
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular recognition of peptides and proteins by cucurbit[n]urils: systems and applications
Lilyanna
Armstrong
a,
Sarah L.
Chang
a,
Nia
Clements
a,
Zoheb
Hirani
a,
Lauren B.
Kimberly
a,
Keturah
Odoi-Adams
 b,
Paolo
Suating
a,
Hailey F.
Taylor
a,
Sara A.
Trauth
a and
Adam R.
Urbach
*a
b,
Paolo
Suating
a,
Hailey F.
Taylor
a,
Sara A.
Trauth
a and
Adam R.
Urbach
*a
aDepartment of Chemistry, Trinity University, San Antonio, TX 78212, USA. E-mail: aurbach@trinity.edu
bDepartment of Chemistry and Physics, Southwestern Oklahoma State University, Weatherford, OK 73096, USA
First published on 17th October 2024
Abstract
The development of methodology for attaching ligand binding sites to proteins of interest has accelerated biomedical science. Such protein tags have widespread applications as well as properties that significantly limit their utility. This review describes the mechanisms and applications of supramolecular systems comprising the synthetic receptors cucurbit[7]uril (Q7) or cucurbit[8]uril (Q8) and their polypeptide ligands. Molecular recognition of peptides and proteins occurs at sites of 1–3 amino acids with high selectivity and affinity via several distinct mechanisms, which are supported by extensive thermodynamic and structural studies in aqueous media. The commercial availability, low cost, high stability, and biocompatibility of these synthetic receptors has led to the development of myriad applications. This comprehensive review compiles the molecular recognition studies and the resulting applications with the goals of providing a valuable resource to the community and inspiring the next generation of innovation.
 From left to right: Lilyanna Armstrong, Nia Clements and Zoheb Hirani | Lilyanna Armstrong, Nia Clements, and Zoheb Hirani did undergraduate research with Prof. Adam R. Urbach at Trinity University. Lilyanna is an undergraduate student at Trinity University studying biochemistry/molecular biology and Spanish. Upon graduation in 2025, she plans to study public and global health throughout the US and Latin America before pursuing an MD. Nia Clements completed a BS in biochemistry and molecular biology at Trinity University, followed by a Master of Public Health degree in infectious disease epidemiology at Cornell and is currently a PhD student in epidemiology at the University of Michigan at Ann Arbor. Her research interests include measuring health disparities related to climate and environmental justice issues caused by chemical exposures. Zoheb completed a BS in chemistry at Trinity University and recently finished his PhD in chemistry at Northwestern University with Prof. William R. Dichtel. His research interests are related to the synthesis and applications of porous, covalent organic frameworks. |
 From left to right: Hailey Taylor, Sarah Chang, Lauren Kimberly, and Sara Trauth | Hailey Taylor, Sarah Chang, Lauren Kimberly, and Sara Trauth received bachelor degrees from Trinity University and did research in biomolecular recognition with Prof. Adam R. Urbach. Hailey earned her MS in chemistry from University of North Carolina at Chapel Hill and is currently a research associate at EpiCypher Inc. Her current research interests lie in developing novel assays to further epigenetic research. Sarah is currently pursuing an MD at the Joe R. and Teresa Lozano Long School of Medicine at the University of Texas Health San Antonio. Lauren is currently pursuing a PhD at the University of Texas at Austin with Prof. Emily Que. Her current research is focused on developing tools for cellular metal sensing. Sara Trauth was a Cottrell Postbaccalaureate Scholar with Prof. Adam R. Urbach for one year, and she is currently a PhD student in pharmacology and molecular sciences at The Johns Hopkins School of Medicine. |
 Keturah Adams | Keturah Adams earned her BS in chemistry from Southwestern Oklahoma State University (SWOSU) and her MS and PhD degrees in chemistry at Texas A&M University. She was a postdoctoral fellow at Trinity University with Prof. Adam R. Urbach before starting as an Assistant Professor of Chemistry (tenure-track) in the Department of Chemistry and Physics at SWOSU. Her research focuses on the functional characterization of the ELOVL4 protein, linked to macular dystrophy and neurodegenerative disorders. She is actively involved in programs supporting STEM education for high school students, rural teachers, and non-traditional students. |
 Paolo Suating | Paolo Suating received his PhD under the supervision of Prof. Bruce Gibb at Tulane University, where he studied the solvation properties of functionalized deep-cavity cavitands. He is currently a postdoctoral fellow with Prof. Adam R. Urbach at Trinity University studying sequence-selective peptide recognition by cucurbit[n]urils. His research interests include the syntheses of novel water-soluble macrocycles, ion–solute and solute–solute interactions in water, and long-range chirality transfer and induction. |
 Adam R. Urbach | Adam R. Urbach earned a BS in chemistry from UT Austin, doing undergraduate research with Profs Jonathan L. Sessler and Thomas J. Kodadek. Following a PhD in organic chemistry at Caltech with Prof. Peter B. Dervan and a postdoc at Harvard with Prof. George M. Whitesides, he moved to Trinity University, where he is Professor of Chemistry. His research has primarily involved studies of biomolecular recognition, with his independent work focused on the sequence-selective molecular recognition of peptides and proteins by cucurbit[n]uril synthetic receptors. Prof. Urbach is a Fellow of the American Association for the Advancement of Science. |
1. Introduction
Protein affinity tags are an essential tool of modern chemical biology with many established uses, including the detection of proteins and their isolation from complex mixtures, the addition of new functionality, and both the enhancement of solubility and the promotion of crystallization.1–4 Affinity tags can be added to proteins via several different approaches, most commonly with genetic engineering but also via posttranslational and chemical modification. Compared to the size of the target protein, a tag may be relatively small (e.g., peptide epitopes His6, FLAG, or Myc) or large (e.g., glutathione-S-transferase or maltose-binding protein) and often alters the structure and/or function of the protein in undesirable ways, necessitating removal of the tag.4–7 The binding partners for an affinity tag (e.g., monoclonal antibodies) can be costly to obtain or develop. His tags are an important exception to these general limitations because they are small and can be bound with commercially available and chemically modifiable metal complexes. Some major disadvantages of His tags are the need for low pH or high concentrations of imidazole to dissociate the complex, the limited binding affinity, and the incompatibility with reducing conditions, EDTA, and certain detergents.6–8 The proven utility, future potential, and general limitations of affinity tags justify their further development. This review focuses on protein tags composed of 1–3 amino acid residues, i.e., “minimal” protein tags, which are bound tightly and selectively by small, stable, inexpensive, and biocompatible synthetic receptors (Fig. 1). A 1–3 residue site cannot form a predictably folded structure, and when installed at the terminus of a protein, the site is even more likely to be structurally disordered and fully solvent-exposed, thus facilitating encapsulation by a synthetic receptor.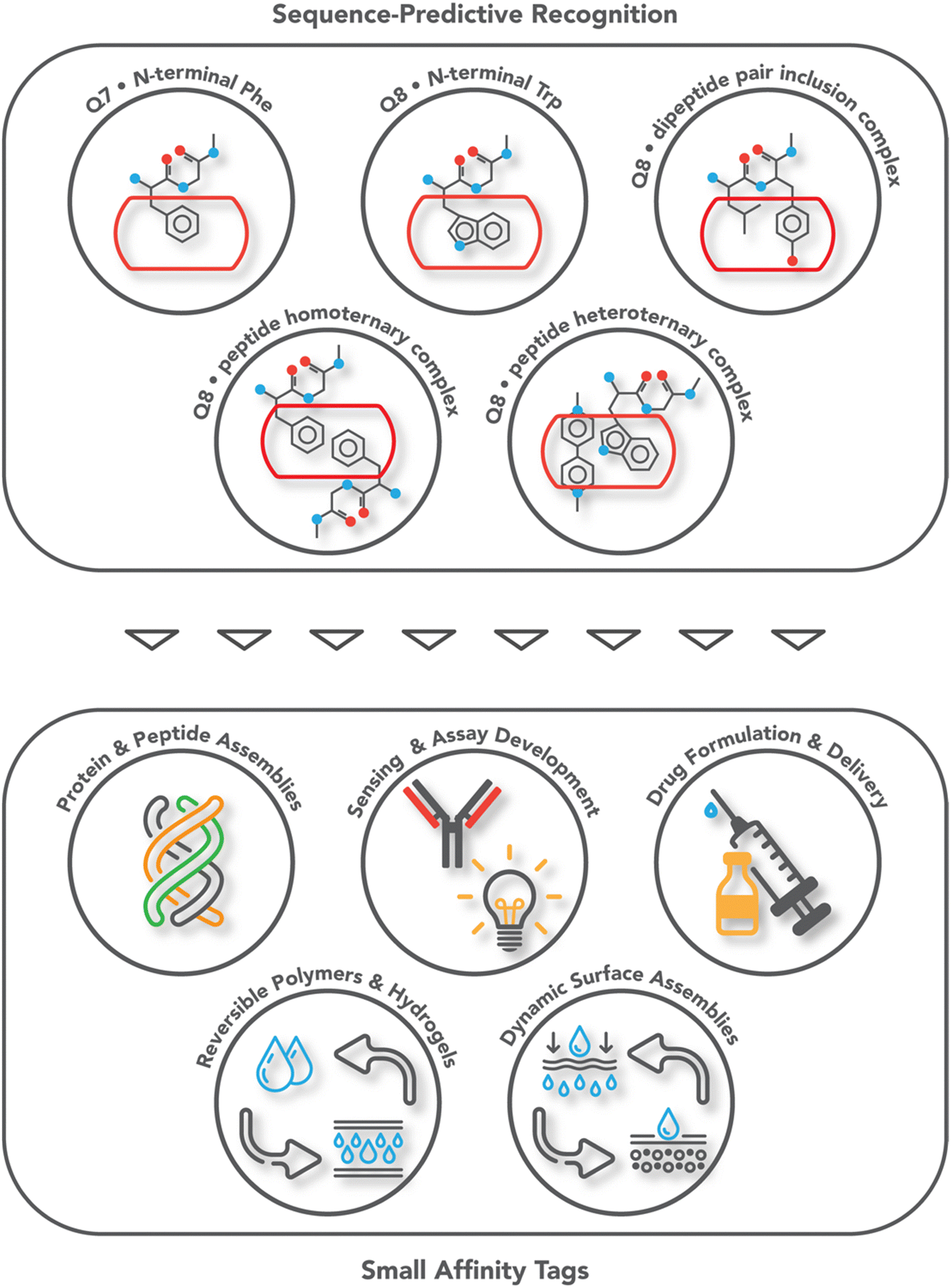 | ||
| Fig. 1 Overview of the systems of sequence-predictive molecular recognition by cucurbit[n]urils and the applications they have enabled. | ||
The complex between an affinity tag and its binding partner requires a substantial interfacial surface area.9 In contrast to a folded protein, which can create a large binding interface by including a small molecule ligand within a well-defined binding cavity (Fig. 2a), the solvent-accessible surface of a minimal affinity tag is convex in shape and requires its binding partner to have a cavity (Fig. 2b). Natural protein/peptide receptors and enzymes, such as antibodies, N-recognins, proteases, and kinases have defined target binding sites within cavities.10
 | ||
| Fig. 2 (a) Binding of a ligand within a protein cavity. Crystal structure of atorvastatin (blue sticks) bound within the active site cavity of HMG CoA reductase, shown as a grey solvent-accessible surface (PDB ID: 1 HWK).11 (b) Binding of a synthetic receptor on the surface of a protein. Crystal structure of three sulfonatocalix[4]arene hosts (blue sticks) bound to lysine residues on the surface of cytochrome c, shown as a grey solvent-accessible surface (PDB ID: 3TYI).12 Reproduced from ref. 10 with permission from the Royal Society of Chemistry. | ||
Small peptides are conformationally disordered and highly solvent-accessible. Therefore, they are good models for intrinsically disordered regions (IDRs) of proteins, while being less expensive to make and modify and easier to characterize by chemical methods. We hypothesize that receptors for small peptides should, in general, translate to intrinsically disordered regions of proteins such as termini and disordered loops.13 Extensive work on developing synthetic receptors for peptides in aqueous solution was pioneered by Breslow, Hamilton, Kelly, Nowick, Schneider, Schmuck, Still, and others and has led to various strategies for targeting the ionic, polar, aromatic, and aliphatic functional groups of peptides.14,15 Having a target site limited to 1–3 amino acid residues dictates that binding must be highly efficient on a per-residue basis. Although binding affinity and selectivity in the early systems was modest when compared with what is possible in biology, the highest affinity noncovalent complexes between synthetic receptors and peptides in aqueous solution have involved the inclusion of aromatic residues,16–19 which are extraordinarily efficient on a per-residue basis, as discussed further below.
Targeting small sites, however, has the inherent problem of selectivity in a proteomic context. As an illustration of this problem, the crystal structure of cytochrome c bound to p-sulfonatocalix[4]calixarene shows multiple receptors bound to the surface of this relatively small protein (Fig. 1b).12 In this review, we describe in detail how the properties of the cucurbit[n]uril (Qn) family of synthetic receptors have allowed them to target peptides and proteins in aqueous solution with sequence-selectively and with high affinity, and how these applications have enabled the development of myriad applications as affinity tags for peptides and proteins.
Cucurbit[n]urils (Qn's) are a family of synthetic macrocyclic receptors composed of bis(methylene)-linked glycoluril units (Fig. 3).20–22 The cavities of Qn's are hydrophobic, which drives the inclusion of nonpolar groups via the exothermic release of water molecules upon guest binding.23 The two identical and constricted entrances to a Qn cavity are lined with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups that bind cations such as organic ammonium groups and metal cations.24 The smaller cavity of cucurbit[6]uril (Q6) can accommodate alkyl groups, whereas the larger cavity of Q7 can accommodate larger guests, with optimal size complementarity for adamantane and ferrocene.25,26 The cavity of Q8 can accommodate even larger guests and, importantly, two guests simultaneously.27,28 Qn's bind to a wide range of guests in aqueous solution with equilibrium association constant (Ka) values up to 1018 M−1,29 which has led to applications in sensing, separations, remediation, catalysis, drug delivery, polymerization, and gelation, among many others.22 Here, we focus on the basic science and applications of the molecular recognition of peptides and proteins by Qn receptors.
O groups that bind cations such as organic ammonium groups and metal cations.24 The smaller cavity of cucurbit[6]uril (Q6) can accommodate alkyl groups, whereas the larger cavity of Q7 can accommodate larger guests, with optimal size complementarity for adamantane and ferrocene.25,26 The cavity of Q8 can accommodate even larger guests and, importantly, two guests simultaneously.27,28 Qn's bind to a wide range of guests in aqueous solution with equilibrium association constant (Ka) values up to 1018 M−1,29 which has led to applications in sensing, separations, remediation, catalysis, drug delivery, polymerization, and gelation, among many others.22 Here, we focus on the basic science and applications of the molecular recognition of peptides and proteins by Qn receptors.
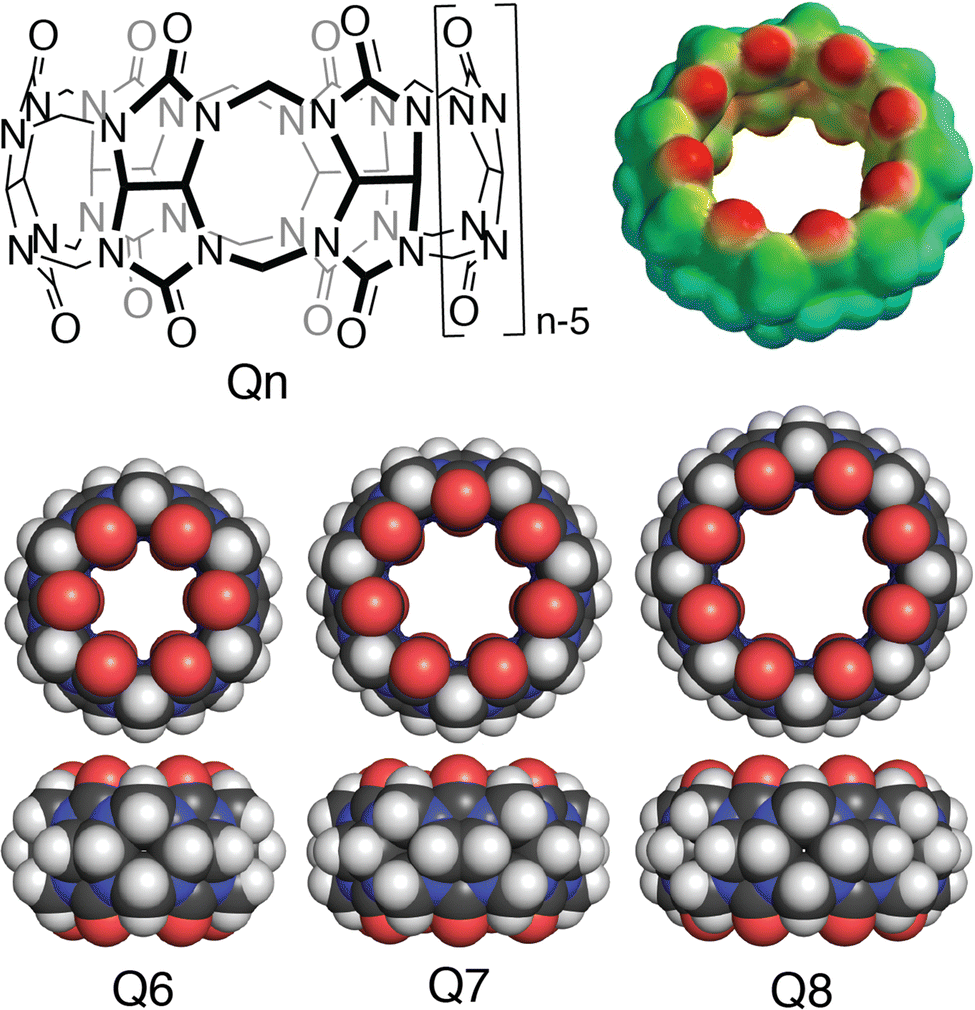 | ||
| Fig. 3 (top left) Structural formula of cucurbit[n]uril; (top right) Electrostatic potential mapped onto the solvent accessible surface of cucurbit[8]uril; (bottom) Space-filling models of cucurbit[n]urils showing the variation in size based on the number of glycoluril units. | ||
The vast majority of the work on peptide recognition by Qn receptors has involved Q7 and Q8, which have an inner cavity volume sufficient to accommodate the side chain(s) of one or two amino acid residue(s). Our first studies in this area followed the pioneering work of Kimoon Kim and coworkers, who reported the formation of ternary complexes in which Q8 binds either two of the same guest (i.e., homoternary)27 or two different guests (i.e., heteroternary).28 They showed that Q8 binds methyl viologen (MV) but not 2,6-dihydroxynaphthalene (HN), and that the Q8·MV complex binds HN as a second guest to form the heteroternary Q8·MV·HN complex (Fig. 4). A crystal structure shows HN and a viologen stacking face-to-face in the cavity of Q8 in the solid state, and NMR data show that both MV and HN are included simultaneously within the Q8 cavity in the solution state. The binding of HN to Q8·MV results in the growth and red-shifting of a visible charge-transfer absorbance and a quenching of naphthalene fluorescence, which corroborates the simultaneous inclusion of both aromatic guests in the Q8 cavity.28
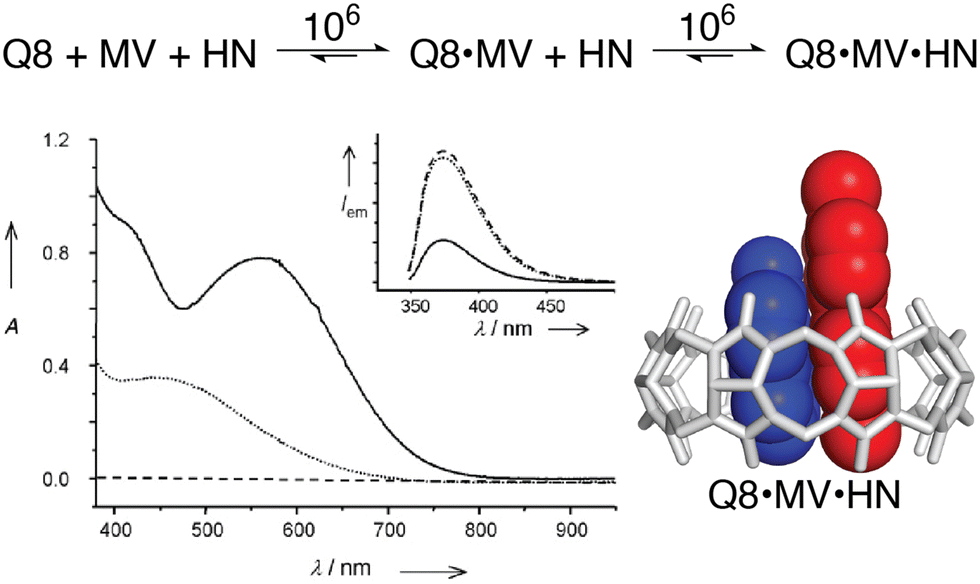 | ||
Fig. 4 (top) Two-step formation of the heteroternary Q8·MV·HN complex with equilibrium association constant (Ka) values (M−1) shown over the arrows (10 mM sodium phosphate, pH 7.0, 300 K).30 (bottom left) UV-visible absorption and emission (inset) spectra of HN (dashed line), a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of HN and MV (dotted line), and a 1:1:1 mixture of Q8, HN, and MV (solid line). (bottom right) Structure of the Q8·MV·HN complex modeled from the crystal structure of an analogue. CCDC ID 154114.28 Adapted with permission from ref. 28. © 2001 Wiley-VCH Verlag GmbH, Weinheim, Fed. Rep. of Germany. :1 mixture of HN and MV (dotted line), and a 1:1:1 mixture of Q8, HN, and MV (solid line). (bottom right) Structure of the Q8·MV·HN complex modeled from the crystal structure of an analogue. CCDC ID 154114.28 Adapted with permission from ref. 28. © 2001 Wiley-VCH Verlag GmbH, Weinheim, Fed. Rep. of Germany. | ||
In parallel with their report on hetero-guest pairs, Kim and coworkers filed a patent claiming that Q8·MV can also bind to aromatic second guests including tryptophan, tyrosine, and dopamine.31 This finding inspired us to further explore the binding of amino acids by Q8·MV.30 We measured the solubility of Q8 in our standard buffer, 10 mM sodium phosphate, pH 7.0 (100 μM), and the Ka value (8.5 × 105 M−1) for the formation of the Q8·MV complex. Isothermal titration calorimetry (ITC) experiments showed that the Q8·MV complex binds to tryptophan with a Ka value of 4.3 × 104 M−1 and with 8-fold selectivity over phenylalanine and 20-fold selectivity over tyrosine.30 No binding was observed for the other 17 amino acids by ITC or by NMR spectroscopy.30,32 Based on the driving forces for cucurbituril binding, we would expect hydrophobic and basic (i.e., cationic) amino acids to be preferred, but only tryptophan, phenylalanine, and tyrosine bind measurably. A comparative analysis of the side chains of the 20 genetically encoded amino acids, in terms of their solvent-exposed surface areas and their transfer energies from water to cyclohexane solution (Fig. 5),33–35 is consistent with a combination of size and hydrophobicity driving the binding of tryptophan, phenylalanine, and tyrosine. Leucine and isoleucine are more hydrophobic but smaller than tryptophan and thus would not displace as many water molecules upon binding. Arginine and lysine are large but too hydrophilic to bind stably within the Q8 cavity.
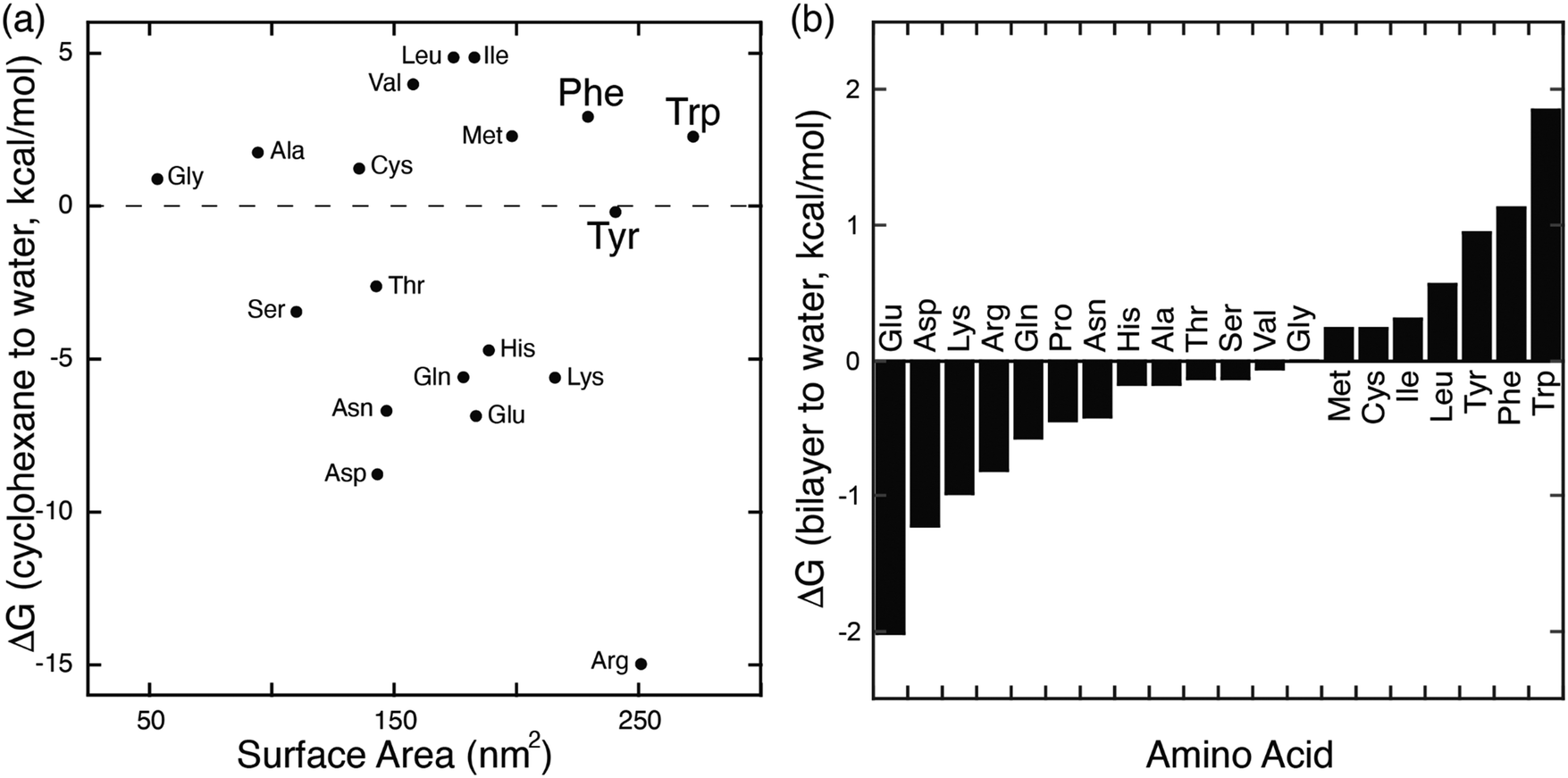 | ||
| Fig. 5 (a) Plot of the transfer energy from cyclohexane solution to neutral aqueous solution versus the solvent-exposed surface area of side chain analogues of the 20 amino acids (i.e., methane for alanine; 3-methylindole for tryptophan). The transfer energies were reported by Radzicka and Wolfenden.34 The surface areas were calculated for tripeptides Gly-X-Gly by Chothia.33 (b) Calculated hydrophobicity at pH 8 for all residues except Arg and Lys, which were calculated at pH 2.35 Extremes on this plot are defined by charged residues at the negative end and aromatic residues at the positive end. Reproduced from ref. 10 with permission from the Royal Society of Chemistry. | ||
Synthetic receptors need efficient ligands, and aromatic residues are particularly efficient. Aromatic residues are especially versatile for intermolecular interactions, with binding driven significantly by electrostatic, van der Waals, and hydrophobic interactions.36 Aromatic groups bind electrostatically to cations, hydrogen bond donors, and other aromatic groups via their quadrupole moments. The large, nonpolar surface areas of aromatic groups allow extensive van der Waals interactions and the release of many water molecules upon binding. The high structural rigidity minimizes the loss of conformational entropy upon binding. The aromatic residues tryptophan (Trp), phenylalanine (Phe), and tyrosine (Tyr) have a unique combination of characteristics, being the largest and most hydrophobic residues. Mutational studies of protein–protein interactions have found that on a per-residue basis, aromatic residues contribute significantly more than non-aromatic residues to the stability of protein–protein interactions.37–39 It is perhaps not surprising, therefore, that aromatic residues can be bound with high affinity by synthetic receptors.
2. Mechanisms of peptide and protein recognition by cucurbit[n]urils
2.1. Discovery of sequence-selective peptide recognition by cucurbit[8]uril
In the Q8·MV·Trp aqueous complex at pH 7.0,30 there is unusually high electrostatic charge density, including the dicationic MV, the zwitterionic tryptophan, and the concentrated negative electrostatic potential of the CO groups of Q8. To probe the influence of charge on binding, we studied the binding of Q8·MV with a series of tryptophan derivatives that vary in the type, number, and location of charge (Fig. 6). Compared to tryptophan, tryptamine (TrpA) and tryptophan methyl ester (Trp-OMe) lack a negative charge, and indole propionic acid (IPA) and N-acetyl tryptophan (N-AcTrp) lack a positive charge. ITC studies showed that Q8·MV binds TrpA, Trp-OMe, and tryptophan with similar affinity and with approximately 20-fold selectivity over IPA and N-AcTrp. It was particularly interesting to learn that the binding affinity of tryptophan correlates more with the cationic derivatives than with the anionic derivatives. These results suggested that binding selectivity is driven by electrostatic stabilization of the cationic ammonium group, most likely via its interaction with the CO groups on Q8.
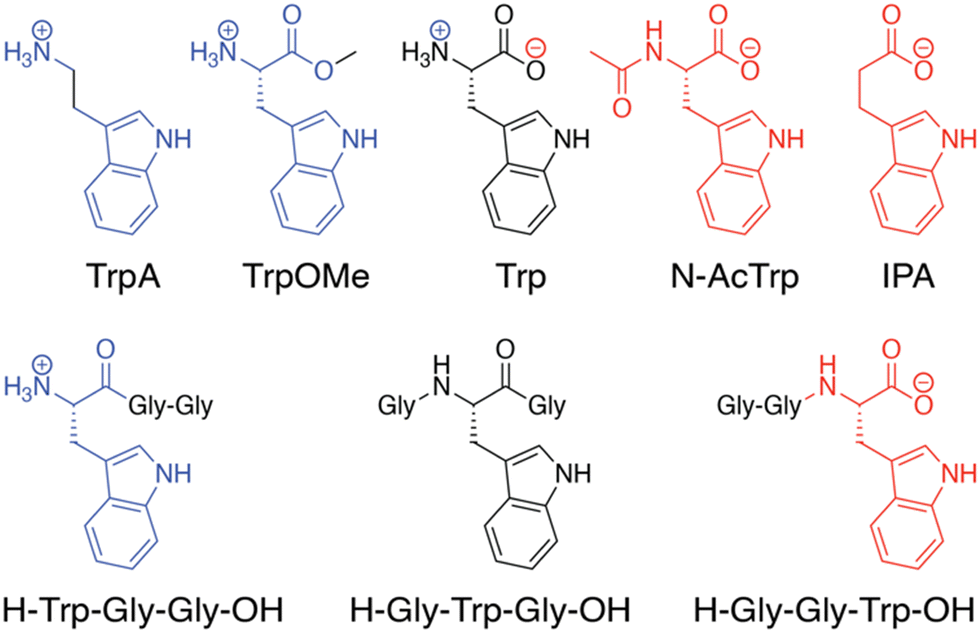 | ||
| Fig. 6 Chemical formulas of tryptophan derivatives and their structurally analogous Trp-containing peptides.30 Reproduced from ref. 10 with permission from the Royal Society of Chemistry. | ||
We reasoned that the singly charged tryptophan derivatives, especially N-AcTrp and Trp-OMe, are structurally analogous to a Trp residue positioned at the N- or C-terminal position of a peptide chain (Fig. 6).30N-AcTrp retains its carboxylate group, akin to a C-terminal Trp, and Trp-OMe retains its ammonium group, akin to an N-terminal Trp. Therefore, we hypothesized that Q8·MV should bind selectively to an N-terminal Trp residue versus a C-terminal Trp residue due to stabilization by the proximal ammonium group. ITC studies showed that Q8·MV binds to the peptide H-Trp-Gly-Gly-OH (Gly = glycine; H- indicates unprotected N-terminal amine; –OH indicates unprotected C-terminal carboxylic acid), which contains an N-terminal Trp residue, with a Ka value of 1.3 × 105 M−1 and with 6-fold selectivity over H-Gly-Trp-Gly-OH and 40-fold selectivity over H-Gly-Gly-Trp-OH (Table 1).30 Both peptides containing a non-terminal Trp residue, H-Gly-Trp-Gly-OH and H-Gly-Gly-Trp-Gly-Gly-OH, bound Q8·MV with similar affinities. NMR data showed that in each complex, both the indole side chain and the viologen group are bound simultaneously within the Q8 cavity, as revealed by upfield chemical shift perturbation of the aromatic signals of both compounds in the presence of Q8. These results demonstrated that Q8·MV can recognize N-terminal Trp in a sequence-selective manner, and that the N-terminal residue, comprising the indole sidechain and the N-terminal ammonium group, comprises a unique epitope for site-selective binding.
| Host | Peptide or protein | K a (M−1) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) |
|---|---|---|---|---|
| a 10 mM sodium phosphate, pH 7.0, 300 K.30 b 10 mM sodium phosphate, pH 7.0, 300 K.40 c 10 mM sodium phosphate, pH 7.0, 303 K.41 d 10 mM sodium phosphate, pH 7.4, 298 K.42 e 10 mM sodium phosphate, 0.5 mM dithiothreitol, pH 7.4, 298 K, Cys* = intramolecular disulfide.42 f 10 mM sodium phosphate, pH 7.0, 300 K.43 g 10 mM HEPES, pH 7.0, temperature not reported.44 h 10 mM sodium phosphate, pH 7.0, 298 K, F′ = 2,3,4,5,6-pentafluorophenylalanyl.45 i Not detected. j Not reported. Putative binding sites are in bold. | ||||
| Q8·MVa | H-Trp-Gly-Gly-OH | 1.3 × 105 | −14.8 | 7.8 |
| Q8·MVa | H-Gly-Trp-Gly-OH | 2.1 × 104 | −11.4 | 5.5 |
| Q8·MVa | H-Gly-Gly-Trp-OH | 3.1 × 103 | −8.8 | 4.0 |
| Q8·MVa | H-Gly-Gly-Trp-Gly-Gly-OH | 2.5 × 104 | −12.1 | 6.1 |
| Q8·MVb | H-Lys-Ala-Trp-Ala-Ala-NH2 | 6.2 × 103 | nrj | nr |
| Q8·MVb | H-Ala-Lys-Trp-Ala-Ala-NH2 | 1.7 × 104 | nr | nr |
| Q8·MVc | H-Trp-Gly-Gly-OH | 3.0 × 105 | −12.6 | 5.1 |
| Q8·MVc | H-Met-Gly-Gly-OH | ndi | nr | nr |
| Q8·MVc | YFP-Trp-Gly-Gly | 2.3 × 105 | −7.2 | −0.3 |
| Q8·MVc | YFP-Met-Gly-Gly | 1.2 × 105 | −4.3 | −2.7 |
| Q8·MVd | H-Ala-Cys-Asn-Thr-Gly-Ser-Pro-Tyr-Glu-Cys-NH2 | 4.3 × 104 | −1.3 | −5.0 |
| Q8·MVd | H-Ala-Cys-Gln-Asn-Pro-Asn-Gln-Lys-Phe-Cys-NH2 | 2.2 × 105 | −11.0 | 3.9 |
| Q8·MVd | H-Ala-Cys-Leu-Lys-Leu-Gly-Glu-Lys-Trp-Cys-NH2 | 4.4 × 104 | −12.0 | 5.5 |
| Q8·MVe | H-Ala-Cys*-Asn-Thr-Gly-Ser-Pro-Tyr-Glu-Cys*-NH2 | nd | nr | nr |
| Q8·MVe | H-Ala-Cys*-Gln-Asn-Pro-Asn-Gln-Lys-Phe-Cys*-NH2 | 5.9 × 104 | −11.0 | 4.6 |
| Q8·MVe | H-Ala-Cys*-Leu-Lys-Leu-Gly-Glu-Lys-Trp-Cys*-NH2 | nd | nr | nr |
| Q8·MVd | Tn3 (Gln-Lys-Phe in BC loop) | 6.1 × 105 | −11.0 | 2.8 |
| Q8·MVd | Tn3 (Gln-Lys-Phe in DE loop) | 8.9 × 104 | −2.1 | −4.7 |
| Q8·MVd | Tn3 (Gln-Lys-Phe in FG loop) | 6.4 × 104 | −1.4 | −5.2 |
| Q8·MBBIf | H-Trp-Gly-Gly-OH | 1.2 × 105 | −15.6 | 8.6 |
| Q8·MBBIf | H-Gly-Trp-Gly-OH | 1.7 × 104 | −16.5 | 10.7 |
| Q8·MBBIf | H-Gly-Gly-Trp-OH | 4.1 × 103 | −13.0 | 8.0 |
| Q8·MDAPg | H-Arg7-OH | nd | nr | nr |
| Q8·MDAPg | H-Leu-Arg-Arg-Trp-Ser-Leu-Gly-OH | nd | nr | nr |
| Q8·MDAPg | H-Leu-Arg-Arg-Trp-pSer-Leu-Gly-OH | nd | nr | nr |
| Q8·MDAPg | H-Trp-Lys-Arg-Thr-Leu-Arg-Arg-Leu-OH | 4.3 × 105 | nr | nr |
| Q8·MDAPg | H-Trp-Lys-Arg-pThr-Leu-Arg-Arg-Leu-OH | 4.1 × 105 | nr | nr |
| Q8·MDAPg | H-Phe-Arg7-OH | 2.0 × 106 | nr | nr |
| Q8·F′GGh | H-Trp-Gly-Gly-OH | 4.6 × 105 | −13.3 | 5.6 |
| Q8·F′GGh | H-Gly-Trp-Gly-OH | 1.0 × 105 | −11.9 | 5.0 |
| Q8·F′GGh | H-Gly-Gly-Trp-OH | 9.0 × 104 | −14.5 | 7.8 |
| Q8·F′GGh | H-Phe-Gly-Gly-OH | 3.6 × 105 | −14.4 | 6.8 |
| Q8·F′GGh | H-Gly-Phe-Gly-OH | 5.8 × 104 | −9.1 | 2.6 |
| Q8·F′GGh | H-Gly-Gly-Phe-OH | 2.2 × 104 | −7.5 | 1.5 |
| Q8·F′GGh | H-Tyr-Gly-Gly-OH | 3 × 103 | −9.6 | 3.6 |
| Q8·F′GGh | H-Gly-Tyr-Gly-OH | 4 × 103 | −4.7 | −0.2 |
| Q8·F′GGh | H-Gly-Gly-Tyr-OH | nd | nd | nd |
| Q8·F′GGh | H-Lys-Gly-Gly-OH | nd | nd | nd |
| Q8·F′GGh | H-Glu-Gly-Gly-OH | nd | nd | nd |
| Q8·F′GGh | H-Leu-Gly-Gly-OH | nd | nd | nd |
Methyl viologen has intrinsic optical and electronic properties that have enabled several applications in the sensing of peptides and proteins, as discussed in detail in Section 3.1. The binding of Trp-containing peptides to Q8·MV induces the growth of a charge-transfer absorbance and the quenching of indole fluorescence.30 Both of these effects are likely due to electron-transfer from indole to viologen in the excited electronic state and do not contribute significantly to the stability of the complexes in the ground state.46 The viologen therefore provides the means for a convenient, built-in optical sensor for peptide binding via absorbance and fluorescence spectroscopy.
With the indole side chain of the Trp residue bound within the Q8 cavity, the neighboring residue would be forced into proximity to Q8·MV. For example, a basic residue could possibly bind to the Q8 portal or be repelled by MV. Therefore, we hypothesized that the identity of the second residue could influence binding. We synthesized a library comprising 104 peptides, each containing a Trp binding site positioned either at the N-terminus of a tripeptide (H-Trp-X-Ala-NH2 and H-Trp-Ala-X-NH2) or at the center of a pentapeptide (H-X-Ala-Trp-Ala-Ala-NH2, H-Ala-X-Trp-Ala-Ala-NH2, H-Ala-Ala-Trp-X-Ala-NH2, and H-Ala-Ala-Trp-Ala-X-NH2), with a single variable position (X varied to all 20 amino acids except Trp and Cys) and with Ala residues at the other positions.40 We used the sensing properties of Trp binding to Q8·MV to facilitate the parallel screening of peptide binding by measuring the relative quenching of Trp fluorescence as a surrogate for relative binding affinity. There were insignificant differences observed among the peptides in each of the two series, with the exception of H-Lys-Ala-Trp-Ala-Ala-NH2versus H-Ala-Lys-Trp-Ala-Ala-NH2. These two peptides were tested by ITC (Table 1) and shown to have only 2-fold difference in affinity. Therefore, the screening assay was sensitive to changes in binding affinity, but in this particular motif (i.e., Q8·MV binding to a peptide with a Trp residue), the sequence context of the target Trp residue does not significantly influence the binding affinity.
2.2. Multivalent peptide binding by self-assembled receptors
In an effort to expand the peptide binding properties of this system, we considered linking Q8 molecules to form multivalent receptors. Although the MV and Q8 work together as co-hosts, the viologen is more synthetically modifiable than the macrocycle. Therefore, we linked viologens together and recruited Q8 molecules to each viologen site to form multivalent receptors (Fig. 7).47 One, two, and three viologen groups were conjugated to flexible, peptide-based scaffolds and used to recruit the same numbers of Q8 groups to form self-assembled monovalent, divalent, and trivalent receptors, which bound to peptides containing the same numbers of Trp residues in monovalent, divalent, and trivalent fashion. The extent of valency in each complex was quantified directly via the additive charge-transfer absorptivities of the Q8·viologen·Trp complexes (Fig. 7). Isothermal titration calorimetry studies showed that the enthalpy of binding was additive with the extent of valency (average ΔH monovalent = −10.9 kcal mol−1, divalent = −24.2 kcal mol−1, trivalent −39.4 kcal mol−1, Table 2), which corroborated the simultaneous formation of all Q8·viologen·indole complexes. The free energies of peptide binding, however, were not additive (average ΔG monovalent = −6.0 kcal mol−1, divalent = −7.8 kcal mol−1, trivalent −9.2 kcal mol−1), which is in contrast to what would be expected for optimal multivalent binding.48 The deviation from optimal binding was due to an entropic penalty for binding that was more than additive with the extent of valency (average −TΔS monovalent = 4.9 kcal mol−1, divalent = 16.4 kcal mol−1, trivalent 30.2 kcal mol−1).47 This resulted in only a modest increase in peptide binding affinity of 30-fold for divalent versus monovalent and 300-fold for trivalent vs. monovalent (Table 2). For the set of four divalent complexes, the length of the oligo(Gly) linker between the two binding sites was varied between four or six Gly units. It is remarkable that the linker length showed no significant influence on the binding thermodynamics, as we would have expected greater loss in entropy for the longer chain. | ||
| Fig. 7 (top) Schematic of the self-assembly of Q8 onto a divalent scaffold and the divalent binding of the resulting receptor to a divalent peptide. (bottom) UV-visible spectra showing the additive molar absorptivities of monovalent, divalent, and trivalent complexes.47 Reproduced with permission from ref. 47. Copyright 2009 American Chemical Society. | ||
| Self-assembled receptor | Peptide | K a (M−1) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) |
|---|---|---|---|---|
| a 10 mM sodium phosphate, pH 7.0, 300 K; Viol-Q8 indicates a viologen-modified residue bound to Q8; Ac-indicates an acetylated N-terminal amine.47 Putative binding sites are in bold. | ||||
| Ac-Gly2-Viol·Q8-Gly2-NH2a | Ac-Gly2-Trp-Gly2-NH2 | 2.2 × 104 | −10.8 | 4.9 |
| Ac-Gly2-Viol·Q8-Gly2-NH2a | Ac-Gly3-Trp-Gly3-NH2 | 2.2 × 104 | −11.0 | 5.0 |
| Ac-Gly3-Viol·Q8-Gly3-NH2a | Ac-Gly2-Trp-Gly2-NH2 | 1.9 × 104 | −10.8 | 4.9 |
| Ac-Gly3-Viol·Q8-Gly3-NH2a | Ac-Gly3-Trp-Gly3-NH2 | 2.2 × 104 | −11.0 | 4.9 |
| Ac-(Gly2-Viol·Q8-Gly2)2-NH2a | Ac-Gly2-Trp-Gly2-NH2 | 1.7 × 104 | −12.8 | 7.0 |
| Ac-(Gly2-Viol·Q8-Gly2)2-NH2a | Ac-Gly3-Trp-Gly3-NH2 | 1.5 × 104 | −13.0 | 7.2 |
| Ac-(Gly3-Viol·Q8-Gly3)2-NH2a | Ac-Gly2-Trp-Gly2-NH2 | 1.8 × 104 | −11.8 | 5.9 |
| Ac-(Gly3-Viol·Q8-Gly3)2-NH2a | Ac-Gly3-Trp-Gly3-NH2 | 1.4 × 104 | −12.3 | 6.6 |
| Ac-(Gly2-Viol·Q8-Gly2)2-NH2a | Ac-(Gly2-Trp-Gly2)2NH2 | 5.0 × 105 | −24.2 | 16.3 |
| Ac-(Gly2-Viol·Q8-Gly2)2-NH2a | Ac-(Gly3-Trp-Gly3)2NH2 | 4.6 × 105 | −24.8 | 17.1 |
| Ac-(Gly3-Viol·Q8-Gly3)2-NH2a | Ac-(Gly2-Trp-Gly2)2NH2 | 5.5 × 105 | −23.4 | 15.6 |
| Ac-(Gly3-Viol·Q8-Gly3)2-NH2a | Ac-(Gly3-Trp-Gly3)2NH2 | 5.0 × 105 | −24.3 | 16.5 |
| Ac-(Gly2-Viol·Q8-Gly2)3-NH2a | Ac-Gly2-Trp-Gly2-NH2 | 1.7 × 104 | −12.2 | 6.4 |
| Ac-(Gly2-Viol·Q8-Gly2)3-NH2a | Ac-Asp2-(Gly2-Trp-Gly2)3-Asp2-NH2 | 4.7 × 106 | −39.4 | 30.2 |
2.3. Dimerization of peptides by cucurbit[8]uril
Early on we were interested to know whether Q8 could bind peptides in the absence of MV and hypothesized that the additional space in the Q8 cavity could accommodate an additional portion of one peptide or part of a second peptide. Twelve peptides of sequence H-X-Gly-Gly-OH, H-Gly-X-Gly-OH, and H-Gly-Gly-X-OH (X = Trp, Phe, Tyr, His), were tested for binding to Q8 by ITC.18 Binding was only measurable for H-Trp-Gly-Gly-OH and H-Phe-Gly-Gly-OH, and binding to these peptides was found to occur in a 2:1 peptide:Q8 stoichiometry (i.e., homodimerization of peptides). NMR data showed that binding occurs site-selectively at the N-terminal aromatic residue. Mass spectrometry confirmed the presence of 2:1 peptide:Q8 complexes. The thermodynamic constants for homoternary complex formation are reported in Table 3. The ternary equilibrium constant (Kter: Q8 + 2 peptide → Q8·(peptide)2) values for Q8·(H-Trp-Gly-Gly-OH)2 and Q8·(H-Phe-Gly-Gly-OH)2 were modest (109 – 1011 M−2) and showed that Q8 prefers Phe to Trp in this context.18 When looking at these values here and elsewhere, it is important to note the molecularity of complexation and not to confuse the magnitude of a Kter value with that of a binary Ka value. For example, a Kter value of 1.5 × 1011 M−2 is roughly equivalent to each molecule of peptide binding with a Ka value of 4 × 105 M−1. Nonetheless, the sequence-selectivity of binding in this system is extraordinary. Due to the lower limit of detection for the ITC instrument of ∼102 M−1, the selectivity of Q8 for H-Phe-Gly-Gly-OH versus H-Gly-Phe-Gly-OH is at least 1000-fold in terms of the affinity per peptide. All twelve peptides had an aromatic side chain, but only Trp and Phe bound measurably, indicating that the identity of the residue is important. All six Trp- and Phe-containing peptides had an N-terminal amine, but only the two peptides containing an N-terminal Trp or Phe bound measurably, indicating that the ammonium group alone is insufficient to drive binding. Instead, the combination of hydrophobic inclusion of a Phe or Trp side chain and electrostatic interaction with the proximal ammonium group is necessary for tight binding. In contrast to our first study,30 these results convinced us that the N-terminal residue could be a highly selective epitope that allows recognition in more complex environments and applications. Additionally, this early study established the foundation for an important technique that would later be used by numerous research groups, discussed in detail in Section 3.3.1: using Q8 to induce the homodimerization of proteins.18
| Host | Amino acid, peptide, or protein | K ter (M−2) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) |
|---|---|---|---|---|
| a 10 mM sodium phosphate, pH 7.0, 300 K.32 b 10 mM sodium phosphate, pH 7.0, 300 K.18 c PBS: 10 mM sodium phosphate, 1.8 mM potassium phosphate, 137 mM NaCl, 2.7 mM KCl, pH 7.4, 298 K.49 d 10 mM sodium phosphate, pH 7.4, 298 K.50 e 10 mM sodium phosphate, pH 7.0, 298 K.51 f Pure water, 298 K.52 g Pure water, 298K.53 h 50 mM sodium acetate, pH 4.74.54 i Monomeric cyan fluorescent protein modified with N-terminal FGG, 10 mM sodium phosphate, pH 7.0, 303 K.55 j Caspase-9 modified with N-terminal FGG, 10 mM sodium phosphate, pH 7.0, 303 K.56 k Glutathione-S-transferase modified with N-terminal FGG, 20 mM sodium phosphate, 1 mM EDTA, pH 7.4, 298 K.57 l 10 mM sodium phosphate, pH 7.4, 298 K.58 m Not detected. n Not reported. Putative binding sites are in bold. | ||||
| Q8a | Tryptophan | 6.9 × 107 | −17.1 | 6.3 |
| Q8a | Phenylalanine | 1.1 × 108 | −15.2 | 4.2 |
| Q8a | All 18 other amino acids | ndm | nrn | nr |
| Q8b | H-Trp-Gly-Gly-OH | 3.6 × 109 | −22.8 | 9.7 |
| Q8b | H-Gly-Trp-Gly-OH | nd | nr | nr |
| Q8b | H-Gly-Gly-Trp-OH | nd | nr | nr |
| Q8b | H-Phe-Gly-Gly-OH | 1.5 × 1011 | −29.6 | 14.2 |
| Q8b | H-Gly-Phe-Gly-OH | nd | nr | nr |
| Q8b | H-Gly-Gly-Phe-OH | nd | nr | nr |
| Q8b | H-Tyr-Gly-Gly-OH | nd | nr | nr |
| Q8b | H-Gly-Tyr-Gly-OH | nd | nr | nr |
| Q8b | H-Gly-Gly-Tyr-OH | nd | nr | nr |
| Q8b | H-His-Gly-Gly-OH | nd | nr | nr |
| Q8b | H-Gly-His-Gly-OH | nd | nr | nr |
| Q8b | H-Gly-Gly-His-OH | nd | nr | nr |
| Q8c | H-Phe-Gly-Gly-OH | 2.3 × 1010 | −25.3 | 11.2 |
| Q8c | H-Phe-Gly6-OH | 4.4 × 109 | −23.0 | 9.8 |
| Q8d | H-Ala-Glu-Phe-Arg-His-NH2 | 3.0 × 1010 | −15.3 | 0.9 |
| Q8d | H-Leu-Val-Phe-Ile-Ala-NH2 | 7.7 × 109 | −9.0 | 4.5 |
| Q8d | H-Val-Ile-Phe-Ala-Glu-NH2 | 1.6 × 1013 | −20.8 | 2.8 |
| Q8e | H-Phe-Leu-NH2 | 1.9 × 1011 | −26.7 | 11.3 |
| Q8e | H-Tyr-Ala-Leu-NH2 | 8.7 × 107 | −18.1 | 7.3 |
| Q8f | H-Phe-Gly-Gly-Gly-Cys-OH | 2.3 × 1013 | −21.8 | 6.2 |
| Q8g | H-Phe-Gly-Gly-OH | 1.7 × 1012 | nr | nr |
| Q8h | H-Tyr-His-OH | 2.4 × 108 | nr | nr |
| Q8i | mCFP-FGG | 2.5 × 1013 | −29.3 | 10.6 |
| Q8j | Caspase-9-FGG | 2.7 × 1012 | −23.7 | 5.3 |
| Q8k | GST-FGG | 2.9 × 1012 | −13.2 | −3.8 |
| Q8l | Aβ 4–16 | 5.5 × 1010 | nr | nr |
| Q8l | Aβ 1–16 | nd | nr | nr |
We attempted to delineate the affinities of each peptide in the stepwise formation of the ternary complexes.18 In the case of H-Trp-Gly-Gly-OH, the observed affinity for the first guest is approximately 4-fold higher than that of the second guest, which is consistent with non-cooperative binding (i.e., the binding of the first guest does significantly influence the stability of binding of the second guest). In the case of H-Phe-Gly-Gly-OH, the data did not yield confident values for the stepwise binding constants. Based on the curvature of the ITC data and on NMR experiments showing the formation of the Q8·(H-Phe-Gly-Gly-OH)2 complex in mixtures containing less than a 2:1 mol ratio of peptide:Q8, we concluded that binding is positively cooperative but were unable to unambiguously determine stepwise binding constants. Subsequently, Huskens, Jonkheijm, and coworkers reported a detailed investigation of cooperativity in the Q8·(H-Phe-Gly-Gly-OH)2 system and concluded that binding is most likely to be non-cooperative.49
In collaboration with the late P. John Hart at UT Health San Antonio, we obtained crystal structures of the ternary Q8·(H-Phe-Gly-Gly-OH)2 complex (Fig. 8) and the binary Q8·H-Trp-Gly-Gly-OH complex.18 In both crystal structures, the aromatic side chains are bound deeply within the Q8 cavity, the N-terminal ammonium groups are bound in close proximity to multiple CO groups on the Q8 portal, the first peptide NH group hydrogen bonds to a CO group on Q8, and the second peptide NH group forms at least one stabilizing dipole–dipole interaction with a CO group on Q8. In addition, the indole NH group forms a bifurcated hydrogen bond with two CO groups on the opposite portal of Q8. These structures provided a molecular basis with atomic detail for the recognition of peptides by Q8. They revealed the shape and electrostatic complementarity of Q8 with N-terminal Phe and Trp, showing that the Q8 cavity is the right size for certain side chains, and that the CO groups of Q8 are preorganized to make multiple stabilizing electrostatic interactions with peptides.
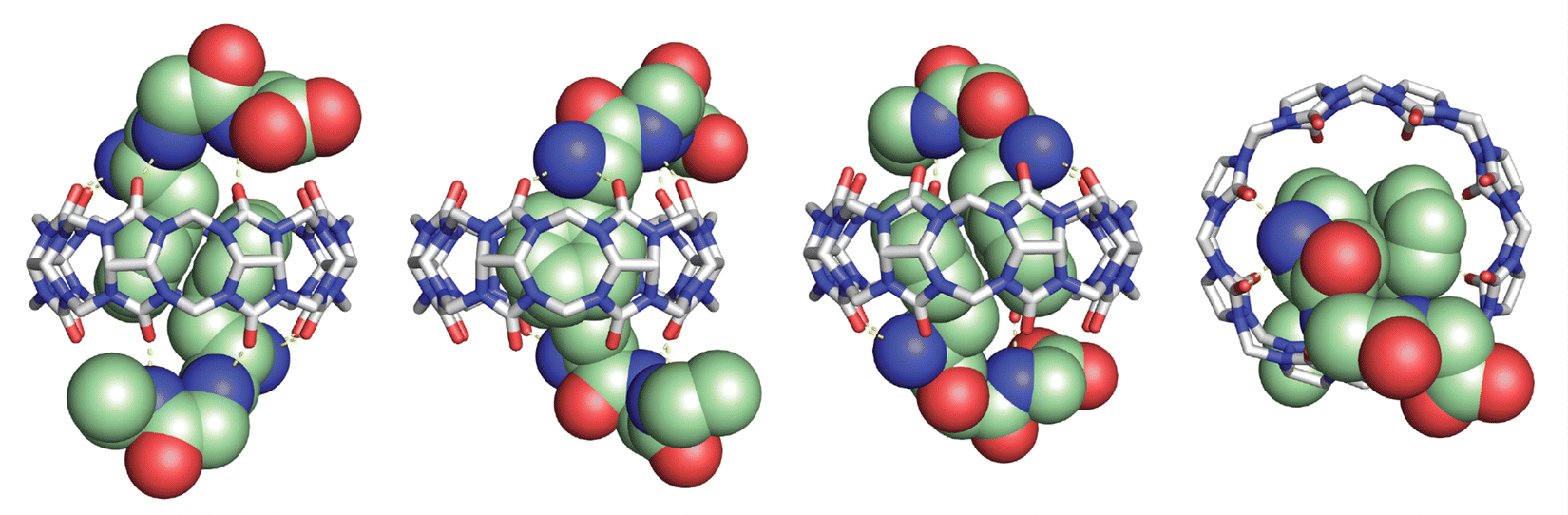 | ||
| Fig. 8 Rendering of the crystal structure of the Q8·(H-Phe-Gly-Gly-OH)2 complex shown from different viewing angles. The peptide is shown as space-filling, and Q8 as sticks. Peptide carbons are green. Q8 carbons are grey. Oxygens are red. Nitrogens are blue. Hydrogens are not shown. Yellow dashes show key intermolecular electrostatic interactions.18 | ||
Recently, Scherman and coworkers demonstrated the ability to direct the heterodimerization of peptides using the noncanonical amino acid, 2,3,4,5,6-pentafluorophenylalanine (PheF5, or F′).45 They showed that Q8 binds to a tripeptide containing N-terminal PheF5 (H-PheF5-Gly-Gly-OH, or F′GG) with a Ka value of 6.6 × 105 M−1, and the resulting Q8·H-PheF5-Gly-Gly-OH complex selectively forms heteroternary complexes with eight different tripeptides, with highest affinity for H-Trp-Gly-Gly-OH and H-Phe-Gly-Gly-OH. The electron-poor PheF5 allows for complementary polar-pi interactions with the relatively electron-rich side chains of Trp, Phe, and Tyr. Shifting the aromatic amino acid away from the N-terminus led to a notable decrease in Ka of the second guest. Heterodimerization was not observed for nonaromatic peptides (H-X-Gly-Gly-OH, X = Lys, Glu, Leu). To demonstrate the utility and selectivity of heteropeptide dimers, they applied the heterodimerization technique to on-resin recognition. Resin functionalized with N-terminal PheF5 showed selectivity for aromatic peptides in a mixture and ∼98% recycling efficiency through multiple cycles of peptide recognition and competitive displacement.
2.4. Recognition of nonterminal Phe by cucurbit[8]uril
Scherman and coworkers investigated the binding of Q8 to peptides containing Phe at non-terminal sites.50 They reported data on three pentapeptides derived from the Aβ1–42 amyloid peptide of sequence H-X1-X2-Phe-X3-X4-NH2 (–NH2C-terminal primary amide). Q8 bound two equivalents of peptide with Kter values comparable to or greater than those observed for H-Trp-Gly-Gly-OH and H-Phe-Gly-Gly-OH.18 The peptide H-Val-Ile-Phe-Ala-Glu-NH2 showed a remarkably high Kter value of 1.6 × 1013 M−2 (Table 3). These results made clear that binding at the N-terminal position may not be as important as originally believed, and that non-terminal Phe is a viable target for micromolar binding of peptides.50
Scherman and coworkers followed this work with a large combinatorial screen of non-terminal sites via a phage-display library of cyclic peptides against a surface-immobilized Q8·viologen complex (Fig. 9).42 From the selection experiments, they reported the most repeated three heptapeptide sequences with the corresponding 3-mer motifs: cAsn-Thr-Gly-Ser-Pro-Tyr-Glu (motif -Ser-Pro-Tyr-), cGln-Asn-Pro-Asn-Gln-Lys-Phe (motif -Gln-Lys-Phe-), and cLeu-Lys-Leu-Gly-Glu-Lys-Trp (motif -Glu-Lys-Trp-). When in cyclic form, all three sequences were shown to bind Q8·MV in a 1:1 stoichiometry and with modest affinities (Ka = 4–22 × 104 M−1, Table 1), but only cGln-Asn-Pro-Asn-Gln-Lys-Phe bound Q8 when in linear form, suggesting a possible role for structural rigidity in peptide binding. The tripeptide epitope from this sequence (-Gln-Lys-Phe-) was incorporated into three different solvent-exposed loops (BC, DE, and FG) in the Tn3 domain derived from the third fibronectin type-III domain of tenascin C. The binding affinities of Q8·MV to these domains was determined by ITC (Ka = 6.4–61 × 104 M−1, Table 1), with one domain giving approximately 7-fold higher affinity than the others. Phe was confirmed as the target site by its mutation to Ala. This study showed that non-terminal Phe is a viable target in the context of a protein, and that disordered loops are viable targets for cucurbit[n]urils. This study also supported the finding that non-terminal Phe is preferred to Trp in a broader sequence context and when not located at the N-terminus.42 These two studies highlight the importance of neighboring sequence context on binding affinity.
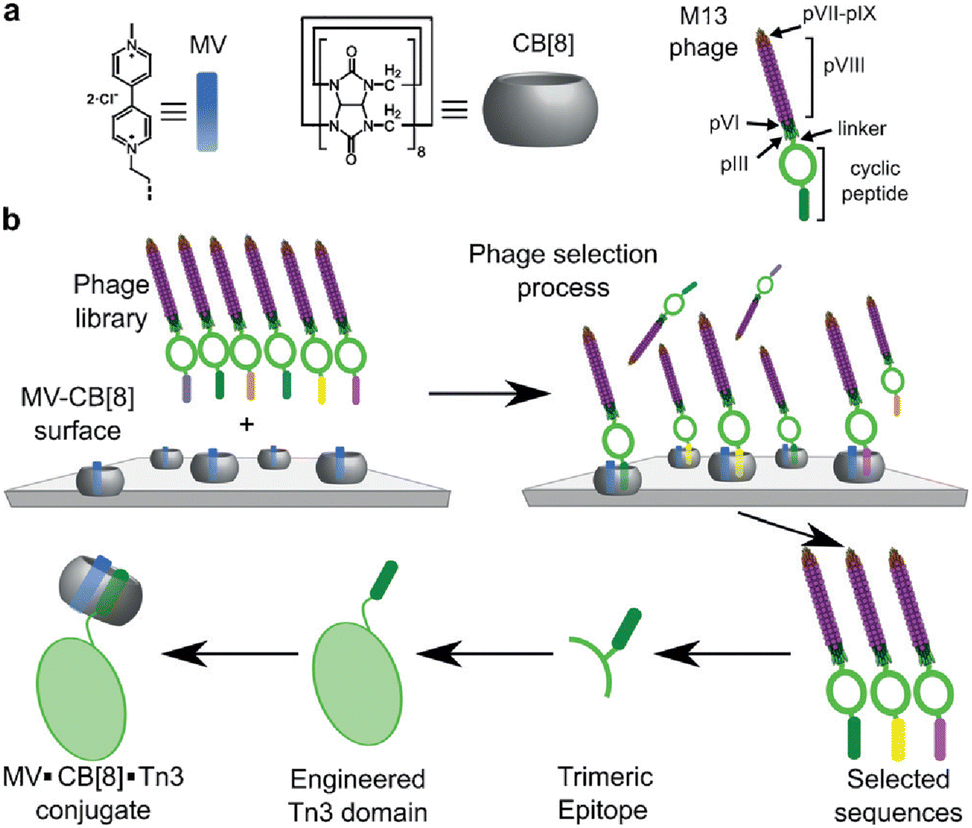 | ||
| Fig. 9 Selection of non-terminal peptide binding sites by a Q8·viologen complex using phage display, followed by engineering abundant epitopes into disordered loops on the surface of a protein. CB[8] is Q8.42 Reproduced with permission from ref. 42. © 2016 Wiley-VCH GmbH & Co. KGaA, Weinheim. | ||
2.5. Dipeptide recognition via pair-inclusion within cucurbit[8]uril
We continued to be interested in exploring the effects of peptide sequence context on the binding of peptides by Q8, but our earlier work required the peptide to be intrinsically fluorescent via a Trp residue.40 In collaboration with the Scherman and Bielawski groups, we replaced MV with an auxiliary guest of similar size and charge, tetramethylbenzobis(imidazolium) (MBBI, Fig. 10),59 which is intrinsically fluorescent. The binding affinities of MBBI and MV for Q8 were shown to be essentially identical, as were the binding affinities of Q8·MBBI and Q8·MV for H-Trp-Gly-Gly-OH, H-Gly-Trp-Gly-OH, and H-Gly-Gly-Trp-OH (Table 1). Therefore, MBBI is an excellent surrogate for MV with respect to targeting Trp. The bright fluorescence of MBBI is quenched slightly in the presence of Q8, and the fluorescence of the Q8·MBBI complex is further quenched upon binding Trp-containing peptides.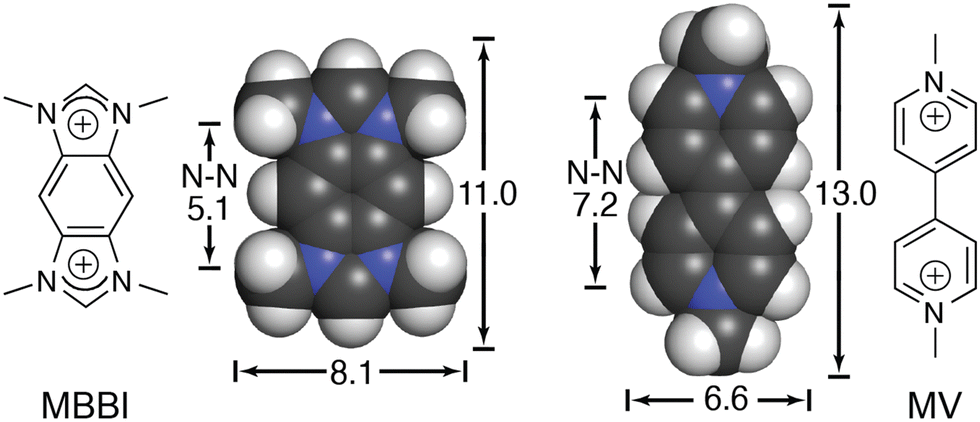 | ||
| Fig. 10 Chemical formulas and space-filling models of MBBI and MV, showing size comparisons in Angstroms.59 Reproduced with permission from ref. 59. © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
As an intrinsically fluorescent component of the sensor, MBBI has the potential to respond to the binding of nonfluorescent peptides. To this end, we synthesized a series of 105 tripeptides of sequence H-Phe-Var1-Ala-NH2, H-Phe-Ala-Var2-NH2, H-Tyr-Var1-Ala-NH2, H-Tyr-Ala-Var2-NH2, H-Trp-Var1-Ala, and H-Trp-Ala-Var2-NH2 (Var1 and Var2 = all 20 amino acids except Trp and Cys).60 The peptides were synthesized using parallel solid-phase synthesis, and the relative extent of binding to Q8·MBBI was analyzed indirectly by comparing the relative change in MBBI fluorescence in the presence of peptide. Fluorescence intensity decreased for almost all of the Trp-containing peptides and increased for almost all of the Phe-containing peptides. We hypothesized that these results were due to the simultaneous inclusion of MBBI and Trp inside the Q8 cavity, which quenches Trp fluorescence, whereas Phe-containing peptides have the ability to dimerize with Q8 with higher overall affinity than a heteroternary complex, and therefore Phe-containing peptides are likely to displace MBBI and increase fluorescence. These hypotheses were not explored further.
The interesting results of this study were in the Tyr series (Fig. 11). We observed that fluorescence increased for certain peptides and decreased for their sequence isomers. For example, the peptide H-Tyr-Leu-Ala-NH2 showed an increase in fluorescence, whereas the peptide H-Tyr-Ala-Leu-NH2 showed a decrease in fluorescence. NMR studies showed that H-Tyr-Leu-Ala-NH2 binds Q8 with slow exchange kinetics on the NMR timescale. This allowed us to conveniently determine a 1:1 Q8:peptide binding stoichiometry and to observe that MBBI is fully displaced upon addition of one equivalent of peptide. This result was surprising given that prior work would predict that Q8 should homodimerize the peptides in a 2:1 peptide:Q8 ratio. The NMR data also revealed large upfield chemical shift perturbations of the signals corresponding to the side chains of both Tyr and Leu residues, which indicates their simultaneous inclusion within the Q8 cavity. ITC data showed a range of binding constants in the low micromolar to high nanomolar range (Table 4). A semiempirical model of the Q8·H-Tyr-Leu-Ala-NH2 complex from NMR-derived distance restraints (Fig. 11) is consistent with a binding mode in which the peptide backbone folds to allow the side chains of Tyr and Leu to bind within the Q8 cavity. Therefore, we termed this binding mechanism the “pair-inclusion motif.” These results showed that Q8 can bind certain dipeptide sites with high affinity, and that the sequence and location of the binding site can have a strong influence on binding affinity. This study also demonstrated the value of MBBI as a critical component of a turn-on peptide sensor.60
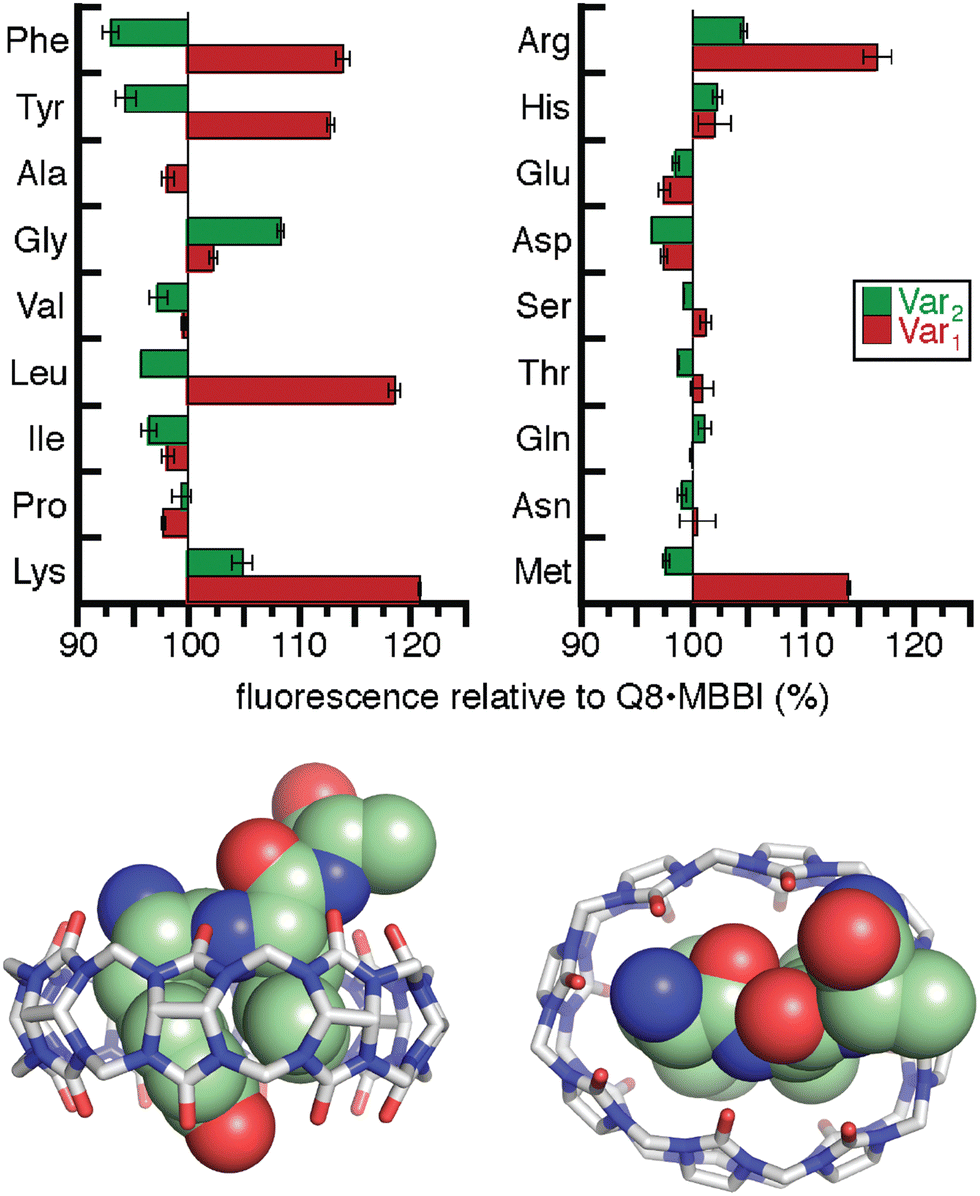 | ||
| Fig. 11 (top) Relative change in fluorescence of 1:1:1 peptide:Q8:MBBI relative to Q8·MBBI under the same condition.60 The peptide sequences are H-Tyr-Var1-Ala-NH2 and H-Tyr-Ala-Var2-NH2. Reproduced with permission from ref. 60. Copyright 2015 American Chemical Society. (bottom) Rendering of the semiempirical NMR structure of the Q8·H-Tyr-Leu-Ala-NH2 complex in two views. The peptide is shown as space-filling, and Q8 as sticks. Peptide carbons are green. Q8 carbons are grey. Oxygens are red. Nitrogens are blue. Hydrogens are not shown. | ||
| Host | Peptide | K a (M−1) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) |
|---|---|---|---|---|
| a 10 mM sodium phosphate, pH 7.0, 300 K.60 b 10 mM sodium phosphate, pH 7.0, 300 K.61 c 10 mM sodium phosphate, pH 7.0, 298 K.51 d 10 mM sodium phosphate, pH 7.0, 298 K.62 e 50 mM sodium acetate, pH 4.74, 298 K.54 f 10 mM sodium phosphate, pH 7.0, 300 K.63 g 10 mM sodium phosphate, pH 7.0.64 h Not detected. i Not reported. Putative binding sites are in bold. | ||||
| Q8a | H-Tyr-Leu-Ala-NH2 | 1.4 × 108 | −15.5 | 4.3 |
| Q8a | H-Tyr-Ala-Leu-NH2 | 2.9 × 104 | −9.5 | 3.4 |
| Q8a | H-Ala-Tyr-Leu-NH2 | 3.2 × 105 | −14.1 | 6.5 |
| Q8a | H-Tyr-Lys-Ala-NH2 | 5.0 × 106 | −15.5 | 6.3 |
| Q8a | H-Tyr-Tyr-Ala-NH2 | 1.4 × 106 | −16.1 | 7.7 |
| Q8a | H-Tyr-Phe-Ala-NH2 | 3.5 × 106 | −16.1 | 7.2 |
| Q8b | H-Met-Phe-Ala-NH2 | 7.1 × 106 | −20.1 | 10.6 |
| Q8b | H-Met-Tyr-Ala-NH2 | 4.0 × 106 | −18.2 | 9.3 |
| Q8b | H-Tyr-Met-Ala-NH2 | 7.7 × 105 | −16.6 | 8.5 |
| Q8b | H-Ala-Met-Tyr-NH2 | 1.6 × 105 | −13.8 | 6.6 |
| Q8b | H-Met-Ala-Tyr-NH2 | ndh | nri | nr |
| Q8b | H-Met-Leu-Ala-NH2 | 1.4 × 106 | −15.8 | 7.4 |
| Q8b | H-Leu-Met-Ala-NH2 | 1.7 × 106 | −12.1 | 3.5 |
| Q8b | H-Ala-Met-Leu-NH2 | nd | nr | nr |
| Q8b | H-Met-Ala-Leu-NH2 | nd | nr | nr |
| Q8b | H-Met-Lys-Ala-NH2 | 3.9 × 105 | −13.7 | 6.0 |
| Q8b | H-Lys-Met-Ala-NH2 | 1.2 × 106 | −10.9 | 2.6 |
| Q8b | H-Ala-Met-Lys-NH2 | nd | nr | nr |
| Q8b | H-Met-Ala-Lys-NH2 | nd | nr | nr |
| Q8b | H-Met-Ala-Ala-NH2 | nd | nr | nr |
| Q8b | H-Met-Tyr-Gly-Gly-Tyr-NH2 | 6.3 × 106 | −19.4 | 10.1 |
| Q8b | H-Met-Leu-Gly-Gly-Tyr-NH2 | 3.3 × 106 | −16.9 | 7.9 |
| Q8b | H-Leu-Met-Gly-Gly-Tyr-NH2 | 6.3 × 106 | −22.9 | 13.5 |
| Q8b | H-Met-Lys-Gly-Gly-Tyr-NH2 | 2.4 × 106 | −16.9 | 8.2 |
| Q8b | H-Met-Lys-Ala-Gly-Tyr-NH2 | 1.1 × 106 | −16.9 | 8.3 |
| Q8b | H-Met-Lys-Val-Gly-Tyr-NH2 | 1.6 × 105 | −15.9 | 8.8 |
| Q8b | H-Met-Ile-Gly-Gly-Tyr-NH2 | nd | nr | nr |
| Q8b | H-Met-Val-Gly-Gly-Tyr-NH2 | nd | nr | nr |
| Q8b | H-Met-Arg-Gly-Gly-Tyr-NH2 | 4.8 × 105 | −12.4 | 4.6 |
| Q8b | H-Met-Ser-Gly-Gly-Tyr-NH2 | nd | nr | nr |
| Q8b | H-Met-Gly-Gly-Gly-Tyr-NH2 | nd | nr | nr |
| Q8c | H-Tyr-Leu-NH2 | 8.2 × 106 | −13.6 | 4.2 |
| Q8c | H-Tyr-Leu-Ala-NH2 | 8.1 × 106 | −12.0 | 2.6 |
| Q8c | H-Tyr-Ala-Leu-NH2 | 2.7 × 104 | −6.2 | 0.2 |
| Q8c | H-Tyr-Leu-Ala-Ala-NH2 | 7.1 × 106 | −11.0 | 1.7 |
| Q8c | H-Ala-Ala-Tyr-Leu-Ala-Ala-NH2 | 1.8 × 105 | −11.6 | 4.4 |
| Q8c | H-Leu-Tyr-NH2 | 1.3 × 107 | −13.7 | 4.0 |
| Q8c | H-Leu-Tyr-Ala-NH2 | 1.3 × 107 | −12.0 | 2.3 |
| Q8c | H-Ala-Leu-Tyr-NH2 | 1.3 × 106 | −11.7 | 3.4 |
| Q8c | H-Phe-Leu-Ala-NH2 | 1.0 × 107 | −12.0 | 2.4 |
| Q8c | H-Ala-Phe-Leu-Ala-NH2 | 2.1 × 106 | −11.4 | 2.8 |
| Q8c | H-Leu-Phe-NH2 | 6.6 × 106 | −12.3 | 3.0 |
| Q8c | H-Phe-Leu-NH2 | 1.3 × 107 | −11.4 | 1.7 |
| Q8d | H-Tyr-Met-Ala-NH2 | 1.0 × 106 | −11.0 | 2.8 |
| Q8d | H-Tyr-Lys-Ala-NH2 | 3.1 × 106 | −11.4 | 2.6 |
| Q8d | H-Tyr-Arg-Ala-NH2 | 1.6 × 106 | −11.1 | 2.7 |
| Q8d | H-Tyr-Met-NH2 | 6.1 × 105 | −12.3 | 4.4 |
| Q8d | H-Tyr-Lys-NH2 | 2.5 × 106 | −12.7 | 4.0 |
| Q8d | H-Tyr-Tyr-NH2 | 8.9 × 105 | −11.6 | 3.5 |
| Q8d | H-Met-Tyr-NH2 | 3.9 × 106 | −19.2 | 10.2 |
| Q8d | H-Lys-Tyr-NH2 | 2.4 × 106 | −11.8 | 3.0 |
| Q8e | H-His-Phe-OH | 3.3 × 103 | nr | nr |
| Q8e | H-His-Leu-OH | 3.3 × 103 | nr | nr |
| Q8e | H-His-Tyr-OH | 1.5 × 103 | nr | nr |
| Q8e | H-Gly-His-OH | < 102 | nr | nr |
| Q8e | H-Leu-His-OH | 9.3 × 104 | nr | nr |
| Q8e | H-Gly-Gly-His-OH | < 102 | nr | nr |
| Q8e | H-His-Gly-Gly-OH | < 102 | nr | nr |
| Q8f | H-Lys-Phe-Gly-Gly-Tyr-OH | 3.0 × 109 | −16.6 | 3.6 |
| Q8f | H-Phe-Lys-Gly-Gly-Tyr-OH | 3.0 × 106 | −13.9 | 5.0 |
| Q8f | H-Leu-Tyr-Gly-Gly-Gly-OH | 8.3 × 108 | −17.0 | 4.7 |
| Q8f | H-Tyr-Leu-Gly-Gly-Gly-OH | 6.7 × 106 | −14.9 | 5.5 |
| Q8g | H-Gly-Gly-Phe-Lys-Gly-Gly-Tyr-OH | 1.2 × 107 | −13.4 | 3.7 |
| Q8g | H-Gly-Gly-Lys-Phe-Gly-Gly-Tyr-OH | 1.7 × 107 | −14.2 | 4.3 |
| Q8g | H-Gly-Gly-Phe-Phe-Gly-Gly-Tyr-OH | 9.1 × 104 | −5.8 | −1.1 |
| Q8g | H-Gly-Gly-Lys-Lys-Gly-Gly-Tyr-OH | 1.1 × 105 | −8.7 | 1.8 |
| Q8g | H-Gly-Gly-Phe-Arg-Gly-Gly-Tyr-OH | 5.9 × 106 | −14.3 | 5.0 |
| Q8g | H-Gly-Gly-Arg-Phe-Gly-Gly-Tyr-OH | 2.9 × 105 | −11.9 | 4.4 |
| Q8g | H-Gly-Gly-Arg-Arg-Gly-Gly-Tyr-OH | < 103 | nd | nd |
| Q8g | H-Gly-Gly-Trp-Lys-Gly-Gly-Tyr-OH | 6.7 × 105 | −12.0 | 4.0 |
| Q8g | H-Gly-Gly-Lys-Trp-Gly-Gly-Tyr-OH | 2.2 × 106 | −13.0 | 4.3 |
| Q8g | H-Gly-Gly-Trp-Trp-Gly-Gly-Tyr-OH | < 103 | nd | nd |
| Q8g | H-Gly-Gly-Tyr-Leu-Gly-Gly-Tyr-NH2 | 1.8 × 106 | −14.9 | 6.3 |
| Q8g | H-Gly-Gly-Leu-Tyr-Gly-Gly-Tyr-NH2 | 4.4 × 106 | −14.9 | 5.8 |
| Q8g | H-Gly-Gly-Tyr-Tyr-Gly-Gly-Tyr-NH2 | < 103 | nd | nd |
| Q8g | H-Gly-Gly-Leu-Leu-Gly-Gly-Tyr-NH2 | 5.6 × 104 | −13.6 | 7.1 |
| Q8g | H-Gly-Ala-Lys-Phe-Ala-Gly-Tyr-NH2 | 1.1 × 106 | −12.0 | 3.7 |
| Q8g | H-Gly-Ala-Phe-Lys-Ala-Gly-Tyr-NH2 | 3.6 × 106 | −11.3 | 5.1 |
| Q8g | H-Gly-Ala-Phe-Lys-Gly-Gly-Tyr-NH2 | 1.0 × 105 | −10.8 | 4.0 |
| Q8g | H-Gly-Gly-Phe-Lys-Ala-Gly-Tyr-NH2 | 2.3 × 105 | −9.0 | 1.6 |
Excited by the discovery that Q8 can bind peptides via inclusion of the side chains of neighboring residues, we considered that the additional complexity of a dipeptide binding site should allow us to identify other possible sequences that bind Q8 with high affinity. The additional binding interface enabled by pair-inclusion should allow inclusion of nonaromatic residues. To this end, we synthesized and screened a series of 144 peptides of sequence H-Var1-Var2-Ala-NH2 (Var1 = Tyr, Phe, Ile, Leu, Met, Pro, Arg, Lys, and Var2 = all 20 amino acids except Trp and Cys).61 The peptides were synthesized using parallel solid-phase synthesis, and the relative extent of binding to Q8·MBBI was analyzed indirectly by comparing the relative change in the fluorescence of Q8·MBBI in the presence vs. absence of peptide. The patterns of fluorescence suggested that certain peptides with N-terminal Met bind tightly to Q8. A detailed structure–activity investigation by ITC and NMR yielded several determinants for sub-micromolar binding via the pair-inclusion motif (Fig. 12) (Table 4). Specifically, when the first residue is Met, then the second residue should be Leu, Tyr, Phe, Lys, or Arg; the first two resides can be reversed in sequence; and the third residue should be Gly or Ala. Therefore, the pair-inclusion motif allowed for targeting N-terminal Met, which is found in all newly translated eukaryotic proteins, as well as entirely nonaromatic binding sites.61
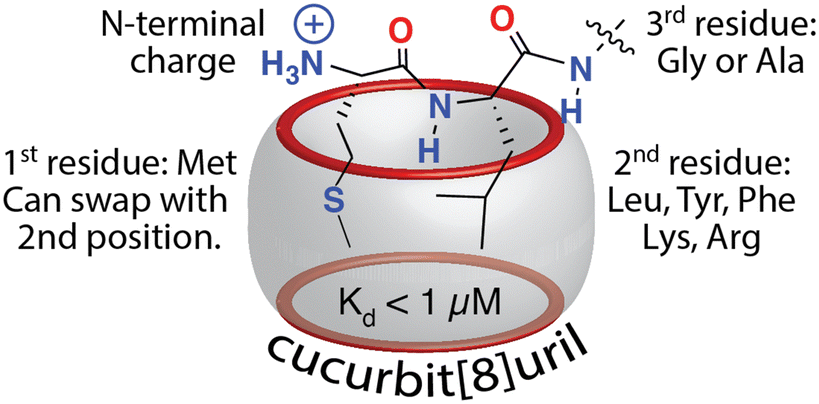 | ||
| Fig. 12 Sequence determinants for high-affinity binding of N-terminal dipeptide sites by Q8.61 Reproduced with permission from ref. 61. Copyright 2018 American Chemical Society. | ||
Scherman and coworkers were also motivated by the 2015 discovery of pair-inclusion binding60 and considered that the proposed mechanism may be inaccurate and that our results could potentially be explained by the formation of 2:2 (Q8:peptide) complexes.51 Using a combination of binding enthalpy and entropy data determined by ITC, 1H NMR chemical shift perturbation (Fig. 13), and diffusion constants determined by diffusion ordered NMR spectroscopy (DOSY), they identified patterns that correlate to 1:1, 2:1, and 2:2 guest:Q8 complexes65 and applied this approach to the study of Q8·peptide complexes in the pair-inclusion motif.51 The data unambiguously corroborated our conclusions on the mechanism of pair-inclusion complexation, including that the binding stoichiometry is 1:1 peptide:Q8, not 2:2, and that the side chains of two neighboring residues in a single peptide molecule include simultaneously within the cavity of a single Q8 molecule.
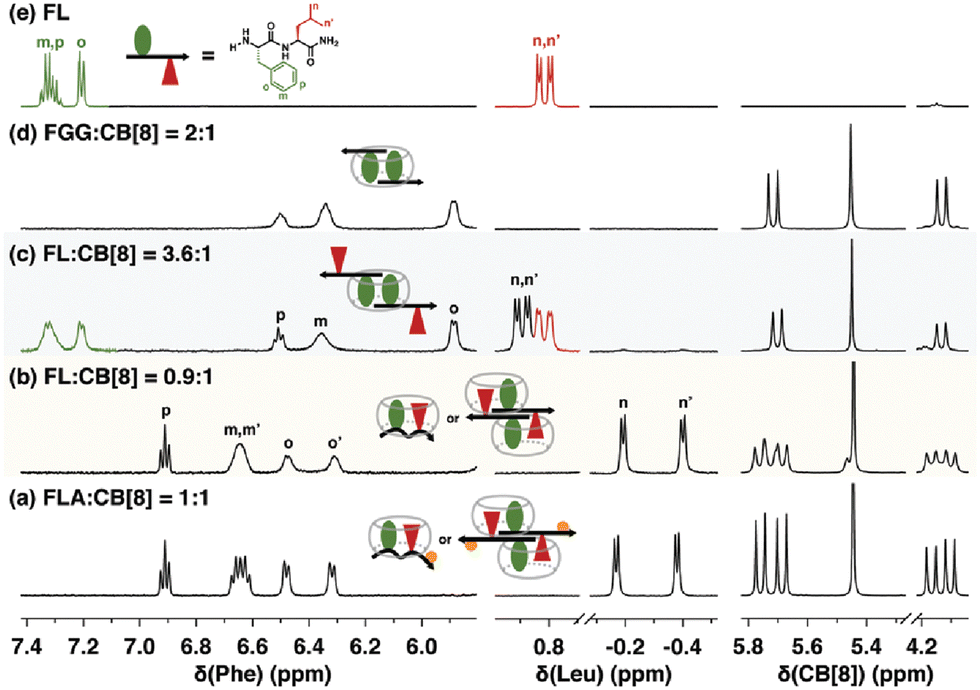 | ||
| Fig. 13 1H NMR spectra of H-Phe-Leu-Ala-NH2 (FLA) and H-Phe-Leu-NH2 (FL) at various ratios with Q8, showing patterns of signal perturbation that indicate the binding modes shown schematically. CB[8] is Q8.51 Reproduced from ref. 51 with permission from the Royal Society of Chemistry. | ||
Recently, Scherman and coworkers investigated the nature of peptide folding induced by Q8 in the pair-inclusion motif.62 A series of dipeptides known to bind Q8 via pair-inclusion was investigated by circular dichroism (CD) spectroscopy, and the CD spectra effectively invert depending on the stereochemistry of the peptide. They proposed a model of “clockwise” or “counterclockwise” folding in order to insert both neighboring side chains into the Q8 cavity (Fig. 14). This concept was used to design a peptide with a central dipeptide binding site that bends upon treatment with Q8, as observed by fluorescence resonance energy transfer.
 | ||
| Fig. 14 Model for sequence-dependent folding of peptides by Q8 as monitored by circular dichroism spectroscopy (right). CB[8] is Q8.62 Reproduced with permission from ref. 62. Copyright 2021 American Chemical Society. | ||
Recently we reported an investigation of Q8 binding at nonterminal sites via the pair-inclusion motif.64 A library of 64 peptides of sequence H-Gly-X1-X2-Gly-NH2 (X = Phe, Leu, Lys, Met, Arg, Tyr, Trp, Pro) was synthesized in parallel and screened by fluorescence for binding to the Q8·MBBI complex (Fig. 15). Relatively high increases in fluorescence were observed for X1-X2 sequences Tyr-Leu, Leu-Tyr, Phe-Leu, Leu-Phe, Tyr-Lys, Lys-Tyr, Phe-Arg, Arg–Phe, Leu-Lys, Lys-Leu, Phe-Lys, Lys-Phe, and Phe-Met. The strongest increase was observed for Phe-Lys. Detailed studies of binding to purified heptapeptides of sequence H-Gly-Gly-X1-X2-Gly-Gly-Tyr-OH using ITC and NMR revealed the highest affinities for X1-X2 = Lys-Phe (1.7 × 107 M −1) and Phe-Lys (1.2 × 107 M−1). Several other submicromolar complexes were identified (Table 4). A study of the effects of the neighboring sequence revealed that while Gly to Ala mutations flanking the Lys-Phe site had only a modest decrease in affinity, the same mutations flanking the Phe-Lys site led to a loss of almost three orders of magnitude in affinity. In all cases where affinity was observable, the binding stoichiometry was 1:1 peptide:Q8, and NMR studies show simultaneous inclusion of both side chains at the target dipeptide site.64
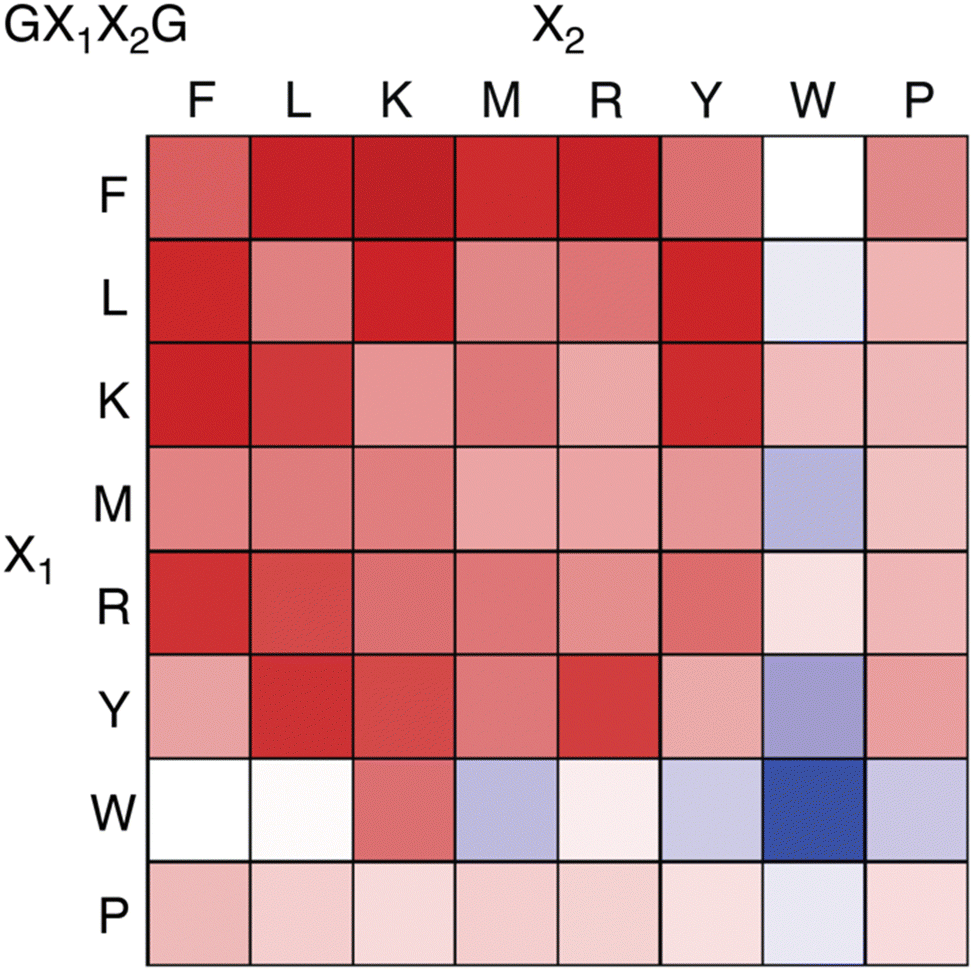 | ||
| Fig. 15 Fluorescence assay heat map, showing the change in fluorescence of the Q8·MBBI complex upon addition of peptide.64 Color saturation correlates to the extent of change in fluorescence, with red indicating an increase in fluorescence, and blue indicating a decrease. Reproduced with permission from ref. 64. Copyright 2024 American Chemical Society. | ||
We obtained a crystal structure of the Q8·H-Gly-Gly-Leu-Tyr-Gly-Gly-Gly-OH complex at 0.79 Å resolution.64 The structure confirmed unambiguously our proposed model for the pair-inclusion motif,60 with the side chains of Leu and Tyr bound within the cavity of Q8 and complementary electrostatic interactions between peptide and Q8. The unanticipated result of this study is the observation that four contiguous residues, Gly-Leu-Tyr-Gly, form a type II β-turn in which the amide NH of Gly5 forms an intramolecular hydrogen bond with the carbonyl oxygen of Gly2 (Fig. 16).
 | ||
| Fig. 16 Crystal structure of the Q8·H-Gly-Gly-Leu-Tyr-Gly-Gly-Gly-OH complex.64 (top) Rendering that illustrates the inclusion of the Leu and Tyr side chains (space-filling models) within the cavity of Q8 (sticks with a solvent-accessible surface), and the deformation of Q8 from D8h symmetry upon binding. (bottom) Rendering that illustrates the Q8-induced β-hairpin turn, with the Gly5 NH⋯Leu3 OC hydrogen bond, Leu3 NH⋯Q8 hydrogen bond, and Leu3-Tyr4 CH⋯π interaction shown as dashed lines. CCDC ID 2312293. Reproduced with permission from ref. 64. Copyright 2024 American Chemical Society. | ||
In a subsequent study, we investigated the binding of Q8 to peptides containing the lead Lys-Phe and Phe-Lys dipeptide sites located at the N-terminus.63 It was found that Q8 binds with extraordinarily high affinity to H-Lys-Phe-Gly-Gly-Tyr-OH (3.0 × 109 M−1) and H-Leu-Tyr-Gly-Gly-Gly-OH (8.3 × 108 M−1) and with 1000-fold and 120-fold selectivity over their sequence isomers H-Phe-Lys-Gly-Gly-Tyr-OH (3.0 × 106 M−1) and H-Tyr-Leu-Gly-Gly-Gly-OH (6.7 × 106 M−1), respectively. We obtained sub-Å resolution crystal structures of the complexes of Q8 with sequence isomers, Q8·H-Leu-Tyr-Gly-Gly-Gly-OH and Q8·H-Tyr-Leu-Gly-Gly-Gly-OH (Fig. 17). These structures correlated well with the structure of Q8 bound at a non-terminal Leu-Tyr site (vide supra) and revealed a few differences that may explain the observed sequence-selectivity. In particular, the side chain of the N-terminal Tyr in Q8·H-Tyr-Leu-Gly-Gly-Gly-OH buries more deeply than that of the non-terminal Tyr in Q8·H-Leu-Tyr-Gly-Gly-Gly-OH, allowing contact between the N-terminal ammonium group and proximal Q8 CO groups. In the Q8·H-Tyr-Leu-Gly-Gly-Gly-OH complex, this effect pushes the Tyr side chain so deeply that its hydroxyl group resides outside of the Q8 portal and needs a water molecule to mediate its electrostatic contact with the Q8 portal. In the Q8·H-Leu-Tyr-Gly-Gly-Gly-OH complex, however, the Tyr hydroxyl group is positioned to form a direct hydrogen bond with Q8.63
 | ||
| Fig. 17 Crystal structure of the isomeric complexes (top) Q8·H-Leu-Tyr-Gly-Gly-Gly-OH and (bottom) Q8· H-Tyr-Leu-Gly-Gly-Gly-OH.63 (left) Rendering that illustrates the inclusion of the Leu and Tyr side chains (space-filling models) within the cavity of Q8 (sticks with a solvent-accessible surface), the relative deformation of Q8 from D8h symmetry upon binding, and the interaction of the Tyr hydroxyl group with a Q8 OC group either directly (Q8·H-Leu-Tyr-Gly-Gly-Gly-OH) or via an ordered water molecule (Q8·H-Tyr-Leu-Gly-Gly-Gly). (right) Rendering that illustrates the relative depth of the peptide within the Q8 cavity and the corresponding alignment of electrostatically complementary groups. CCDC ID 2313004 and 2314758. Reproduced from ref. 63 with permission from the Royal Society of Chemistry. | ||
2.6. Aromatic peptide recognition by cucurbit[7]uril
Q7 has a smaller cavity than Q8 and can only accommodate the side chain of one aromatic residue. Kim, Inoue and coworkers first reported the high-affinity and selective binding of peptides by Q7 in 2006.66 They reported a Ka value of 1.4 × 107 M−1 for H-Phe-Leu-NH2 in 0.1 M NaCl with 2.6-fold selectivity over H-Phe-Phe-NH2 18-fold selectivity over H-Phe-Ala-NH2, and 480-fold selectivity over H-Phe-Pro-NH2 (Table 5). The decrease in affinity for a Pro residue at the second position is likely due to limited conformational freedom and steric interactions, but it remains unclear to us why Leu at the second position significantly increases affinity. Diastereomeric sequences (e.g., H-Phe-D-Ala-OH) were compared, and Q7 bound with modest 2–10-fold diastereoselectivity, sometimes favoring the native sequence and sometimes favoring the diastereomer. This study demonstrated that N-terminal Phe is also a high-affinity site for Q7, and that the identity of the second residue can significantly influence binding affinity.66| Host | Peptide | K a (M−1) | ΔH (kcal mol−1) | -TΔS (kcal mol−1) |
|---|---|---|---|---|
| a 0.1 M NaCl, 298 K, dPhe is the D-isomer.66 b Pure water, 298 K.67 c 0.1 M NaCl, 298 K.67 d 10 mM sodium phosphate, pH 7.0, 300 K.68 e 10 mM sodium phosphate, pH 7.0, 300 K, tBuPhe is 4-tert-butylphenylalanine, AmPhe is 4-aminomethylphenylalanine.69 f 10 mM sodium phosphate, pH 7.0, 300 K.70 g Pure water, 298 K.52 h 10 mM ammonium phosphate, pH 7.2, 310 K.71 i 10 mM sodium phosphate, pH 7.0, 298 K.72 j 10 mM ammonium phosphate, pH 7.2, 300 K.73 k 50 mM sodium phosphate, pH 4.74, 298 K.54 l 10 mM ammonium phosphate, pH 7.0, temperature not reported, pSer is phosphoserine, pThr is phosphothreonine.44 m Not detected. n Not reported. Putative binding sites are in bold. | ||||
| Q7a | H-Phe-Ala-OH | 7.9 × 105 | −7.3 | −0.7 |
| Q7a | H-dPhe-Ala-OH | 1.3 × 106 | −7.8 | −0.6 |
| Q7a | H-Phe-Pro-OH | 2.9 × 104 | −5.9 | −0.2 |
| Q7a | H-Phe-dPro-OH | 5.0 × 104 | −6.5 | 0.1 |
| Q7a | H-Phe-Phe- NH2 | 5.3 × 106 | −8.8 | −0.4 |
| Q7a | H-dPhe-Phe- NH2 | 1.3 × 106 | −6.9 | −1.4 |
| Q7a | H-Phe-Leu-NH2 | 1.4 × 107 | −8.7 | −1.1 |
| Q7a | H-Phe-dLeu-NH2 | 1.7 × 106 | −5.8 | −2.7 |
| Q7b | H-Phe-Gly-OH | 3.0 × 107 | −11.3 | 1.1 |
| Q7b | H-Gly-Phe-OH | 1.3 × 103 | −7.2 | 2.9 |
| Q7b | H-Tyr-Gly-OH | 3.6 × 106 | −10.6 | 1.6 |
| Q7b | H-Gly-Tyr-OH | 2.0 × 102 | −5.5 | 2.4 |
| Q7b | H-Trp-Gly-OH | 5.6 × 105 | −10.7 | 2.8 |
| Q7b | H-Gly-Trp-OH | 2.8 × 102 | −4.3 | 1.0 |
| Q7b | H-Phe-Ala-OH | 1.9 × 107 | −11.8 | 1.9 |
| Q7b | H-Ala-Phe-OH | 1.3 × 103 | −8.3 | 4.0 |
| Q7c | H-Phe-Gly-OH | 1.7 × 106 | −7.7 | −0.9 |
| Q7c | H-Phe-Ala-OH | 7.9 × 105 | −7.3 | −0.7 |
| Q7c | H-Phe-Gly-NH2 | 3.7 × 106 | −7.4 | −1.6 |
| Q7c | H-Gly-Phe-NH2 | 4.4 × 104 | −7.3 | 1.0 |
| Q7c | H-Phe-Ala-NH2 | 8.1 × 106 | −9.9 | 0.5 |
| Q7c | H-Ala-Phe-NH2 | 4.5 × 104 | −7.7 | 1.4 |
| Q7c | H-Tyr-Gly-NH2 | 1.1 × 107 | −10.2 | 0.6 |
| Q7c | H-Gly-Tyr-NH2 | 7.5 × 104 | −9.4 | 2.7 |
| Q7d | H-Phe-Gly-Gly-OH | 2.8 × 106 | −17.5 | 8.7 |
| Q7d | H-Gly-Phe-Gly-OH | 2.2 × 104 | −9.3 | 3.3 |
| Q7d | H-Gly-Tyr-Gly-OH | 2.7 × 103 | −2.2 | −2.5 |
| Q7e | H-Phe-Gly-Gly-NH2 | 3.2 × 106 | −13.4 | 4.4 |
| Q7e | H-Gly-Phe-Gly-NH2 | 2.3 × 105 | −9.8 | 2.4 |
| Q7e | H-tBuPhe-Gly-Gly-NH2 | 4.8 × 106 | −16.2 | 7.1 |
| Q7e | H-AmPhe-Gly-Gly-NH2 | 1.1 × 109 | −14.2 | 1.8 |
| Q7e | H-Gly-AmPhe-Gly-NH2 | 2.0 × 106 | −8.2 | −0.5 |
| Q7f | H-AmPhe-Gly-Asn-Gln-NH2 | 2.0 × 107 | −16.9 | 6.8 |
| Q7f | H-Phe-Gly-Asn-Gln-NH2 | 7.1 × 106 | −15.6 | 6.1 |
| Q7f | H-AmPhe-Val-Asn-Gln-NH2 | 1.3 × 107 | −15.7 | 6.0 |
| Q7f | H-Phe-Val-Asn-Gln-NH2 | 4.0 × 106 | −14.5 | 5.3 |
| Q7g | H-Phe-Gly-Gly-Gly-Cys | 1.2 × 107 | −13.8 | 4.15 |
| Q7h | H-Thr-Gly-Ala-Phe-Met-NH2 | 1.3 × 104 | nrn | nr |
| Q7h | H-Thr-Gly-dAla-Phe-Met-NH2 | 2.6 × 104 | nr | nr |
| Q7h | H-Thr-Gly-Ala-Phe-Leu-NH2 | 3.5 × 103 | nr | nr |
| Q7h | H-Thr-Gly-Ser-Phe-Met-NH2 | 1.9 × 104 | nr | nr |
| Q7h | H-Thr-Gly-Gly-Phe-Met-NH2 | 1.4 × 104 | nr | nr |
| Q7h | H-Thr-Gly-Ala-Phe-Leu-OH | 1.8 × 103 | nr | nr |
| Q7h | H-Phe-Met-NH2 | 1.5 × 107 | nr | nr |
| Q7h | H-Phe-Leu-NH2 | 2.7 × 107 | nr | nr |
| Q7h | H-Phe-Leu-OH | 2.1 × 106 | nr | nr |
| Q7i | H-Thr-Gly-Ala-Phe-Met-NH2 | 3.2 × 103 | nr | nr |
| Q7i | H-Thr-Gly-Ala-Phe-Leu-NH2 | 1.4 × 104 | nr | nr |
| Q7i | H-Thr-Gly-Ala-Phe-Leu-OH | 2.4 × 103 | nr | nr |
| Q7i | H-Phe-Met-NH2 | 2.6 × 107 | nr | nr |
| Q7i | H-Phe-Leu-NH2 | 1.4 × 107 | nr | nr |
| Q7i | H-Phe-Leu-OH | 2.1 × 106 | nr | nr |
| Q7j | H-Phe-Met-NH2 | 1.4 × 107 | −22.7 | 12.9 |
| Q7j | H-Tyr-Met-NH2 | 6.4 × 105 | −20.0 | 12.1 |
| Q7j | H-Trp-Met-Gly-NH2 | 2.3 × 105 | −20.1 | 12.7 |
| Q7j | H-AmPhe-Met-NH2 | 5.3 × 108 | −10.5 | −1.8 |
| Q7k | H-His-Phe-OH | 5.0 × 102 | nr | nr |
| Q7k | H-His-Leu-OH | 5.0 × 102 | nr | nr |
| Q7k | H-His-Tyr-OH | 1.1 × 102 | nr | nr |
| Q7k | H-Gly-His-OH | 3.3 × 102 | nr | nr |
| Q7k | H-Leu-His-OH | 2.2 × 104 | nr | nr |
| Q7k | H-Tyr-His-OH | 1.2 × 105 | nr | nr |
| Q7k | H-Gly-Gly-His-OH | <102 | nr | nr |
| Q7l | H-Pro-Leu-Ile-Tyr-Leu-Arg-Leu-Leu-Arg-Gly-Gln-Phe-OH | ndm | nr | nr |
| Q7l | H-Arg7-OH | nd | nr | nr |
| Q7l | H-Leu-Arg-Arg-Trp-Ser-Leu-Gly-OH | nd | nr | nr |
| Q7l | H-Leu-Arg-Arg-Trp-pSer-Leu-Gly-OH | nd | nr | nr |
| Q7l | H-Trp-Lys-Arg-Thr-Leu-Arg-Arg-Leu-OH | 4.1 × 104 | nr | nr |
| Q7l | H-Trp-Lys-Arg-pThr-Leu-Arg-Arg-Leu-OH | 4.1 × 104 | nr | nr |
| Q7l | H-Phe-Arg7-OH | 2.9 × 107 | nr | nr |
Kim, Inoue, and coworkers followed this work with a study of Q7 binding to dipeptides containing Phe, Tyr, or Trp residues.67 In pure water, Q7 was reported to bind H-Phe-Gly-OH with a Ka value of 3.0 × 107 M−1, with 8-fold selectivity over H-Tyr-Gly-OH and with 54-fold selectivity over H-Trp-Gly-OH (Table 5). This finding was analogous to the selectivity of Q8 for N-terminal Phe versus N-terminal Trp,18 and it showed that Q7 binds well to N-terminal Tyr, whereas Q8 does not bind measurably to H-Tyr-Gly-Gly-OH. NMR spectra showed the selective upfield perturbation of aromatic signals upon binding of H-Phe-Gly-OH or H-Tyr-Lys-OH to Q7, which corroborated Q7 binding at the N-terminal Phe or Tyr, respectively. Q7 was shown to bind H-Phe-Gly-OH with 23000-fold selectivity over its sequence isomer, H-Gly-Phe-OH. Similarly, N-terminal Tyr and N-terminal Trp were preferred over their sequence isomers by 18000-fold and 2000-fold, respectively.67 This degree of sequence-selectivity is truly remarkable. The thermodynamic data presented in the supporting information for that paper showed that substituting Gly with Ala does not significantly influence binding affinity.62 This is important because although Gly is the simplest amino acid residue, Ala has a β carbon and is therefore more representative of the other amino acids. These data also showed that the observed binding affinity is reduced by ∼20-fold for H-Phe-Gly-OH and H-Phe-Ala-OH in the presence of 0.1 M NaCl. This result is consistent with the competition of Na+ for binding at the Q7 portals. Q7 binds Na+ with a Ka value of 2600 M−1,74 and a simple competition model (Q7·Na+ + peptide → Q7·peptide + Na+) would predict a reduction of 258-fold in binding affinity at this concentration. Therefore, these data suggest that Na+ does not fully compete with organic guests for binding to cucurbit[n]urils. This assertion would be consistent with the availability of multiple sites and binding geometries for metal cations at the CO portals, and this discrepancy signifies a fundamental complication in the analysis of all Qn binding data determined under different experimental conditions.
In the time since this study, there have been many binding constants reported for Q7 in complex with aromatic peptides,44,68,71,73,75 and there is general agreement in the trends observed (Table 5). One point of interest is the comparison of Q7 binding to H-Phe-Gly-Gly-NH2versus H-Gly-Phe-Gly-NH2,69 in which the binding site has moved to the center of a peptide versus a terminal position. In this case, the selectivity for the N-terminal versus non-terminal position is only 14-fold. This result is in contrast to the 84-fold selectivity of Q7 for H-Phe-Gly-NH2versus Gly-Phe-NH2 and the 23000-fold selectivity of H-Phe-Gly-OH versus H-Gly-Phe-OH.67 The differences between these data sets suggest that the continuing peptide chain is involved in complexation and works to mitigate sequence-selectivity. This is a minor caution against the use of dipeptides to model the behavior of longer peptides or proteins.
In a pair of molecular dynamics computational studies, Li and coworkers predicted that Q7 should bind best to an aromatic residue at the N-terminus, and that placing a basic residue at the third position of the H-X-Gly-Z-OH tripeptide (X = Phe, Tyr, Trp; Z = His, Lys, Arg) should significantly increase binding affinity due to interaction of the basic side chain with the portal CO groups of Q7.76,77 Although there are not yet experimental studies to verify these predictions, they are interesting to consider.
2.7. Molecular recognition of insulin by cucurbit[7]uril
It was clear that cucurbit[n]urils could bind strongly and selectively to N-terminal aromatic sites on peptides, but it was not clear early on whether this binding would translate to folded proteins. In contrast to peptides, the folded structures of proteins have limited solvent accessibility and constrained conformational mobility, which are of particular concern when a ligand needs to fully encapsulate a portion of the protein. This translation from peptide to protein would validate the use of Qn-mediated peptide recognition as affinity tags for proteins. Brunsveld and coworkers genetically engineered proteins to contain the Phe-Gly-Gly (i.e., FGG) sequence at the N-terminus and showed that this modification did not significantly influence the binding affinity to Q8, as discussed in detail below (Table 3).78 This study was the first to demonstrate the use of Qn–peptide interactions as protein affinity tags.In parallel with these studies, we were also learning that Q7 can bind to a native protein containing an N-terminal Phe residue.68 Human insulin comprises a 21-mer A-chain and 30-mer B-chain linked covalently via two disulfide bonds. We reported a structure and activity study showing that Q7 binds site-selectively to the Phe residue at the N-terminus of the B-chain (i.e., PheB1).68 ITC data showed that Q7 binds regular human insulin in 10 mM sodium phosphate, pH 7.0, with a Ka value of 1.5 × 106 M−1 and with more than 10000-fold selectivity versus a GluB1, GluB27 mutant (Ka < 100 M−1) (Table 6). The similarity in Q7 binding affinity for insulin versus H-Phe-Gly-Gly-OH (2.8 × 106 M−1) also supported the translation of peptide recognition to protein recognition. In essence, human insulin has a built-in affinity tag for Q7. The sub-micromolar affinity of this complex, however, would be insufficient to bind effectively at the picomolar to nanomolar physiological concentrations of insulin.
| Host | protein | K a (M−1) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) |
|---|---|---|---|---|
| a 10 mM sodium phosphate, pH 7.0, 300 K.68 b 10 mM sodium phosphate, pH 7.0, 298 K.79 c PBS: 10 mM sodium phosphate, 1.8 mM potassium phosphate, 137 mM NaCl, 2.7 mM KCl, pH 7.4, 298 K.75 d 20 mM PBS, 298 K.80 e 0.1% formic acid, pH 2.7, 298 K.81 f 10 mM sodium phosphate, pH 7.0, 300 K.70 g 10 mM sodium phosphate, pH 3.5, 300 K.80 h Pure water.81 i 20 mM PBS, 298 K.82 j 20 mM sodium phosphate, 150 mM NaCl pH 7.0, 298 K.58 k 20 mM PBS, 298 K.83 l Not detected. m Not reported. | ||||
| Q7a | Human insulin | 1.5 × 106 | −10.8 | 2.3 |
| Q7a | Human insulin PheB1Glu ThrB27Glu | ndl | nrm | nr |
| Q7b | Human growth hormone | 5.9 × 105 | nr | nr |
| Q7c | Glutathione-S-transferase | nd | nr | nr |
| Q7c | Glutathione-S-transferase Gln207tBuPhe | 2.3 × 105 | nr | nr |
| Q7c | Glutathione-S-transferase Arg108tBuPhe | 4.1 × 104 | nr | nr |
| Q7d | Human insulin | 4.4 × 106 | nr | nr |
| Q7e | Human insulin | 3.8 × 107 | nr | nr |
| Q7e | Human insulin B1–13 | 1.1 × 106 | nr | nr |
| Q7e | Human insulin B1–13 PheB1Ala | 1.7 × 104 | nr | nr |
| Q7e | Human insulin B14–30 | 5.4 × 103 | nr | nr |
| Q7e | Human insulin B14–30 PheB24Ala | 5.5 × 103 | nr | nr |
| Q7e | Human insulin B14–30 PheB25Ala | 7.9 × 104 | nr | nr |
| Q7e | Human insulin B14–30 PheB24Ala PheB25Ala | 6.6 × 103 | nr | nr |
| Q7f | Human insulin PheB1AmPhe | 1.0 × 107 | −10.8 | 2.3 |
| Q7g | Human insulin | 2.3 × 106 | nr | nr |
| Q7g | Human insulin PheB1Ala N-terminal benzoic acid | 1.1 × 104 | nr | nr |
| Q7g | human insulin PheB1Ala N-terminal phenylacetic acid | 8.2 × 104 | nr | nr |
| Q7g | Human insulin | 2.8 × 106 | nr | nr |
| Q7g | Human insulin PheB1Ala N-terminal benzoic acid | <103 | nr | nr |
| Q7g | Human insulin PheB1Ala N-terminal phenylacetic acid | <103 | nr | nr |
| Q7h | Human insulin Pro28Asp | 1.9 × 106 | nr | nr |
| Q7h | Pramlintide | 2.6 × 104 | nr | nr |
| Q7i | Aβ 1–40 | 7.1 × 104 | nr | nr |
| Q7i | Aβ 1–40 Phe4Ala | 5.2 × 104 | nr | nr |
| Q7i | Aβ 1–40 Phe19Ala | 5.1 × 104 | nr | nr |
| Q7i | Aβ 1–40 Phe20Ala | 5.1 × 104 | nr | nr |
| Q7i | Aβ 1–40 Phe4Ala Phe19Ala Phe20Ala | 6.8 × 103 | nr | nr |
| Q7i | Aβ 1–42 | 2.9 × 104 | nr | nr |
| Q7j | Aβ 4–16 | 1.1 × 106 | nr | nr |
| Q7j | Aβ 1–16 | 1.0 × 104 | nr | nr |
| Q7k | Human calcitonin | 6.7 × 104 | nr | nr |
| Q7k | Human calcitonin Tyr12Ala | 2.6 × 104 | nr | nr |
| Q7k | Human calcitonin Phe16Ala | 2.0 × 104 | nr | nr |
| Q7k | Human calcitonin Phe19Ala | 2.7 × 104 | nr | nr |
| Q7k | Human calcitonin Phe22Ala | 3.2 × 104 | nr | nr |
| Q7k | Human calcitonin Tyr12Ala Phe16Ala Phe19Ala Phe22Ala | nd | nr | nr |
An interesting observation of this study involves one disparity between peptide and protein binding. Q7 binds weakly to non-terminal Phe and Tyr residues in the context of small peptides, H-Gly-Phe-Gly-OH and H-Gly-Tyr-Gly-OH, with Ka values of 2.2 × 104 M−1 and 2.7 × 103 M−1, respectively. However, Q7 binding was not measurable to the GluB1, GluB27 mutant, which has two surface-exposed, non-terminal Phe residues and four surface-exposed, non-terminal Tyr residues.68 This disparity is perhaps due to the neighboring sequence context or, more likely, to the difference in the accessibility of the side chain of a target site to encapsulation by Q7 when located on a peptide versus on the surface of a folded protein. A high-resolution crystal structure of the Q7·insulin complex shed some light on this issue.68 The asymmetric unit comprised two molecules of insulin, one of which is bound to one molecule of Q7 at the PheB1 position (Fig. 18). The aromatic side chain of PheB1 is buried within the Q7 cavity, and the N-terminal ammonium group is proximal to Q7 CO groups, with a binding interface of ∼200 Å2. Therefore, the molecular basis for the recognition of N-terminal Phe by Q7 is congruent with that of Q8.18
 | ||
| Fig. 18 Crystal structure of the Q7·insulin complex.68 Q7 is shown as a stick model. The Q7-bound insulin molecule is shown in green. The insulin molecule not bound to Q7 is shown in black. The PheB1 position is rendered as space-filling. (a) Superposition with minimized RMSD over all alpha carbons. This rendering shows the N-terminal region of the B-chain unfolding from an alpha helix upon binding to Q7 and unfolding to separate from the rest of the protein. (b) This rendering shows the proximity of the N-terminal ammonium group with Q7 CO groups. PDB ID 3Q6E. | ||
Superposition of the two insulin molecules in the asymmetric unit was useful for studying the relationship between insulin structure and Q7 binding (Fig. 17).68 Structural homology between the two insulins is extensive, with a 0.37 Å root-mean-square deviation (RMSD) over 35 alpha carbons. The largest deviation is at residues B1–B4, which unfold in order to present a fully solvent-accessible site for Q7 to encapsulate. Akin to short peptides, it is common for protein termini to be more disordered and solvent-exposed than other regions of the protein.84 This observation suggested a more general principle, which is that extensive solvent accessibility is an essential feature of protein recognition by cucurbit[n]urils. This feature of cucurbit[n]urils is perhaps an advantage for targeting intrinsically disordered regions of proteins, which are known to be essential for the function of many proteins and to be implied in the etiology of many diseases.85 The observation by Scherman and coworkers that Q8 can bind to sites on disordered loops (vide supra)42 supports this hypothesis.
Like cucurbit[n]urils, natural sequence-selective receptors for proteins also require those sites to be fully solvent-accessible. For example, N-recognin proteins are part of the N-degron pathway of protein homeostasis that defines the biological lifetime of a protein by the identity of its N-terminal residue (i.e., the N-end rule).10,86 N-recognins in both prokaryotes and eukaryotes include the unfolded N-terminal residue of their protein targets within their active sites (Fig. 19). This is also true for many proteases, kinases, and other biomolecular receptors that target proteins sequence-selectively.10 In this context, cucurbit[n]urils behave as synthetic N-recognins.
 | ||
| Fig. 19 Crystal structures of N-recognins bound to their cognate peptide termini. (a) ClpS bound to Trp-Leu-Phe, showing burial of the N-terminal indole side chain deep within a cavity (PDB ID: 3GQ1).87 (b) UBR box of UBR2 bound to Arg-Ile-Phe, showing several electrostatic contacts (<3 Å, red dashed lines) with Asp side chains in the binding site (PDB ID: 3NY3).88 N-recognins are shown as grey solvent exposed surfaces. Peptides are shown as sticks with green carbons, red oxygens, and blue nitrogens. | ||
We were concerned that although Q7 binds site-selectively to PheB1, it may not bind with selectivity for insulin versus other proteins. In an initial test of protein selectivity, we used a fluorescent indicator displacement assay to measure the relative extent of binding of Q7 to native human insulin, the GluB1, GluB27 mutant of human insulin, human serum albumin, human immunoglobulin G, and bovine carbonic anhydrase.68 Q7 was highly selective for native human insulin versus the other four proteins, but this result did not imply selectivity in a biological context. Therefore, we carried out a selectivity study using a multiplex method. In collaboration with the Isaacs group at University of Maryland, we covalently conjugated Q7 to sepharose beads and used the resulting “Q7-resin” to selectively isolate human insulin and human growth hormone (hGH, Ka = 5.9 × 105 M−1) from a simple mixture of proteins.79 The insulin and hGH were released by treating the resulting resin with a high-affinity competitor, N,N′-diethyl-1,6-diaminohexane, but not a low-affinity competitor, diethylamine, thus demonstrating selective recognition in a mixture. We repeated this experiment in human serum with both insulin and hGH added at 10–100 μM and showed selective enrichment of these proteins. This remarkable result shows that the N-terminal residue is a highly selective epitope capable of conferring protein recognition in the presence of high concentrations (500–700 μM) of other proteins.
2.8. Recognition of lysine and methylated lysine
Receiving less attention until recently is the Lys residues in peptides and proteins. Kim, Inoue, and coworkers first showed that Q6 can bind N-terminal Lys.67 Buried in the supporting information of their 2008 paper is the fascinating result that Q6 binds N-terminal Lys in the peptide H-Lys-Ala-NH2 with a Ka value of 1.6 × 104 M−1 (Table 7). Nau and coworkers reported weak affinity of Q7 for the amino acid lysine (Ka = 800 M−1).89 Gamal-Eldin and Macartney then showed that methylating the ε-amino group of Lys, which is used to regulate DNA transcription, increases the affinity for Q7 by 3.4-fold, 110-fold, and 3600-fold for mono-, di-, and trimethylation, respectively.90 Crowley and coworkers engineered a dimethyl Lys residue (LysMe2) into a disordered loop of the Ralstonia solanacearum lectin protein (RSL*) and estimated a Ka value of ∼103 M−1 in 20 mM potassium phosphate, 50 mM NaCl, pH 6.0, via chemical shift perturbation of the side chain resonances of the LysMe2 residue.91 The crystal structure showed Q7 binding in different modes, but the dimethylammonium group is buried in the cavity of Q7 and exists in two states in the crystal, showing that it is free to move within the Q7 cavity (Fig. 20). This group engineered an additional solvent-accessible LysMe2 binding site into RSL* and showed Q7 binding at both sites in solution and in the solid state.92 Additional work with this system was used for engineering crystalline protein architectures.93–95 Zhong, Hooley, and coworkers showed that Q7 shifts the electrophoretic mobility of methylated histone peptides, allowing for more efficient separation by capillary electrophoresis.96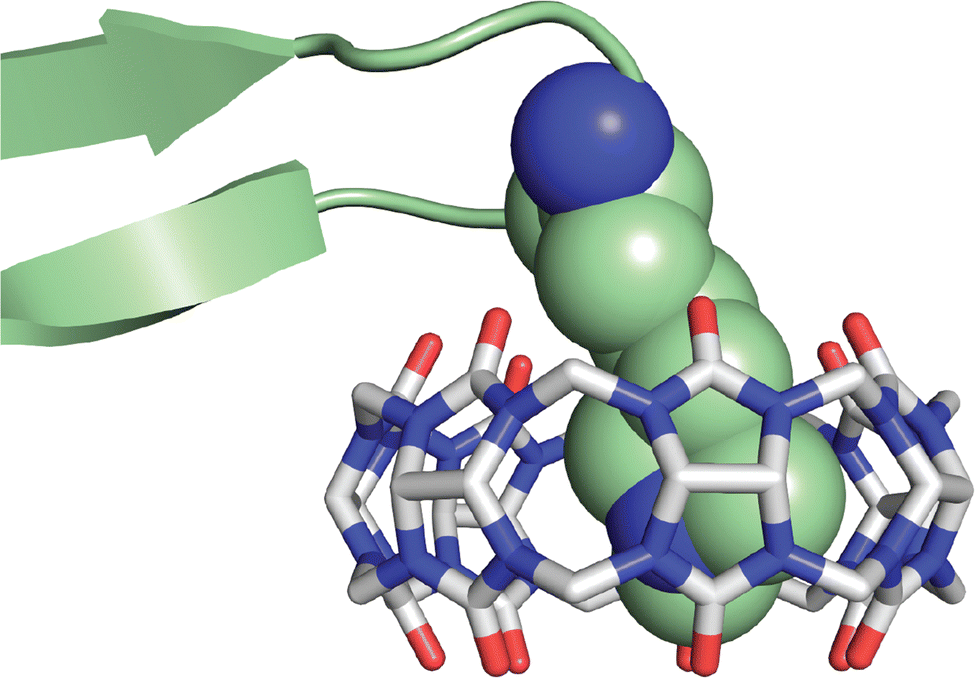 | ||
| Fig. 20 Close-up rendering of the crystal structure of the Q7·RSL* complex, showing Q7 bound to the LysMe2 residue.91 Only one of the two conformations of the LysMe2 side chain is shown, and it is rendered as space-filling with green carbons. Q7 is rendered as sticks with grey carbons. Oxygens are red. Nitrogens are blue. PDB ID 6F7W. | ||
In contrast to the work on Q7 and methylated lysines, Crowley recently investigated Q6 binding to unmodified Lys residues in several proteins and found that Q6 can recognize Lys, but not LysMe2, and that recognition depends heavily on the sequence context.97 High-affinity binding of Q6 at N-terminal Met-Lys, but not at non-terminal Met-Lys, was inferred by observation of slow exchange kinetics on the NMR timescale. Binding of Q8 to N-terminal Met-Lys in the context of a protein demonstrates that the pair-inclusion motif within short peptides translates to folded proteins.
2.9. Recognition of noncanonical residues
Excited by the development of techniques for incorporating noncanonical amino acids into proteins, we were interested to explore the capacity of Q7 to bind chemically modified Phe residues. To this end, we screened a small library of modified phenylalanines for binding to Q7 and found that 4-tert-butyl-phenylalanine (tBuPhe) and 4-aminomethyl-phenylalanine (AmPhe) bind Q7 with 20–30-fold increased binding affinity (Table 5).69 N-terminal tBuPhe is not a significant improvement over N-terminal Phe, but N-terminal AmPhe binds Q7 with a Ka value of 1.1 × 109 M−1 in 10 mM sodium phosphate, pH 7.0. The extraordinary 340-fold enhancement in affinity is perhaps due to the deep burial of the side chain within the Q7 cavity, enabling both the side chain ammonium group and the N-terminal ammonium group to bind to opposite portals of Q7, as supported by NMR data.69 In a subsequent study, we confirmed the relatively high affinity of Q7 for the AmPhe residue in the peptide H-AmPhe-Met-NH2 (5.3 × 108 M−1).73Encouraged by these results, we pursued the incorporation of the AmPhe residue into a protein. Human insulin was chosen as the target due to the presence of an N-terminal Phe residue at the B1 position. By mutating this residue to AmPhe, we hoped to increase the affinity of Q7 by orders of magnitude while only adding an aminomethyl group to the protein. Aminomethyl insulin (i.e., human insulin FB1AmPhe) was produced in a four-step semisynthesis starting from commercially available, recombinant regular human insulin70 and found to bind Q7 with a Ka value of 1.0 × 107 M−1, a modest 7-fold increase over regular human insulin. Surprised by this result, we evaluated the binding of Q7 to four tetrapeptide analogues of the N-terminal region of the B-chain of human insulin, including the native B1–4 sequence if H-Phe-Val-Asn-Gln-NH2 (4.0 × 106 M−1) as parent, the less sterically hindered ValB2Gly mutant, H-Phe-Gly-Asn-Gln-NH2 (7.1 × 106 M−1), and the PheB1AmPhe mutants of both, i.e., H-AmPhe-Val-Asn-Gln-NH2 (1.3 × 107) and H-AmPhe-Gly-Asn-Gln-NH2 (2.0 × 107 M−1). While we were disappointed that the affinity of Q7 for AmPhe in these contexts is weaker than in H-AmPhe-Gly-Gly-NH2 and H-AmPhe-Met-NH2, it was interesting to observe that the binding of Q7 to aminomethyl insulin is remarkably similar to its B1–4 tetrapeptide analogue, H-AmPhe-Val-Asn-Gln-NH2.
Recently, Liu and coworkers reported a general method for inhibiting protein activity site-selectively by incorporating a single tBuPhe residue and blocking activity with Q7.75 A single tBuPhe was incorporated at carefully chosen sites near the active sites of glutathione-S-transferase, protein tyrosine kinase, and tumor necrosis factor alpha. Q7 inhibited the activities of these proteins, while adding H-Phe-Gly-Gly-OH as a competitor restored protein activity.
2.10. Lessons learned from molecular recognition studies
These molecular recognition studies have yielded several principles for targeting small sites on peptides and proteins with cucurbit[n]urils in a site-selective and sequence-predictive manner and with affinities that are compatible with a range of possible applications, as detailed in Section 3. Targeting the N-terminal residue, as a unique epitope, offers the most straightforward approach to high affinity and selectivity. N-terminal Phe can be targeted by Q7 in a 1:1 (Q7:polypeptide) stoichiometry and by Q8 in a 1:2 (Q8:polypeptide) stoichiometry, both with sub-micromolar affinities. Most residues can be tolerated at the second position, but Pro significantly destabilizes binding. N-terminal Tyr can be bound by Q7 in a 1:1 (Q7:polypeptide) stoichiometry and with low-micromolar affinity, whereas N-terminal Lys-Phe, and Leu-Tyr can be bound in a 1:1 (Q8:polypeptide) stoichiometry and with low- to sub-nanomolar affinities. Q8 can also target N-terminal Met with submicromolar affinity, in which the identities of the second (Leu, Met, Lys, Arg, Phe, and Tyr preferred) and third residues (Gly and Ala preferred) are important. Entirely nonaromatic sequences are possible in the pair-inclusion motif. In some cases, the first and second residues of an N-terminal dipeptide site can be swapped, while in other cases (e.g., Lys-Phe vs. Phe-Lys and Leu-Tyr vs. Tyr-Leu), there is sequence selectivity of several orders of magnitude. The molecular basis for recognition in these motifs is supported by extensive NMR and crystallographic data. N-terminal sites are likely to be tolerated by proteins due to their propensity to unfold and become fully accessible to encapsulation by the host. Similarly, sites located in disordered loops are also likely to be accessible. Non-terminal sites containing Phe or AmPhe are accessible to Q7 at low micromolar affinity, whereas numerous dipeptide sequences are accessible to Q8 binding, with affinities reaching mid-nanomolar for the sequences Lys-Phe and Phe-Lys. N-terminal and non-terminal Trp can be targeted by preformed complexes including Q8 and a cofactor, and binding is essentially independent of the neighboring sequence context.
The studies described in this section also demonstrate several properties of Qn·peptide complexes that make them particularly well suited to applications involving affinity tags for proteins and peptides: (1) binding occurs in purely aqueous solution (i.e., no organic solvent) and is compatible with a wide range of buffer conditions, including salts of different types, salt concentrations up to physiologic level (e.g., PBS, serum), and pH from neutral to acidic. (2) The binding affinities are mostly in the range of 105–109 M−1, which enable working concentrations to be in the useful high micromolar to low nanomolar range. (3) Binding is observable and quantifiable by numerous methods. (4) Site-selectivity is relatively high for certain sequences (e.g., Q7 binding to N-terminal Phe, or Q8 binding to N-terminal Lys-Phe) and relatively low for others (e.g., Q8·MV binding to non-terminal Trp), which allows for adaptation to a diverse range of applications. (5) The rules for sequence recognition and the various modes of binding are sufficiently well understood to accurately predict binding. (6) Target binding sites can be engineered into proteins with minimal to no loss in affinity while maintaining site-selectivity. (7) N-terminal and non-terminal sites can be targeted with high affinity and selectivity. (8) Q8 can noncovalently couple peptides and proteins to an auxiliary guest that can add functionality via heteroternary complex formation. (9) Q8 can reversibly homodimerize peptides and proteins. (10) Q7 and Q8 can bind peptides and proteins with high affinity in a 1:1 ratio. 11) Q7 retains its recognition properties when attached to a solid support. These favorable characteristics have enabled the demonstration of a broad range of applications, as described in detail in Section 3.
3. Applications of peptide and protein recognition by cucurbit[n]urils
The molecular recognition properties detailed in Section 2 have facilitated the development of numerous applications involving the site-specific binding of cucurbit[n]urils, particularly Q7 and Q8, to peptides and proteins, as reviewed comprehensively in this section. One can view these applications as ways to create or improve affinity tags for polypeptides, or to extend the classical idea of an affinity tag to new applications such as protein dimerization, oligomerization, and polymerization, or dynamic surfaces and reversible materials. We hope these examples will inspire the next generation of scientists to build programmable and functional devices and materials.3.1. Sensor development
Chemical sensors interact with a target chemical and, upon interaction, transduce an observable signal that can be amplified and read out by the user.98 Sensors that are selective for peptides and proteins are highly useful for the detection and quantification of desired polypeptide targets in vitro, in vivo (e.g., probes), and ex vivo (e.g., medical diagnostics). The selective recognition of polypeptides by Q7 and Q8 satisfies the first requirement of a chemical sensor. This section reviews the work of numerous groups to couple this binding to a signal transduction mechanism in order to create functional sensors.Methyl viologen is limited as an optical sensor due to its lack of intrinsic fluorescence. Several alternative auxiliary guests for Q8 have been developed with intrinsic fluorescence (Fig. 21). Kaifer and coworkers replaced MV as a first guest for Q8 with 2,7-dimethyldiazaphenanthrenium (DPT2+) and 2,7-dimethyldiazapyrenium (DAP2+), which have limited rotational freedom, compared to MV, and are intrinsically fluorescent.99 The resulting Q8·guest complexes bound to indole, tryptophan, and serotonin, which quenched the fluorescence of the first guest. We followed this work with a report on the use of tetramethylbenzobis(imidazolium) (MBBI) as an intrinsically fluorescent sensing component for peptides, as described in more detail in Section 2.5.43
 | ||
| Fig. 21 Chemical formulas of guests used for sensor development with cucurbit[n]urils. | ||
Biedermann, Nau, and coworkers reported the use of complexes of Q8 with various aromatic chromophores, including DAP, MDPP, and MVE (Fig. 21), to sense by CD spectroscopy the binding and reaction of chiral guests including aromatic amino acids and peptides as well as the proteins somatostatin and insulin.100 Biedermann and coworkers recently demonstrated enhanced sensitivity using fluorescence-detected CD to detect amino acids, peptides, and proteins.101
Aryal, Huang, Hunter, and coworkers reported a sensor for aromatic amino acids and peptides based on a water-soluble perylene-monimide, PMI (Fig. 20).102 The Q8·PMI complex has a Ka value of 1.3 × 104 M−1 and causes PMI to deaggregate and increase in fluorescence by up to 19-fold. Upon competitive displacement of PMI with phenylalanine-containing peptides, the fluorescence decreases in a concentration-dependent manner. Schmuck, Nau, and coworkers demonstrated a ratiometric fluorescence sensor based on Q8 and a bis(pyrene), AP-1 (Fig. 17).103 Q8 interrupts the intramolecular pyrene excimer, which is restored upon the competitive binding of phenylalanine-containing peptides and insulin to Q8. The ratio of pyrene monomer fluorescence emission to excimer emission was used as the sensing metric.
Hennig, Nau, and coworkers applied Qn-based optical sensing to measure the transport of cell-penetrating peptides across phospholipid membranes.44 Q8·DAP and Q7·berberine complexes were trapped within liposomes and used to detect the permeation of unlabeled Phe- and Trp-containing peptides into the liposome at effective concentrations equal to or higher than the Kd value for the peptides. Biedermann, Nau, and coworkers extended this method to the time-resolved, quantitative determination of membrane permeability for a wide array of analytes using Q8 in complex with DAP, MVE, and MBBI.104
Li and coworkers developed a sensor based on the Q8-induced flocculation of Au nanoparticles.105 The nanoparticles were modified with peptides containing a C-terminal Cys for covalent attachment to the Au surface and an N-terminal Trp to induce nanoparticle aggregation via the Q8-mediated dimerization of Trp residues. Because the aggregation of Au nanoparticles modulates their plasmon absorbance, introducing a protein that targets the surface-bound peptide, competitively displaces Q8, and drives disaggregation of the nanoparticles allows for optical detection of the protein with a lower limit of detection of 0.2 nM. Hou and coworkers used the H-Phe-Gly-Gly-OH peptide as an example analyte to demonstrate a novel detection platform in which Q8 is bound to surfactant molecules at an interface, and sensing is activated by addition of the peptide and competitive displacement of the surfactant.106
Cao and coworkers reported the formation and adaptive chirality of an achiral Q8-based supramolecular organic framework (SOF) and its use as a chiroptical sensor for polypeptides.107 SOFs are formed upon the complexation of tetra(6-coumarinylmethyl-pyridinium)tetraphenylethylene (TPE) units with Q8. Through the addition of Phe or Trp containing dipeptides with L- or D- stereochemistry, the TPE units can be induced to have a rotational conformation of M- or P-, producing mirror-image circular dichroism and circularly polarized luminescence signals upon binding. Taking advantage of adaptive chirality, SOFs were able to distinguish dipeptides H-Phe-Ala-OH, H-Ala-Phe-OH, and H-Phe-Phe-OH, as well as proteins somatostatin and regular human insulin, with CD spectra characteristic of either M- or P-SOFs. They then exchanged the TPE unit with a hexaphenylbenzene (HPB) derivative, tetra(6-coumarinylmethyl-pyridinium)hexaphenylbenzene, and distinguished L-Trp-X from L-Phe-X dipeptides based on the adaptive chirality of the SOF.108 Importantly, chiral induction of the SOF was only observed when Trp and Phe were placed at the N-terminus, allowing distinction between L-Trp/Phe-X and L-X-Trp/Phe. The adaptive chirality and chiroptical sensing properties of the supramolecular organic framework may be applied to determine enantiopurity of amino acids and distinguish biological chiral molecules.
 | ||
| Fig. 22 Protein sensing technique through electrochemical readout at a gold surface. CB[8] is Q8.109 Reproduced with permission from ref. 109. Copyright 2012 American Chemical Society. | ||
Yuan, Chia, and coworkers described the incorporation of Q8·MV into a sensor for microRNA by binding to Trp-labeled DNA strands used in a target cycling and strand displacement amplification method.110 Trp-labeled oligonucleotide strands produced in the reaction cycling bind to a sensor electrode coated with Q8·MV groups and are released by single-electron reduction of the viologen, followed by oxidative regeneration of the sensor.
O portals of Q7 stabilizing positive charges and thus increasing proton affinities of the bound guests, which improves ionization efficiency. The addition of Q7 also resulted in increased fractionalization of peptides, which was also attributed to charge stabilization and interactions with the carbonyl portals.
The selectivity of Q7 for N-terminal Phe has also been applied to signal enhancement by surface enhanced Raman scattering (SERS).112 Camden, Webber, and coworkers demonstrated how the addition of Q7 to samples can improve the sensitivity and detection limits of peptides and proteins with N-terminal Phe via SERS by increasing the concentration of analyte in proximity to the nanoparticle surface.
:1 binding of NVoc-FGG with Q8 was confirmed by ITC, NMR spectroscopy, and mass spectrometry. Photodeprotection of the NVoc-FGG·Q8 complex triggered the dimerization of H-Phe-Gly-Gly-OH in Q8, as confirmed by NMR spectroscopy. This approach points toward the use of stimuli-responsive supramolecular chemistry in biological contexts.
3.2. Measuring and inhibiting protease activity
Proteases are enzymes that catalytically hydrolyze peptide bonds and are involved in digestion, protein maturation, protein homeostasis, viral processing, and many other processes. As such, proteases are important targets for measurement and control using synthetic ligands. The selective recognition of peptides and proteins by cucurbit[n]urils therefore provides fertile ground for the development of applications involving proteases.In collaboration with the Nau group at Jacobs University Bremen, we reported a method for monitoring proteolysis by the endopeptidase thermolysin, which cleaves on the N-terminal side of hydrophobic residues such as Phe and Leu, using Q7 and acridine orange as the reporter pair (Fig. 23).71 The substrate peptides contained a non-terminal Phe residue, which binds relatively weakly to Q7, whereas the product peptides contained an N-terminal Phe, which competitively displaces acridine orange and results in a decrease in fluorescence intensity. This method was used to rapidly determine the Michaelis Menten kcat/KM values for the proteolysis of a series of unlabeled enkephalin-based peptides, thereby profiling the substrate selectivity of this enzyme. We also used the assay to measure the inhibitory constant of a known protease inhibitor.
 | ||
| Fig. 23 Supramolecular tandem enzyme assay for real-time measurement of the proteolysis of enkephalin-type peptides with thermolysin, using Q7 and acridine orange as the reporter pair. (top right) Fluorescence intensity as a function of time at different concentrations of substrate, used to measure kcat/KM. (bottom right) Reaction rate as a function of inhibitor concentration, used to measure the inhibitory constant.71 Reproduced with permission from ref. 71. Copyright 2011 American Chemical Society. | ||
Hennig, Nau, and coworkers combined this method for monitoring enzyme kinetics with their aforementioned technique for detecting peptide membrane permeation to demonstrate the dual-color chemosensing of a compartmentalized reaction network.116 The compartmentalized liposomal system was designed to detect the enzymatic cleavage of enkephalin-related peptides in the extravesicular space by a Q7·methylene blue reporter pair, and to detect the membrane permeation of the cleaved product in the intravesicular space by either Q8·2-anilinonaphthalene-6 sulfonic acid or Q7·berberine reporter pairs. The dyes enabled selective optical excitation and emission, providing a way to kinetically discriminate between two sequential reactions, cleavage and permeation.
 | ||
| Fig. 24 Schematic for the sequence-specific inhibition of a nonspecific protease.73 Reproduced with permission from ref. 73. Copyright 2013 American Chemical Society. | ||
Gong, Cao, and coworkers used this approach to measure the activity of APN.118 Au nanoparticles coated with H-Phe-Gly-Gly-Phe-Glu-Leu-Leu-Cys-OH peptides were flocculated upon addition of Q8, akin to the method reported by Li and coworkers, vide supra.105 APN was introduced to remove the N-terminal Phe, which prevents flocculation and allows for the measurement of enzymatic activity. This approach was reported to have improved detection limit and response time compared to other methods for measuring APN activity.
3.3. Protein assembly
The formation of protein dimers, oligomers, polymers, discrete assemblies, and indistinct aggregates is ubiquitous in nature. Therefore, the ability to measure or control these processes has significant potential value in bioscience and biotechnology. This section illuminates the practicality of applying cucurbit[n]urils toward these ends. In the context of affinity tags, this is an area of new development made possible by the dimerizing capability of Q8.:2 FGGGC-Pt/Q8 stoichiometry in head-to-head and head-to-tail arrangements at the terpyridyl units and Phe residues, respectively, a structure with the same stoichiometry but in dual head-to-head arrangements, and a head-to-tail dimer with a 4:4 FGGGC-Pt/Q8 stoichiometry.
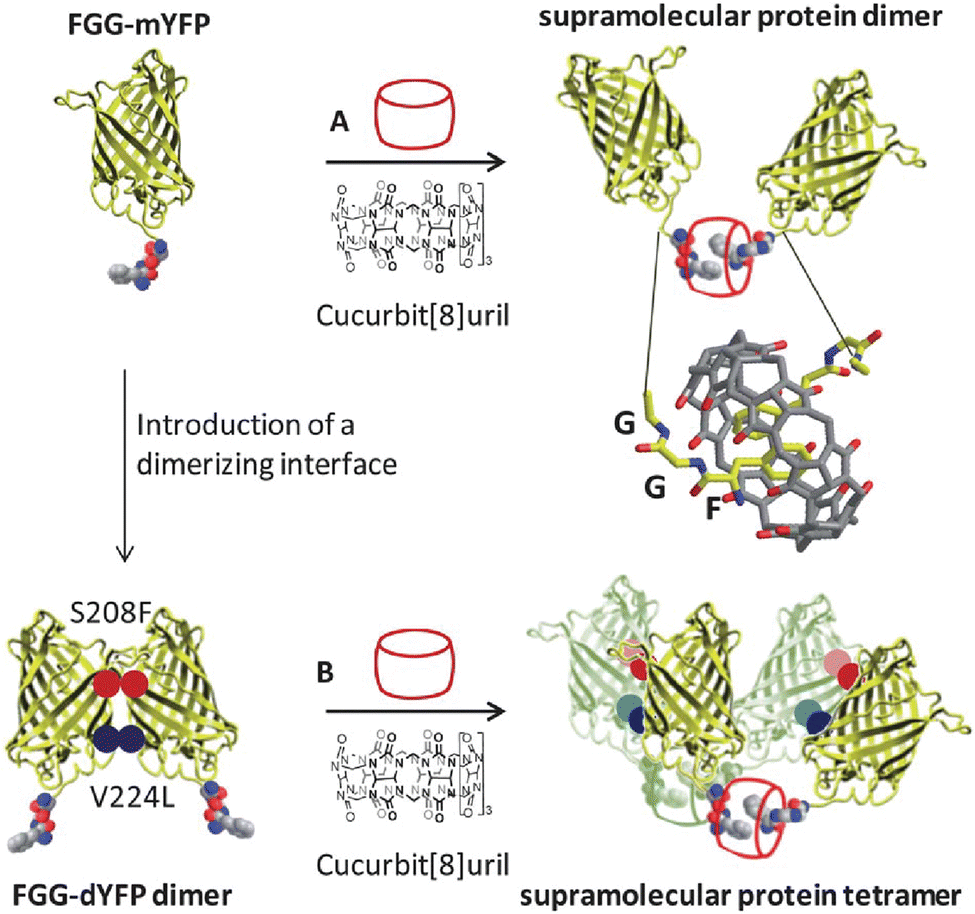 | ||
| Fig. 25 Design of a protein tetramer by Q8-mediated dimerization of an FGG-modified protein dimer.120 Reproduced from ref. 120 with permission from the Royal Society of Chemistry. | ||
Brunsveld and coworkers reported the structure of a Q8-mediated protein dimer using dynamic light scattering and solution-based small angle X-ray scattering to show that the Q8-mediated dimer of FGG-mYFP forms a Z-shaped structure that retains the size and shape of the individual YFP subunits and with a relatively compact contact area between the proteins.121 The parallel, coplanar alignment of the beta barrels helps to explain the efficient FRET between the proteins.
Q8-mediated protein dimerization was then applied to controlling the activity of the enzyme caspase-9, which normally exists in monomeric form and is activated by dimerization. Brunsveld and coworkers engineered a mutant of caspase-9 to contain an N-terminal FGG-tag (i.e., FGG-casp-9) and showed that enzyme activity was increased 50-fold upon dimerization with Q8.122 This enhancement was dependent on the concentration of Q8 and could be reversed in a concentration-dependent fashion by addition of the tripeptide H-Phe-Gly-Gly-OH as a competitive inhibitor. The activity of a control caspase-9 mutant with N-terminal H-Met-Gly-Gly- was not affected by Q8. They recently extended supramolecular control of caspase-9 activity by engineering a light-sensitive peptide cage for Q8.123 The cage contained two N-terminal FGG-tags and two photo-cleavable amino-3-(2-nitrophenyl)propanoic acid residues. Upon photocleavage, the affinity of the cage for Q8 substantially decreases, thus liberating Q8 to drive the dimerization of FGG-casp-9 monomers. The activity of caspase-9 activated by photocleavage of the cage was comparable to that of caspase-9 activated by uncaged Q8. Notably, this approach to enzyme activation avoids modification of the enzyme active site, thereby providing an additional measure of control.
Brunsveld and coworkers reported the use of Q8 to drive split-enzyme complementation and restore enzymatic activity for the heterocomplexation of split-luciferase fragment pairs.124 An N-terminal FGG-tag was incorporated into the two protein fragments, and Q8-mediated combination of the NFluc437 and CFluc398 fragments resulted in enzymatic activity in a concentration-dependent manner. Memantine, a high-affinity guest for Q8, was shown to be an effective competitor to reverse this process. Ottmann, Brunsveld, and coworkers then reported the use of Q8 to mediate supramolecular protein assembly with a 14–3–3 protein dimer.125 Estrogen receptor α (ERα) has a 14–3–3 protein-binding domain at its C-terminus. An N-terminal FGG-tag was added to ERα, and addition of Q8 induced protein dimerization. The affinity of ERα for 14–3–3 protein was enhanced significantly in the presence of Q8. When the FGG-ERα peptide was acetylated at the N-terminal amine (Ac-FGG), no binding to Q8 was observed. Importantly, they were able to determine the crystal structure of the Q8-bound protein tetramer (Fig. 26), showing stacking of Phe side chains within the Q8 cavity and chelation of the N-terminal ammonium groups by Q8 CO groups, in a manner identical to the Q8·(H-Phe-Gly-Gly-OH)2 structure.18 Akin to the Q7·insulin crystal structure,63 the binding site on FGG-ERα is fully solvent-accessible. Brunsveld and coworkers then used Q8-mediated activation of FGG-modified caspase-8 to overcome limitations in studying the mechanism of dimerization and activation by circumventing the need to mutate the dimerization interface.126 Utilizing Q8-mediated activation of caspase-9, Ottman, Brunsveld, Merkx, and coworkers designed a synthetic self-activating protease signaling network in which dimerization and activation of FGG-caspase-9 by Q8 was shown to activate a 14–3–3 scaffold inhibited by ExoS peptides, thus triggering 14–3–3-templated split-luciferase complementation.127 This method demonstrates the control of common mechanisms for intracellular signaling by a modular supramolecular platform.
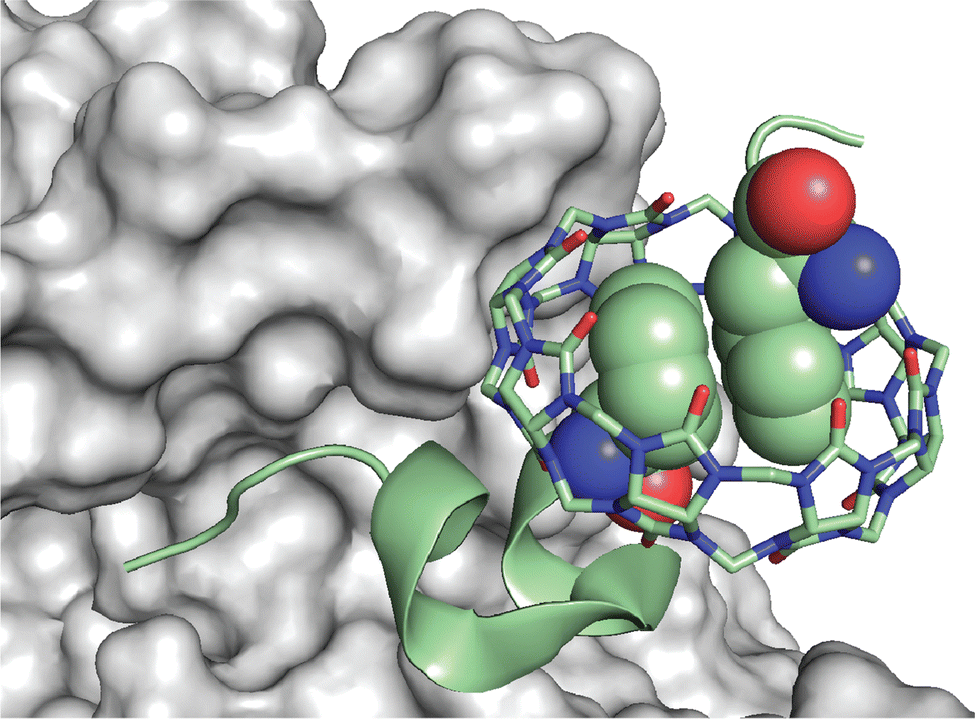 | ||
| Fig. 26 Rendering of the crystal structure of the Q8·(14–3–3·FGG-ERα)2 complex.125 The solvent-accessible surface of 14–3–3 is shown in grey. The backbone of FGG-ERα is shown as a green ribbon. The N-terminal Phe residues are shown as space-filling. Q8 is shown as sticks. Carbons are green, oxygens are red, and nitrogens are blue. PDB ID 5N10. | ||
Dankers, Brunsveld, and coworkers reported the reversal of Q8-mediated dimerization of FGG-mYFP using a bivalent guest presenting two N-terminal Phe-Gly-Gly-Gly peptides linked with a penta(ethylene glycol).128 They measured a Ka value of 9.0 × 106 M−1 for Q8 binding to the bivalent guest, and they proposed the formation of an interesting pseudocatenane structure. Using fluorescence polarization anisotropy, FGG-modified yellow fluorescent protein was dimerized when bound to Q8. When the FGG-modified penta(ethylene glycol) was introduced as a competitor, it reversed protein dimerization.
Liu, Li and coworkers then fused GST to a redox-sensitive mutually exclusive protein (MEP) and fused an N-terminal FGG-tag.129 The tagged protein spontaneously formed homodimers that could be induced to form larger assemblies by addition of Q8. The morphologies of the assemblies could be controlled reversibly via the redox properties of the protein in order to assemble into nanowires or nanorings. To circumvent the need for genetic engineering, Liu and coworkers then conjugated a maleimide group to the C-terminus of H-Phe-Gly-Gly- in order to make a compound that can be conjugated to the side chain of any solvent-exposed Cys residue.130 They demonstrated the utility of this approach by attaching the conjugate to the Cys side chains of a variant GST. The Cys groups on the GST homodimer projected in a V shape, and Q8 induced the assembly of the homodimers into linear supramolecular polymers, rings, and spirals in a concentration-dependent fashion (Fig. 27). Liu, Hou, and coworkers conjugated a pyridinium derivative to a protein engineered with a single Cys residue and formed two-dimensional protein arrays in the presence of Q8.131 Addition of the tripeptide H-Phe-Gly-Gly-OH competitively reversed assembly formation.
Hugh Kim, Kimoon Kim, and coworkers reported the inhibition of the amyloidogenesis of insulin and β-amyloid (Aβ1–40 and Aβ1–42) using Q7 (Fig. 28).132 An insulin:Q7 ratio of 2:1 or higher showed inhibition of fibrillation based on a thioflavin T spectroscopic assay, which reports the formation of β-sheets. Although Q7 bound selectively to the PheB1 position, ITC experiments at pH 2.7 showed binding of a second equivalent of Q7 to PheB24 with lower affinity. PheB24 binding was corroborated by molecular dynamics simulations with a partially unfolded insulin structure and suggests that Q7 binding to PheB24 inhibits insulin·insulin dimerization along the interface comprising B11–17 and B24–26. In parallel, Q7 was also shown to suppress the fibrillation of Aβ peptide at high Q7:peptide ratios. Titration studies revealed a Ka value of 7.1 × 104 M−1 in sodium phosphate buffer (Table 6). Binding studies with point mutants of Aβ1–40 revealed equal accessibility of Q7 to Phe4, Phe19, and Phe20, and Q7 binding was postulated to inhibit the clustering of these residues and the formation of the partially folded intermediate on the path to amyloid assembly. Q7 binds Aβ1–42 and Aβ1–40 with similar affinity and suppresses fibrillation. de Oliveira and coworkers used molecular dynamics simulations to suggest that the inhibition of Aβ1–42 by Q7 is due to binding of Q7 to Asp1, Lys 16, and Val36, with prevention of protofibril elongation.133 Bowers and coworkers reported the binding of one and two molar equivalents of Q7 to monomers and dimers, respectively, of the Aβ25–35 fragment (Gly-Ser-Asn-Lys-Gly-Ala-Ile-Ile-Gly-Leu-Met), presumably via the Lys residue, leading to the suppression of oligomer formation.134 Li, Hong, and coworkers reported the Q7- and Q8-mediated inhibition of amyloid fibrillation of Aβ1–16 and the truncated Aβ4–16, which contains an N-terminal Phe.58 They reported Ka values for Q7 with Aβ4–16 of 1.1 × 106 M−1 and with Aβ1–16 of 1 × 104 M−1 (Table 6). Q8 binds two equivalents of Aβ4–40 with a ternary binding constant Kter value of 5.5 × 1010 M−2 (Table 3), whereas no binding was observed for Q8 with the Aβ1–16 peptide. They showed that Q7 and Q8 slow the aggregation of Aβ4–40 at a lower concentration than Aβ1–40, presumably by binding to the ends of short oligomers, and that Q7 and Q8 reduce the cytotoxicity of Aβ4–40. Q7, but not Q8, reduces the cytotoxicity of Aβ1–40.
 | ||
| Fig. 28 Schematic for the inhibition of fibrillation using Q7 to stabilize the monomeric forms of (top) insulin and (bottom) Aβ1–40. CB[7] is Q7.132 Reproduced with permission from ref. 132. © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Hugh Kim, Kimoon Kim, and coworkers reported that Q6 can be used to control the length of amyloid fibrils of human insulin.135 They showed that the length of low polydispersity (1.3) oligomers of insulin can be increased by increasing the Q6 concentration above its solubility limit. They proposed that length control is mediated by the coprecipitation of the Q6·insulin complex, which controls the concentration of available insulin monomer for fibrillation. This approach was extended to the control of the fibrillation for several other proteins, including human islet amyloid polypeptide, hen egg white lysozyme, Aβ1–40, and Aβ1–42.
Building on their early work showing that Q8 can dimerize fragments of Aβ by binding non-terminal Phe sites, Scherman and coworkers took a different approach to inhibiting the toxicity of amyloid fibrils by promoting fibrillation of Aβ1–42 with Q8 (Fig. 29).136 ITC analysis with Aβ1–42versus a Phe4Ala Phe19Ala Phe20Ala triple mutant suggested binding of Q8 at the three Phe sites, and CD data suggested pi-stacking between Phe side chains. An excess of Q8 accelerated elongation, and the Aβ1–42 aggregates had lower toxicity and cell uptake in the presence versus absence of Q8, thus introducing a possible new approach to reducing amyloid toxicity.
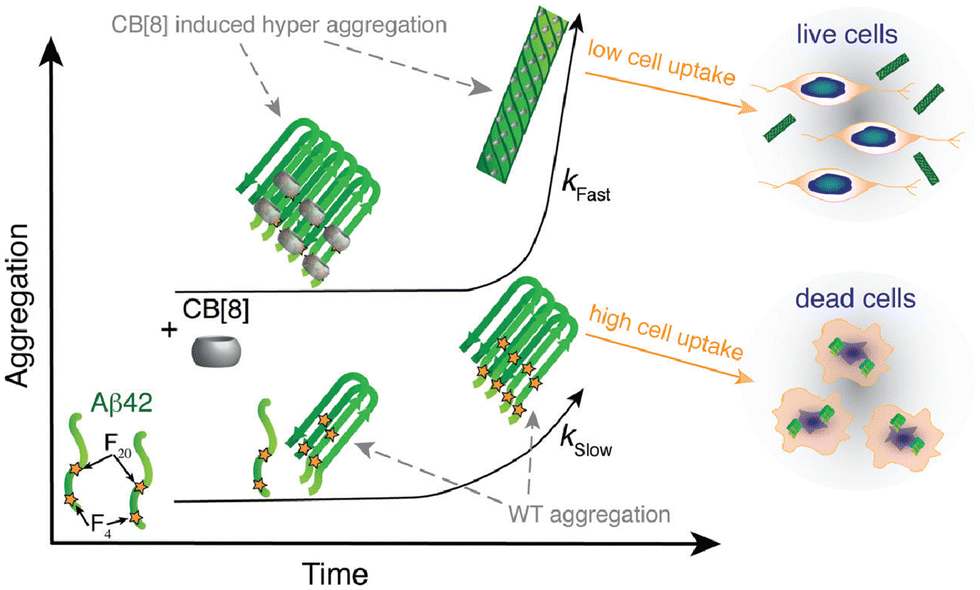 | ||
| Fig. 29 Time-based aggregation of Aβ1–42 in the presence and absence of Q8. Q8 induces the aggregates to grow faster and larger, and cell uptake and toxicity are reduced. CB[8] is Q8.136 Reproduced from ref. 136 with permission from the Royal Society of Chemistry. | ||
Li, Zhu, Chen, and coworkers reported the inhibition of the fibrillation of human calcitonin (hCT) using Q7.83 Alanine screening of the four aromatic residues showed a reduction in affinity in all mutants. Q7 increased L929 cell viability until it caused cytotoxicity. The addition of Q7 with hCT promoted proliferation, osteogenic differentiation, and osteogenesis of MC3T3 cells. The presence of Q7 was hypothesized to promote the activity of hCT, which would lead to increased cell proliferation. Q7 and hCT administered subcutaneously also resulted in calcitonin release and reduced serum calcium levels, which was thought to be due to the stabilization of calcitonin from fibrillation/aggregation. Q7 also decreased the immunogenicity of hCT. This paper sheds mechanistic light on a prior study by this group that reports a sustained release of hCT from a poly(D,L-lactic acid-co-glycolic acid)-b-poly(ethylene glycol)-b-poly(D,L-lactic acid) block copolymer hydrogel in the presence of excess Q7.137
Derrick, van der Walle, and coworkers reported that the colloidal stability of monoclonal antibodies containing an aggregation prone region (APR) is improved in the presence of Q7.138 Using a combination of dynamic light scattering, intrinsic Trp fluorescence, ITC, and NMR spectroscopy, a comparative analysis of an antibody and its mutant deleting two aromatic residues from the APR makes the case for colloidal stability induced by binding of Q7 to the nonterminal aromatic residues. This group then reported the modulation of fibrillation of the HIV fusion inhibitor enfuvirtide (ENF) using Q7.139 Alanine screening of the C-terminal Phe suggested single-site binding of Q7 at the C-terminus. Q7 delayed the onset of fibrillation of wild-type ENF. ENF/Q7 and mutant ENF fibrils exhibited comparable morphologies, differing from wild-type ENF fibrils, suggesting the potential for Q7 in determining fibril morphology. Hamilton and coworkers reported the inhibition of islet amyloid polypeptide (IAPP) aggregation through the recognition of N-terminal “hot segments” by Q7.140 IAPP “hot segments” contain two Phe residues known to drive peptide aggregation. Q7 completely inhibited de novo and membrane-catalyzed fibril formation at 10 molar equivalents. Q7 also inhibited elongation and secondary nucleation caused by toxic preformed oligomers and rescued rat insulinoma cells from IAPP assembly-mediated cytotoxicity.
3.4. Supramolecular polymers and hydrogels
The synthesis of materials with dynamic properties is a major aim of modern chemistry. Such materials would have the advantages of being responsive to external stimuli, being self-healing, and having other properties that change in time. This section describes the use of cucurbit[n]urils to induce the reversible formation of polymer and hydrogel materials via interaction with materials modified with N-terminal aromatic amino acid residues. In a sense, this work could be viewed as utilizing affinity tags for reversible polymerization and gelation.:monomer ratio of 1:1 induced supramolecular polymerization in aqueous solution at concentrations higher than the observed Kd (0.27 μM) (Fig. 30). This group then reported a two-step supramolecular polymerization based on a conjugate comprising an FGG-tag linked to diazobenzene.142 Two equivalents of the conjugate bind to Q8 via the N-terminal Phe, and the resulting bivalent supramonomer polymerizes upon addition of a bivalent bis-β-cyclodextrin. Polymerization was reversible both by addition of a competitive guest for Q8 and by photoisomerization of the diazobenzene groups.
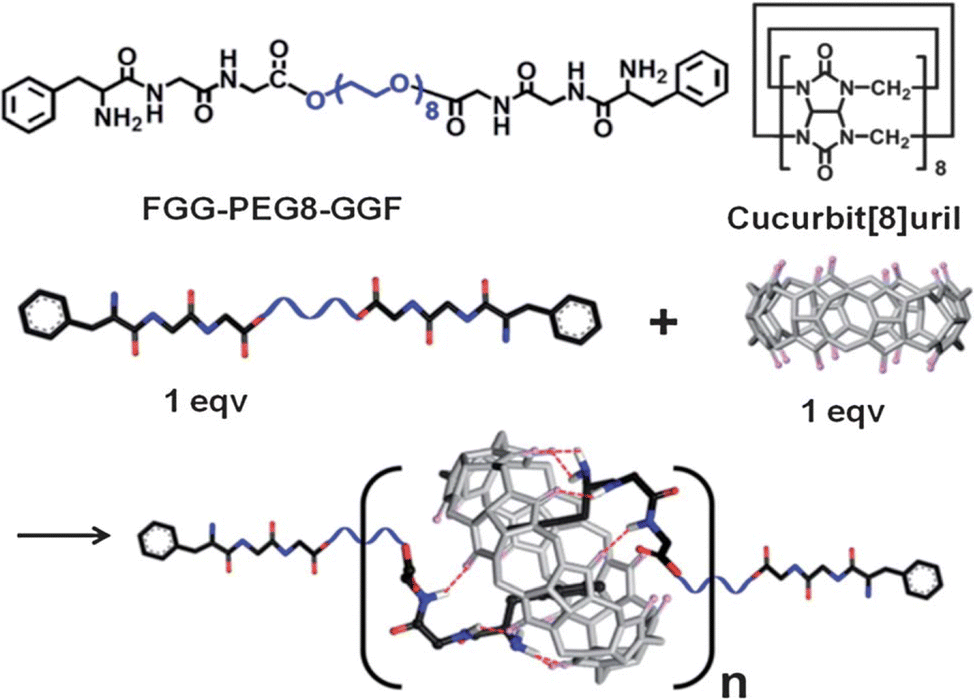 | ||
| Fig. 30 Supramolecular polymerization of a bifunctional monomer with Q8 in aqueous solution.141 Reproduced from ref. 141 with permission from the Royal Society of Chemistry. | ||
Liu, Dong, and coworkers reported the formation of supramolecular polymers from supramonomers comprising a 1:2 complex between Q8 and the hexapeptide H-Phe-Gly-Gly-Gly-Gly-Tyr-OH (Fig. 30).143 Horseradish peroxidase (HRP) was used to induce the covalent cross-coupling of the tyrosine side chains to yield polymers. Liu, Xu, and coworkers added an extra Tyr residue, H-Phe-Gly-Gly-Gly-Tyr-Tyr-OH, which allowed for the crosslinking of polymer chains via HRP-mediated cross-coupling to form highly monodisperse, spherical “nanocapsules” (Fig. 31).144 This hydrogel material was used to deliver doxorubicin antitumor agent and indocyanine green indicator to human breast cancer cells. Several other examples of supramolecular hydrogels are discussed in the next section.
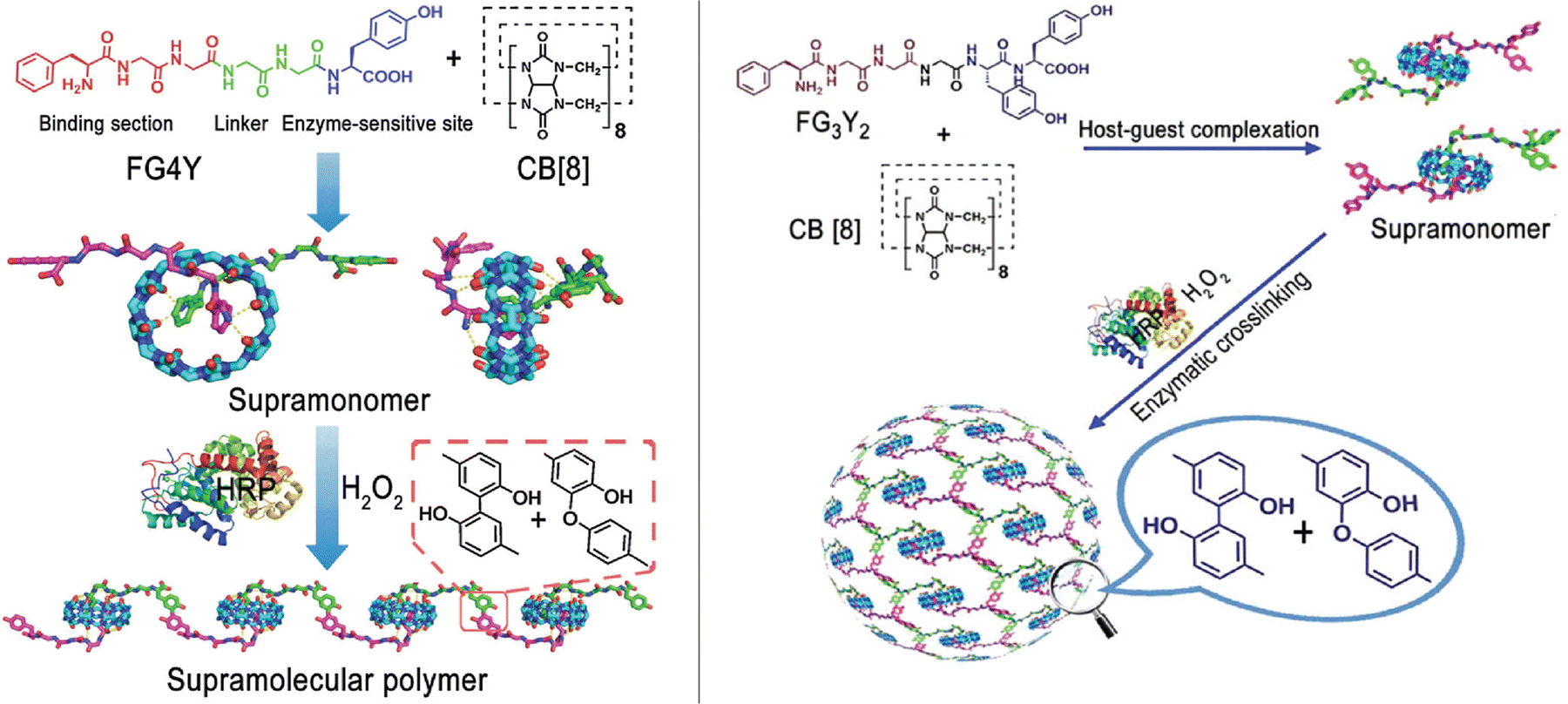
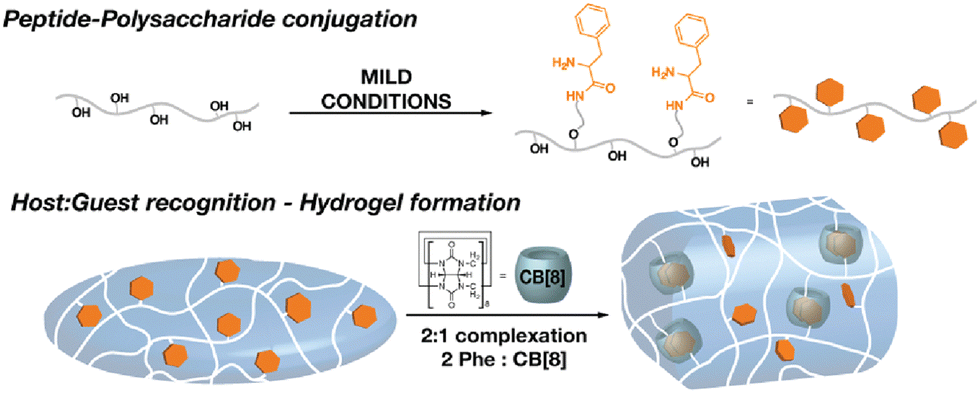 | ||
| Fig. 32 Supramolecular hydrogels comprising polysaccharides grafted with N-terminal Phe and cross-linked by Q8. CB[8] is Q8.148 Reproduced with permission from ref. 148. Copyright 2015 American Chemical Society. | ||
Supramolecular hydrogels have been used to demonstrate several applications in biomedical science. Scherman and coworkers reported the sustained release of bovine serum albumin from a Q8-mediated hydrogel comprising polyvalent copolymers presenting viologen and naphthyl groups.151 Wang and coworkers reported a strategy for in situ activation of an antibiotic using Q8 as the activator (Fig. 33).152 A polymer comprising a branched poly(ethyleneimine) was modified on the branches with N-terminal-Phe groups and cross-linked in the presence of Q8. The resulting hydrogel showed antibiotic activity with four bacterial cell lines, which was eliminated in the presence of a competitive guest for Q8.
 | ||
| Fig. 33 An eco-friendly, in situ antibiotic that is reversibly activated by Q8-induced supramolecular crosslinking. CB[8] is Q8.152 Reproduced from ref. 152 with permission from the Royal Society of Chemistry. | ||
In parallel with the report on supramolecular nanocapsules for drug delivery, as described in the previous section, Wang and coworkers reported the synthesis of supramolecular nanogels for stimulus-responsive drug delivery.153 N-terminal Phe groups were grafted onto a chitosan scaffold and crosslinked using Q8 to form highly monodisperse and biocompatible hydrogels. The hydrogels released doxorubicin upon treatment with high-affinity competitors and were shown to be cell permeable and to reduce the toxicity of doxorubicin in two cell lines. Hu, Xu, Di, and coworkers reported an alternative approach to stimulus-responsive oral drug delivery for targeting intestinal microbiota (Fig. 34).154 Mesoporous silica nanoparticles were loaded with the small molecule agent hydrocortisone and then capped with a supramolecular multilayer comprising chitosan grafted with N-terminal Trp groups and hyaluronic acid grafted with azobenzene groups, which were held together by Q8-mediated heteroternary complexes. In vitro, the multilayers disassembled in the presence of dithionite, a surrogate for azoreductase that reduces azobenzene to aniline, and released Cy5 fluorescent dye. In mice, the particles accumulated and released hydrocortisone in the colon, where azoreductase is released by intestinal microbiota.
 | ||
| Fig. 34 Mesoporous silica nanoparticles capped with Q8-mediated multilayers that release cargo upon azobenzene reduction. CB[8] is Q8.154 Reproduced from ref. 154 with permission from the Royal Society of Chemistry. | ||
Wang and coworkers reported a stimulus-responsive material that makes use of a specific peptide sequence for multimodal self-assembly and targeted drug delivery (Fig. 35).155 The peptide H-Phe-Phe-Val-Leu-Lys-OH was designed to have a binding site for Q7 at the N-terminus and a sequence that would self-assemble in the absence of Q7. This peptide was conjugated via the Lys side chain to the antitumor agent camptothecin. Q7 complexed with the amphiphilic camptothecin conjugates, forming monodisperse micellar structures and entering cells. The structures decomposed when spermine, which was overexpressed in the cancer cells, competitively released the camptothecin conjugate from Q7. The conjugate then self-assembled into fibrils with improved accumulation, retention, and sustained release of camptothecin.
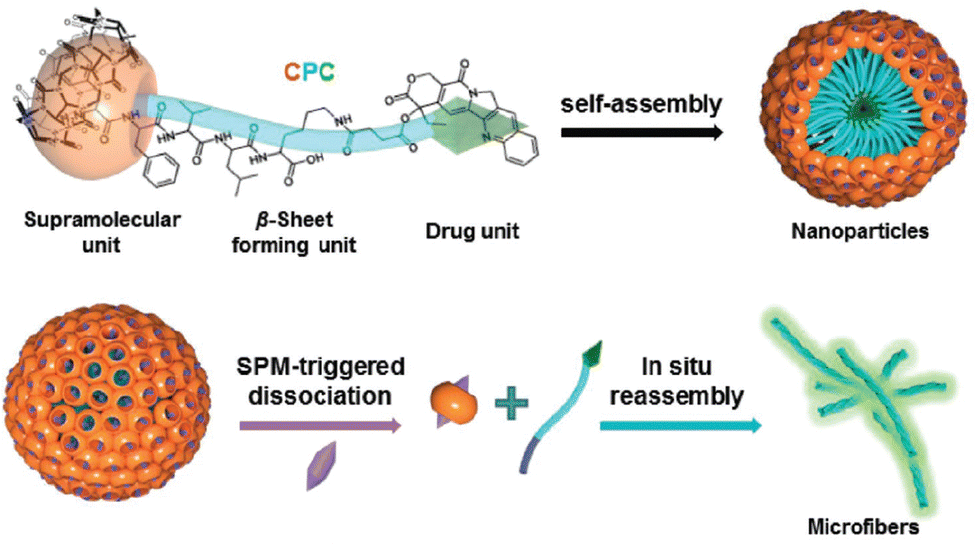 | ||
| Fig. 35 Drug delivery strategy involving a drug conjugated to a pentapeptide designed to bind Q7, enter cells, release Q7, self-assemble into microfibers, and release the drug.155 Reproduced with permission from ref. 155. © 2021 Wiley-VCH GmbH. | ||
Das and coworkers reported the synthesis of a supramolecular hydrogel that promotes efficient cell adhesion and proliferation.156 A peptide of sequence H-Phe-Gly-Gly-Lys-Tyr-Cys-Cys-Tyr-Arg-Gly-Asp-Ser-NH2 was designed to contain a site for Q8-mediated supramolecular cross-linking via the N-terminal Phe, a binding site for integrins at the C-terminus, two Cys residues for covalent cross-linking via disulfide bond formation, and two Tyr residues for cross-linking via horseradish peroxidase (Fig. 36). The three mechanisms for cross-linking allowed for fine-tuning of particle size. Stable hydrogels presented integrin-binding sites on the surface. The particles were noncytotoxic and promoted efficient cell adhesion.
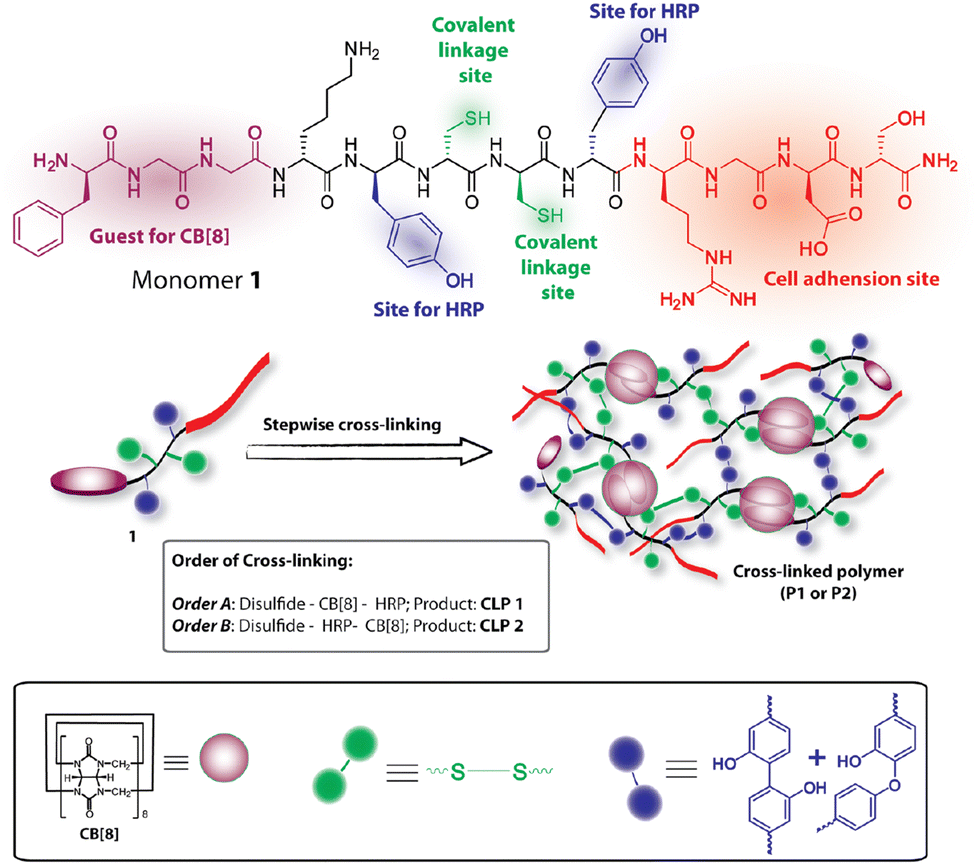 | ||
| Fig. 36 Highly cross-linked, peptide-based, hydrogel capable of promoting cell adhesion and proliferation. CB[8] is Q8.156 Reproduced with permission from ref. 156. Copyright 2018 American Chemical Society. | ||
Myung and coworkers reported the synthesis of supramolecular hydrogels that encapsulate cells and are injectable.157 Norbornene groups were grafted onto gelatin, and the ternary complex Q8·(H-Phe-Gly-Gly-Cys-OH)2 was used to crosslink the polymers into a hydrogel via thiol–ene click chemistry with the Cys side chains. The resulting materials were optically transparent and injectable through an 18-gauge needle. Human fibroblasts grown in the hydrogel remained viable long after injection.
Supramolecular hydrogels based on self-assembling peptide amphiphiles (PA) are promising materials for modeling and engineering biological tissue. Mata, Azevedo, and coworkers reported on the Q8-mediated hydrogelation of peptide amphiphile nanofibers as an alternative to ion-based equivalents.158 PA units comprising an N-terminal palmitoyl tail, a β-sheet-promoting amino acid sequence (V3A3), and three ionizable glutamic acid residues were engineered with either a Phe or Trp C-terminal amide. Upon self-assembly, the Phe and Trp residues are presented at the surface of the nanofibers, allowing recognition and tunable gelation by Q8. Compared with Ca2+-based PA hydrogels, Q8-based hydrogels formed more efficiently and required less Q8 than Ca2+ to achieve similar mechanical properties. Cell culture studies demonstrated the cell viability of the hydrogels and point toward the possibility of embedding cells during the gelation process.
3.5. Dynamic surfaces
Dynamic chemistry occurring at solid–liquid interfaces is ubiquitous in living systems and is necessary for many technologies such as heterogeneous catalysis, affinity purification, and sensors. Surfaces presenting ligands or receptors are effectively two-dimensional polyvalent materials. The capability of Q8 to bind two guests presents an opportunity to form polyvalent surfaces that dynamically trap and release ligands. One guest can be tethered to the surface covalently while the other can be the target solution-phase ligand. This section describes several applications involving dynamic surfaces that were made possible by the binding of cucurbit[n]urils to polypeptides.Scherman and coworkers first described the use of dynamic supramolecular surface chemistry involving Q8-peptide interactions (Fig. 37).159 Viologen groups were patterned on a self-assembled monolayer of alkanethiolates on Au and bound to Q8. Peptides containing an N-terminal Trp were captured selectively by heteroternary complex formation with the surface-bound Q8-viologen complexes and released selectively by single-electron reduction of the viologen. Capture and release were achieved for numerous cycles. This group then reported the synthesis of silica nanoparticles that present catenanes in which viologen axles are threaded through Q8 molecules and then covalently anchored on both ends to the surface.160 The functional surfaces of these superparamagnetic nanoparticles bound to aromatic compounds, including amino acids and peptides, via the Q8 groups, with selectivity for Phe- and Trp-containing peptides versus nonaromatic peptides. The captured peptides could then be released from the nanoparticles by single-electron reduction of the viologens. They also used a dynamic surface presenting Q8·viologen groups to select for high-affinity peptides via phage display, as mentioned in Section 2.4.42
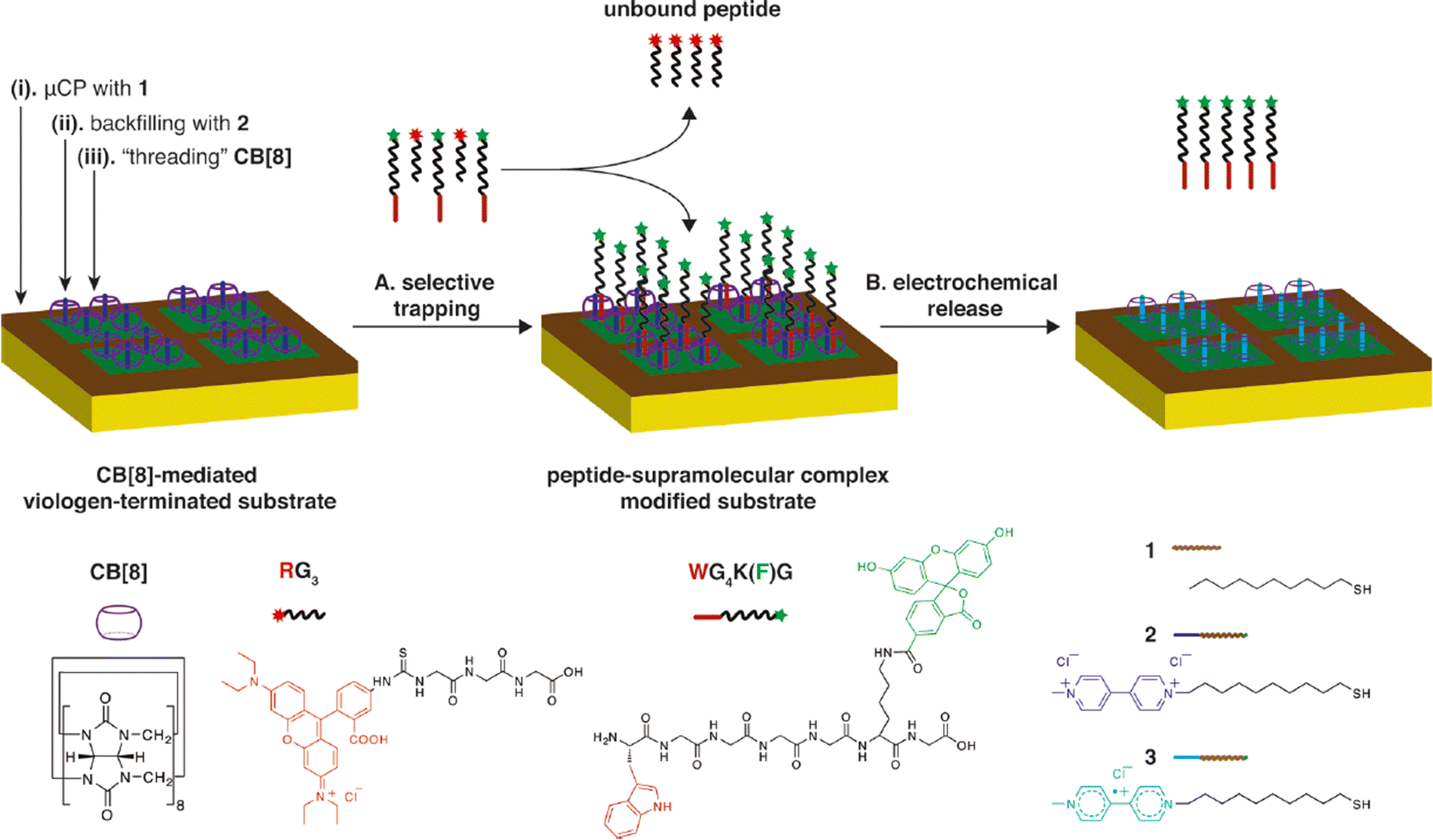 | ||
| Fig. 37 Reversible, supramolecular capture and release of peptides mediated by the binding and redox properties of the surface-bound viologen. CB[8] is Q8.159 Reproduced with permission from ref. 159. Copyright 2010 American Chemical Society. | ||
Li and coworkers demonstrated the synthesis of native protein multilayers mediated by Q8.161 Q8 spontaneously coated a silica surface presenting ammonium groups, and multilayers of hemoglobin and/or catalase were applied, with Q8 adhering the layers. Catalase retained its catalytic activity when assembled. Lysozyme, glucose oxidase, bovine serum albumin, and β-lactoglobulin did not assemble in this fashion, suggesting the necessity of selective binding. Free Q8 was also shown to aggregate solutions of hemoglobin, catalase, glutathione-S-transferase, and insulin in a process that was reversible upon the addition of methyl viologen. Q6 and Q7 did not aggregate proteins, suggesting the necessity of the dimerization capacity of Q8.
Jonkheijm and coworkers reported a series of studies demonstrating the power of supramolecular chemistry for making dynamic surfaces that can manipulate living cells. They first reported a bioactive monolayer for cell adhesion with a reversible electrochemical switch.162 Viologen groups were presented on a self-assembled monolayer of alkanethiolates on Au, which recruited Q8. Peptides of sequence H-Trp-Gly-Gly-Arg-Gly-Asp-Ser-OH were designed to contain an N-terminal Trp binding site for the surface-bound Q8·viologen complexes and an RGD site for integrin binding in order to drive cell adhesion. In the presence of Q8, the peptides bound to the surface, and cell adhesion was observed. Cells could then be released electrochemically via reduction of the viologen groups. They then engineered bacterial cells to present on their surfaces miniproteins containing a disordered loop with the sequence Gly-Gly-Trp-Gly-Gly.163 Q8 mediated the assembly of these cells in a concentration-dependent and sequence-selective manner and was inhibited by the competitive binding of H-Phe-Gly6-OH. These cells also bound to a surface presenting viologen groups and showed enhanced motility, presumably mediated by reversible multivalent interactions at the surface. They then developed a strategy to immobilize knottin miniproteins on a self-assembled monolayer and capture its cognate binding partner, β-trypsin (Fig. 38).164 Knottins were genetically engineered to contain one or two Gly-Gly-Trp-Gly-Gly sites for binding to surface-immobilized Q8·viologen complexes. Binding was improved for the divalent versus monovalent knottins, and surface adhesion of trypsin only occurred for the divalent knottin. All knottins were strong inhibitors of trypsin in solution. In a subsequent study, they produced knottins containing one, two, three, or four Gly-Gly-Trp-Gly-Gly binding sites and found that the binding affinity to surfaces presenting Q8·viologen complexes correlated with the valency of the knottins.165 The knottins also contained an RGD site for cell adhesion, and the higher valency knottins mediated greater cell elongation and more pronounced focal adhesion.
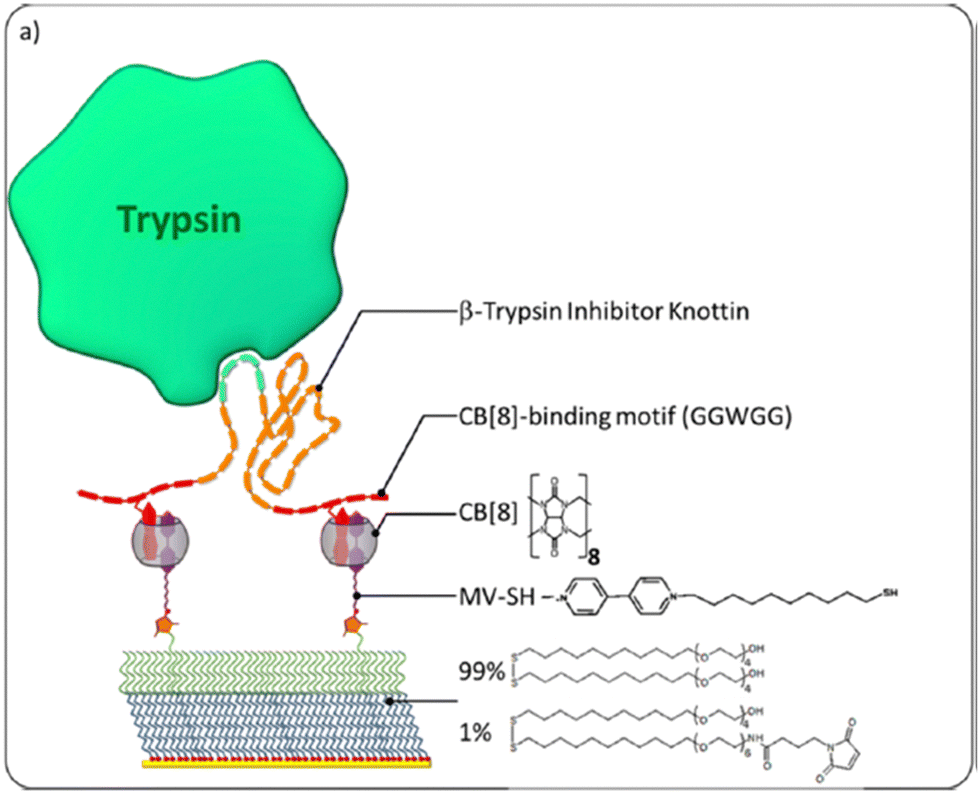 | ||
| Fig. 38 Surface for reversibly immobilizing functional mini-proteins for enzyme inhibition. CB[8] is Q8.164 Reproduced with permission from ref. 164. Copyright 2015 American Chemical Society. | ||
Brunsveld and coworkers demonstrated the reversible immobilization of a protein on lipid bilayers.166 A supported bilayer comprising dioleylphosphatidylcholine on silica-coated quartz was prepared and treated with a cholesterol-viologen conjugate, which incorporated into the bilayer to present viologen groups on the surface (Fig. 39). Q8 was used to immobilize yellow fluorescent proteins (YFP) tagged with N-terminal H-Trp-Gly-Gly (WGG-YFP) or H-Met-Gly-Gly (MGG-YFP). Both proteins bound to the surfaces, but MGG-YFP bound more weakly than FGG-YFP. The binding of WGG-YFP vs. MGG-YFP was also studied in solution, and although MGG did not bind Q8, MGG-YFP bound Q8 with similar affinity as WGG-YFP, indicating that the Q8·MGG-YFP complex is likely mediated by a nonterminal binding site. Protein binding was reversible upon addition of Q8. Brunsveld, Dankers, and coworkers then used Q8 to reversibly immobilize proteins on the surface of a thermoplastic elastomer.167 H-Phe-Gly-Gly-Lys was conjugated to a 2-uriedo-4-pyrimidinone (UPy) group via the Lys side chain to generate FGGK(UPy). This conjugate was mixed with a thermoelastic polymer and drop-coated or spin-coated onto surfaces. In the presence of Q8, the FGG-containing surfaces pulled down FGG-YFP selectively versus MGG-YFP. The complex disassembled when washed.
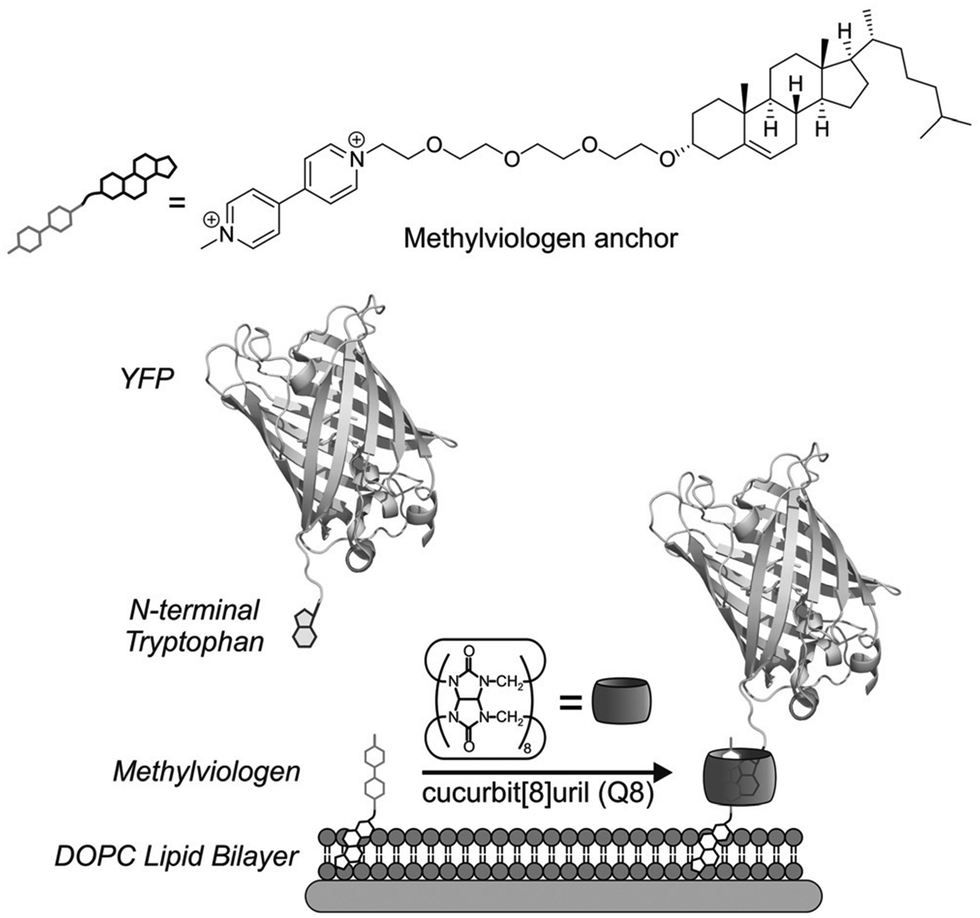 | ||
| Fig. 39 Reversible immobilization of WGG-labeled proteins on a supported bilayer.166 Reproduced with permission from ref. 166. © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Wang and coworkers used the Q7·Phe interaction to prepare a dynamic material capable of selectively isolating proteins containing methylated Lys via affinity purification.168 A resin presenting covalently bound Q7 groups was coated with the peptide H-Phe-Gly-Gly-Ala-Ala-Pro-Gly-Phe-pTyr-X-Glu-Ala-Gln (pTyr is phosphotyrosine, X is a noncanonical residue presenting a chloroacetyl group). The C-terminal domain of this peptide binds the protein PLCγ1 and covalently attaches to a Cys residue via coupling with the chloroacetyl group. A PLCγ1 domain was inserted into the HP1β CD-PLCγ1-c-SH2 fusion protein, which was then conjugated to the peptide-coated resin. The HP1β CD domain binds selectively to methylated Lys residues, and this material was used to enrich cell extract with methyl Lys-containing proteins, allowing for the identification of 255 proteins via mass spectrometric analysis of the trypsinized extract. A gentle elution of the protein of interest was achieved by adding amantadine to competitively release the peptide.
3.6. Protein drug formulation
Conjugating pharmaceutical proteins with poly(ethylene glycol) (PEG) is known to stabilize the proteins, inhibit aggregation, and limit proteolytic degradation. However, the covalent modification complicates isolation and purification of the protein after labeling and may alter protein function. Anderson, Langer, Isaacs, and coworkers demonstrated a novel supramolecular approach to stabilizing biopharmaceuticals by reversibly attaching Q7-PEG conjugates to the native PheB1 affinity tag in human insulin.169 Q7 was covalently conjugated to PEG chains of varying length (Mn = 5, 10, or 30 kDa) via copper-free click chemistry, and the conjugates spontaneously bound to insulin (Fig. 40). PEGylation did not significantly change the secondary structure of insulin or the affinity of Q7 for insulin. With agitation, insulin aggregated in the presence and absence of Q7 and PEG, but not in the presence of Q7-PEG, even after 100 days. The 100-day aged insulin stabilized by Q7-PEG retained its full activity. At pharmaceutical concentrations, Q7-PEG was also able to stabilize proteins without an N-terminal Phe residue. Q7-PEG stabilized an antibody, which was fully active after 24 hours, and Q7-PEG stabilized glucagon, which was fully soluble after 24 h. In vivo, insulin and Q7-PEG delivered subcutaneously to diabetic mice prolonged normoglycemia for time durations that correlated with the length of the PEG chain. This observation was believed to be due to delayed absorption via lymphatic circulation of the larger molecular weight complexes.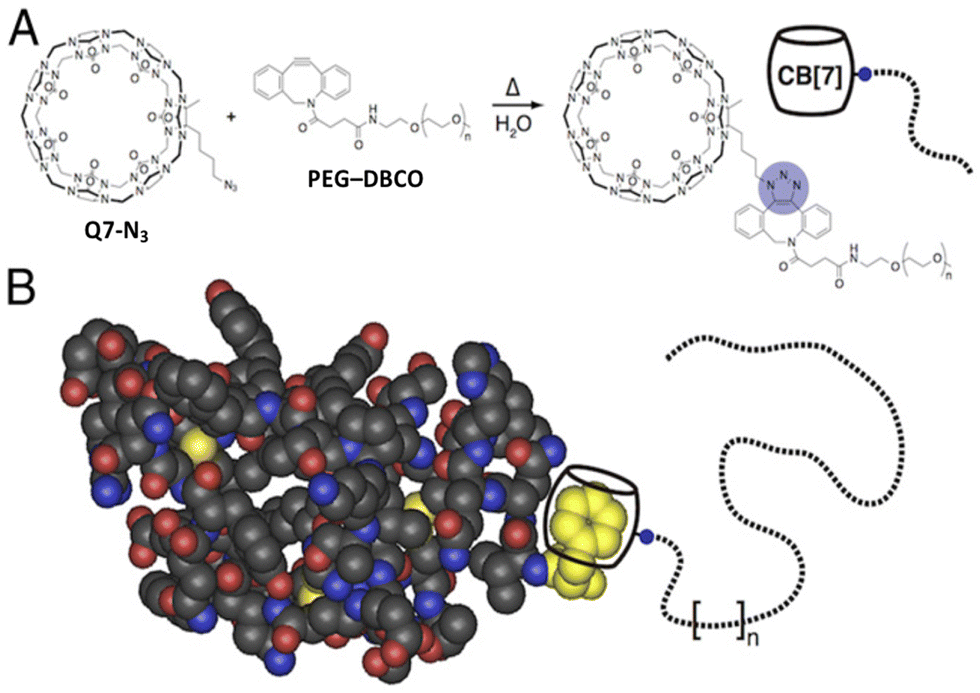 | ||
| Fig. 40 (A) Copper-free click reaction carried out between azidobutyl-Q7 and PEG polymer with dibenzocyclooctyne-modified PEG chains. (B) Supramolecular PEGylation of insulin via the PheB1 residue (yellow). CB[7] is Q7.169 Reproduced with permission from ref. 169. Copyright 2016 National Academy of Sciences. | ||
Continuing this line of inquiry, Webber, Appel, and coworkers demonstrated that Q7-PEG stabilizes the fast-acting insulin aspart and lispro analogues in their monomeric states and reduces aggregation.170 They also demonstrated that Q7-PEG can stabilize co-formulations of insulin and pramlintide and improve the pharmacokinetics of this dual hormone therapy.171 They then conjugated Q7 to proteins and demonstrated that the affinity of guest-conjugated PEG chains to Q7-protein correlates directly to the time of total insulin exposure, which is likely governed by the absorption rate.172
One concern in the supramolecular PEGylation of insulin is the prolonged in vivo duration of action due to sustained complex formation in the subcutaneous depot. Webber, Chou, and coworkers demonstrated the ability to tune the duration of action while preserving the stabilizing effects of PEGylation by engineering N-terminal acid-modified insulin analogs, which bind with weaker affinity for Q7.80 Another challenge with supramolecular PEGylation is biodegradability and intracellular accumulation. Seeking a more biocompatible method, Kramer and coworkers demonstrated the stabilization of insulin and calcitonin by Q7 conjugated with zwitterionic polypeptides.173 Inhibition of protein aggregation was directly correlated with the length of the zwitterionic chain. Treatment of the zwitterionic chain with natural proteases led to steady biodegradation.
3.7. Facilitating polypeptide modification
Site-selective modification of proteins is an area of intense current interest, with broad application in biochemistry and biomedical science. The ability of cucurbit[n]urils to bind site-selectively to peptides and proteins has inspired work in this area. Appel and coworkers demonstrated the use of Q7 as a protecting group for protein modification.174 Q7 was used to bind the PheB1 position of insulin and effectively protect the N-terminal amine from electrophilic attack by keeping it protonated, while a PEG chain was coupled site-selectively and covalently to the unprotected N-terminal amine of the A-chain. Li, Li, and coworkers demonstrated regioselective coupling via Michael addition to a Cys residue with a proximal Trp.175 A peptide presenting a viologen group and a dehydroalanine residue was bound to Q8 and then to a series of peptides, with varying distance between Trp and Cys residues. The efficiency of coupling was shown to depend on the sequence order and the distance between the Trp and Cys residues.4. Outlook
The applications described in Section 3 exemplify the extraordinary capabilities of cucurbit[n]urils in the context of bioscience and biotechnology. From sensing and separations to effectors and inhibitors of protein assembly, the interactions of Q7 and Q8 with peptides and proteins are strong, predictably selective, and compatible with a wide range of useful conditions, including protein mixtures, living cells, and mice. This system is practical in part because Q7 and Q8 are commercially available and readily accessible synthetically and function in neutral aqueous buffer at working concentrations that are compatible with many measurement techniques. This system is also practical in part because much is known about the chemistry of peptides and proteins, including their chemical synthesis, biosynthesis, and posttranslational modification, and the engineering and use of affinity tags. Q8 provides the powerful capability of reversible, noncovalent dimerization of peptides and proteins with one another and with a functional guest. The principles for molecular recognition in this system described in Section 2 are well understood and enable highly predictive, programmable, and switchable binding. Taken together, these properties are extraordinary and should enable the development of many more applications. We are particularly excited by the recent work on stabilizing protein drug formulations. We envision further development of methods for the selective enrichment and analysis of complex mixtures, the synthesis of functional hydrogels for tissue engineering, the synthesis of probes for biological processes, the creation of selective agents for biological imaging, and the creation of catalysts for site-selective protein modification. It will be exciting to see what the next decade of creativity brings to bear.Data availability
This review article contains no new data. All presented data are reproduced from other published sources, which are cited within the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is dedicated to Prof. George M. Whitesides on the occasion of his 85th birthday and in appreciation for his mentorship and his extraordinary contributions to science. We gratefully acknowledge financial support from the National Institutes of Health (GM141708), the Welch Foundation (W-1640 and W-0031), and Trinity University.References
- C. L. Young, Z. T. Britton and A. S. Robinson, Biotechnol. J., 2012, 7, 620–634 CrossRef CAS.
- J.-C. Janson, Protein Purification: Principles, High Resolution Methods, and Applications, Wiley, Hoboken, USA, 3rd edn, 2011 Search PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS.
- V. Mishra, Affinity Tags for Protein Purification, Curr. Protein Pept. Sci., 2020, 21, 821–830 CrossRef CAS.
- P. N. Hengen, Trends Biochem. Sci., 1995, 20, 285–286 CrossRef CAS.
- C. Zhao, L. M. Hellman, X. Zhan, W. S. Bowman, S. W. Whiteheart and M. G. Fried, Anal. Biochem., 2010, 399, 237–245 CrossRef CAS.
- J. Arnau, C. Lauritzen, G. E. Petersen and J. Pedersen, Protein Expression Purif., 2006, 48, 1–13 CrossRef CAS.
- M. H. Hefti, J. G. Caroline, V. V. der Toorn, R. Dixon and J. A. Vervoort, Anal. Biochem., 2001, 295, 180–185 CrossRef CAS.
- E. S. Day, S. M. Cote and A. Whitty, Biochemistry, 2012, 51, 9124–9136 CrossRef CAS.
- E. Cha, N. Clements, C. Hofman, B. Lavoie, A. Van Zile, L. Warden and A. R. Urbach, in Supramolecular Protein Chemistry, ed. P. Crowley, Royal Society of Chemistry, Cambridge, UK, 2020 Search PubMed.
- E. S. Istvan and J. Deisenhofer, Science, 2001, 292, 1160–1164 CrossRef CAS.
- R. E. McGovern, H. Fernandes, A. R. Khan, N. P. Power and P. B. Crowley, Nat. Chem., 2012, 4, 527–533 CrossRef CAS.
- J. Habchi, P. Tompa, S. Longhi and V. N. Uversky, Chem. Rev., 2014, 114, 6561–6588 CrossRef CAS.
- M. W. Peczuh and A. D. Hamilton, Chem. Rev., 2000, 100, 2479–2494 CrossRef CAS.
- M. Giese, J. Niemeyer and J. Voskuhl, ChemPlusChem, 2020, 85, 985–997 CrossRef CAS.
- S. Tashiro, M. Kobayashi and M. Fujita, J. Am. Chem. Soc., 2006, 128, 9280–9281 CrossRef CAS.
- C. Dolain, Y. Hatakeyama, T. Sawada, S. Tashiro and M. Fujita, J. Am. Chem. Soc., 2010, 132, 5564–5565 CrossRef CAS.
- L. M. Heitmann, A. B. Taylor, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2006, 128, 12574–12581 CrossRef CAS.
- M. V. Rekharsky, H. Yamamura, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Chem. Commun., 2008, 2236–2238 RSC.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS.
- S. J. Barrow, S. Kasera, M. Cao, J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS.
- K. Kim, Cucurbiturils and Related Macrocycles, Royal Society of Chemistry, Croydon, UK, 1st edn, 2020 Search PubMed.
- F. Biedermann, W. M. Nau and H.-J. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11158–11171 CrossRef CAS.
- A. E. Kaifer, Isr. J. Chem., 2018, 58, 244–249 CrossRef CAS.
- S. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959–15967 CrossRef CAS.
- M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737–20742 CrossRef CAS.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- H.-J. Kim, J. Heo, W. S. Jeon, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 1526–1529 CrossRef CAS.
- L. Cao, M. Śekutor, P. Y. Zavalij, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS.
- M. E. Bush, N. D. Bouley and A. R. Urbach, J. Am. Chem. Soc., 2005, 127, 14511–14517 CrossRef CAS.
- K. Kim, J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee and J.-K. Kang, Cucurbituril derivatives, their preparation methods and uses, US Pat., US6365734, 2000 Search PubMed.
- P. Rajgariah and A. R. Urbach, J. Inclusion Phenom. Macrocyclic Chem., 2008, 62, 251–254 CrossRef CAS.
- C. Chothia, J. Mol. Biol., 1976, 105, 1–12 CrossRef CAS.
- A. Radzicka and R. Wolfenden, Biochemistry, 1998, 27, 1664–1670 CrossRef.
- W. C. Wimley and S. H. White, Nat. Struct. Biol., 1996, 3, 842–848 CrossRef CAS.
- D. W. Johnson and F. Hof, Aromatic Interactions: Frontiers in Knowledge and Application in Supramolecular Chemistry, Royal Society of Chemistry, Croydon, UK, 2017 Search PubMed.
- L. M. Espinoza-Fonseca, Mol. BioSyst., 2012, 8, 237–246 RSC.
- T. Clackson and J. A. Wells, Science, 1995, 267, 383–386 CrossRef CAS.
- A. A. Bogan and K. S. Thorn, J. Mol. Biol., 1998, 280, 1–9 CrossRef CAS.
- O. A. Ali, E. M. Olson and A. R. Urbach, Supramol. Chem., 2013, 25, 863–868 CrossRef CAS.
- R. P. G. Bosmans, W. E. Hendriksen, M. Verheijden, R. Eelkema, P. Jonkheijm, J. H. van Esch and L. Brunsveld, Chem. – Eur. J., 2015, 21, 18466–18473 CrossRef CAS.
- S. Sonzini, A. Marcozzi, R. J. Gubeli, C. F. van der Walle, P. Ravn, A. Herrmann and O. A. Scherman, Angew. Chem., Int. Ed., 2016, 55, 14000–14004 CrossRef CAS.
- F. Biedermann, U. Rauwald, M. Cziferszky, K. A. Williams, L. D. Gann, B. Y. Guo, A. R. Urbach, C. W. Bielawski and O. A. Scherman, Chem. – Eur. J., 2010, 16, 13716–13722 CrossRef CAS.
- X. Chen, Z. Huang, R. L. Sala, A. M. McLean, G. Wu, K. Sokolowski, K. King, J. A. McCune and O. A. Scherman, J. Am. Chem. Soc., 2022, 144, 8474–8479 CrossRef CAS.
- A. Barba-Bon, Y.-C. Pan, F. Biedermann, D.-S. Guo, W. M. Nau and A. Hennig, J. Am. Chem. Soc., 2019, 141, 20137–20145 CrossRef CAS.
- F. Biedermann and O. A. Scherman, J. Phys. Chem. B, 2012, 116, 2842–2849 CrossRef CAS.
- J. J. Reczek, A. A. Kennedy, B. T. Halbert and A. R. Urbach, J. Am. Chem. Soc., 2009, 131, 2408–2415 CrossRef CAS.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- E. Cavatorta, P. Jonkheijm and J. Huskens, Chem. – Eur. J., 2017, 23, 4046–4050 CrossRef CAS.
- S. Sonzini, S. T. J. Ryan and O. A. Scherman, Chem. Commun., 2013, 49, 8779 RSC.
- G. Wu, D. E. Clarke, C. Wu and O. A. Scherman, Org. Biomol. Chem., 2019, 17, 3514–3520 RSC.
- H. Barbero and E. Masson, Chem. Sci., 2021, 12, 9962–9968 RSC.
- R. J. Fernandes, P. Remón, A. J. Moro, A. Seco, A. S. D. Ferreira, U. Pischel and N. Basílio, J. Org. Chem., 2021, 86, 8472–8478 CrossRef CAS.
- E. Zaorska and M. Malinska, Chem. – Eur. J., 2024, 30, e202302250 CrossRef CAS.
- H. D. Nguyen, D. T. Dang, J. L. J. van Dongen and L. Brunsveld, Angew. Chem., Int. Ed., 2010, 49, 895–898 CrossRef CAS.
- D. T. Dang, H. D. Nguyen, M. Merkx and L. Brunsveld, Angew. Chem., Int. Ed., 2013, 52, 2915–2919 CrossRef CAS.
- C. Hou, J. Li, L. Zhao, W. Zhang, Q. Luo, Z. Dong, J. Xu and J. Liu, Angew. Chem., Int. Ed., 2013, 52, 5590–5593 CrossRef CAS.
- G. Li, W.-Y. Yang, Y.-F. Zhao, Y.-X. Chen, L. Hong and Y.-M. Li, Chem. – Eur. J., 2018, 24, 13647–13653 CrossRef CAS.
- F. Biedermann, U. Rauwald, M. Cziferszky, K. A. Williams, L. D. Gann, B. Y. Guo, A. R. Urbach, C. W. Bielawski and O. A. Scherman, Chem. – Eur. J., 2010, 7 Search PubMed.
- L. C. Smith, D. G. Leach, B. E. Blaylock, O. A. Ali and A. R. Urbach, J. Am. Chem. Soc., 2015, 137, 3663–3669 CrossRef CAS.
- Z. Hirani, H. F. Taylor, E. F. Babcock, A. T. Bockus, C. D. Varnado, C. W. Bielawski and A. R. Urbach, J. Am. Chem. Soc., 2018, 140, 12263–12269 CrossRef CAS.
- D. E. Clarke, G. Wu, C. Wu and O. A. Scherman, J. Am. Chem. Soc., 2021, 143, 6323–6327 CrossRef CAS.
- P. Suating, M. B. Ewe, L. B. Kimberly, H. D. Arman, D. J. Wherritt and A. R. Urbach, Chem. Sci., 2024, 15, 5133–5142 RSC.
- P. Suating, L. B. Kimberly, M. B. Ewe, S. L. Chang, J. M. Fontenot, P. R. Sultane, C. W. Bielawski, D. A. Decato, O. B. Berryman, A. B. Taylor and A. R. Urbach, J. Am. Chem. Soc., 2024, 146, 7649–7657 CrossRef CAS.
- G. Wu, M. Olesinska, Y. Wu, D. Matak-Vinkovic and O. A. Scherman, J. Am. Chem. Soc., 2017, 139, 3202–3208 CrossRef CAS.
- M. V. Rekharsky, H. Yamamura, C. Inoue, M. Kawai, I. Osaka, R. Arakawa, K. Shiba, A. Sato, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2006, 128, 14871–14880 CrossRef CAS.
- M. V. Rekharsky, H. Yamamura, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Chem. Commun., 2008, 2236 RSC.
- J. M. Chinai, A. B. Taylor, L. M. Ryno, N. D. Hargreaves, C. A. Morris, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2011, 133, 8810–8813 CrossRef CAS.
- L. A. Logsdon, C. L. Schardon, V. Ramalingam, S. K. Kwee and A. R. Urbach, J. Am. Chem. Soc., 2011, 133, 17087–17092 CrossRef CAS.
- H. R. Anderson, W. L. Reeves, A. T. Bockus, P. Suating, A. G. Grice, M. Gallagher and A. R. Urbach, Bioconjugate Chem., 2023, 34, 212–217 CrossRef CAS.
- G. Ghale, V. Ramalingam, A. R. Urbach and W. M. Nau, J. Am. Chem. Soc., 2011, 133, 7528–7535 CrossRef CAS.
- R. Jiang, M. Nilam, A. Hennig and W. Nau, Adv. Mater., 2024, 36, 2306922 CrossRef CAS.
- L. A. Logsdon and A. R. Urbach, J. Am. Chem. Soc., 2013, 135, 11414–11416 CrossRef CAS.
- S. Zhang, L. Grimm, Z. Miskolczy, L. Biczok, F. Biedermann and W. M. Nau, Chem. Commun., 2019, 55, 14131–14134 RSC.
- W. Cao, W. Qin, Y. Wang, Z. Dai, X. Dai, H. Wang, W. Xuan, Y. Zhang, Y. Liu and T. Liu, Angew. Chem., Int. Ed., 2021, 60, 11196–11200 CrossRef CAS.
- F. Ma, X. Zheng and Z. Li, Phys. Chem. Chem. Phys., 2021, 23, 13724–13733 RSC.
- F. Ma, X. Zheng, L. Xie and Z. Li, J. Mol. Liq., 2021, 328, 115479 CrossRef CAS.
- H. D. Nguyen, D. T. Dang, J. L. J. van Dongen and L. Brunsveld, Angew. Chem., Int. Ed., 2010, 49, 895–898 CrossRef CAS.
- W. Li, A. T. Bockus, B. Vinciguerra, L. Isaacs and A. R. Urbach, Chem. Commun., 2016, 52, 8537–8540 RSC.
- R. Meudom, Y. Zhang, M. A. VandenBerg, L. Zou, Y. W. Zhang, M. J. Webber and D. H.-C. Chou, Acta Pharm. Sin. B, 2023, 13, 2281–2290 CrossRef CAS.
- C. L. Maikawa, A. A. A. Smith, L. Zou, G. A. Roth, E. C. Gale, L. M. Stapleton, S. W. Baker, J. L. Mann, A. C. Yu, S. Correa, A. K. Grosskopf, C. S. Liong, C. M. Meis, D. Chan, M. Troxell, D. M. Maahs, B. A. Buckingham, M. J. Webber and E. A. Appel, Nat. Biomed. Eng., 2020, 4, 507–517 CrossRef CAS.
- H. H. Lee, T. S. Choi, S. J. C. Lee, J. W. Lee, J. Park, Y. H. Ko, W. J. Kim, K. Kim and H. I. Kim, Angew. Chem., Int. Ed., 2014, 53, 7461–7465 CrossRef CAS.
- H. Shang, A. Zhou, J. Jiang, Y. Liu, J. Xie, S. Li, Y. Chen, X. Zhu, H. Tan and J. Li, Acta Biomater., 2018, 78, 178–188 CrossRef CAS.
- E. Jacob and R. Unger, Bioinformatics, 2007, 23, e225–e230 CrossRef CAS.
- V. N. Uversky, V. Dave, L. M. Iakoucheva, P. Malaney, S. J. Metallo, R. P. Pathak and A. C. Joerger, Chem. Rev., 2014, 114, 6844–6879 CrossRef CAS.
- A. Varshavsky, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 358–366 CrossRef CAS.
- G. Roman-Hernandez, R. A. Grant, R. T. Sauer and T. A. Baker, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8888–8893 CrossRef CAS.
- E. Matta-Camacho, G. Kozlov, F. F. Li and K. Gehring, Nat. Struct. Mol. Biol., 2010, 17, 1182–1187 CrossRef CAS.
- D. M. Bailey, A. Hennig, V. D. Uzunova and W. M. Nau, Chem. – Eur. J., 2008, 14, 6069–6077 CrossRef CAS.
- M. A. Gamal-Eldin and D. H. Macartney, Org. Biomol. Chem., 2013, 11, 488–495 RSC.
- F. Guagnini, P. M. Antonik, M. L. Rennie, P. O’Byrne, A. R. Khan, R. Pinalli, E. Dalcanale and P. B. Crowley, Angew. Chem., Int. Ed., 2018, 57, 7126–7130 CrossRef CAS.
- F. Guagnini, S. Engilberge, K. O. Ramberg, J. Perez and P. B. Crowley, Chem. Commun., 2020, 56, 360–363 RSC.
- F. Guagnini, S. Engilberge, R. J. Flood, K. O. Ramberg and P. B. Crowley, Cryst. Growth Des., 2020, 20, 6983–6989 CrossRef CAS.
- K. O. Ramberg, F. Guagnini, S. Engilberge, M. A. Wronska, M. L. Rennie, J. Perez and P. B. Crowley, Chem. – Eur. J., 2021, 27, 1–10 CrossRef.
- K. O. Ramberg and P. B. Crowley, J. Struct. Biol., 2023, 215, 107969 CrossRef CAS.
- J. Lee, L. Perez, Y. Liu, H. Wang, R. J. Hooley and W. Zhong, Anal. Chem., 2018, 90, 1881–1888 CrossRef CAS.
- K. O. Ramberg, S. Engilberge, F. Guagnini and P. B. Crowley, Org. Biomol. Chem., 2021, 19, 837–844 RSC.
- L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS.
- Y. Ling, W. Wang and A. E. Kaifer, Chem. Commun., 2007, 610–612 RSC.
- F. Biedermann and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 5694–5699 CrossRef CAS.
- A. Prabodh, Y. Wang, S. Sinn, P. Albertini, C. Spies, E. Spuling, L.-P. Yang, W. Jiang, S. Bräse and F. Biedermann, Chem. Sci., 2021, 12, 9420–9431 RSC.
- G. H. Aryal, L. Huang and K. W. Hunter, RSC Adv., 2016, 6, 82566–82570 RSC.
- D. Maity, K. I. Assaf, W. Sicking, C. Hirschhäuser, W. M. Nau and C. Schmuck, Chem. – Eur. J., 2019, 25, 13088–13093 CrossRef CAS.
- F. Biedermann, G. Ghale, A. Hennig and W. M. Nau, Commentat. Biol., 2020, 3, 1–10 CrossRef.
- L. Wei, X. Wang, C. Li, X. Li, Y. Yin and G. Li, Biosens. Bioelectron., 2015, 71, 348–352 CrossRef CAS.
- H. Wang, Y. Fan, B. Chen, J. Lei, S. Yu, X. Chen and X. Hou, Nat. Commun., 2022, 13, 1906–1914 CrossRef CAS.
- Y. Li, Q. Li, X. Miao, C. Qin, D. Chu and L. Cao, Angew. Chem., Int. Ed., 2021, 60, 6744–6751 CrossRef CAS.
- C. Yan, Q. Li, X. Miao, Y. Zhao, Y. Li, P. Wang, K. Wang, H. Duan, L. Zhang and L. Cao, Angew. Chem., Int. Ed., 2023, 62, e202308029 CrossRef CAS.
- H. Li, H. Xie, Y. Cao, X. Ding, Y. Yin and G. Li, Anal. Chem., 2013, 85, 1047–1052 CrossRef CAS.
- Y. Chang, Y. Zhuo, Y. Chai and R. Yuan, Anal. Chem., 2017, 89, 8266–8272 CrossRef CAS.
- J. W. Lee, M. H. Shin, W. Mobley, A. R. Urbach and H. I. Kim, J. Am. Chem. Soc., 2015, 137, 15322–15329 CrossRef CAS.
- J. E. Olson, A. S. Braegelman, L. Zou, M. J. Webber and J. P. Camden, Appl. Spectrosc., 2020, 74, 1374–1383 CrossRef CAS.
- G. Ghale and W. M. Nau, Acc. Chem. Res., 2014, 47, 2150–2159 CrossRef CAS.
- A. Hennig, H. Bakirci and W. M. Nau, Nat. Methods, 2007, 4, 629–632 CrossRef CAS.
- Y. Cai and G. Li, Dyes Pigm., 2021, 195, 109734 CrossRef CAS.
- R. Jiang, M. Nilam, A. Hennig and W. M. Nau, Adv. Mater., 2024, 36, 2306922 CrossRef CAS.
- A. Hennig, G. Ghale and W. M. Nau, Chem. Commun., 2007, 1614–1616 RSC.
- X. Mao, Y. Li, P. Han, X. Wang, S. Yang, F. Zhang, X. Gong and Y. Cao, Sens. Actuators, B, 2018, 267, 336–341 CrossRef CAS.
- H. Barbero and E. Masson, Chem. Sci., 2021, 12, 9962–9968 RSC.
- D. T. Dang, J. Schill and L. Brunsveld, Chem. Sci., 2012, 3, 2679 RSC.
- D. T. Dang, R. P. G. Bosmans, C. Moitzi, I. K. Voets and L. Brunsveld, Org. Biomol. Chem., 2014, 12, 9341–9344 RSC.
- D. T. Dang, H. D. Nguyen, M. Merkx and L. Brunsveld, Angew. Chem., Int. Ed., 2013, 52, 2915–2919 CrossRef CAS.
- P. J. De Vink, T. Van Der Hek and L. Brunsveld, Chem. Sci., 2021, 12, 6726–6731 RSC.
- R. P. G. Bosmans, J. M. Briels, L.-G. Milroy, T. F. A. de Greef, M. Merkx and L. Brunsveld, Angew. Chem., Int. Ed., 2016, 55, 8899–8903 CrossRef CAS.
- P. J. de Vink, J. M. Briels, T. Schrader, L.-G. Milroy, L. Brunsveld and C. Ottmann, Angew. Chem., 2017, 129, 9126–9130 CrossRef.
- D. T. Dang, A. H. A. M. van Onzen, Y. L. Dorland and L. Brunsveld, ChemBioChem, 2018, 19, 2490–2494 CrossRef CAS.
- S. J. A. Aper, A. Den Hamer, S. F. A. Wouters, L. J. M. Lemmens, C. Ottman, L. Brunsveld and M. Merkx, ACS Synth. Biol., 2018, 7, 2216–2225 CrossRef CAS.
- M. Ramaekers, S. P. W. Wijnands, J. L. J. van Dongen, L. Brunsveld and P. Y. W. Dankers, Chem. Commun., 2015, 51, 3147–3150 RSC.
- R. Wang, S. Qiao, L. Zhao, C. Hou, X. Li, Y. Liu, Q. Luo, J. Xu, H. Li and J. Liu, Chem. Commun., 2017, 53, 10532–10535 RSC.
- X. Li, Y. Bai, Z. Huang, C. Si, Z. Dong, Q. Luo and J. Liu, Nanoscale, 2017, 9, 7991–7997 RSC.
- Y. Li, L. Zhao, H. Chen, R. Tian, F. Li, Q. Luo, J. Xu, C. Hou and J. Liu, Chem. Commun., 2021, 57, 10620–10623 RSC.
- H. H. Lee, T. S. Choi, S. J. C. Lee, J. W. Lee, J. Park, Y. H. Ko, W. J. Kim, K. Kim and H. I. Kim, Angew. Chem., Int. Ed., 2014, 53, 7461–7465 CrossRef CAS.
- O. V. de Oliveira, A. da Silva Gonçalves and N. E. Castilho de Almeida, J. Biomol. Struct. Dyn., 2022, 40, 9602–9612 CrossRef CAS.
- N. E. C. de Almeida, T. D. Do, M. Tro, N. E. LaPointe, S. C. Feinstein, J.-E. Shea and M. T. Bowers, ACS Chem. Neurosci., 2016, 7, 218–226 CrossRef CAS.
- T. S. Choi, H. H. Lee, Y. H. Ko, K. S. Jeong, K. Kim and H. I. Kim, Sci. Rep., 2017, 7, 5710 CrossRef.
- S. Sonzini, H. F. Stanyon and O. A. Scherman, Phys. Chem. Chem. Phys., 2017, 19, 1458–1465 RSC.
- H. Shang, X. Chen, Y. Liu, L. Yu, J. Li and J. Ding, Int. J. Pharm., 2017, 527, 52–60 CrossRef CAS.
- M. Martinez Morales, M. Zalar, S. Sonzini, A. P. Golovanov, C. F. van der Walle and J. P. Derrick, Mol. Pharmaceutics, 2019, 16, 3100–3108 CrossRef CAS.
- M. Martinez Morales, C. F. van der Walle and J. P. Derrick, Mol. Pharmaceutics, 2023, 20, 3559–3569 CrossRef CAS.
- D. Maity, Y. Oh, L. Gremer, W. Hoyer, M. Magzoub and A. D. Hamilton, Chem. – Eur. J., 2022, 28, e202200456 CrossRef CAS.
- X. Tan, L. Yang, Y. Liu, Z. Huang, H. Yang, Z. Wang and X. Zhang, Polym. Chem., 2013, 4, 5378 RSC.
- Q. Song, F. Li, X. Tan, L. Yang, Z. Wang and X. Zhang, Polym. Chem., 2014, 5, 5895–5899 RSC.
- Z. Huang, Y. Fang, Q. Luo, S. Liu, G. An, C. Hou, C. Lang, J. Xu, Z. Dong and J. Liu, Chem. Commun., 2016, 52, 2083–2086 RSC.
- S. Liu, Z. Huang, F. Li, T. Yan, S. Fu, R. Tian, C. Hou, Q. Luo, J. Xu and J. Liu, Polym. Chem., 2019, 10, 3566–3570 RSC.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS.
- E. A. Appel, X. J. Loh, S. T. Jones, F. Biedermann, C. A. Dreiss and O. A. Scherman, J. Am. Chem. Soc., 2012, 134, 11767–11773 CrossRef CAS.
- M. J. Rowland, E. A. Appel, R. J. Coulston and O. A. Scherman, J. Mater. Chem. B, 2013, 1, 2904 RSC.
- M. J. Rowland, M. Atgie, D. Hoogland and O. A. Scherman, Biomacromolecules, 2015, 16, 2436–2443 CrossRef CAS.
- Q. Song, Y. Gao, J.-F. Xu, B. Qin, M. J. Serpe and X. Zhang, ACS Macro Lett., 2016, 5, 1084–1088 CrossRef CAS.
- J. Zhang, S. Hou, Y. Chen, J. Zhou, H. Chen and Y. Tan, Soft Matter, 2019, 15, 9797–9804 RSC.
- E. A. Appel, X. J. Loh, S. T. Jones, C. A. Dreiss and O. A. Scherman, Biomaterials, 2012, 33, 4646–4652 CrossRef CAS.
- S. Li, N. Jiang, W. Zhao, Y.-F. Ding, Y. Zheng, L.-H. Wang, J. Zheng and R. Wang, Chem. Commun., 2017, 53, 5870–5873 RSC.
- Y.-F. Ding, J. Wei, S. Li, Y.-T. Pan, L.-H. Wang and R. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 28665–28670 CrossRef CAS.
- S. Cheng, H. Shen, S. Zhao, Y. Zhang, H. Xu, L. Wang, B. Di, L. Xu and C. Hu, Nanoscale, 2020, 12, 15348–15363 RSC.
- C. Sun, Z. Wang, K. Yang, L. Yue, Q. Cheng, Y.-L. Ma, S. Lu, G. Chen and R. Wang, Small, 2021, 17, e2101139 CrossRef.
- P. Dowari, S. Saha, B. Pramanik, S. Ahmed, N. Singha, A. Ukil and D. Das, Biomacromolecules, 2018, 19, 3994–4002 CrossRef CAS.
- A. C. Madl, C. M. Madl and D. Myung, ACS Macro Lett., 2020, 9, 619–626 CrossRef CAS.
- C. Redondo-Gómez, S. Padilla-Lopátegui, A. Mata and H. S. Azevedo, Bioconjugate Chem., 2022, 33, 111–120 CrossRef.
- F. Tian, M. Cziferszky, D. Jiao, K. Wahlström, J. Geng and O. A. Scherman, Langmuir, 2011, 27, 1387–1390 CrossRef CAS.
- X. Ren, Y. Wu, D. E. Clarke, J. Liu, G. Wu and O. A. Scherman, Chem. – Asian J., 2016, 11, 2382–2386 CrossRef CAS.
- H. Yang, Q. An, W. Zhu, W. Li, Y. Jiang, J. Cui, X. Zhang and G. Li, Chem. Commun., 2012, 48, 10633 RSC.
- Q. An, J. Brinkmann, J. Huskens, S. Krabbenborg, J. de Boer and P. Jonkheijm, Angew. Chem., Int. Ed., 2012, 51, 12233–12237 CrossRef CAS.
- S. Sankaran, M. C. Kiren and P. Jonkheijm, ACS Nano, 2015, 9, 3579–3586 CrossRef CAS.
- S. Sankaran, M. de Ruiter, J. J. L. M. Cornelissen and P. Jonkheijm, Bioconjugate Chem., 2015, 26, 1972–1980 CrossRef CAS.
- S. Sankaran, E. Cavatorta, J. Huskens and P. Jonkheijm, Langmuir, 2017, 33, 8813–8820 CrossRef CAS.
- R. P. G. Bosmans, W. E. Hendriksen, M. Verheijden, R. Eelkema, P. Jonkheijm, J. H. van Esch and L. Brunsveld, Chem. – Eur. J., 2015, 21, 18466–18473 CrossRef CAS.
- O. J. G. M. Goor, R. P. G. Bosmans, L. Brunsveld and P. Y. W. Dankers, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3607–3616 CrossRef CAS.
- L. Li, M. Liu, L. Yue, R. Wang, N. Zhang, Y. Liang, L. Zhang, L. Cheng, J. Xia and R. Wang, Anal. Chem., 2020, 92, 9322–9329 CrossRef CAS.
- M. J. Webber, E. A. Appel, B. Vinciguerra, A. B. Cortinas, L. S. Thapa, S. Jhunjhunwala, L. Isaacs, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14189–14194 CrossRef CAS.
- C. L. Maikawa, A. A. A. Smith, L. Zou, C. M. Meis, J. L. Mann, M. J. Webber and E. A. Appel, Adv. Ther., 2020, 3, 1900094 CrossRef.
- C. L. Maikawa, A. A. A. Smith, L. Zou, G. A. Roth, E. C. Gale, L. M. Stapleton, S. W. Baker, J. L. Mann, A. C. Yu, S. Correa, A. K. Grosskopf, C. S. Liong, C. M. Meis, D. Chan, M. Troxell, D. M. Maahs, B. A. Buckingham, M. J. Webber and E. A. Appel, Nat. Biomed. Eng., 2020, 4, 507–517 CrossRef CAS.
- C. L. Maikawa, A. I. d’Aquino, E. T. Vuong, B. Su, L. Zou, P. C. Chen, L. T. Nguyen, A. A. A. Autzen, J. L. Mann, M. J. Webber and E. A. Appel, Biomacromolecules, 2021, 22, 3565–3573 CrossRef CAS.
- Z. S. Clauss, R. Meudom, B. Su, M. A. VandenBerg, S. S. Saini, M. J. Webber, D. H. C. Chou and J. R. Kramer, Biomacromolecules, 2023, 24, 481–488 CrossRef CAS.
- A. A. A. Smith, C. L. Maikawa, G. A. Roth and E. A. Appel, Org. Biomol. Chem., 2020, 18, 4371–4375 RSC.
- G. Li, J. Hu, H. Chen, Y.-X. Chen and Y.-M. Li, Chem. Commun., 2021, 57, 6086–6089 RSC.
| This journal is © The Royal Society of Chemistry 2024 |