocyanates.319–321 The seemingly endless choice of polyols, combined with a couple of isocyanates and various additives (e.g., chain extenders, flame retardants, plasticizers) results in a diverse set of PUR materials. The structure of PUR spans from compact to foamed, from soft to rigid and comprises everything in between, whereas their applications extend from furniture and bedding to automotive components, construction materials, clothing, and thermal insulation.317,322,323
The polyaddition reaction of isocyanate and polyol leads to the linear urethane (or carbamate) link characteristic for PUR. In addition, isocyanate can undergo a plethora of alternative reactions with other active hydrogen compounds present in the reaction system (Fig. 10A). A second essential linear polyaddition reaction yields urea through the condensation of isocyanate with a primary or secondary amino group. The presence of these amines in the system can be deliberate or result from the reaction of isocyanate with water. When isocyanate reacts with water, carbamic acid is initially formed, which then spontaneously decomposes into an amine accompanied by the elimination of CO2. The formed CO2 expands and thereby promotes the formation of the porous structure in foams. In this regard, the formation of urea and CO2 is known as the blowing reaction, while the formation of the rigid urethane bond is often referred to as the gelling reaction. The formed urethane and urea groups retain active hydrogen atoms in their structure and are therefore capable of engaging in crosslinking reactions with isocyanate, yielding allophanates and biurets, respectively. In addition to polyaddition reactions, isocyanate can also oligomerize, resulting in cyclic structures such as uretdione, isocyanurate and uretonimine.317,318,322 Consequently, catalysis plays a crucial role in selectively controlling each of these reactions as they all distinctively contribute to the final properties of the polymer. In general, two categories of catalysts are used: organometallic Lewis acid catalysts, with organotin compounds as most prominent class, and Lewis base tertiary amines.324 The latter will be discussed in more detail in the scope of this Review.
 | ||
| Fig. 10 (A) Overview of possible polymerization reactions in the synthesis of polyurethane. The two primary polymerization reactions, namely gelling (urethane formation) and blowing (urea formation) are highlighted in blue and red, respectively. Examples of (B) traditional PUR catalysts and (C) novel reactive PUR catalysts which have been (green frame) or could potentially be (green dashed frame) produced from (hemi)cellulose-derived oxygenates. | ||
4.2.2.2. Tertiary amine PUR catalysts. Tertiary amines catalyze both the isocyanate-polyol and isocyanate-water reactions (i.e., the gelling and blowing reactions), as well as some side reactions such as the trimerization reaction forming isocyanurate. As Lewis bases, tertiary amines catalyze the polymerization reactions by activating the hydroxyl group of the polyol or water for nucleophilic addition (Farka's mechanism) and/or facilitating the proton transfer between hydroxyl and isocyanate by activating the isocyanate (Baker's mechanism).325 Therefore, the catalytic activity of tertiary amines mainly depends on their basicity, steric hindrance and number of catalytic sites. Nonetheless, predicting the catalytic selectivity of tertiary amines a priori based on these properties remains challenging. Tertiary amines that preferentially interact with water molecules catalyze the isocyanate-water reaction and are called blowing catalysts, while those that favor the isocyanate-polyol reaction are referred to as gelling catalysts.326–328
In recent years, the use of traditional tertiary amines, such as triethylenediamine (TriEDA), DMCHA and bis-(2-dimethylaminoethyl)ether (Fig. 10B), has progressively declined in industrial applications. These amines possess relatively high volatility and are susceptible to oxidative degradation over time, which combined results in significant emissions of harmful volatile organic compounds (VOCs) and associated undesirable odors.318,329 This limitation has prompted the development of nonvolatile catalysts such as reactive catalysts and autocatalytic polyols, which are both designed to chemically bind into the polymer matrix using isocyanate-reactive groups. Reactive catalysts are tertiary amines functionalized with a hydroxyl or primary/secondary amino group.324,327 In contrast to traditional tertiary amine catalysts, larger quantities of reactive catalysts are required as they become less effective when incorporated into the polymer network. Furthermore, the choice of the reactive group strongly affects the catalytic activity. For example, reactive catalysts with a secondary hydroxyl group will exhibit a prolonged catalytic activity compared to those containing a primary hydroxyl group as they react more slowly with the polymer matrix. However, this advantage is offset by the risk of never being incorporated into the network. Conversely, a reactive catalyst with a primary or secondary amino group is highly likely to integrate into the polymer network, but it may rapidly lose its catalytic effectiveness. Some examples of commercially important reactive catalysts are given in Fig. 10C.330–332 Autocatalytic polyols go a step further as they are polyols functionalized with catalytic sites. Similarly to traditional polyols, autocatalytic polyols are typically produced by reacting a cyclic ether or dicarboxylic acid with an initiator, in this case being a reactive tertiary amine catalyst.317,333,334
4.2.3.1. Introduction. Reversible-deactivation radical polymerization (RDRP) is an umbrella term for well-controlled polymerization techniques that are based on establishing a dynamic equilibrium between a small number of actively propagating radicals and a large number of dormant species incapable of propagation and termination. In this way, the lifetime of the growing polymer chains is significantly extended from mere seconds to several hours or even days, allowing to precisely control of the composition, topology, functionality and complex architecture of the synthesized polymer.335 IUPAC recently described this technology as a revolution in polymer synthetic chemistry and has included it in its top 10 emerging chemical innovations that can change the world.336
Atom transfer radical polymerization (ATRP), independently developed by Matyjaszewski337 and Sawamoto338 in 1995, is one of the most robust and widely used methods of RDRP. In ATRP, a broad range of monomers (e.g., styrenes, acrylonitrile, (meth)acrylates, (meth)acrylamides) can be polymerized into well-defined and uniform polymers applicable in various applications. Over the years, several companies have successfully commercialized ATRP-derived polymers.339
From a mechanistic perspective, ATRP is a catalytic reversible-deactivation method wherein the activation and deactivation of radicals occur through a concurrent atom and electron transfer reaction regulated by a catalyst (Fig. 11A). A successful ATRP reaction requires a minimal amount of termination reactions, uniform growth of all chains, and fast initiation coupled with rapid reversible deactivation. These three requirements all heavily rely on the choice of catalyst as it regulates the equilibrium dynamics between active and dormant species (i.e., kact, kdeact and KATRP). The catalyst is typically a homogeneous complex consisting of a transition metal and a ligand. While Cu is frequently employed as the transition metal, other transition metals (e.g., Ti, Fe, Ru, Co, Pd) have also been explored. In combination with Cu, nitrogen-containing ligands, particularly aliphatic tertiary polyamines and pyridine derivatives, stand out as the most active ligands.340,341 The aliphatic polyamine ligands connect to the interest of this review and will be addressed in more detail.
 | ||
| Fig. 11 (A) General principles of atom transfer radical polymerization. (B) Examples of ATRP catalytic ligands which have been (green frame) or could potentially be (green dashed frame) produced from (hemi)cellulose-derived oxygenates. | ||
4.2.3.2. ATRP aliphatic polyamine ligands. Although already reported in 1997,342 traditional linear tertiary polyamines, namely N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETriA) and HMTriETA, are still regarded as robust and active ATRP ligands, demonstrating an excellent control over molecular weight and molecular weight distribution. As a rule of thumb, the performance of these linear aliphatic polyamine ligands generally improves with increasing number of nitrogen atoms and decreasing length of carbon linker (i.e., C2 ≫ C3 > C4). Later, the branched aliphatic polyamine tris(2-dimethylaminoethyl)amine (3(DMAE)A) was developed to further improve the catalytic performance. To date, the Cu-3(DMAE)A complex is one of the most active and selective ATRP catalysts reported in the literature (Fig. 11B).343,344
4.3. Quaternary ammonium compounds (QACs)
Quaternary ammonium compounds (QACs) constitute a fourth distinct type of functional amino group, in addition to primary, secondary and tertiary amines. Unlike the other three amino groups, QACs lack basicity and nucleophilicity due to the absence of a lone electron pair at the nitrogen atom. Instead, QACs are cationic as the nitrogen center contains four substituents (R4N+).345 QACs are generally formed through the exhaustive alkylation of an amine with an electrophilic alkylation agent. In this way, an extensive scope of QACs can be obtained from various amines and alkylation agents, thereby broadening their utility in a wide range of applications.346 For example, QACs can be used as phase-transfer catalysts in organic synthesis,347 as framework templates in supramolecular chemistry such as zeolite synthesis,348 as cationic amphiphilic surfactants in detergents and fabric softeners,199,349 and as biocides in pesticide and antimicrobial formulations.350 The latter application will be discussed in more detail to highlight the potential of QACs. | ||
| Fig. 12 Traditional monoQACs employed as antibacterial agents. | ||
Consequently, the development of next-generation antibacterial QACs that are less susceptible to resistance development, is of utmost importance. An ideal next-generation QAC should meet four criteria: (i) high antibacterial activity, particularly against resistant bacteria, (ii) minimal toxicity, i.e., exhibiting high selectivity towards microbial cells over eukaryotic cells, (iii) resilience against the development of resistance, and (iv) biodegradability to avoid accumulation in the environment. In this regard, the research groups of Minbiole and Wuest have synthesized and assessed more than 200 QACs in the search for these next-generation QACs. Their research efforts have primarily focused on the development of multiQACs, characterized by the presence of more than one charged nitrogen atom.353,354 Among all tested multiQACs, aliphatic multiQACs are regarded as the most promising next-generation QACs. These multiQACs are obtained by straightforward quaternization of aliphatic tertiary polyamines such as TMEDA, PMDETriA, HMTriETA, 3(DMAE)A and tris(2-dimethylaminopropyl)amine (3(DMAP)A), and fulfill multiple of the predetermined criteria. First, various tested multiQACs outperformed the traditional monoQACs in terms of antibacterial activity. Notably, activity did not increase with the number of cationic centers, as multiple bis-, tri- and tetraQACs resulted in comparably excellent activities. Furthermore, multiQACs containing symmetric alkyl side chains with a carbon length between 12 and 14 exhibited the highest activity as this length is required to successfully penetrate and disrupt bacterial cell membranes. However, the most active compounds also displayed the highest toxicity.355,356 Second, experimental results indicated that aliphatic multiQACs, in contrast to monoQACs and aryl-containing QACs, were active against bacterial strains carrying QAC resistant genes,357 which is likely at least partly attributable to their increased affinity to the bacterial cell surface.358 Furthermore, no de novo resistance development was observed against aliphatic multiQACs after more than 500 generations. In contrast, resistance development was observed after a few hundred generations for mono- and bisQACs possessing aryl substituents.359 These results indicate that to counteract aliphatic multiQACs, bacteria need to evolve a resistance mechanism that differs from the mechanism used against conventional QACs.353 Third, these multiQACs can become “soft” antibacterials that are designed to biodegrade after a certain time or trigger by incorporating cleavable amide- or ester-containing side chains in their structure. All of these soft compounds were stable in water, but the ester-QACs rapidly decomposed in any sort of buffered solution while the amide-QACs only decomposed in acidic media. Interestingly, several of these soft multiQACs retained their excellent antibacterial activity.360 Finally, these multiQACs are among the most potent QAC-based biofilm eradicators published to date. Both the polycationic character and the presence of the alkyl chains are vital for this activity.359,361 Biofilms are complex three-dimensional communities of microorganisms and contribute to over 80% of all microbial infections. Furthermore, bacteria in biofilms are inherently more tolerant to antibacterial treatments, making it extremely difficult to treat and eradicate the pathogenic biofilm effectively.362 The development of multiQACs with biofilm-eradicating properties is very promising but still requires more insights into the mechanism of action and a significant reduction in their toxicity. Overall, mutiQACs possess the potential to become the next-generation antibacterial QACs.354 A summary of the most promising antibacterial multiQACs and their preparation methods are given in Table 2.
| Component | Yielda | Antibacterial activityb | Toxicityc | Biofilm eradicationd | ||
|---|---|---|---|---|---|---|
| a QAC product yield after purification. b Minimum inhibitory concentrations (MIC) against Pseudomonas aeruginosa (PA), Staphylococcus aureus (SA) and methicillin-resistant Staphylococcus aureus (MRSA). c Lysis20 values represent the compound concentration at which 20% or less of red blood cells are lysed. d Minimum biofilm elimination concentration (MBEC) against MRSA. N.R. = not reported. Results obtained from Minbiole et al.355,356,359–361,363 Amine multiQACS that have been produced from (hemi)cellulose-derived oxygenates are framed in green, whereas amines that potentially could be produced from these oxygenates are framed in dashed green. | ||||||
| [%] | MIC [μM] | Lysis20 [μM] | MBEC [μM] | |||
| PA | SA | MRSA | MRSA | |||
| Common monoQAC | ||||||
| BAC | 63 | 8 | 32 | 63 | >200 | |
| MultiQACs | ||||||
| TMEDA |
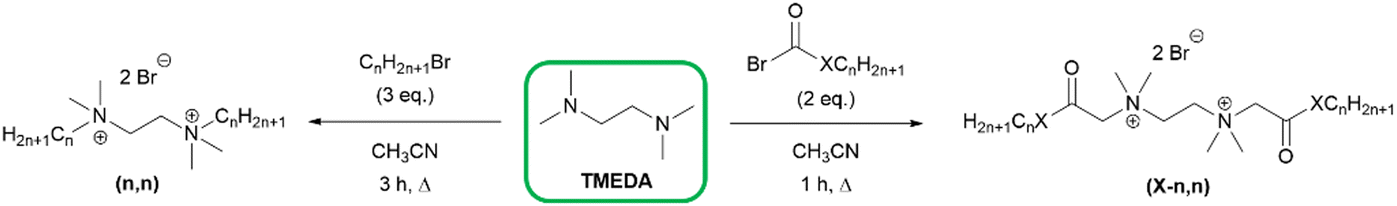
|
|||||
| 12,12 | 94 | 4 | 1 | 0.5 | 8 | 75–100 |
| E-12,12 | 77 | 32 | 2 | 4 | 16 | N.R. |
| A-13,13 | 44 | 2 | 1 | 0.5 | 8 | N.R. |
| PMDETriA |
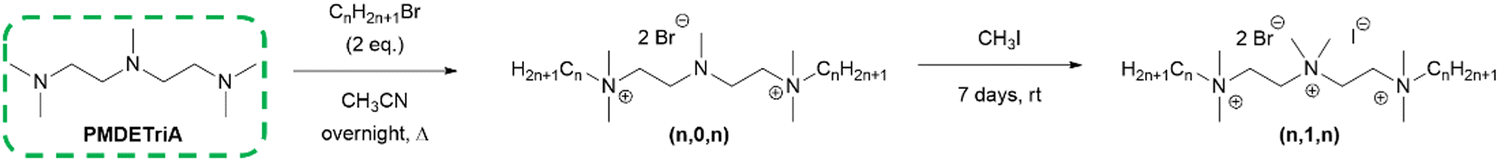
|
|||||
| 12,0,12 | 95 | 4 | 1 | 0.5 | 8 | 75–100 |
| 12,1,12 | 90 | 1 | 1 | 1 | 16 | 50 |
| HMTriETA |
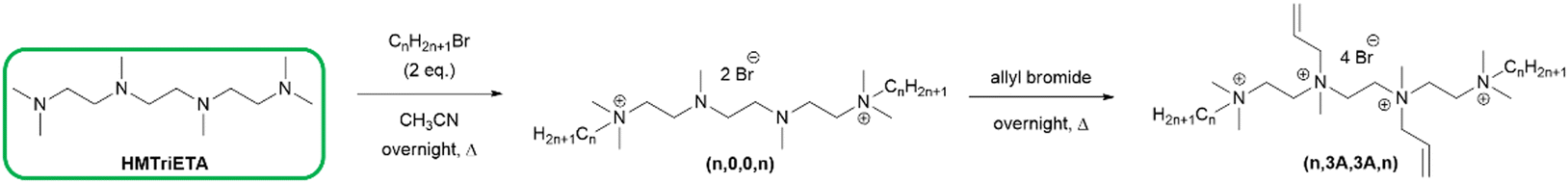
|
|||||
| 12,0,0,12 | 93 | 4 | 1 | 1 | 4 | 100 |
| 12,3A,3A,12 | 91 | 2 | 1 | 1 | 8 | 150 |
| 3(DMAE)A |
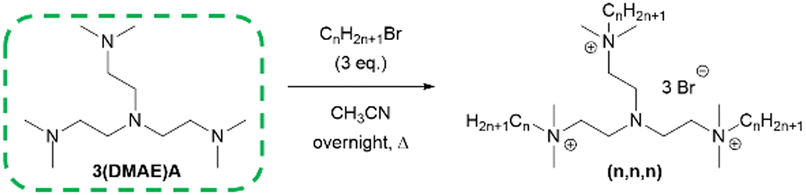
|
|||||
| 12,12,12 | 82 | 8 | 1 | 2 | 8 | >200 |
| 3(DMAP)A |
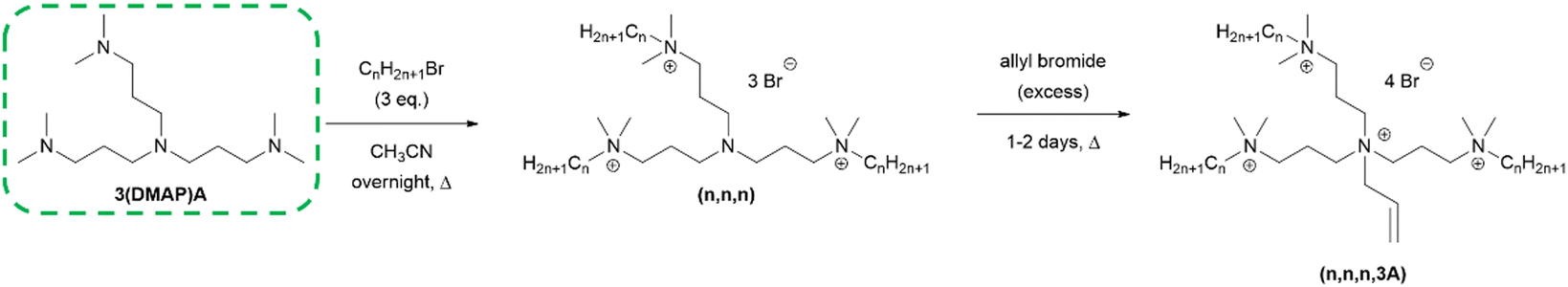
|
|||||
| 12,12,12 | 99 | 4 | 0.5 | 0.5 | 4 | 200 |
| 12,12,12,3A | 94 | 2 | 1 | 1 | 4 | 100 |
5. Critical discussion and conclusive remarks
Aliphatic amines encompass a versatile class of amines including alkyl polyamines, heterocyclic amines and alkanolamines. The characteristic amino group in aliphatic amines is connected via a linear or branched aliphatic carbon chain to either another amino group, in the case of alkyl polyamines and piperazine-related heterocyclic amines, or to a hydroxyl group, in the case of alkanolamines. Current aliphatic amine production heavily relies on fossil feedstock and is facing pressing sustainability, health and safety issues. These issues can be fundamentally resolved by shifting from fossil feedstock to biomass. Consequently, the academic and industrial interest in bio-based aliphatic amines has systematically increased over the past years. Similar to the current aliphatic amine industry, the production of these bio-based amines will be the middle part of a larger, ideally circular, industrial value chain. This value chain additionally includes, as the first part, the refinery of biomass to suitable substrates and, as the final part, the utilization of these amines across various applicative domains. Therefore, applying a holistic perspective already in the research stage enables to acknowledge the requirements and limitations of each part and to efficiently bridge knowledge gaps between the different parts (Fig. 13).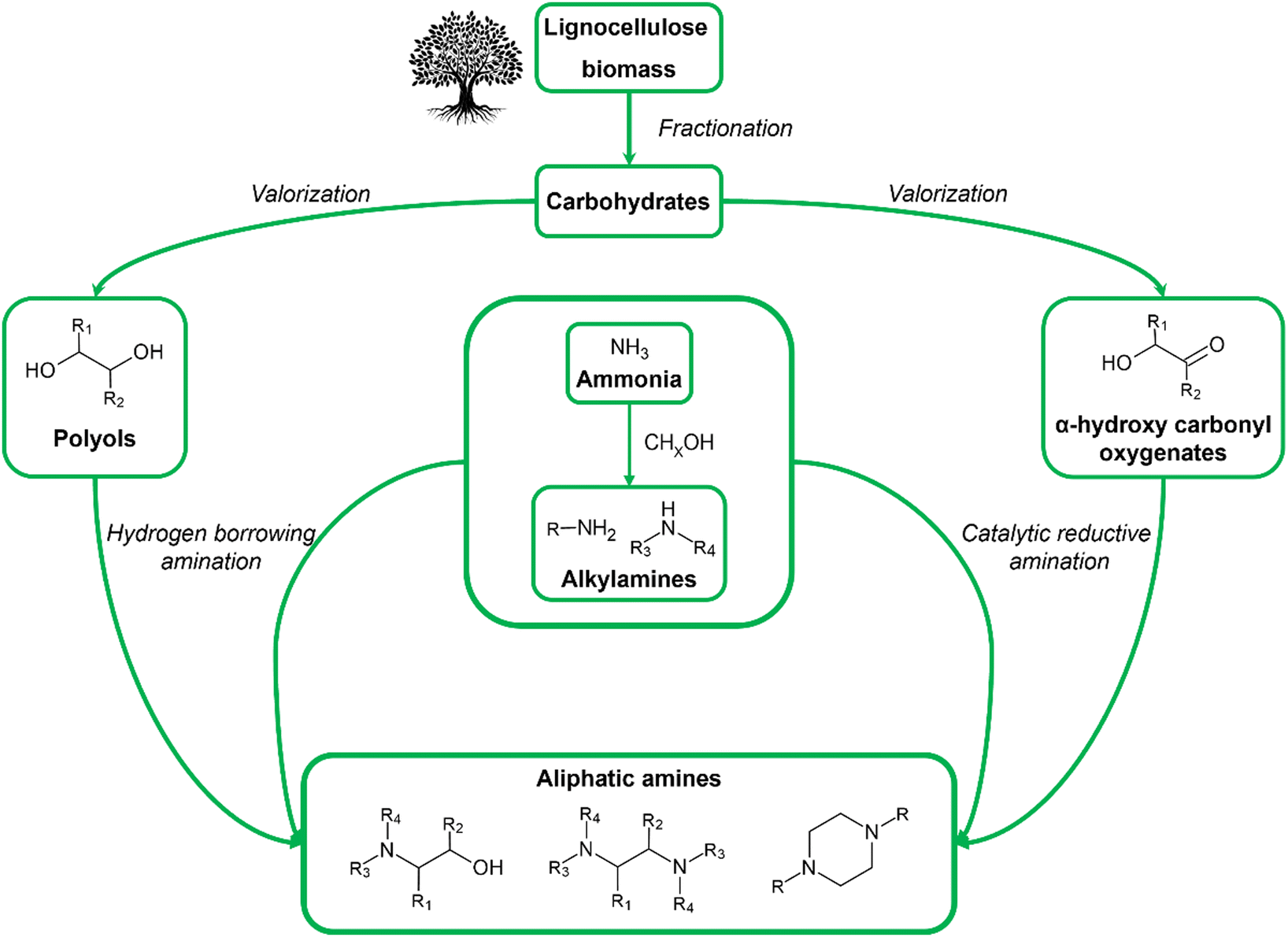 | ||
| Fig. 13 Schematic overview of the holistic aliphatic amine industry. The carbon part of the aliphatic amines originates from oxygenates obtained via valorization of lignocellulose-derived carbohydrates. The nitrogen part originates from green ammonia and bio-alcohol-derived alkylamines. | ||
5.1. Chemocatalytic (hemi)cellulose valorization
Lignocellulose, the major structural constituent of plant cell walls, is universally recognized as an alternative feedstock for the production of chemicals due to its abundance, low cost and susceptibility to chemical modifications. In the context of the holistic bio-based aliphatic amine value chain, the carbohydrate fraction of lignocellulose, namely the aliphatic polymers cellulose and hemicellulose, emerges as a suitable feedstock. Carbohydrates circumvent many health and safety concerns associated with the current fossil-based amine process as they are non-toxic and safe to handle. Hence, the chemocatalytic transformation of lignocellulose-derived carbohydrate feedstock into suitable substrates is a heavily studied and well-known process that passes through two stages. In the first stage, the biopolymers are depolymerized into their corresponding monomers (e.g., glucose and xylose) via acid-catalyzed hydrolysis. In the second stage, these carbohydrate monomers are further upgraded into multiple smaller hydroxyl- and carbonyl-functionalized oxygenates. This chemocatalytic valorization involves a multi-step process comprising four elementary key reactions, namely isomerization, retro-aldol condensation, dehydration and (de)hydrogenation. Since these four elementary reactions occur consecutively, various oxygenates can be derived by chemocatalytic valorization from a single substrate. For example, glucose can simultaneously be converted into the sugar alcohol sorbitol, multiple polyols (e.g., ethylene glycol (EG), 1,2-propylene glycol (1,2 PG), glycerol) and hydroxy carbonyl oxygenates (e.g., glyceraldehyde, glycolaldehyde (GA), acetol).In the state of the art, this product selectivity challenge is managed by consciously fine-tuning two aspects of the reaction system: (i) the reaction conditions and (ii) the catalytic system. On the one hand, reaction conditions such as temperature and H2 pressure strongly affect product selectivity. For example, retro-aldol condensation and dehydrogenation, both endothermic reactions, benefit from an elevated reaction temperature, whereas hydrogenation is favored under an increased H2 pressure. On the other hand, the intrinsic properties of the catalytic system determine which competitive key reactions are promoted. For instance, the presence of transition metals in the catalytic system is essential to facilitate (de)hydrogenation, which require noble (e.g., Ru, Pt) or non-noble metals (e.g., Cu, Ni), as well as retro-aldol condensation, which is catalyzed by tungsten-group metals. In addition, the presence of acid or base sites strongly influences the selectivity of the catalytic system. Acid sites favor dehydration, while basic sites facilitate both isomerization and retro-aldol condensation. Furthermore, acid–base pair sites improve the dehydrogenation activity of the catalytic system. In the state of the art, two successful approaches have been described in terms of catalytic system. It can either consist of one integrated multifunctional catalyst containing all required properties or a binary system in which two catalysts possess complementary properties.
These mechanism-derived handles can be grouped in a set of general strategies to selectively steer carbohydrate upgrading toward a variety of oxygenates.
5.2. Bio-based aliphatic amines via catalytic reductive amination
Carbohydrate-derived oxygenates, ranging from bifunctional O2 substrates such as GA and EG to O6 substrates such as glucose, are chemocatalytically converted into aliphatic amines via catalytic reductive amination. Notably, reductive amination of oxygenates with at least two oxygen-containing functional groups is a multi-step process that yields all three types of aliphatic amines, encompassing alkanolamines, alkyl polyamines and aliphatic heterocyclic amines. In this way, a striking parallel emerges between carbohydrate upgrading and reductive amination. Both chemocatalytic processes encounter the same challenge of achieving a desirable product selectivity amidst multiple potential products originating from a single substrate. Furthermore, both multi-step processes are influenced by the same four elementary reactions, namely isomerization, retro-aldol condensation, dehydration and (de)hydrogenation. From a mechanistic point of view, reductive amination inherently includes dehydration and (de)hydrogenation as elementary steps, while all four key reactions can take place as substrate-modifying, pre-amination reactions.In the state of the art, five main control handles have been investigated: (i) the catalytic system (i.e., catalyst and co-catalyst selection), (ii) process design (i.e., one-step or two-step processes), (iii) reaction temperature, (iv) amine-to-substrate (ATS) molar ratio, and (v) solvent choice. While the first three handles are also pivotal in the selectivity challenge of (hemi)cellulose valorization, the latter two are specific to the selectivity challenge in catalytic reductive amination. To date, this bottom-up methodology has been applied to the reductive amination of GA and acetol. In other words, cross-pollination of information between the two first parts of the holistic value chain remains limited to these two substrates, indicating a substantial opportunity to broaden the substrate scope.
Another notable future research direction relates to further expanding the aliphatic amine product scope. Of the three types of aliphatic amines, only aliphatic heterocyclic amines, such as piperazine and its derivatives, have not been systematically studied. From an academic standpoint, their selective synthesis would contribute to an even more profound understanding of the reaction pathway, while from an industrial standpoint, these compounds are indispensable in various applicative domains.
A final future research direction, which is also valid for lignocellulose valorization, involves the evolution from lab-scale reactions to industrialization by upscaling the process. This transition is accompanied by a set of new priorities and research topics. Some potential topics include the use of highly concentrated industrial feedstock solutions, catalyst development with emphasis on catalyst recycling and stability, reactor configuration (e.g., batch, fed-batch, or a continuous set-up), downstream processing, etc. Furthermore, the development of an industrial process introduces new perspectives and evaluation criteria. In addition to selectivity and productivity, a potentially viable industrial process will be assessed using a multitude of criteria, including economics, sustainability, health and safety, work-up requirements, etc. underscoring the importance of a techno-economic analysis and life cycle assessment as decision-making tools when upscaling the process.
5.3. Applicative domains of (bio-based) aliphatic amines
Selectively produced bio-based aliphatic amines are indispensable compounds in a range of applications, the final part of the holistic bio-based aliphatic amine value chain. The pivotal role of aliphatic amines is exemplified by three diverse applicative domains that all require distinct characteristics of the employed (bio-based) aliphatic amine: (i) CO2-reactive applications, (ii) polymerization and (iii) quaternary ammonium compounds.5.3.1.1. Carbon capture. On the one hand, these characteristic interactions can be exploited in carbon capture, in which anthropogenic CO2 is removed from industrial flue gas streams using amine-based absorption. Traditional capturing systems demonstrate either fast absorption kinetics or favorable desorption. Consequently, research efforts have centered around the design of novel amine systems that integrate rapid absorption with facile desorption, while considering additional criteria such as resistance to degradation and low volatility.
5.3.1.2. Carbon capture: structural requirements. The most promising amine capture systems, both water-rich and water-lean, contain at least one sterically hindered primary or secondary amino group, as these amines generally combine fast absorption and favorable desorption thermodynamics.
5.3.1.3. Switchable solvents. On the other hand, the reactivity of aliphatic amines toward CO2 can be exploited in switchable solvents, in which the presence of CO2 either alters the ionic strength (i.e., switchable water, SW) or interchanges the hydrophilic–hydrophobic nature of the amine solvent (i.e., switchable hydrophilicity solvents, SHSs). Compared to traditional systems, switchable solvents are regarded as energy-efficient separation methods suitable for extraction, product purification and catalyst recovery.
5.3.1.4. Switchable solvents: structural requirements. Tertiary and sterically hindered secondary polyamines are preferentially used as switchable solvents due to their facile reversibility. Furthermore, each amino group of the polyamine can interact with CO2 resulting in a more pronounced switchable effect. As SW and SHS require a hydrophilic and hydrophobic aliphatic amine, respectively, the water-miscibility of the amine can be modulated through its C/N ratio and the incorporation of alkyl substituents.
5.3.2.1. Epoxy resins. Epoxy resins are irreversibly transformed into a three-dimensional epoxy polymer by using curing agents such as aliphatic amines. Primary and secondary polyamines act as co-reactive curing agents by reacting with the epoxide functionalities of the resins, accelerating both polymerization and crosslinking. These co-reactive curing agents are incorporated into the polymer network and, hence, strongly influence the physicochemical properties of the resulting epoxy polymer.
5.3.2.2. Epoxy resins: structural requirements. Commercial ethylene polyamines have been extensively used as co-reactive curing agents, but they suffer from health- and safety-related drawbacks. In this regard, ethanolpolyamines seem to be promising alternatives since preliminary research indicates that they mitigate these shortcomings while remaining highly reactive.
5.3.2.3. Polyurethanes (PUR). PUR chemistry is characterized by a multitude of possible polymerization and crosslinking reactions. Therefore, catalysis is essential to consciously control each of these reactions. In this way, the choice of catalyst strongly affects the final properties of the polymer product. Tertiary aliphatic amines are an important class of PUR catalysts that affect the polymerization reactions via their Lewis basicity.
5.3.2.4. PUR: structural requirements. Over the past years, the high vapor pressure and susceptibility to oxidative degradation of traditional tertiary amines have decreased their use and stimulated the development of non-emissive, reactive tertiary amines. These reactive catalysts are functionalized with an additional hydroxyl or primary/secondary amino group that chemically binds into the polymer network.
5.3.2.5. Atom transfer radical polymerization (ATRP). ATRP is a robust reversible-deactivation radical polymerization method yielding well-defined and uniform polymers by the activation and deactivation of radicals regulated by a homogeneous catalytic complex. This catalytic complex consists of a transition metal, generally Cu, supported by a linear or branched tertiary alkyl polyamine ligand.
5.3.2.6. ATRP: structural requirements. To date, the most active and selective ATRP catalysts contain a polyamine ligand with three to four tertiary amino groups separated by two carbon atom linkers.
5.3.3.1. QACs: structural requirements. Recently, aliphatic multiQACs (i.e., linear and branched alkyl polyamines with at least two charged nitrogen atoms) have emerged as promising next-generation antibacterial agents. Compared with current antibacterial QACs, these multiQACs are more active, less susceptible to resistance development and sometimes even possess promising biofilm-eradicating properties. Moreover, preliminary results indicate that the biodegradability of these multiQACs can be enhanced by incorporating cleavable ester or amide side chains.
The growing significance of sustainability within the chemical industry has led to increased awareness regarding health and safety requirements. For example, (eco)toxicity has become an important evaluation criterion that should be considered in the development of novel aliphatic amines in various domains since it is a genuine concern for certain prevailing commercial amines. Furthermore, the development of bio-based monomers has gained profound research interest in recent years. However, a polymer can only be labeled fully bio-based if every component originates from biomass, including (reactive) aliphatic amine catalysts.
In this part of the holistic value chain, cross-pollination of information between different domains could also be valuable. Challenges of a certain domain could already be well-established structure–activity principles in another domain. For example, while amine blush, oxidative degradation and volatility of aliphatic amines are emerging and unresolved topics in PUR chemistry, they have been extensively investigated as fundamental principles in carbon capture.
5.4. Closing note
From a holistic perspective, bio-based aliphatic amines represent a versatile and valuable group of chemicals. Through catalytic reductive amination, a variety of non-toxic, safe-to-handle, carbohydrate-derived substrates can be transformed into numerous alkyl polyamines, alkanolamines and aliphatic heterocyclic amines. The disparate structural properties of these three aliphatic amines make them fundamental components across a broad spectrum of applicative domains.Abbreviations
| 1,2DA3P | 1,2-Diamino-3-propanol |
| 1,2PDA | 1,2-Propylenediamine |
| 1,2PG | 1,2-Propylene glycol |
| 1,2TMPDA | N,N,N′,N′-Tetramethyl-1,2-propylenediamine |
| 1,3PG | 1,3-Propylene glycol |
| 1,3TEPDA | N,N,N′,N′-Tetraethyl-,1,3-propylenediamine |
| 1,3TMPDA | N,N,N′,N′-Tetramethyl-1,3-propylenediamine |
| 1A2P | 1-Amino-2-propanol |
| 1DMA2P | N,N-Dimethyl-1-amino-2-propanol |
| 2A1,3PG | 2-Amino-1,3-propylene glycol |
| 2A1P | 2-Amino-1-propanol |
| 2EEDiPEDA | N-(2-Ethoxyethyl)-N′,N′-diisopropylethylenediamine |
| 2EEMPA | N-(2-Ethoxyethyl)-3-morpholinopropyleneamine |
| 2iPA1P | N-Isopropyl-2-amino-1-propanol |
| 3(DMAE)A | Tris(2-dimethylaminoethyl)amine |
| 3(DMAP)A | Trsi(2-dimethylaminopropyl)amine |
| 3A1,2PG | 3-Amino-1,2-propylene glycol |
| 3DEA1,2PG | N,N-Diethyl-3-amino-1,2-propylene glycol |
| 3DEA1P | N,N-Diethyl-3-amino-1-propanol |
| 3DMA1P | N,N-Dimethyl-3-amino-1-propanol |
| AMP | 2-Amino-2-methyl-1-propanol |
| AMT | Ammonium metatungstate |
| ATRP | Atom transfer radical polymerization |
| ATS molar ratio | Amine-to-substrate molar ratio |
| BAC | Benzalkonium chloride |
| BHEDMEDA | N,N′-Bis(2-hydroxyethyl)-N,N′-dimethylethylenediamine |
| CCUS | Carbon capture utilization and/or storage |
| CPC | Cetylpyridinium chloride |
| CTAB | Cetyltrimethylammonium bromide |
| DBEA | N,N-Dibutylethanolamine |
| DDAC | Dimethyldodecylammonium chloride |
| DEA | Diethanolamine |
| DEEA | N,N-Diethylethanolamine |
| DEGA | N,N-Diethylglucamine |
| DETriA | Diethylenetriamine |
| DHA | Dihydroxyacetone |
| DMA | Dimethylamine |
| DMAMP | N,N-Dimethyl-2-amino-2-methyl-1-propanol |
| DMCHA | N,N-Dimethylcyclohexylamine |
| DMEA | N,N-Dimethylethanolamine |
| DMPZ | N,N′-Dimethylpiperazine |
| DMSO | Dimethyl sulfoxide |
| EDA | Ethylenediamine |
| EDC | Ethylene dichloride |
| EG | Ethylene glycerol |
| EO | Ethylene oxide |
| EPOL | 1-Ethyl-3-pyrrolidinol |
| GA | Glycolaldehyde |
| HMF | 5-Hydroxymethylfurfural |
| HMTriETA | N,N,N′,N′′,N′′′,N′′′-Hexamethyltriethylenetetramine |
| HMTriPTA | N,N,N′,N′′,N′′′,N′′′-Hexamethyltripropylenetetramine |
| MBEA | N-Monobutylethanolamine |
| MBEC | Minimum biofilm elimination concentration |
| MDEA | N-Methyldiethanolamine |
| MEA | Monoethanolamine |
| MEEA | N-Monoethylethanolamine |
| MeOH | Methanol |
| MHEDETriA | N-(2-Monohydroxyethyl)-diethylenetriamine |
| MIC | Minimum inhibitory concentration |
| MiPEA | N-Monoisopropylethanolamine |
| MMA | Monomethylamine |
| MMEA | N-Monomethylethanolamine |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| PA | Pseudomonas aeruginosa |
| PMDETriA | N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine |
| PTriA | 1,2,3-Propylenetriamine |
| PUR | Polyurethane |
| PZ | Piperazine |
| QAC | Quaternary ammonium compound |
| RDRP | Reversible-deactivation radical polymerization |
| SA | Staphylococcus aureus |
| SHS | Switchable hydrophilicity solvent |
| SW | Switchable water |
| THF | Tetrahydrofuran |
| TMA | Trimethylamine |
| TMEDA | N,N,N′,N′-Tetramethylethylenediamine |
| TriEA | Triethanolamine |
| TriEDA | Triethylenediamine |
| TriEPA | Triethylenepentamine |
| TriETA | Triethylenetetramine |
| TriMAEEA | N,N,N′-Trimethylaminoethylethanolamine |
| TriMEDA | N,N,N′-Trimethylethylenediamine |
Data availability
All data is obtained from peer-reviewed articles, books, and patents as reported in the References list. No other datasets have been used.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. V. and B. F. S. acknowledge the C2 project of KU Leuven on bio-based amines. S. V. P. and B. F. S. acknowledge the Flemish government for their financial support in the A3 and Mimosa 2.0 project (Vlaio). W. A. and B. F. S. thank internal funding from KU Leuven (IDN-FFASSD). T. N. acknowledges the BioApp C3/20/120 – Industrial Research Fund (IOF) KU Leuven for the doctoral fellowship. C. Z. thanks the Chinese Scholarship Council (no. 201906310137) for financial support.References
- S. A. Lawrence, Amines: synthesis, properties, and applications, Cambridge University Press, 2004 Search PubMed.
- P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2015, pp. 1–55 Search PubMed.
- V. Pattabathula, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, 2019, pp. 1–33 Search PubMed.
- M. Appl, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2011, pp. 107–137 Search PubMed.
- M. Appl, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2011, vol. 3, pp. 139–225 Search PubMed.
- G. J. Leigh, in Catalysts for Nitrogen Fixation, ed. B. E. Smith, R. L. Richards and W. E. Newton, Springer, 2004, pp. 33–54 Search PubMed.
- P. Roose, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2015, pp. 1–10 Search PubMed.
- D. R. Corbin, S. Schwarz and G. C. Sonnichsen, Catal. Today, 1997, 37, 71–102 CrossRef CAS.
- K. S. Hayes, Appl. Catal., A, 2001, 221, 187–195 CrossRef CAS.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, 4th edn, 2003, pp. 51–52 Search PubMed.
- H. Zimmermann and R. Walzl, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2009, pp. 465–529 Search PubMed.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, 4th edn, 2003, pp. 63–66 Search PubMed.
- Y. Gao, L. Neal, D. Ding, W. Wu, C. Baroi, A. M. Gaffney and F. Li, ACS Catal., 2019, 9, 8592–8621 CrossRef CAS.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, 4th edn, 2003, pp. 145–152 Search PubMed.
- S. Rebsdat and D. Mayer, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2001, pp. 547–572 Search PubMed.
- E.-L. Dreher, K. K. Beutel, J. D. Myers, T. Lübbe, S. Krieger and L. H. Pottenger, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2014, pp. 1–81 Search PubMed.
- M. Frauenkron, J.-P. Melder, G. Ruider, R. Rossbacher and H. Höke, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2001, pp. 405–431 Search PubMed.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, 4th edn, 2003, pp. 159–162 Search PubMed.
- C. Jones, M. R. Edens and J. F. Lochary, Kirk-Othmer Encyclopedia of Chemical Technology, Wiley-VCH, 2004, pp. 122–147 Search PubMed.
- S. Sridhar and R. G. Carter, Kirk-Othmer Encyclopedia of Chemical Technology, Wiley-VCH, 2001, vol. 8, pp. 485–519 Search PubMed.
- E. E. Sergeev, L. L. Gogin, T. B. Khlebnikova, E. G. Zhizhina and Z. P. Pai, Catal. Ind., 2022, 14, 218–230 CrossRef.
- Maximize Market Research, Aliphatic Amines Market – Global Industry Analysis and Forecast (2024–2030), https://www.maximizemarketresearch.com/market-report/global-aliphatic-amines-market/69637/, (accessed 17 July 2024).
- S&P Global, Ethanolamines, https://www.spglobal.com/commodityinsights/en/ci/products/ethanolamines-chemical-economics-handbook.html, (accessed 17 July 2024).
- S&P Global, Ethyleneamines, https://www.spglobal.com/commodityinsights/en/ci/products/ethyleneamines-chemical-economics-handbook.html, (accessed 17 July 2024).
- Organization of the Petroleum Exporting Countries (OPEC), 2022 World Oil Outlook 2045, 2022.
- International Energy Agency, The future of petrochemicals – Towards more sustainable plastics and fertilizers, 2018.
- E. De Jong, H. Stichnothe, G. Bell and H. Jorgensen, Bio-Based Chemicals: A 2020 Update, 2020 Search PubMed.
- J. Harnisch, C. Jubb, A. Nakhutin, V. C. S. Cianci, R. Lanza, T. Martinsen, A. K. W. Mohammad, M. M. O. Santos, A. McCulloch and B. T. Mader, in 2006 IPCC Guidelines for National Greenhouse Gas Inventories. V, Intergovernmental Panel on Climate Change, 2006, p. chapter 3.
- A. Boulamanti and J. A. Moya, Energy efficiency and GHG emissions: Prospective scenarios for the Chemical and Petrochemical Industry, 2017 Search PubMed.
- W. H. Faveere, S. Van Praet, B. Vermeeren, K. N. R. Dumoleijn, K. Moonen, E. Taarning and B. F. Sels, Angew. Chem., 2021, 133, 12312–12331 CrossRef.
- European Chemicals Agency, Ethylene oxide: substance infocard, https://echa.europa.eu/substance-information/-/substanceinfo/100.000.773.
- European Chemicals Agency, 1,2-dichloroethane: substance infocard, https://echa.europa.eu/substance-information/-/substanceinfo/100.003.145.
- M. Pera-Titus and F. Shi, ChemSusChem, 2014, 7, 720–722 CrossRef CAS PubMed.
- V. Froidevaux, C. Negrell, S. Caillol, J. P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- M. Pelckmans, T. Renders, S. Van De Vyver and B. F. Sels, Green Chem., 2017, 19, 5303–5331 RSC.
- S. Hameury, H. Bensalem and K. D. O. Vigier, Catalysts, 2022, 12, 1–35 CrossRef.
- N. K. Gupta, P. Reif, P. Palenicek and M. Rose, ACS Catal., 2022, 12, 10400–10440 CrossRef CAS.
- L. Krietsch Boerner, Industrial ammonia production emits more CO2 than any other chemical-making reaction. Chemists want to change that, https://cen.acs.org/environment/green-chemistry/Industrial-ammonia-production-emits-CO2/97/i24.
- S. Ghavam, M. Vahdati, I. A. G. Wilson and P. Styring, Front. Energy Res., 2021, 9, 1–19 Search PubMed.
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8, 1–25 Search PubMed.
- K. H. R. Rouwenhorst, P. M. Krzywda, N. E. Benes, G. Mul and L. Lefferts, Ullmann's Encyclopedia of Industrial Chemistry, 2020, pp. 1–20 Search PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- M. J. Biddy, C. J. Scarlata and C. M. Kinchin, Chemicals from biomass: A market assessment of bioproducts with near-term potential, 2016 Search PubMed.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Advances in Catalysis, Elsevier, 2017, vol. 60, pp. 59–123 Search PubMed.
- G. Hayes, M. Laurel, D. Mackinnon, T. Zhao, H. A. Houck and C. R. Becer, Chem. Rev., 2023, 123, 2609–2734 CrossRef CAS PubMed.
- M. Christou and E. Alexopoulou, in Biorefinery: From Biomass to Chemicals and Fuels, ed. M. Aresta, A. Dibenedetto and F. Dumeignil, De Gruyter, 2012, pp. 49–80 Search PubMed.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- W. Deng, Y. Feng, J. Fu, H. Guo, Y. Guo, B. Han, Z. Jiang, L. Kong, C. Li, H. Liu, P. T. T. Nguyen, P. Ren, F. Wang, S. Wang, Y. Wang, Y. Wang, S. S. Wong, K. Yan, N. Yan, X. Yang, Y. Zhang, Z. Zhang, X. Zeng and H. Zhou, Green Energy Environ., 2023, 8, 10–114 CrossRef CAS.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed.
- X. Chen, H. Yang and N. Yan, Chem. – Eur. J., 2016, 22, 13402–13421 CrossRef CAS PubMed.
- M. J. Hülsey, Green Energy Environ., 2018, 3, 318–327 CrossRef.
- M. Kaisler, L. A. M. van den Broeck and C. Boeriu, Chitin and chitosan: properties an applications, Elsevier, 1st edn, 2020, pp. 229–244 Search PubMed.
- T. T. Pham, X. Chen, T. Söhnel, N. Yan and J. Sperry, Green Chem., 2020, 22, 1978–1984 RSC.
- W. Schutyser, T. Renders, S. Van Den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- A. M. Raspolli Galletti and C. Antonetti, in Biorefinery: From Biomass to Chemicals and Fuels, ed. M. Aresta, A. Dibenedetto and F. Dumeignil, De Gruyter, 2012, pp. 101–121 Search PubMed.
- Z. Sun and K. Barta, Chem. Commun., 2018, 54, 7725–7745 RSC.
- X. Shen and R. Sun, Carbohydr. Polym., 2021, 261, 117884 CrossRef CAS PubMed.
- M. Ragnar, G. Henriksson, M. E. Lindström, M. Wimby, J. Blechschmidt and S. Heinemann, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2014, pp. 1–92 Search PubMed.
- T. Renders, S. Van Den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed.
- Z. Sun, A. De Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- A. A. Ahmad, N. A. Zawawi, F. H. Kasim, A. Inayat and A. Khasri, Renewable Sustainable Energy Rev., 2016, 53, 1333–1347 CrossRef CAS.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- C. B. Schandel, M. Høj, C. M. Osmundsen, A. D. Jensen and E. Taarning, ChemSusChem, 2020, 13, 688–692 CrossRef CAS PubMed.
- A. J. J. Straathof, Chem. Rev., 2014, 114, 1871–1908 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- J. A. Geboers, S. Van De Vyver, R. Ooms, B. Op De Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 RSC.
- I. Delidovich and R. Palkovits, ChemSusChem, 2016, 9, 547–561 CrossRef CAS PubMed.
- I. Delidovich, ACS Catal., 2023, 13, 2250–2267 CrossRef CAS.
- H. Li, S. Yang, S. Saravanamurugan and A. Riisager, ACS Catal., 2017, 7, 3010–3029 CrossRef CAS.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- J. M. de Bruijn, A. P. G. Kieboom and H. van Bekkum, Starch/Stärke, 1987, vol. 39, pp. 23–28 Search PubMed.
- C. Liu, J. M. Carraher, J. L. Swedberg, C. R. Herndon, C. N. Fleitman and J.-P. Tessonnier, ACS Catal., 2014, 4, 4295–4298 CrossRef CAS.
- C. Moreau, R. Durand, A. Roux and D. Tichit, Appl. Catal., A, 2000, 193, 257–264 CrossRef CAS.
- P. Zhu, H. Li and A. Riisager, Catalysts, 2022, 12, 1–19 Search PubMed.
- E. Peeters, S. Calderon-Ardila, I. Hermans, M. Dusselier and B. F. Sels, ACS Catal., 2022, 12, 9559–9569 CrossRef CAS.
- P. Sun, C. Liu, H. Wang, Y. Liao, X. Li, Q. Liu, B. F. Sels and C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215737 CrossRef CAS PubMed.
- L. Petruš, M. Petrušova and Z. Hricovíniová, in Glycoscience, ed. A. E. Stütz, Springer, Berlin, 2001, vol. 215, pp. 15–41 Search PubMed.
- F. Ju, D. Vander Velde and E. Nikolla, ACS Catal., 2014, 4, 1358–1364 CrossRef CAS.
- M. Zheng, J. Pang, R. Sun, A. Wang and T. Zhang, ACS Catal., 2017, 7, 1939–1954 CrossRef CAS.
- C. B. Schandel, M. Høj, C. M. Osmundsen, M. J. Beier, E. Taarning and A. D. Jensen, ACS Sustainable Chem. Eng., 2021, 9, 305–311 CrossRef CAS.
- J. Zhang, B. Hou, A. Wang, Z. Li, H. Wang and T. Zhang, Am. Inst. Chem. Eng., 2014, 60, 3804–3813 CrossRef CAS.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–606 CrossRef CAS PubMed.
- A. Bayu, A. Abudula and G. Guan, Fuel Process. Technol., 2019, 196, 106162 CrossRef CAS.
- H. Baniamerian, H. Martin, M. Josef and A. D. Jensen, Appl. Catal., B, 2023, 330, 122650 CrossRef CAS.
- Y. Liu, C. Luo and H. Liu, Angew. Chem., Int. Ed., 2012, 51, 3249–3253 CrossRef CAS PubMed.
- Y. Liu, W. Zhang, C. Hao, S. Wang and H. Liu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, 1–12 Search PubMed.
- C. Dussenne, T. Delaunay, V. Wiatz, H. Wyart, I. Suisse and M. Sauthier, Green Chem., 2017, 19, 5332–5344 RSC.
- F. Delbecq, M. R. Khodadadi, D. Rodriguez Padron, R. Varma and C. Len, Mol. Catal., 2020, 482, 110648 CrossRef CAS.
- E. Cousin, K. Namhaed, Y. Pérès, P. Cognet, M. Delmas, H. Hermansyah, M. Gozan, P. A. Alaba and M. K. Aroua, Sci. Total Environ., 2022, 847, 157599 CrossRef CAS PubMed.
- S. P. Teong, G. Yi and Y. Zhang, Green Chem., 2014, 16, 2015–2026 RSC.
- B. R. Caes, R. E. Teixeira, K. G. Knapp and R. T. Raines, ACS Sustainable Chem. Eng., 2015, 3, 2591–2605 CrossRef CAS.
- C. Chatterjee, F. Pong and A. Sen, Green Chem., 2015, 40–71 RSC.
- M. J. Ahmed and B. H. Hameed, J. Taiwan Inst. Chem. Eng., 2019, 96, 341–352 CrossRef CAS.
- S. Nishimura, Handbook of heterogeneous catalytic hydrogenation for organic synthesis, John Wiley & Sons, 2001 Search PubMed.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- N. H. R. Annuar, Z. A. Alexzman, A. R. M. Daud, A. F. N. Alias, H. M. Hairi and H. D. Setiabudi, J. Environ. Chem. Eng., 2022, 10, 107229 CrossRef CAS.
- Y. Huang, B. Wang, H. Yuan, Y. Sun, D. Yang, X. Cui and F. Shi, Catal. Sci. Technol., 2021, 11, 1652–1664 RSC.
- G. Liang, L. He, H. Cheng, W. Li, X. Li, C. Zhang, Y. Yu and F. Zhao, J. Catal., 2014, 309, 468–476 CrossRef CAS.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- H. N. Anchan, N. S. Bhat, N. Vinod, P. S. Prabhakar and S. Dutta, Biomass Convers. Biorefin., 2024, 14, 9915–9948 CrossRef CAS.
- H. Zhao, J. E. Holladay, Y. Wang, J. M. White and Z. C. Zhang, J. Biobased Mater. Bioenergy, 2017, 1, 210–215 CrossRef.
- Q. Xiang, Y. Y. Lee, P. O. Pettersson and R. W. Torget, Appl. Biochem. Biotechnol., 2003, 105–108, 505–514 CrossRef CAS PubMed.
- L. Hu, L. Lin, Z. Wu, S. Zhou and S. Liu, Appl. Catal., B, 2015, 174–175, 225–243 CrossRef CAS.
- R. Zhong and B. F. Sels, Appl. Catal., B, 2018, 236, 518–545 CrossRef CAS.
- J. Zhong, J. Pérez-Ramírez and N. Yan, Green Chem., 2021, 23, 18–36 RSC.
- L. Shuai and X. Pan, Energy Environ. Sci., 2012, 5, 6889–6894 RSC.
- N. S. Kapu and H. L. Trajano, Biofuels, Bioprod. Biorefin., 2014, 8, 857–870 CrossRef.
- H. Kobayashi, H. Kaiki, A. Shrotri, K. Techikawara and A. Fukuoka, Chem. Sci., 2016, 7, 692–696 RSC.
- V. I. Sharkov, Angew. Chem., Int. Ed. Engl., 1963, 2, 405–492 CrossRef.
- J. M. Robinson, C. E. Burgess, M. A. Bently, C. D. Brasher, B. O. Horne, D. M. Lillard, J. M. Macias, H. D. Mandal, S. C. Mills, K. D. O’Hara, J. T. Pon, A. F. Raigoza, E. H. Sanchez and J. S. Villarreal, Biomass Bioenergy, 2004, 26, 473–483 CrossRef CAS.
- R. Palkovits, K. Tajvidi, M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS PubMed.
- W. Deng, X. Tan, W. Fang, Q. Zhang and Y. Wang, Catal. Lett., 2009, 133, 167–174 CrossRef CAS.
- J. Geboers, S. Van De Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC.
- A. Negoi, K. Triantafyllidis, V. I. Parvulescu and S. M. Coman, Catal. Today, 2014, 223, 122–128 CrossRef CAS.
- L. S. Ribeiro, J. J. de, M. Órfão and M. F. R. Pereira, Bioresour. Technol., 2017, 244, 1173–1177 CrossRef CAS PubMed.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Acc. Chem. Res., 2018, 51, 761–768 CrossRef CAS PubMed.
- Y. Zhou, R. L. Smith and X. Qi, Green Chem., 2024, 26, 202–243 RSC.
- S. Saravanamurugan and A. Riisager, Catal. Sci. Technol., 2014, 4, 3186–3190 RSC.
- F. Carly and P. Fickers, Yeast, 2018, 35, 455–463 CrossRef CAS PubMed.
- D. A. Rzechonek, A. Dobrowolski, W. Rymowicz and A. M. Mirończuk, Crit. Rev. Biotechnol., 2018, 38, 620–633 CrossRef CAS PubMed.
- S. Tolborg, S. Meier, S. Saravanamurugan, P. Fristrup, E. Taarning and I. Sádaba, ChemSusChem, 2016, 9, 3054–3061 CrossRef CAS PubMed.
- E. Santacesaria, G. M. Vicente, M. Di Serio and R. Tesser, Catal. Today, 2012, 195, 2–13 CrossRef CAS.
- M. Checa, S. Nogales-Delgado, V. Montes and J. M. Encinar, Catalysts, 2020, 10, 1–41 CrossRef.
- P. F. F. Amaral, T. F. Ferreira, G. C. Fontes and M. A. Z. Coelho, Food Bioprod. Process., 2009, 87, 179–186 CrossRef CAS.
- B. Katryniok, H. Kimura, E. Skrzyńska, J. S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960–1979 RSC.
- J. Sun and H. Liu, Green Chem., 2011, 13, 135–142 RSC.
- C. Heisig, C. Glotzbach, S. Schirrmeister and T. Turek, Chem. Eng. Technol., 2021, 44, 761–772 CrossRef CAS.
- Y. Wang, J. Zhou and X. Guo, RSC Adv., 2015, 5, 74611–74628 RSC.
- Avantium Knowledge Centre, US10131600 B2, 2018.
- G. Zhao, M. Zheng, R. Sun, Z. Tai, J. Pang, A. Wang, X. Wang and T. Zhang, AIChE J., 2016, 63, 2072–2080 CrossRef.
- R. Ooms, M. Dusselier, J. A. Geboers, B. Op De Beeck, R. Verhaeven, E. Gobechiya, J. A. Martens, A. Redl and B. F. Sels, Green Chem., 2014, 16, 695–707 RSC.
- Y. Li, Y. Liao, X. Cao, T. Wang, L. Ma, J. Long, Q. Liu and Y. Xua, Biomass Bioenergy, 2015, 74, 148–161 CrossRef CAS.
- M. K. Wong, S. S. M. Lock, Y. H. Chan, S. J. Yeoh and I. S. Tan, Chem. Eng. J., 2023, 468, 143699 CrossRef CAS.
- A. N. Marchesan, M. P. Oncken, R. Maciel Filho and M. R. Wolf Maciel, Green Chem., 2019, 21, 5168–5194 RSC.
- M. Główka and T. Krawczyk, ACS Sustainable Chem. Eng., 2023, 11, 7274–7287 CrossRef.
- Y. Hirano, K. Sagata and Y. Kita, Appl. Catal., A, 2015, 502, 1–7 CrossRef CAS.
- Z. Xiao, S. Jin, G. Sha, C. T. Williams and C. Liang, Ind. Eng. Chem. Res., 2014, 53, 8735–8743 CrossRef CAS.
- C. Liu, C. Zhang, S. Hao, S. Sun, K. Liu, J. Xu, Y. Zhu and Y. Li, Catal. Today, 2016, 261, 116–127 CrossRef CAS.
- C. Liu, Z. Zhang, X. Zhai, X. Wang, J. Gui, C. Zhang, Y. Zhu and Y. Li, New J. Chem., 2019, 43, 3733–3742 RSC.
- A. A. B. Kirali, S. Sreekantan and B. Marimuthu, Catal. Commun., 2022, 165, 106447 CrossRef.
- T. Deng and H. Liu, J. Mol. Catal. A: Chem., 2014, 388–389, 66–73 CrossRef CAS.
- S. Basu and A. K. Sen, ChemBioEng Rev., 2021, 8, 633–653 CrossRef CAS.
- T. Rajkhowa, G. B. Marin and J. W. Thybaut, Appl. Catal., B, 2017, 205, 469–480 CrossRef CAS.
- R. A. Sheldon, D. Brady and M. L. Bode, Chem. Sci., 2020, 11, 2587–2605 RSC.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- T. Knaus, W. Böhmer and F. G. Mutti, Green Chem., 2017, 19, 453–463 RSC.
- S. D. Mürtz, N. Kurig, F. J. Holzhäuser and R. Palkovits, Green Chem., 2021, 23, 8428–8433 RSC.
- T. Kim, D. Il Park, S. Kim, D. Yadav, S. Hong, S. H. Kim, H. J. Yoon and K. Jin, Chem. Commun., 2023, 59, 4818–4821 RSC.
- S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037–1057 CrossRef CAS.
- K. Murugesan, T. Senthamarai, V. G. Chandrashekhar, K. Natte, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Chem. Soc. Rev., 2020, 49, 6273–6328 RSC.
- E. Podyacheva, O. Afanasyev, A. Tsygankov, M. Makarova and D. Chusov, Synthesis, 2019, 2667–2677 CAS.
- E. W. Baxter and A. B. Reitz, Organic Reactions, Wiley-VCH, 2004, pp. 1–57 Search PubMed.
- M. L. Moore, Organic Reactions, Wiley-VCH, 1949, vol. 5, pp. 301–324 Search PubMed.
- M. Kitamura, D. Lee, S. Hayashi, S. Tanaka and M. Yoshimura, J. Org. Chem., 2002, 67, 8685–8687 CrossRef CAS PubMed.
- A. De, N. C. Ghosal, S. Mahato, S. Santra, G. V. Zyryanov and A. Majee, ChemistrySelect, 2018, 3, 4058–4066 CrossRef CAS.
- D. Chusov and B. List, Angew. Chem., Int. Ed., 2014, 53, 5199–5201 CrossRef CAS PubMed.
- A. A. Tsygankov, M. Makarova and D. Chusov, Mendeleev Commun., 2018, 28, 113–122 CrossRef CAS.
- Y. Wang, S. Furukawa, X. Fu and N. Yan, ACS Catal., 2020, 10, 311–335 CrossRef CAS.
- T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS PubMed.
- J. P. Adams, C. M. Alder, I. Andrews, A. M. Bullion, M. Campbell-Crawford, M. G. Darcy, J. D. Hayler, R. K. Henderson, C. A. Oare, I. Pendrak, A. M. Redman, L. E. Shuster, H. F. Sneddon and M. D. Walker, Green Chem., 2013, 15, 1542–1549 RSC.
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- A. Baiker, Stud. Surf. Sci. Catal., 1988, 41, 283–290 CrossRef CAS.
- G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed.
- K. I. Shimizu, Catal. Sci. Technol., 2015, 5, 1412–1427 RSC.
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS.
- J. M. Carraher, C. N. Fleitman and J.-P. Tessonnier, ACS Catal., 2015, 5, 3162–3173 CrossRef CAS.
- C. Li, Y. Wang, Y. Zhang, M. Wang, X. Sun, H. Cui and Y. Xie, ChemistrySelect, 2020, 5, 270–279 CrossRef CAS.
- J. Tang, X. Guo, L. Zhu and C. Hu, ACS Catal., 2015, 5, 5097–5103 CrossRef CAS.
- Z. Zhou, X. Li, T. Zeng, W. Hong, Z. Cheng and W. Yuan, Chin. J. Chem. Eng., 2010, 18, 384–390 CrossRef CAS.
- J. E. Hodge, Agricultural and Food Chemistry, 1953, vol. 1, pp. 928–943 Search PubMed.
- H. Nursten, The Maillard Reaction: Chemistry, biochemistry and implications, Royal Society of Chemistry, 2005, pp. 5–30 Search PubMed.
- D. M. Roundhill, Chem. Rev., 1992, 92, 1–27 CrossRef CAS.
- J. He, L. Chen, S. Liu, K. Song, S. Yang and A. Riisager, Green Chem., 2020, 22, 6714–6747 RSC.
- D. Ruijten, T. Narmon, H. De Weer and R. Van Der Zweep, ChemSusChem, 2022, 15, e202200868 CrossRef CAS PubMed.
- D. Ruijten, T. Narmon, K. Van Aelst, H. De Weer, R. Van Der Zweep, T. Hendrickx, C. Poleunis, L. Li, K. M. Van Geem, D. P. Debecker and B. F. Sels, ACS Sustainable Chem. Eng., 2023, 11, 4776–4788 CrossRef CAS.
- X. Wu, M. V. Galkin and K. Barta, Chem Catal., 2021, 1, 1466–1479 CrossRef CAS.
- X. Wu, M. De Bruyn and K. Barta, ChemSusChem, 2022, 15, e202200914 CrossRef CAS PubMed.
- J. Ma, D. Le and N. Yan, Chem, 2023, 9, 1–12 Search PubMed.
- H. Li, H. Guo, Z. Fang, R. L. Smith and M. Aida, Green Chem., 2020, 22, 582–611 RSC.
- A. C. Fernandes, Methodologies in Amine Synthesis: Challenges and Applications, Wiley-VCH, 2021, pp. 341–376 Search PubMed.
- W. Deng, Y. Wang, S. Zhang, K. M. Gupta, M. J. Hülsey, H. Asakura, L. Liu, Y. Han, E. M. Karp, G. T. Beckham, P. J. Dyson, J. Jiang, T. Tanaka, Y. Wang and N. Yan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5093–5098 CrossRef CAS PubMed.
- R. Coeck and D. E. De Vos, Green Chem., 2020, 22, 5105–5114 RSC.
- A. Garcia-Ortiz, J. D. Vidal, M. J. Climent, P. Concepcion, A. Corma and S. Iborra, ACS Sustainable Chem. Eng., 2019, 6243–6250 CrossRef CAS.
- J. Sha, B. T. Kusema, W. Zhou, Z. Yan, S. Strei and M. Pera-titus, Green Chem., 2021, 23, 7093–7099 RSC.
- T. A. Gokhale and B. M. Bhanage, in Production of N-containing Chemicals and Materials from Biomass, ed. Z. Fang, R. L. Smith and L. Xu, Springer, 2023, pp. 21–71 Search PubMed.
- J. F. Lin, C. C. Wu and M. H. Lien, J. Phys. Chem., 1995, 99, 16903–16908 CrossRef CAS.
- P. Pérez and A. Toro-Labbé, Theor. Chem. Acc., 2001, 105, 422–430 Search PubMed.
- V. A. Yaylayan, A. H. Despointes and M. S. Feather, Crit. Rev. Food Sci. Nutr., 1994, 34, 321–369 CrossRef CAS PubMed.
- T. M. Wrodnigg and B. Eder, Glycoscience, Springer, 2001, pp. 115–152 Search PubMed.
- K. U. Leuven, WO2019/193117 A1, 2019.
- W. Faveere, T. Mihaylov, M. Pelckmans, K. Moonen, F. Gillis-D’Hamers, R. Bosschaerts, K. Pierloot and B. F. Sels, ACS Catal., 2020, 10, 391–404 CrossRef CAS.
- S. Van Praet, G. Preegel, F. Rammal and B. F. Sels, ACS Sustainable Chem. Eng., 2022, 10, 6503–6508 CrossRef CAS.
- Rhodia Operations, WO2021/114166 A1, 2021.
- S. Van Praet, F. Rammal, B. Vermeeren, T. Nicolaï, K. Dumoleijn, T. Rottiers and B. F. Sels, Chem. Eng. J., 2023, 475, 146316 CrossRef CAS.
- G. Liang, A. Wang, L. Li, G. Xu, N. Yan and T. Zhang, Angew. Chem., Int. Ed., 2017, 56, 3050–3054 CrossRef CAS PubMed.
- BASF SE, US2012/0271068 A1, 2012.
- BASF SE, US2009/0240084 A1, 2009.
- BASF SE, US11208373 B2, 2018.
- BASF SE, WO2020/178085A1, 2020, 1–42.
- Z. Xie, H. An, X. Zhao and Y. Wang, Mol. Catal., 2022, 528, 112492 CrossRef CAS.
- H. An, J. Li, G. Zheng, G. Wang, X. Zhao and Y. Wang, ChemistrySelect, 2022, 7, 1–11 CrossRef.
- BASF SE, WO2020/249427 A1, 2020.
- BASF SE, WO2020/249426 A1, 2020.
- J. Runeberg, A. Baiker and J. Kijenski, Appl. Catal., 1985, 17, 309–319 CrossRef CAS.
- M. Pelckmans, T. Mihaylov, W. Faveere, J. Poissonnier, F. Van Waes, K. Moonen, G. B. Marin, J. W. Thybaut, K. Pierloot and B. F. Sels, ACS Catal., 2018, 8, 4201–4212 CrossRef CAS.
- M. Pelckmans, W. Vermandel, F. Van Waes, K. Moonen and B. F. Sels, Angew. Chem., Int. Ed., 2017, 56, 14540–14544 CrossRef CAS PubMed.
- B. Vermeeren, S. Van Praet, W. Arts and B. F. Sels, Chem Catal., 2023, 3, 100602 CrossRef CAS.
- T. Trégner, J. Trejbal, N. Ruhswurmová and M. Zapletal, Chem. Biochem. Eng. Q., 2018, 31, 455–470 CrossRef.
- T. Takanashi, Y. Nakagawa and K. Tomishige, Chem. Lett., 2014, 43, 822–824 CrossRef CAS.
- C. Yue, K. Di, L.-P. Gu, Z.-W. Zhang and L.-L. Ding, Mol. Catal., 2019, 477, 110539 CrossRef.
- Wanhua Chemical Group, CN112125814A, 2020.
- B. Vermeeren, S. Van Praet, A. De Ridder and B. F. Sels, ACS Sustainable Chem. Eng., 2024, 12, 10209–10220 CrossRef CAS.
- Jefferson Chemical Company, US3448153, 1969.
- J. Boulos, F. Goc, T. Vandenbrouck, N. Perret, J. Dhainaut, S. Royer and F. Rataboul, ChemSusChem, 2024, 17, e202400540 CrossRef CAS PubMed.
- Procter & Gamble Company, US2007/0287865 A1, 2007.
- Merck, US5023379, 1991.
- Penn A Kem LLC, US8653306 B1, 2014.
- BASF SE, US2010/0240894A1, 2010.
- W. Wayne and H. Adkins, J. Am. Chem. Soc., 1940, 62, 3314–3316 CrossRef CAS.
- T. Seddig, T. Naundorf, H. Kipphardt and W. Maison, Mater. Corros., 2023, 74, 394–402 CrossRef CAS.
- BASF SE, US2010/0311973 A1, 2010.
- M. Safariamin, S. Paul, K. Moonen, D. Ulrichts, F. Dumeignil and B. Katryniok, Catal. Sci. Technol., 2016, 6, 2129–2135 RSC.
- J. Ding, M. Cui, T. Ma, R. Shao, W. Xu and P. Wang, Mol. Catal., 2018, 457, 51–58 CrossRef CAS.
- Dalian Institute, CN107011194 B, 2019.
- G. Drake and Y. Roman-Leshkov, Chem Catal., 2023, 3, 1–3 Search PubMed.
- A. Fischer, T. Mallat and A. Baiker, Catal. Today, 1997, 37, 167–189 CrossRef CAS.
- Jefferson Chemical Company, US 3151115, 1964.
- BASF SE, US 3270059, 1966.
- V. A. Yaylayan, S. Harty-Majors and A. A. Ismail, J. Agric. Food Chem., 1999, 47, 2335–2340 CrossRef CAS PubMed.
- J. Sun, S. So and G. da Silva, Int. J. Quantum Chem., 2017, 117, 1–5 CrossRef.
- J. Niemeier, R. V. Engel and M. Rose, Green Chem., 2017, 19, 2839–2845 RSC.
- F. Du, X. Jin, W. Yan, M. Zhao, P. S. Thapa and R. V. Chaudhari, Catal. Today, 2018, 302, 227–232 CrossRef CAS.
- X. Dai, J. Rabeah, H. Yuan, A. Brückner, X. Cui and F. Shi, ChemSusChem, 2016, 9, 3133–3138 CrossRef CAS PubMed.
- DuPont de Nemours & Company, US 2016962, 1935.
- Cerestar Holding BV, EP0536939A1, 1993.
- J. Poissonnier, M. Pelckmans, F. Van Waes, K. Moonen, B. F. Sels, J. W. Thybaut and G. B. Marin, Appl. Catal., B, 2018, 227, 161–169 CrossRef CAS.
- Taminco bvba, EP3290401 A1, 2018.
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A. C. Roger, R. Amal, H. He and S. E. Park, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- J. C. M. Pires, F. G. Martins, M. C. M. Alvim-Ferraz and M. Simões, Chem. Eng. Res. Des., 2011, 89, 1446–1460 CrossRef CAS.
- T. Wilberforce, A. G. Olabi, E. T. Sayed, K. Elsaid and M. A. Abdelkareem, Sci. Total Environ., 2021, 761, 143203 CrossRef CAS PubMed.
- M. Bui, C. S. Adjiman, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- F. de Meyer and S. Jouenne, Curr. Opin. Chem. Eng., 2022, 38, 1–7 Search PubMed.
- I. M. Bernhardsen and H. K. Knuutila, Int. J. Greenhouse Gas Control, 2017, 61, 27–48 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- E. Kayahan, U. Di Caprio, A. Van den Bogaert, M. N. Khan, M. Bulut, L. Braeken, T. Van Gerven and M. E. Leblebici, Chem. Eng. Process., 2023, 184, 109285 CrossRef CAS.
- C. Zhou, I. Khalil, F. Rammal, M. Dusselier, P. Kumar, M. Lacroix, E. Makshina, Y. Liao and B. F. Sels, ACS Catal., 2022, 12, 11485–11493 CrossRef CAS.
- F. de Meyer and C. Bignaud, Chem. Eng. J., 2022, 428, 1–12 CrossRef.
- F. Bougie and M. C. Iliuta, J. Chem. Eng. Data, 2012, 57, 635–669 CrossRef CAS.
- F. A. Chowdhury, H. Okabe, H. Yamada, M. Onoda and Y. Fujioka, Energy Procedia, 2011, 4, 201–208 CrossRef CAS.
- N. El Hadri, D. V. Quang, E. L. V. Goetheer and M. R. M. Abu Zahra, Appl. Energy, 2017, 185, 1433–1449 CrossRef CAS.
- S. Martin, H. Lepaumier, D. Picq, J. Kittel, T. De Bruin, A. Faraj and P. L. Carrette, Ind. Eng. Chem. Res., 2012, 51, 6283–6289 CrossRef CAS.
- F. Vega, A. Sanna, B. Navarrete, M. M. Maroto-Valer and V. Cortés, Greenhouse Gases: Sci. Technol., 2014, 4, 707–733 CrossRef CAS.
- P. Singh and G. F. Versteeg, Process Saf. Environ. Prot., 2008, 86, 347–359 CrossRef CAS.
- P. Singh, J. P. M. Niederer and G. F. Versteeg, Chem. Eng. Res. Des., 2009, 87, 135–144 CrossRef CAS.
- G. Rochelle, E. Chen, S. Freeman, D. Van Wagener, Q. Xu and A. Voice, Chem. Eng. J., 2011, 171, 725–733 CrossRef CAS.
- F. A. Chowdhury, H. Yamada, T. Higashii, K. Goto and M. Onoda, Ind. Eng. Chem. Res., 2013, 52, 8323–8331 CrossRef CAS.
- M. Xiao, H. Liu, H. Gao and Z. Liang, J. Chem. Thermodyn., 2018, 122, 170–182 CrossRef CAS.
- Q. Yang, G. Puxty, S. James, M. Bown, P. Feron and W. Conway, Energy Fuels, 2016, 30, 7503–7510 CrossRef CAS.
- G. Puxty, R. Rowland, A. Allport, Q. Yang, M. Bown, R. Burns, M. Maeder and M. Attalla, Environ. Sci. Technol., 2009, 43, 6427–6433 CrossRef CAS PubMed.
- E. F. da Silva and H. F. Svendsen, Int. J. Greenhouse Gas Control, 2007, 1, 151–157 CrossRef CAS.
- X. Rozanska, E. Wimmer and F. de Meyer, J. Chem. Inf. Model., 2021, 61, 1814–1824 CrossRef CAS PubMed.
- A. A. Orlov, A. Valtz, C. Coquelet, X. Rozanska, E. Wimmer, G. Marcou, D. Horvath, B. Poulain, A. Varnek and F. de Meyer, Commun. Chem., 2022, 5, 1–7 CrossRef PubMed.
- F. Vega, F. M. Baena-Moreno, L. M. Gallego Fernández, E. Portillo, B. Navarrete and Z. Zhang, Appl. Energy, 2020, 260, 114313 CrossRef CAS.
- P. Muchan, C. Saiwan, J. Narku-Tetteh, R. Idem, T. Supap and P. Tontiwachwuthikul, Chem. Eng. Sci., 2017, 170, 574–582 CrossRef CAS.
- N. Mirza and D. Kearns, State of the Art: CCS Technologies 2022, 2022.
- R. R. Wanderley, D. D. D. Pinto and H. K. Knuutila, Sep. Purif. Technol., 2021, 260, 118193 CrossRef CAS.
- J. Leclaire and D. J. Heldebrant, Green Chem., 2018, 20, 5058–5081 RSC.
- D. J. Heldebrant, P. K. Koech, V. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117, 9594–9624 CrossRef CAS PubMed.
- F. Barzagli, F. Mani and M. Peruzzini, Environ. Sci. Technol., 2016, 50, 7239–7246 CrossRef CAS PubMed.
- D. C. Cantu, D. Malhotra, M. T. Nguyen, P. K. Koech, D. Zhang, V. A. Glezakou, R. Rousseau, J. Page, R. Zheng, R. J. Perry and D. J. Heldebrant, ChemSusChem, 2020, 13, 3429–3438 CrossRef CAS PubMed.
- R. F. Zheng, D. Barpaga, P. M. Mathias, D. Malhotra, P. K. Koech, Y. Jiang, M. Bhakta, M. Lail, A. Rayer, G. A. Whyatt, C. J. Freeman, A. J. Zwoster, K. K. Weitz and D. J. Heldebrant, Energy Environ. Sci., 2020, 13, 4106–4113 RSC.
- D. Barpaga, Y. Jiang, R. F. Zheng, D. Malhotra, P. K. Koech, A. Zwoster, P. M. Mathias and D. J. Heldebrant, ACS Sustainable Chem. Eng., 2022, 10, 4522–4528 CrossRef CAS.
- C. J. Clarke, W. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102 CrossRef CAS PubMed.
- P. G. Jessop and M. F. Cunningham, CO2-switchable materials: solvents, surfactants, solutes and solids, Royal Society of Chemistry, 2020 Search PubMed.
- P. G. Jessop, Aldrichimica Acta, 2015, 48, 18–21 CAS.
- J. R. Vanderveen, S. Burra, J. Geng, A. Goyon, A. Jardine, H. E. Shin, T. Andrea, P. J. Dyson and P. G. Jessop, ChemPhysChem, 2018, 19, 2093–2100 CrossRef CAS PubMed.
- S. M. Mercer and P. G. Jessop, ChemSusChem, 2010, 3, 467–470 CrossRef CAS PubMed.
- S. M. Mercer, T. Robert, D. V. Dixon, C. S. Chen, Z. Ghoshouni, J. R. Harjani, S. Jahangiri, G. H. Peslherbe and P. G. Jessop, Green Chem., 2012, 14, 832–839 RSC.
- A. K. Alshamrani, J. R. Vanderveen and P. G. Jessop, Phys. Chem. Chem. Phys., 2016, 18, 19276–19288 RSC.
- S. Püschel, J. Sadowski, T. Rösler, K. R. Ehmann, A. J. Vorholt and W. Leitner, ACS Sustainable Chem. Eng., 2022, 10, 3749–3756 CrossRef PubMed.
- Y. Bazel, M. Rečlo and Y. Chubirka, Microchem. J., 2020, 157, 105115 CrossRef CAS.
- P. G. Jessop, L. Kozycz, Z. G. Rahami, D. Schoenmakers, A. R. Boyd, D. Wechsler and A. M. Holland, Green Chem., 2011, 13, 619–623 RSC.
- J. R. Vanderveen, J. Durelle and P. G. Jessop, Green Chem., 2014, 16, 1187–1197 RSC.
- C. Samorì, D. López Barreiro, R. Vet, L. Pezzolesi, D. W. F. Brilman, P. Galletti and E. Tagliavini, Green Chem., 2013, 15, 353–356 RSC.
- C. Russell and C. Rodriguez, Energy, 2023, 278, 127983 CrossRef CAS.
- S. Cheng, K. Rathnakumar and S. I. Martínez-Monteagudo, Foods, 2019, 8, 265 CrossRef CAS PubMed.
- V. Sapone, A. Iannone, A. Alivernini, A. Cicci, P. G. Jessop and M. Bravi, Sep. Purif. Technol., 2023, 308, 122843 CrossRef CAS.
- C. Samorì, D. Cespi, P. Blair, P. Galletti, D. Malferrari, F. Passarini, I. Vassura and E. Tagliavini, Green Chem., 2017, 19, 1714–1720 RSC.
- J. R. Vanderveen, J. Geng, S. Zhang and P. G. Jessop, RSC Adv., 2018, 8, 27318–27325 RSC.
- J. Cheng, H. Guo, Y. Qiu, Z. Zhang, Y. Mao, L. Qian, W. Yang and J. Y. Park, Bioresour. Technol., 2020, 312, 123607 CrossRef CAS PubMed.
- X. Li, Z. Yang, H. Sui, A. Jain and L. He, Fuel, 2018, 221, 303–310 CrossRef CAS.
- K. J. Viner, H. M. Roy, R. Lee, O. He, P. Champagne and P. G. Jessop, Green Chem., 2019, 21, 4786–4791 RSC.
- H. Q. Pham and M. J. Marks, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2005, pp. 156–244 Search PubMed.
- W. Brostow, S. H. Goodman and J. Wahrmund, Handbook of Thermoset Plastics, Elsevier, 3rd edn, 2014, pp. 191–252 Search PubMed.
- G. Gibson, Brydson's Plastics Materials, 8th edn, 2017, pp. 773–797 Search PubMed.
- F. L. Jin, X. Li and S. J. Park, J. Ind. Eng. Chem., 2015, 29, 1–11 CrossRef CAS.
- R. Auvergne, S. Caillol, G. David, B. Boutevin and J. P. Pascault, Chem. Rev., 2014, 114, 1082–1115 CrossRef CAS PubMed.
- E. A. Baroncini, S. Kumar Yadav, G. R. Palmese and J. F. Stanzione, J. Appl. Polym. Sci., 2016, 133, 44103 CrossRef.
- J. Liu, S. Wang, Y. Peng, J. Zhu, W. Zhao and X. Liu, Prog. Polym. Sci., 2021, 113, 101353 CrossRef CAS.
- K. Van Aelst, E. Van Sinay, T. Vangeel, Y. Zhang, T. Renders, S. Van den Bosch, J. Van Aelst and B. F. Sels, Chem. Commun., 2021, 57, 5642–5645 RSC.
- C. Pappa, E. Feghali, K. Vanbroekhoven and K. S. Triantafyllidis, Curr. Opin. Green Sustainable Chem., 2022, 38, 100687 CrossRef CAS.
- Y. Zhang, S. Stepanova, K. Van Aelst and B. F. Sels, Curr. Opin. Green Sustainable Chem., 2023, 40, 100750 CrossRef CAS.
- ThreeBond, Curing agents for epoxy resin, 1990.
- B. L. Burton, Epoxy Resin Formulators’ Meeting, 2006, pp. 1–17 Search PubMed.
- A. S. Mora, R. Tayouo, B. Boutevin, G. David and S. Caillol, Eur. Polym. J., 2020, 123, 1–14 CrossRef.
- A. S. Mora, R. Tayouo, B. Boutevin, G. David and S. Caillol, Green Chem., 2018, 20, 4075–4084 RSC.
- A. K. Ingberman, R. K. Walton, C. F. Pitt and M. N. Paul, Ind. Eng. Chem., 1957, 49, 1105 CrossRef CAS.
- M. F. Sonnenschein, Polyurethanes: Science, Technology, Markets, and Trends, John Wiley & Sons, 2nd edn, 2021 Search PubMed.
- G. Wegener, M. Brandt, L. Duda, J. Hofmann, B. Klesczewski, D. Koch, R. J. Kumpf, H. Orzesek, H. G. Pirkl, C. Six, C. Steinlein and M. Weisbeck, Appl. Catal., A, 2001, 221, 303–335 CrossRef CAS.
- B. Nohra, L. Candy, C. Guerin and Y. Raoul, Macromolecules, 2013, 46, 3771–3792 CrossRef CAS.
- P. Furtwengler and L. Avérous, Polym. Chem., 2018, 9, 4258–4287 RSC.
- A. Tenorio-Alfonso, M. C. Sánchez and J. M. Franco, J. Polym. Environ., 2020, 28, 749–774 CrossRef CAS.
- G. Brereton, R. M. Emanuel, R. Lomax, K. Pennington, T. Ryan, H. Tebbe, M. Timm, P. Ware, K. Winkler, T. Yuan, Z. Zhu, N. Adam, G. Avar, H. Blankenheim, W. Friederichs, M. Giersig, E. Weigand, M. Halfmann, F.-W. Wittbecker, D.-R. Larimer, U. Maier, S. Meyer-Ahrens, K.-L. Noble and H.-G. Wussow, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2019, pp. 1–76 Search PubMed.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- A. L. Silva and J. C. Bordado, Catal. Rev.: Sci. Eng., 2004, 46, 31–51 CrossRef.
- S. Dworakowska, D. Bogdał, F. Zaccheria and N. Ravasio, Catal. Today, 2014, 223, 148–156 CrossRef CAS.
- N. Malwitz, S. W. Wong, K. C. Frisch and P. A. Manis, J. Cell. Plast., 1987, 23, 461–502 CrossRef CAS.
- R. Van Maris, Y. Tamano, H. Yoshimura and K. M. Gay, J. Cell. Plast., 2005, 41, 305–322 CrossRef CAS.
- M. Muuronen, P. Deglmann and Ž. Tomović, J. Org. Chem., 2019, 84, 8202–8209 CrossRef CAS PubMed.
- B. Eling, Ž. Tomović and V. Schädler, Macromol. Chem. Phys., 2020, 221, 1–11 CrossRef.
- Huntsman Corporation, Performance Products JEFFCAT catalysts, 2012.
- BASF, Intermediates for the PUR Industry, 2015.
- Evonik, Additives for spray polyurethane foam (SPF) applications, 2017.
- A. Strachota, B. Strachotová and M. Špírková, Mater. Manuf. Processes, 2008, 23, 566–570 CrossRef CAS.
- Dow Global Technologies Inc, WO03/016372 A1, 2004.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- F. Gomollón-Bel, Chem. Int., 2019, 41, 12–17 Search PubMed.
- J. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimuras, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- M. Destarac, Polym. Chem., 2018, 9, 4947–4967 RSC.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- S. Dworakowska, F. Lorandi, A. Gorczyński and K. Matyjaszewski, Adv. Sci., 2022, 9, 2106076 CrossRef CAS PubMed.
- J. Xia and K. Matyjaszewski, Macromolecules, 1997, 30, 7697–7700 CrossRef CAS.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS PubMed.
- T. G. Ribelli, F. Lorandi, M. Fantin and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, 1–44 CrossRef PubMed.
- E. B. Walker, in Handbook of Topical Antimicrobials, ed. D. S. Paulson, CRC Press, 2002 Search PubMed.
- F. Bureš, Top. Curr. Chem., 2019, 377, 14 CrossRef PubMed.
- A. Brändström, Adv. Phys. Org. Chem., 1977, 15, 267–330 CrossRef.
- M. Dusselier and M. E. Davis, Chem. Rev., 2018, 118, 5265–5329 CrossRef CAS PubMed.
- S. Mishra and V. K. Tyagi, J. Oleo Sci., 2007, 56, 269–276 CrossRef CAS PubMed.
- A. N. Vereshchagin, N. A. Frolov, K. S. Egorova, M. M. Seitkalieva and V. P. Ananikov, Int. J. Mol. Sci., 2021, 22, 6793 CrossRef CAS PubMed.
- C. P. Gerba, Appl. Environ. Microbiol., 2015, 81, 464–469 CrossRef PubMed.
- M. C. Jennings, K. P. C. Minbiole and W. M. Wuest, ACS Infect. Dis., 2015, 1, 288–303 CrossRef CAS PubMed.
- K. P. C. Minbiole, M. C. Jennings, L. E. Ator, J. W. Black, M. C. Grenier, J. E. LaDow, K. L. Caran, K. Seifert and W. M. Wuest, Tetrahedron, 2016, 72, 3559–3566 CrossRef CAS.
- K. R. Morrison, R. A. Allen, K. P. C. Minbiole and W. M. Wuest, Tetrahedron Lett., 2019, 60, 1–13 CrossRef PubMed.
- T. J. Paniak, M. C. Jennings, P. C. Shanahan, M. D. Joyce, C. N. Santiago, W. M. Wuest and K. P. C. Minbiole, Bioorg. Med. Chem. Lett., 2014, 24, 5824–5828 CrossRef CAS PubMed.
- M. E. Forman, M. C. Jennings, W. M. Wuest and K. P. C. Minbiole, ChemMedChem, 2016, 11, 1401–1405 CrossRef CAS PubMed.
- M. A. Mitchell, A. A. Iannetta, M. C. Jennings, M. H. Fletcher, W. M. Wuest and K. P. C. Minbiole, ChemBioChem, 2015, 16, 2299–2303 CrossRef CAS PubMed.
- M. C. Jennings, M. E. Forman, S. M. Duggan, K. P. C. Minbiole and W. M. Wuest, ChemBioChem, 2017, 18, 1573–1577 CrossRef CAS PubMed.
- M. C. Jennings, B. A. Buttaro, K. P. C. Minbiole and W. M. Wuest, ACS Infect. Dis., 2015, 1, 304–309 CrossRef CAS PubMed.
- R. A. Allen, M. C. Jennings, M. A. Mitchell, S. E. Al-Khalifa, W. M. Wuest and K. P. C. Minbiole, Bioorg. Med. Chem. Lett., 2017, 27, 2107–2112 CrossRef CAS PubMed.
- M. C. Jennings, L. E. Ator, T. J. Paniak, K. P. C. Minbiole and W. M. Wuest, ChemBioChem, 2014, 15, 2211–2215 CrossRef CAS PubMed.
- A. D. Verderosa, M. Totsika and K. E. Fairfull-Smith, Front. Chem., 2019, 7, 1–17 CrossRef PubMed.
- J. W. Black, M. C. Jennings, J. Azarewicz, T. J. Paniak, M. C. Grenier, W. M. Wuest and K. P. C. Minbiole, Bioorg. Med. Chem. Lett., 2013, 24, 99–102 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cs00244j |
| This journal is © The Royal Society of Chemistry 2024 |