
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLocal structures and robust oxygen reduction performances of TiN-supported bimetallic Pt–Cu electrocatalysts for fuel cells†
Hirosuke
Matsui
 *a,
Asako
Shoji
a,
Chaoqi
Chen
a,
Xiao
Zhao
b,
Tomoya
Uruga
bc and
Mizuki
Tada
*a
*a,
Asako
Shoji
a,
Chaoqi
Chen
a,
Xiao
Zhao
b,
Tomoya
Uruga
bc and
Mizuki
Tada
*a
aDepartment of Chemistry, Graduate School of Science & Research Center for Materials Science (RCMS) & Integrated Research Consortium on Chemical Science (IRCCS) & Institute for Advanced Study, Nagoya University, Furo, Chikusa, Nagoya, Aichi 464-8602, Japan. E-mail: matsui.hirosuke.x1@f.mail.nagoya-u.ac.jp; tada.mizuki.u6@f.mail.nagoya-u.ac.jp
bInnovation Research Center for Fuel Cells, The University of Electro-Communications, Chofu, Tokyo, 182-8585 Japan
cJapan Synchrotron Radiation Research Center, SPring-8, Koto, Sayo, Hyogo 679-5198, Japan
First published on 7th February 2024
Abstract
The local structures and oxygen reduction reaction (ORR) performances of Pt–Cu fuel-cell electrocatalysts on robust TiN nanoparticles were investigated. The structures of the PtCu/TiN catalysts were fully characterized by XRF, XRD, TEM, STEM-EDS, XAFS, and XPS, which revealed that loaded Cu was partially alloyed with Pt and fine Pt–Cu alloy nanoparticles (2–3 nm) were formed on TiN. The alloying of Cu to Pt was saturated at the Cu/Pt molar ratio (X) of 1.0 and the fraction of Cu in the alloy structure was estimated to be 40%. The ORR activity was found to be affected by the formation of the Pt–Cu alloy structure, and the electrocatalytic activity enhanced with increasing X. Notably, the best ORR performance was obtained at X = 1 and further Cu loading produced cationic Cu2+ species, which did not improve the ORR activity. We also evaluated the performance of the PtCu/TiN catalyst (X = 1) on a PEFC MEA, which showed 1.5 times higher mass-specific power density, compared to an MEA with a commercial Pt/C catalyst, and significant durability (70% of ECSA after 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles of the typical accelerated degradation test of the PEFC).
000 cycles of the typical accelerated degradation test of the PEFC).
Introduction
Polymer electrolyte fuel cells (PEFCs) are one of the essential technologies for the local production and consumption of electronic energy for low-carbon or carbon-neutral society.1 Supplied fuel (H2) at the anode in a membrane electrode assembly (MEA) is converted to H+ ions and electrons by an electrocatalyst translating to the cathode side through a polymer electrolyte membrane between the anode and cathode with an external circuit. Compared to the anode, there are still various challenges in cathode catalysts for the further improvement of PEFC activity and durability.2 One of the typical cathode catalysts, carbon-supported Pt catalysts (Pt/C), working in acidic environments often deteriorates under PEFC operating conditions with high electrode potential and high oxygen concentration, resulting in serious dissolution and degradation of the cathode catalyst and support.3–5 The corrosion of the carbon support occurs at a low electrode potential below 0.2 V, leading to the detachment and migration of supported electrocatalysts.6 There have been many reports to improve the oxygen reduction reaction (ORR) performance of Pt catalysts by alloying with secondary transition metals,7–9 controlling nanostructures,10–12 functionalizing with polymers and oxides,13,14 capsulizing into porous materials,15,16 and developing alternative materials.17–19 However, further improvements in the weak interaction between Pt catalysts and the C support and the robustness of the catalyst support are still essential in the enhancement of the activity and durability of electrocatalysts for the development of PEFC catalysts.Recently, transition metal nitrides (TMNs) have been explored as an alternative to the conventional carbon support due to their chemical tolerance, high electroconductivity, and high thermal stability.20,21 It has been reported that the Pt nanoparticles prepared on robust TMN supports facilitate the mobility of electron transfer and also induce strong metal–support interaction through N-termination on TMN surfaces.22,23 The series of TiN-supported Pt catalysts with added second metals (PtM/TiN or Pt/TiMN) have shown high ORR performances.24–27 In particular, Pt–Cu nano-frames on unique TiN nanoarchitectures exhibited significantly increased ORR performances compared to Pt/C catalysts. The role of nitride has been discussed from the viewpoints of the strain effects by the insertion of nitrides to the Pt catalysts, the modulation of the electronic state of the Pt catalysts, and the ligand effects with the nitride support surface.24–28 X-ray photoelectron spectroscopy (XPS) suggested the existence of Pt0+ species on TiN, but the local structures of the active Pt species in the Pt–Cu alloy catalysts on TiN and their working states under electrocatalytic conditions were not reported yet.24–27
Here, we prepared Pt–Cu alloy catalysts supported on commercial TiN nanoparticles with different Cu/Pt loadings and systematically investigated the structures and ORR performances of the PtCu-X/TiN (X: loaded Cu/Pt molar ratio) catalysts. It was suggested that some of the Cu was incorporated in the Pt–Cu alloy, which was a key factor for the enhancement of the ORR activity on the series of the PtCu-X/TiN catalysts. A membrane electrode assembly (MEA) using the prepared PtCu-1.0/TiN catalyst as the cathode catalyst showed high power density and significant durability for a typical accelerated degradation test (ADT).
Results and discussion
Preparation of Pt–Cu alloy catalysts supported on TiN particles
Commercial TiN nanoparticles with an average diameter of 20 nm (catalog spec) were used for the support, whose surface area was estimated to be 55 m2 g−1 by N2 adsorption analysis (Fig. S1†). Cu(CH3COO)2·H2O and H2PtCl6 were impregnated in a mixture of distilled water and ethanol (3/2 = v/v) and reduced by a NaBH4 solution (Fig. 1(a)). The Pt loading was fixed, while the Cu loading was varied in the range of 0–12 wt% by changing the amount of the Cu precursor for the catalyst preparation. The X-ray fluorescence (XRF) analysis of the prepared samples suggested that around 60–90% of the used Pt and Cu precursors were loaded on TiN, and Cu/Pt molar ratios in the obtained catalysts (denoted as X) were estimated in the range from 0 to 2.4 (Table S1†). In the following discussions, the prepared samples are denoted as PtCu-X/TiN (X: loaded Cu/Pt molar ratio, not alloy composition). | ||
| Fig. 1 (a) Preparation scheme of PtCu-X/TiN by an impregnation method using Pt and Cu precursors. By changing the amount of the Cu precursor, the set of PtCu-X/TiN catalysts with different Cu/Pt molar ratios (X = 0–2.4) was systematically synthesized. (b) Powder XRD patterns of the PtCu-X/TiN catalysts with different Cu loadings (X = 0–2.4). The XRD profiles of the TiN support (data) and Pt (ICSD180981) and Cu (ICSD180109) references are also presented. (c) Relationship between the Pt(111) peak positions of the PtCu-X/TiN catalysts (left) and the estimated alloy composition (Cu/Pt ratio) by considering Vegard's rule with the (111) peak positions of Pt(111) (2θ = 39.8°, blue dotted line) and Cu(111) (2θ = 43.3°, red dotted line) (right) against X. | ||
The powder XRD profiles of the PtCu-X/TiN catalysts (X = 0–2.4) showed two sharp peaks attributed to TiN and a broad peak attributed to Pt(111) as shown in Fig. 1(b). The Pt(111) peak of a Pt/C catalyst was similar to that of Pt metal, while it was little shifted to a higher angle for the Pt/TiN catalyst without Cu, which may indicate the lattice distortion of Pt with N-termination of the TiN support. With increasing Cu loading from 0 to 12 wt% (X = 0–2.4), the observed Pt(111) peak at 2θ of around 39.8° was shifted to a higher angle with decreasing signal intensity as shown in Fig. 1(b). Compared with those of pure Pt (ICSD180981) and pure Cu (ICSD180109), these results suggested the alloying of Cu into the framework of the Pt fcc lattice. Regarding Vegard's rule that explains linearity between lattice constants and alloy composition,29 the composition of the formed PtCu alloy in PtCu-X/TiN was estimated to be 0.15 (X = 0.3), 0.40 (X = 1.0), and 0.41 (X = 2.4).
The estimated compositions of the Pt–Cu alloys by Vegard's rule were plotted against the loaded Cu/Pt molar ratio (X) determined by XRF (Fig. 1(c)). Accompanied by the increase in X, the Cu composition in the Pt–Cu alloy almost linearly increased in the X range of 0–1.0, until saturation occurred at around X of 1.0 with an alloy composition of 0.4 at the maximum. Further loading of Cu (X > 1.0) did not increase the alloying of Cu into Pt. The maximum alloy ratio of 0.4 in the PtCu-1.0/TiN catalyst suggested that 40% of Cu was incorporated into the Pt–Cu alloy. There were no distinct peaks attributed to other Cu species (Cu and Cu oxides) in the XRD profiles, indicating the negligible formation of aggregates of such Cu species in the catalyst. Similar intensities and positions of the XRD peaks of the TiN support suggested negligible changes in the TiN support affected by the alloy structure (Fig. 1(b)).
The transmission electron microscopy (TEM) images of the PtCu-1.0/TiN catalyst suggested the formation of nanoparticles deposited on the surface of the TiN support (Fig. 2(a-1)). The size distribution of the nanoparticles was estimated from the TEM images, and the average particle size was estimated to be 2.3 ± 0.8 nm. In the cases of low Cu loading, the formation of large Pt particles was observed by TEM (Fig. S2(a)†), and the higher intensity of the XRD Pt (111) peak at the low Cu loading would be caused by the aggregation of the Pt particles. The lattice spacing of the PtCu-1.0/TiN catalyst estimated from the fringe pattern of the highly magnified TEM image was 0.218 ± 0.008 nm (Fig. 2(a-2)), which is between that of Pt (111) (0.227 nm) and that of Cu (111) (0.209 nm). The expected Pt–Cu alloy composition derived from the lattice parameter was almost 50%, whereas negligible changes in the lattice spacing of TiN (200) (0.212 ± 0.008 nm) were observed for the samples with and without Cu loading. The lattice fringes attributed to Cu (111) were not observed. Scanning transmission electron microscopy-energy dispersive X-ray spectroscopy (STEM-EDS) analysis was also conducted for the PtCu-1.0/TiN catalyst (Fig. 2(b)). The STEM-EDS images showed the overlapping of Pt and Cu distributions, suggesting the formation of Pt–Cu alloy nanoparticles on the TiN support. Cu species dispersed on the TiN surface were also observed independent of the Pt–Cu alloy, indicating that the excess Cu species were not incorporated into the Pt–Cu alloy nanoparticles but highly dispersed on the TiN surface. Similar trends were observed for both PtCu-0.2/TiN and PtCu-2.4/TiN with different Cu loadings (Fig. S2(b and c)†).
 | ||
| Fig. 2 (a-1) TEM image with particle size distribution and (a-2) highly magnified TEM images of the PtCu-1.0/TiN catalyst. The averaged particle size was estimated by the Gaussian curve fitting of the particle size distribution (red line). (b) STEM-EDS mappings for Pt Lα, Cu Kα, and Ti Kα of the PtCu-1.0/TiN catalyst with (1) low and (2) high magnifications. | ||
The local structures of Pt, Cu, and Ti in the PtCu-X/TiN catalysts were characterized by X-ray absorption fine structure (XAFS) analysis. Fig. 3(a) and S3(a)† show the Pt LIII-edge X-ray absorption near-edge structure (XANES) spectra of the PtCu-X/TiN catalysts with different Cu loadings (X = 0–2.3). The Pt LIII-edge white-line height of Pt/TiN without Cu was similar to that of Pt foil, indicating minor electronic charges between Pt and TiN. It was found that the XANES spectrum of PtCu-1.0/TiN showed a slight positive energy shift of the white-line peak and broadening at the shoulder around 11570 eV compared to that of Pt/TiN. This finding can be explained by the modulation of the electronic state and local coordination structure by incorporating Cu onto the Pt lattice.30–32 Similar trends were observed for the PtCu-X/TiN catalysts (X > 0) as shown in Fig. S3(a).†
 | ||
| Fig. 3 (a) Normalized Pt LIII-edge XANES spectra of PtCu-1.0/TiN (blue line), Pt/TiN (red line), and Pt foil (black dotted line). (b) Pt LIII-edge k3-weighted EXAFS-Fourier transforms (k = 3.0–14.5 × 10 nm−1, black line) with curve-fitting results (R = 1.9–3.1 × 10−1 nm, red line) of PtCu-1.0/TiN and Pt/TiN. Other XANES and EXAFS spectra and the summary of fitting results are presented in Fig. S3 and Table S2.† (c) Normalized Cu K-edge XANES spectra of PtCu-1.0/TiN (red line), Cu foil (black dotted line), and CuO (blue dotted line). (d) The interatomic distances of Pt–Pt (red circle) and Pt–Cu bonds (white square) and (e) the coordination numbers (CNs) of Pt–Pt (red circle) and Pt–Cu bonds (white square) estimated by the curve-fitting analysis of the Pt LIII-edge EXAFS Fourier transforms. (f) Relationship between the fraction of CNPt–Cu to CNTotal (CNPt–Pt + CNPt–Cu) and the estimated alloy composition by the XRD-Vegard's rule. | ||
The curve-fitting analysis of Pt LIII-edge k3-weighted extended X-ray absorption fine structure (EXAFS)-Fourier transforms (FTs) revealed two major contributions of Pt–Pt bonds and Pt–Cu bonds, whose fitting results are summarized in Table S2.† The EXAFS-FT of Pt/TiN without Cu was fitted with a single scattering shell of Pt–Pt at 2.76 × 10−1 nm (Fig. 3(d)). When Cu was incorporated, the EXAFS-FT major peak was shifted to a shorter R-range than that of Pt/TiN without Cu as shown in Fig. 3(b) and S3(c),† and the EXAFS fitting analysis did not converge by the use of the Pt–Pt scattering shell only. The EXAFS-FTs of PtCu-X/TiN (X > 0) were successfully fitted by the use of two shells of Pt–Pt and Pt–Cu bonds, whose results are summarized in Fig. 3(d and e). With increasing X, the Pt–Pt bond distance decreased from 2.76 × 10−1 nm (X = 0) to 2.73 × 10−1 nm (X = 0.3), 2.69 × 10−1 nm (X = 1.0), and 2.70 × 10−1 nm (X = 2.3) as shown in Fig. 3(d) and Table S2.† On the other hand, the Pt–Cu bond distances were almost constant at around 2.65–2.68 × 10−1 nm. The incorporation of Cu with a shorter ionic radius relative to Pt made a compressed distortion in the Pt lattice, thus resulting in changes in the Pt–Pt bond distance.
We also plotted changes in the coordination numbers (CNs) of the Pt–Pt and Pt–Cu bonds (hereafter, denoted as CNPt–Pt and CNPt–Cu) in Fig. 3(e). The CNPt–Pt of Pt/TiN was fitted as 8.3 ± 0.4 (X = 0), and the CNPt–Pt and CNPt–Cu of PtCu-X/TiN were 7.0 ± 0.6 and 1.3 ± 0.2 (X = 0.3), 5.8 ± 1.3 and 2.5 ± 0.4 (X = 1.0), 6.3 ± 1.3 and 2.4 ± 0.8 (X = 1.3), and 6.7 ± 1.6 and 2.2 ± 0.6 (X = 2.3), respectively (Table S2†). The summation of CNPt–Pt and CNPt–Cu, which presents the total of metal–metal bonds on the metal particle (CNTotal), was found to be almost constant at around 8–9 for all the PtCu-X/TiN catalysts. Accompanied by the increase in X, CNPt–Cu increased and saturated at 2–2.5, also consistent with the results of XRD. The calculated ratios of CNPt–Cu/CNTotal, which indicate the incorporated Cu ratios in the Pt–Cu alloy of the samples, were plotted against the ratio of Cu in the Pt–Cu alloy structure as estimated by the XRD-Vegard's rule, showing an almost linear relationship as shown in Fig. 3(f).
The Cu K-edge XANES spectra suggested the mixture of metallic Cu and high-valent Cu in PtCu-X/TiN (Fig. 3(c) and S4(a)†). With increasing Cu loading, the high valent Cu species with the main peak at around 8998 eV increased, whose shape was similar to CuO. Considering the facts that some of the Cu was alloyed into Pt particles forming Pt–Cu alloy and excess Cu species would be dispersed on the surface of the TiN support, metallic Cu in the Pt–Cu alloy and dispersed Cu oxide may co-exist in the PtCu-X/TiN catalysts. The Ti K-edge XANES spectra of TiN, Pt/TiN, and PtCu-1.0/TiN were similar to each other, suggesting that the loaded Pt and Cu did not affect the local structure of TiN (Fig. S4(b)†). These results were consistent with the XRD, TEM, and XAFS analyses.
There were many reports on the preparation of Pt bimetallic electrocatalysts for the ORR such as Pt–Co and Pt–Ni.33 We investigated the surface electronic state of the Pt and Cu species in the PtCu-1.0/TiN catalyst by XPS. After subtracting the background by the Shirley method, Pt 4f peaks (EB = 70–80 eV) were fitted by Gaussian functions, considering the spin–orbit splitting between 4f7/2 and 4f5/2 and the overlapped peak of Cu 3p (EB = 76.0 eV) as shown in Fig. 4(a). We fitted the XPS spectrum using five Gaussian curves, which were 4f7/2 and 4f5/2 peaks of metallic Pt at EB = 71.3 eV and 74.6 eV (area percentage of 72%), Pt2+ at EB = 74.0 eV and 77.3 eV (23%), and overlapped Cu 3p (5%) (Table S3†). The mean free path length of excited photoelectrons of Pt 4f was estimated to be 1.7 nm,34 allowing signal detection from the total volume of Pt particles, thus metallic Pt was a major species in the catalyst. The observed high-valent species can be assigned to Pt2+ from the references (Fig. S5(a)†),34 which would be surface Pt species exposed to air. The binding energy assigned to Pt0+ species was observed at a similar binding energy to that of Pt metal (71.3 eV),35 suggesting negligible electronic contribution of TiN and Cu to Pt in the PtCu-X/TiN catalysts.
 | ||
| Fig. 4 (a) Pt 4f and (b) Cu 2p3/2 XPS spectra with the Gaussian fitting results of the PtCu-1.0/TiN sample. | ||
The Cu 2p3/2 XPS spectrum of PtCu-1.0/TiN, showed two peaks at 931.8 eV and 934.2 eV, which were attributed to Cu0+ and Cu2+ (Fig. 4(b) and S5(b)†).35 No obvious peak shift of Pt 4f of the Pt–Cu alloy system has been reported, whereas a linear peak shift of Cu 2p3/2 has been observed in the Pt–Cu alloy composition.36 The linear relationship between the Cu/Pt alloy composition and the core-level shift of Cu 2p3/2 suggested that the composition of the Pt–Cu alloy particles in the PtCu-1.0/TiN catalyst could be estimated to be 0.3–0.4, in agreement with the results of the XRD analysis using Vegard's rule and the Pt LIII-edge EXAFS-fitting analysis. The observed Cu2+ species was supposed to originate from the excess Cu species dispersed on the exterior of the Pt–Cu alloy particles.
Based on these results, the structures of the series of PtCu-X/TiN catalysts can be explained as follows.
(1) Some of the loaded Cu was alloyed to form the Pt–Cu alloy nanoparticles at 2–3 nm size. Accompanied by the increase in X, the fraction of Cu in the alloy increased and saturated at 0.3–0.4 at X = 1. Further Cu loading (X > 1.0) did not increase the Cu/Pt alloy composition. Since a clear charge transfer from Cu to Pt was not observed by XANES and XPS, it is suggested that the major role of Cu is distortion of the Pt lattice as well as the regulation of Pt dispersion and particle size on the TiN surface. In the phase diagram of Pt–Cu intermetallics, there was a phase boundary between a disordered fcc-lattice and the fcc-based layer-by-layer lattice of Pt and Cu planes (L11 phase) at the lower Cu content region of the PtCu alloy system.37 To increase the Cu content in the PtCu alloy, the re-arrangement of atoms is required at higher thermal energy above 1000 K and hence the PtCu alloy composition prepared at room temperature reached a plateau at around 0.3–0.4.
(2) Excess Cu species were distributed at the exterior of the Pt–Cu alloy nanoparticles and dispersed on the surface of the TiN support as high-valent Cu2+.
Evaluation of ORR activity by the RDE method
To evaluate the ORR activity of the prepared catalysts, the rotating disk electrode (RDE) method was employed. 30 wt% of Ketjen black was added to improve the dispersion of catalyst ink and electronic conductivity among the TiN nanoparticles.38 The cyclic voltammograms (CVs) of the PtCu-X/TiN catalysts (X = 0–2.4) are presented in Fig. 5(a-1) and S6(a).† Positive shifts of O adsorption peaks (60 mV shift for the anodic scan) and O desorption peaks (30 mV shift for the cathodic scan) were observed on PtCu-1.0/TiN as compared to a commercial Pt/C catalyst (TKK, TEC10E50E) (Fig. 5(a-2)). With increasing the Cu loading, the electrochemical surface areas (ECSAs) of the PtCu-X/TiN catalysts enhanced significantly from 20.4 m2 gPt−1 (X = 0) to 54.6 m2 gPt−1 (X = 1.0), suggesting that the high dispersion of the Pt catalysts with Cu also affects electrochemical results (Table S4†). Further increasing Cu loading (X > 1) did not improve the ECSA (Fig. 5(b)). The XRD and Pt LIII-edge XAFS results showed that excess Cu (X > 1.0) did not increase the Cu ratios of the Pt–Cu alloy nanoparticles, which was consistent with the trend in ECSA.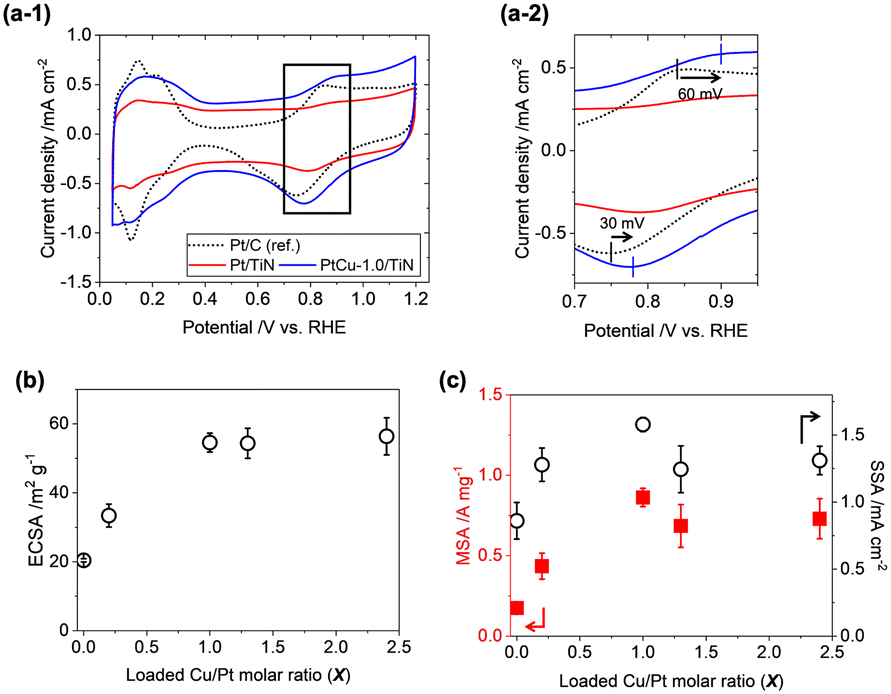 | ||
| Fig. 5 (a-1) CV profiles for Pt/TiN (red), PtCu-1.0/TiN (blue), and commercial Pt/C (TKK, TEC10E50E; black dotted line). (a-2) The expanded figure of the black square area in (a-1). (b) Electrochemical surface area (ECSA) and (c) mass specific activity (MSA, red square, left axis) and surface specific activity (SSA, white circle, right axis) for PtCu-X/TiN catalysts (X = 0–2.4). | ||
Linear sweep voltammograms (LSVs) in O2-saturated HClO4 solutions were measured with a rotating working electrode (WE) at 400, 900, 1600, and 2500 rpm speeds (Fig. S6(b)†). The Koutecký–Levich (KL) plots showed a nearly linear relationship between oxygen diffusion (ω−1/2) and reaction kinetics (Ik−1). From the slope of KL plots at 0.9 VRHE, the electron transfer number of the prepared catalysts was estimated to be 3.8 ± 0.4, suggesting that the 4e− transfer ORR process is dominant (Fig. S6(c)†). Mass-specific activity (MSA) and surface-specific activity (SSA) were estimated by normalizing the intercept of kinetic current at ω−1/2 = 0 (jk) with the loaded Pt weight and ECSA. The plots of the MSA and SSA variations of PtCu-X/TiN (X = 0–2.4) showed similar trends to ECSA (Fig. 5(c) and Table S4†).
The fact that the ORR performance is highly related to the incorporated ratio of Cu into the Pt–Cu alloy suggests that Pt–Cu alloy nanoparticles are active for the ORR. The slight decrease in the MSA and SSA (X > 1) may be caused by the excess Cu2+ species dispersed on the catalyst surface. The best ORR performance was observed on PtCu-1.0/TiN; 0.86 A mgPt−1 (MSA) and 1.58 mA cmPt−2 (SSA), which were 5.1 times and 6.3 times larger than those of the commercial Pt/C catalyst (TEC10E50E, TKK). Compared to reported Pt/TiN catalysts (TiNiN@Pt: MSA 0.83 A mgPt−1, SSA 0.49 mA cmPt−2;24 Ti0.9Cu0.1N@Pt/NCNT: 1.06 A mgPt−1, SSA 0.69 mA cmPt−2;25 Pt3Cu nano-frame/TiN architecture: 5.32 A mgPt−1, 2.43 mA cmPt−2;26 Pt/Ti0.9Co0.1N: 0.46 A mg−1, 0.54 mA cm−2)27 presented in Table S5,† the present PtCu-1.0/TiN catalyst exhibited higher MSA and SSA than Ti0.9Cu0.1N@Pt/NCNT and Pt/Ti0.9Co0.1N and lower ORR activity than the Pt3Cu nano-frame/TiN architecture, which had a unique skeleton Pt structure on TiN nano-frames.
Electrochemical performance of the MEA with PtCu-1.0/TiN
The ORR performances of the above mentioned Pt–Cu–TiN catalysts were evaluated by the RDE method, but there were no reports on the evaluation of electrochemical performances in the PEFC membrane electrode assembly (MEA) with Pt–Cu/TiN catalysts. We prepared a practically available MEA for PEFC testing, based on a three-layered structure with an anode catalyst consisting of Pt/C (TEC10E50E, TKK; 0.5 mgPt cm−2) or Pd/C (TECPd(ONLY)E50E, TKK; 0.5 mgPd cm−2; used for operando XAFS measurement) and the prepared PtCu-1.0/TiN (0.11 mgPt cm−2) mixed with 30 wt% of Ketjen black (used as a conductive aid) as a cathode catalyst. A 3 cm × 3 cm-sized catalyst layer was hot-pressed onto a Nafion membrane (Sigma-Aldrich, NR-212).IV profiles were recorded supplying 1200 ccm of 20% O2/N2 at the cathode and the ORR activity of the MEA was evaluated. In the low current density area above 0.8 V, the slopes of the IV profiles of the commercial Pt/C and PtCu-1.0/TiN were similar to each other (Fig. 6(a)). The power plot showed the mass-specific maximum power density to be 1.5 W mgPt−1, which was 1.5 times larger than that of the commercial Pt/C catalyst (Fig. 6(a) and Table S6†). Incorporation of secondary transition metals can improve the oxygen reduction activity.7–9 Our PtCu-1.0/TiN catalyst exhibited a similarly increased ORR activity by incorporating Cu into Pt.
 | ||
| Fig. 6 (a) IV profiles for PtCu-1.0/TiN (○) and Pt/C (□) and mass specific power plots for PtCu-1.0/TiN (●) and Pt/C (■). (b) CV profiles of the MEA with PtCu-1.0/TiN before (after initial conditioning) and after 80000 cycles of the accelerated degradation test (ADT). (c) ECSA variation against the number of the ADT cycles for PtCu-1.0/TiN and Pt/C (TEC10E50E, TKK). | ||
Although the PEFC performance of the MEA highly depends on the PEFC structure and operating conditions, the prepared MEA with the PtCu-1.0/TiN cathode catalyst exhibited a comparable ECSA and MSA to the previously reported MEA with a Pt/TiN catalyst.38,39 Further investigation of MEA preparation methods will improve the electronic resistance and mass diffusion of the catalyst membrane, resulting in the improvement of the PEFC performance.
The durability of the prepared MEA with PtCu-1.0/TiN was tested by a typical accelerated degradation test (ADT), consisting of repeated voltage steps between 0.6 V and 1.0 V (for each 3 s) under a N2 flow at the cathode.40 The ECSA estimated from the CV profile was originally 60.3 m2 gPt−1 after the initial conditioning (Fig. 6(b and c) and S7†), which was almost comparable to that measured by RDE (54.6 m2 gPt−1). The ECSA of the MEA with the commercial Pt/C catalyst showed 72.4 m2 gPt−1 after the initial conditioning.
It is well known that the standard Pt/C catalyst significantly degrades under PEFC voltage operating conditions, and the dissolution and migration of Pt are severe in MEAs. The ADT, which accelerates the degradation of the cathode catalyst by typical voltage operation cycles, causes a significant decrease in ECSA, and the ECSA of the commercial Pt/C catalyst significantly decreased as shown in Fig. 6(c). After applying 20000 ADT cycles, the ECSA of the commercial Pt/C catalyst was decreased to 29.9 m2 gPt−1, which corresponds to 40% of the initial ECSA. It is to be noted that the PtCu-1.0/TiN catalyst exhibited remarkable stability in the ADT compared to Pt/C as shown in Fig. 6(c). After applying 80000 ADT cycles, the ECSA remained at 70% of the initial value. Considering the rapid decrease in the ECSA of the commercial Pt/C catalyst, the high tolerance of the PtCu-1.0/TiN catalyst was obvious under real PEFC operating conditions.
The structures of the PtCu-1.0/TiN catalyst under PEFC working conditions and after the PEFC use
The use of hard X-rays with high transmission properties enables us to reveal the real structures of working catalysts in PEFC MEAs.41 The operando Pt LIII-edge and Cu K-edge XANES spectra of the PtCu-1.0/TiN catalyst in the MEA were measured under PEFC operating conditions (Fig. 7(a and b)). The MEA with Pd/C as the anode catalyst instead of Pt/C was used to avoid interference of X-ray absorption and conditioned under a N2 flow at the cathode by applying 0.6 V for 1 h. The cell voltage was operated at 0.4 V and 1.0 V, and the operando XAFS spectra under both voltages were successfully recorded. The Pt LIII-edge XANES spectrum measured at 0.4 V suggested that the Pt was metallic as shown in Fig. 7(a). At 1.0 V, a slight increase in the Pt LIII-edge white-line height was observed on the MEA with the PtCu-1.0/TiN cathode, and the difference between the white-line height at 1.0 V and that at 0.4 V, which is the index of the redox properties of the Pt catalyst, was smaller than that of the commercial Pt/C catalyst. These results indicate that the oxidation of Pt in the Pt–Cu alloy nanoparticles was widely inhibited compared to Pt/C. Similar trends were observed on Pt-alloy catalysts with higher activity than pure Pt, and the positive suppression of unfavorable oxidation on the active Pt surface by controlling the oxygen binding energy is considered one of the key factors for ORR activity.25,26,42,43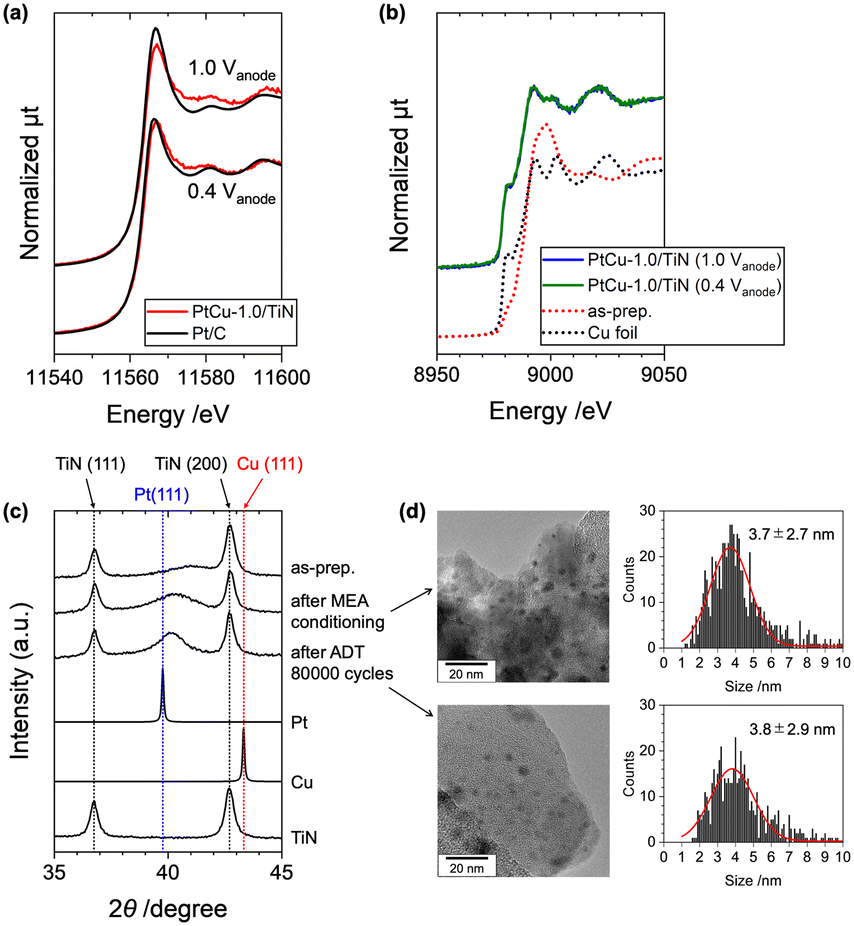 | ||
| Fig. 7 (a) Operando Pt LIII-edge XANES spectra of the MEAs with PtCu-1.0/TiN (red) and Pt/C (black) cathodes at PEFC cell voltages of 1.0 V and 0.4 V. The data of samples were taken in transmission mode. (b) Operando Cu K-edge XANES spectra of the MEA with the PtCu-1.0/TiN cathode at PEFC cell voltages of 1.0 V (blue) and 0.4 V (green) with those of as-prepared PtCu-1.0/TiN (powder, red dotted line) and Cu foil (black dotted line) as references. The data of samples were taken in XRF yield. (c) Powder-XRD profiles of PtCu-1.0/TiN collected from the cathode catalyst layer of the used MEA. The as-prepared catalyst (powder), after the MEA conditioning and (c) after 80000 cycles of the ADT. XRD reference profiles of Pt (ICSD180981) and Cu (ICSD180109) from the ICSD database are presented. (d) TEM images with the particle size distributions of the PtCu-1.0/TiN catalyst collected from the MEA cathode catalyst layer (after the MEA conditioning and after 80000 cycles of the ADT). The red line in the particle size distribution shows Gaussian fitting to estimate the average particle size. | ||
In contrast, the Cu K-edge XANES showed different features (Fig. 7(b)). In the as-prepared PtCu-1.0/TiN catalyst, oxidized Cu2+ species were observed in addition to metallic Cu, but metallic Cu became major in the conditioned MEA at 0.4 V. We did not find any difference between the Cu K-edge XANES spectra at 0.4 V and 1.0 V as shown in Fig. 7(b), indicating negligible oxidation on the loaded Cu in the catalyst. The synchronized structure–ORR performance relationship suggested that the alloying of Cu into Pt was the key factor for the ORR activity and Cu in the Pt–Cu alloy affected the suppression of the unfavorable oxidation of Pt under the ORR conditions.
After the conditioning and ADT cycles, the MEAs were removed from the PEFC cell, and the cathode catalysts were scratched from the MEA and were analyzed by XRD and TEM. The Pt(111) peak of the collected catalyst after the MEA conditioning was negatively shifted as shown in Fig. 7(c), indicating the partial dealloying of Cu during the MEA conditioning process. However, the (111) peak was observed at 2θ = 40.2°, which was higher than that of Pt(111) (2θ = 39.8°), and the Pt–Cu alloy composition after the electrochemical conditioning was found to be 28% to the initial alloy composition, suggesting that the loaded Cu in the Pt–Cu alloy still remained after the electrochemical operation. It is to be noted that the XRD Pt(111) peak after the 80000 cycles of the ADT was similar to that before the ADT (after the conditioning), suggesting that the Pt–Cu alloy catalyst was stable under the ADT process. The estimated particle sizes of the PtCu catalysts from the TEM images were not changed after the ADT cycles (3.7 ± 2.7 nm ⇒ 3.8 ± 2.9 nm; Fig. 7(d)). In contrast, the commercial Pt/C catalyst (TKK TEC10E50E) exhibited significant particle aggregation with large changes in the particle size distribution (2.6 ± 0.8 nm ⇒ 5.4 ± 1.8 nm).44 These results suggested the high stability of the PtCu/TiN catalyst for the ADT cycles.
The electrochemical operation often accelerates the transformation of a Pt alloy to a thermally stable structure by changing the chemical composition of the alloy.45–48 The initial conditioning by the cell voltage operation would prepare the stable Pt–Cu alloy nanoparticles and the formed Pt–Cu alloy nanoparticles (11% of Cu was in the alloy structure) exhibited not only favorable ORR activity but also stable ORR performances for the ADT process. On conventional Pt/C catalysts, weak interaction between the Pt catalyst and carbon support and carbon corrosion under operation have been considered as some of the major reasons for catalyst degradation.49 The high acidic tolerance and high electronic conductivity of the TiN support and the unique Pt–Cu alloy particles formed on the TiN surface were considered to affect the high activity and durability of the PtCu-1.0/TiN catalyst in the MEA.
Conclusion
In conclusion, a set of PtCu/TiN catalysts with different Cu/Pt loadings was systematically synthesized by a simple impregnation method and complementarily characterized by XRF, XRD, TEM, STEM-EDS, XAFS, and XPS techniques. The RDE experiments showed the increases in MSA and SSA, with increasing loaded Cu/Pt ratio. The loaded Cu was partially alloyed with the Pt nanoparticles, resulting in compressed lattice strain and controlling the particle size and distribution. The best performing electrocatalyst PtCu-1.0/TiN used as a cathode material in a PEFC MEA showed 1.5 times higher mass-specific power density, compared to the commercial Pt/C. Additionally, outstanding durability was observed, maintaining over 70% of the initial ECSA after 80000 ADT cycles. Operando Pt LIII-edge XANES analysis showed a small response of the Pt oxidation state relative to the applied cell voltages, suggesting that the chemical interaction with the loaded Cu and the use of the TiN surface as the support accelerated the facile H2O desorption during the ORR, which is considered as one of the major factors that control the high activity and durability in this sample system.
Experimental
Preparation of PtCu-X/TiN catalysts
Commercial TiN nanoparticles (EMJapan Co., NI-TIN-2, purity > 99.2%, particle size: 20 nm, manufacturer data) were used as a catalyst support without any pretreatment. Other chemicals were purchased from Wako and Sigma-Aldrich and used as received. PtCu-X/TiN catalysts were prepared by a simple impregnation method. 0 to 0.11 g of Cu(II) acetate monohydrate (Wako) was dissolved in 50 mL of a distilled water/ethanol (30/20 = v/v) mixed solution and 1.31 g of 8 wt% H2PtCl6 solution in water (Sigma-Aldrich, 20 wt% of Pt to the TiN support) was added and stirred at ambient temperature for 1 h. Then, 0.2 g of TiN was added and stirred at ambient temperature for 1 h. 50 mL of 0.1 M NaBH4 (Wako) solution in distilled water was prepared and added to the mixed solution at once and stirred at ambient temperature for 1 h. After filtration, the obtained sample was washed with distilled water/ethanol solution (3/2 = v/v) and dried overnight.Characterization of PtCu-X/TiN catalysts
The N2 adsorption of the purchased TiN was recorded on a Shimadzu, Micromeritics ASAP-2020 at 77 K. 1.21 g of a sample taken in a glass cell was pretreated at 473 K under vacuum for 2 h before a BET measurement. The amounts of loaded Pt and Cu on TiN were estimated by using XRF on a JEOL, JSX-1000S. The emission lines of Pt Lα and Cu Kα relative to Ti Kα were analyzed by using standard curves. The calibration samples were prepared by an impregnation method in ethanol using 8 wt% of H2PtCl6 solution in water (Sigma-Aldrich), Cu acetate monohydrate (Wako), and rutile TiO2 (JRC-TIO-3).The powder-XRD patterns were measured on PANalytical Empyrean equipment using Cu Kα as an X-ray source. After subtracting the background, the positions, heights, and FWHMs of the XRD peaks were analyzed by curve-fitting using Gaussian functions. The alloy compositions of the prepared PtCu-X/TiN catalysts were evaluated by Vegard's rule, which explains the linearity of the lattice constant and composition in the alloy structure,29 considering the peak positions of Pt(111) (2θ = 39.8°, Pt (not Pt/C supported catalyst), ICSD180981) and Cu(111) (2θ = 43.3°, ICSD180109). TEM and STEM-EDS images were recorded on a TEM microscope (JEOL, JEM-ARM200F Cold, 200 kV) and the sample was supported on a Mo grid for the measurements. The sizes of 200–300 isolated particles were counted, and the average of the long and short diameters was defined as the particle size. The averaged particle size and distribution range were estimated by the Gaussian fitting of the particle size distribution and defined as the peak position and full width at half maximum (FWHM) of the fitted Gaussian curve.
XAFS spectra at Pt LIII, Cu K, and Ti K-edges were measured in a transmission mode at the BL36XU and BL37XU stations of SPring-8, and at the BL5S1 and BL11S2 stations of Aichi-SR. A sample was formed into a pellet with φ1 cm size and placed in an X-ray optical path. The incident and transmission X-ray signals were monitored by a set of two ion chambers. The XAFS spectra were processed and analyzed using the FEFF8.5 code (ifeffit).50 The absorption edge was defined as the inflection point of the first derivation for the standard samples (Pt, Cu, and Ti foil), and the X-ray energies were calibrated to 11559 eV at the Pt LIII-edge, 8979 eV at the Cu K-edge, and 4966 eV at the Ti K-edge, respectively. The k3-weighted EXAFS oscillations were Fourier-transformed into R-space and fitted with single-scattering shells in R-space. The coordination number (CN), interatomic distance (R), Debye–Waller factor, (σ2), and edge energy (ΔE0) were estimated (Table S2†).
XPS was performed by using an R4000 electron spectrometer (VG Scienta). The monochromatized Al Kα X-rays (hν = 1486.6 eV, SAX100 X-ray source with an XM780 monochromator, VG Scienta) were irradiated to the pellet of the PtCu-1.0/TiN sample mounted on a Mo-made sample holder during neutralizing an electron (VG Scienta, FG300 flood gun). The background of a measured spectrum was subtracted by the Shirley method, and the Pt 4f and Cu 2p peaks were fitted by multiple Gauss functions, after the calibration of binding energy using an Au 4f reference at 84.0 eV. The energy gap and the peak ratio between Pt 4f7/2 and Pt 4f5/2 were fixed at 3.3 eV and 4/3, respectively.
RDE measurements
The rate of mass diffusion was controlled by the rotation speed of the working electrode (WE) with a coated catalyst in a closed batch cell. A glassy carbon tip (φ = 5 mm) with a Teflon shaft (AFE2M050GC, PINE Research Inst.) was used as the WE and was polished in agglomerated alumina suspension (0.05 μm, AP-A, Struers) before use. The catalyst ink was prepared by dispersing a defined amount of catalyst and electrolyte onto the WE to make a thin-film electrode. The prepared catalyst was mixed with 30 wt% of Ketjen black (EC300J, Lion Specialty Chemicals Co., Ltd.) by using an agate mortar. The suspension of 10 mg of the mixed solid in 10 mL of Nafion dispersion (2-propanol (Wako)/Milli-Q water/5% Nafion (DE520 CS type, Wako) at a volume ratio of 8/10/0.1) was ultrasonicated in an ice bath for 20 min. The prepared catalyst ink was deposited several times and dried at 333 K for 20 min until a Pt loading of 12 μg cm−2. Commercial Pt/C (TEC10E50E, TKK) was also deposited on the WE and the Pt loading was 16.9 μg cm−2.Electrochemical measurements were performed using a three-electrode system with the WE, counter electrode (Pt sheet, 1 cm × 1 cm), and reversible hydrogen electrode (RHE) in 0.1 M HClO4 solution (70%, purity: 99.999% trace-metal basis, Sigma-Aldrich) at 293 K. For conditioning of the catalyst, 35 CV cycles between 0.05 and 1.2 VRHE at 50 mV s−1 scan speed were performed in N2 (99.99995%)-saturated HClO4 solution. ECSA was estimated by the averaged charge of H+ adsorption and desorption in the range of 0.05–0.4 VRHE considering the area of an electronic double layer as the background. The ORR activity was evaluated by LSV in the potentio-range of 0.05–1.0 VRHE at 10 mV s−1 speed in O2 (99.99995%)-saturated HClO4 solution. The WE rotation speeds were 400, 900, 1600, and 2500 rpm. The background current, which was measured in a similar potential range in N2-saturated HClO4 solution, and the ohmic iR drop compensation were considered. The inversed intercept of the Koutecký–Levich plot at 0.9 VRHE, which had good linearity of diffusion limiting current (A−1) and the square root of the WE rotation rate ((rad s−1)−1/2), gave the kinetic current density (jk). SSA and MSA were estimated by normalizing jk with ECSA and the loaded Pt amount on the WE, respectively.51
MEA preparation
A three-layered MEA was prepared with anode and cathode catalysts as follows. Pt/C (TEC10E50E, TKK, 50 Pt wt%) or Pd/C (TKK, TECPd(ONLY)E50E, 50 Pd wt%) was loaded at 0.5 mgPt, Pd cm−2 for an anode catalyst. Pd/C was used instead of Pt/C for the MEA for operando XAFS measurements to avoid interference of signals from the cathode and the anode Pt catalysts. The prepared PtCu-1.0/TiN mixed with 30 wt% of Ketjen black was loaded at 0.11 mgPt cm−2 at the cathode. The catalyst ink was prepared using an equivalent weight ratio of the catalyst and Nafion with 30 wt% of Ketjen black as a conductive aid. The prepared ink was deposited by spray coating in a 3 cm × 3 cm size of the catalyst-coated membrane, which was hot-pressed onto the Nafion membrane (t = 50 μm, NR-212, Sigma-Aldrich). For comparison of electrochemical experiments, an MEA with 0.5 mgPt cm−2 of commercial Pt/C (TEC10E50E, TKK) at both the anode and the cathode was also prepared without a carbon filler. At both sides of the MEA, carbon paper (t = 100 μm, TGP-H-030, Toray Ind., Inc.) was inserted as gas diffusion layers (GDLs).PEFC operation and durability test
The MEA with the GDLs was inserted into an in situ PEFC cell. The PEFC cell consists of carbon-made gas-flow channel plates (1 mm × 1 mm, serpentine), current collectors (Au-coated Cu plate), Kapton-made film heaters, and Mg–Al alloy-made separators at the anode and cathode sides.41 The cell temperature was monitored using a K-type thermocouple and heated at 353 K during operation. The cell voltage between the anode and cathode was controlled with a potentio/galvano stat (VSP, BioLogic Science Instruments Co. Ltd.) with a current amplifier (VMP3B-20, BioLogic Science Instruments), using the anode as a pseudo-reference and counter electrode in a three electrode configuration.The MEA sample was conditioned under the constant potential of 0.6 V for 1 h, supplying humidified H2 at the anode (RH = 93%, 150 mL min−1, purity: 99.99995%) and humidified N2 at the cathode (RH = 93%, 600 mL min−1, purity: 99.99995%). CV was measured between 0.05 and 1.0 V at 50 mV s−1 speed by stopping the gas flow at the cathode. ECSA was similarly estimated as described in the RDE measurements section. Then, the cathode gas was changed to 20% O2/N2 (99.99995%, RH = 93%, 1200 mL min−1), and the IV profile and its power plot were measured by a galvanic scan between 0.0 and 6.0 A mg−1 in 43 steps for 15 s each. Note that the power density was shown in the normalized value to the Pt weight because the loading amounts of Pt were different between the PtCu-1.0/TiN catalyst and the Pt/C reference catalyst. To investigate the durability of the prepared MEAs, the ADT with a rectangular voltage operation at 0.6 and 1.0 V for 3 s each was employed under a N2 flow at the cathode (RH = 93%, 600 mL min−1, purity: 99.99995%).
Operando XAFS analysis of the prepared MEA under PEFC operation conditions
The operando Pt LIII-edge and Cu K-edge XANES spectra of the MEA (Pd/C anode) were measured under PEFC operating conditions at the BL36XU station of SPring-8.52 The in situ PEFC cell with a channel and window for the X-ray transmission and emission was set on the X-ray optical path and was connected to a PEFC operating system (CNF52742, NF Co., Ltd.). For the XRF-yield XAFS measurement, the XRF signals emitted from the PEFC cell were monitored using a 21-ch Ge detector (EGPX 40 × 40 × 7-21PIX, Canberra).EX situ characterization of the catalyst used as the MEA
The used MEAs were removed from the PEFC cell and dried under a vacuum overnight. The catalyst was removed using a spatula and characterized by powder-XRD and TEM. For TEM analysis, the recovered catalysts were ultrasonicated in isopropanol for 30 min and the dispersion was dropped onto a Mo-made TEM grid.Author contributions
H. M. conceived the project and wrote the manuscript. A. S. synthesized the materials and carried out the electrochemical experiments and structural characterization. C. C. carried out the electrochemical analysis. X. Z. proposed the sample preparation for RDE measurements. T. U. contributed to the setup of XAFS measurements. M. T. directed this study.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was partially supported by a JSPS KAKENHI Grant-in-Aid for Young Scientists (20K15023), Kiban B (18H01940), NEDO, the Kao Foundation, the Foundation of Public Interest of Tatematsu, and the R-ing (Reaction Infography) World Research Unit (B-1) at Nagoya University. XAFS measurements were performed at SPring-8 (No. 2017B7821, 2018A1332, and 2018B7821) and Aichi-SR (No. 201903108). TEM, STEM-EDS, and SEM-EDS were measured at the High Voltage Electron Microscope Laboratory, Institute of Materials and Systems for Sustainability, Nagoya University, supported by the “Advanced Characterization Nanotechnology Platform” of MEXT, Japan. We thank Assistant Prof. T. Koitaya (IMS) and Prof. G. Samjeske (Nagoya Univ.) for their assistance with XPS measurements and English proofing.Notes and references
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- I. E. Stephens, J. Rossmeisl and I. Chorkendorff, Science, 2016, 354, 1378–1379 CrossRef CAS PubMed.
- L. Mølmen, K. Eiler, L. Fast, P. Leisner and E. Pellicer, APL Mater., 2021, 9, 040702 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. C. Meier, C. Galeano, I. Katsounaros, A. A. Topalov, A. Kostka, F. Schüth and K. J. J. Mayrhofer, ACS Catal., 2012, 2, 832–843 CrossRef CAS.
- J. Wu, X. Z. Yuan, J. J. Martin, H. Wang, J. Zhang, J. Shen, S. Wu and W. Merida, J. Power Sources, 2008, 184, 104–119 CrossRef CAS.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D.-J. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS PubMed.
- C.-Y. Ahn, J. E. Park, S. Kim, O.-H. Kim, W. Hwang, M. Her, S. Y. Kang, S. Park, O. J. Kwon, H. S. Park, Y.-H. Cho and Y.-E. Sung, Chem. Rev., 2021, 121, 15075–15140 CrossRef CAS PubMed.
- X. Zhao, S. Takao, T. Kaneko and Y. Iwasawa, Chem. Rec., 2019, 19, 1337–1353 CrossRef CAS PubMed.
- M. Kiu, Z. Zhao, X. Duan and Y. Huang, Adv. Mater., 2019, 31, 1802234 CrossRef PubMed.
- M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C.-Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov, Z. Lin, E. Zou, T. Yu, Q. Jia, J. Guo, L. Zhang, W. A. Goddard III, Y. Huang and X. Duan, Science, 2016, 354, 1414–1419 CrossRef CAS PubMed.
- G. Shi, T. Tano, D. A. Tryk, A. Iiyama, M. Uchida, Y. Kuwauchi, A. Masuda and K. Kakinuma, J. Catal., 2022, 407, 300–311 CrossRef CAS.
- K. Ichihashi, S. Muratsugu, S. Miyamoto, K. Sakamoto, N. Ishiguro and M. Tada, Dalton Trans., 2019, 48, 7130–7137 RSC.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. Hübner, U. Gernert, M. Gliech, M. Lerch and P. Strasser, Nat. Mater., 2019, 19, 77–85 CrossRef PubMed.
- L. Zhang, J. M. T. A. Fischer, Y. A. Jia, X. Yan, W. Xu, X. Wang, J. Chen, D. Yang, H. Liu, L. Zhuang, M. Hankel, D. J. Searles, K. Huang, S. Feng, C. L. Brown and X. Yao, J. Am. Chem. Soc., 2018, 140, 10757–10763 CrossRef CAS PubMed.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 CrossRef CAS PubMed.
- N. Wang, B. Lu, L. Li, W. Niu, Z. Tang, X. Kang and S. Chen, ACS Catal., 2018, 8, 6827–6836 CrossRef CAS.
- S. T. Oyama, The Chemistry of Transition Metal Carbides and Nitrides, 1st edn, Springer, 1996 Search PubMed.
- J. Zheng, W. Zhang, J. Zhang, M. Lv, S. Li, H. Song, Z. Cui, L. Du and S. Liao, J. Mater. Chem. A, 2020, 8, 20803–20818 RSC.
- B. Avasarala, T. Murray, W. Li and P. Halder, J. Mater. Chem., 2009, 19, 1803–1805 RSC.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS PubMed.
- X. Tian, J. Luo, H. Nan, H. Zou, R. Chen, T. Shu, X. Li, Y. Li, H. Song, S. Liao and R. R. Adzic, J. Am. Chem. Soc., 2016, 138, 1575–1583 CrossRef CAS PubMed.
- X. Tian, H. Tang, J. Luo, H. Nan, T. Shu, L. Du, J. Zeng, S. Liao and R. R. Adzic, ACS Catal., 2017, 7, 3810–3817 CrossRef CAS.
- Z. Wu, D. Dang and X. Tian, ACS Appl. Mater. Interfaces, 2019, 11, 9117–9124 CrossRef CAS PubMed.
- Y. Xiao, G. Zhan, Z. Fu, Z. Pan, C. Xiao, S. Wu, C. Chen, G. Hu and Z. Wei, J. Power Sources, 2015, 284, 296–304 CrossRef CAS.
- K. A. Kuttiyiel, K. Sasaki, Y. Choi, D. Su, P. Liu and R. R. Adzic, Nano Lett., 2012, 12, 6266–6271 CrossRef CAS PubMed.
- A. R. Denton and N. W. Ashcroft, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 43, 3161 CrossRef CAS PubMed.
- H. Matsui, S. Takao, K. Higashi, T. Kaneko, G. Samjeské, T. Uruga, M. Tada and Y. Iwasawa, ACS Appl. Mater. Interfaces, 2022, 14, 6762–6776 CrossRef CAS PubMed.
- T. Kaito, H. Tanaka, H. Mitsumoto, S. Sugawara, K. Shinohara, H. Ariga, H. Uehara, S. Takakusagi and K. Asakura, J. Phys. Chem. C, 2016, 120, 11519–11527 CrossRef CAS.
- Y.-F. Hsieh, Y.-C. Hsieh, Y.-C. Tseng, P.-W. Wu, G. G. Chao, P. Lin and J.-F. Lee, J. Alloys Compd, 2013, 552, 329–335 CrossRef CAS.
- J. Greeley, I. E. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- W. Smekal, W. S. M. Werner and C. J. Powell, Surf. Interface Anal., 2005, 37, 1059–1067 CrossRef CAS.
- NIST X-ray Photoelectron Spectroscopy Database, NIST Standard Reference Database Number 20, National Institute of Standards and Technology, Gaithersburg, MD, 20899, 2000, DOI:10.18434/T4T88K.
- E.-J. Cho and S.-J. Oh, J. Korean Phys. Soc., 1997, 31, 323–328 CAS.
- T. Abe, B. Sundman and H. Onodera, J. Phase Equilib. Diffus., 2006, 27, 5–13 CrossRef CAS.
- H. Shintani, K. Kakinuma, H. Uchida, M. Watanabe and M. Uchida, J. Power Sources, 2015, 280, 593–599 CrossRef CAS.
- A. Perego, G. Giuffredi, P. Mazzolini, M. Colombo, R. Brescia, M. Prato, D. C. Sabarirajan, I. V. Zenyuk, F. Bossola, V. D. Santo, A. Casalegno and F. D. Fonzo, ACS Appl. Energy Mater., 2019, 2, 1911–1922 CrossRef CAS.
- A. Ohma, K. Shinohara, A. Iiyama, T. Yoshida and A. Daimaru, ECS Trans., 2011, 41, 775 CrossRef CAS.
- H. Matsui, N. Ishiguro, T. Uruga, O. Sekizawa, K. Higashi, N. Maejima and M. Tada, Angew. Chem., Int. Ed., 2017, 56, 9371–9375 CrossRef CAS PubMed.
- S. Ozawa, H. Matsui, N. Ishiguro, Y. Tan, N. Maejima, M. Taguchi, T. Uruga, O. Sekizawa, T. Sakata, K. Nagasawa, K. Higashi and M. Tada, J. Phys. Chem. C, 2018, 122, 14511–14517 CrossRef CAS.
- N. Ishiguro and M. Tada, Catal. Lett., 2018, 148, 1597–1609 CrossRef CAS.
- N. Ishiguro, S. Kityakarn, O. Sekizawa, T. Uruga, H. Matsui, M. Taguchi, K. Nagasawa, T. Yokoyama and M. Tada, J. Phys. Chem. C, 2016, 120, 19642–19651 CrossRef CAS.
- S. Takao, O. Sekizawa, K. Higashi, G. Samjeské, T. Kaneko, T. Sakata, T. Yamamoto, T. Uruga and Y. Iwasawa, ACS Appl. Mater. Interfaces, 2020, 12, 2299–2312 CrossRef CAS PubMed.
- Y. Tan, H. Matsui, N. Ishiguro, T. Uruga, D. N. Nguyen, O. Sekizawa, T. Sakata, N. Maejima, K. Higashi, H.-C. Dam and M. Tada, J. Phys. Chem. C, 2019, 123, 18844–18853 CrossRef CAS.
- Y. Cui, Y. Wu, Z. Wang, X. Yao, Y. Wei, Y. Kang, H. Du, J. Li and L. Gan, J. Electrochem. Soc., 2020, 167, 064520 CrossRef CAS.
- T. Đukić, L. J. Moriau, L. Pavko, M. Kostelec, M. Prokop, F. Ruiz-Zepeda, M. Šala, G. Dražić, M. Gatalo and N. Hodnik, ACS Catal., 2022, 12, 101–115 CrossRef PubMed.
- K. Nagasawa, S. Takao, K. Higashi, S. Nagamatsu, G. Samjeské, Y. Imaizumi, O. Sekizawa, T. Yamamoto, T. Uruga and Y. Iwasawa, Phys. Chem. Chem. Phys., 2014, 16, 10075–10087 RSC.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- W.-K. Wan, G. Samjeske, H. Matsui, C. Chen, S. Muratsugu and M. Tada, Dalton Trans., 2021, 50, 6811–6822 RSC.
- T. Uruga, M. Tada, O. Sekizawa, Y. Takagi, T. Yokoyama and Y. Iwasawa, Chem. Rec., 2019, 19, 1444–1456 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: BET profile, chemical composition analysis by XRF, STEM-EDS mapping, Pt LIII-edge, Cu K-edge, and Ti K-edge XAFS analysis, a summary of XPS fitting results, the relationship of the Pt 4f7/2 and Cu 2p3/2 core-level shift relative to valence states, electrochemical analysis by the RDE technique and practical PEFC operations. See DOI: https://doi.org/10.1039/d3cy01438j |
| This journal is © The Royal Society of Chemistry 2024 |