
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the error imbalance of large-scale machine learning potentials in catalysis†
Kareem
Abdelmaqsoud
 a,
Muhammed
Shuaibi
b,
Adeesh
Kolluru
a,
Raffaele
Cheula
ac and
John R.
Kitchin
*a
a,
Muhammed
Shuaibi
b,
Adeesh
Kolluru
a,
Raffaele
Cheula
ac and
John R.
Kitchin
*a
aDepartment of Chemical Engineering, Carnegie Mellon University, 5000 Forbes Avenue, Pittsburgh, Pennsylvania 15213, USA. E-mail: jkitchin@andrew.cmu.edu
bMeta Fundamental AI Research, Menlo Park, California 94025, USA
cDepartment of Physics and Astronomy, Aarhus University, Nordre Ringgade 1, 8000 Aarhus C, Denmark
First published on 14th August 2024
Abstract
Machine learning potentials (MLPs) have greatly accelerated atomistic simulations for material discovery. The Open Catalyst 2020 (OC20) dataset is one of the largest datasets for training MLPs for heterogeneous catalysis. The mean absolute errors (MAE) of the MLPs on the energy target of the dataset have asymptotically approached about 0.2 eV over the past two years with increasingly sophisticated models. The errors were found to be imbalanced between the different material classes with non-metals having the highest errors. In this work, we investigate several potential sources for the imbalanced distribution of errors. We examined material class-specific convergence errors in the density functional theory (DFT) calculations including k-point sampling, plane wave cutoff and smearing width. Significant DFT convergence errors with a mean absolute value of ∼0.15 eV were found on the total energies of non-metals, higher than the other material classes. However, as a result of cancellation of errors, convergence errors on adsorption energies have a mean absolute value of ∼0.05 eV across all material classes. Moreover, we found that the MAEs of the MLPs are not affected by these convergence errors. Second, we show that calculations with surface reconstruction can introduce inconsistencies to the adsorption energy referencing scheme that cannot be fit by the MLPs. Nonmetals and halides were found to have the highest fraction of calculations with surface reconstructions. Removing calculations with surface reconstructions from the validation sets, without re-training, significantly lowers the MAEs by ∼35% and reduces the imbalance of the MAEs. Alternatively, MLPs trained on total energies provide a solution to the surface reconstruction inconsistencies since they eliminate the referencing issue, and have comparable MAEs to MLPs trained on adsorption energies.
1 Introduction
Catalyst discovery is essential for enabling sustainable clean energy generation and conversion. The challenge in discovering new catalysts is that the space of the possible catalytic materials is too large to be explored experimentally.1 Atomic-scale simulation based on density functional theory (DFT)2 has proven to be a successful tool in modeling the properties of catalytic materials. DFT can be used to screen materials based on desired properties such as high activity and selectivity and suggest catalysts for experimental testing.1,3–6 However, the computational cost of such ab initio methods becomes prohibitive when screening a large number of materials.7,8 Machine learning potentials (MLPs) trained on DFT data have shown to be effective surrogate models for DFT simulations.9–11 Once trained, these MLPs can predict catalytic properties with a speed more than 104 faster than DFT calculations without a significant loss in accuracy.12 Due to their low computational cost, MLPs expand the number of materials that can be screened computationally, enabling more efficient and broader exploration of the catalytic material space.The generalization ability of the MLPs is heavily dependent on the size and the diversity of the datasets used for training.13 The Open Catalyst 2020 dataset (OC20) is an example of a large and diverse dataset that is used to train MLPs for heterogeneous catalysis.14 The dataset was gathered by running 1.28 million DFT relaxations with ∼260 M single-point calculations of energy and forces. OC20 spans 55 unique elements that make up unary, binary and ternary materials and 82 adsorbates. The materials in the dataset can be categorized into intermetallics, metalloids, non-metals and halides based on the elements they contain.15 The OC20 dataset presented several tasks for benchmarking the accuracy of the MLPs. The most general task evaluates the ability of an MLP to predict the energy and the per-atom forces of a given material at any configuration along the relaxation trajectory. This task is referred to as structure to energy and forces (S2EF). The energy target is referenced following an adsorption energy referencing scheme according the eqn (1). The DFT total energy of the surface + adsorbate system (Esystem) can be the relaxed or intermediate structures along the relaxation trajectory. The reference energies of the relaxed surface (i.e., slab) without an adsorbate (Eslab) and the energy of the adsorbate in the gas phase (Egas) are subtracted from the system total energy to yield the adsorption energy. Following the referencing scheme used in the OC20 dataset,14 the value of Egas for each adsorbate was computed as a linear combination of N2, H2O, CO, and H2 atomic energies.
| Eadsorption = Esystem − Eslab − Egas | (1) |
Fitting results for a series of increasingly sophisticated MLPs13,16–19 are shown in Fig. 1. Initially, the energy MAE of the different model architectures submitted decrease significantly by over 50% from OC20's initial release. However, the energy MAE of the models appears to plateau at ∼0.2 eV for the past two years. The target accuracy is 0.1 eV which is the estimated error level of the DFT calculations.20 One hypothesis that attempts to explain this plateauing behavior is that there are non-systematic errors that come from the DFT calculations used to generate the dataset that prevent the models from achieving lower errors. As shown by Kolluru et al.,15 the ML errors on nonmetals are the highest while the errors on intermetallics are the lowest. Nonmetals are thus hypothesized to have a larger error distribution compared to intermetallics and other material classes. In this work, we investigate the DFT settings and the adsorption energy referencing scheme used in the dataset as possible sources of this error imbalance.
 | ||
| Fig. 1 Mean absolute errors (MAEs) of the different MLPs submitted for evaluation on the structure to energy and forces (S2EF) task of the OC20 dataset.21 The MAEs are computed between the predicted energy of a given material at any configuration along the structure relaxation trajectory and the corresponding DFT energy. The reported values are the average MAEs across the in-domain and out-of-domain OC20 test sets. The MAEs appear to plateau at ∼0.2 eV for the past two years, signaling an inherent error distribution that can not be fit by the MLPs. | ||
While the DFT settings are often curated on a system-by-system basis, the scale and diversity of OC20 make such a task infeasible. As a result, OC20 uses a conservative set of settings with appropriate trade-offs between accuracy and efficiency. This work investigates the choice of the DFT settings used to generate the OC20 dataset as a possible source of this error imbalance. The OC20 dataset was run at the same DFT settings of all material classes. Although these settings might lead to converged calculations for intermetallics, they might not lead to converged calculations for nonmetals. The non-convergence of the calculations of the nonmetals with respect to the DFT settings could lead to non-systematic errors that are difficult to learn by the ML models, leading to inconsistently higher errors for nonmetals.
Beyond DFT convergence errors, DFT calculations with surface reconstruction can introduce inconsistencies in the adsorption energy referencing scheme used in the dataset that cannot be fit by the MLPs. The presence of an adsorbate on the surface during a relaxation can induce surface reconstruction that might not be observed in the case of the relaxing the surface without the adsorbate. To make an accurate adsorption energy calculation, the corresponding relaxed slab reference needs to be identical or similar to that of the relaxed adsorbate + slab. Therefore, whenever there is surface reconstruction, the slab reference is no longer a consistent reference, an ill-posed problem for the MLP. Thus, we hypothesize that removing these surface reconstructions would remove inconsistencies from the dataset and thus reduce ML errors.
In this work, we first show that the DFT convergence errors on total energies are significant and non-metals have total energy convergence errors with MAE of ∼0.15 eV. However, due to the cancellation of errors, we find the convergence errors on the adsorption energies are less than 0.05 eV across all material classes. Therefore, the OC20 dataset adsorption energies are converged with respect to the k-points sampling, plane-wave energy cutoff and smearing width DFT settings. Second, we show that removing the systems with surface reconstructions from the validation sets without re-training significantly decreases the ML energy MAEs by ∼35%. Lastly, we show that the total energy models can offer an alternative to adsorption energy models since they eliminate the referencing issue of calculations with surface reconstruction and reduce the imbalance of the MAEs between the different material classes. Moreover, due to cancellation of ML errors, total energy models have comparable MAEs to adsorption energy models.
2 Methods
2.1 Investigating the choice of the DFT settings
To determine if the DFT settings of the OC20 dataset were converged for non-metals, we ran DFT convergence studies on the relaxed structures of one hundred non-metallic systems. We focus the convergence studies on the hundred nonmetals with the highest ML errors since we hypothesize that these systems would require the tightest DFT settings. All DFT calculations were run using the Vienna ab initio simulation package (VASP).22–24 The calculations were run at the same settings as the OC20 dataset.14 The convergence studies were run on three settings: the KPOINTS setting which specifies the 3D sampling grid, the ENCUT setting which defines the energy cutoff for the plane-wave basis set and the SIGMA setting which specifies the smearing width. Calculations that are not converged with respect to these settings are hypothesized to lead to inconsistencies in the dataset that are difficult to fit by the MLPs.In the Open Catalyst 2020 (OC20) dataset, the KPOINTS mesh was set to be inversely proportional to the unit cell size and is calculated based using eqn (2). The symbol a0 denotes the norm of the first lattice vector of the unit cell, and b0 denotes the norm of the second lattice vector of the unit cell. The method calculates the mesh size by dividing a predetermined multiplier by the norm of the unit cell vectors along the reciprocal lattice directions, effectively adjusting the k-point density according to the dimensions of the structure. The default value of this parameter is 40 which leads to (4 × 4 × 1) grid for a (4 × 4) copper (100) surface for instance. To test the convergence of the KPOINTS setting, we calculate the total energy at the current OC20 value of 40 and a much higher value of 80. Using a multiplier value of 80 leads to (8 × 8 × 1) grid for the copper (100) surface. To determine the convergence errors over KPOINTS, the total energy was calculated at a multiplier value of 40 and was subtracted from the total energy value at a multiplier of 80. Other multiplier values were not explored because there was not a significant difference in the energies calculated between the 40 and 80 multiplier values.
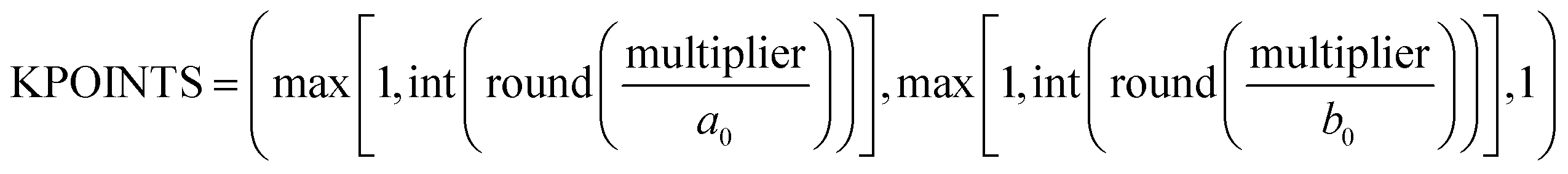 | (2) |
To determine the convergence errors over the ENCUT setting, the total energies of the hundred systems were calculated at ENCUT values of 400, 450, 500, and 550 eV. These total energies were then compared to the total energies calculated at an ENCUT value of 350 eV which is the setting used in the OC20 dataset. To calculate the convergence errors over the SIGMA, the total energies calculated at SIGMA values of 0.15, 0.1, 0.05, and 0.01 eV were compared to the total energies calculated at a SIGMA value of 0.2 eV which is the OC20 setting. It was found that decreasing the SIGMA value leads to calculations that are not converged electronically. Therefore, we increased the number of electronic steps to 120 steps whereas the default value is 60 steps. These studies are used to determine the tighter KPOINTS, ENCUT and SIGMA DFT settings. We refer to these settings as the “tighter settings” in this paper.
To test the effect of using the tighter settings on a larger scale, we ran an experiment on the OC20-200k training set and the OC20-30k in-domain validation set. These two sets contain 200k and 30k structures that represent the distribution of materials in the full OC20 training and in-domain validation set respectively. These are S2EF sets which not only include the initial and the final structures but also include intermediate structures along the DFT relaxation trajectory. DFT single-point calculations were run at the tighter DFT settings on these structures. The relaxed slab energies at the tighter settings were obtained by taking the already relaxed OC20 slabs and relaxing them again at the tighter settings to avoid the computational cost of relaxing the slabs from scratch. The adsorbate reference energies in the gas phase were calculated from the atomic energies calculated at the tighter settings. DFT calculations that did not reach electronic convergence at the tighter settings were removed from this experiment. The difference in energy and forces between the data at the original OC20 settings and the tighter settings were quantified as convergence errors. To test the effect of using tighter DFT settings on the MLP MAEs, we compare the MAEs of 1) a Gemnet-OC13 model trained and evaluated on data at the original OC20 settings, 2) a Gemnet-OC model trained and evaluated on data at the tighter settings.
2.2 Effect of removing surface reconstructions on the MAEs
The energies in the dataset are referenced to the energy of the relaxed slab and the gas-phase adsorbate energy to calculate the adsorption energy. To accurately calculate the adsorption energy, the corresponding relaxed slab reference needs to be identical or similar to that of the relaxed adsorbate + slab. In the case of adsorbate-induced surface reconstruction, the relaxed slab is no longer similar to the relaxed adsorbate + slab, as shown in Fig. 2. For systems with no surface reconstructions, the target is the adsorption energy. However, for systems with surface reconstructions, the target is not the adsorption energy, but it is the summation of the adsorption energy and the surface reconstruction energy. Therefore, computing adsorption energy for calculations with surface reconstructions results in an ill-posed, noisy target. This is one of the reasons why the energy target in the Open Catalyst 2022 (OC22) dataset is total energy, not adsorption energy.25 This inconsistency cannot be fit by the MLPs, leading to higher MAEs for systems with surface reconstructions. | ||
| Fig. 2 Shows the inconsistency in the energy referencing introduced by adsorbate-induced surface reconstructions. Without surface reconstructions, the target is the adsorption energy (Eads). With surface reconstruction, the relaxed slab is no longer similar to the relaxed adsorbate + slab. Thus, the target is not the adsorption energy, but it is the summation of the adsorption energy and the surface reconstruction energy. This inconsistency can not be fit by the MLPs. | ||
The DFT relaxation calculations were determined to be anomalous using an anomaly detection method developed in the AdsorbML paper.26 This method classifies an anomalous DFT relaxation calculation as having one or multiple of the following anomalies: adsorbate-induced surface reconstruction, adsorbate dissociation and adsorbate desorption. In this section, we focus only on the surface reconstruction detection method. The adsorbate dissociation and desorption anomalies do not affect the adsorption energy referencing and thus are not expected to affect the MAEs as shown in Section 4 of the ESI.† A slab + adsorbate system is determined to have an adsorbate-induced surface reconstruction based on the connectivity of the atoms in the system after being relaxed with and without an adsorbate. A connectivity matrix is constructed for the relaxed slab without an adsorbate. Another connectivity matrix is constructed for the relaxed adsorbate-slab configuration after removing the adsorbate. Atoms are considered connected if there is any overlap of the atomic radii with a small cushion. This cushion is a hyperparameter and was chosen to be a 1.5 multiplier of the covalent radii. If the connectivity matrices of the relaxed adslab surface and the relaxed slab surface are different, the system is flagged as a reconstructed surface.
After detecting the surface reconstructions, we run ML experiments to show the effect of removing these reconstructions from the validation set. We focus the ML experiments on three MLPs: GemNet-OC-large,13 eSCN16 and Equiformer-V2,17 the current top performing models on the OC20 dataset. We randomly selected 30k systems from each of the in-domain (ID) and out-of-domain (OOD) OC20 validation sets. The ID validation set is sampled from the same distribution as the training set. The OOD validation sets contain element compositions that were not seen by the MLPs in the training set. There are three OOD validation sets: unseen adsorbates (OOD Ads), unseen element compositions for catalysts (OOD Cat), and unseen both adsorbates and catalysts (OOD Both). To determine the effect of removing surface reconstructions, we compare the validation energy MAEs of pre-trained MLPs before and after removing the surface reconstructions from the validation sets. All pre-trained ML models used in this work are publicly available at the Open Catalyst Project (OCP) GitHub repository.
To investigate the effect of removing the surface reconstructions from both training and validation sets, we trained two models from scratch with and without the surface reconstructions. This experiment was done on the OC20-200k training set. After removing the surface reconstructions from this split, the size of this split becomes 156k structures. To enable meaningful comparisons, we randomly selected 156k data points from the 200k split to train a model on data that includes surface reconstructions. The performance of the two models trained on data with and without surface reconstructions is evaluated on the OC20-30k in-domain validation set after removing the surface-reconstructed systems.
2.3 Adsorption energy from total energy models
While surface reconstructions pose challenges for MLPs trained on adsorption energy, they are still valuable data that we can leverage for training models. One way to go about this is by training models to predict the total energy instead of the adsorption energy. To explore this, we used the GemNet-OC OC20 + OC22 pretrained model, the only publicly available model on total energies which was trained on both OC20 and the Open Catalyst 2022 (OC22)25 datasets. It is important to mention that this model is trained on the raw DFT total energy without any normalization or referencing. We test whether removing the calculations with surface reconstructions from the OC20-30k in-domain validation set has an impact on the total energy MAEs. We also explore if using total energy models impacts the imbalance in the ML MAEs of the different material classes.Furthermore, we compare adsorption energy metrics using a total energy model and a baseline adsorption energy model on 25k in-domain and out-of-domain validation sets. These validation sets are the OC20 IS2RE (initial structure to relaxed energy) benchmark validation sets. For this comparison, we relax both the adsorbate + slab and slab structures using the total energy model. On the other hand, we can only relax the adsorbate + slab structure using the adsorption energy model since it assumes you have access to the DFT relaxed structure of the slab to be used for referencing. The structures were relaxed using ML until all per-atom forces were less than 0.03 eV Å−1 or up to 300 relaxation steps. For the total energy model, the adsorption energies are calculated using eqn (3). The total energy of the relaxed structure of adsorbate + slab structure (EMLsystem) and the energy of the relaxed structure of the slab (EMLslab) are both predicted using the total energy model. The energy of adsorbate in the gas-phase, (Egas), was calculated as a linear combination of N2, H2O, CO, and H2 atomic energies. The MAEs are calculated between the ML predicted adsorption energy energy and the DFT adsorption energy.
| Eadsorption = EMLsystem − EMLslab − Egas | (3) |
3 Results & discussion
3.1 DFT convergence errors
In this section, we focus only on the energy convergence with respect to the DFT settings since the forces were found converged within 0.03 eV Å−1 as shown in Fig. S1.† The convergence errors with respect to the k-points multiplier were calculated by subtracting the total energy at a multiplier value of 40 from the total energy at 80. The differences in total energies were found converged within 0.05 eV. The differences in adsorption energies were found converged within 0.02 eV due to cancellation of errors. Therefore, we do not focus on the convergence errors across the k-points settings. The convergence errors over the ENCUT setting were calculated by subtracting the total energy at 350, 400, 450, and 500 eV from the total energy at 550 eV, as shown in Fig. 3a. Large convergence errors (up to 2 eV) are found with respect to the OC20 default of ENCUT = 350 eV. An ENCUT setting of 500 eV is shown to reduce the convergence errors to have a mean absolute value of ∼0.03 eV and thus is chosen as the new converged value.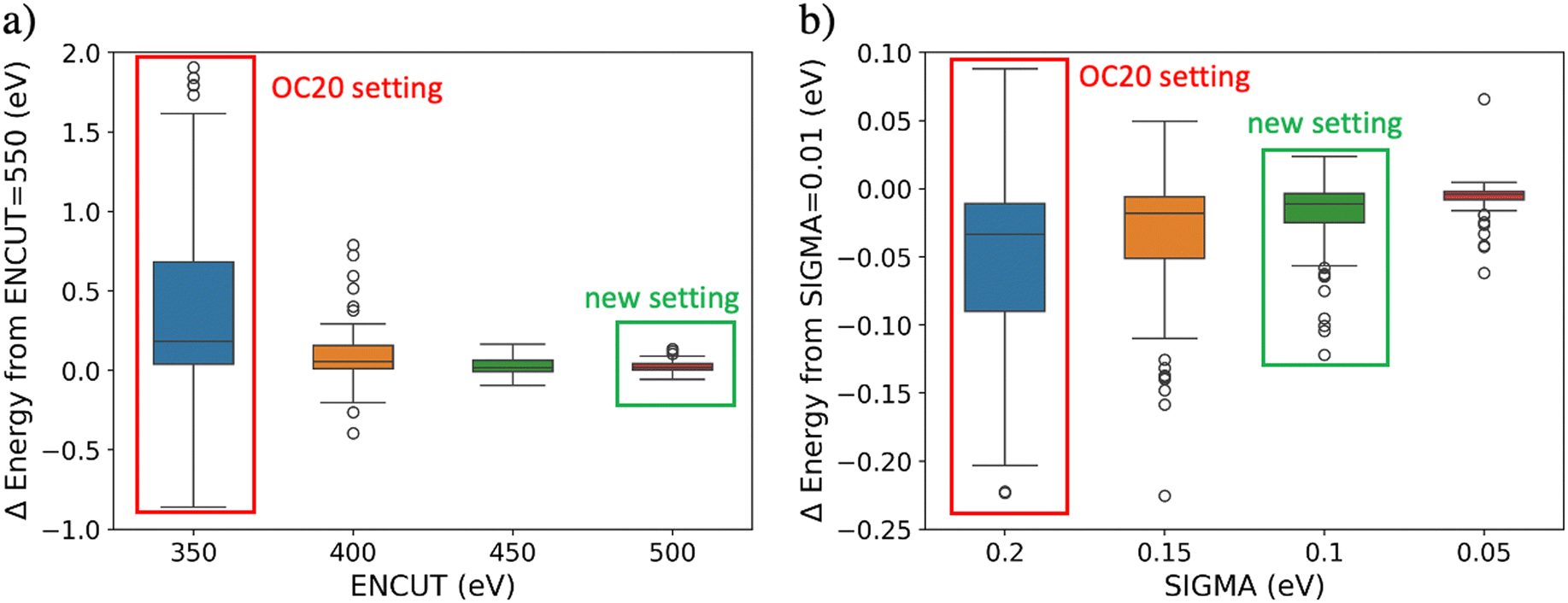 | ||
| Fig. 3 Determining the converged plane-wave energy cutoff (ENCUT) and the smearing width (SIGMA) DFT settings for a hundred non-metallic systems from the OC20 dataset. We selected the hundred non-metals with the highest ML errors since we hypothesize that these systems would require the tightest DFT settings. The original OC20 setting value is highlighted in red and the selected converged setting is highlighted in green. a) A plot of the difference in energy between each value of the ENCUT setting and the highest value tested (ENCUT = 550 eV). b) A plot of the difference in energy between each value of the SIGMA setting and the highest value tested (SIGMA = 0.01 eV). | ||
The convergence errors over SIGMA were calculated by subtracting the total energy calculated at SIGMA values of 0.2, 0.15, 0.1, and 0.05 from the total energy at SIGMA of 0.01 eV as shown in Fig. 3b. The convergence errors with respect to SIGMA are smaller than the ENCUT convergence errors, yet they are significant (up to 0.15 eV). In choosing a SIGMA value, a trade-off was observed where decreasing the SIGMA value reduces the convergence errors but increases the number of calculations that are not converged electronically. Calculations that did not reach electronic convergence were removed from this study. A SIGMA value of 0.1 eV was chosen because it reduced the error distribution while only causing 2.5% of the calculations to be not converged electronically. SIGMA of 0.05 eV on the other hand led to 6% of the calculations being not converged electronically. Thus, the converged DFT settings are k-points multiplier = 40, ENCUT = 500 eV, SIGMA = 0.1 eV.
After determining the converged DFT settings, an experiment was run to determine the magnitude of the convergence errors on a large subset of the dataset. The convergence error results shown in Fig. 4 are computed based on the OC20-200k training split. Systems with convergence errors that lie outside the whiskers of the box plots with energy differences that lie in the top and bottom 5% of the energy difference distribution. These systems are considered outliers and are described in Section 2 of the ESI.† The magnitude of the convergence errors shown before in Fig. 3 is larger because we selected the nonmetallic systems that are expected to have the largest convergence errors. On the other hand, in Fig. 4, we compute the convergence errors on a larger and more representative sample of the full dataset, not just non-metals. The convergence errors in the slab + adsorbate total energies are significant (>0.1 eV) and nonmetals have the highest convergence errors as shown in Fig. 4a. The convergence errors in the slab energies are also significant and nonmetals have the highest convergence errors as shown in Fig. 4b. The slab energies at the tighter settings were computed by taking the relaxed slabs at the original settings and relaxing them again at the tighter settings. This leads to systematically negative slab energy differences (Etighter − EOC20) as shown in Fig. 4b. Due to cancellation of errors, the magnitudes of the convergence errors on the adsorption energy are significantly smaller than those on total energies as shown in Fig. 4c. Moreover, nonmetals have comparable convergence errors in the adsorption energies to other material classes. The adsorption energy differences are shifted to be more positive because of the negative systematic shift of the slab energies at the tighter settings. Overall, the adsorption energies appear to be mostly converged within 0.1 eV with respect to the KPOINTS, ENCUT and SIGMA.
 | ||
| Fig. 4 Differences between the energies calculated at tighter and OC20 DFT settings (Etighter − EOC20) on a larger scale of OC20-200k dataset. a) Significant differences in the total energies are observed with a mean absolute value greater than 0.1 eV. Non-metals have the highest differences matching the trend in the ML errors. b) The differences in the slab energy are also significant with non-metals having the highest differences. c) Due to cancellation of errors, differences in the adsorption energy converged within 0.1 eV with non-metals having comparable differences to other material classes. | ||
Interestingly, we found that the energy MAE of a model trained on data with the tighter DFT settings is comparable to the MAE of a model trained on the data with the OC20 settings. As shown in Fig. 5a, the total energy MAEs are not significantly affected by using tighter DFT settings. The reduction of halides MAEs is larger than other material classes, but since the percentage of halides in the dataset is small (1.4%), the overall MAEs are not affected significantly. As shown in Fig. 5b, for all the material classes, the adsorption energy MAEs do not change significantly as a result of using the tighter DFT settings. Therefore, the ML errors are not affected by the DFT convergence errors. Moreover, for MLPs trained on the data with the tighter DFT settings, nonmetals and halides still have inconsistently higher errors compared to other material classes. Therefore, it can be concluded that the DFT convergence errors are not the cause of non-metals having inconsistently higher MAEs than other material classes.
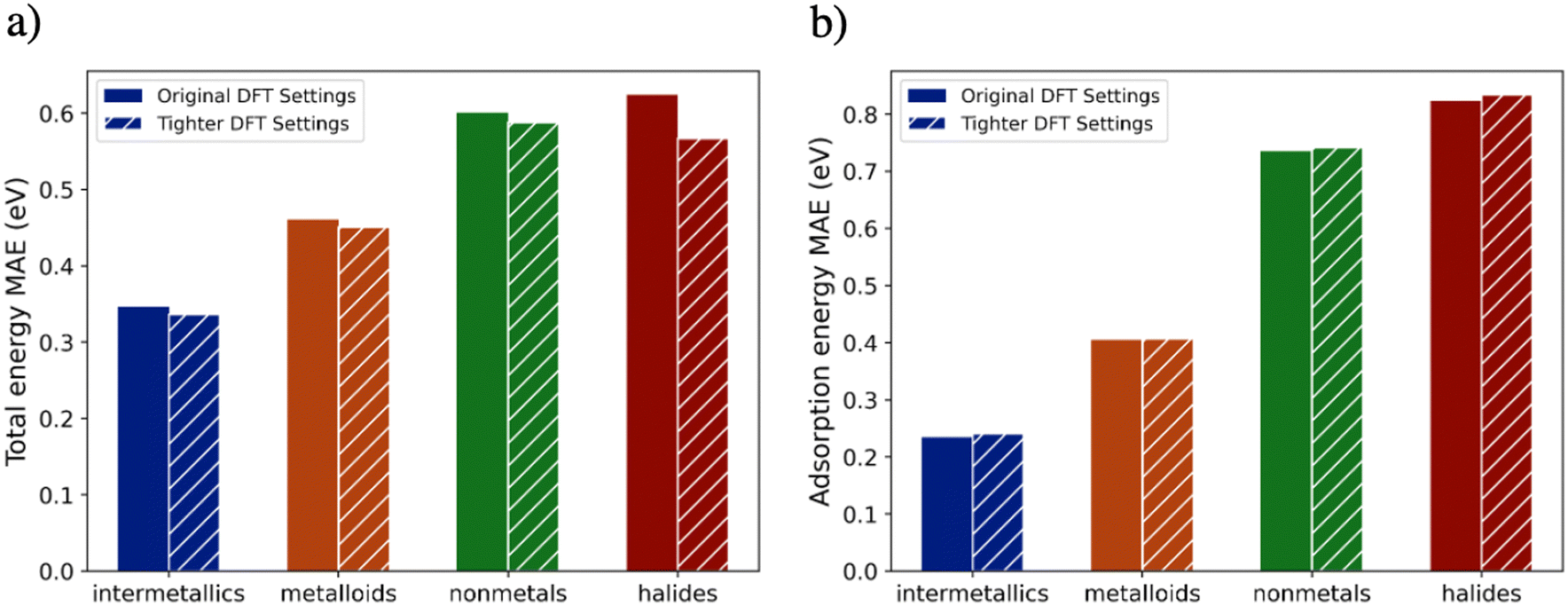 | ||
| Fig. 5 A comparison of the MAEs of two GemNet-OC models 1) trained and evaluated on data at the original OC20 settings, 2) trained and evaluated on data at the tighter settings. The training set is the OC20-200k dataset and the evaluation set is the 30k in-domain validation set. The figure shows a comparison of a) total energy MAEs and b) adsorption energy MAEs for the different material classes. No significant change in the MAEs as a result of using the tighter DFT settings. | ||
The results of this section could be useful to future efforts focusing on generating large-scale DFT datasets. The OC20 dataset, for example, uses a conservative set of settings with appropriate trade-offs between accuracy and efficiency. We show that the cancellation of DFT errors reduces the effect of the convergence errors on the adsorption energy target of the OC20 dataset. However, one should not completely disregard the convergence errors in generating a DFT dataset assuming that the cancellation of errors would always lead to accurate energies. Instead, the effect of convergence errors should be quantified on a sufficiently large and representative subset of the dataset to determine the appropriate DFT settings needed. It is also important to mention that other settings besides the 3 DFT settings studied here could cause convergence issues. One example is the VASP ISMEAR parameter which determines how the partial occupancies are set for each orbital. The Methfessel–Paxton scheme was used in the OC20 dataset. This scheme could cause issues for semiconductors and insulators in the dataset. Therefore, if the MLPs trained on the OC20 dataset are used to study semiconductor or insulator systems, one should investigate the convergence errors of these systems with respect to the ISMEAR parameter.
3.2 Effect of removing surface reconstructions on the MAEs
To investigate another possible source of non-systematic errors in the dataset, we identify the systems having adsorbate-induced surface reconstructions using the anomaly detection method described before. A large percentage (22%) of the calculations in the OC20 dataset were identified as having surface reconstruction. Fig. 6a shows the percentage of calculations with surface reconstructions for each of the four material classes in the dataset. Nonmetals and halides have higher percentages of calculations with surface reconstructions compared to the intermetallics and metalloids calculations. Fig. 6b shows the effect of removing the systems with surface reconstruction from the validation set, without retraining, on the adsorption energy MAEs. The reduction in the MAEs of the non-metals and halides is larger than the reduction of intermetallics and metalloids, matching the fractions of surface reconstructions. | ||
| Fig. 6 a) Fractions of systems that have surface reconstructions in the four material classes on randomly selected 30k systems from the in-domain validation set. Non-metals and halides have a much higher ratio of calculations with surface reconstructions compared to intermetallics and metalloids. b) The validation MAEs of a pre-trained GemNet-OC model before (solid bars) and after (hashed bars) removing the surface reconstructions from the validation set without retraining. The reduction in the MAEs of the non-metals and halides is larger than the reductions for intermetallics and metalloids, matching the fractions of surface reconstructions. | ||
Table 1 shows the effect of removing the calculations with surface reconstructions from the validation sets, without retraining, on the MAEs. Consistently across all the three model architectures investigated, removing the surface reconstructions significantly reduces the MAEs. This finding supports the hypothesis that the surface reconstructions introduce inconsistencies in the adsorption energy referencing scheme used in the OC20 dataset which cannot be fit by the MLPs. Moreover, the forces MAEs did not change significantly as a result of removing the surface reconstructions as shown in Table S1.† This is expected since these reconstructions only affect the referencing of the energy whereas the forces are not referenced.
| OC20-S2EF validation energy MAE [eV] | ||||||
|---|---|---|---|---|---|---|
| ID | OOD ads | OOD cat | OOD both | Average | ||
| GemNet-OC | With reconstructions | 0.164 | 0.191 | 0.286 | 0.353 | 0.248 |
| Without reconstructions | 0.100 | 0.136 | 0.168 | 0.234 | 0.160 | |
| eSCN | With reconstructions | 0.169 | 0.213 | 0.255 | 0.344 | 0.245 |
| Without reconstructions | 0.097 | 0.153 | 0.142 | 0.231 | 0.156 | |
| EqV2 | With reconstructions | 0.159 | 0.172 | 0.257 | 0.317 | 0.226 |
| Without reconstructions | 0.090 | 0.112 | 0.146 | 0.200 | 0.137 | |
Next, we ran an experiment that tested the effect of removing the surface reconstructions from both training and validation sets. A Gemnet-OC model trained on a dataset that includes surface reconstructions has an energy MAE of 0.31 eV whereas a model trained on a dataset without surface reconstructions has an MAE of 0.29 eV when both are evaluated on an in-domain validation set without surface reconstructions. Since there is no significant reduction (∼0.02 eV) in the model errors as a result of removing the surface reconstructions from the training set, retraining the models on datasets without surface reconstructions might not be necessary. This result is similar to what has been observed by Vita et al.27 where they showed that models trained on a dataset with added noise to the energy and force labels have comparable errors to models trained on non-noisy data given the models are trained on large datasets and evaluated on a non-noisy test set. In our case, this added noise is represented by the added reconstruction energy to the adsorption energy label. The models have comparable errors whether they are trained on data with or without this added noise since they are trained on a large dataset and evaluated on a non-noisy dataset.
Removing calculations with surface reconstructions is not an ideal solution since these calculations are valid DFT calculations. It is only the adsorption energy referencing for these systems that is ill-posed. Training models to predict the total energy instead of the adsorption energy eliminates the referencing issue of the calculations with surface reconstructions. Using a model trained on total energy, we show that removing the calculations with surface reconstructions does not affect the total energy MAEs, as shown in Fig. S3.† Therefore, MLPs trained on total energies instead of adsorption energies resolve the issue of calculations with surface reconstructions.
3.3 Adsorption energy from total energy models
To determine if models trained on total energies can be used as alternatives to models trained on adsorption energy, we compare their adsorption energy MAEs on the OC20-IS2RE validation sets. The MAEs in Table 2 are computed between the energy of the ML relaxed structure and the DFT relaxed structure which could be different structures. Thus, these MAEs are higher than the MAEs reported in Table 1 which compares the energy of the same structure. If we ensure that the MAEs are computed on the same structure by predicting the energy of the DFT relaxed structures, we get adsorption energy MAEs ∼0.1 eV as shown in Table S5.† In predicting the energy of the adsorbate + slab structures, the total energy model has higher MAEs on the out-of-domain catalyst (OOD Cat) compared to the in-domain (ID) and out-of-domain adsorbate (OOD Ads) datasets. The same trend is also observed in the MAEs of the slab energy prediction. Interestingly, these errors do not add up in computing the adsorption energy. Instead, the errors cancel out, reducing the adsorption energy MAEs on OOD Cat and OOD Both significantly (∼50%). For the OOD Cat dataset, the total energy model appears to have systematic errors on systems with new element compositions that the model was not trained on. These systematic errors in the adsorbate + slab and the slab energy predictions cancel out, leading to lower adsorption energy MAEs. On the other hand, since the errors on the ID and OOD Ads datasets are smaller, they are more likely to be randomly distributed errors. Therefore, we observe smaller cancellation of errors on these two datasets. Ock et al. previously showed that MLPs trained on the OC20 dataset show significant error cancellation between chemically similar systems up to 77%.28 Herein, we show that cancellation of MLP errors can improve the generalization of the MLPs on out-of-domain catalysts datasets.| OC20-IS2RE validation energy MAE [eV] | ||||||
|---|---|---|---|---|---|---|
| ID | OOD ads | OOD cat | OOD both | Average | ||
| Total energy model | Adsorbate + slab energy | 0.344 | 0.371 | 0.630 | 0.637 | 0.495 |
| Slab energy | 0.206 | 0.207 | 0.530 | 0.541 | 0.371 | |
| Adsorption energy | 0.384 | 0.408 | 0.384 | 0.351 | 0.382 | |
| Adsorption energy model | Adsorption energy | 0.334 | 0.364 | 0.385 | 0.354 | 0.359 |
Moreover, we find that the adsorption energy MAEs of the total energy model are slightly higher, but still comparable to the MAEs of the adsorption energy model. It is worth re-emphasizing that we relaxed both the adsorbate + slab and the slab structures using the total energy model. Whereas we relaxed the adsorbate + slab structure only using the adsorption energy model and relaxed the slab structure using DFT. Therefore, although the total energy model has slightly higher MAEs, it saves the cost of relaxing the slab structure using DFT. Even after removing calculations with surface reconstructions, we also show that the total energy model has comparable MAEs to the adsorption energy model as shown in Table S3.† The MAEs in Table 2 can be reduced significantly by using an active learning framework such as the FINETUNA29 framework which accelerates the DFT relaxations accurately by leveraging pre-trained MLPs on the OC20 dataset. The framework was found to reduce the number of DFT single-point calculations during a relaxation by 91% while maintaining an accuracy threshold of 0.02 eV in 93% of cases.29 Besides the magnitude of the MAEs, the total energy model shows a more uniform distribution of MAEs across the different classes as shown in Fig. S4.† Therefore, besides resolving the issue with calculations with surface reconstructions, total energy models reduce the MAE imbalance between the different material classes in the OC20 dataset.
4 Conclusions
The energy MAEs of the different MLPs trained and evaluated on the OC20 dataset were found to be plateauing at around 0.2 eV. The MAEs of the MLPs on the non-metallic systems were consistently higher than other material classes. A hypothesis that attempts to explain this behavior is that there are non-systematic errors in the dataset that prevent the models from having MAEs lower than 0.2 eV, and that non-metals have a larger error distribution. This work narrows down two of the possible sources of non-systematic errors in the dataset. First, we investigate the convergence of the DFT calculations across the k-point sampling, plane-wave energy cutoff, and smearing-width DFT settings. We show that due to cancellation of errors, convergence errors on adsorption energies to be converged within 0.1 eV, and that the convergence errors of nonmetals are comparable to the other material classes. We further show that the DFT convergence errors do not affect the MAEs of the MLPs. Therefore, DFT convergence errors are not the source of this plateau and the MAE imbalance. Second, we showed that systems with surface reconstructions introduce inconsistencies to the adsorption energy referencing scheme that cannot be fit by the MLPs. Non-metals and halides systems were found to include a higher fraction of calculations with surface reconstructions than intermetallics and metalloids. We showed that removing these calculations from the validation dataset, without retraining, significantly decreases the energy MAEs by ∼36% and reduces the error imbalance.This work highlights the importance of considering the cancellation of DFT errors in building large datasets for heterogeneous catalysis applications. It also shows the importance of having a consistent reference scheme for the performance of MLPs. Similarly to DFT, total energy models show cancellation of errors of the adsorbate + slab and slab energy predictions. Before using an adsorption energy model, one should be careful about predicting the energy of systems with surface reconstructions as they can hurt the model performance. Alternatively, total energy models are more robust models to surface reconstructions that still work on par with existing adsorption energy models, albeit with slightly worse performance. Total energy models, however, do provide access to predictions on clean slabs, a limitation of current adsorption energy models.
The findings of this work can be useful in investigating other large datasets for heterogeneous catalysis such as the Open Catalyst 2022 (OC22) dataset.25 Although the convergence errors with respect to the DFT settings were not found to be a significant issue for the OC20 adsorption energies, they can cause significant issues for the OC22 dataset. The selection of Hubbard U corrections and the magnetic moment initialization can cause significant inconsistencies in the OC22 dataset. An interesting future direction is to explore the effect of changing these DFT settings on the accuracy of the MLPs trained on the OC22 dataset.
Data availability
Data for this article, including notebooks data and csv files of data used in the analysis are available at Kareem Abdelmaqsoud. (2024). kareem-Abdelmaqsoud/error_imbalance_mlps: initial release (0.1.0). Zenodo. https://doi.org/10.5281/zenodo.12704843.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the computing resources used in this work was provided by the National Energy Research Scientific Computing Center (NERSC). Author R. C. acknowledges support from European Union's Horizon Europe (Marie Skłodowska-Curie grant No. 101108769).Notes and references
- F. Ren, L. Ward, T. Williams, K. J. Laws, C. Wolverton, J. Hattrick-Simpers and A. Mehta, Sci. Adv., 2018, 4, eaaq1566 CrossRef PubMed.
- D. S. Sholl and J. A. Steckel, Density Functional Theory: A Practical Introduction, John Wiley & Sons, 2022 Search PubMed.
- R. Jose, N. U. Zhanpeisov, H. Fukumura, Y. Baba and M. Ishikawa, J. Am. Chem. Soc., 2006, 128, 629–636 CrossRef CAS PubMed.
- S. Rahali, M. A. B. Aissa, L. Khezami, N. Elamin, M. Seydou and A. Modwi, Langmuir, 2021, 37, 7285–7294 CrossRef CAS PubMed.
- N. D. Rode, I. Abdalghani, A. Arcadi, M. Aschi, M. Chiarini and F. Marinelli, J. Org. Chem., 2018, 83, 6354–6362 CrossRef CAS PubMed.
- L. Xu, X. Meng, M. Li, W. Li, Z. Sui, J. Wang and J. Yang, Chem. Eng. J., 2019, 361, 1511–1523 CrossRef CAS.
- A. J. Medford, M. R. Kunz, S. M. Ewing, T. Borders and R. Fushimi, ACS Catal., 2018, 8, 7403–7429 CrossRef CAS.
- S. Matera, W. F. Schneider, A. Heyden and A. Savara, ACS Catal., 2019, 9, 6624–6647 CrossRef CAS.
- C. M. Clausen, J. Rossmeisl and Z. W. Ulissi, Adapting OC20-trained EquiformerV2 Models for High-Entropy Materials, arXiv, 2024, preprint, arXiv:2403.09811 [cond-mat, physics:physics], DOI:10.48550/arXiv.2403.09811, https://arxiv.org/abs/2403.09811.
- K. Broderick, E. Lopato, B. Wander, S. Bernhard, J. Kitchin and Z. Ulissi, Appl. Catal., B, 2023, 320, 121959 CrossRef CAS.
- R. Tran, D. Wang, R. Kingsbury, A. Palizhati, K. A. Persson, A. Jain and Z. W. Ulissi, J. Chem. Phys., 2022, 157, 074102 CrossRef CAS PubMed.
- D. Chen, C. Shang and Z.-P. Liu, npj Comput. Mater., 2023, 9, 1–9 CrossRef.
- J. Gasteiger, M. Shuaibi, A. Sriram, S. Gunnemann, Z. Ulissi, C. L. Zitnick and A. Das, GemNet-OC: Developing Graph Neural Networks for Large and Diverse Molecular Simulation Datasets, arXiv, 2022, preprint, arXiv:2204.02782 [cond-mat, physics:physics], DOI:10.48550/arXiv.2204.02782, https://arxiv.org/abs/2204.02782.
- L. Chanussot, A. Das, S. Goyal, T. Lavril, M. Shuaibi, M. Riviere, K. Tran, J. Heras-Domingo, C. Ho, W. Hu, A. Palizhati, A. Sriram, B. Wood, J. Yoon, D. Parikh, C. L. Zitnick and Z. Ulissi, ACS Catal., 2021, 11, 6059–6072 CrossRef CAS.
- A. Kolluru, M. Shuaibi, A. Palizhati, N. Shoghi, A. Das, B. Wood, C. L. Zitnick, J. R. Kitchin and Z. W. Ulissi, ACS Catal., 2022, 12, 8572–8581 CrossRef CAS.
- S. Passaro and C. L. Zitnick, Reducing SO(3) Convolutions to SO(2) for Efficient Equivariant GNNs, arXiv, 2023, preprint, arXiv:2302.03655 [physics], DOI:10.48550/arXiv.2302.03655, https://arxiv.org/abs/2302.03655.
- Y.-L. Liao, B. Wood, A. Das and T. Smidt, EquiformerV2: Improved Equivariant Transformer for Scaling to Higher-Degree Representations, arXiv, 2024, preprint, arXiv:2306.12059 [physics], DOI:10.48550/arXiv.2306.12059, https://arxiv.org/abs/2306.12059.
- W. Hu, M. Shuaibi, A. Das, S. Goyal, A. Sriram, J. Leskovec, D. Parikh and C. L. Zitnick, ForceNet: A Graph Neural Network for Large-Scale Quantum Calculations, arXiv, 2021, preprint, arXiv:2103.01436 [cs], DOI:10.48550/arXiv.2103.01436, https://arxiv.org/abs/2103.01436.
- M. Shuaibi, A. Kolluru, A. Das, A. Grover, A. Sriram, Z. Ulissi and C. L. Zitnick, Rotation Invariant Graph Neural Networks using Spin Convolutions, arXiv, 2021, preprint, arXiv:2106.09575 [cs], DOI:10.48550/arXiv.2106.09575, https://arxiv.org/abs/2106.09575.
- E. Romeo, M. F. Lezana-Muralles, F. Illas and F. Calle-Vallejo, ACS Appl. Mater. Interfaces, 2023, 15, 22176–22183 CrossRef CAS PubMed.
- OC20 Leaderboard, https://opencatalystproject.org/leaderboard.html#task_s2ef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- R. Tran, J. Lan, M. Shuaibi, B. M. Wood, S. Goyal, A. Das, J. Heras-Domingo, A. Kolluru, A. Rizvi, N. Shoghi, A. Sriram, F. Therrien, J. Abed, O. Voznyy, E. H. Sargent, Z. Ulissi and C. L. Zitnick, ACS Catal., 2023, 13, 3066–3084 CrossRef CAS.
- J. Lan, A. Palizhati, M. Shuaibi, B. M. Wood, B. Wander, A. Das, M. Uyttendaele, C. L. Zitnick and Z. W. Ulissi, npj Comput. Mater., 2023, 9, 1–9 CrossRef.
- J. A. Vita and D. Schwalbe-Koda, Mach. Learn.: Sci. Technol., 2023, 4, 035031 Search PubMed.
- J. Ock, T. Tian, J. Kitchin and Z. Ulissi, J. Chem. Phys., 2023, 158, 214702 CrossRef CAS PubMed.
- J. Musielewicz, X. Wang, T. Tian and Z. Ulissi, Mach. Learn.: Sci. Technol., 2022, 3, 03LT01 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data supporting the conclusions. See DOI: https://doi.org/10.1039/d4cy00615a |
| This journal is © The Royal Society of Chemistry 2024 |