
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A practical approach to quantitative analytical surface-enhanced Raman spectroscopy
Yikai
Xu
 *a,
Wafaa
Aljuhani
b,
Yingrui
Zhang
b,
Ziwei
Ye
a,
Chunchun
Li
*bc and
Steven E. J.
Bell
*b
*a,
Wafaa
Aljuhani
b,
Yingrui
Zhang
b,
Ziwei
Ye
a,
Chunchun
Li
*bc and
Steven E. J.
Bell
*b
aKey Laboratory for Advanced Materials and Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobiology and Dynamic Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, 200237, Shanghai, P. R. China. E-mail: yikaixu@ecust.edu.cn
bSchool of Chemistry and Chemical Engineering, Queen's University Belfast, BT9 5AG, Belfast, UK. E-mail: s.bell@qub.ac.uk
cInstitute of Photochemistry and Photofunctional Materials, University of Shanghai for Science and Technology, 516 Jungong Road, 200093, Shanghai, P. R. China. E-mail: cli13@qub.ac.uk
First published on 21st November 2024
Abstract
Many of the features of SERS, such as its high sensitivity, molecular specificity and speed of analysis make it attractive as an analytical technique. However, SERS currently remains a specialist technique which has not yet entered the mainstream of analytical chemistry. Therefore, this review draws out the underlying principles for analytical SERS and provides practical tips and tricks for SERS quantitation. The aim is to show the readers how to rationally design their SERS experiments to improve quantitation performance. We begin by introducing the three core components in SERS analysis: (1) the enhancing substrate material, (2) the Raman instrument and (3) the processed data that is used to establish a calibration curve. This is followed by discussion of the analytical figures of merit relevant to SERS. In the following sections each of the three essential components in SERS quantitation and how they affect the quality of the analysis are described in more detail using examples from the literature. Finally, we highlight the current challenges in applying SERS to the analysis of complex real-life samples and briefly introduce the state-of-the-art developments on multifunctional substrates, digital SERS and AI-assisted data processing, which will help SERS rise to the challenge of moving out into routine real-world analysis.
 Yikai Xu | Dr Yikai Xu is a tenure-track professor at East China University of Science and Technology. Before this he was a Leverhulme Early Career Fellow (PI) at Queen's University Belfast. Dr Xu was the recipient of the 2019 Kathleen Lonsdale Royal Irish Academy Prize for the most outstanding PhD research in chemical science in Ireland. His work has been featured in the Emerging Investigator Series by Journal of Materials Chemistry C and Analyst. His research interest lies in surface chemistry, SERS, and the bottom-up synthesis of colloidal nanomaterials. |
 Yingrui Zhang | Dr Yingrui Zhang obtained her PhD in Chemistry and Chemical Engineering from Queen's University Belfast in 2023. She is currently a postdoctoral Researcher under the supervision of Professor Steven Bell at Queen's University Belfast. Her research specializes in the development of surface-accessible nanomaterials, including core–shell nanoparticles, plasmonic 2D films, Pickering emulsions, and 3D colloidosomes, and focuses on their innovative applications in surface-enhanced Raman spectroscopy. |
 Ziwei Ye | Dr Ziwei Ye received his Phd in Chemistry at Queen's University Belfast. After that he joined East China University of Science and Technology as a postdoctoral fellow. He was selected into the Shanghai Post-doctoral Excellence Program in 2021 and the Shanghai Pujiang Talent Program in 2023. He is now a tenure-track associate professor at East China University of Science and Technology. His research interests are in the synthesis of plasmonic/semiconductor nanomaterials and their applications in (photo)catalysis and surface-enhanced Raman spectroscopy. |
 Chunchun Li | Dr Chunchun Li is an associate professor at the University of Shanghai for Science and Technology and an honorary lecture at Queen's University Belfast. She obtained her PhD and performed postdoctoral research at Queen's University Belfast in Prof Steven Bell's research group. Dr Li was the recipient of the 2023 Kathleen Lonsdale Royal Irish Academy Prize for the most outstanding PhD research in chemical science in Ireland. Her research focuses on understanding molecular adsorption and related phenomenon on noble metal nanomaterial surfaces via SE(R)RS for the construction of nanomaterials with tailored surface properties. |
 Steven E. J. Bell | Prof. Steven E. J. Bell received his PhD from Queen's University Belfast and worked at the Rutherford-Appleton Laboratory and the University of York before returning to QUB where he is a Chair Professor of Physical Chemistry in the School of Chemistry and Chemical Engineering. His research centres on nanomaterials and Raman spectroscopy. He was founder/director of Avalon Instruments Ltd. He is a Fellow of the RSC, a Fellow and Vice-president of the Institute of Chemistry of Ireland. He is an elected member and the Vice President for Research of the Royal Irish Academy. He is also the recipient of the 2024 RSC Theophilus Redwood Prize. |
Key learning points1. Quantitative SERS measurements depend on three core components: (1) the enhancing substrate material, (2) the Raman instrument and (3) the processed data that is used to establish the calibration.2. Aggregated Ag and Au colloids are easily accessible and provide robust performance so they are a good starting point for non-specialists. 3. Since plasmonic enhancement falls off steeply with distance, substrate-analyte interactions are critical in determining successful SERS detection of an analyte. 4. SERS quantitation is subject to numerous sources of variance associated with the instrument, enhancing substrate and sample matrix but many of these can be minimized by use of internal standards. 5. The precision of SERS measurements is often indicated by quoting the standard deviation of the signal but it is the standard deviation in the recovered concentration which is useful in assessing the precision of the analysis and can be compared to other techniques. |
1. Introduction
Surface-enhanced Raman spectroscopy (SERS) is a vibrational spectroscopic technique that exploits the plasmonic and/or chemical properties of nanomaterials to dramatically amplify the intensity of Raman scattered light from molecules present on the surface of these materials. In the time since its discovery 50 years ago,1 SERS has grown from a niche technique to one which is in the mainstream of academic research. As an extension of Raman spectroscopy, the growth of SERS was greatly enabled by the rise in the availability of Raman instruments, ranging from sophisticated microscopes to simple handheld devices.2,3 This, coupled with significant advances in nanotechnology and a deeper theoretical understanding of SERS and plasmonic effects, has enabled the development of SERS methods for detecting, identifying and quantitating chemical targets in samples ranging from bacteria4 to batteries.5Many techniques for the analysis of low concentration samples are already well established, although each has their own particular strengths and weaknesses. For example gas chromatography-mass spectrometry (GC-MS) is often used because it provides high sensitivity and has a level of molecular specificity which allows measurements to be made with an excellent level of confidence. In addition, it can be used to analyze samples containing mixtures of chemical compounds, which is particularly useful for real-life samples.6 However, GC-MS does have some important disadvantages in that it requires expensive specialist equipment, is time-consuming and is not generally considered to be field portable. In contrast, SERS has the potential to offer sensitivity and molecular specificity that matches GC-MS but in a cheaper, faster and portable fashion, which could provide unique solutions for many challenging analytical problems, such as bedside diagnostics7 and in-field forensic analysis.8
The clear potential of SERS in analytical chemistry has led to an explosion of research, and it has recently been noted that a keyword search of “SERS + Analytical” over 2013–2023 gives ca. 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hits.9 Despite this, the reality is that most quantitative SERS measurements up to this point have been carried out by SERS specialists whose primary interest is in method development, rather than using SERS for routine analytical measurements. This is often the case with any new analytical technique but with SERS the gap between the amount of research activity and the number of non-specialists who have adopted the technique is particularly notable. With this in mind, this Tutorial Review is aimed towards non-specialist users to promote SERS so that it can sit alongside more established approaches as a general-purpose analytical tool. Here, we focus particularly on SERS quantitation since this is arguably one of the most useful but also challenging aspects of SERS analysis, particularly to non-specialists who wish to adopt this technique. In order to make this Tutorial Review useful to a wide body of researchers with varying technical backgrounds and analytical problems, this paper draws out the aspects of SERS that are specifically relevant to quantitative measurements and are important, irrespective of the detailed nature of the analytical problem. We would encourage readers who are interested in obtaining further technical details to explore the very informative references provided throughout this review. In addition, there are also numerous excellent recent reviews on SERS which discuss experimental methods,10,11 state-of-the-art research trends,12,13 substrate design,14,15 fundamental theory,16,17 and portability.18,19
000 hits.9 Despite this, the reality is that most quantitative SERS measurements up to this point have been carried out by SERS specialists whose primary interest is in method development, rather than using SERS for routine analytical measurements. This is often the case with any new analytical technique but with SERS the gap between the amount of research activity and the number of non-specialists who have adopted the technique is particularly notable. With this in mind, this Tutorial Review is aimed towards non-specialist users to promote SERS so that it can sit alongside more established approaches as a general-purpose analytical tool. Here, we focus particularly on SERS quantitation since this is arguably one of the most useful but also challenging aspects of SERS analysis, particularly to non-specialists who wish to adopt this technique. In order to make this Tutorial Review useful to a wide body of researchers with varying technical backgrounds and analytical problems, this paper draws out the aspects of SERS that are specifically relevant to quantitative measurements and are important, irrespective of the detailed nature of the analytical problem. We would encourage readers who are interested in obtaining further technical details to explore the very informative references provided throughout this review. In addition, there are also numerous excellent recent reviews on SERS which discuss experimental methods,10,11 state-of-the-art research trends,12,13 substrate design,14,15 fundamental theory,16,17 and portability.18,19
As shown in Fig. 1, the most basic description of a quantitative SERS experiment is that a laser is used to irradiate an enhancing substrate material to generate enhanced Raman scattering signals of the chemical species on the substrate material at various concentrations. The SERS signal intensity of the analyte can be plotted against its concentration to obtain a calibration curve which can be then used for quantitation. As such, the essential components of a SERS quantitation experiment can be categorized as: (1) the enhancing substrate material, (2) the Raman instrument and (3) the processed data that is used to establish a calibration curve. Therefore, in this review, we start off by introducing the analytical figures of merit in SERS (Section 2). This is followed by a general introduction to the typical types of SERS enhancing substrates (Section 3), Raman instruments (Section 4) and data processing methods (Section 5) that are used in SERS measurements. Importantly, we discuss how the properties of the enhancing substrate and Raman instrument, as well as the selection of different data processing methods, affect the figures of merit in SERS quantitation. The aim is to show the readers how to rationally design their experiments to improve quantitation performance. After discussing the present state of quantitative SERS research, we move on to discuss the main research trends in literature on improving the performance of SERS in quantitative analysis of complex real-life samples (Section 6). More specifically, we introduce the development of smart multifunctional SERS sensors, digital SERS and AI-assisted data processing methods. Finally, we present an outlook (Section 7) which discusses the barriers to adoption of SERS within the wider analytical community and the areas where more research is needed. We also speculate on how the field might develop as it rises to the challenge of moving out of the research lab into routine real-world analysis.
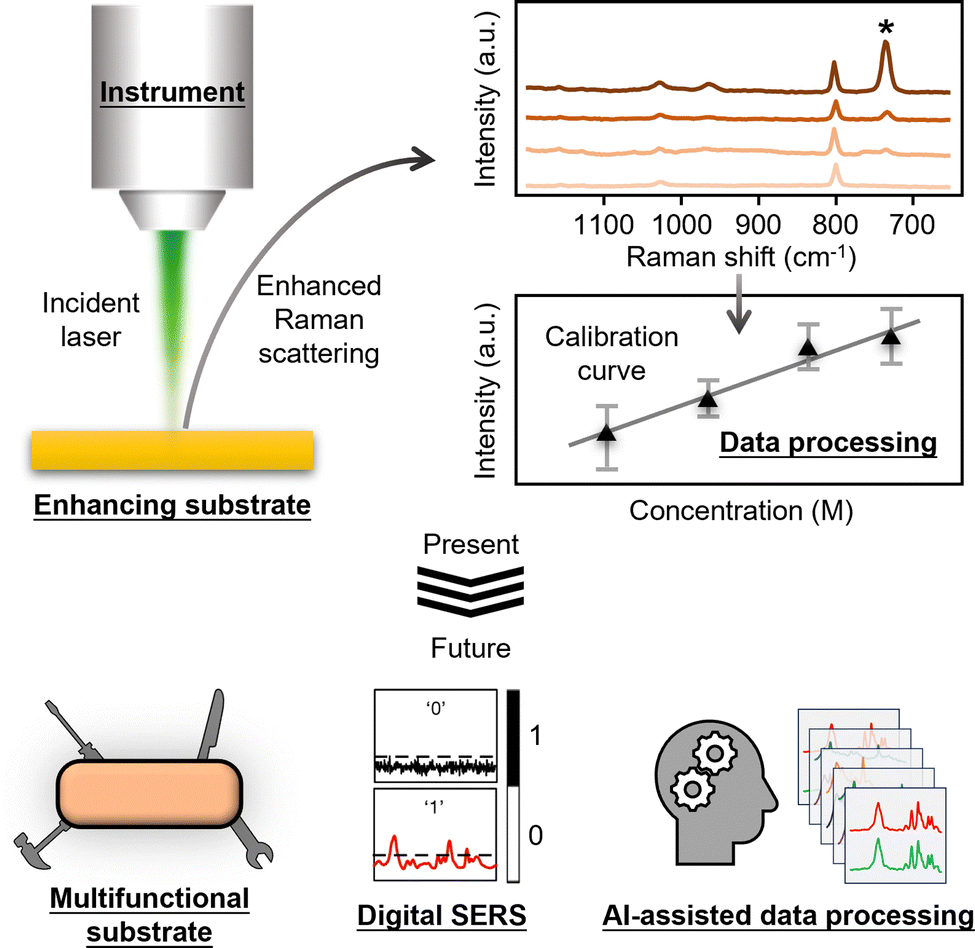 | ||
| Fig. 1 Schematic illustrations which show the Raman instrument, the enhancing substrate, the processed data as the three essential components in SERS quantitation, as well as how each component will likely develop in the near future. Adapted with permission from ref. 20, copyright Springer Nature 2024. | ||
2. Analytical figures of merit in SERS quantitation: precision, accuracy, LOQ, LOD
In quantitative SERS, the concentration (the recovered value) is typically determined from calibration plots of the SERS signal (the response) versus concentration. The signal is normally calculated as the height of a relevant band, rather than the area since this is less susceptible to interference from adjacent, partly overlapping bands. As shown in Fig. 2A, unlike techniques such as HPLC, where linear calibrations of peak area against concentration are expected, in SERS the fact that there are a finite number of enhancing sites within any substrate means that the signal will reach a limit as the surface becomes saturated. This leads to calibration curves that rise approximately linearly at low concentration but then start to plateau at higher concentrations (Fig. 2B). In simple cases, where the adsorption of the analyte is ideal, the response will follow a Langmuir model but other isotherms may be used to include interactions between analytes or other effects. In quantitative SERS analyses, a common approach is to use a limited section of the calibration curve which is approximately linear over the working range selected. This region of the calibration curve is often referred to as the “quantitation range”, as shown in Fig. 2C. The precision of the SERS measurement is typically expressed by calculating the relative standard deviation, or “RSD” in short, of the SERS signal intensity for multiple repeated experiments, while the linearity of the calibration curve, which is an indicator of the accuracy that the SERS measurement will provide, is typically expressed through the “R2” value.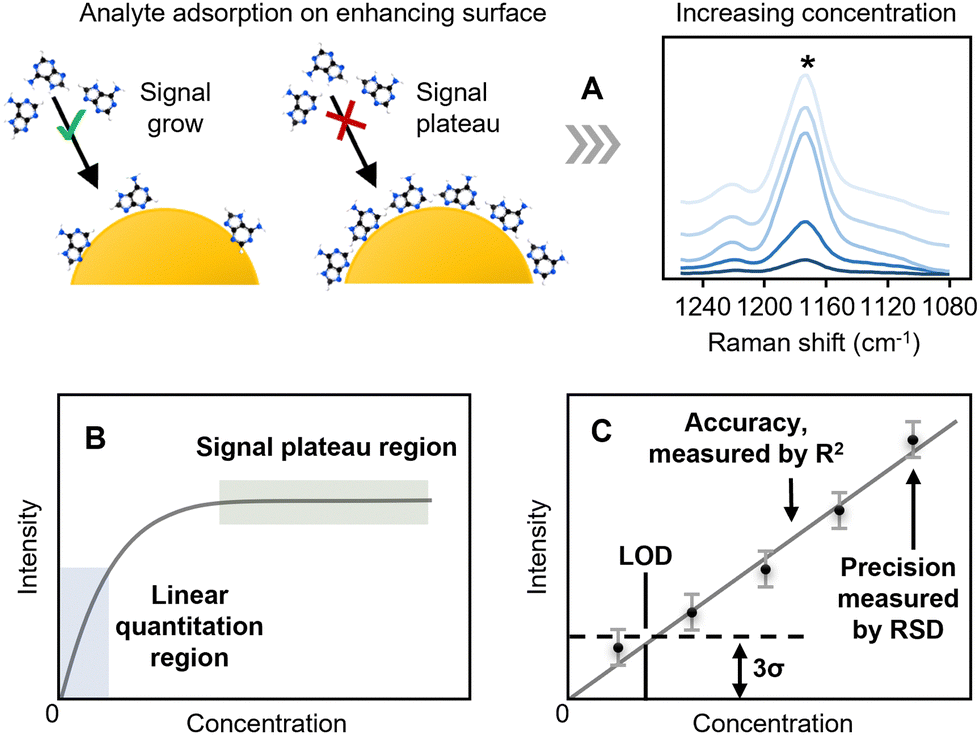 | ||
| Fig. 2 The key analytical figures of merit in SERS quantitation. (A) Schematic illustrations of the adsorption of analyte molecules on the enhancing substrate surface and the corresponding change in the SERS signal of the analyte observed. (B) and (C) Schematic illustration of a typical calibration curve between analyte concentration and signal intensity from SERS analysis. (C) Schematic illustration of the linear region of the calibration curve. | ||
Two other important analytical figures of merit in SERS quantitation are the “Limit of Quantitation” and the “Limit of Detection”, or “LOQ” and “LOD”. As illustrated in Fig. 2C, the LOD is formally defined as the concentration where the signal is calculated to be 3× the standard deviation of the blank (3σ). The LOQ is the lowest analyte concentration in the quantitation range, defined as the concentration where the signal is 10σ. However, it is also common in the literature to see the lowest concentration which gives a SERS spectrum with discernible sample peaks described as the LOD. It is useful to note here that the extremely large fluctuations in SERS signals observed at low analyte concentrations, due to analyte molecules randomly entering plasmonic hot-spots, mean that detectable SERS signals may occasionally be detected even when the sample concentration is below the formal LOD. For example, in Fig. 2C the lowest concentration point will, on average, give a signal which is undetectable because it is lower than the noise level set by the LOD but the random fluctuations in the signal intensity may mean that in some rare measurements it may nevertheless be larger than the noise and therefore detectable. This effect has been exploited with the recent development of digital SERS, which is discussed in Section 6.
In summary, this section discussed the key figures of merit that can be used to determine the success of SERS quantitation. This leads naturally to the following sections, where we will discuss the typical enhancing substrates, Raman instruments and data processing methods that are presently used in quantitative SERS measurements, as well as how these affect the key figures of merit in SERS quantitation.
3. Enhancing substrates for SERS quantitation: standard substrates and design principles
Arguably, the most important component in SERS is the enhancing substrate, which is most commonly a plasmonic Ag and Au nanomaterial. At the simplest level, these Ag and Au nanomaterials can be regarded as providing SERS enhancement through charge-transfer induced resonance effects (known as “chemical enhancement”) and near-field electromagnetic effects (known as “electromagnetic enhancement”). These effects are not discussed in detail here since they have already been covered in many excellent reviews.17,21 The crucial role which enhancing substrates play in SERS means that they have been extensively studied and, as a result, one challenge facing a new potential user of SERS is the bewildering variety of minutely-differentiated approaches/substrates which have been reported. In this regard, the area has become extremely fragmented with numerous solutions proposed to meet the challenges of achieving the required sensitivity, reproducibility, selectivity, stability of the substrates etc. This is particularly apparent in the reporting of novel enhancing substrates, which often claim without rigorous justification that they are an improvement on existing SERS approaches because they give higher sensitivity and/or reproducibility for a particular analyte molecule. Unfortunately, this can lead to confusion, not least because of the lack of a widely agreed standard method for characterizing the performance of SERS enhancing substrates.11 Therefore, in this section we discuss the current classes of SERS substrates available, and introduce the use of Ag and Au colloids as a good starting point for non-specialists who wish to experiment with SERS quantitation. Importantly, we discuss how the plasmonic properties and surface chemistry of the enhancing substrate affect the analytical figures of merit in SERS quantitation, which can serve as the rationale for customizing the enhancing substrate to achieve optimal performance for a given sample with its own specific properties. Particular emphasis is placed on discussing the use of internal standards, since this is the most straightforward way of obtaining robust quantitative SERS measurements.3.1. Typical types of SERS substrates: liquid media, solid chips
The current generation of SERS substrates can be generally categorized into liquid- and solid-nanostructured materials. As shown in Fig. 3A, liquid-based enhancing substrates are typically colloidal nanoparticles and their assemblies. The technology for preparing colloidal nanoparticles has developed enormously in the past years, so that particles with complex morphologies and nanostructures are now accessible through bottom-up chemical synthesis that can be carried out in standard research laboratories without the need for specialised equipment.22,23 In addition, plasmonic colloidal nanoparticles including Au nanospheres, Ag nanocubes and Au nanorods are also readily available from numerous commercial sources, such as BBI Solutions and Beijing Zhongkeleiming Technology. These colloidal particles have been used as the enhancing media for SERS quantitation of a very diverse set of target analytes, ranging from cells to pesticides.24,25 In addition, colloidal nanoparticles have also been used as plasmonic building-blocks for the construction of liquid-based nanoparticle-assemblies ranging from dimers and trimers26,27 to 2-dimensional arrays28,29 and colloidosomes,30,31 which typically provide improved plasmonic enhancement in SERS quantitation. | ||
| Fig. 3 The typical types of plasmonic SERS substrates. These are liquid substrates which include (A) colloidal Ag and Au nanoparticles and (B) nanoparticle assemblies; solid substrates which include (C) chips carrying plasmonic nanostructures created via top-down approaches and (D) materials coated with chemically synthesized plasmonic nanomaterials. The scale bars in panels (A) and (B) (i)–(iii) correspond to 100 nm. The scale bars in panels (B) (iv) and (C) (i)–(iii) correspond to 1 μm. The scale bar in panel (C) (iv) corresponds to 50 μm. The scale bars in the insets of panel (D) correspond to 50 nm. Panels (A) (i)–(iv) were reproduced with permissions from ref. 32 (copyright 2015 American Chemical Society), ref. 33 (copyright 2010 American Chemical Society), ref. 23 (copyright 2023 American Chemical Society) and ref. 34 (copyright 2019 The Authors), respectively. Panels (B) (i)–(iv) were reproduced with permissions from ref. 35 (copyright 2017 Springer Nature), ref. 26 (copyright 2010 American Chemical Society), ref. 36 (copyright 2018 Elsevier Ltd) and ref. 37 (copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim), respectively. Panels (C) (i)–(iv) were reproduced with permissions from ref. 38 (copyright 2021 Elsevier B. V.), ref. 39 (copyright 2010 IOP Publishing Ltd), ref. 40 (copyright 2018 Elsevier B. V.), and ref. 41 (copyright 2023 Wiley-VCH GmbH), respectively. Panels (D) (i)–(iv) were reproduced from ref. 42 (copyright 2016 The Royal Society of Chemistry), ref. 43 (copyright 2019 Elsevier Ltd), ref. 44 (copyright 2022 The Royal Society of Chemistry), and ref. 45 (copyright 2014 American Chemical Society), respectively. | ||
As shown in Fig. 3B, solid enhancing materials typically come in the form of films or chips with plasmonic metal nanostructures on the surface. These materials are mostly made in-house but increasing numbers of commercial substrates are also becoming available, although Klarite,46,47 the commercial substrate most widely discussed in the literature, is no longer manufactured. In the preparation of solid SERS substrates, the metal nanostructures can be formed via bottom-up approaches, such as the deposition of pre-formed colloidal nanoparticles35,48 and in situ particle growth,49,50 or top-down approaches, such as chemical etching51,52 and lithography.40,41 Depending on the synthetic approach, a variety of support materials have been used for the fabrication of solid SERS substrates which range from paper42 and polymer43 to superhydrophobic needles53 and electrodes.54
In general, the advantage of liquid SERS substrates, particularly Ag and Au colloids, is that they are straightforward to obtain (both commercially and through in-house synthesis), are relatively inexpensive and can give large enhancements. The disadvantage of using colloidal substrates for SERS quantitation is that they are mostly limited to studying of analyte molecules which adsorb spontaneously to the metal surface. The fact that colloids are liquids also makes them less convenient to use outside the laboratory in the real-life applications that are often envisaged for SERS. In contrast, solid substrates are easy to handle and deploy in field measurements. Moreover, solid substrates can be used for a wider range of analyte molecules since the sample solution can be deposited directly onto the enhancing surface to create the substrate-target interaction that is necessary for SERS enhancement. However, the disadvantage of solid substrates is that they are much more challenging to prepare, especially with good reproducibility, which make them less accessible for non-experts. For the reasons listed above, we recommend Ag and Au colloids as a good starting point for non-specialists who can use them to carry out at least preliminary experiments without the need to decide which of the many other possible enhancing particles might be appropriate, since they are easily accessible and provide robust performance in SERS quantitation.
3.2. Ag and Au colloids for SERS quantitation: standard procedures and common pitfalls
Despite the huge variety of enhancing materials which have now been prepared and tested for SERS, the chemically-reduced Ag and Au colloids, particularly citrate-reduced55,56 and hydroxylamine-reduced colloids,57 remain the most widely applied SERS substrates. An indication of their popularity is that Lee and Meisel's original paper on the preparation of citrate-reduced silver colloids currently has ca. 4500 citations (Web of Science). The original method for preparing citrate-reduced colloids is extremely simple, and involves boiling an aqueous solution of sodium citrate and the appropriate Ag or Au salt. The colloids can be used in SERS simply by mixing them with an aqueous metal salt solution, which acts as the aggregating agent, and a solution of analyte.58 The average diameter of the product nanoparticles can be roughly controlled by altering the relative amounts of the reducing agent and/or metal salt. Of course, the crudeness of the preparation method means that the product particles are not uniform at the nanometer scale, both in morphology and size, which is often discussed as a major issue in SERS quantitation in literature. However, since bulk analytical measurements of colloidal suspensions typically involve recording signals which are averaged over large numbers of particles, the spectra obtained from different regions of even quite heterogeneous colloids may be highly reproducible.59,60 Similarly, while some batch-to-batch variation is inevitable, it has been shown that the batch-to-batch variation in SERS enhancement factors of simple colloids as low as 5–10% can be routinely achieved.58For a given colloid and sample, the main source of variation in the intensity of the SERS signals between measurements of the same sample is normally the extent of aggregation. This is an intrinsic problem, for individual quasi-spherical nanoparticles the increase in the electromagnetic field near their surface given by plasmonic resonance is small which leads to low enhancement, so they need to be aggregated to form plasmonic hot spots which give the necessary high enhancement factor.61 Typically, the particles in the as-prepared colloid are stabilized by the electrostatic repulsion created by the surface layer of charged capping ligands they carry which, in the case of citrate-reduced Ag and Au colloids for example, is mostly residual citrate. Addition of metal salts such as NaCl, KBr or MgSO4 perturbs the electrical double layer and shields the charge, allowing the particles to approach each other sufficiently closely that short range attractive van der Waals forces bring them together. The structure and surface chemistry of the nanoparticle aggregates significantly affects their performance as SERS enhancing materials. Therefore, as shown in Fig. 4A, the SERS enhancement provided by aggregated colloids changes with the nature and concentration of the salt, as well as the surface chemistry and concentration of the nanoparticles.62 However, irrespective of the details, the general pattern of the salt-induced nanoparticle aggregation process remains the same in that the particles initially come together to form small aggregates, which will continue to grow, so that they eventually become so large that they sediment out of the suspension (Fig. 4B). For SERS measurements, this means that the enhancement provided by the colloid will rise during the early stages of the aggregation process as increasing numbers of particles join to form hot spots, but it will then drop as the aggregates grow too large so that they start to precipitate out of solution.63
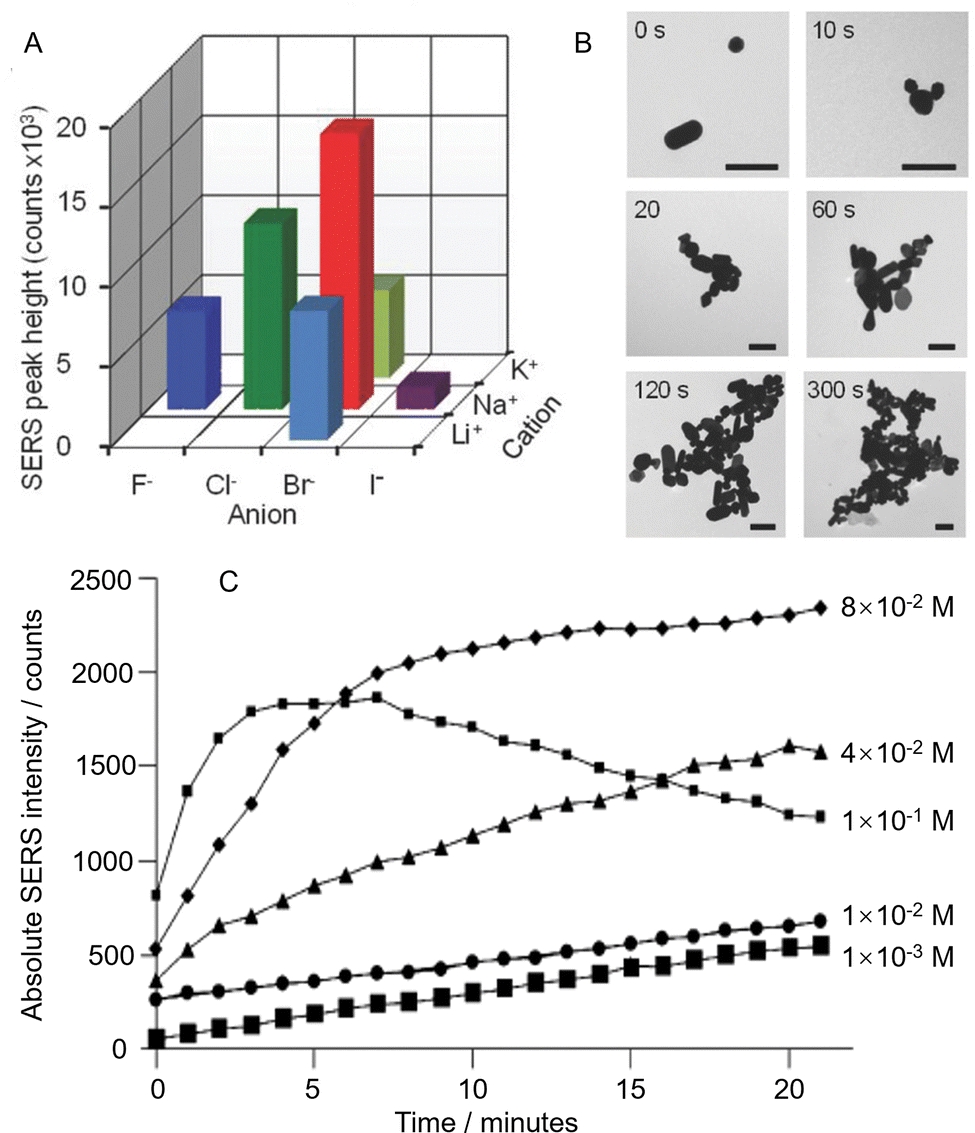 | ||
| Fig. 4 The effect of colloid aggregation on SERS enhancement. (A) Variation in SERS signal intensity of a test molecule obtained from Au colloid aggregated with the same concentration of six different salts. (B) TEM images of Au nanoparticles before aggregation and after aggregation for 10, 20, 60, 120, and 300 s with NaBr. The scale bar is 200 nm. (C) Time dependence of the SERS signal intensities of citrate obtained by aggregating citrate-reduced silver colloid with different concentrations of MgSO4. Panels (A) and (B) were reproduced with permission from ref. 62, copyright 2018 Wiley-VCH Verlag GmbH & Co. kGaA, Weinheim. Panel (C) was reproduced with permission from ref. 63, copyright 2013 American Chemical Society. | ||
The obvious result of having a dynamic aggregation process is that for consistent signal enhancement the spectra should always be recorded at the same time after aggregation is initiated, otherwise the random changes in enhancement due to variation in the extent of aggregation will decrease the precision and accuracy of SERS quantitation. The effect of this time-dependence may be minimized by choosing a salt concentration which maximizes the time window during which the signal remains relatively constant. For example, Fig. 4C shows time-dependent SERS intensity data for citrate-reduced Ag colloid aggregated with different concentrations of MgSO4, where low salt concentrations show a slow rise in the SERS signal while the highest salt concentration gives signals that rise and decay rapidly.63 From the data set, it can be seen that the optimum concentration of salt gives a near-plateau region which extends for >10 minutes, which is ample time for several SERS measurements.
It should be noted that even subtle differences in experimental conditions, such as whether the same amount of sample is added as a small volume of concentrated solution or if it is added in a more dilute form can change the dynamics of the aggregation and the absolute signal.64 Therefore, the development and adoption of a robust experimental protocol is very important when using aggregated colloids for quantitative SERS. For example, the sequence of steps where analyte and aggregating agent are added into colloidal nanoparticles followed by placing the sample in the compartment of the instrument should be practiced so that the processes always take a similar amount of time. This experimental approach is not commonly highlighted within the literature but can have a crucial effect on the reproducibility of the SERS measurements. The levels of reproducibility which can be achieved by carefully controlling experimental conditions, including the size of the nanogaps and aggregation time is very striking, for example Grys et al. have demonstrated sample-to-sample variation in intensity from aggregated colloids as low as <1% relative standard deviation (RSD),65 although this values is exceptional and under standard conditions 5–10% is more usual.66,67
3.3. Rational substrate design for improved quantitation performance: plasmonics and surface chemistry
Aggregated colloids were recommended above as being a good route for non-specialist users to start carrying out quantitative SERS measurements. This approach is particularly robust for samples which contain a single type of analyte molecule dispersed in aqueous solution. However, as the complexity of the sample and/or application increases, it might become necessary to customize the SERS enhancing substrate to achieve adequate quantitation performance. There are limitless possibilities for this, depending on the specific application and enhancing material at hand. Regardless, there are some general rules of thumb in substrate design that can be considered, which will be highlighted in this section.Overall, the main approaches to improving substrate design in SERS quantitation are fine-tuning the surface chemistry and the plasmonic properties, as shown in Fig. 5. Since the main contribution to SERS enhancement originates from the localized surface plasmon resonance (LSPR) effect, this means that the plasmonic properties of the substrate will significantly affect SERS signal intensity, and in turn the LOD, LOQ and sensitivity of the quantitation. As shown in Fig. 5A, the plasmonic properties of the enhancing material can be improved through engineering nanostructures which create strong electric field localization. More specifically, this includes the creation of nanoparticles with tips,68,69 or internal gaps,70,71 or the creation of nanoparticle assemblies which contain interparticle gaps.72,73 One challenge that comes with engineering complex nanostructures to improve plasmonic properties is that it is difficult to achieve high levels of reproducibility, particularly at the nanoscale. This results in variation in the plasmonic properties of the substrate and in turn the precision of SERS quantitation. In some cases, this issue can be largely resolved by choosing the appropriate Raman instrument, which will be discussed in the next section of this review. In other cases, it may be helpful to fine-tune the synthetic procedure to create highly mono-disperse nanoparticles74,75 and/or uniform superlattice assemblies,76,77 as shown in Fig. 5B. However, even with very regular structures, although the non-uniformity may be reduced, some residual variation (RSD ≥2–3%) is still commonly observed.
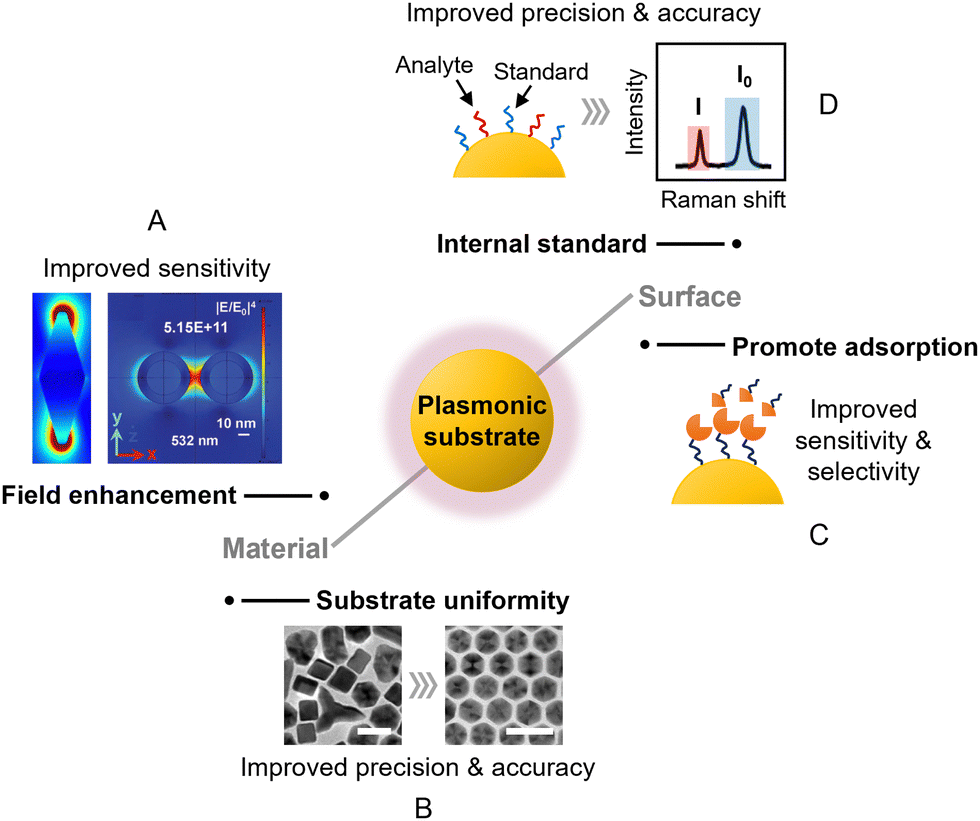 | ||
| Fig. 5 The main approaches to improving substrate design in SERS quantitation. These include (A) designing new nanostructures to improve field enhancement; (B) increasing the structural uniformity of the substrate material to improve SERS reproducibility; (C) modifying the surface of the substrate to promote analyte adsorption; (D) introducing internal standards to compensate for experimental variations. The scale bars in panel (B) correspond to 25 nm. Panel (A) was reproduced with permission from ref. 68 (copyright 2020 Elsevier Ltd) and ref. 72 (copyright 2015 the Owner Societies). Panel (B) was reproduced with permission from ref. 74, copyright 2013 American Chemical Society. | ||
The surface chemistry of the substrate material is important since it dictates the interaction between the target analyte and the enhancing surface, as shown in Fig. 5C. This is often overlooked in SERS experiments, but can be critical in determining whether an analyte gives SERS signals at all, since SERS enhancement is a short-range effect that is localized to within tens of nanometers of the enhancing surface. For example, recently we have shown that polyaromatic hydrocarbons can adsorb spontaneously to Ag and Au nanosurfaces through dispersive π–metal interactions to generate strong SERS signals, but this interaction is prohibited by surface oxidation or the presence of strongly binding ligands such as thiols on Ag/Au.78 This is one of many examples where having a clean and accessible metal surface promotes the adsorption of analyte molecules which improves the sensitivity of SERS quantitation.79,80 Conversely, for analyte molecules which do not interact strongly with Ag and Au, adsorption to the enhancing surface can be promoted by functionalizing the surface of the metal with ligands that capture the target molecules via intermolecular forces (Fig. 5C). This either allows detection of the non-adsorbing analyte directly via SERS or indirectly through systematic changes in the SERS signals of the ligand molecules.81,82 For example, metal–organic frameworks (MOFs) have been used as a functional coating to adsorb and localize volatile aromatic molecules near the enhancing surface for SERS.83 Other typical examples of functional ligands that can be used to promote the adsorption of analyte molecules include supramolecular hosts,82 self-assembled monolayers of small molecules,84 and aptamers.85
In some cases, the target may be too large to fit into the plasmonic region of the enhancing substrate for direct SERS analysis. This can be overcome via indirect SERS approaches where the plasmonic nanoparticles are functionalized with a mixture of Raman reporter molecules and modifier molecules that selectively bind to the target.86 Using these nanoparticles (often referred to as “SERS tags”) allows the presence and quantity of the targets to be indirectly analyzed via the SERS signal intensity of the Raman reporter and has been demonstrated as an effective approach for the analysis of biological targets, such as proteins87 and extracellular vesicles.88
3.4. Internal standards for improved accuracy and precision in quantitation
Besides dictating the adsorption of analytes, the surface of the enhancing substrate can also be functionalized with SERS active molecules, which act as internal standards to improve the precision of quantitation using ratiometric methods. As shown in Fig. 5D, this is typically achieved by pre-adsorbing a SERS active component to the enhancing substrate, which allows the SERS intensity of the target molecule (I) to be normalized against the internal standard molecule (I0). This provides a standard which tracks the enhancement provided by the substrate and can therefore compensate for variations in intensity caused by changes in the experimental conditions between measurements. Commonly used internal standard molecules are strongly adsorbing/binding compounds that generate strong Raman scattering, such as dyes,89 thiols90 and aromatic amines.91 The extent to which such pre-adsorbed standards can improve robustness by correcting for both random variations in the enhancing medium and changes arising from instrumental factors was demonstrated by Chen et al. who prepared two-dimensional close-packed films of Ag nanoparticles with an octadecanethiolate internal standard surface layer with good signal uniformity (RSD = 4.3%, see Fig. 6A).90 With this substrate, using crystal violet embedded in a spin-on glass layer as the target (Fig. 6B), normalisation of the crystal violet signals to the signals of the internal standard gave a linear calibration plot with R2 > 0.999 with samples containing from 5–5000 molecules per μm2. Notably, to demonstrate the effectiveness of the internal standard in the calibration of experimental errors, the authors established the calibration plot mentioned above deliberately using data obtained from two different Raman instruments (Fig. 6C).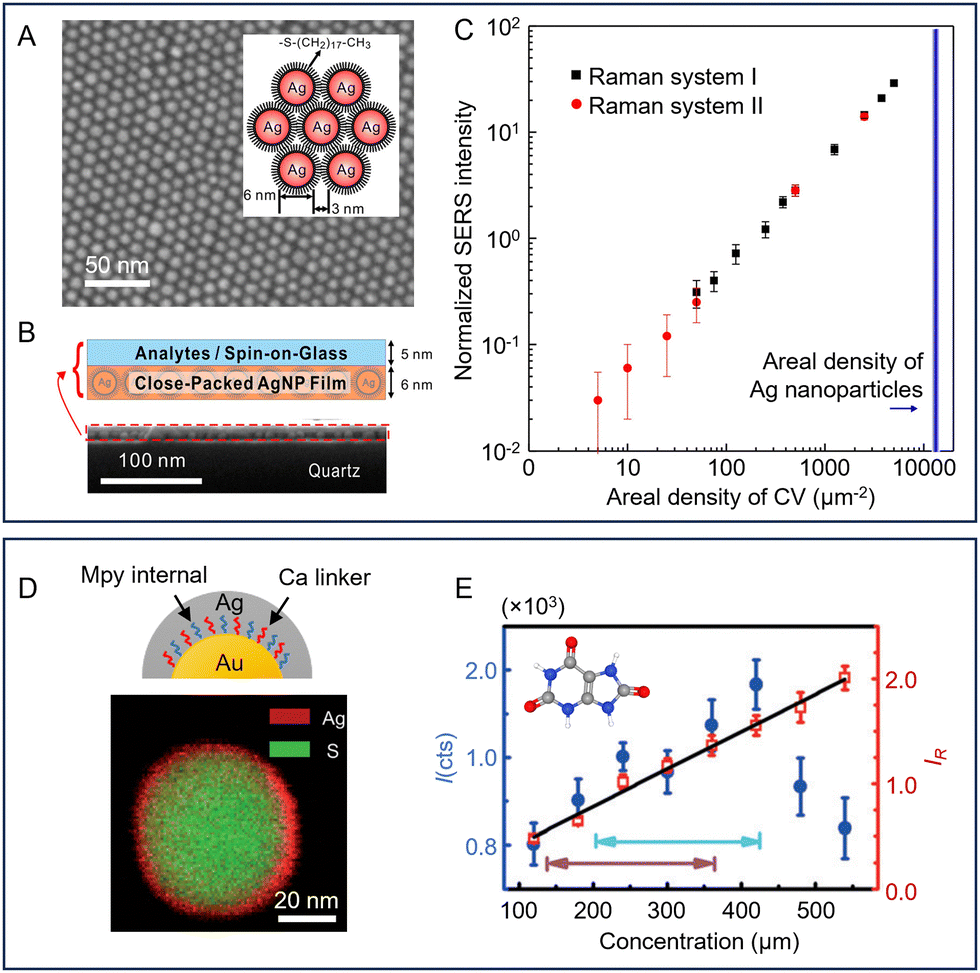 | ||
| Fig. 6 Examples of pre-adsorbed internal standard molecules used to improve the reproducibility of SERS quantitation. (A) SEM image of a close-packed monolayer AgNP film on quartz. Inset shows a schematic diagram of the octadecanethiolate-covered NPs. (B) Diagram of the structure of the SERS substrate and the analyte layer. Lower SEM image shows a cross section of the substrate and sample. (C) Log/log calibration plot of crystal violet SERS intensity normalized to thiolate signal against areal density. The black and red points were obtained using different Raman spectrometers. (D) Schematic diagram of Au@Ca + Mpy@Ag NPs (Ca = cysteamine, Mpy = 4-mercaptopyridine). STEM image showing the composition of a single particle. (E) Calibration plots for detection of uric acid. The normal human concentration ranges are marked with turquoise (male) and blue (female) arrows. Panels (A)–(C) were reproduced with permission from ref. 90, copyright 2015 American Chemical Society. Panels (D) and (E) were reproduced with permission from ref. 92, copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
One potential issue with directly adsorbed internal standards is that they may interfere with target binding. This can be addressed by including the SERS-active component as a constituent of the enhancing material itself.93,94 For example, Lin et al. prepared sensors composed of Au core particles coated with a layer of 4-mercaptobenzoic acid and with an outer Ag shell where the 1073 cm−1 band of 4-mercaptobenzoic acid could be used as an internal standard.95 These could then be used to construct paper-based sensors. Importantly, it was shown that this approach corrected for physical changes in the experiment, since the intensities of 1617 cm−1 band from the crystal violet test analyte and the 1073 cm−1 internal standard band both changed in the same way when the focus position was changed, so their relative intensity remained fixed within the overall 3.7% RSD even when the focus was changed so that absolute signal level fell by a factor of 4×. Similarly, non-uniformity in the distribution of nanoparticles on the paper support was reduced by normalising the intensity of the crystal violet analyte band to the standard at each point.
This approach has been extended to one where the intermediate layer is composed of two types of molecules, one which acts as the internal standard and one which acts as a linker to the outer shell. For example, as shown in Fig. 6D, Shen et al. prepared core/shell particles with gold nanosphere cores surrounded by a mixed layer of cysteamine with 4-mercaptopyridine as the internal standard and an Ag outer shell.92 In this case, since the internal standard molecules were encapsulated within the nanoparticles, the surface of the particles was accessible to a wider range of analyte molecules. With these particles a plot of the SERS intensities of uric acid over the normal human concentration range showed significant non-linearity in absolute intensity but improved to a good linear response once the signal of uric acid was normalized against the 4-mercaptopyridine internal standard (Fig. 6E).
The disadvantage of using pre-adsorbed SERS internal standards is that while they can compensate for non-uniform distribution of the plasmonic hot-spots of the enhancing material in the probe volume and other physical variations in experimental conditions, they cannot be used to correct for changes in the way which the target interacts with the surface. This means that subtle changes to the surface chemistry of the SERS substrate between the calibration process and the analytical measurement can perturb the relative intensities of the target and standard and therefore the apparent concentration i.e. accuracy of the quantitation. This is a particularly challenging problem since the fact that the surface has changed may not be obvious from the SERS spectra. These changes in surface chemistry can arise during storage due to oxidation of the surface (particularly for dry Ag substrates with exposed surfaces)96,97 or more subtle changes, such as those associated with surface restructuring.98 In addition, changes in the sample matrix, which are often encountered in biological samples for example, can perturb the adsorption of the target onto the surface by introducing compounds which bind competitively with the surface.99
This issue can be resolved by adding a known amount of an internal standard compound, which has chemical properties similar to those of the target molecules, to co-adsorb with the target molecules during SERS quantitation. In this case, any change in the enhancing medium will affect the adsorption and Raman scattering signals of both the target molecules and the standard in a similar way.100 This has the advantage over pre-adsorbed standards that it can compensate for not only variations in physical parameters but also any changes in chemical properties of the substrate. In this regard, the ultimate internal standards are isotopologues of the analyte because both the target and standard will respond in essentially identical ways to any perturbation in the experimental conditions.101,102 This has been demonstrated by Behabib et al. in a study of amines that are important in refinery processes which used d3- and d4-monoethanolamine (MEA-d4) as internal standards for the SERS quantification of MEA.103 As shown in Fig. 7A, the test spectra were scaled to the characteristic MEA-d4 band at 870 cm−1 which gave excellent results for both standard laboratory solutions and real-world refinery process water samples. Notably, the distribution in the results of SERS measurements of a standard MEA solution was reported for data which were collected over 4.5 years on 25 different Raman spectrometers (Fig. 7B). In addition, it was reported that the calibration developed with the isotopically substituted standards was extremely robust and was used over a period of 7 years with thousands of samples and hundreds of nanoparticle batches without adjustment or recalibration. This data, which was validated using ion chromatography, is shown in Fig. 7C.
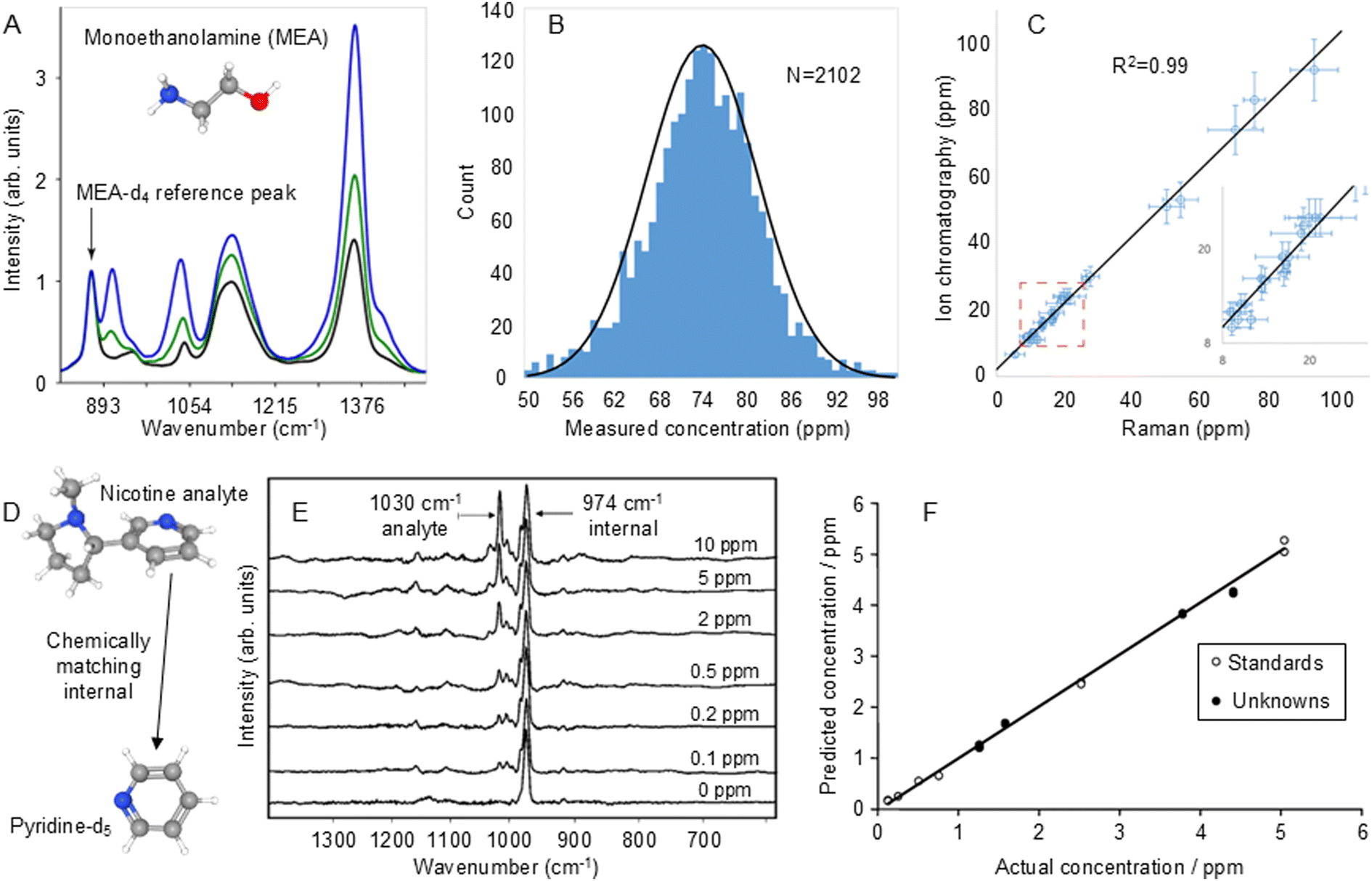 | ||
| Fig. 7 Examples of isotopologue internal standards used to improve the reproducibility of SERS quantitation. (A) SERS spectra of 50 ppm MEA-d4 internal standard with 0 (black), 25 ppm (green) and 75 ppm (blue) of the MEA-d0 target analyte. (B) The distribution in the results of SERS measurements of 75 ppm laboratory standard MEA solutions that were collected over 4.5 years on 25 different Raman spectrometers. The RSD is 10.3%. (C) Comparison of SERS measurement of MEA in refinery process water samples with ion chromatography. The error bars on the Raman axis are the 95% confidence interval on the measured value. The error bars on the ion chromatography axis are 10%. The average relative error above 20 ppm is 9%. Inset shows an expanded view of the data at the low end of the concentration range. (D) Chemical structure of the nicotine analyte and deuterated pyridine internal standard. (E) SERS spectra of samples containing d5-pyridine internal standard (974 cm−1) and 0–10 ppm nicotine (1030 cm−1). (F) Calibration plot of predicted vs actual concentration of nicotine obtained from spectra normalised to the internal standard band. Open circles show data used to build the PLS-1 calibration model and closed circles are blind unknowns. Panels (A)–(C) were reproduced with permission from ref. 103, copyright 2023 American Chemical Society. Panels (E) and (F) were reproduced with permission from ref. 104, copyright 2004 The Royal Society of Chemistry. | ||
The main difficulty in using isotopologues of the target molecules as internal standards is obtaining suitable isotopologues and their high cost. However, if isotopologues are not available, standards which chemically match the target and therefore respond in the same way to perturbations may be used instead. One problem for selection of suitable chemically-matched standards is that the chemical similarity of target and standard may also lead to similarities in the spectra and in particular, overlap in the target and standard bands. For example, this approach was used in SERS quantitation of nicotine where pyridine was chosen as a chemically-matched standard, since it would be expected to bind to the enhancing surface through its aromatic amine group in the same way as nicotine.104 However, the strongest pyridine band overlapped that of the nicotine target at 1030 cm−1. This problem was resolved by switching to d5-pyridine, whose strongest band is shifted to 974 cm−1 but is easier to obtain than isotopically substituted nicotine (Fig. 7D and E). This process gave excellent analytical results, the calibration plot of actual vs. predicted concentrations obtained by PLS-1 analysis of linearised data had R2 ∼ 0.998 over the concentration range 1–10 ppm (Fig. 7F). A blind test of the method with unknown samples gave results where the root mean square (RMS) error in the concentration was 0.10 ppm. Interestingly, a subsequent experiment of quantification of nicotine in e-liquids with d4-nicotine as the standard, gave spectra which looked very similar to those with d5-pyridine but the linear calibration was even better, with R2 = 0.9996.105
Overall, if internal standards are required, the choice is typically between the experimental convenience of having pre-adsorbed, strongly bound standards, which are inexpensive and applicable across a wide range of analytes, and chemically-matched/isotopically substituted standards, which are less flexible but make the calibrations more robust. With samples in well-controlled and understood matrices, the possibility of random perturbations to binding caused by the matrix is low and the condition of the enhancing materials can be checked before the measurement, so pre-added standards may be sufficient to overcome the effects of varying focus or amounts of substrate in the probe volume. With uncontrolled real-life samples, such as potentially contaminated water, soils, biofluids etc., the risk of matrix interference is larger. One possibility of dealing with this is to use some form of sample pretreatment, such as LC-SERS,106,107 to reduce matrix interference. However, since there is no easy method of detecting if the sample pretreatment has been entirely effective, confidence in the result may be set by how much trust can be placed in the pre-treatment method, rather than the SERS measurement. Conversely, using isotopologues or chemically-matched standards, which can cope with these effects, can give a high level of robustness and assurance that the result is accurate, despite any possible changes in the sample matrix and this may be the level of confidence that is required for quantitative measurements of critical analytes. Indeed, it is notable that methods which use this approach underpin some of the few commercially-available SERS sensors intended for routine quantitative analysis of real-life targets by non-specialists.103,108
4. Raman instrument selection for SERS quantitation: instrument selection and instrument parameters
One aspect of SERS which is often not discussed in detail is the effect of the instrumentation on the overall success of the analytical measurements which are being reported, presumably because modern instruments have evolved to the extent that the commercial systems which most researchers now use give at least adequate performance. However, the fact that the performance of a given substrate in SERS quantitation can be significantly improved by selecting the appropriate Raman instrument and experimental parameters should not be ignored. For example, specific laser wavelengths can be used to couple with the LSPR of the enhancing substrate to significantly boost its plasmonic response109 while large laser spot sizes can be used to improve signal uniformity by averaging out the nonuniformity of the plasmonic nanostructures.110 Therefore, this section discusses the selection of Raman instruments and Raman instrument parameters as well as their effect on different analytical figures of merit used in SERS quantitation. The aim is to provide general guidelines which readers can use in determining the optimal parameters for different applications.4.1. Selection of Raman instrument for improved quantitation
In general, commercial Raman spectrometers can be broadly divided into large instruments where excitation and collection are achieved through optical microscopes and “macro” systems which have poorer spatial resolution but can be portable or use fibre probes. The Raman signals acquired from the same sample can vary dramatically in signal-to-noise (S/N) ratio, spectral background and spectral range depending on the instrument used. For example, a state-of-the-art confocal Raman microscope which efficiently collects a large fraction of the Raman scattered light and is equipped with a high throughput spectrometer and cooled detector to reduce thermal noise will give spectra with significantly higher S/N ratios for the same sample and laser power than a compact portable Raman spectrometer with an uncooled detector. This is one of the reasons to why it is difficult to assess the relative performance of enhancing substrates from different groups by simply comparing the LOD of a standard test molecule, since the detection limit can be pushed lower, even for the same enhancing substrate, by using a spectrometer which gives better S/N. Overall, since the SERS signal is determined by the combination of the SERS scattering and the instrument used to detect it, the best quantitative analysis will be obtained with research grade instruments. However, if a substrate which gives high SERS scattering with the target analytes can be found this may compensate for the lower performance of compact or low-cost Raman instruments, which could be useful for in-field applications.4.2. Selection of Raman instrument parameters for improved quantitation performance
It is important to note that even with the same Raman instrument and sample, different instrument parameters including laser power, laser wavelength, accumulation time, spot size and probe depth can all significantly affect the success of quantitative SERS measurements. Here, we focus our discussions on benchtop microscope systems, not only because they are widely used in current SERS research, but also because they provide more options for varying instrument parameters than compact low-cost systems. The first instrument parameter that is typically set at the start of the SERS measurement is the wavelength of the laser, with the most common being 532, 633 and 785 nm. While many samples and substrates will give adequate signals using any of the three laser wavelengths, the best signal intensity is typically achieved when the frequency of the incident laser matches the energy of an electronic transition in the target molecule and/or the resonant frequency of the LSPR of the enhancing material, which can bring about additional enhancement in signal intensity, and in turn improvement in the LOD and LOQ by several orders of magnitude.111During SERS measurements, the most obviously adjustable parameters are the laser power and accumulation time. Since the S/N ratio increases as the square root of the accumulation time, increasing the accumulation time from 1 s to 16 s will give a 4× improvement in S/N and therefore a 4× improvement in the LOD, even if all other factors are held constant. Similarly, increasing the power of the probe laser will obviously lead to a corresponding increase in the Raman scattering signal and therefore an improved LOD as the signal at any concentration will grow while the noise remains constant.
Irrespective of any differences in performance associated with thermal noise and throughput, there are more fundamental differences between highly confocal Raman microscopes and macroscopic Raman spectrometers with their larger spot sizes and depth of focus, which are particularly important in SERS quantitation. While it might appear that microscopes with high numerical aperture objectives and small spot size would be superior for SERS measurements, this is only partly correct. It is true for imaging experiments or if measurements need to focus on specific areas of the sample, such as coffee ring deposits of analyte crystals on solid enhancing substrates. However, if the sample is non-uniform due to spatial variations in the plasmonic properties of the enhancing material or differences in the distribution of the analyte on the substrate, larger spot sizes may be preferable for improving the precision of the quantitation, since they can average over variations in signal intensity. This is important since point-to-point variations in EF are a common feature of solid substrates; in a recently published list of values for 49 substrates the point-to-point variation was shown to be 5–15% RSD.65 For spatial averaging, the important parameter is the size of the sampled area compared to the length scale of the non-uniformity. For example, Fig. 8A shows a substrate composed of nanoparticle aggregates dispersed in a sprayed polymer film on a paper support.112 This substrate is clearly extremely heterogeneous on the sub-micrometer scale but with a laser probe spot diameter of 60 μm the signals of numerous aggregates are included in each measurement, so the point-to-point signal variation is reduced to 19.1%, which allowed SERS quantitation of thiram to be performed with adequate reproducibility (Fig. 8B and C).
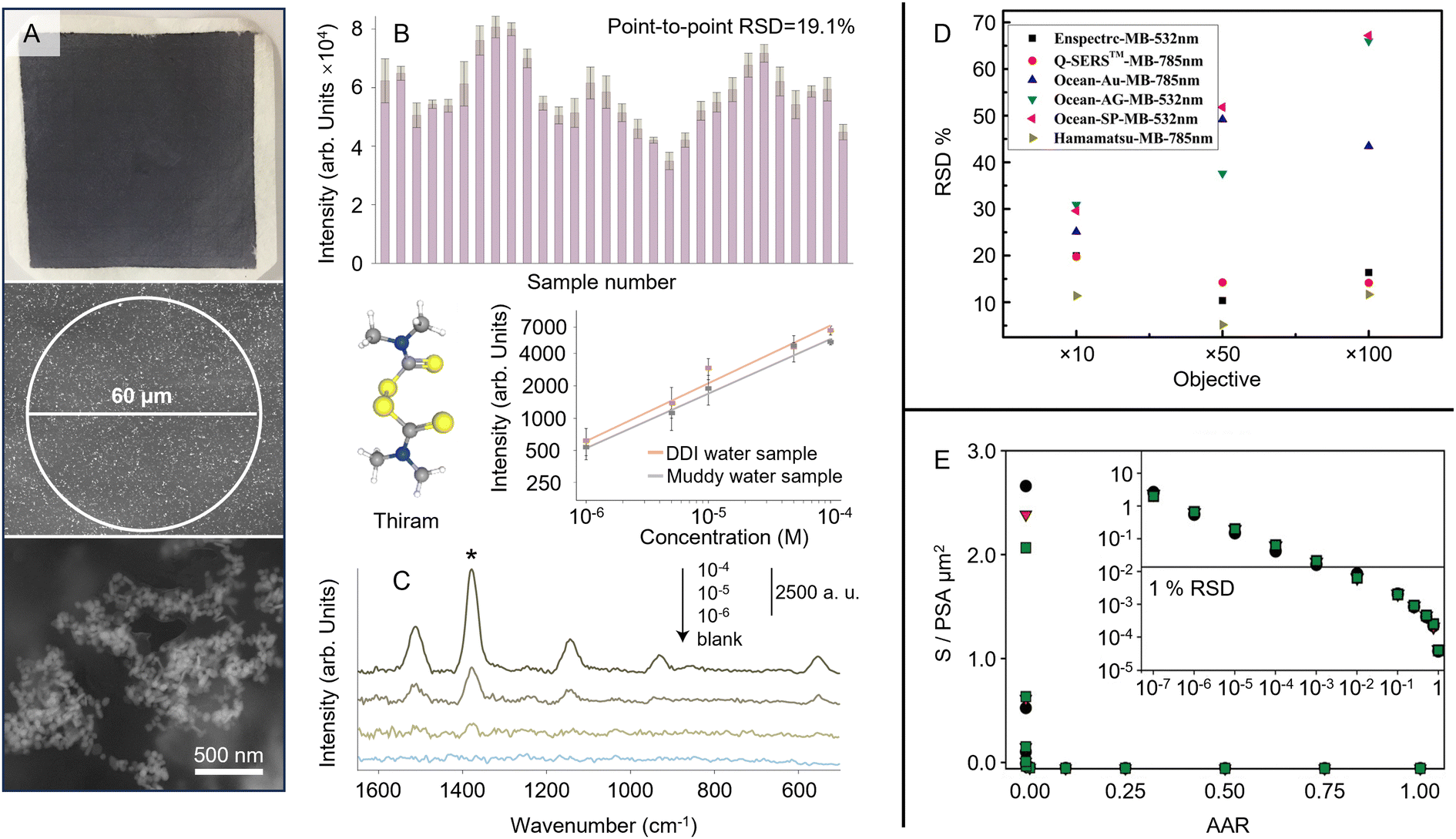 | ||
| Fig. 8 Demonstration of the effect of probe area on the reproducibility of SERS quantitation. (A) Optical and scanning electron microscopy image of the substrate. The diameter of the Raman laser spot is 60 μm. (B) The SERS signal intensity of crystal violet obtained on 30 random points on a typical sample. (C) The SERS spectra of several solutions of thiram contaminated with sand. The inset shows Semi-log plot of the SERS intensity of different concentrations of aqueous thiram solutions with (grey) and without (orange) sand contamination. Data points are an average over 5 measurements and error bars represent ±1 SD. (D) Illustration of the effect of laser spot size on the RSD of SERS measurements taken with different substrates. (E) Results of Monte Carlo simulations aimed at predicting the effect of sampling on the precision (RSD) of SERS measurements of labelled antibodies. Panels (A)–(C) were reproduced with permission from ref. 112, copyright 2021 The Royal Society of Chemistry. Panel (D) was reproduced with permission from ref. 113, copyright 2020 Springer. Panel (E) was reproduced with permission from ref. 114, copyright 2016 American Chemical Society. | ||
A more systematic study of the effect of the laser spot size of Raman microscopes on the RSD of the SERS measurement was presented by Liu et al.113 In their experiments, they changed the spot size of the probe laser by altering the numerical aperture of the objective lens used in the Raman microscope. Fig. 8D shows data obtained for several different commercial substrates with 10×, 50× and 100× objectives (numerical aperture = 0.25, 0.75 and 0.9, respectively). The data show that the change in spot diameter with numerical aperture had a significant effect on the RSD of the SERS measurements, and led to the RSD of the signal increasing from 30% to 70% in the most dramatic case. The issue of sampling error has been treated in detail by Crawford et al. in the context of quantitative SERS immunoassays.114Fig. 8E shows the results of Monte Carlo simulations aimed at predicting the effect of sampling on the precision of SERS measurements of labelled antibodies. These simulations allowed the RSD of the measurement of a random distribution of point-sized adsorbates (PSAs) to be predicted under various conditions. The plot in the inset shows how the RSD is predicted to change as a function of the area analysis ratio (AAR), which is the proportion of the sample probed in each experiment. Under the conditions chosen for the simulation, sampling errors rose above 1% when the probed area was decreased to the point that the AAR dropped below 10−3.
For instruments which do not offer the option to increase probe area, multipoint sampling can be used, since the uncertainty in the measured signal intensity will decrease as the square root of the number of points sampled (assuming the intensities are normally distributed). Alternatively, the laser beam may be rastered over the surface of a stationary sample, an option which is available in some commercial systems (Orbital Raster Scan (ORS™), Metrohm Inc.). The important advantage of probing over a larger area of the sample is that it can reduce the sampling error associated with point-to-point non-uniformity of the substrate or non-uniform analyte distribution even more effectively than increasing the laser spot size. This is common in quantitative Raman measurements, where samples which are heterogeneous on length scales larger than the probe beam diameter have been studied using rastered beams,115 multipoint sampling110,116 or large beam diameters, up to 6 mm.105,117 Indeed, for many SERS applications, it may be more efficient to use or develop sampling methods that increase the sampled area rather than concentrating on attempts to further improve the uniformity of the enhancing substrate.
4.3. Effect of instrument parameters on sample integrity
Finally, it is important to note that the appropriate choice of instrument parameters is also crucial for preserving the integrity of the sample, particularly for organic and biological samples which are more susceptible to laser damage. Obviously, increasing the laser power and length of accumulation will increase the chance of damaging the sample. In addition, decreasing the spot size also increases the possibility of sample degradation due to photochemical effects or simple sample heating due to high laser power/unit area.118,119 For example, Zeng et al. used photothermal heterodyne imaging (PHI) combined with SERS and scanning electron microscopy (SEM) to study local heating of nanoparticle aggregates used for SERS.118Fig. 9A and B show bubble formation due to photothermal heating of Au NP aggregates supported on an indium tin oxide (ITO) support covered in ethanol. Heating for just 5 s with 1.8 mW of 532 nm laser irradiation was sufficient to cause bubble formation, which was unsurprising since simulations of the system predicted that the aggregates could reach a maximum temperature of 390 °C. Apart from causing disruption to the sample during analysis, photothermal effects can also lead to degradation of organic materials on the surface of the SERS substrates. As shown in Fig. 9C, this can often be detected by the appearance of a broad doublet of bands centered at ca. 1200 and 1600 cm−1, which can be attributed to the D and G bands of amorphous/graphitic carbon.120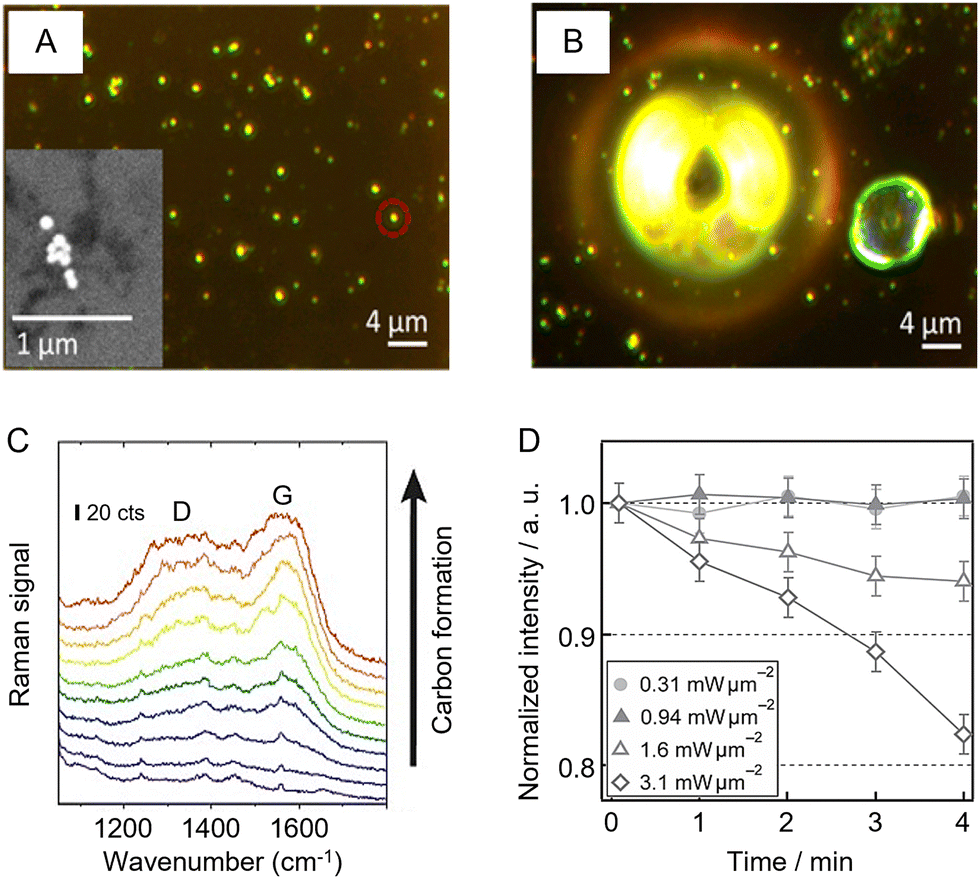 | ||
| Fig. 9 Examples of photothermal effects in SERS analysis. (A) Dark field image of Au NP aggregates in ethanol. Inset shows the scanning microscopy image of a typical Au aggregate in the sample. (B) Bubble formation following 5 s heating with 1.8 mW of 532 nm laser which simulations predict would give a maximum temperature of 390 °C. (C) SERS signals of consecutive scans over an area of 170 × 190 μm2 with 60 nm silver dimers, demonstrating the gradual increase of the amorphous carbon signal with characteristic D and G bands. (D) Monitoring laser heating of a SERS substrate using thermal desorption of thiophenol from a Au island film. Panels (A) and (B) were reproduced with permission from ref. 118, copyright 2017 American Chemical Society. Panel (C) was reproduced with permission from ref. 120, copyright 2019 The Authors. Panel (D) was reproduced with permission from ref. 119, copyright 2016 The Chemical Society of Japan. | ||
Obviously, any sample degradation will reduce the accuracy of quantitative measurements and needs to be avoided if possible. Indeed, the need to reduce the laser power so that it lies below the damage threshold may partly cancel out the advantages of high collection efficiency given by high numerical aperture objective lenses in microscope-based systems. Apart from increasing the size of the probe area, another way to reduce localized heating is to perform the SERS measurements in aqueous solution so that the water acts as a heat sink. For example, Mochizuki et al. have used thermal desorption of thiols to track temperature increases in SERS substrates experimentally (Fig. 9D) and showed that the effect of plasmonic heating common in dry substrates is mitigated to some extent in wet samples due to the higher thermal conductivity around the excited region.119
The examples above illustrate how selection of the Raman instrument and instrument parameters can significantly impact the performance of SERS quantitation. In particular, we highlight the effect of sampling area on SERS reproducibility which is often overlooked in literature. More specifically, sampling over larger areas of a given substrate can significantly increase the precision in SERS quantitation. This would suggest that macro systems with large spot sizes would be preferable to use of microscope-based instruments (particularly since they may also reduce sample damage) but there is a trade-off because microscopes typically have much more efficient collection of the Raman scattered light. This, coupled with the high-quality detectors such research instruments are normally fitted with, means that the best compromise will vary with the sample and substrate being used for the application. With very heterogeneous substrates the averaging given by macro instruments will be most important, while for weak signals or studies aimed at detecting signals arising from small volumes, such as single molecule studies, microscope-based systems will be a better choice. In addition to the probe area, a variety of other Raman instrument parameters can also be tuned to improve the performance of SERS quantitation. These parameters should be tested and set at the beginning of the SERS experiments and be kept constant throughout the calibration and measurement process.
5. Classical manual data processing methods in SERS quantitation: calibration curves and logarithmic data processing
Like any other analytical technique, the processing of data to establish a calibration curve is an essential part of quantitation in SERS. In general, this typically involves extracting the signal intensity of the target analyte from the raw SERS data and plotting it against the analyte concentration. However, this process is not always straightforward, extracting the right information and plotting the data in a way that does not compromise the true physical meaning of the data can be challenging, and if not performed correctly, could easily lead to the wrong conclusion. Therefore, this section discusses the data processing methods that are most commonly used for SERS quantitation, including discussion of the use of logarithmic data in calibration curves and calculating uncertainty. The aim is to give practical advice to the reader through a quick overview using examples in literature to show the strengths and weaknesses of the various approaches which are available. The simplest case, where the signal falls off at high concentration simply due to saturation of the surface, is well described by the Langmuir isotherm,121 which gives a linear response at low concentrations, as illustrated in Fig. 2B. Cases where the adsorption is more complex may mean that data are better fitted by other models, such as the Hill122 or Frumkin123 isotherms. However, while fitting experimental data to appropriate physical models has the potential to provide insights into the mechanism of adsorption which is not possible in the same way with log/log or semi-log plots, the simplicity of logarithmic data processing has meant that it has become predominant in SERS research. For more advanced SERS applications where multiple peaks need to be measured quantitatively, it is often necessary to involve chemometrics and multivariate methods such as partial least squares (PLS) regression.124 However, these methods require some knowledge not just of SERS but also of machine learning, which is why they are not covered in this Section but are briefly introduced in Section 6.5.1. Log/log plots in SERS quantitation
Log/log calibration plots (which can also be regarded as fitting to the empirical Freundlich equation) in SERS are calibration curves obtained by plotting log [concentration] versus log [intensity] (for an example see Fig. 6C). They are most often used for target analytes with a small binding coefficient to the enhancing surface since they allow the effect on the signal of changing the concentration by several orders of magnitude to be displayed clearly. In addition, it is often found from experiment that log/log plots calibration plots are reasonably linear, even if the underlying physical reasons for this are unclear.125,126 Considering how prevalent log/log calibration plots have become, it is useful to discuss the effect which using such plots has on the apparent precision of the analysis. As a simple example, if the precision is limited by the point-to-point variation of the SERS measurement which gives a RSD of 10%, in a simple calibration plot, the absolute size of the error bars will be larger at higher concentrations than at the lower concentrations, since their magnitude follows that of the signals. However, in the log/log plot of the same data the error bars will have similar size across the whole concentration range. This can be misleading because it might appear that the uncertainty in the concentration which can be determined from intensity values is similar at all concentrations while in fact, although the relative uncertainty in the concentration remains fixed, the absolute values vary dramatically across the concentration range. This is much more obvious if data are plotted as simple signal versus concentration curves. In addition, at high concentration even relatively small uncertainties in the signal result in large uncertainties in the concentration, due to the lower slope. This is illustrated in Fig. 10A and B which shows SERS quantitation data for melamine in milk samples.127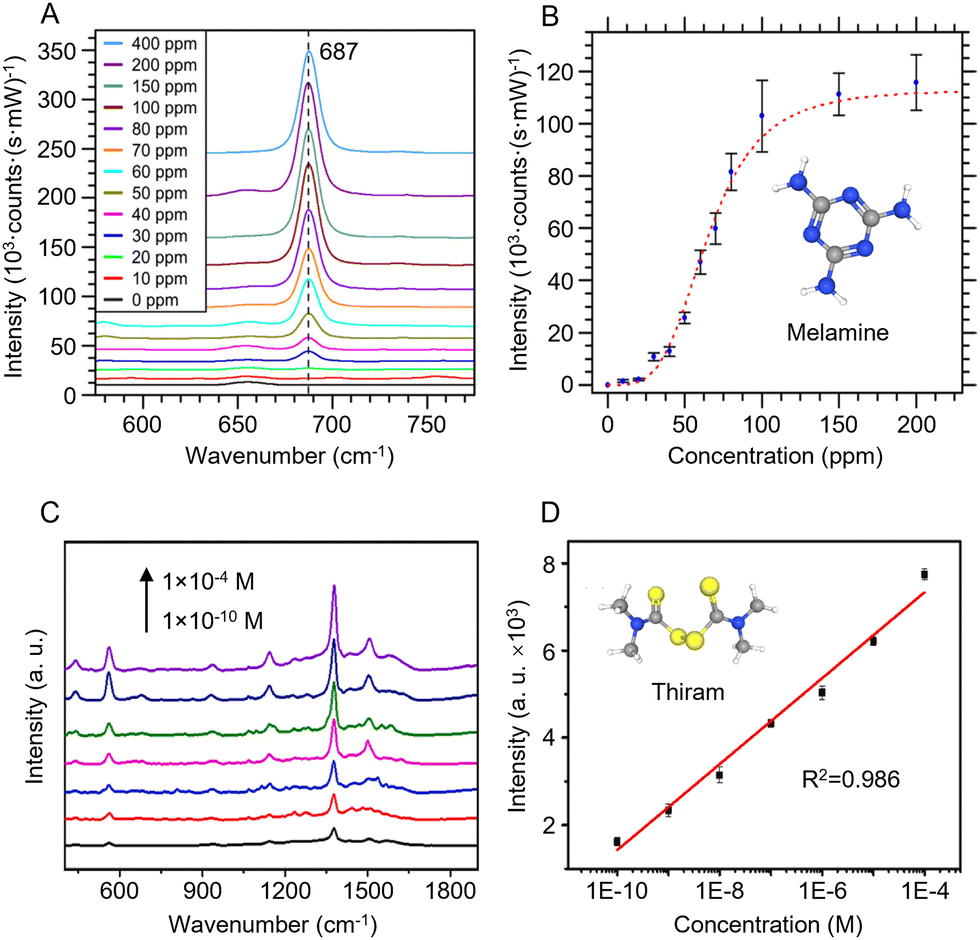 | ||
| Fig. 10 Examples of quantitative SERS calibration plots. (A) SERS spectra for various melamine concentrations in milk solutions obtained using an integrated Au-coated Si nanopillar SERS substrate. (B) Calibration plot of the intensity of the melamine peak at 687 cm−1 against concentration. (C) SERS spectra for various concentrations of thiram in juice obtained using Au@Ag NPs in a hydrogel matrix as the enhancing substrate. (D) Semi-log calibration plot of signal intensity against log[thiram]. Panels (A) and (B) were reproduced with permission from ref. 127, copyright 2017 American Chemical Society. Panel (C) and (D) were reproduced with permission from ref. 128, copyright 2023 Elsevier B. V. | ||
More generally, the purpose of the calibration is to allow the recovered concentration of analyte molecules to be determined from the response (signal height). The absolute uncertainty in the concentration obtained from a given signal level depends on the uncertainty in the response combined with the sensitivity of the measurement, which is the change in the response given by a change in concentration. In simple linear signal versus concentration SERS calibrations, the sensitivity is the slope of the regression line. Of course, even in these linear calibrations the sensitivity may well change when different target analytes and experimental conditions are used. However, irrespective of the value of the sensitivity (slope), a 5% variation in the response will indeed correspond to 5% variation in the recovered concentration. In contrast, for log–log calibration plots, the uncertainty in the recovered concentration will scale with the reciprocal of the slope (m) i.e. 1/m. So, in a typical log–log plot which has a slope of 0.5, the fact that a 100× increase in concentration gives only a 10× increase in the response means that the uncertainty in the recovered concentrations will be much larger than is the case for samples giving simple linear signal-concentration plots. For example, for a plot with m = 0.5 a RSD of 5% in the response (not the Log response) will result in a 5%/0.5 = 10% RSD in the recovered concentration. It is not unusual for log/log plots with slopes of <0.3 to be reported and in this case the uncertainty in the concentration derived from the signals will be correspondingly larger again since the multiplier changes from 1/0.5 to >1/0.3.
5.2. Semi-log plots in SERS quantitation
The largest difference between the precision of the measured intensities and the precision of the concentration measurement is found in cases where relatively small changes in the signal occur over a wide concentration range, which are typically displayed as semi-log plots in SERS. In this case the measurement has the advantage of covering a wide concentration range but the low sensitivity means that even a relatively small deviation in signal height gives a much larger uncertainty in the recovered concentration.125 For example, Fig. 10C and D shows a sensor which covers 6 orders of magnitude and uses a substrate which has a good signal RSD of <4%.128 However, using the regression line fitted to the data in the figure (y = 11274 + 985x) shows that if the signal is 6000 a.u. (corresponding to a recovered concentration of 10−5.355 = 4.41 × 10−6 M), then a random 4% change in the signal would take it to 6240 a.u., which gives a recovered concentration of 10−5.111 = 7.73 × 10−6 M. This means that although this is a good substrate with a small RSD, even a change in the signal of just +1 standard deviation is enough to almost double the recovered concentration. It is important to emphasize that this result is not due to any type of error in the method, some reduction in the precision of recovered values is inevitable if signals with a limited dynamic range (the difference between the lowest and highest signals recorded) have to cover a large concentration range. Of course, if the objective of a measurement is simply to determine whether the analyte exceeds a threshold value in the sample, lower precision may be acceptable. Nonetheless, it is useful to be aware of the extent to which the precision may be reduced in measurements which appear acceptable when displayed as semi-log plots but in fact have low sensitivity.
5.3. Calculation of uncertainty in SERS quantitation
Since the uncertainty in the recovered concentration is critical, it is important to discuss more rigorous treatments which include not only the uncertainty in the response (SERS signal) but also uncertainty in the calibration plot, which was neglected in the preceding paragraphs. It is obvious that if the slope and the intercept of the calibration line have their own associated variance this will reduce the precision, compared to the case where the calibration line was composed of a set of points lying in a perfectly straight line. The effect can be calculated as follows.If the concentration value (x0) is determined from a measured SERS response (y0) using a calibration line which has been fitted to n (xi, yi) values, then the standard deviation in that concentration value (Sx0) can be calculated from:
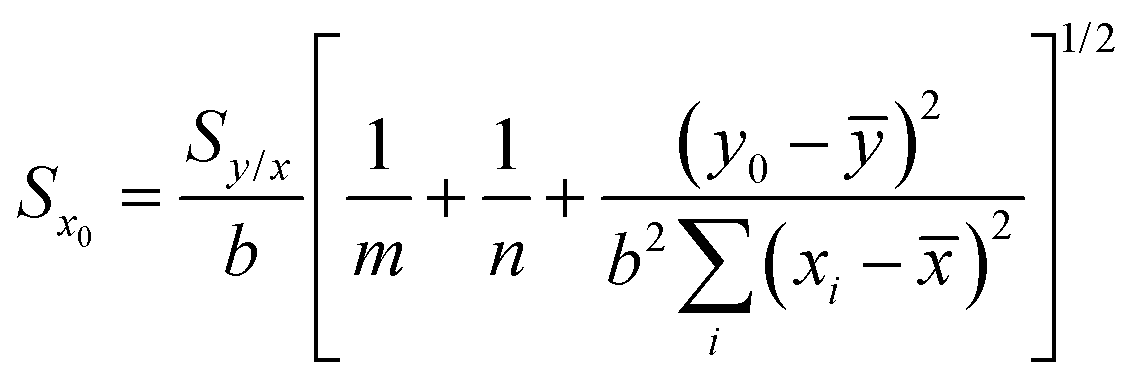 | (1) |
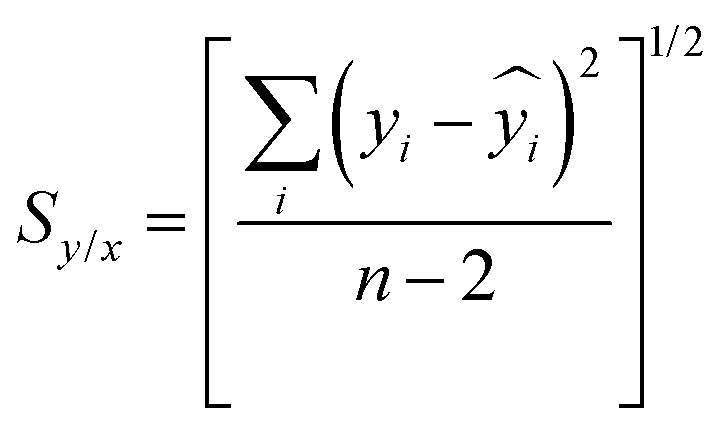 | (2) |
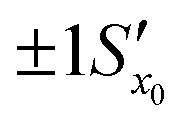 . However, after the additional uncertainty is included, the recovered value changes to
. However, after the additional uncertainty is included, the recovered value changes to 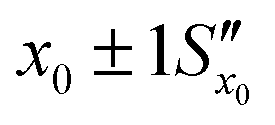 which is a significantly larger range. Furthermore, if these values need to be expressed as confidence limits, e.g. for 95% confidence, then the range needs to be further multiplied by the appropriate t95 value.
which is a significantly larger range. Furthermore, if these values need to be expressed as confidence limits, e.g. for 95% confidence, then the range needs to be further multiplied by the appropriate t95 value.
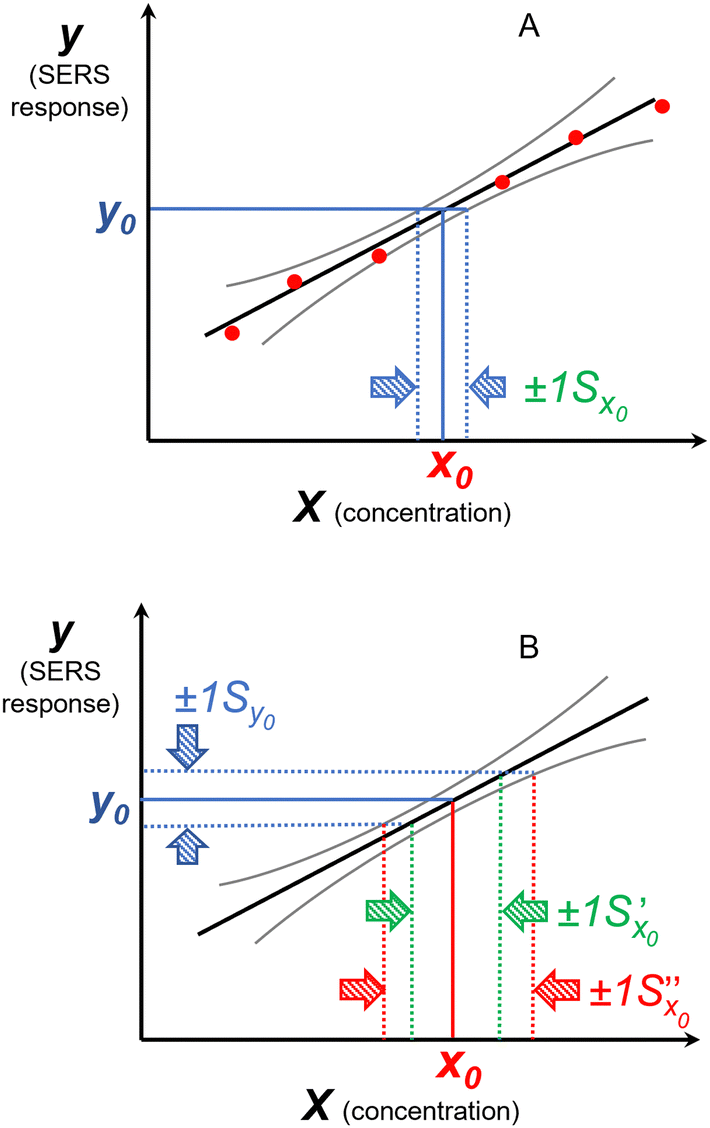 | ||
Fig. 11 The effect of including uncertainty in the slope and intercept of a calibration plot obtained by linear regression. (A) The relationship between a single y0 value and the corresponding x0 and Sx0 (recovered concentration and standard deviation) obtained from a plot of the regression line (black) and the ±1Sx interval lines (grey) calculated from eqn (1). (B) An example where the measured y0 value (SERS signal) itself has uncertainty, with a standard deviation Sy0. Green lines show the result obtained from y0 when the best fit regression line (black) is used, which is that the recovered concentration is 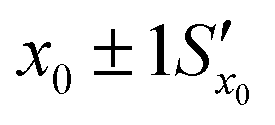 . Red lines show the result of including the uncertainty in the calibration (grey lines) which gives the recovered value as x0 . Red lines show the result of including the uncertainty in the calibration (grey lines) which gives the recovered value as x0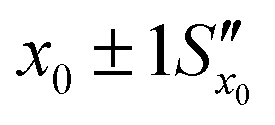 , a significantly larger range than , a significantly larger range than 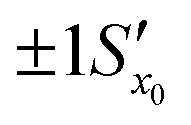 . . | ||
These are not abstract considerations, determining the precision of the recovered concentration values which are provided by SERS experiments is important because the ultimate objective of the measurement is to determine the concentration, not the SERS intensity. However, since these calculations are not straightforward, it may be useful to take a simpler approach to determine the uncertainty in the recovered concentration. This can be achieved by carrying out repeat measurements of a known sample at a given concentration (preferably one which is selected because it is relevant to the analysis) and determining the RSD of the recovered values from the measured responses and the best fit regression calibration curve. An example showing the RSD in the recovered concentrations obtained for a standard monoethanolamine sample was shown in Fig. 7B.103 Measuring the RSD in the recovered concentrations is an excellent way to assess the success of quantitative measurements but unfortunately it is not used as frequently as it ought to be in SERS measurements. In contrast, it is normal for chromatographic analysis, where the literature shows that established methods can routinely achieve high levels of accuracy and precision in their recovered concentrations. For example, HPLC is one of the most common analytical methods used to measure active pharmaceutical ingredient (API) content in medicines and the precision of recovered concentrations obtained with compendial HPLC methods is generally 0.5–1.0% RSD,132 This precision would be very challenging to match with even what are regarded as good SERS calibrations. However, the levels of precision which are regarded as acceptable are normally larger for chromatographic measurements when more challenging samples are being studied and these may be closer to what can typically be achieved with SERS. For example, the target RSD for detecting pesticide residues using GC-MS at the low end of the working range is 20%, which is set at this high value because it is measured in the region where the combination of low signal sizes and highest uncertainty in the slope come together.133 Even within these constraints, in a study of 155 different pesticide residues in milk using GC-MS, at the low concentration of 5 ppb almost all the recovered concentrations had <10% RSD.134 These levels of precision are a challenging but achievable target for SERS although typically, if RSD in recovered values at these levels of precision are reported (i.e. <10%), they are for values near the centre of the calibration range rather than at the more difficult extremes.135,136
Of course, the critical comparison is between the accuracy and precision which is required and that which the analysis method can provide and there are many applications where the acceptable levels are more relaxed, in particular for screening measurements, which only need an indication that the concentration of the target has exceeded a threshold value. Nonetheless, even in these cases, it is important to be able to quantify the confidence that can be placed in any recovered concentrations.
In summary, while monitoring the standard deviation in the response signal and the magnitude of sensitivity is useful and meaningful in the context of comparing different Raman measurements, it may also underestimate the corresponding uncertainty in the recovered concentration. Relying on the response RSD alone may also make it difficult to compare the analytical performance of the SERS measurements with other approaches where the precision of the results is typically reported as the more meaningful RSD in the recovered concentrations.
6. Future research trends for SERS quantitation of complex real-life samples: multifunctional SERS sensors, digital SERS and AI-assisted chemometrics
The previous sections highlighted the enhancing substrate material, the instrument and instrument parameters, and data processing methods as the three key components in a quantitative SERS experiment. The discussions focused mainly on how each component fundamentally affected the different analytical figures of merit in SERS quantitation. Understanding these factors should allow a non-specialist to take on simple quantitative SERS studies of single types of analyte molecules dispersed in an ideal solution, which is the most common analytical problem addressed by SERS in the literature. However, the aim of many expert SERS research groups is to continue to develop SERS as a sensing technique that so that it may be used for more advanced applications where the sample is much more complex and may contain several different chemical constituents. This is clearly a much more challenging and complex issue, nevertheless it can still be broken down into a smaller sub-set of problems that can be tackled via innovations in substrate design, methodology development and data processing. Within this context, this section discusses three future research trends in the field, namely the development of multifunctional SERS sensors (substrate design), digital SERS (methodology development) and AI-assisted data processing (data processing), that could potentially enable SERS quantitation of complex real-life samples. The aim is not to provide a comprehensive summary of these areas but rather to give the readers a taste of the state-of-the-art technologies in analytical SERS research.6.1. Multifunctional SERS sensors
A growing trend in SERS substrate design for improving quantitation performance is to exploit the huge advances in methods for synthesising and assembling of nanomaterials to produce multifunctional materials.15 For example, particles which combine plasmonic enhancement with built-in internal standards were discussed above (Section 3), while other combinations of functions such as filtration137,138 and regeneration139,140 have also been successfully demonstrated. The next step up in complexity is to create multifunctional SERS substrates which combine several different functions simultaneously and can therefore address multiple commonly encountered problems in a single package. It is easy to envisage materials which have pre-formed hotspots, carry out some sort of physical separation/filtration, contain internal standards, and are assembled into a ready-to-use single “smart-SERS” sensor, similar to the “lab-on-a-chip” approach. If this approach could be successfully implemented it might ultimately reduce the fragmentation in the field by converging towards a smaller number of multipurpose, “smart-SERS” sensors which could be used across a range of different samples. Of course, developing such multifunctional materials is challenging but many of the methods for adding different functions orthogonally, i.e. in a way that does not interfere with other properties of the system, have been developed. For example, assembly of nanoparticles into larger arrays without modifying their surfaces is now routine.141 Similarly, methods for adding porous shells to metallic cores often do not depend on the shape of the core.142,143 Essentially, many of the individual processes needed for step-by-step construction of multifunctional “smart SERS” materials are already known. Moreover, there are clear examples of the potential of this approach from other areas of nanotechnology, for example in cancer therapy, theranostic particles which combine a biocompatible surface with simultaneous support for multiple imaging modes (fluorescence/photoacoustic/magnetic resonance) and tumour phototherapy have been developed.144As discussed in Section 3, the surface chemistry of the SERS substrate plays a key role in SERS quantitation since SERS is a surface-specific analytical technique and so in most cases it is only the molecules which are directly adsorbed on the surface of the enhancing material which give SERS signals. As a result, a barrier to developing universal “smart-SERS” sensors is the challenge of creating surfaces which can adsorb all the potential target molecules that might be encountered. The various different ways of promoting adsorption were discussed above, but some of these are only applicable to very narrow ranges of chemical compounds. Also, it is important to note that even if a universal substrate were to be developed, it would be very vulnerable to having non-target sample components dominate the signal. This means there is a tension between generality and effectiveness. Nonetheless, it is possible to envisage a middle ground where different substrates, which each show sensitivity across a broad range of targets of a particular class, could be prepared, giving a family of sensors that together could cover most samples. This would mean that different sensors would need to be selected for different classes of analytes, which would move away from an ideal “universal” substrate but may be a more feasible route towards the real-life application of SERS in the near future. For example, this has been shown by Ling et al. using deposited Ag nanocubes functionalized with a thioguanine self-assembled monolayer, which acted as Raman reporters that allowed indirect detection of several different classes of analyte molecules. More specifically, as shown in Fig. 12A, the thioguanine reporter interacted differently with different types of functional groups which allowed differentiation and quantification of weakly adsorbing analyte molecules with small Raman cross sections using SERS.145
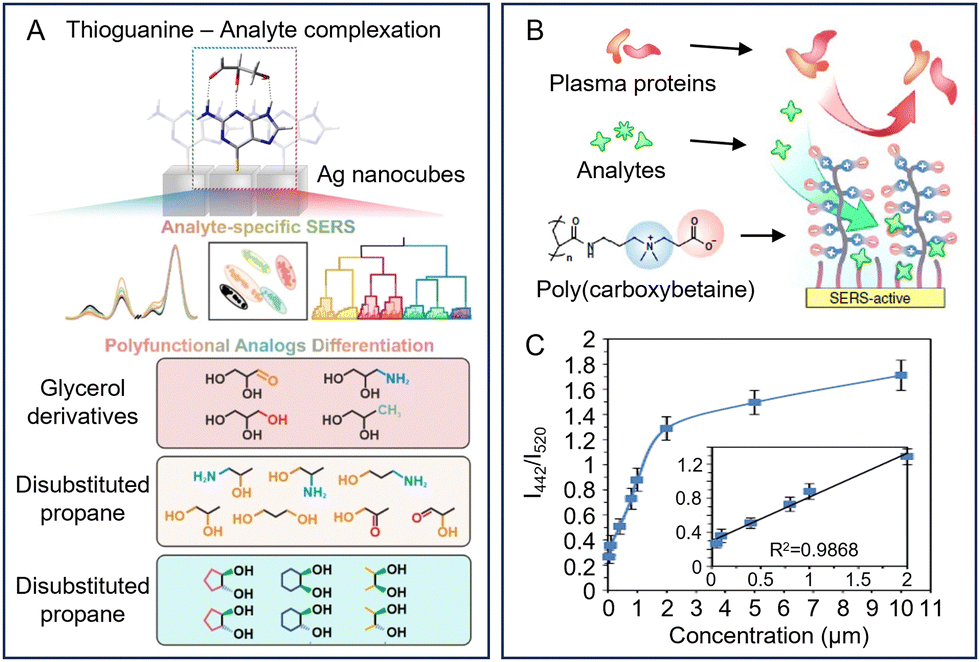 | ||
| Fig. 12 Multifunctional SERS sensors for complex real-life samples. (A) Schematic illustration of Ag nanocubes modified with thioguanine which acts as a tridentate receptor for indirect SERS discrimination and quantitation of several classes of analytes with similar molecular structures. (B) SERS substrate modified with hierarchical zwitterionic self-assembled monolayers which provides antifouling properties which allows the substrate to retain its function in plasma. The silicon-based substrate also generates a Raman signal which can be used as an internal standard in SERS quantitation. (C) Calibration curve established by measuring the signal ratio between doxorubicin and Si standard versus the concentration of doxorubicin in ultra-filtrated plasma. Panel (A) was reproduced with permission from ref. 145, copyright 2024 Wiley-VCH GmbH. Panel (B) was reproduced with permission from ref. 146, copyright 2016 The Authors. | ||
One problem with promoting adsorption using adsorbed ligand layers is that it is difficult to carry out surface modification in a way which is orthogonal to the other functions required, since these often also involve modifying the surface. One approach is to add functionality within the core, as in the case of core–shell nanoparticles with the internal standard layer encapsulated beneath the outer shell as discussed in Section 3.4. As shown in Fig. 12B, another example was published by Sun et al. who demonstrated a hierarchical surface modification strategy where a gold surface was modified with a mixed self-assembled monolayer that contained molecular reporters and antifouling zwitterionic polymer brushes, which attracted analytes, including different types of drugs and prevented protein adsorption. In addition, the silicon-based substrate also generated a consistent and intense Raman band at 520 cm−1, which could be used as an internal standard to calibrate SERS signals in quantitative measurements performed in human plasma (Fig. 12C).146 This system is “smart” in the sense that it combines several functions and can be applied across a range of chemically different analytes.
6.2. Digital SERS
Another useful approach to enhancing quantitation performance in SERS is to develop novel experimental methodologies, such as the use of orbital raster scanning in Section 4.2. Within this context, a promising new method in SERS quantitation is digital SERS, which was designed to address the challenge of performing SERS quantitation at analyte concentrations in the single-molecule region.The ability of SERS to detect molecules at single-molecule levels was first reported in 1997 and is now well-established in literature.147,148 However, at such low analyte concentration levels, the non-uniform distribution of the electromagnetic field in the enhancing substrate material becomes a major limiting issue in quantitative measurements. For example, even a slight variation of the position of the molecule in the plasmonic hot-spot by a single nanometer could alter the intensity of the SERS signals by order(s) of magnitude.72 This results in enormous signal fluctuations in the single-molecule concentration region which makes quantitation measurements based directly on tracking SERS signal intensity nearly impossible. As shown in Fig. 13, to combat this issue, digital SERS uses the idea that under single molecule conditions, the observation of SERS signals at different hotspots can be classified simply as a “yes” or “no” event, so the number of SERS events can be used to determine concentration, rather than the total intensity of the signals.149
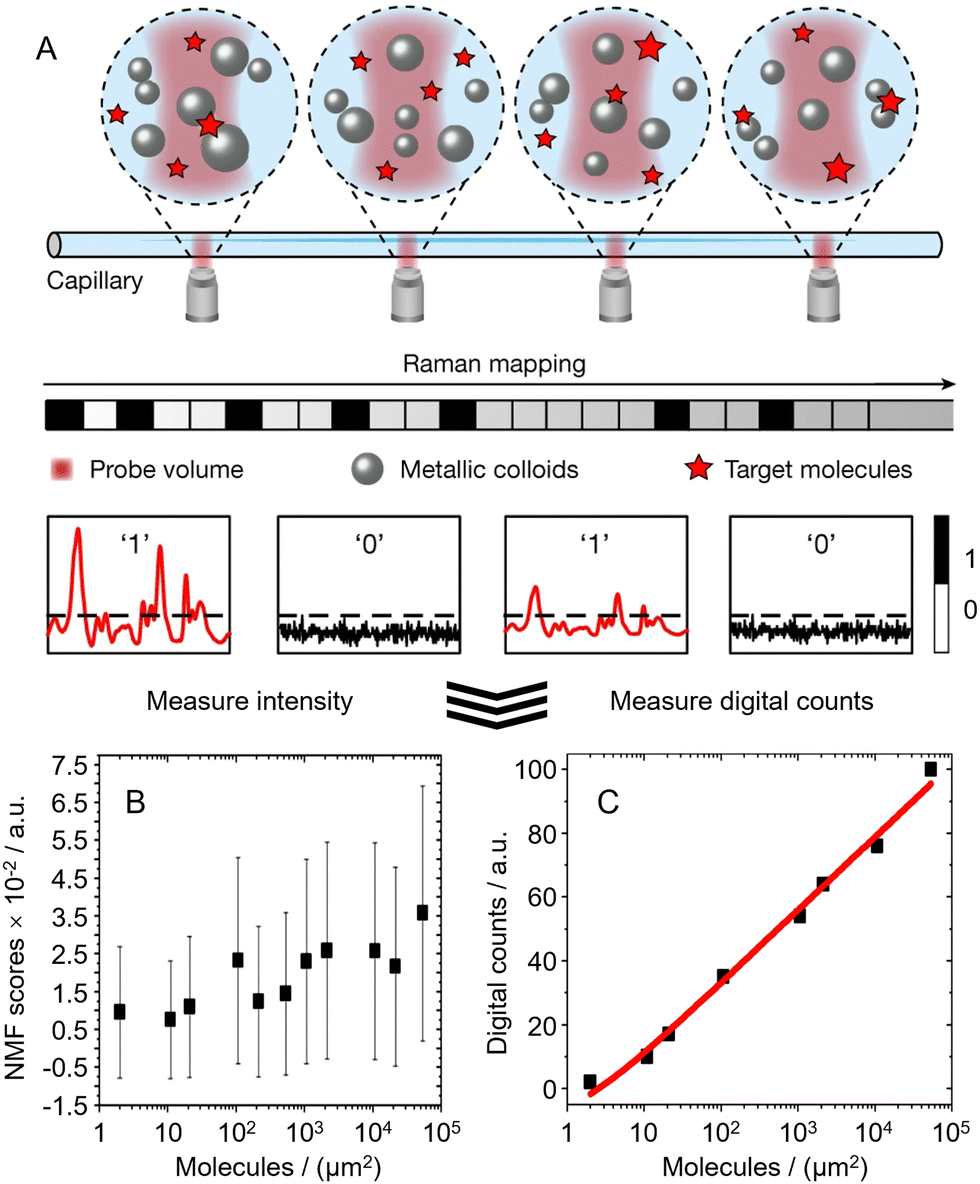 | ||
| Fig. 13 The working principles of digital SERS. (A) Schematic illustrations of how digital SERS is performed. In short Raman mapping of a sample spiked with analyte molecules at the single-molecule concentration region is performed. (B) If the SERS signal is processed directly using signal intensity, large fluctuations are observed which leads to a loss of linearity in the calibration curve. (C) If the signal is processed digitally a linear calibration curve can be obtained. Panel (A) was reproduced with permission from ref. 20 (copyright 2024 The Authors). | ||
In practice, this means that digital SERS is performed by mapping of the sample to obtain a large number of data points, which are analysed to determine the number of SERS events. At very low concentrations, the precision of the SERS analysis becomes limited by the statistical fluctuations in the number of events detected. These follow a Poisson distribution, which means that at the low concentration end of the calibration plot the error bars are proportional to 1/√N, where N is the number of events, so the precision can be significantly improved by increasing the amount of sampling points.149 For example, in a recent study by Ye et al., it was found that the RSD of the measurements could be improved from 14.2% to 5.3% when the total number of acquired voxels was increased from 1200 to 5400.20 Importantly, it has been shown that digital SERS allows quantitation of important analytes including thiram, dopamine, enrofloxacin, cytokines at sub-nanomolar levels.20,149–151
The concept of digital SERS should be readily transferable to a wide range of SERS substrates and samples. Indeed, up to now digital SERS has been applied to both solid and liquid enhancing substrates for quantitation of complex samples including lake water20 and clinical samples.150 The current limitation with digital SERS is that it inevitably requires long signal acquisition times, which poses a major challenge in kinetic studies, but this could be resolved in the near future with the development of more plasmonically active enhancing substrates, high-speed mapping Raman instruments and AI-assisted automated spectra processing software packages.
6.3. AI-assisted chemometrics in SERS quantitation
Chemometrics is the science of extracting information from chemical data via mathematical, statistical or other methods that employ formal logic. The key feature that separates chemometrics from the classical analytical approach discussed in Section 5 is that the classical approach is reductionist and aims to examine one factor at a time, while chemometric approaches are multivariate and are designed to examine multiple variables at the same time. The nature of chemometric approaches means that they require more computational power than simple univariate data analysis methods. In addition, chemometric methods are often used to process large SERS data sets that are generated with mapping or online measurements and would be impossibly time consuming with the classical manual approach.12,152 For example, when an artificial neural network (ANN) based on supervised machine learning was used to process >1000 SERS spectra it was possible to simultaneously quantify of caffeine and its xenometabolites theobromine and paraxanthine between 10−5–10−7 M using simple aggregated silver colloid and a portable Raman spectrometer.153In addition, the multivariate nature of chemometric methods means that it can be used to make sense of not just large data sets but also data with high-dimensionality. This feature is particularly useful in SERS, since even subtle changes in the vibrational signature of the target molecule, such as relative peak height, peak position, bandwidth et al., carry chemical information that could be used to realize or improve quantitation. For example, Ling et al. designed a machine-learning driven “SERS taster”, which used support vector machine (SVM) regression for SERS quantitation of flavoring molecules in an artificial wine mixture.154 As shown in Fig. 14, the SERS taster measured subtle changes in the SERS spectra generated mostly by noncovalent intermolecular interactions between various Raman reporters and flavouring molecules, which was used to construct a super-profile that could be used to train the SVM regression model. Ultimately, this allowed multiplex quantification of two wine flavoring molecules with near 100% accuracy.
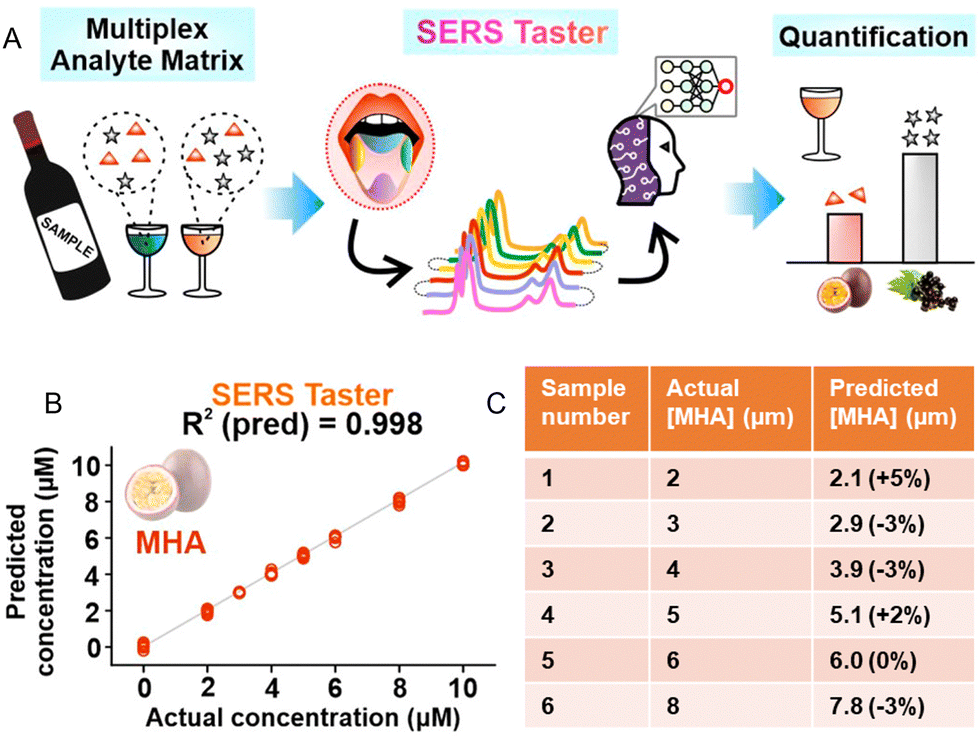 | ||
| Fig. 14 A machine learning assisted SERS taster. (A) Schematics showing the working principles of the SERS taster. (B) Calibration curves obtained using SVM-R for 3-mercaptohexylacetate (MHA) using the SERS taster. (C) Data showing the quantification accuracy of the SERS taster using six artificial wine samples with varying concentrations of MHA. For each sample, the predicted flavour concentration, and its deviation from the actual concentration (% difference) is shown. Panels (A)–(C) were reproduced with permission from ref. 154, copyright 2021 American Chemical Society. | ||
In summary, AI-assisted data processing techniques present many crucial advantages over classic manual data processing in SERS quantitation since they allow large amounts of high-dimensional data to be processed. Apart from the chemometric methods directly related to quantitation introduced above, AI based approaches have also been developed for spectra denoising,155 classification,156 substrate design157 in SERS. One major challenge in SERS that AI could be particularly useful in addressing is the assignment of SERS signals for the identification of unknown chemical compounds. Similar to the case with normal Raman and infrared spectroscopies, while the bands observed in SERS are characteristic of the molecular structure, the complexity of SERS spectra has made it extremely challenging for non-specialists to determine the structure of molecules on the basis of their SERS bands alone. This has largely limited SERS quantitation to well-defined samples in which the target analytes’ molecular structure and associated SERS signature are already established. Clearly the use of AI in data processing should not only allow more physical information to be obtained from the experimental data, but also make spectral interpretation more accessible, which would simplify data analysis for non-expert users, especially once user-friendly software is developed.155 Therefore, it seems inevitable that AI will increasingly be used in data processing as we continue to generalize SERS and move from ideal test samples in the laboratory to complex real-life samples in the field. However, challenges in deploying AI in this context still remain. For example, pre-processing steps, such as baseline correction, are currently required before regression modelling of the data, which increases the complexity of the method. Moreover, there is still a lack of commercially available software packages that can be easily employed by non-experts. As a result, current AI-assisted approaches for data processing are only accessible to researchers with considerable knowledge of both SERS and chemometrics.
7. Outlook
This Tutorial Review contrasts with a previous one published in this journal in 2008.100 At that time, obtaining even simple, quantitative calibrations with standard colloids was regarded as extremely challenging by the analytical community.158 The significant advances that have been made in preparation of substrates with excellent plasmonic enhancement and at least reasonable reproducibility/uniformity means that quantitative measurements carried out under ideal conditions, have now become routine.159 Indeed, quantitative SERS analysis has now been demonstrated for hundreds of target compounds. In parallel, researchers have continued to push the boundaries of what is possible and dramatically expanded the sensitivity, accuracy and range of measurements that can be carried out. In this sense, the first phase of the development of quantitative SERS, where good data can be obtained by researchers with some experience and understanding of the technique is now complete. Of course, there are still many improvements which could be made. Notably there is a need for better methods for modifying surfaces that can be systematically tailored to promote adsorption of target analytes. This is particularly important since SERS is a surface-specific technique, which has largely limited its use to quantitative analysis of those analyte molecules which adsorb spontaneously to noble metals. A related issue is that methods for treating mixtures of analytes are still at an early stage of development, these have not been discussed here but are critical to the employment of SERS in real-life applications where the samples are complex and typically contain multiple types of analytes of interest and/or interfering species. Finally, it would be useful to have more systematic studies of internal standards both as a way of correcting for the sample/substrate variability and to increase the robustness of measurements. In many cases, this could be more practical and efficient than attempting to achieve yet higher uniformity in the structural and plasmonic properties of the enhancing substrate. Although none of these developments is expected to be easy to achieve, it would be expected that they will be addressed in the near future since they are a natural extension of current SERS research areas.A broader challenge for the future of quantitative SERS is to also take the next step of extending the user community to those who have no interest in developing methodology, but simply want to use the advantages which SERS provides to improve their ability to measure their own particular samples. This may need to be under conditions which are far from ideal, because of possible matrix effects or the need to carry out field measurements using untrained personnel. There are huge opportunities for SERS to play to its strengths by filling the gap between existing analytical technologies, such as GC-MS, which give excellent results but are confined to the laboratory and field-deployable tests of various kinds, which often lack the sensitivity/specificity that SERS can provide. This potential is already being pursued in areas as diverse as bedside clinical measurements and the detection of contaminants in food but the range of applications is only limited by our imagination. However, the need to make these measurements in a routine and robust way pushes the onus back on specialists to both use their expertise to develop substrates and protocols which can make this possible and also to find ways to make these widely available to the broader user community. Fortunately, huge advances have already been made in the preparation and exploitation of complex substrates, with particles and nanostructures of different size, shape and composition, as well as methods to introduce internal standards, build particles into larger bulk scale assemblies and suppress matrix interference. These developments in material synthesis can pave the way for the design of new multifunctional “smart SERS” sensors which can provide the ease of use and robustness needed for the real world. In parallel with advances in SERS methodology, we can also expect the cost of equipment to continue falling and the implementation of new ubiquitous technologies to make data acquisition and processing easy, for example by wirelessly linking portable instruments to the internet.160 Similarly, AI will inevitably have a large impact on data processing and interpretation.161 More generally, all the above shows that there is already a well-stocked toolbox of approaches which can be used to develop existing methods to either make it easier to solve existing problems or address new challenges.
In conclusion, after 50 years of SERS research, we are now starting to see simple SERS sensors aimed at solving important individual challenges appear.162 Moving forward, the next generation of SERS sensors can build on this success by integrating the latest developments in nanotechnology, computer science and instrumentation to deliver the sensitive, accurate and robust analytical solutions for complex and challenging samples which can be used by non-specialists in real life applications.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Chunchun Li acknowledges the Shanghai Pujiang Program for support (23PJ1409000). Yikai Xu acknowledges the Young Scientist Fund of the National Natural Science Foundation of China (22402060). Ziwei Ye acknowledges the Shanghai Pujiang Program (23PJ1401900) and Shanghai Post-doctoral Excellence Program (2021111) for funding support. Wafaa Aljuhani acknowledges the support of the Ministry of Education in Saudi Arabia. Steven Bell acknowledges support of the Department of the Economy (N.I.), US-Ireland R&D Partnership Grant USI157. The authors would like to thank Prof. He Tian, Prof. Yi Cheng and Prof. Xiang Ma for support.References
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- B. M. Cullum, J. Mobley, Z. Chi, D. L. Stokes, G. H. Miller and T. Vo-Dinh, Rev. Sci. Instrum., 2000, 71, 1602–1607 CrossRef CAS.
- N. J. Everall, Appl. Spectrosc., 2009, 63, 245A–262A CrossRef CAS PubMed.
- X. Zhou, Z. Hu, D. Yang, S. Xie, Z. Jiang, R. Niessner, C. Haisch, H. Zhou and P. Sun, Adv. Sci., 2020, 7, 2001739 CrossRef CAS PubMed.
- M. J. Piernas-Munoz, A. Tornheim, S. Trask, Z. Zhang and I. Bloom, Chem. Commun., 2021, 57, 2253–2256 RSC.
- F. Akoueson, C. Chbib, S. Monchy, I. Paul-Pont, P. Doyen, A. Dehaut and G. Duflos, Sci. Total Environ., 2021, 773, 145073 CrossRef CAS PubMed.
- Y. Goksel, E. Dumont, R. Slipets, S. T. Rajendran, S. Sarikaya, L. H. E. Thamdrup, K. Schmiegelow, T. Rindzevicius, K. Zor and A. Boisen, ACS Sens., 2022, 7, 2358–2369 CrossRef CAS.
- D. S. Burr, W. L. Fatigante, J. A. Lartey, W. Jang, A. R. Stelmack, N. W. McClurg, J. M. Standard, J. R. Wieland, J. H. Kim, C. C. Mulligan and J. D. Driskell, Anal. Chem., 2020, 92, 6676–6683 CrossRef CAS.
- P. Dey, ACS Meas. Sci. Au, 2023, 3, 434–443 CrossRef CAS PubMed.
- R. Goodacre, D. Graham and K. Faulds, TrAC, Trends Anal. Chem., 2018, 102, 359–368 CrossRef CAS.
- S. E. J. Bell, G. Charron, E. Cortés, J. Kneipp, M. L. de la Chapelle, J. Langer, M. Procházka, V. Tran and S. Schlücker, Angew. Chem., Int. Ed., 2020, 59, 5454–5462 CrossRef CAS.
- X. Bi, L. Lin, Z. Chen and J. Ye, Small Methods, 2024, 8, e2301243 CrossRef PubMed.
- M. Fan, G. F. S. Andrade and A. G. Brolo, Anal. Chim. Acta, 2020, 1097, 1–29 CrossRef CAS.
- X. Wang and L. Guo, Angew. Chem., Int. Ed., 2020, 59, 4231–4239 CrossRef CAS.
- C. Li, Y. Huang, X. Li, Y. Zhang, Q. Chen, Z. Ye, Z. Alqarni, S. E. J. Bell and Y. Xu, J. Mater. Chem. C, 2021, 9, 11517–11552 RSC.
- N. Valley, N. Greeneltch, R. P. Van Duyne and G. C. Schatz, J. Phys. Chem. Lett., 2013, 4, 2599–2604 CrossRef CAS.
- S.-Y. Ding, E.-M. You, Z.-Q. Tian and M. Moskovits, Chem. Soc. Rev., 2017, 46, 4042–4076 RSC.
- D. Bhowmik, J. J. S. Rickard, R. Jelinek and P. G. Oppenheimer, Sustainable Food Technol., 2024 10.1039/d4fb00192c.
- C. Liu, D. Xu, X. Dong and Q. Huang, Trends Food Sci. Technol., 2022, 128, 90–101 CrossRef CAS.
- X. Bi, D. M. Czajkowsky, Z. Shao and J. Ye, Nature, 2024, 628, 771–775 CrossRef CAS.
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguie, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. Garcia de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Kall, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J. F. Li, X. Y. Ling, S. A. Maier, T. Mayerhofer, M. Moskovits, K. Murakoshi, J. M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlucker, L. L. Tay, K. G. Thomas, Z. Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzan, ACS Nano, 2020, 14, 28–117 CrossRef CAS.
- L. Zhao, Y. Wang, S. Jin, N. An, M. Yan, X. Zhang, Z. Hong and S. Yang, Nat. Synth., 2024, 3, 867–877 CrossRef.
- S. Kwon, M. J. Oh, S. Lee, G. Lee, I. Jung, M. Oh and S. Park, J. Am. Chem. Soc., 2023, 145, 27397–27406 CrossRef CAS PubMed.
- C. M. MacLaughlin, N. Mullaithilaga, G. Yang, S. Y. Ip, C. Wang and G. C. Walker, Langmuir, 2013, 29, 1908–1919 CrossRef CAS.
- B. Wang, Y. Liu, C. Ai, R. Chu, M. Chen, H. Ye, H. Wang and F. Zhou, Opt. Express, 2022, 30, 15846–15857 CrossRef CAS.
- K. L. Wustholz, A.-I. Henry, J. M. McMahon, R. G. Freeman, N. Valley, M. E. Piotti, M. J. Natan, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2010, 132, 10903–10910 CrossRef CAS.
- W. Li, P. H. Camargo, L. Au, Q. Zhang, M. Rycenga and Y. Xia, Angew. Chem., Int. Ed., 2010, 49, 164–168 CrossRef CAS PubMed.
- A. B. Serrano-Montes, D. Jimenez de Aberasturi, J. Langer, J. J. Giner-Casares, L. Scarabelli, A. Herrero and L. M. Liz-Marzan, Langmuir, 2015, 31, 9205–9213 CrossRef CAS PubMed.
- M. P. Cecchini, V. A. Turek, J. Paget, A. A. Kornyshev and J. B. Edel, Nat. Mater., 2013, 12, 165–171 CrossRef CAS PubMed.
- G. C. Phan-Quang, H. K. Lee, H. W. Teng, C. S. L. Koh, B. Q. Yim, E. K. M. Tan, W. L. Tok, I. Y. Phang and X. Y. Ling, Angew. Chem., Int. Ed., 2018, 57, 5792–5796 CrossRef CAS PubMed.
- Z. Ye, C. Li, Q. Chen, Y. Xu and S. E. J. Bell, Angew. Chem., Int. Ed., 2019, 58, 19054–19059 CrossRef CAS.
- L. Scarabelli, A. Sanchez-Iglesias, J. Perez-Juste and L. M. Liz-Marzan, J. Phys. Chem. Lett., 2015, 6, 4270–4279 CrossRef CAS PubMed.
- Q. Zhang, W. Li, C. Moran, J. Zeng, J. Chen, L.-P. Wen and Y. Xia, J. Am. Chem. Soc., 2010, 132, 11372–11378 CrossRef CAS PubMed.
- Y. Zhang, Y. Gu, J. He, B. D. Thackray and J. Ye, Nat. Commun., 2019, 10, 3905 CrossRef PubMed.
- V. Flauraud, M. Mastrangeli, G. D. Bernasconi, J. Butet, D. T. Alexander, E. Shahrabi, O. J. Martin and J. Brugger, Nat. Nanotechnol., 2017, 12, 73–80 CrossRef CAS.
- Y. Xu, Z. Ye, C. Li, H. McCabe, J. Kelly and S. E. J. Bell, Appl. Mater. Today, 2018, 13, 352–358 CrossRef.
- D. Liu, F. Zhou, C. Li, T. Zhang, H. Zhang, W. Cai and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 9596–9600 CrossRef CAS.
- J. Cai, R. Liu, S. Jia, Z. Feng, L. Lin, Z. Zheng, S. Wu and Z. Wang, Opt. Mater., 2021, 122, 111779 CrossRef CAS.
- M. R. Gartia, Z. Xu, E. Behymer, H. Nguyen, J. A. Britten, C. Larson, R. Miles, M. Bora, A. S. Chang, T. C. Bond and G. L. Liu, Nanotechnology, 2010, 21, 395701 CrossRef.
- X. Zhang, W. J. Salcedo, M. M. Rahman and A. G. Brolo, Surf. Sci., 2018, 676, 39–45 CrossRef CAS.
- T. Zhang, Y. Li, X. Lv, S. Jiang, S. Jiang, Z. Sun, M. Zhang and Y. Li, Adv. Funct. Mater., 2023, 34, 2315668 CrossRef.
- A. L. Schmucker, S. Tadepalli, K.-K. Liu, C. J. Sullivan, S. Singamaneni and R. R. Naik, RSC Adv., 2016, 6, 4136–4144 RSC.
- Z. Ye, C. Li, N. Skillen, Y. Xu, H. McCabe, J. Kelly, P. Robertson and S. E. J. Bell, Appl. Mater. Today, 2019, 15, 398–404 CrossRef.
- K. Sugawa, Y. Hayakawa, Y. Aida, Y. Kajino and K. Tamada, Nanoscale, 2022, 14, 9278–9285 RSC.
- Z. Gong, H. Du, F. Cheng, C. Wang, C. Wang and M. Fan, ACS Appl. Mater. Interfaces, 2014, 6, 21931–21937 CrossRef CAS.
- S. Botti, L. Cantarini, S. Almaviva, A. Puiu and A. Rufoloni, Chem. Phys. Lett., 2014, 592, 277–281 CrossRef CAS.
- M. A. Tahir, X. Zhang, H. Cheng, D. Xu, Y. Feng, G. Sui, H. Fu, V. K. Valev, L. Zhang and J. Chen, Analyst, 2019, 145, 277–285 RSC.
- W. Zhao, Y. Zhang, J. Yang, J. Li, Y. Feng, M. Quan, Z. Yang and S. Xiao, Nanoscale, 2020, 12, 18056–18066 RSC.
- G. A. Vinnacombe-Willson, Y. Conti, A. Stefancu, P. S. Weiss, E. Cortes and L. Scarabelli, Chem. Rev., 2023, 123, 8488–8529 CrossRef CAS PubMed.
- D. Chen, L. Zhang, P. Ning, H. Yuan, Y. Zhang, M. Zhang, T. Fu and X. He, Nano Res., 2021, 14, 4885–4893 CrossRef CAS.
- F. Wang, J. Zheng, X. Li, Y. Ji, Y. Gao, W. Xing and T. Lu, J. Electroanal. Chem., 2003, 545, 123–128 CrossRef CAS.
- W. Wang, Y.-F. Huang, D.-Y. Liu, F.-F. Wang, Z.-Q. Tian and D. Zhan, J. Electroanal. Chem., 2016, 779, 126–130 CrossRef CAS.
- C. Li, L. Chai, Q. Chen, Z. Ye, Y. Xu and S. E. J. Bell, J. Raman Spectrosc., 2020, 52, 386–393 CrossRef.
- J. Liu, Z. Y. Cai, W. X. Sun, J. Z. Wang, X. R. Shen, C. Zhan, R. Devasenathipathy, J. Z. Zhou, D. Y. Wu, B. W. Mao and Z. Q. Tian, J. Am. Chem. Soc., 2020, 142, 17489–17498 CrossRef CAS PubMed.
- G. Frens, Nature (London), Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- P. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- N. Leopold, M. Haberkorn, T. Laurell, J. Nilsson, J. R. Baena, J. Frank and B. Lendl, Anal. Chem., 2003, 75, 2166–2171 CrossRef CAS PubMed.
- C. Li, Y. Zhang, Z. Ye, S. E. Bell and Y. Xu, Nat. Protoc., 2023, 18, 2717–2744 CrossRef CAS PubMed.
- S. E. Bell and M. R. McCourt, Phys. Chem. Chem. Phys., 2009, 11, 7455–7462 RSC.
- J. E. Park, Y. Jung, M. Kim and J. M. Nam, ACS Cent. Sci., 2018, 4, 1303–1314 CrossRef CAS PubMed.
- P. G. Etchegoin and E. C. Le Ru, Phys. Chem. Chem. Phys., 2008, 10, 6079–6089 RSC.
- V. Turzhitsky, L. Zhang, G. L. Horowitz, E. Vitkin, U. Khan, Y. Zakharov, L. Qiu, I. Itzkan and L. T. Perelman, Small, 2018, 14, e1802392 CrossRef.
- M. P. Konrad, A. P. Doherty and S. E. Bell, Anal. Chem., 2013, 85, 6783–6789 CrossRef CAS.
- B. L. Darby and E. C. Le Ru, J. Am. Chem. Soc., 2014, 136, 10965–10973 CrossRef CAS PubMed.
- D. B. Grys, R. Chikkaraddy, M. Kamp, O. A. Scherman, J. J. Baumberg and B. de Nijs, J. Raman Spectrosc., 2020, 52, 412–419 CrossRef.
- W. Zhu, B. Y. Wen, L. J. Jie, X. D. Tian, Z. L. Yang, P. M. Radjenovic, S. Y. Luo, Z. Q. Tian and J. F. Li, Biosens. Bioelectron., 2020, 154, 112067 CrossRef CAS.
- J. Liu, J. Li, F. Li, Y. Zhou, X. Hu, T. Xu and W. Xu, Anal. Bioanal. Chem., 2018, 410, 5277–5285 CrossRef CAS PubMed.
- Q. Ke, L. Chen, B. Fang, Y. Chen and W. Zhang, Mater. Today Commun., 2021, 26, 101953 CrossRef CAS.
- Z. Ye, C. Li, M. Celentano, M. Lindley, T. O'Reilly, A. J. Greer, Y. Huang, C. Hardacre, S. J. Haigh, Y. Xu and S. E. J. Bell, JACS Au, 2022, 2, 178–187 CrossRef CAS.
- M. J. Oh, S. Kwon, S. Lee, I. Jung and S. Park, ACS Nano, 2024, 18, 7656–7665 CrossRef CAS PubMed.
- D. K. Lim, K. S. Jeon, J. H. Hwang, H. Kim, S. Kwon, Y. D. Suh and J. M. Nam, Nat. Nanotechnol., 2011, 6, 452–460 CrossRef CAS PubMed.
- D. Radziuk and H. Moehwald, Phys. Chem. Chem. Phys., 2015, 17, 21072–21093 RSC.
- S. Sergiienko, K. Moor, K. Gudun, Z. Yelemessova and R. Bukasov, Phys. Chem. Chem. Phys., 2017, 19, 4478–4487 RSC.
- W. Zhou, J. Wu and H. Yang, Nano Lett., 2013, 13, 2870–2874 CrossRef CAS.
- L. Xing, Y. Xiahou, P. Zhang, W. Du and H. Xia, ACS Appl. Mater. Interfaces, 2019, 11, 17637–17646 CrossRef CAS.
- C. Song, B. Ye, J. Xu, J. Chen, W. Shi, C. Yu, C. An, J. Zhu and W. Zhang, Small, 2022, 18, e2104202 CrossRef.
- J. Peng, P. Liu, Y. Chen, Z.-H. Guo, Y. Liu and K. Yue, Nano Res., 2022, 16, 5056–5064 CrossRef.
- C. Li, Z. Chen, Y. Huang, Y. Zhang, X. Li, Z. Ye, X. Xu, S. E. J. Bell and Y. Xu, Chem, 2022, 8, 2514–2528 CAS.
- Q. Fan, K. Liu, Z. Liu, H. Liu, L. Zhang, P. Zhong and C. Gao, Part. Part. Syst. Charact., 2017, 34, 1700075 CrossRef.
- Y. Hu, Y. Li, L. Yu, Y. Zhang, Y. Lai, W. Zhang and W. Xie, Chem. Sci., 2022, 13, 11792–11797 RSC.
- L. Jiang, C. H. He, H. Y. Chen, C. Y. Xi, E. K. Fodjo, Z. R. Zhou, R. C. Qian, D. W. Li and M. E. Hafez, Anal. Chem., 2021, 93, 12609–12616 CrossRef CAS PubMed.
- R. W. Taylor, T.-C. Lee, O. A. Scherman, R. Esteban, J. Aizpurua, F. M. Huang, J. J. Baumberg and S. Mahajan, ACS Nano, 2011, 5, 3878–3887 CrossRef CAS.
- X. Liu, Z. Ye, Q. Xiang, Z. Xu, W. Yue, C. Li, Y. Xu, L. Wang, X. Cao and J. Zhang, Chem, 2023, 9, 1464–1476 CAS.
- A. B. Sánchez-Alvarado, J. Zhou, P. Jin, O. Neumann, T. P. Senftle, P. Nordlander and N. J. Halas, ACS Nano, 2023, 17, 25697–25706 CrossRef.
- M. Su, S. Yang, M. Xu, S. Du, L. Zheng, X. Wang, C. Qu and H. Liu, Anal. Chem., 2023, 95, 12398–12405 CrossRef CAS PubMed.
- Y. Hang, J. Boryczka and N. Wu, Chem. Soc. Rev., 2022, 51, 329–375 RSC.
- S. Sloan-Dennison, M. R. Bevins, B. T. Scarpitti, V. K. Sauve and Z. D. Schultz, Analyst, 2019, 144, 5538–5546 RSC.
- J. Wang, A. Wuethrich, A. A. I. Sina, R. E. Lane, L. L. Lin, Y. Wang, J. Cebon, A. Behren and M. Trau, Sci. Adv., 2020, 6, eaax3223 CrossRef CAS PubMed.
- A. M. Fales and T. Vo-Dinh, J. Mater. Chem. C, 2015, 3, 7319–7324 RSC.
- H.-Y. Chen, M.-H. Lin, C.-Y. Wang, Y.-M. Chang and S. Gwo, J. Am. Chem. Soc., 2015, 137, 13698–13705 CrossRef CAS.
- X. Yan, P. Li, B. Zhou, X. Tang, X. Li, S. Weng, L. Yang and J. Liu, Anal. Chem., 2017, 89, 4875–4881 CrossRef CAS PubMed.
- W. Shen, X. Lin, C. Jiang, C. Li, H. Lin, J. Huang, S. Wang, G. Liu, X. Yan, Q. Zhong and B. Ren, Angew. Chem., Int. Ed., 2015, 54, 7308–7312 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, W. Wang, K. Sun and L. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 28105–28115 CrossRef CAS PubMed.
- N. G. Khlebtsov, L. Lin, B. N. Khlebtsov and J. Ye, Theranostics, 2020, 10, 2067–2094 CrossRef CAS.
- S. Lin, X. Lin, S. Han, Y. Liu, W. Hasi and L. Wang, Anal. Chim. Acta, 2020, 1108, 167–176 CrossRef CAS.
- N. Michieli, R. Pilot, V. Russo, C. Scian, F. Todescato, R. Signorini, S. Agnoli, T. Cesca, R. Bozio and G. Mattei, RSC Adv., 2017, 7, 369–378 RSC.
- M. Erol, Y. Han, S. K. Stanley, C. M. Stafford, H. Du and S. Sukhishvili, J. Am. Chem. Soc., 2009, 131, 7480–7481 CrossRef CAS PubMed.
- D. B. Grys, B. de Nijs, A. R. Salmon, J. Huang, W. Wang, W. H. Chen, O. A. Scherman and J. J. Baumberg, ACS Nano, 2020, 14, 8689–8696 CrossRef CAS PubMed.
- D. Zhang, S. M. Ansar, K. Vangala and D. Jiang, J. Raman Spectrosc., 2009, 41, 952–957 CrossRef.
- S. E. Bell and N. M. Sirimuthu, Chem. Soc. Rev., 2008, 37, 1012–1024 RSC.
- A. Subaihi, H. Muhamadali, S. T. Mutter, E. Blanch, D. I. Ellis and R. Goodacre, Analyst, 2017, 142, 1099–1105 RSC.
- S. Zakel, O. Rienitz, B. Guttler and R. Stosch, Analyst, 2011, 136, 3956–3961 RSC.
- M. Benhabib, S. L. Kleinman and M. C. Peterman, Energy Fuels, 2023, 37, 1881–1886 CrossRef CAS.
- S. E. Bell and N. M. Sirimuthu, Analyst, 2004, 129, 1032–1036 RSC.
- N. Itoh and S. E. Bell, Analyst, 2017, 142, 994–998 RSC.
- A. Subaihi, D. K. Trivedi, K. A. Hollywood, J. Bluett, Y. Xu, H. Muhamadali, D. I. Ellis and R. Goodacre, Anal. Chem., 2017, 89, 6702–6709 CrossRef CAS.
- L. Xiao, C. Wang, C. Dai, L. E. Littlepage, J. Li and Z. D. Schultz, Angew. Chem., Int. Ed., 2020, 59, 3439–3443 CrossRef CAS PubMed.
- S. L. Kleinman, M. C. Peterman, M. Benhabib, M. T. Cheng, J. D. Hudson and R. E. Mohler, Anal. Chem., 2017, 89, 13190–13194 CrossRef CAS PubMed.
- R. A. Alvarez-Puebla, J. Phys. Chem. Lett., 2012, 3, 857–866 CrossRef CAS PubMed.
- A. L. P. Queiroz, B. M. Kerins, J. Yadav, F. Farag, W. Faisal, M. E. Crowley, S. E. Lawrence, H. A. Moynihan, A. M. Healy, S. Vucen and A. M. Crean, Cellulose, 2021, 28, 8971–8985 CrossRef CAS PubMed.
- B. K. Sahu, A. Dwivedi, K. K. Pal, R. Pandian, S. Dhara and A. Das, Appl. Surf. Sci., 2021, 537, 147615 CrossRef CAS.
- H. S. Siebe, Q. Chen, X. Li, Y. Xu, W. R. Browne and S. E. J. Bell, Analyst, 2021, 146, 1281–1288 RSC.
- Y. Liu, Y. Zhang, M. Tardivel, M. Lequeux, X. Chen, W. Liu, J. Huang, H. Tian, Q. Liu, G. Huang, R. Gillibert, M. L. de la Chapelle and W. Fu, Plasmonics, 2019, 15, 743–752 CrossRef.
- A. C. Crawford, A. Skuratovsky and M. D. Porter, Anal. Chem., 2016, 88, 6515–6522 CrossRef CAS PubMed.
- J. L. Abell, J. M. Garren and Y. Zhao, Appl. Spectrosc., 2011, 65, 734–740 CrossRef CAS PubMed.
- S. Stewart, R. J. Priore, M. P. Nelson and P. J. Treado, Annu. Rev. Anal. Chem., 2012, 5, 337–360 CrossRef CAS PubMed.
- S. E. J. Bell, J. R. Beattie, J. J. McGarvey, K. L. Peters, N. M. S. Sirimuthu and S. J. Speers, J. Raman Spectrosc., 2004, 35, 409–417 CrossRef CAS.
- Z. C. Zeng, H. Wang, P. Johns, G. V. Hartland and Z. D. Schultz, J. Phys. Chem. C, 2017, 121, 11623–11631 CrossRef CAS PubMed.
- M. Mochizuki, S. Asatyas, K. Suthiwanich and T. Hayashi, Chem. Lett., 2016, 45, 1207–1209 CrossRef CAS.
- C. Heck, Y. Kanehira, J. Kneipp and I. Bald, Molecules, 2019, 24, 2324 CrossRef CAS PubMed.
- W. L. Chen, C. Y. Lo, Y. C. Huang, Y. C. Wang, W. H. Chen, K. J. Lin and Y. M. Chang, J. Raman Spectrosc., 2021, 53, 33–39 CrossRef.
- G. Barbillon, O. Graniel and M. Bechelany, Nanomaterials, 2021, 11, 2174 CrossRef CAS.
- P. Mosier-Boss and S. J. A. s Lieberman, Appl. Spectrosc., 2003, 57, 1129–1137 CrossRef CAS PubMed.
- N. Siraj, D. K. Bwambok, P. N. Brady, M. Taylor, G. A. Baker, M. Bashiru, S. Macchi, A. Jalihal, I. Denmark, T. Le, B. Elzey, D. A. Pollard and S. O. Fakayode, Appl. Spectrosc. Rev., 2021, 56, 615–672 CrossRef CAS.
- P. L. Urban, Anal. Chem., 2020, 92, 10210–10212 CrossRef CAS PubMed.
- D. Kong, J. Zhao, S. Tang, W. Shen and H. K. Lee, Anal. Chem., 2021, 93, 12156–12161 CrossRef CAS PubMed.
- O. Durucan, T. Rindzevicius, M. S. Schmidt, M. Matteucci and A. Boisen, ACS Sens., 2017, 2, 1400–1404 CrossRef CAS PubMed.
- G. Qi, Y. Wang, T. Liu and D. Sun, Anal. Chim. Acta, 2023, 1282, 341903 CrossRef CAS PubMed.
- J. N. Miller, Analyst, 1991, 116, 3–14 RSC.
- E. J. Billo, Excel for Chemists: A Comprehensive Guide, John Wiley & Sons, New York, 2004 Search PubMed.
- D. A. Skoog, D. M. West, F. J. Holler and S. R. Crouch, Fundamentals of analytical chemistry, Saunders College Pub, Fort Worth, 1996 Search PubMed.
- J. D. Hofer, B. A. Olsen and E. C. Rickard, J. Pharm. Biomed. Anal., 2007, 44, 906–913 CrossRef CAS PubMed.
- A. R. F.-A. Eurl-Fv, M. Gamón, G. V. Eurl-Fv, C. F. A. Eurl-Fv, M. E. Poulsen, D. Eurl-Cf, R. Lippold, C. Eurl-Ao, M. Anastassiades and C. Eurl-Srm, SANTE/11813/2017, European Commission Directorate General For Health And Food Safety, 2017.
- S. P. Singh, S. K. Budakoti and D. Oulkar, Large-scale screening and quantitation of pesticide residues in milk using GC-(EI)-MS/MS, Thermo Fisher Scientific, 2019 Search PubMed.
- X. Fang, Y. Song, Y. Huang, G. Yang, C. Han, H. Li and L. Qu, Analyst, 2020, 145, 7421–7428 RSC.
- S. Hernandez, J. V. Perales-Rondon, A. Arnaiz, M. Perez-Estebanez, E. Gomez, A. Colina and A. Heras, J. Electroanal. Chem., 2020, 879, 114743 CrossRef CAS.
- C. Wang, C. Wang, X. Wang, K. Wang, Y. Zhu, Z. Rong, W. Wang, R. Xiao and S. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 19495–19505 CrossRef CAS PubMed.
- Q. Ding, J. Wang, X. Chen, H. Liu, Q. Li, Y. Wang and S. Yang, Nano Lett., 2020, 20, 7304–7312 CrossRef CAS PubMed.
- O. Guselnikova, R. Samant, P. Postnikov, A. Trelin, V. Svorcik and O. Lyutakov, J. Mater. Chem. C, 2019, 7, 14181–14187 RSC.
- B. Qiu, M. Xing, Q. Yi and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 10643–10647 CrossRef CAS PubMed.
- Y. Xu, M. P. Konrad, W. W. Lee, Z. Ye and S. E. Bell, Nano Lett., 2016, 16, 5255–5260 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou and D. Y. J. N. Wu, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- Y. Guo, Y. Li, R. Fan, A. Liu, Y. Chen, H. Zhong, Y. Liu, H. Chen, Z. Guo and Z. Liu, Nano Lett., 2023, 23, 8761–8769 CrossRef CAS PubMed.
- K. Wu, H. Zhao, Z. Sun, B. Wang, X. Tang, Y. Dai, M. Li, Q. Shen, H. Zhang, Q. Fan and W. Huang, Theranostics, 2019, 9, 7697–7713 CrossRef CAS.
- L. B. T. Nguyen, E. X. Tan, S. X. Leong, C. S. L. Koh, M. Madhumita, I. Y. Phang and X. Y. Ling, Angew. Chem., Int. Ed., 2024, 63, e202410815 CAS.
- F. Sun, H. C. Hung, A. Sinclair, P. Zhang, T. Bai, D. D. Galvan, P. Jain, B. Li, S. Jiang and Q. Yu, Nat. Commun., 2016, 7, 13437 CrossRef CAS.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef PubMed.
- C. D. L. de Albuquerque, R. G. Sobral-Filho, R. J. Poppi and A. G. Brolo, Anal. Chem., 2018, 90, 1248–1254 CrossRef CAS PubMed.
- J. Li, A. Wuethrich, A. A. I. Sina, H. H. Cheng, Y. Wang, A. Behren, P. N. Mainwaring and M. Trau, Nat. Commun., 2021, 12, 1087 CrossRef CAS PubMed.
- W. Nam, W. Kim, W. Zhou and E. A. You, Nanoscale, 2021, 13, 17340–17349 RSC.
- R. Cardoso Rial, Talanta, 2024, 274, 125949 CrossRef CAS PubMed.
- O. Alharbi, Y. Xu and R. Goodacre, Anal. Bioanal. Chem., 2015, 407, 8253–8261 CrossRef CAS PubMed.
- Y. X. Leong, Y. H. Lee, C. S. L. Koh, G. C. Phan-Quang, X. Han, I. Y. Phang and X. Y. Ling, Nano Lett., 2021, 21, 2642–2649 CrossRef CAS PubMed.
- S. H. Luo, X. J. Zhao, M. F. Cao, J. Xu, W. L. Wang, X. Y. Lu, Q. T. Huang, X. X. Yue, G. K. Liu, L. Yang, B. Ren and Z. Q. Tian, Anal. Chem., 2024, 96, 6550–6557 CrossRef CAS PubMed.
- J. Zhang, P. L. Xin, X. Y. Wang, H. Y. Chen and D. W. Li, J. Phys. Chem. A, 2022, 126, 2278–2285 CrossRef CAS.
- E. X. Tan, Y. Chen, Y. H. Lee, Y. X. Leong, S. X. Leong, C. V. Stanley, C. S. Pun and X. Y. Ling, Nanoscale Horiz., 2022, 7, 626–633 RSC.
- M. Sackmann and A. Materny, J. Raman Spectrosc., 2006, 37, 305–310 CrossRef CAS.
- S. Sloan-Dennison, G. Q. Wallace, W. A. Hassanain, S. Laing, K. Faulds and D. Graham, Nano Convergence, 2024, 11, 1–30 CrossRef PubMed.
- H. Zhang, N. Zhao, H. Li, M. Wang, X. Hao, M. Sun, X. Li, Z. Yang, H. Yu, C. Tian and C. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 51253–51264 CrossRef CAS.
- Y. X. Leong, E. X. Tan, S. X. Leong, C. S. Lin Koh, L. B. Thanh Nguyen, J. R. Ting Chen, K. Xia and X. Y. Ling, ACS Nano, 2022, 16, 13279–13293 CrossRef CAS PubMed.
- S. X. Leong, Y. X. Leong, E. X. Tan, H. Y. F. Sim, C. S. L. Koh, Y. H. Lee, C. Chong, L. S. Ng, J. R. T. Chen, D. W. C. Pang, L. B. T. Nguyen, S. K. Boong, X. Han, Y. C. Kao, Y. H. Chua, G. C. Phan-Quang, I. Y. Phang, H. K. Lee, M. Y. Abdad, N. S. Tan and X. Y. Ling, ACS Nano, 2022, 16, 2629–2639 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |