
Reaction mechanism and regioselectivity of uridine diphosphate glucosyltransferase RrUGT3: a combined experimental and computational study†
Mengsha
Li‡
 ab,
Cai
You‡
cde,
Fei
Guo‡
c,
Qingfang
Han
ab,
Xixian
Xie
a,
Lijuan
Ma
a,
Shengying
Li
c,
Lei
Du
*c,
Xiang
Sheng
*bf and
Hao
Su
*bf
ab,
Cai
You‡
cde,
Fei
Guo‡
c,
Qingfang
Han
ab,
Xixian
Xie
a,
Lijuan
Ma
a,
Shengying
Li
c,
Lei
Du
*c,
Xiang
Sheng
*bf and
Hao
Su
*bf
aCollege of Biotechnology, Tianjin University of Science & Technology, Tianjin 300457, P. R. China
bTianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, P. R. China. E-mail: shengx@tib.cas.cn; suhao@tib.cas.cn
cState Key Laboratory of Microbial Technology, Shandong University, Qingdao 266237, Shandong, P. R. China. E-mail: lei.du@sdu.edu.cn
dShandong Provincial Key Laboratory of Synthetic Biology, CAS Key Laboratory of Biofuels, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, Shandong, P. R. China
eShandong Energy Institute, Qingdao 266101, P. R. China
fNational Center of Technology Innovation for Synthetic Biology, National Engineering Research Center of Industrial Enzymes and, Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin 300308, P. R. China
First published on 9th July 2024
Abstract
Uridine diphosphate glucosyltransferase (UGT) from Rhodiola rosea shows high selectivity towards the phenolic hydroxyl group in the glucosylation of p-hydroxybenzyl alcohol (HBA). However, the detailed catalytic mechanism, including the substrate binding mechanism and the origins of the regioselectivity remain unclear. In this work, we solved the crystal structure of the enzyme and elucidated the molecular basis of the catalytic mechanism using site-directed mutagenesis and multiscale computational studies. Our results indicate that the conformational change of a loop region from the “open” to the “closed” state is crucial for the formation of the active enzyme–substrate ternary complex, and the loop conformation change is favored by substrate binding. The quantum mechanical/molecular mechanical (QM/MM) and quantum chemical calculation results suggest that the catalytic power of RrUGT3 mainly originates from the preorganized protein environment that stabilizes the catalytically active state of the sugar donor uridine-5′-diphosphate-glucose (UDPG). Moreover, the favored substrate binding and lower activation barrier of the phenolic glycosylation dictate the regiopreference of the enzyme. These results expand our knowledge on the catalytic mechanism of UGTs, and will facilitate the rational modification and design of highly active and regioselective UGT variants for the practical production of glycosides.
1. Introduction
Glycosyltransferases (GTs) are widely spread in animals, plants, fungi, and bacteria, and play key roles in polysaccharide biosynthesis, cell wall formation, and glycosylation of various metabolites. So far, at least 135 families have been classified according to the carbohydrate-active enzyme database CAZy. Among them, a subgroup of GTs from the GT1 family utilizes the uridine diphosphate (UDP) sugars, such as UDP-glucose, -galactose, -xylose, and -rhamnose, as the donor substrate to glycosylate diverse sugar acceptors, including saccharides, lipids, proteins and secondary metabolites, with high regioselectivity and substrate specificity.1–6 Structural and mechanistic studies on GT1s revealed that these enzymes usually utilize a His-Asp catalytic dyad to abstract a proton from the acceptor to initiate the reaction. Subsequently, the reaction takes place via an “SN2” mechanism, resulting in the formation of a product with the anomeric configuration inverted.6–8 Depending on the glycosylation site of acceptors, UGTs can be classified into O-, N-, C- and S-GTs by anchoring the sugar donor to acceptor substrates through O-, N-, C- and S-glycosidic bonds.9–11 As the presence of a sugar moiety can significantly increase the chemical stability and water solubility of molecules,12–14 the glycosylation of bioactive molecules has attracted great attention in the pharmaceutical industry.13,15The O-glycosides are particularly attractive in biomedical and pharmaceutical research due to their abundance in nature and their unique functional properties.16–18 However, the structural complexity of O-glycosides makes the chemical synthesis of these glycoconjugates challenging and costly. In contrast, enzymatic glycosylation offers a promising alternative strategy. Recent studies showed that RrUGT3 from Rhodiola rosea is capable of catalyzing the formation of phenolic aglycons. The experimental results revealed that RrUGT3 shows a broad substrate spectrum toward phenolic aglycons and also displays activity in forming N- and S-glucosides.19 Interestingly, using uridine-5′-diphosphate-glucose (UDPG) as the sugar donor, RrUGT3 catalyzes the glucosylation of p-hydroxybenzyl alcohol (HBA) with a high regioselectivity of 98.5% towards the phenolic group, generating phenolic O-glycoside (gastrodin), which shows diverse pharmacological activities such as sedative, hypnotic, anticonvulsive and neuroprotective properties (Fig. 1).19–22 The broad substrate spectrum, high activity and regioselectivity make RrUGT3 a highly promising biocatalyst for producing unnatural glycosides containing an alkylphenol structural moiety. However, the lack of a crystal structure of RrUGT3 and further mechanistic study has hindered the elucidation of the factors controlling the catalytic activity and regioselectivity of RrUGT3.
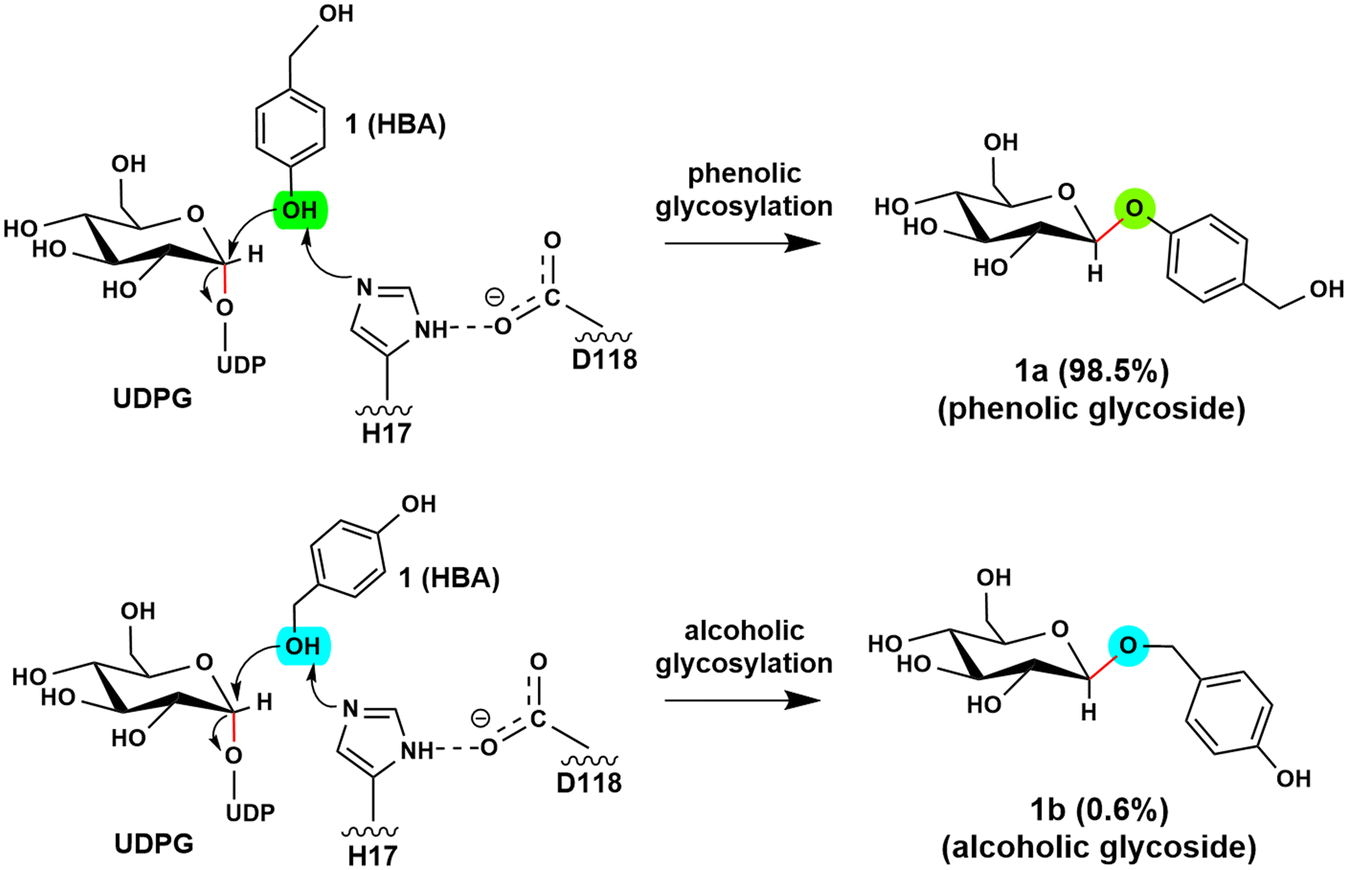 | ||
| Fig. 1 RrUGT3-catalyzed glycosylation of p-hydroxybenzyl alcohol with high regioselectivity to the phenolic hydroxyl group: phenolic O-glycoside (1a) as the main product (98.5%) and alcoholic O-glycoside (1b) as the minor product (0.6%). | ||
By comparing and analyzing the various crystal structures of highly homologous UGT enzymes,6,8,9,23 the overall structure of these enzymes show high similarity, however, a loop region (denoted as the Ω-1 loop) displays large flexibility (Fig. 2 and S1a†). As shown in Fig. 2, the crystal structures of the apo-form of PaGT2 (PDB ID: 6JEL),23 the binary complex of PtUGT1 (PDB ID: 6SU6)6 and the binary complex of NtUGT1 (chain A, PDB ID: 8CHD) are in their “open” state (Fig. 2a–c), with their corresponding Ω-1 loop located far away from the active site. In contrast, in the other structures, namely the TcCGT1 binary complex (PDB ID: 6JTD),9 the NtUGT1 (chain B) binary complex (PDB ID: 8CHD) and the UGT72B1 ternary complex with a donor analog and acceptor (PDB ID: 2VCE),8 are in their “closed” state with their loop region close to the active site (Fig. 2d–f). Specifically, in the structures of the NtUGT1 (chain B) and UGT72B1 complex, a tyrosine residue (Y319 in NtUGT1 and Y315 in UGT72B1) on the Ω-1 loop is within a hydrogen-bonding distance to the main chain of a loop region nearby the substrate binding site (Fig. S1b†). Previous mutagenesis study revealed that the mutation of Y315 led to a significant decrease in the enzyme activity.8 Thus, Y315 was proposed to play an important role in maintaining the catalytic dyad in a catalytic active position.8 Although the loop conformation flexibility is common among the GT families,24 insights into the effect of the conformation change on the catalytic reaction and the substrate-binding process still remain elusive.
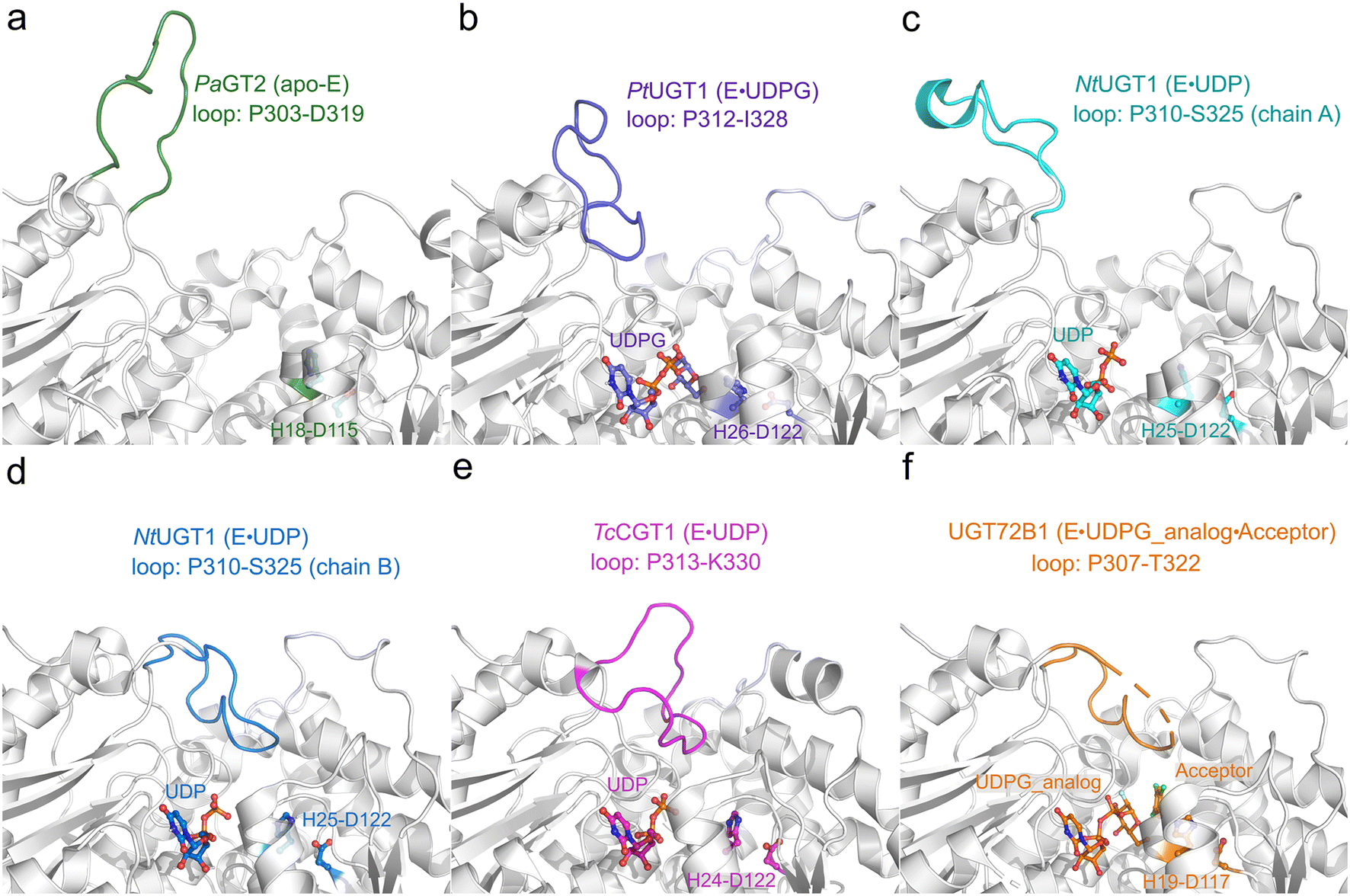 | ||
| Fig. 2 Crystal structures of various homologous enzymes of RrUGT3, including PaGT2, PtUGT1, NtUGT1, TcCGT1 and UGT72B1. (a) apo-PaGT2 (PDB ID: 6JEL)23 with the Ω-1 loop region (P303-D319) shown in green. (b) PtUGT1 binary complex (PDB ID: 6SU6)6 with the Ω-1 loop region (P312-I328) shown in purple. (c) NtUGT1 (chain A) binary complex with the Ω-1 loop region (P310-S325) shown in cyan. (d) NtUGT1 (chain B) binary complex (PDB ID: 8CHD) with the Ω-1 loop region (P310-S325) shown in blue. (e) TcCGT1 binary complex (PDB ID: 6JTD)9 with the Ω-1 loop region (P313-K330) shown in pink. (f) The ternary complex of UGT72B1 (PDB ID: 2VCE)8 with the Ω-1 loop region (P307-T322) shown in orange. The catalytic dyad (His-Asp) and substrates are shown as balls and sticks. | ||
In this work, combined structural, mutational, and multidimensional modelling studies are conducted to shed light on the key factors dictating the activity and the regioselectivity of RrUGT3-catalyzed glycosylation reactions. Our site-directed mutation experiments reveal that the Y314A mutation abolishes the activity of RrUGT3, indicating that Y314, which is far from the active site in the crystal structure of apo-enzyme, plays a vital role in the catalytic process. By using MD simulations, QM/MM and DFT calculations, the conformational change of the Ω-1 loop, the catalytic mechanism and the main factors controlling the regioselectivity of RrUGT3 were studied. Our results indicate that the “open-to-closed” conformational change of the Ω-1 loop is favored by substrate binding, and the active enzyme–substrate ternary complex remains stable only in the “closed” conformation. Furthermore, the regioselectivity of the RrUGT3-catalyzed HBA glycosylation reaction results from the ability of enzymes to stabilize different substrate orientations, and the difference in the activation energies of the phenolic and alcoholic glycosylation reactions.
2. Materials and methods
2.1 Strains, plasmids, and culture conditions
Strains and plasmids used in this study are listed in Table S1.†Escherichia coli DH5α was used as a host for gene cloning and plasmid construction using Luria–Bertani (LB) agar plates or LB liquid media. E. coli BL21(DE3) was used for protein expression and purification, and cultured in LB or terrific broth (TB: 1.2% tryptone, 2.4% yeast extract, 0.94% K2HPO3, 0.22% KH2PO3, 4% glycerol) media. All E. coli strains were grown at 37 °C unless otherwise specified. When required, appropriate antibiotics were added to the broth.2.2 Molecular manipulation, biochemical reagents, and chemicals
Gene cloning, plasmid transformation, agarose gel electrophoresis, and other standard techniques of molecular cloning were performed according to general protocols. Primers were synthesized by Sangon Biotech (Shanghai, China), and are listed in Table S2.† An E.Z.N.A.™ Plasmid Miniprep Kit (Omega Biotek, Norcross, GA) was used for plasmid isolation. An E.Z.N.A.™ Gel Extraction Kit (Omega Biotek, Norcross, GA) was used for DNA fragment purification. A T5 Super PCR Mix (Tsingke Biotechnology, Beijing, China) was used for colony PCR. A PrimeSTAR (Takara Bio) and Phanta Max Super-Fidelity DNA Polymerase (Vazyme Biotech, Nanjing, China) were used for routine PCR amplifications. A ClonExpress Ultra One Step Cloning Kit (Vazyme Biotech, Nanjing, China) was used for plasmid construction. HBA was purchased from Shanghai Aladdin.2.3 Protein expression and purification
The gene sequence encoding RrUGT3 was synthesized by BGI Genomics (Shenzhen, China), inserted into the expression vector pETM11 (EMBL)/pET-28b, and transformed into E. coli BL21(DE3) for overexpression of the recombinant protein. The bacteria were grown in Luria–Bertani (LB) medium at 37 °C until the value of OD600 reached about 0.6, and then 0.2 mM IPTG was added to induce protein expression, with further culturing at 16 °C overnight.Cells were harvested and disrupted by sonication in lysis buffer containing 50 mM Tris-HCl, 300 mM NaCl, pH 8.0. The lysates were clarified by centrifugation (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 60 min) at 4 °C and the supernatant was applied onto a nickel-chelating Sepharose affinity chromatography column (GE Healthcare). The N-terminal His6-tag was removed by TEV protease and further purification was performed by size-exclusion chromatography using a HiLoad 16/60 Superdex 200 column (GE Healthcare) in a buffer containing 50 mM Tris-HCl, 300 mM NaCl, 10 mM β-mercaptoethanol, pH 8.0. The purified proteins were concentrated to about 20 mg ml−1 as determined by absorbance at 280 nm and stored in aliquots at −80 °C after flash freezing in liquid nitrogen.
000g, 60 min) at 4 °C and the supernatant was applied onto a nickel-chelating Sepharose affinity chromatography column (GE Healthcare). The N-terminal His6-tag was removed by TEV protease and further purification was performed by size-exclusion chromatography using a HiLoad 16/60 Superdex 200 column (GE Healthcare) in a buffer containing 50 mM Tris-HCl, 300 mM NaCl, 10 mM β-mercaptoethanol, pH 8.0. The purified proteins were concentrated to about 20 mg ml−1 as determined by absorbance at 280 nm and stored in aliquots at −80 °C after flash freezing in liquid nitrogen.
2.4 Enzymatic assays and analytical methods
The in vitro enzymatic reactions were performed for characterizing the activity of RrUGT3 and its mutants. The reaction system contained 5 μM purified UGT enzyme, 0.5 mM HBA, 10 mM MgCl2, and 1 mM UDPG. All enzymatic assays were carried out in 100 μL of 50 mM Tris·HCl buffer (pH 7.5) at 30 °C for 2 h. Reactions were quenched with two volumes of methanol before HPLC analysis.Samples were analyzed on a Thermo Infinity HPLC system with a photodiode array detector. For characterization of UGT mutants, enzymatic samples were separated on a YMC Triart-C18 column by using a linear mobile phase gradient ranging from 5% acetonitrile in 0.1% TFA aqueous solution to 70% acetonitrile in 0.1% TFA aqueous solution over 25 min. The flow rate was set to 1 mL min−1, and the injection volume was 20 μL. Full-wavelength scans (210–400 nm) were recorded.
2.5 Crystallization and structure determination
The crystallization experiments were performed with a sitting-drop vapor-diffusion method at 20 °C. The drops contained an equal volume of protein sample and reservoir solution. After optimization, the best crystals of RrUGT3 were obtained in reservoir solution containing 0.05 M citric acid and 0.05 M bis-tris propane at pH 4.5 using 19% PEG3350. The crystallization solution added with 15% glycerol served as the cryoprotectant. Suitable crystals were quickly soaked in the cryo-protectant solution and frozen directly in liquid nitrogen.The diffraction data were collected on the BL19U1 beamlines at the Shanghai Synchrotron Radiation Facility (SSRF).25 The data were processed by XDS.26 The structure of RrUGT3 was solved by molecular replacement with the structure of homologous protein UGT72B1 (PDB ID: 2VCE) as the search model using experimental Phaser.27,28 The structures were further refined with Refmac 5 (ref. 29 and 30) based on the model built using Coot.31 The crystallographic data are summarized in Table S3.†
2.6 Model setup
MODELLER32 was used to add the missing non-terminal residues of RrUGT3 (74–81, 160–167 and 246–252, as shown in Fig. S2†). Then, the ternary complex was obtained by placing ligands UDPG and HBA into the binding pocket of RrUGT3 using the substrate-binding pattern of the ternary complex of UGT72B1·UDP-2-deoxy-2-fluoro glucose·2,4,5-trichlorophenol (PDB ID: 2VCE) as the reference. The protonation state of all titratable residues (His, Asp, Glu) were assessed by using the APBS server,33 and the position of the hydrogen atoms on histidine was inspected taking into account the H-bonding network around. The residue H363 was set to be double protonated while other histidine residues were in their neutral state. For the acidic residues, only Glu213 was set to its protonated state while others were set as deprotonated. The AMBER force fields FF19SB and GAFF2 were used for protein and substrates, respectively. To obtain the force field parameters for substrates, the structure of the substrates were firstly optimized at the B3LYP/6-31G* level of theory, then the partial atomic charges were obtained using a restraint electrostatic potential (RESP) method at the HF/6-31G* level of theory. All quantum mechanical calculations were performed using Gaussian 16 software.34Following the same protocol as in previous studies,35,36 we used the cluster approach with an active site model to explore the preferred protonation state of the diphosphate group of UDPG in the active site of RrUGT3. By analyzing the crystal structures of homologous enzymes of RrUGT3 in complex with cofactors (PtUGT1, PDB ID: 6SU6; UGT72B1, PDB ID: 2VCE; TcCGT1, PDB ID: 6JTD; NtUGT1, PDB ID: 8CHD), two water molecules are observed near the diphosphate group of UDPG in most of these structures. Therefore, the same number of water molecules were added around the diphosphate group of UDPG in our QC model. The final built QC models consist of the truncated UDPG, the two added water molecules, and the residues (H17, G275, S276, G277, Q348, H363, S368, E387, Q388) that show hydrogen bonding and steric interactions with the diphosphate moiety of UDPG, and HBA. The solvation effect of the protein environment was approximated by using the SMD37 solvation model with a dielectric constant of ε = 4. The evaluated pKa of different oxygen atoms of the diphosphate of UDPG is shown in Table S4.† Based on the evaluated pKa, two diphosphate oxygen atoms may be in the protonated state. However, the calculated energies of different models with the protonated diphosphate group at different oxygen atoms suggest that the model has the lowest energy when the phosphate group directly linked to the glucose moiety (Op1 position, Fig. S3a†) is protonated. Thus, our calculation results indicated that the diphosphate of UDPG prefers to be in the singly protonated state at the Op1 position. And the diphosphate moiety of UDPG was accordingly modeled as singly protonated in the following simulations. Then, all the missing hydrogen atoms in the system were added using the LEaP program. Subsequently, the built systems were solvated in a TIP3P water38,39 box which extended from the protein boundary of 12.0 Å, and then Na+ ions were added to neutralize the system.
2.7 Molecular dynamics simulations
After the system setup, the resulting systems were subjected to two-step minimization to remove the unreasonable interatomic contacts. At first, the protein–substrate complex was restrained using a force constant of 20 kcal (mol−1 Å−2), while the positions of the solvent and Na+ ions in the system were fully optimized (2500 steps for the steepest descent and 2500 steps for the conjugate gradient, respectively). For the second step of minimization, the employed parameters are the same as the first step, but the simulation was performed without any restraint. Subsequently, the system was slowly heated from 0 to 300 K under the NVT ensemble for 200 ps with a 2 fs time step, with a restraint constant of 50 kcal (mol−1 Å−2) on the protein–substrate complex to avoid any unreasonable movement.After that, the density of the system was equilibrated for 250 ps under the NPT ensemble at a constant temperature of 300 K and 1.0 bar pressure. In this procedure, a Langevin thermostat40 with a collision frequency of 2 ps−1 and a Berendsen barostat41 with a pressure relaxation time of 1 ps were used. Then, a productive MD simulation under the NPT ensemble was performed with a time step of 2.0 fs. The periodic boundary conditions were employed throughout the simulations. A Particle Mesh Ewald (PME)42 method with a cutoff distance of 10 Å for non-bonded interactions was used, and the bonds involving hydrogen were constrained by using a SHAKE43 method. The molecular mechanics/Poisson–Boltzmann surface area (MM-PBSA)44 calculations were performed on intervals of 5 frames of all the relaxed MD simulation snapshots for the “open” and “closed” state ternary complex of the phenolic glucosylation (the “open” state named as Model-AO and the “closed” state named as Model-AC) and the alcoholic glycosylation (the “open” state named as Model-BO and the “closed” state named as Model-BC). All the productive MD simulations were conducted using the GPU version of the AMBER 20 software package.45
2.8 Umbrella sampling simulations
Based on the crystal structure of RrUGT3, three models were built to explore the influence of substrate binding on the Ω-1 loop conformational change using the classical MD simulations combined with umbrella sampling (US).46,47 The constructed models include the RrUGT3·UDPG·HBA ternary complex model (named as Model-A) in which the phenolic hydroxyl group of HBA is located near the anomeric carbon of UDPG, the apo-RrUGT3 model (Model-1), and the RrUGT3·UDPG binary complex model (Model-2). Before US simulations, 50 ns restricted MD simulations were carried out for all systems with a weak force of 5 kcal (mol−1 Å−2) on UDPG, HBA and the surrounding residues (H17, E85, D118, S276, H363, Y385) to maintain them in the active center. The last snapshot from the equilibrated trajectories were then selected as the starting point of US simulations. In the US simulations, the collective variables (CVs) vary from 25.75 to 2.50 Å for Model-A, 29.00 to 2.00 Å for Model-1 and 25.25 to 2.00 Å for Model-2. The interval between the two adjacent windows was set to be 0.25 Å. To obtain a smooth free energy profile, the restraint MD simulations of each window were carried out successively, that is using the restart file of the last window as the starting point for the next window. To ensure a sufficient overlap between two adjacent windows, 20 ns MD simulation with a harmonic potential of 20 kcal (mol−1 Å−2) on the selected CV was performed for each window (Fig. S4†). Meanwhile, to maintain the proper binding of HBA, a force constraint of 5 kcal (mol−1 Å−2) was set on the HBA, UDPG and the residues H17 and E85. The dump frequency for the data collection of the following potential of mean force (PMF) analysis was set to be 5 ps−1, then PMF was calculated using the data from the last 10 ns data of each window by a weighted histogram analysis method (WHAM).48,49 All the US simulations were performed using the GPU accelerated version of PMEMD implemented in AMBER 20.45 Then, the last snapshot of the trajectory from the window corresponding to the “closed” state (∼4.60 Å on the CV of Model-A) was used as the starting point for the MD simulations. To understand the molecular basis of the regioselectivity of the enzyme, both the phenolic glucosylation and alcoholic glycosylation models were considered in our MD simulations. Based on the snapshot of the “closed” state of Model-A, the “closed” state of Model-B was constructed by flapping the HBA molecule in a way that the alcoholic hydroxyl group is near the anomeric carbon of UDPG. To further equilibrate the obtained “closed” state ternary complexes, 20 ns restricted MD simulations were performed for each system, in which the position of UDPG and the key distances involved in the catalytic reaction (O1HBA–CαUDPG, O1HBA–OaY314, H1HBA–NεH17 in Model-AC; O2HBA–CαUDPG, O2HBA–OaY314, H2HBA–NεH17 in Model-BC) were restrained using force constants of 10 kcal (mol−1 Å−2) and 5 kcal (mol−1 Å−2), respectively. After that, 50 ns multi-trajectory fully relaxed MD simulations were carried out with 5 independent 10 ns MD simulations. Then, clustering was carried out for each system to group the trajectories into 10 clusters on the basis of the RMSD of residues within 5 Å of substrates in the first snapshot as the distance metric. The most representative snapshot from each system was taken as the starting point (Fig. 7c and d) for the following quantum mechanics/molecular mechanics (QM/MM) calculations.2.9 Quantum mechanical/molecular mechanical calculations
The additive QM/MM calculations were performed using ChemShell v7.3.1,50 which combines ORCA v5.0.2 (ref. 51) for the QM region and DL_POLY52 for the MM region. The MM region was described by using the AMBER FF19SB force field. For the phenolic glucosylation model (Model-AC), the QM region included the side chain of residues (H17, D118, S141, S276, Y314, H363, E387, Q388 and Y385), the diphosphate-glucose group of UDPG, HBA and six water molecules that directly form hydrogen bonds with substrates and Y314 (Fig. 3a). Meanwhile, for the alcoholic glucosylation model (Model-BC), only two water molecules were observed forming direct hydrogen bonds with the substrates and Y314 (Fig. 3b). The resulting QM region for Model-AC and Model-BC contains 163 atoms and 151 atoms, respectively, and both systems have an overall charge of −2. The active region consists of the residues and waters within 8 Å from the QM region. The electrostatic embedding scheme53 was employed to describe the polarization effect of enzymes on the QM region. The QM/MM boundary was treated by using hydrogen link atoms with the charge shift model.54,55 For geometry optimizations and frequency calculations the B3LYP-D3(BJ)/def2-SVP level of theory was used for the QM region. All the minimum searching was performed by using the hybrid delocalised internal coordinates (HDLC) and limited-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) optimisation algorithm using the DL-FIND module.56 To reduce the computational cost, all the water molecules with a distance larger than 10 Å from the RrUGT3·UDPG·HBA complex were removed from the system.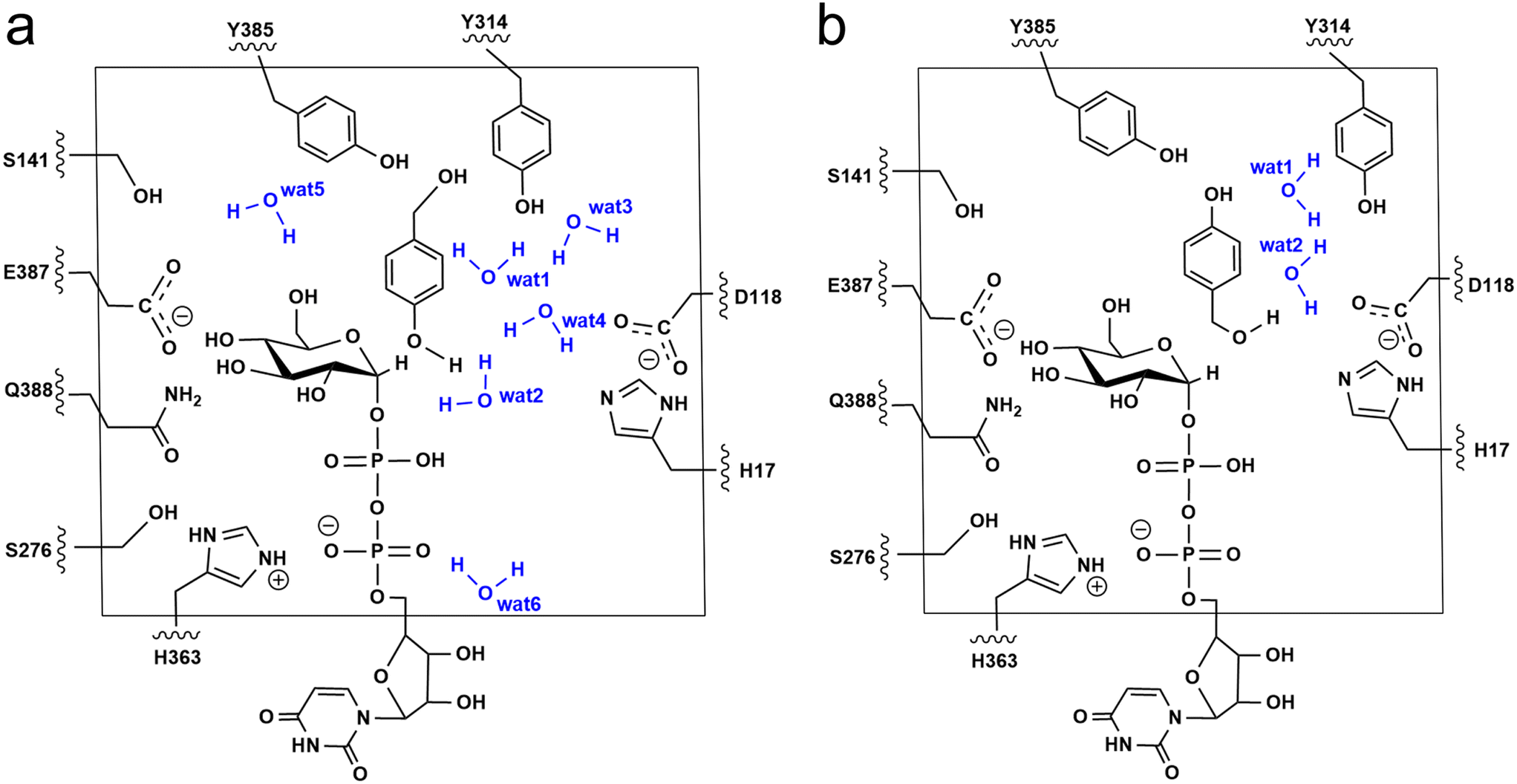 | ||
| Fig. 3 QM regions used in our QM/MM calculations for (a) Model-AC and (b) Model-BC. | ||
The initial geometry of the transition states were obtained by scanning the potential energy surface (PES), which was further optimized by the P-RFO algorithm implemented in the HDLC code.57 All the stationary points were validated by frequency calculations with a single imaginary frequency for the transition states, and no imaginary frequency for the intermediates. The single-point calculations were performed to obtain more accurate energies with a larger basis set of def2-TZVP on the basis of the optimized geometries. The final reported QM/MM energies were single-point energies with the large basis set corrected by zero-point energies (ZPE).
2.10 Quantum chemical calculations
To further investigate the inherent driving force of the glycosylation, the reaction energy profiles of the “core” reaction region of the phenolic-glycosylation and alcoholic-glycosylation processes were calculated in a water environment using the Gaussian 16 software.34 In this calculation, only the catalytic dyad, the truncated UDPG, and the sugar acceptor were considered as shown in Fig. S5 and S6.† Truncations of residues were made at the β-carbons of the amino acids and hydrogen atoms were added to saturate the β-carbons. To avoid unreasonable movements during the geometry optimizations, the truncated carbons and some hydrogens were fixed. Geometries were optimized at the B3LYP-D3(BJ)/def2-SVP level of theory. Based on the optimized structures, frequency calculations were carried out at the same theoretical level to confirm the properties of the corresponding stationaries and to obtain the ZPE. At the same theoretical level with geometry optimization, the solvation effects of the water environment was approximated by using the SMD37 solvation model with a dielectric constant of ε = 78. The single-point energies were calculated to obtain more accurate energies with a larger basis set def2-TZVP. The reported QC energies for these models were single-point energies corrected by ZPE and solvation.3. Results and discussion
3.1 Crystal structure of RrUGT3
Enzymatic structural information is important for understanding the molecular mechanism. The crystal structure of substrate-free RrUGT3 was solved at 1.87 Å resolution in the space group P212121 with one molecule in the asymmetric unit forming one monomer (PDB ID code: 8YP7). Partial loop regions including residues 74–81, 160–167, 246–252, and the N-terminal residues 1–2, and C-terminal residues 471–472 lacked defined electron density, supposedly due to their flexibility. The majority of modelled residues exhibited excellent electron density and geometry. Despite numerous attempts, we were unable to obtain the complex structure of RrUGT3 with UDPG, UDP, or both UDP and HBA.The overall structure of RrUGT3 exhibits a typical GT-B folding mode, consisting of two Rossmann-folded β/α/β domains at N- and C-terminals, respectively. A long and narrow cleft is formed between the two domains, where the ligand-binding sites are supposed to be located (Fig. S7†). In the structure, the C-terminal domain is composed of five β-strands and ten α-helices and has highly conserved plant secondary product glucosyltransferase (PSPG) motif43 (W345-Q388). Upon superimposition of the obtained structure of RrUGT3 with the previously reported homologous UGTs (Fig. 2 and S1†), it is evident that the flexible Ω-1 loop region is also present in RrUGT3 (P306-K321). A tyrosine residue (Y314) corresponding to Y315 in UGT72B1, which has been demonstrated to be crucial for catalysis,8 is also observed at the Ω-1 loop region. Furthermore, the catalytic dyad in RrUGT3 is confirmed to be composed of H17 and D118.
3.2 Site-directed mutagenesis analysis
To verify the importance of Tyr314, located at the Ω-1 loop region, and the residues composing the catalytic dyads (H17 and D118), a series of site-directed mutational analysis was performed. Additionally, two active site residues potentially interacting with HBA (F120 and E85) were also considered. The effects of the mutations on catalytic activities were evaluated by HPLC analysis (Fig. 4). For the wild-type (WT) enzyme of RrUGT3, the conversion ratio of HBA has been previously determined to be 97.7% to 1a, with only 0.6% 1b as the minor product. The two catalytic dyad mutants, H17A and D118A, showed no activity, confirming their essential roles as the general base and charge balance residue, respectively. Interestingly, the Y314A mutant completely lost the catalytic activity, indicating an important role of Y314 in the effective catalysis of RrUGT3. This finding is consistent with that reported for UGT72B1.8 Mutation of F120 to alanine caused a decrease of the conversion ratio to 74.7%, implying its role in forming the sugar acceptor binding pocket. For E85, the replacement by an alanine resulted in only a slight decrease of the activity.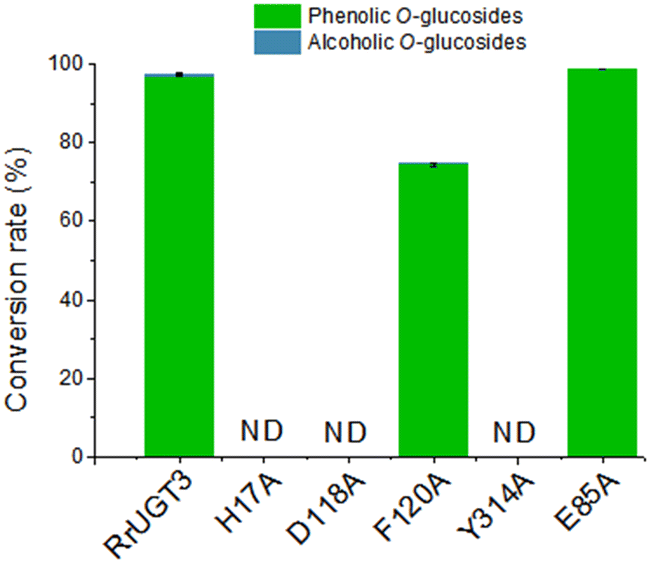 | ||
| Fig. 4 The structure-guided mutagenesis analysis with the relative catalytic activities of RrUGT3 wild-type (WT) and mutants (H17A, D118A, F120A, Y314A, and E85A). The ratio of both phenolic O-glycoside and alcoholic O-glycoside products are given. “ND” means no products detected. | ||
3.3 Ligand induced “open-to-closed” conformational change
The obtained crystal structure of apo-RrUGT3 revealed that the key residue Y314 for the enzyme's activity is located in the Ω-1 loop region far from the active center (Fig. S7†). Moreover, as discussed above, among homologous GTs of RrUGT3, both the “open” and “closed” conformations of the Ω-1 loop region were observed. These results imply that the “open-to-closed” state change of the loop region may play an important role in catalysis. To gain further insights into the catalytic state of RrUGT3, a series of MD simulations were performed to study the RrUGT3·UDPG·HBA ternary complex.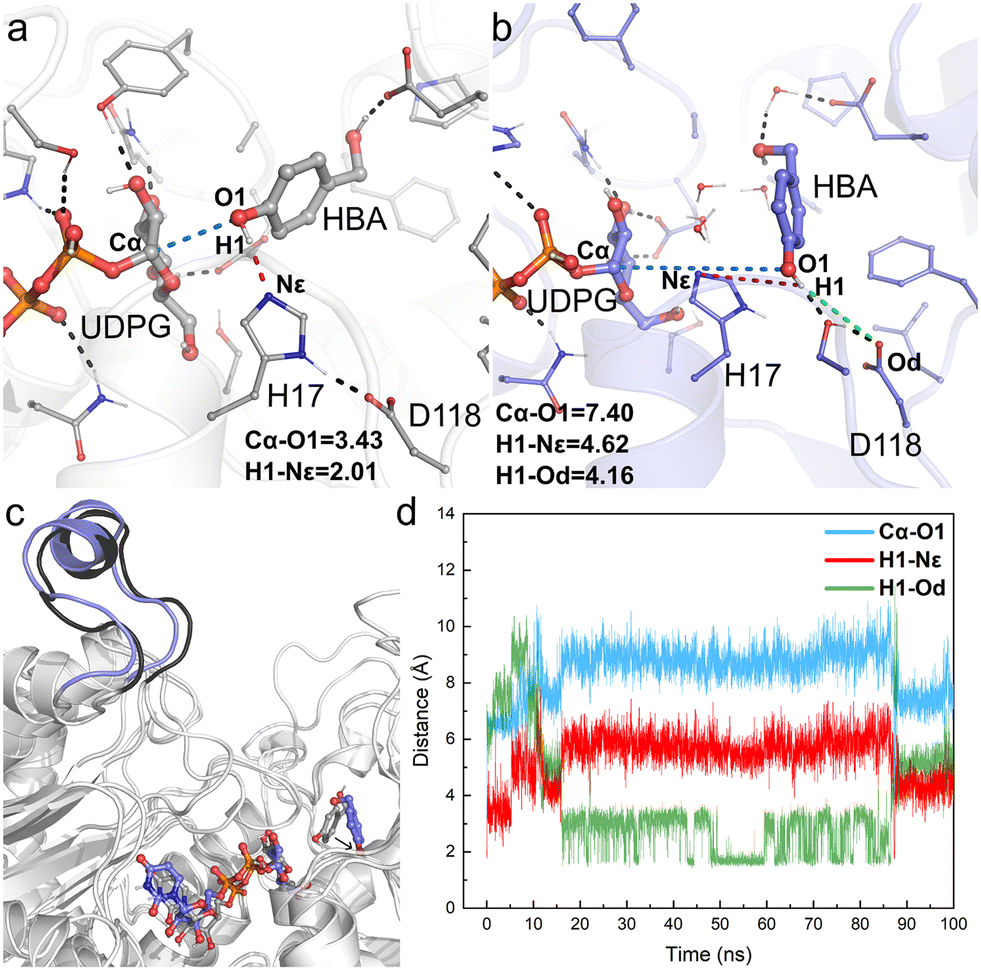 | ||
| Fig. 5 Key geometry information obtained from MD simulations of Model-AO. (a) Details of the active center of the initial structure of MD simulations; (b) details of the active center of the final snapshot; (c) structural alignment of the initial structure and the final snapshot; (d) fluctuation of the key distances involved in the catalytic process during 100 ns MD simulations, including Cα–O1 (blue line), H1–Nε (red line) and H1–Od (green line). Hydrogen bonds and key distances (in angstrom) are shown as dashed lines. | ||
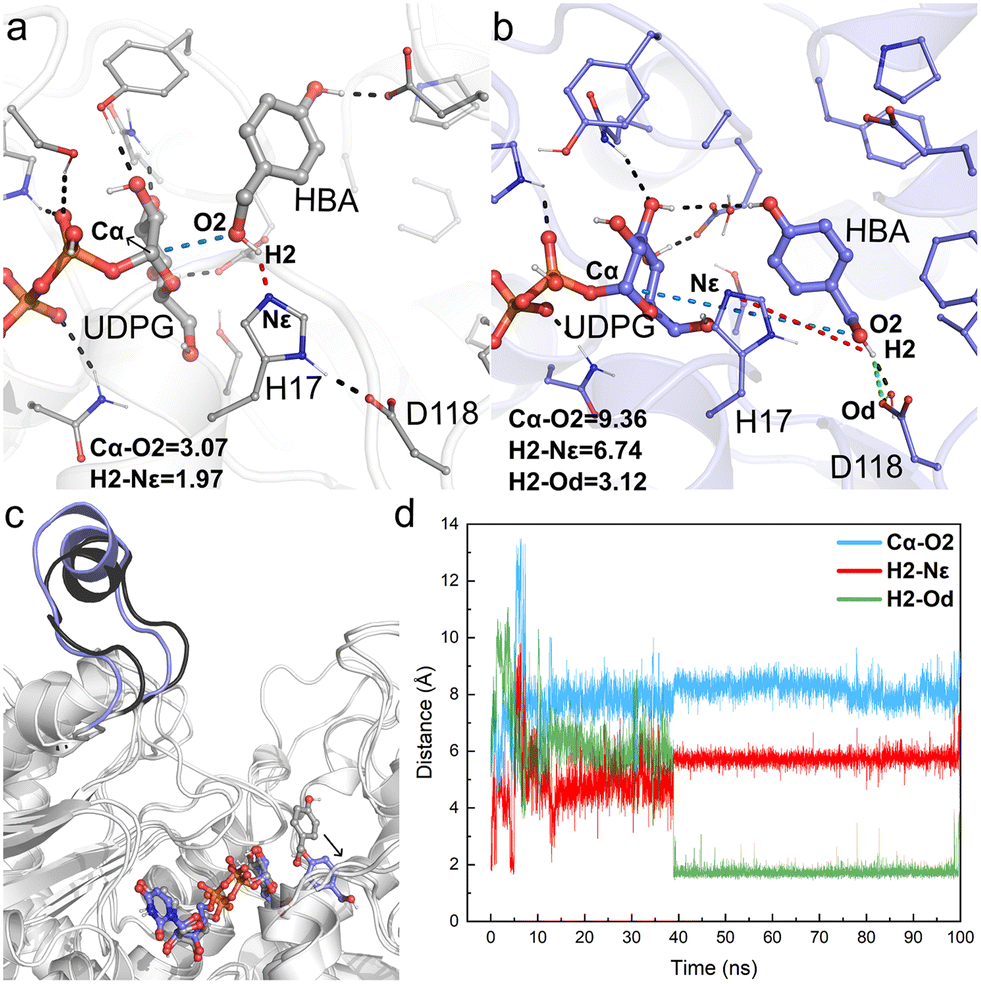 | ||
| Fig. 6 Key geometry information obtained from MD simulations of Model-BO. (a) Details of the active center of the initial structure of MD simulations; (b) details of the active center of the final snapshot; (c) structural alignment of the initial model and the final snapshot; (d) fluctuation of the key distances involved in the catalytic process during 100 ns MD simulations, including Cα–O2 (blue line), H2–Nε (red line), and H2–Od (green line). Hydrogen bonds and key distances (in angstrom) are shown as dashed lines. | ||
For Model-AO, as shown in Fig. 5, the influence of water molecules on the binding state of HBA is observed at the onset of the MD simulations. This breaks the hydrogen bond between HBA and H17 residues, as evidenced by the measured increase of d(H1–Nε) to ca. 3.5 Å within 5 ns and further to ca. 5 Å (Fig. 5d). After 15 ns MD simulation, the phenolic hydroxyl group forms a hydrogen bond with D118 of the catalytic dyad and this interaction is maintained in the simulation. Meanwhile, as shown in Fig. 5b, the H17 residue together with several water molecules are stacked between HBA and the anomeric carbon of the UDPG sugar donor. In such a binding mode, the nucleophilic attack pathway of the phenolic hydroxyl group on the anomeric carbon is blocked, and this mode is thus not favorable for the reaction to occur. A similar binding mode is observed in Model-BO (Fig. 6), in which the H17 residue blocks the nucleophilic attack route between the alcoholic hydroxyl group and the anomeric carbon.
Furthermore, we performed a two-step simulation consisting of an initial MD simulation with constrains on the reaction site to first minimize the surroundings, which is followed by a regular simulation without constrains. The constrains were run for 50 ns, with a weak force of 5 kcal (mol−1 Å−2) on UDPG, HBA and the surrounding residues (H17, D118, E85, S276, Y385, H363) to maintain them at a pre-reaction mode. However, upon removing constrains, the HBA molecules quickly move far away from the anomeric carbon of UDPG in both Model-AO and Model-BO (Fig. S8†).
Therefore, the simulation results show that the enzyme conformation in the “open” state fails to stabilize the substrate in the catalytically favorable mode. Specifically, the conformation in the “open” state is not conducive for the reaction to occur.
The umbrella sampling (US) technique has been used to study the free energy profile of the conformational change of various proteins,58–62 and is employed in the present study to investigate the effect of the substrate binding on the Ω-1 loop configuration change. In total, three models were constructed, namely Model-1 for apo-RrUGT3, Model-2 for the RrUGT3·UDPG binary complex, and Model-A for the ternary complex. Previous study revealed that the phenolic glycoside is the main product of RrUGT3;19 therefore, for the ternary complex of RrUGT3, only the phenolic glycosylation preferred binding mode with the phenolic hydroxyl group of HBA located near the anomeric carbon of UDPG (Model-A) is considered here. The CVs of our US simulations were set based on the reported crystal structures of homologous enzymes. As observed in the binary complex of NtUGT1 (PDB ID: 8CHD; chain B), the Ω-1 loop region in its “closed” state with tyrosine (Y319) is located near the carbonyl group of the main chain of a proline residue (P21). Therefore, for Model-1 and Model-2 of RrUGT3, the CVs were set to be the distance between Oa of Y314 and Ob of P13 (the black dashed line in Fig. 7a). For the ternary enzyme–substrate complex, as observed in the crystal structure of UGT72B1 (PDB ID: 2VCE),8 the tyrosine (Y315) on the Ω-1 loop region is located within the hydrogen bond distance to the sugar acceptor; therefore for Model-A of RrUGT3, the CV was set to be the distance between Oa of Y314 and O1 of HBA (the red dashed line in Fig. 7a).
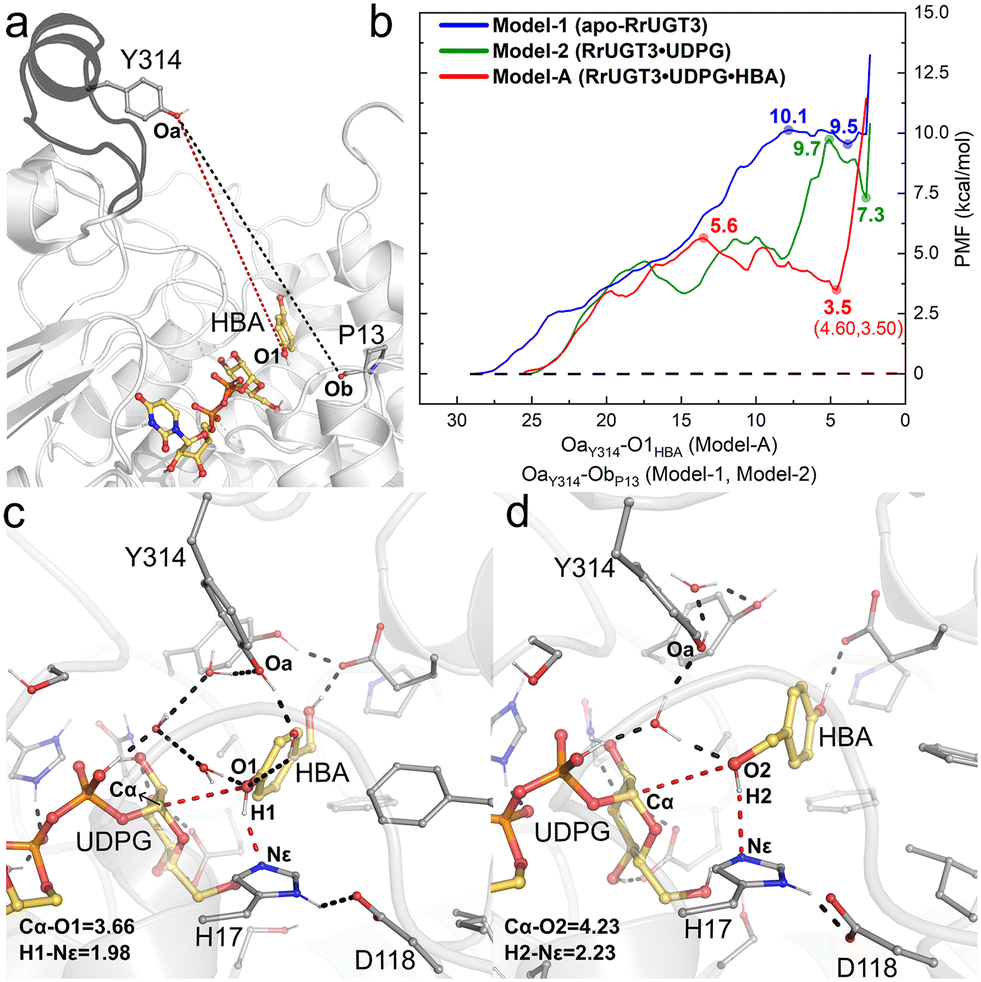 | ||
| Fig. 7 (a) Collective variables used in the umbrella sampling simulations for Model-1 and Model-2 (black dashed line), and Model-A (red dashed line). (b) The calculated PMF for the “open-to-closed” transformation of the Ω-1 loop region in the three models. (c) The representative conformation of Model-AC. (d) The representative conformation of Model-BC. Hydrogen bonds and key distances are shown as dashed lines. Key distances are given in angstrom. | ||
Our results show that for Model-1, the calculated free energy of the “closed” state is 9.5 kcal mol−1 higher than that of the “open” state and the conformational transition barrier is 10.1 kcal mol−1 (Fig. 7b). Upon binding of UDPG (Model-2), the “open-to-closed” transition barrier slightly decreased to 9.7 kcal mol−1, and the relative energy between the “closed” state and the “open” state decreased to 7.3 kcal mol−1 (Fig. 7b). However, in the case with both UDPG and HBA bound (Model-A), the energy of the “closed” state is only 3.5 kcal mol−1 higher than that of the “open” state and the barrier is decreased to 5.6 kcal mol−1 (Fig. 7b). Evidently, the binding of the substrates could facilitate the “open-to-closed” conformational change. This finding is consistent with the previous study on retaining glucosyl-3-phosphoglycerate synthase (GpgS) from Mycobacterium tuberculosis, where the substrate binding also decreased the conformational transition energy barrier and the relative energy between the “closed” and “open” conformation.63 Analyzing the hydrogen bond networks around the loop region at the “closed” state of the three models (Fig. S9†) revealed that there are more hydrogen bond interactions between the Ω-1 loop region and the surrounding residues in Model-A. Specifically, we observed additional hydrogen bond interactions between N311 and S312 on the Ω-1 loop region with H81 and P82 in Model-A. These hydrogen bond interactions may be induced by the binding of UDPG and HBA, which causes a different binding conformation of the Ω-1 loop compared to the other two models. Consequently, the Ω-1 loop in the ternary complex (Model-A) can form more hydrogen bonds with surrounding residues and thus forms a more stable “closed” state than the apo-enzyme or binary complex. Overlapping the “closed” state of Model-A with the reported crystal structure of the highly homologous enzyme (TcCGT1, Fig. S10†) reveals that the backbone of the Ω-1 loop region shows good alignment between the structures.
Then, the last snapshot of the window corresponding to the “closed” state of Model-A (∼4.60 Å of the CV) was taken as the initial geometry for the MD simulations of the ternary complexes of RrUGT3 with substrates. Two models were considered in the simulations, where the HBA substrate is oriented to facilitate the phenolic glucosylation (Model-AC) and the alcoholic glycosylation (Model-BC). The enzyme–substrate binding energies were evaluated by using MM-PBSA calculations. The results indicate that the binding affinity of the substrates in the “closed” state (−36.2 kcal mol−1 and −39.1 kcal mol−1 for Model-AC and Model-BC, respectively, Table S5†) is stronger than that in the “open” state (−24.5 kcal mol−1 and −13.7 kcal mol−1 for Model-AO and Model-BO, respectively, Table S5†). Upon analyzing the obtained trajectories of the “closed” state, it was observed that in most of the sampled trajectories of Model-AC, the phenolic hydroxyl group can reach the catalytically active conformation, with the d(O1–Cα) shorter than 4.0 Å, while larger d(O2–Cα) distance fluctuation is observed in Model-BC (Fig. S11†). Thus, the substrate binding pocket of RrUGT3 favors the binding mode of the HBA substrate for the phenolic glycosylation over the alcoholic glycosylation. This is consistent with the experimental result, which shows that the phenolic O-glycoside is the main product.19 To further elucidate the main factors controlling the regioselectivity, we investigated the detailed catalytic mechanism of the RrUGT3-catalyzed HBA glycosylation reactions using QM/MM calculations on the basis of the most representative snapshots in MD simulations of Model-AC (Fig. 7c) and Model-BC (Fig. 7d).
3.4 Catalytic mechanism and regioselectivity
The QM/MM optimized geometries of the reactants for phenolic glycosylation (ES) and alcoholic glycosylation (ES′) are shown in Fig. 8. In ES, the glucose moiety of UDPG forms hydrogen bonds with the residues S141, E387, and Q388, while the uracil moiety interacts with W345 by π-stacking. For the HBA substrate, its phenolic and alcoholic hydroxyl groups form hydrogen bonds with H17 and E85, respectively. In ES′, the binding mode of the UDPG substrate in the alcoholic glycosylation model is similar to that in ES. However, the orientation of HBA in ES′ is reversed, with the phenolic and alcoholic hydroxyl groups of HBA hydrogen-bonded with E85 and H17, respectively. Furthermore, stable hydrogen bonding networks involving the diphosphate group of UDPG, the hydroxyl groups of HBA, Y314 and a number of water molecules were observed. These interactions may play important roles in constraining the glucose ring of the sugar donor and in orientating the sugar acceptor in a catalytically favorable position. Note that, during the MD simulations, a stable hydrogen bonding interaction between Y385 and E85 was observed (Fig. S12†); this hydrogen bonding interaction anchors the position of E85 and further stabilizes the orientation of the sugar acceptor HBA. Therefore, our simulations suggested that E85 together with Y385 may play a role in substrate recognition and orientation, thus explaining why the E85A mutant shows only a slight decrease in the catalytic activity.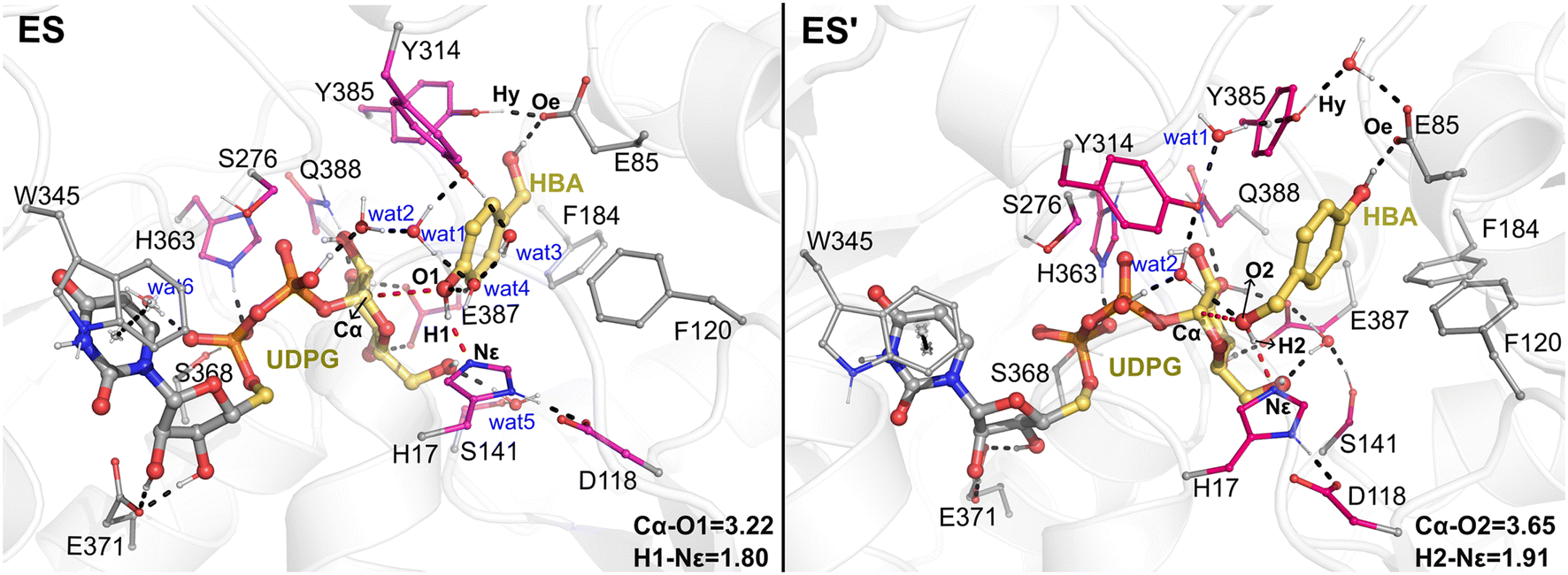 | ||
| Fig. 8 The enzyme–substrate interaction analysis of the QM/MM optimized structures of the reactants for phenolic glycosylation (ES) and alcoholic glycosylation (ES′). Hydrogen bonds and key distances are shown as dashed lines. The QM region is highlighted in pink and yellow while the MM region is shown in gray. Key distances are given in angstrom. | ||
Then, from the optimized reactants, the detailed reaction mechanism of the RrUGT3-catalyzed glycosylations was investigated. Given the fact that the acidity of phenolic hydroxyl is stronger than alcoholic hydroxyl, the barrier of phenolic glycosylation would be expected to be lower than the alcoholic glycosylation. After testing various potential reaction mechanisms, including the proton transfer and nucleophilic attack stepwise reactions, our QM/MM calculation results show that these glycosylation reactions follow an “SN2” type reaction mechanism, with only one transition state located for each reaction. The calculated barriers of the phenolic glycosylation and alcoholic glycosylation reactions are 15.5 kcal mol−1 and 19.8 kcal mol−1, respectively (Fig. 9, Table S6†). The transition states of these glycosylation reactions involve three processes, including the proton transfer from the OH-group of HBA to the catalytic dyad, the nucleophilic attack of the deprotonated OH-group to the sugar anomeric Cα, and the breaking of the Cα–Op glycosidic bond of UDPG. Analyzing the geometries of the optimized transition states reveals that, in Model-AC (TS, Fig. 9) the proton of the phenolic hydroxyl group has already transferred to H17 and the nucleophilic attack distance (Cα–O1) is 2.20 Å, while in Model-BC (TS′, Fig. 9) the proton transfer from the alcoholic hydroxyl group to H17 has not happened yet and the nucleophilic attack distance (Cα–O2) is 1.86 Å. Thus, the transition state in Model-AC is fairly earlier than that of the Model-BC along the reaction coordinate.
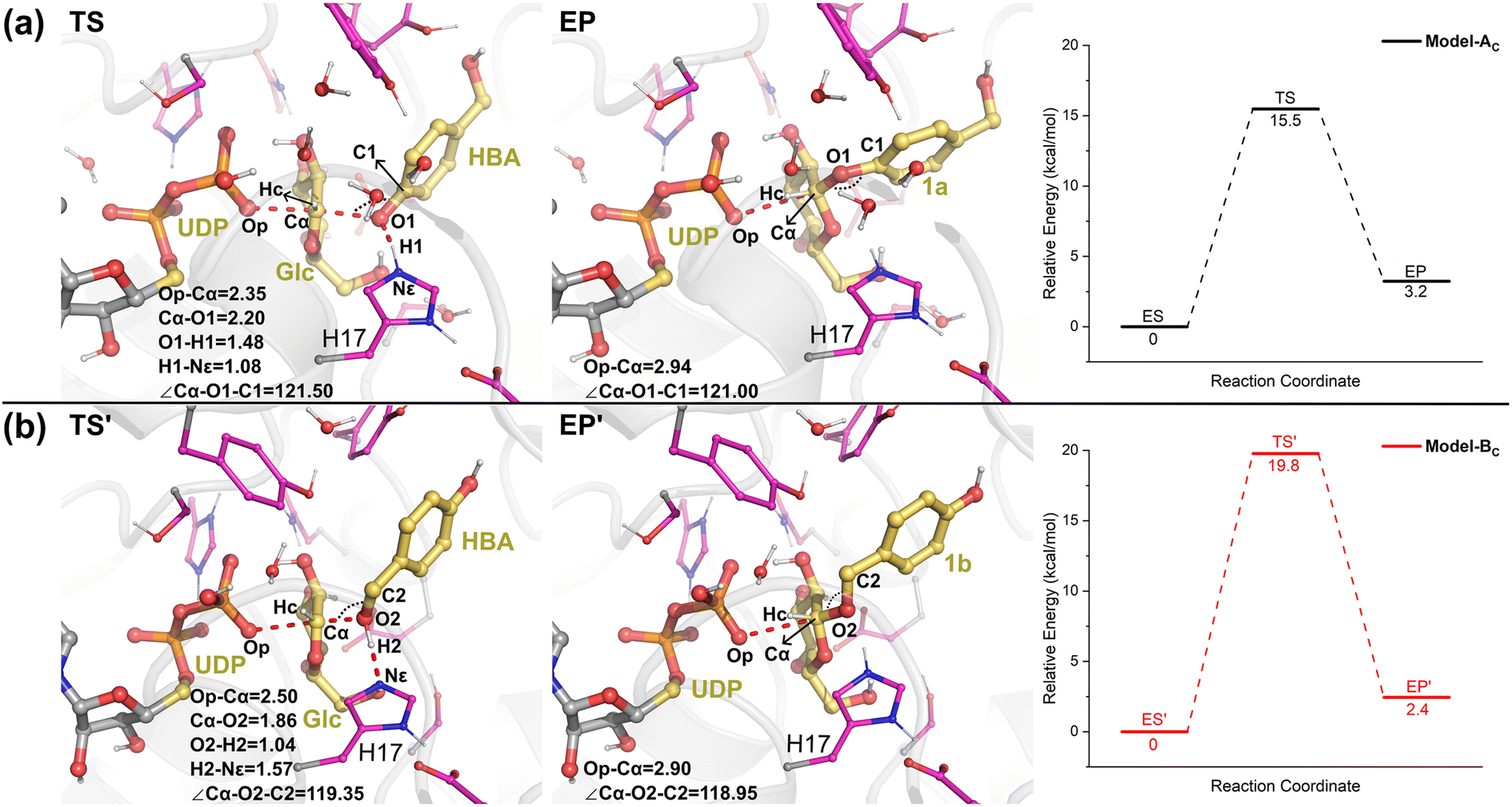 | ||
| Fig. 9 The QM/MM optimized geometries of the transition states, the enzyme-product complexes, and the calculated energy profiles for the (a) phenolic glycosylation (TS and EP) and (b) alcoholic glycosylation (TS′ and EP′). Key distances and angles are given in angstrom and degree, respectively. | ||
The calculated energy barrier for the phenolic glycosylation reaction is 4.3 kcal mol−1 lower than that of the alcoholic glycosylation reaction. Experimentally, the RrUGT3-catalyzed reaction between HBA and UDPG produced phenolic O-glycoside as the main product with a regioselectivity of 98.5%,19 showing that the phenolic glycosylation is much more favored. The calculation results are thus in good agreement with the experimental data. As discussed above, the MD simulations reveal that the enzyme accommodates the conformation for phenolic glycosylation more stably than for alcoholic glycosylation (Fig. S11†). Therefore, the observed regioselectivity towards phenolic glycosylation in RrUGT3 can be attributed to the favorable substrate binding and the lower activation energy barrier compared to the alcoholic glycosylation.
3.5 The effect of the protein environment on glycosylation reactions
To investigate the influence of the protein environment on the energetics of the RrUGT3-catalyzed glycosylation of HBA, quantum chemical (QC) calculations were further performed using a model consisting of the catalytic dyad, HBA, and the truncated UDPG.Because the pKa value of the diphosphate moiety of UDP-GlcNAc was predicted to be 6.5 by the previous 31P nuclear magnetic resonance (NMR) study,64 thus the diphosphate moiety should be in its deprotonated state in water. Using a model containing deprotonated diphosphate UDPG and the catalytic triad, the calculated energy barriers for phenolic and alcoholic glycosylation in aqueous solution are as high as 28.8 kcal mol−1 and 52.6 kcal mol−1 (Fig. S5, Table S7†), respectively. Meanwhile, different from that in water, the diphosphate prefers to be in its singly protonated state in the active site of RrUGT3 (Fig. S3, Table S4†); thus we calculated the reaction with a singly protonated diphosphate. The barriers are lowered to 12.2 kcal mol−1 and 25.1 kcal mol−1 for the phenolic glycosylation and alcoholic glycosylation, respectively (Fig. S6, Table S8†). These results indicated that the protonation state of the diphosphate moiety of UDPG is vital for the reaction to occur. Thus, it can be concluded that the protein environment of RrUGT3 increases the pKa value of the diphosphate moiety of UDPG by a preorganized binding pocket, which significantly reduces the barriers of glycosylation reactions.
4. Conclusions
In the present work, we employed crystallographic, site-directed mutagenesis, and computational studies to shed light on the catalytic mechanism of RrUGT3. The MD simulations based on the obtained crystal structure reveal that although the Ω-1 loop (residues 306 to 321) of RrUGT3 tends to be in the “open” state, the “closed” state of the loop region is essential for the catalysis. This conformational change is induced by the binding of the substrates (UDPG and HBA), and only when the RrUGT3 at the “closed” conformation can the substrates bind steadily in the catalytic mode. Furthermore, our calculation results indicate that RrUGT3 catalyzes the glycosylation reaction via an “SN2” type mechanism. The catalytic power of RrUGT3 can be attributed to the preorganized protein environment, which alters the preferred protonation state of the diphosphate of UDPG from deprotonated in water to singly protonated in RrUGT3. Finally, the regioselectivity of the enzyme is mainly controlled by the favorable substrate binding and the lower activation barrier of the phenolic glycosylation compared to the alcoholic glycosylation. Overall, the obtained information provides valuable insights for the rational design of UGTs and is helpful in developing new strategies for synthesizing unnatural O-glucosides.Data availability
The Cartesian coordinates of key stationaries along the reaction mechanism are provided in the ESI.† All other data underlying this work will be shared on reasonable request to the corresponding authors.Author contributions
H. S., X. S. and L. D. designed the project and supervised the project. C. Y. solved the structure of RrUGT3. F. G. performed experimental mutagenesis. M. L. performed computational calculations and the corresponding data analysis. Q. H. is also involved in the data analysis. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFA0911500 to Xiang Sheng and Lei Du), the National Natural Science Foundation of China (22203102 to Hao Su; 32200030 to Cai You; 32370032 and 32170088 to Lei Du), and the Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-CXRC-063 to Hao Su; TSBICIP-CXRC-026 to Xiang Sheng). A portion of the calculations in this study were performed on the ORISE Supercomputer.References
- S. Paquette, B. L. Møller and S. Bak, On the origin of family 1 plant glycosyltransferases, Phytochemistry, 2003, 62(3), 399–413 CrossRef CAS PubMed.
- J. R. Rich, A. Szpacenko, M. M. Palcic and D. R. Bundle, Glycosyltransferase-Catalyzed Synthesis of Thiooligosaccharides, Angew. Chem., Int. Ed., 2004, 43(5), 613–615 CrossRef CAS PubMed.
- P. Tiwari, R. S. Sangwan and N. S. Sangwan, Plant secondary metabolism linked glycosyltransferases: An update on expanding knowledge and scopes, Biotechnol. Adv., 2016, 34(5), 714–739 CrossRef CAS.
- Y. Yang, Y. Liang, F. Cui, Y. Wang, L. Sun, X. Zan and W. Sun, UDP-Glycosyltransferases in Edible Fungi: Function, Structure, and Catalytic Mechanism, Fermentation, 2023, 9(2), 164 CrossRef CAS.
- K. Yonekura-Sakakibara and K. Hanada, An evolutionary view of functional diversity in family 1 glycosyltransferases, Plant J., 2011, 66(1), 182–193 CrossRef CAS PubMed.
- D. Teze, J. Coines, F. Fredslund, K. D. Dubey, G. N. Bidart, P. D. Adams, J. E. Dueber, B. Svensson, C. Rovira and D. H. Welner, O-/N-/S-Specificity in Glycosyltransferase Catalysis: From Mechanistic Understanding to Engineering, ACS Catal., 2021, 11(3), 1810–1815 CrossRef CAS.
- J. Ross, Y. Li, E.-K. Lim and D. J. Bowles, Higher plant glycosyltransferases, Genome Biol., 2001, 2(2), reviews3004.3001 CrossRef PubMed.
- M. Brazier-Hicks, W. A. Offen, M. C. Gershater, T. J. Revett, E.-K. Lim, D. J. Bowles, G. J. Davies and R. Edwards, Characterization and engineering of the bifunctional N- and O-glucosyltransferase involved in xenobiotic metabolism in plants, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(51), 20238–20243 CrossRef CAS.
- J.-B. He, P. Zhao, Z.-M. Hu, S. Liu, Y. Kuang, M. Zhang, B. Li, C.-H. Yun, X. Qiao and M. Ye, Molecular and Structural Characterization of a Promiscuous C-Glycosyltransferase from Trollius chinensis, Angew. Chem., Int. Ed., 2019, 58(33), 11513–11520 CrossRef CAS.
- J. Paz-Ares, M. I. Puga, M. Rojas-Triana, I. Martinez-Hevia, S. Diaz, C. Poza-Carrión, M. Miñambres and A. Leyva, Plant adaptation to low phosphorus availability: Core signaling, crosstalks, and applied implications, Mol. Plant, 2022, 15(1), 104–124 CrossRef CAS PubMed.
- C. J. Thibodeaux, C. E. Melançon and H.-W. Liu, Unusual sugar biosynthesis and natural product glycodiversification, Nature, 2007, 446(7139), 1008–1016 CrossRef CAS.
- D. Bowles, J. Isayenkova, E.-K. Lim and B. Poppenberger, Glycosyltransferases: managers of small molecules, Curr. Opin. Plant Biol., 2005, 8(3), 254–263 CrossRef CAS PubMed.
- V. Kren and L. Martinkova, Glycosides in Medicine: “The Role of Glycosidic Residue in Biological Activity”, Curr. Med. Chem., 2001, 8(11), 1303–1328 CrossRef CAS.
- S. J. Thorson, J. T. Hosted Jr, J. Jiang, B. J. Biggins and J. Ahlert, Natures Carbohydrate Chemists The Enzymatic Glycosylation of Bioactive Bacterial Metabolites, Curr. Org. Chem., 2001, 5(2), 139–167 CrossRef.
- T. Mijatovic, E. Van Quaquebeke, B. Delest, O. Debeir, F. Darro and R. Kiss, Cardiotonic steroids on the road to anti-cancer therapy, Biochim. Biophys. Acta, Rev. Cancer, 2007, 1776(1), 32–57 CrossRef CAS PubMed.
- N. J. Brooks, H. A. A. Hamid, R. Hashim, T. Heidelberg, J. M. Seddon, C. E. Conn, S. M. Mirzadeh Husseini, N. I. Zahid and R. S. D. Hussen, Thermotropic and lyotropic liquid crystalline phases of Guerbet branched-chain -D-glucosides, Liq. Cryst., 2011, 38(11–12), 1725–1734 CrossRef CAS.
- V. Vill and R. Hashim, Carbohydrate liquid crystals: structure–property relationship of thermotropic and lyotropic glycolipids, Curr. Opin. Colloid Interface Sci., 2002, 7(5), 395–409 CrossRef CAS.
- T. Z. Rizvi, Liquid crystalline biopolymers: A new arena for liquid crystal Research, J. Mol. Liq., 2003, 106(1), 43–53 CrossRef CAS.
- F. Guo, X. Zhang, C. You, C. Zhang, F. Li, N. Li, Y. Xia, M. Liu, Z. Qiu, X. Zheng, L. Ma, G. Zhang, L. Luo, F. Cao, Y. Feng, G.-R. Zhao, W. Zhang, S. Li and L. Du, Diversification of phenolic glucosides by two UDP-glucosyltransferases featuring complementary regioselectivity, Microb. Cell Fact., 2022, 21(1), 208 CrossRef CAS PubMed.
- Y. Bai, H. Yin, H. Bi, Y. Zhuang, T. Liu and Y. Ma, De novo biosynthesis of Gastrodin in Escherichia coli, Metab. Eng., 2016, 35, 138–147 CrossRef CAS PubMed.
- Z. Peng, S. Wang, G. Chen, M. Cai, R. Liu, J. Deng, J. Liu, T. Zhang, Q. Tan and C. Hai, Gastrodin Alleviates Cerebral Ischemic Damage in Mice by Improving Anti-oxidant and Anti-inflammation Activities and Inhibiting Apoptosis Pathway, Neurochem. Res., 2015, 40(4), 661–673 CrossRef CAS PubMed.
- G. Xiao, R. Tang, N. Yang and Y. Chen, Review on pharmacological effects of gastrodin, Arch. Pharmacal Res., 2023, 46(9), 744–770 CrossRef CAS.
- R. Maharjan, Y. Fukuda, N. Shimomura, T. Nakayama, Y. Okimoto, K. Kawakami, T. Nakayama, H. Hamada, T. Inoue and S.-I. Ozaki, An Ambidextrous Polyphenol Glycosyltransferase PaGT2 from Phytolacca americana, Biochemistry, 2020, 59(27), 2551–2561 CrossRef CAS PubMed.
- D. Albesa-Jové and M. E. Guerin, The conformational plasticity of glycosyltransferases, Curr. Opin. Struct. Biol., 2016, 40, 23–32 CrossRef PubMed.
- W.-Z. Zhang, J.-C. Tang, S.-S. Wang, Z.-J. Wang, W.-M. Qin and J.-H. He, The protein complex crystallography beamline (BL19U1) at the Shanghai Synchrotron Radiation Facility, Nucl. Sci. Tech., 2019, 30(11), 170 CrossRef.
- W. Kabsch, Integration, scaling, space-group assignment and post-refinement, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(2), 133–144 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, Phaser crystallographic software, J. Appl. Crystallogr., 2007, 40(4), 658–674 CrossRef CAS PubMed.
- L. Potterton, J. Agirre, C. Ballard, K. Cowtan, E. Dodson, P. R. Evans, H. T. Jenkins, R. Keegan, E. Krissinel, K. Stevenson, A. Lebedev, S. J. McNicholas, R. A. Nicholls, M. Noble, N. S. Pannu, C. Roth, G. Sheldrick, P. Skubak, J. Turkenburg, V. Uski, F. von Delft, D. Waterman, K. Wilson, M. Winn and M. Wojdyr, CCP4i2: the new graphical user interface to the CCP4 program suite, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74(2), 68–84 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubak, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, REFMAC5 for the refinement of macromolecular crystal structures, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67(4), 355–367 CrossRef CAS PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Refinement of Macromolecular Structures by the Maximum-Likelihood Method, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53(3), 240–255 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Features and development of Coot, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(4), 486–501 CrossRef CAS PubMed.
- N. Eswar, D. Eramian, B. Webb, M.-Y. Shen and A. Sali, Protein Structure Modeling with MODELLER, Methods Mol. Biol., 2008, 426, 145–159 CrossRef CAS PubMed.
- E. Jurrus, D. Engel, K. Star, K. Monson, J. Brandi, L. E. Felberg, D. H. Brookes, L. Wilson, J. Chen, K. Liles, M. Chun, P. Li, D. W. Gohara, T. Dolinsky, R. Konecny, D. R. Koes, J. E. Nielsen, T. Head-Gordon, W. Geng, R. Krasny, G.-W. Wei, M. J. Holst, J. A. McCammon and N. A. Baker, Improvements to the APBS biomolecular solvation software suite, Protein Sci., 2018, 27(1), 112–128 CrossRef CAS.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al., Gaussian 16, Revision C.01, Gaussian, Inc., 2016 Search PubMed.
- M. Rupp, R. Korner and I. V. Tetko, Predicting the pKa of Small Molecules, Comb. Chem. High Throughput Screening, 2011, 14(5), 307–327 CrossRef CAS PubMed.
- X. Sheng and F. Himo, Mechanism of 3-methylglutaconyl CoA decarboxylase AibA/AibB:Pericyclic reaction versus direct decarboxylation, Angew. Chem., Int. Ed., 2020, 59(51), 22973–22977 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- A. D. MacKerell, , Jr., D. Bashford, M. Bellott, R. L. Dunbrack Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins, J. Phys. Chem. B, 1998, 102(18), 3586–3616 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79(2), 926–935 CrossRef CAS.
- J. A. Izaguirre, D. P. Catarello, J. M. Wozniak and R. D. Skeel, Langevin stabilization of molecular dynamics, J. Chem. Phys., 2001, 114(5), 2090–2098 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81(8), 3684–3690 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98(12), 10089–10092 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23(3), 327–341 CrossRef CAS.
- B. R. Miller, T. D. McGee, , Jr., J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, MMPBSA.py: An efficient program for end-state free energy calculations, J. Chem. Theory Comput., 2012, 8(9), 3314–3321 CrossRef CAS PubMed.
- D. A. Case, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, A. Kovalenko, R. Krasny, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, V. Man, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, A. Onufriev, F. Pan, S. Pantano, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, L. Wilson, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue, D. M. York and P. A. Kollman, AMBER 2020, University of California, San Francisco, CA, 2020 Search PubMed.
- J. Kästner, Umbrella sampling, WIREs Comput. Mol. Sci., 2011, 1(6), 932–942 CrossRef.
- G. M. Torrie and J. P. Valleau, Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling, J. Comput. Phys., 1977, 23(2), 187–199 CrossRef.
- M. Souaille and B. T. Roux, Extension to the weighted histogram analysis method: combining umbrella sampling with free energy calculations, Comput. Phys. Commun., 2001, 135(1), 40–57 CrossRef CAS.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method, J. Comput. Chem., 1992, 13(8), 1011–1021 CrossRef CAS.
- S. Metz, J. Kästner, A. A. Sokol, T. W. Keal and P. Sherwood, ChemShell—a modular software package for QM/MM simulations, WIREs Comput. Mol. Sci., 2014, 4(2), 101–110 CrossRef CAS.
- F. Neese, The ORCA program system, WIREs Comput. Mol. Sci., 2012, 2(1), 73–78 CrossRef CAS.
- W. Smith and T. R. Forester, DL_POLY_2.0: A general-purpose parallel molecular dynamics simulation package, J. Mol. Graphics, 1996, 14(3), 136–141 CrossRef CAS PubMed.
- D. Bakowies and W. Thiel, Hybrid Models for Combined Quantum Mechanical and Molecular Mechanical Approaches, J. Phys. Chem., 1996, 100(25), 10580–10594 CrossRef CAS.
- A. H. De Vries, P. Sherwood, S. J. Collins, A. M. Rigby, M. Rigutto and G. J. Kramer, Zeolite Structure and Reactivity by Combined Quantum-Chemical−Classical Calculations, J. Phys. Chem. B, 1999, 103(29), 6133–6141 CrossRef CAS.
- P. Sherwood, A. H. de Vries, S. J. Collins, S. P. Greatbanks, N. A. Burton, M. A. Vincent and I. H. Hillier, Computer simulation of zeolite structure and reactivity using embedded cluster methods, Faraday Discuss., 1997, 106, 79–92 RSC.
- J. Kästner, J. M. Carr, T. W. Keal, W. Thiel, A. Wander and P. Sherwood, DL-FIND: An Open-Source Geometry Optimizer for Atomistic Simulations, J. Phys. Chem. A, 2009, 113(43), 11856–11865 CrossRef.
- S. R. Billeter, A. J. Turner and W. Thiel, Linear scaling geometry optimisation and transition state search in hybrid delocalised internal coordinates, Phys. Chem. Chem. Phys., 2000, 2(10), 2177–2186, 10.1039/A909486E.
- J. Wang, Q. Shao, Z. Xu, Y. Liu, Z. Yang, B. P. Cossins, H. Jiang, K. Chen, J. Shi and W. Zhu, Exploring Transition Pathway and Free-Energy Profile of Large-Scale Protein Conformational Change by Combining Normal Mode Analysis and Umbrella Sampling Molecular Dynamics, J. Phys. Chem. B, 2014, 118(1), 134–143 CrossRef CAS PubMed.
- P. Wu, F. Fan, J. Song, W. Peng, J. Liu, C. Li, Z. Cao and B. Wang, Theory Demonstrated a “Coupled” Mechanism for O2 Activation and Substrate Hydroxylation by Binuclear Copper Monooxygenases, J. Am. Chem. Soc., 2019, 141(50), 19776–19789 CrossRef CAS PubMed.
- W. Cui, Y.-H. Cheng, L.-L. Geng, D.-S. Liang, T.-J. Hou and M.-J. Ji, Unraveling the Allosteric Inhibition Mechanism of PTP1B by Free Energy Calculation Based on Umbrella Sampling, J. Chem. Inf. Model., 2013, 53(5), 1157–1167 CrossRef CAS.
- J. Li, Y. Zhou, Y. Tang, W. Li and Y. Tu, Dissecting the Structural Plasticity and Dynamics of Cytochrome P450 2B4 by Molecular Dynamics Simulations, J. Chem. Inf. Model., 2020, 60(10), 5026–5035 CrossRef CAS PubMed.
- Z. Wang, S. Shaik and B. Wang, Conformational Motion of Ferredoxin Enables Efficient Electron Transfer to Heme in the Full-Length P450TT, J. Am. Chem. Soc., 2021, 143(2), 1005–1016 CrossRef CAS.
- D. Albesa-Jové, J. Romero-García, E. Sancho-Vaello, F. X. Contreras, A. Rodrigo-Unzueta, N. Comino, A. Carreras-González, P. Arrasate, S. Urresti, X. Biarnés, A. Planas and M. E. Guerin, Structural Snapshots and Loop Dynamics along the Catalytic Cycle of Glycosyltransferase GpgS, Structure, 2017, 25(7), 1034–1044.e1033 CrossRef.
- I. Jancan and M. A. Macnaughtan, Acid dissociation constants of uridine-5′-diphosphate compounds determined by 31phosphorus nuclear magnetic resonance spectroscopy and internal pH referencing, Anal. Chim. Acta, 2012, 749, 63–69 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic statistics of RrUGT3; strains, plasmids and primers used in this study; structural overlap between RrUGT3 and its homologous UGT enzymes; analysis of MD simulations; geometry information, energies and Cartesian coordinates of the optimized structures; additional data as noted in the text. See DOI: https://doi.org/10.1039/d4cy00721b |
| ‡ M. L., C. Y., and F. G. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |