
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in selective methanol oxidation electrocatalysts for the co-production of hydrogen and value-added formate†
Jiaxin
Li
,
Hongmei
Yu
 *,
Jingchen
Na
,
Senyuan
Jia
,
Yutong
Zhao
,
Kaiqiu
Lv
,
Wenzhuo
Zhang
,
Jun
Chi
and
Zhigang
Shao
*
*,
Jingchen
Na
,
Senyuan
Jia
,
Yutong
Zhao
,
Kaiqiu
Lv
,
Wenzhuo
Zhang
,
Jun
Chi
and
Zhigang
Shao
*
Fuel Cell System and Engineering Laboratory, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning, China. E-mail: hmyu@dicp.ac.cn; zhgshao@dicp.ac.cn
First published on 27th August 2024
Abstract
Traditional water splitting is significantly impeded by the sluggish kinetics and large overpotential of the anodic oxygen evolution reaction (OER). Accordingly, replacing the OER with a more thermodynamically favorable organic substance oxidation reaction to combine with the hydrogen evolution reaction (HER) is an innovative strategy to obtain green hydrogen. In this case, the electro-reforming of methanol coupled with the electrochemical HER can realize the energy-saving co-generation of value-added formate and hydrogen. Therefore, controlling the process of methanol oxidation and making it selectively transform to formate have become a worthy topic. Thus far, various catalysts and modification strategies have been developed for the selective methanol oxidation reaction (SMOR). Transition metal-based materials are the most studied catalysts because their moderate catalytic ability can better control the process of methanol oxidation. Electronic structure modulation is the most efficient strategy to improve the SMOR performance of catalysts. However, few systematic reviews on the SMOR have been reported. In light of significant advances achieved recently, herein, we reviewed the recent advances in SMOR electrocatalysts for the co-production of value-added formate and green hydrogen. In particular, the mechanism of the SMOR is initially introduced, including the traditional surface adsorption mechanism and the newly developed lattice oxygen participation mechanism. Subsequently, strategies for catalyst design are analyzed from the aspects of chemical bond activation/inhibition, electronic structure manipulation, dual active site construction, and increasing the number of active sites. Thereafter, performance descriptors involving electrochemical measurements and product detection are discussed to show the basic evaluation criterion, and various catalysts for the SMOR are categorized according to their composition to display the development of catalysts. Finally, conclusions and perspectives are presented. We hope that this comprehensive effort will be helpful in the literature survey of the SMOR and provide inspiration to the SMOR research community, attracting more attention to the electro-upgradation of organic substances coupled with green hydrogen generation.
1. Introduction
Generating “green hydrogen” from water electrolysis powered by renewable energy is considered an ideal hydrogen production method that is devoid of energy depletion and environmental pollution.1–3 However, the sluggish kinetics and large overpotential of the anodic oxygen evolution reaction (OER) largely impede the cost-efficiency of overall water splitting (OWS).4,5 Although the theoretical equilibrium potential of OWS is 1.23 V, the applied voltage in practical use may increase to as high as 1.8–2.0 V to overcome the thermodynamic barrier and extra resistance.6,7 It has been reported that 94.5% of the total energy input for OWS is consumed by the OER.8 Consequently, various catalysts have been explored to achieve higher reaction efficiency.9,10 However, another concern is that the anodic product of O2 is less valuable and will inevitably mix with H2 in the cathode, leading to the risk of explosion.11 Thus, to alleviate these limitations, several oxidation reactions of organic substrates have been explored for substituting the OER, such as alcohols, aldehydes, and urea.12–16 These biomass oxidations are more thermodynamically favorable and can lower overpotential while presenting additional benefits such as high value-added products and pollutant degradation, which have become research hotspots in recent years.17–20 Guo's group reported a comprehensive review on chemical-assisted water splitting with the concurrent evolution of H2 and high value-added chemicals.21 Their review introduced different alternative chemicals at the anode, especially for small molecules that can be produced from biomass.Among the alternative organic substrates, methanol is the most important liquid C1 resource, which has attracted significant attention due to its simple structure, large aqueous solubility, high volumetric energy density (15.9 MJ L−1), good oxidation reactivity, and extensive resources.22–24 The methanol oxidation reaction (MOR) has long been used in direct methanol fuel cells (DMFCs)25–28 and methanol-assisted water splitting.29–31 However, the focus is mainly on enhancing the power output and catalytic performance by completely oxidizing methanol, whose product is the environmentally unfriendly CO2. In recent years, increasing attention has been paid to partially oxidizing methanol to formate, which has a higher economic value of ca. 1300 $ per ton than that of methanol (350 € per ton).32 The standard potential for the electro-oxidation of methanol to formate is reported to be as low as 0.103 V vs. SHE, which is much lower than that of OER.32 Besides, the MOR proceeding via the formate pathway still yields an energy density of 9.2 MJ L−1, which is slightly higher than that of liquid hydrogen (8 MJ L−1).33 Moreover, the methanol-to-formate route offers several economic and practical benefits, particularly in the fields of chemical production and environmental sustainability. Formate is one of the basic organic chemical raw materials and has been widely used in the pharmaceutical, leather, rubber, textile, and pesticide industries.34,35 The conversion of methanol to formate also can be integrated into carbon capture and utilization (CCU) strategies. Methanol can be synthesized from captured CO2, which is then transformed into formate, thus providing a sustainable loop for carbon utilization. Therefore, it is a promising way to replace OER with the selective methanol oxidation reaction (SMOR) to formate for anodic chemical upgrading and cathodic hydrogen production. Moreover, SMOR provides a green sustainable method for chemical synthesis superior to industrial manufacturing with complex procedures and high energy consumption.36–39
In 2003, the formate pathway in the methanol electro-oxidation process was discovered.40In situ surface-enhanced IR absorption spectroscopy revealed the symmetric stretching of formate species on the surface of Pt. However, the systematic study on SMOR to formate is less than ten years. It was only in 2020 that the number of relevant reports started to rapidly increase, with various catalysts and modification methods reported. Pt-, Ru-, and Pd-based materials are the most active catalysts for MOR,41,42 but their resource scarcity, high cost, and CO poisoning issues severely restrict their widespread application.43 Moreover, these noble metals tend to completely oxidize methanol to CO2. Alternatively, non-noble metal-based catalysts have been widely studied for SMOR. Because of their mild catalytic activity, the process of SMOR can be easily modulated. Among the non-noble metals, Ni is the most frequently used, followed by Co, Fe, Cu, and Mo. For example, an NiCo layered double hydroxide (LDH) was reported to have a high faradaic efficiency for methanol to formate (FEformate) of over 95% at a wide potential range.44 The Ni3+ species formed during the electrochemical oxidation were believed to be the active sites. Moreover, the ultrathin morphology was beneficial for the creation of oxygen vacancy, which could lower the energy barrier for methanol oxidation. In another study on Fe-incorporated NiCo-LDH for SMOR,45 Co was proven to reduce the potential required for the Ni2+/Ni3+ redox via the coordinating effect. Most importantly, Fe was found to act as an “electron pump”, where it underwent valence changes and accelerated the electron transfer. As a result, a current density of 10 mA cm−2 was achieved at only 1.349 V vs. RHE and the FEformate reached about 95%.
Considering the great potential of methanol upgrading to value-added chemicals coupled with green hydrogen generation in energy and environmentally relevant application, many technology advances have been made including catalyst synthesis, design strategy, catalytic mechanism, and practical application. Recently, Zhu's group reviewed the study of simultaneous production of hydrogen and formate by coupling the hydrogen evolution reaction (HER) and methanol oxidation reaction (MOR).46 They summarized the concept, fundamental electrochemistry of MOR, materials design and development of the methanol–water hybrid electrolysis. However, they only analyzed Ni-based electrocatalysts in alkaline medium. Herein, we mainly focus on the selectivity and process control of SMOR. In addition, all types of catalysts reported in recent years and the prevailing evaluation descriptors are included to make our work more comprehensive. Firstly, we present the mechanism and catalyst design strategies of SMOR with a focus on the reaction process control of methanol oxidation and the selective production of formate. Then, the catalytic performance descriptors and catalyst development are summarized to give an overview of the advancement in research. Finally, the problems, challenges, and perspectives are provided on this significant topic. We believe this review can be instructive for SMOR research and provide reference to the community devoted to the co-production of value-added chemicals and green hydrogen.
2. Mechanism of selective methanol oxidation reaction
To better control the SMOR and achieve higher selectivity, it is necessary to understand the reaction process of MOR and identify the intermediates. In fact, many pioneering research articles and critical reviews have conducted sufficient studies on the mechanism of methanol oxidation.47–50 Thus, we will not repeat it in detail and just give a brief description. The complete oxidation of methanol at the anode can be described by the following equation: CH3OH + 6OH− → CO2 + 5H2O + 6e−.51 The specific reaction mechanism can be broadly classified into two routes, namely the indirect way with CO* as a poisoning intermediate and the direct way without CO* formation (Fig. 1). It can be seen that the diversion of these two ways originates from the different dehydrogenation sites on methanol molecules. Thus, facilitating the fracture of the O–H bond of methanol to undergo the direct way at the initial stage and suppress the further transformation to CO2 at the final stage will be helpful for the generation of formate and improve its selectivity.52 SMOR is actually the partial oxidation of methanol. The electrooxidation of methanol to formate is a four-electron transfer process, as follows: CH3OH + 5OH− → HCOO− + 4H2O + 4e−.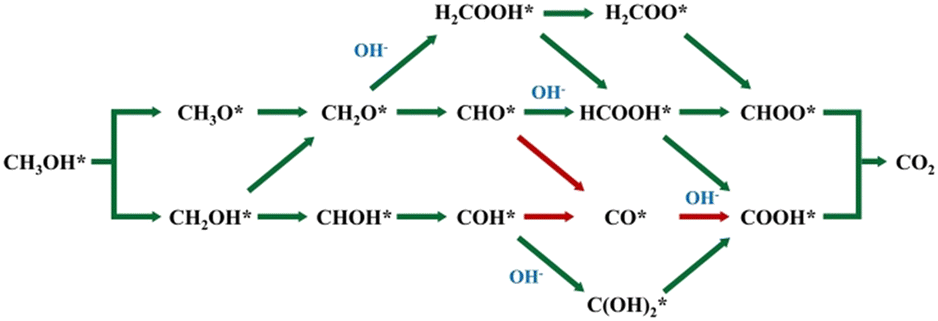 | ||
| Fig. 1 Possible reaction pathways and intermediates of the MOR, where * represents the active site of the catalyst. | ||
It is known that noble metals such as Pt and Pd generally facilitate the complete oxidation of methanol to form CO2; conversely, transition metals are more suitable to electro-catalyze methanol to formate due to their moderate activity. Therefore, the catalytic mechanism of SMOR on transition metals is mainly introduced here. The in situ-formed high valent transition metal species are believed to be the active phases in many anodic organic upgrading reactions,53–55 which is also applicable for SMOR. In a study on SMOR catalyzed by CoO–Ni(OH)2 nanosheets grown on MOF-derived carbon nanotube arrays (CNTs@CoO–Ni(OH)2),56in situ Raman spectroscopy detected two peaks located at 475 and 560 cm−1, corresponding to the Ni–O bending and Ni–O stretching vibration of Ni3+OOH in the background KOH solution, respectively (Fig. 2a). However, Ni3+OOH peaks were not found in the SMOR process, indicating its fast consumption in the presence of methanol. Thus, an indirect mechanism of SMOR involving the interaction between Ni3+OOH and methanol was assumed, where Ni3+OOH species was first electrochemically generated from the pre-catalyst, and then chemically oxidized the methanol molecules (Fig. 2b). A similar conclusion that Ni3+OOH is the active site for the oxidation of alcohols can also be seen in other reports.57,58 Zhu et al. further proposed the combined active sites in Ni3+OOH, where Ni3+ and the nearby electrophilic oxygen species cooperatively promoted SMOR.32 In particular, the exposed Ni3+ species provided adsorption sites for methanol, while the nearby electrophilic oxygen acted as a hydrogen acceptor, resulting in concerted hydrogen-transfer from methanol to the catalyst. Fu's group claimed the significant synergistic effect of foreign species with NiOOH.59 They synthesized hollow NiSe supported on carbon nanotubes (h-NiSe/CNTs) for the co-production of H2 and formate via electrocatalytic methanol upgrading. The in situ-formed NiOOH still acted as the active site to break the O–H bond to form the Ni–OCH3 intermediate. Meanwhile, SeOx and NiOOH were revealed to collaboratively tune the d-band center of the catalyst to optimize the adsorption energy during the SMOR process and inhibit the further oxidation of formate to CO2, thereby achieving high selectivity. In addition to SeOx, the incorporation of Co and FeCo in NiOOH was also proven to be efficient to SMOR with Ni0.9Co0.1Se and FeCoNi disulfide as pre-catalysts, respectively.60,61 In fact, the phase transformation on the catalyst surface during the electrocatalytic process is well recognized as surface reconstruction. Especially in the case of transition metals with multiple valences, high valent species are usually formed during the surface reconstruction and act as the real active phases. The surface reconstruction is indeed significant to improve the catalytic activity of pre-catalysts, and different pre-catalysts have different abilities for surface reconstruction, thus varying catalytic performances were observed.62 Therefore, the pre-catalysts should be designed to be more inclined to undergo surface reconstruction. Strategies should be performed to accelerate the redox reactions between low and high valent species, reduce the energy barrier for the formation of active phases, and then promote surface reconstruction.
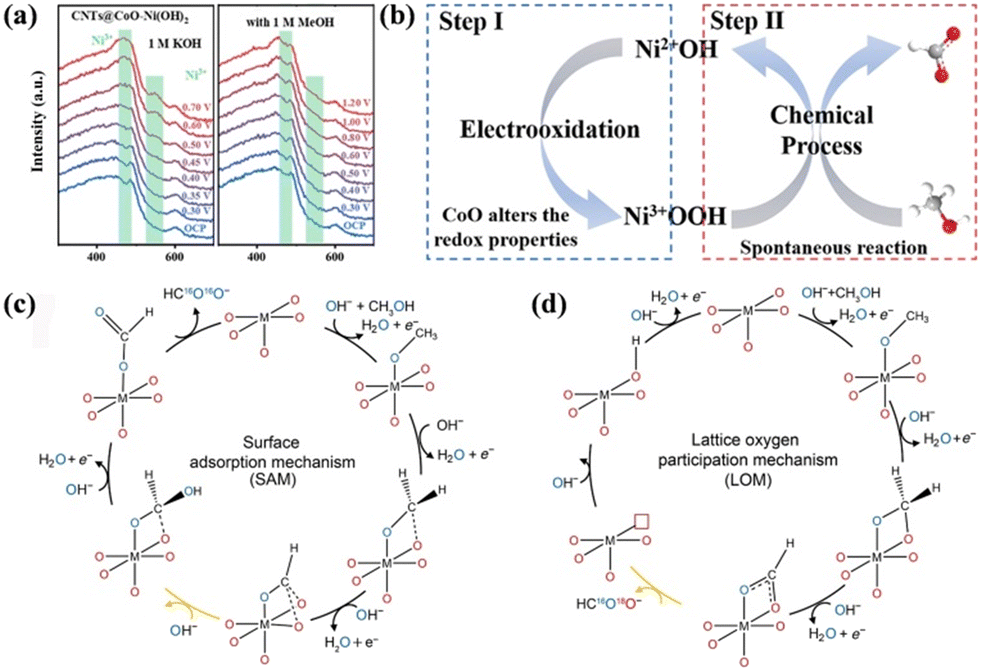 | ||
| Fig. 2 (a) In situ Raman spectra of the OER and MOR for CNTs@CoO–Ni(OH)2. (b) Dynamic reconstruction of the active Ni3+OOH layer.56 Reproduced from ref. 56 with permission from the Royal Society of Chemistry, Copyright 2023. Possible reaction mechanisms for (c) SAM and (d) LOM.63 Reproduced from ref. 63 with permission from American Association for the Advancement of Science, Copyright 2023. | ||
The above-mentioned mechanism was explained by the adsorption/desorption relationship between reactants and active sites. Recently, Meng et al. proposed a novel mechanism involving the participation of lattice oxygen.63 The authors fabricated 18O isotope-labeled SrCoO3−σ and NiOxHy catalysts to explore the origin of the O atoms in formate. Gas chromatography-mass spectrometry (GCMS) measurement detected the presence of HC16O18OH with one oxygen atom from the oxide lattice, which required the formation of oxygen vacancies and the activation of lattice oxygen during SMOR. Thus, the lattice oxygen participation mechanism (LOM) was proven, which is different from the surface adsorption mechanism (SAM). In the SAM mechanism, deprotonation occurred in the first three steps to form the *CHO intermediate, and then successive attack of hydroxide happened in the following two steps to generate HC16O16O− (Fig. 2c). In the LOM mechanism, the first three steps were the same as that in SAM, but then the attacking species shifted from OH− to lattice oxygen, accompanied with the formation of *16O18OCH and the creation of oxygen vacancies (Fig. 2d). Moreover, the lower rate-determining step (RDS) energy barrier for the LOM was confirmed by density functional theory (DFT) calculation, which was superior to the SAM.
3. Strategies for catalyst design
3.1 Chemical bond activation and inhibition
As mentioned in the part on the mechanism, whether the C–H bond or the O–H bond breaks at the beginning of the reaction results in the complicated catalytic kinetics and process of SMOR. It is beneficial for realizing highly efficient SMOR to formate by proceeding through the non-CO pathway by facilitating the cleavage of the O–H bond on the methanol molecule. Thus, exploring catalysts that tend to break the O–H bond is significant for improving the SMOR performance. In the research by Zhou et al., a partial pyrolyzation treatment was conducted to modify Ni-MOFs for methanol chemical upgradation, presenting insight into the variation in the reaction energy.64 Different from the fully carbonized products, the partially pyrolyzed Ni-MOFs obtained at 350 °C (Ni-MOFs@350) not only well inherited the porous properties, but also exhibited good conductivity. Most importantly, the Ni–O coordination number decreased from 6 (Ni-MOFs) to 3.92 (Ni-MOFs@350). By computing the Gibbs free energy for each fundamental step in the direct way (O–H activation, OHA) and the indirect way (C–H activation, CHA) (Fig. 3a–c), the energy steps in the OHA route were found to have a mild fluctuation in relation to CHA, indicating its more favorable thermodynamics under the catalysis of Ni-MOFs@350. As a result, only 1.37 V vs. RHE was required to achieve 100 mA cm−2 for SMOR, and the FEformate reached up to 98.4% benefiting from the modulation of O–H activation.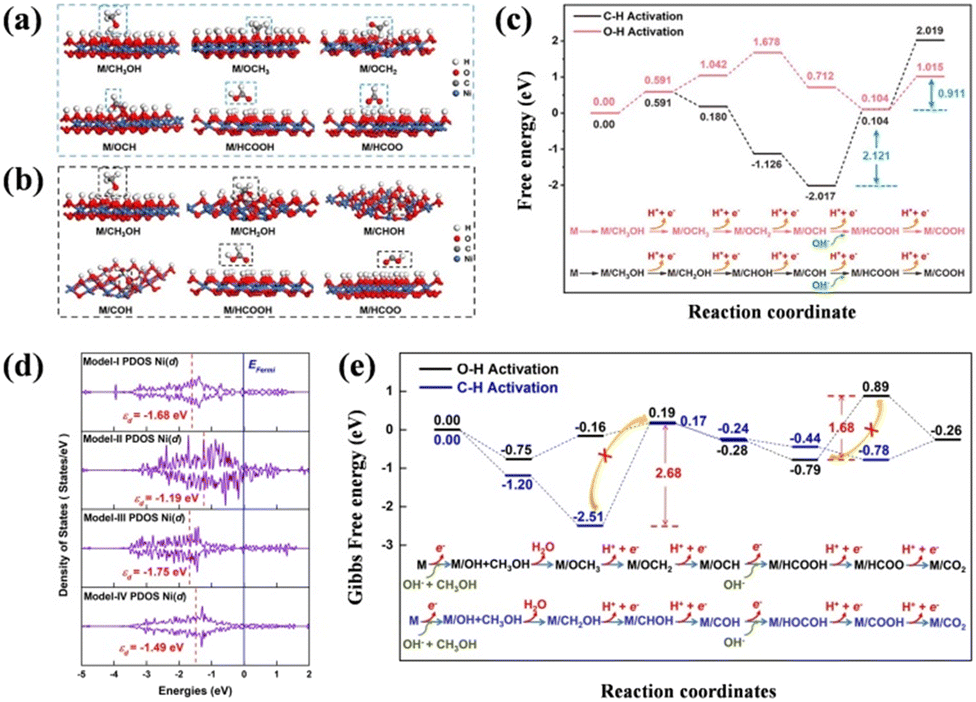 | ||
| Fig. 3 Intermediate adsorption configurations on NiOOH in (a) OHA and (b) CHA pathways. (c) Free energy diagram through the OHA and CHA routes.64 Reproduced from ref. 64 with permission from the American Chemical Society, Copyright 2023. (d) Corresponding projected density of states (PDOS) of model-I-SOx/NiOOH, model-II-Ni, model-III-SOx, and model-IV-SOx/Ni. (e) Gibbs free energy diagrams for methanol conversion on the modeled Ni3S2 surface.65 Reproduced from ref. 65 with permission from Elsevier, Copyright 2021. | ||
In addition to promoting the generation of formate, preventing its further transformation is also important to control the methanol oxidation process. Generally, it is challenging to balance high activity and high selectivity because highly efficient catalysts prefer to fully oxidize methanol to CO2 rather than an intermediate. Thus, suppressing the overoxidation of formate to CO2 is significant for SMOR. To achieve the two targets in one stroke, Fu's group constructed a defect-rich Ni3S2-CNF catalyst to improve methanol-to-formate without CO2 production at large current densities.65 The FEformate was kept beyond 90% in the wide potential range of 1.32–2.02 V with the current density above 700 mA cm−2. The synergism of the in situ formed NiOOH and SOx tuned the d-band center of Ni3S2, which inhibited the further oxidation to CO2 and gained high selectivity. As shown by the projected density of states (PDOS) (Fig. 3d), the Ni d-band center of SOx/NiOOH (−1.68 eV) was between that of Ni (−1.19 eV) and SOx (−1.75 eV). According to the Sabatier principle,66,67 the active sites with neither too strong nor too weak adsorption of intermediates are more favorable, and this adsorption behavior can be affected by adjusting the d-band center.68 Thus, SOx/NiOOH with a moderate adsorption energy well maintained the balance of reducing the energy barrier and facilitating the intermediate product release. Regarding the step of electron loss from M/HCOOH to M/HCOO, an uphill energy barrier of 1.68 eV was needed, as proven by DFT calculation (Fig. 3e), which made it difficult to further transfer to the final product of CO2, leading to high SMOR activity and selectivity. In another study by Fu's group, the unique NiSe (102) facets were selectively exposed in the presence of carbon nanofibers (CNFs@NiSe) through one-pot hot injection.69 The increased exposure of NiSe (102) was confirmed to prevent the further oxidation of formate by requiring a large energy barrier (1.21 eV). Thus, a high FEformate of 97.9% was achieved.
It can be seen that the activation and inhibition of chemical bonds during methanol oxidation were mainly realized by regulating the coordination number, d-band center, and crystal facet orientation. However, the interaction between them is ambiguous, and thus a more specific and quantitative descriptor such as the “volcano plot” of OER should be proposed to better guide the design and prediction of catalysts for SMOR.
3.2 Electronic structure manipulation
Acting as a surface-sensitive reaction, SMOR has a close relationship with the electron distribution and chemical environment of the catalyst. It has been reported that the tuning of the electron structure of the catalyst can improve its electrical conductivity, optimize the binding energy of intermediates, and even reconstruct the active centre.70–72 According to the d-band centre theory, the position of the d-band centre is relative to the Fermi level, which plays a crucial role in determining the adsorption properties of the catalyst.73 By tuning the d-band centre, the binding energies of the intermediates can be optimized for enhanced catalytic activity. The electron redistribution and electron density shift can be modulated by integrating elements with different electronic properties or by modifying the catalyst structure.74 Moreover, geometric effects that involve the arrangement and coordination of atoms on the catalyst surface can also tune the electron configuration.75 In the case of SMOR, electronic structure manipulation is a frequently used strategy to generate an appropriate adsorption energy, reduce the energy barrier, and improve the catalytic performance. Techniques such as hetero-atom doping, vacancy/defect creation, and heterostructure construction are all effective strategies to manipulate the electronic structure.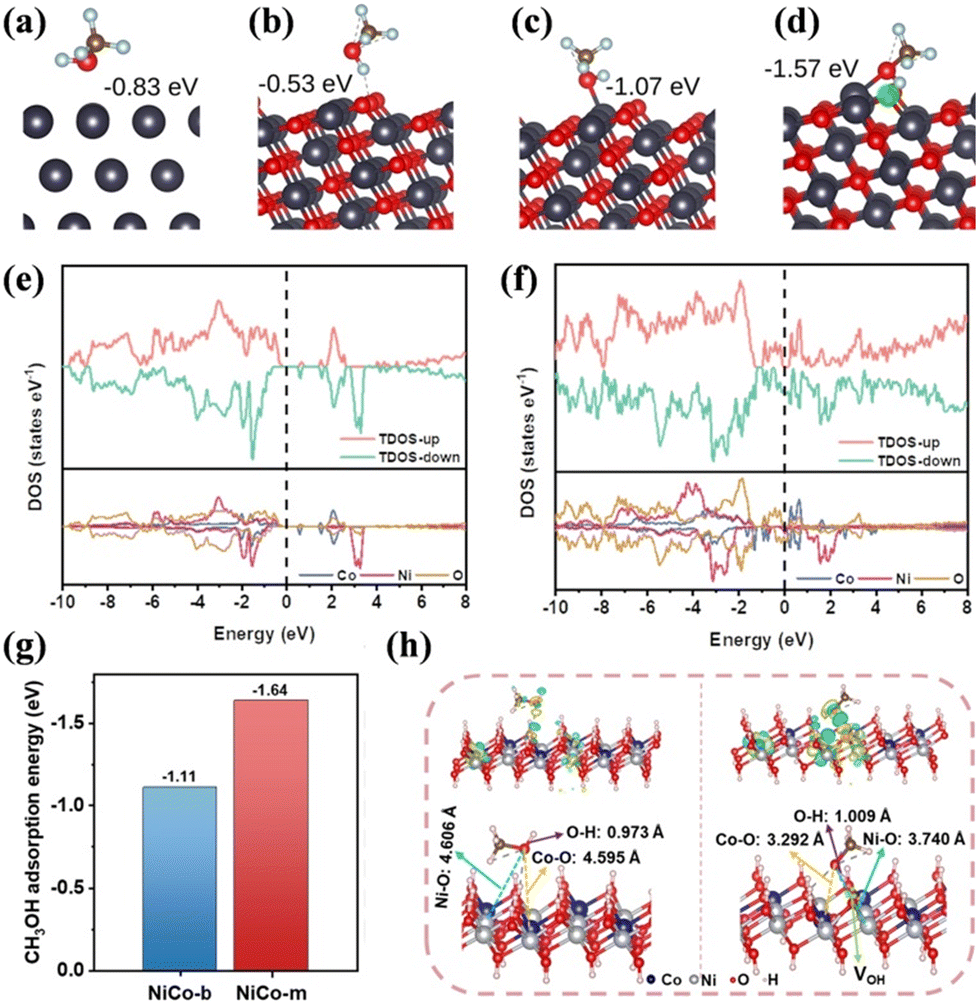 | ||
| Fig. 4 (a) Cu(111), (b) pristine CuO (111) for interaction with the H atom, (c) pristine CuO (111) for interaction with the O atom, and (d) O-vacant CuO (111) for interaction with the O atom. The position of the VO is highlighted in green. The binding energy is given in each case.86 Reproduced from ref. 86 with permission from Springer Nature, Copyright 2022. Total and partial density of states (TDOS and PDOS) for (e) NiCo-b and (f) NiCo-m. (g) DFT-calculated adsorption energies of CH3OH on the surfaces of the NiCo-LDHs with and without VOH and (h) models of CH3OH adsorbed on the surfaces of the NiCo-LDHs with and without VOH, and the differential charge density images of NiCo-LDHs with and without VOH adsorbing CH3OH.87 Reproduced from ref. 87 with permission from Elsevier, Copyright 2022. | ||
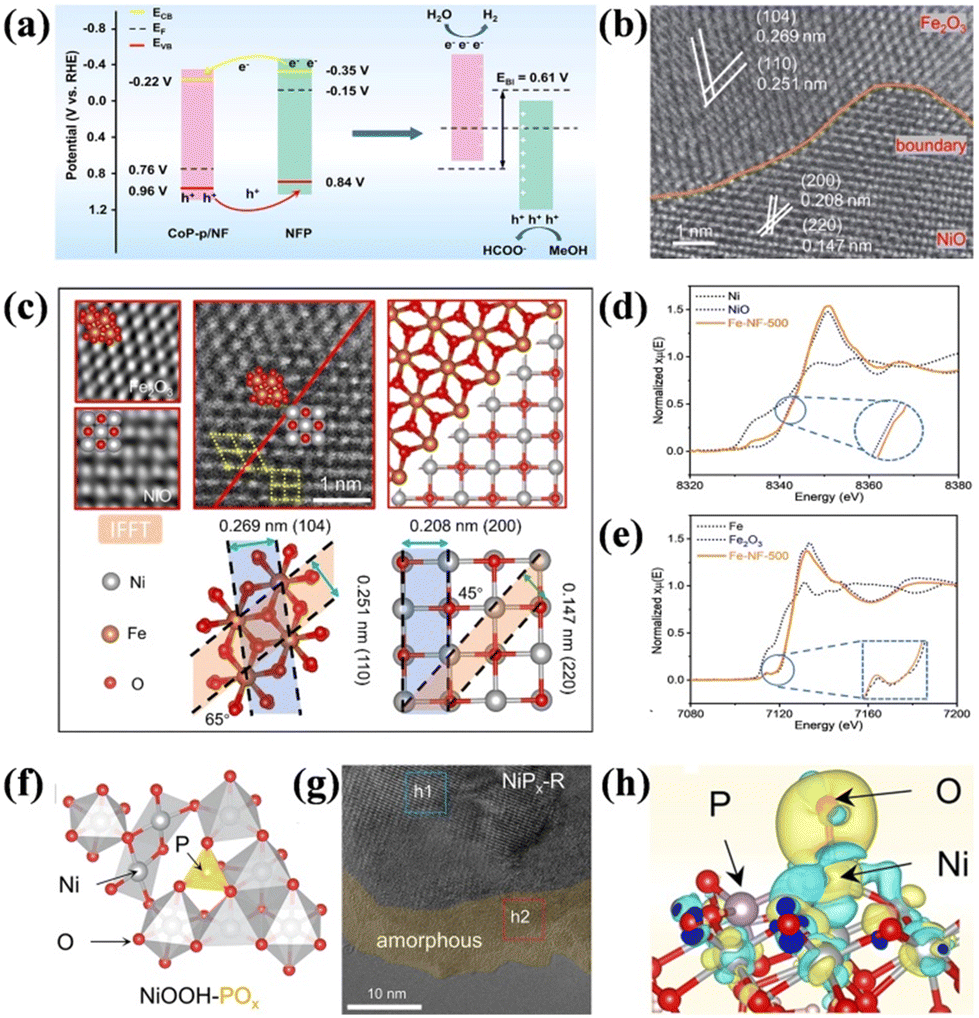 | ||
| Fig. 5 (a) Energy diagrams of CoP-p and NFP before and after contact.97 Reproduced from ref. 97 with permission from the American Chemical Society, Copyright 2023. (b) HAADF-STEM images of Fe-NF-500. (c) Atomic-resolution STEM partial enlargement images and structure analysis images. (d) Normalized XANES spectra of Ni in Fe-NF-500, NiO, and Ni foil. (e) Normalized XANES spectra of Fe in Fe-NF-500, Fe2O3, and Fe foil.98 Reproduced from ref. 98 with permission from the Royal Society of Chemistry, Copyright 2023. (f) Optimized structural models of NiOOH-POx. (g) HRTEM image of NiPx-R. (h) Charge density difference of OH intermediates adsorption on NiOOH-POx.100 Reproduced from ref. 100 with permission from Springer Nature, Copyright 2022. | ||
3.3 Constructing dual active sites
SMOR is a four-electron process with various intermediates and possible reaction pathways. In theory, a catalyst with a single active site cannot provide the optimal conditions (activity, adsorption energy, anti-poisoning ability, etc.) for all the fundamental steps. Alternatively, the construction of dual active sites may ensure each step occurs at their preferred reactive site, thus accelerating the overall reaction kinetics. Furthermore, the existence of dual active sites is conducive to fabricating bifunctional catalysts for SMOR-assisted water splitting. For instance, a spinel catalyst of NiFe2O4 loaded on nickel foam (NiFe2O4/NF) was synthesized as a bifunctional catalyst for SMOR and HER.24 DFT calculation revealed that the adsorption energy of the C1 intermediates including CO*, CH3O*, CH2O*, H2COOH*, and HCOOH* on the Fe site was larger than that on the Ni site. In contrast, *OH was more strongly adsorbed on the Ni site (−3.567 eV) in relation to the Fe site (−2.431 eV) (Fig. 6a and b). This phenomenon suggested that the methanol activation preferentially occurred on the Fe site, and the Ni site was more favorable for H2O activation. Under the action of dual active sites, a current density of more than 800 mA cm−2 was achieved at 2.0 V in a two-electrode system with a high FEformate (>95%) and FEhydrogen (>96%). It is known that Pt catalysts have high intrinsic activity for methanol oxidation but are vulnerable to CO poisoning.101 Thus, a dual-site strategy was used to modify the Pt single-atom catalyst for SMOR by anchoring Pt1 on dual-doped TiO2 (Pt1/Ti0.8W0.2NxOy).102 Therein, the activation and dehydrogenation of CH3O* were likely to happen on the W sites due to the lower free energy (Fig. 6c). Moreover, the resultant CHO* was transferred to the Pt1 site for further oxidation to formate. In the meantime, the OH− adsorbed on the W–O site favored the oxidation of the CO toxic intermediate, alleviating the poisoning effect. Finally, a high mass activity of 560 mA mgPt−1 at 0.82 V vs. RHE and formate selectivity of 90% were achieved due to the P1–WO3−x dual active sites.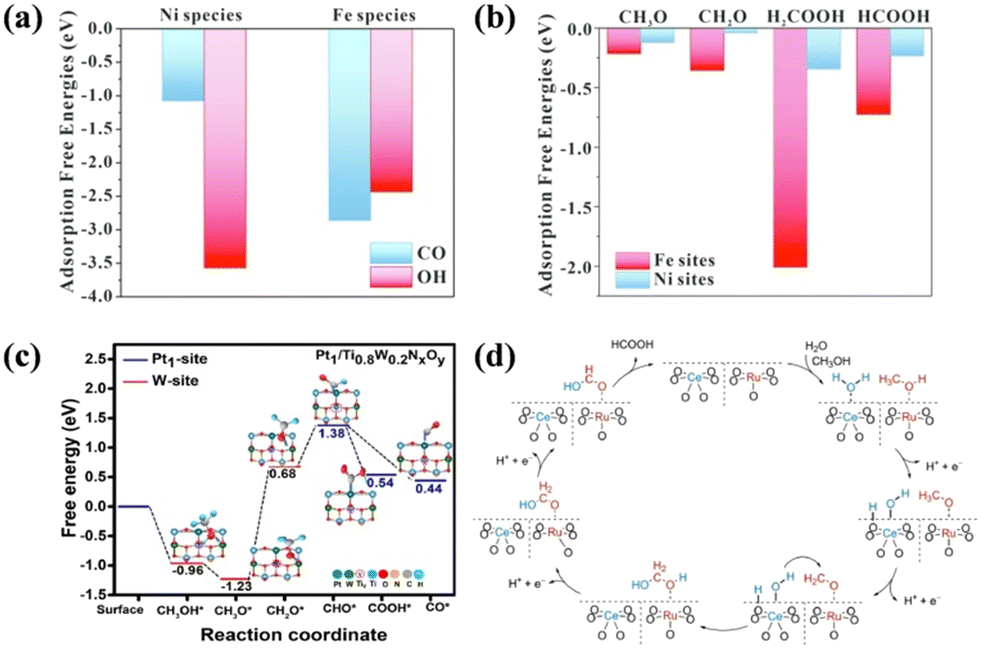 | ||
| Fig. 6 (a) Adsorption energies of *OH and *CO on the surface of NiFe2O4. (b) Adsorption energies of C1 intermediates on the Fe sites and Ni sites.24 Reproduced from ref. 24 with permission from Elsevier, Copyright 2023. (c) Theoretical free energy calculations of reaction intermediates on dual active sites of Pt1/Ti0.8W0.2NxOy.102 Reproduced from ref. 102 with permission from Elsevier, Copyright 2022. (d) Schematic illustration of reaction pathway for the SMOR to HCOOH on CeO2/RuO2.103 Reproduced from ref. 103 with permission from Elsevier, Copyright 2022. | ||
The construction of dual active sites is even more crucial for acidic SMOR. Unlike alkaline media with sufficient OH−, which is more favorable for SMOR, the acidic environment is not conducive to the progress of SMOR because only a small amount of OH− can be obtained from the dissociation of water. In addition, the carbon monoxide pathway is more likely to occur in an acidic solution instead of the formate pathway.104 Liu et al. performed innovative research in terms of the performance and mechanism of methanol-to-formate in 0.5 M H2SO4 solution by integrating CeO2 with RuO2 (CeO2/RuO2).103 The presence of CeO2 induced oxygen vacancies and accelerated water dissociation to provide OH− for the hydration of the intermediate in the methanol oxidation process. The electronic interaction between CeO2 and RuO2 promoted the formation of high-valent Ru species, which were beneficial for methanol oxidation. As shown in the reaction pathway (Fig. 6d), methanol oxidation and water activation occurred at the RuO2 site and CeO2 site, respectively. After two successive dehydrogenation steps on the O–H and C–H bonds, the obtained HCHO* was hydrated with the nearby dissociated water molecule to generate  . The resultant HCH(OH)2 further underwent two dehydrogenation processes and produced HCOOH*, which became free formate after desorption. Though the collaborative two active sites of RuO2 and CeO2 promoted the selective methanol oxidation in H2SO4 electrolyte, and the highest FEformate reported in this article was only 53.7%, which is much lower than that in alkaline media. Thus, more efficient modification methods should be adopted to further improve the acidic SMOR.
. The resultant HCH(OH)2 further underwent two dehydrogenation processes and produced HCOOH*, which became free formate after desorption. Though the collaborative two active sites of RuO2 and CeO2 promoted the selective methanol oxidation in H2SO4 electrolyte, and the highest FEformate reported in this article was only 53.7%, which is much lower than that in alkaline media. Thus, more efficient modification methods should be adopted to further improve the acidic SMOR.
3.4 Increasing the available active sites
The active site is the place where the electrocatalytic reaction occurs and it only works when exposed to the electrolyte. The more available active sites, the higher the catalytic activity under similar reaction conditions. Generally, there are two ways to obtain more available active sites, namely enlarging the surface area and increasing the density of active sites. The former can be achieved by morphology engineering, while the latter can be realized by coordination tuning. Wang et al. fabricated N-doped carbon-supported Co/CoO grown on cobalt foam (Co–N–C/CoO/CF),105 and the high porosity and conductive microchannels provided a large Cdl value of 4.50 mF cm−2 (specific surface area of 83.30 m2 g−1), largely exposing the Co active sites and improving the activity with 50 mA cm−2 at 1.309 V vs. RHE. A self-supporting heterostructure catalyst of MoN/Mo3N/NF was fabricated.106 The calculated Cdl value for SMOR of MoN/Ni3N/NF was 43.8 mF cm−2, which was higher than that of Ni3Mo/NF (7.8 mF cm−2), Ni3N/NF (7.0 mF cm−2), and NF (1.1 mF cm−2). The results showed that the heterostructure can expose more active sites compared to a single component. To further expose the active sites, a core–shell nanosheet array catalyst of NiFexP@NiCo-LDH/CC was prepared.107 The Cdl of NiFexP@NiCo-LDH/CC was calculated to be 43.9 mF cm−2, which is 2.6 times larger than that of NiCo-LDH (17.1 mF cm−2). The increased available surface area derived from the unique morphology greatly enhanced the SMOR activity.Regarding the coordination modulation, it is known that when the density of actives sites increases to a certain degree, the interaction within adjacent atoms or moieties cannot be ignored, which may induce better catalytic properties.108 Thus, the design of coordination modulation is beneficial to largely increase the density of active sites. Especially for the surface atoms, they usually have a low coordination number and unsaturated bonds, which make them more chemically reactive.109 Meanwhile, they can more readily adsorb and react with electrochemical species. In summary, the coordination number affects the ECSA by influencing the density and reactivity of the active sites. For example, 3D Mo-doped Ni(OH)2 with low Ni–Ni coordination was designed.110 DFT and X-ray absorption spectroscopy revealed that the low coordination of Ni–Ni not only increased the density of active sites but also introduced porosity, thus increasing the contact area and facilitating the catalytic reaction.
4. Catalysts and catalytic performance
To provide an intuitive overview and comparison of the catalytic performance of SMOR, nearly all the catalysts reported in the recent literature are listed in Table S1,† including their composition, electrolyte, scan rate, potential at certain current density, faradaic efficiency, and stability. It can be seen that the vast majority of research is conducted under alkaline conditions, the commonly used background solution is 1 M KOH or NaOH, and the concentration of CH3OH ranges from 0.5 to 4 M. Some researchers used simulated seawater containing 3.5% NaCl as the electrolyte to selectively oxidize methanol. Only one study on acid SMOR was performed in 0.5 M H2SO4 and 2.5 M CH3OH. The potential at a certain current density is widely regarded as the standard for comparing catalytic ability. It has been found that lowering the potential at 10 mA cm−2 below 1.3 V vs. RHE is a challenge for non-noble metal catalysts. Regarding catalytic selectivity, the faradaic efficiency of formate of most catalysts can exceed 90% and even reach close to 100%. In the following part, initially we will briefly introduce the catalytic performance descriptors, and then discuss the catalyst development in detail according to the elemental composition.4.1 Catalytic performance descriptors
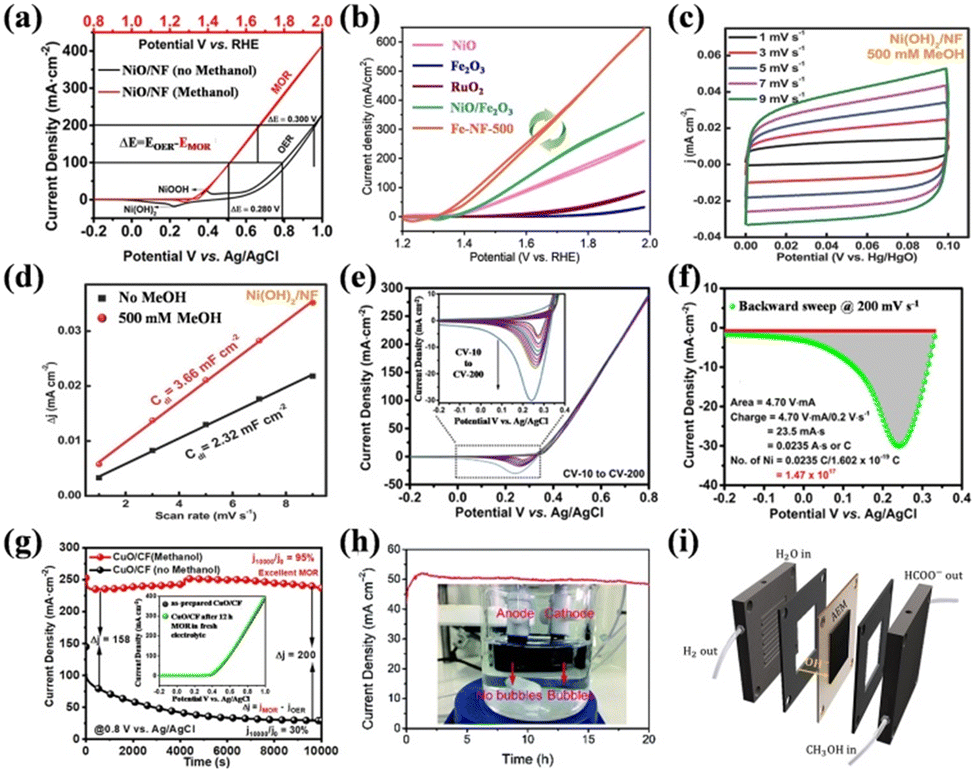 | ||
| Fig. 7 (a) CV curves for NiO/NF in the presence and absence of methanol in 1.0 M KOH.113 Reproduced from ref. 113 with permission from the American Chemical Society, Copyright 2021. (b) CV curves for all the as-prepared samples in 1.0 M KOH and 1 M CH3OH.98 Reproduced from ref. 98 with permission from the Royal Society of Chemistry, Copyright 2023. (c) CV curves of Ni(OH)2/NF in 1.0 M KOH with MeOH at different scan rates. (d) Corresponding Cdl value comparison of Ni(OH)2/NF in 1 M KOH with and without MeOH.118 Reproduced from ref. 118 with permission from Elsevier, Copyright 2021. (e) CVs of NiO/NF in 1 M KOH and 1 M methanol obtained at different scan rates. (f) Scale-up of reverse CV curve of NiO/NF.113 Reproduced from ref. 113 with permission from the American Chemical Society, Copyright 2021. (g) Chronoamperometric curves recorded in N2-purged 1 M KOH with and without 1 M MeOH.86 Reproduced from ref. 86 with permission from Springer Nature, Copyright 2022. (h) Stability test for the Pt–Co3O4/CP towards the electrocatalytic HER coupled with methanol oxidation at 1.0 V (inset is the photograph of the electrolytic cell).120 Reproduced from ref. 120 with permission from the Royal Society of Chemistry, Copyright 2021. (i) Illustration of the MEA cell.119 Reproduced from ref. 119 with permission from Elsevier, Copyright 2022. | ||
The catalytic kinetics can be approximately estimated by the Tafel slope calculated from the CV curves and electrochemical impedance spectroscopy (EIS).121–124 A small Tafel slope means that a smaller potential change will cause a larger increase in current density,125 indicating fast kinetics. By fitting the EIS Nyquist plot with an equivalent circuit, the charge transfer resistance (Rct) can be obtained, where the smaller the Rct, the easier the reaction is to proceed. Catalytic stability is an important indicator for evaluating whether a catalyst can be put into practical use. The durability in SMOR is mainly probed by long-term steady-state stability tests using chronoamperometry (CA) or chronopotentiometry (CP) techniques and dynamic stability evaluation by consecutively scanning LSV or CV curves (Fig. 7g).86
The catalytic performance of a catalyst in SMOR can be explored via the above-mentioned tests in a three-electrode system, which is the half-reaction for SMOR-assisted water splitting. Therefore, a two-electrode system is utilized to investigate the performance of selective methanol electrolysis (Fig. 7h).120 Therein, the as-prepared catalyst can serve as the anode or both the anode and cathode to investigate its bifunctional catalytic activity. In some reports, the absence of bubbles on the anode is considered evidence of good selectivity for methanol oxidation without oxygen evolution.118,126 To further probe the feasibility of practical use, SMOR also can be tested in a membrane electrode assembly (MEA) (Fig. 7i).119 Typically, MEA is assembled with an anion exchange membrane (AEM) and bipolar plate. The anolyte and catholyte are CH3OH-containing KOH solution and pure KOH solution, respectively. Polarization curves and steady-state stability tests can be conducted to evaluate the SMOR and HER performance.
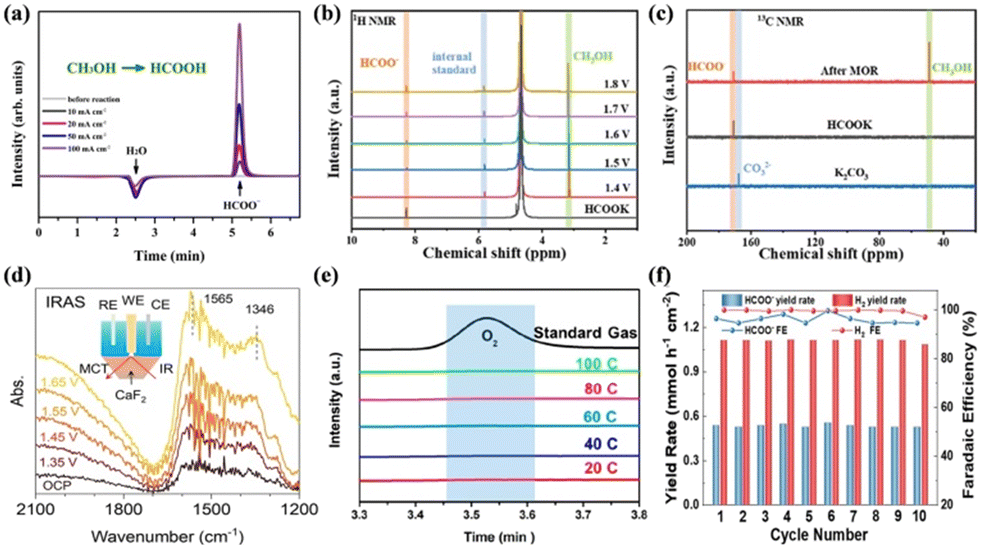 | ||
| Fig. 8 (a) IC traces of the electrolyte.127 Reproduced from ref. 127 with permission from John Wiley and Sons, Copyright 2021. (b) 1H NMR results of the electrolyte after MOR. (c) 13C NMR spectra after MOR.56 Reproduced from ref. 56 with permission from the Royal Society of Chemistry, Copyright 2023. (d) IRAS spectra of MOR on the surface of NiPx-R in the electrolyte of 0.1 M KOH with 0.5 M methanol.100 Reproduced from ref. 100 with permission from Springer Nature, Copyright 2022. (e) GC traces from the anode at different passed charges at 0.6 V.128 Reproduced from ref. 128 with permission from the American Chemical Society, Copyright 2021. (f) FE and yield rate for HCOO− and H2 during the cycling test at 60 mA cm−2.129 Reproduced from ref. 129 with permission from John Wiley and Sons, Copyright 2023. | ||
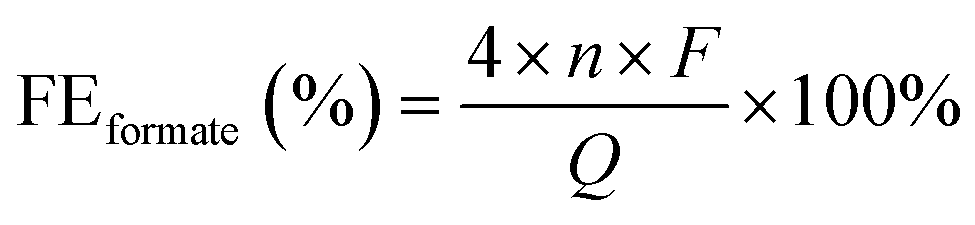
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), and Q is the total charge passed (C).
485 C mol−1), and Q is the total charge passed (C).
4.2 Catalyst development
Compared with the single metal catalyst, metal alloys containing different metallic elements usually yield intriguing physicochemical properties such as improved conductivity and variable valence state.135 When alloys are formed, the redistribution of electrons will occur owing to the different chemical properties of multi-components, which can lower the energy barrier for the generation of intermediates on the active sites.136 The most commonly used elements in metal alloy catalysts for SMOR are Ni, Co, Cu, and Mo. A porous NiCo alloy with oxygen vacancies supported on NF (Ov–NiCo@NF) was used methanol upgradation.137 The bimetallic synergism and the formation of Ov were believed to alter the electron distribution of Ni–Co, resulting in the optimum chemisorption energy of intermediates during the SMOR process. Moreover, the metallic porous structure was conducive to the penetration of the electrolyte and release of gaseous products, which accelerated the reaction kinetics. Finally, enhanced activity in SMOR-assisted hydrogen production was achieved. Only a cell voltage of 1.49 V was required to reach the current density of 50 mA cm−2. Likewise, an NiCu interconnected porous nanostructure was prepared for SMOR via the hydrogen bubble templating electrodeposition method.138 The insertion of Cu into Ni was claimed to improve the electronic conduction and charge transfer ability. The sacrificial template method of bubble-assisted electrodeposition enlarged the active surface area and enhanced the contact between the electrolyte and active sites, thereby promoting the hybrid electrolysis of SMOR and HER with a low cell voltage of 1.45 V at 10 mA cm−2. In addition to binary alloys, ternary alloys have also been studied. Chen et al. fabricated NiCoMo alloy arrays on carbon cloth (NiCoMo/CC) with a nanowire/nanosheet morphology by combined the hydrothermal and annealing methods for methanol–water co-electrolysis.139 Under the catalysis of NiCoMo/CC, the input cell voltage decreased by 208 mV by replacing OER with SMOR. Meanwhile, the FEformate remained above 85% after the 50 h long-term stability test. The authors proved that Mo not only acted as an electronic structure modulator but also played the role of surface reconstruction promoter. At the anode where a positive bias was applied, substantial Mo was leached, and the residual amount of Mo was substituted in NiCoOOH, which promoted the formation of high-valent Ni3+/Co3+ active species and optimized the adsorption/desorption energy for the intermediates. Besides binary and ternary metal alloys, high-entropy alloys have become a research hot-spot, which may be useful for SMOR in the future.140
000 s durability test. Another study introduced oxygen vacancies (Vo) in Co3O4−x/NF to improve SMOR.144 Initially, the authors electrodeposited Co(OH)2 on NF, and then conducted O2 plasma and air calcination to synthesize the target Co3O4−x/NF-P and its counterpart of Co3O4/NF-C, respectively. The Vo-enriched Co3O4−x/NF-P merely needed 1.318 V vs. RHE to deliver 10 mA cm−2 in SMOR. When coupled with HER, the cell voltage at 10 mA cm−2 for methanol-assisted water splitting was only 1.540 V. The experimental and calculation results demonstrated that the Vo promoted the phase transformation of Co3O4−x/NF-P, which accelerated the deprotonation of methanol and suppressed the further oxidation of HCOO*. Therefore, the FE of methanol to formate exceeded 95% at the passed charges from 50 to 250 C. Furthermore, copper oxide was also proven to be efficient for SMOR. Wei et al. used CuO nanosheets grown on copper foam (CuO NS/CF) as an anodic catalyst for methanol oxidation.8 The largest FEformate of CuO NS/CF for SMOR reached 97% and an ultra-low potential of 1.47 V vs. RHE at 100 mA cm−2 was achieved. Besides the commonly used first-row 3d transition metal oxides, amorphous Nb2O5 was reported as a new type of electrocatalyst for methanol electro-reforming.145In situ Raman revealed that the short Nb–O bond in the amorphous structure was the origin of the enhanced activity, and a near 100% of FEformate was obtained. Perovskite is a type of mixed oxide used in many catalytic reactions due to its low cost, diversity of element composition, and the outstanding properties of being substituted and doped.146,147 An iron-substituted lanthanum cobaltite (LaCo1−xFexO3) was employed to realize the mild oxidation of methanol to formate.119 The catalytic performance was optimized by tuning the Fe/Co ratio, and LaCo0:5Fe0:5O3 was found to have the highest formate production rate of 24.5 mmol h−1 goxide−1.
Hydroxide is the most studied catalyst for SMOR owing to its simple synthesis, facile active phase formation, and high intrinsic activity. To date, Ni(OH)2, NiCo-LDH, NiFe-LDH, and their derivatives have been developed. The pure phase of Ni(OH)2 on NF can be employed as a bifunctional catalyst for SMOR and HER via a simple hydrothermal or ultrasonic synthesis process.118,148 Its activity can be further improved by the cation doping strategy. Ming et al. prepared Co2+-doped Ni(OH)2 nanosheets on NF via the facile electrodeposition approach.149 The optimal catalyst with the Co2+ doping content of 10 mol% exhibited the best activity with 1.32 V vs. RHE at 100 mA cm−2. The FEformate was improved to above 96.5%. Moreover, doping chromium into α-Ni(OH)2 was also reported to realize the efficient selective conversion of methanol to formate.150 The Cr dopant increased the conductivity of Ni(OH)2, accelerated the adsorption of methanol, and optimized the kinetics for the formation of NiOOH, thereby achieving a faradaic efficiency of 92.1% for formate. Unlike the single hydroxide with one metal cation, LDHs are comprised layers of divalent and trivalent metal cations coordinated to hydroxide anions and the intercalated charge compensating anions between metal layers.151,152 Because of their flexible chemical composition and exchangeable interlaminar anions, LDHs display attractive electrocatalytic performances.153 For example, NixCo1−x(OH)2/NF was reported to have approximately 100% FEformate and the optimal Ni0.33Co0.67(OH)2/NF catalyst displayed the highest activity and robustness for the co-generation of formate and H2via the assistance of SMOR.126 In another study, Liu et al. incorporated Cu2+ in Co LDH to obtain a novel SMOR catalyst (CuxCoCo-LDHs/CF) with an interconnected nanosheet array morphology through the combined method of liquid deposition and pseudomorphic transformation (Fig. 9a and b).154 In the anodic reaction, Cu0.33CoCo-LDHs/CF yielded a current density of 10 mA cm−2 at 1.28 V vs. RHE for SMOR, which was 270 mV lower than that of OER (Fig. 9c). When coupling SMOR with HER, the catalyst could stably run for 24 h and no bubbles were observed on the anode electrode (Fig. 9d). The FEformate and FEhydrogen both reached about 99% (Fig. 9e). DFT calculations revealed that the adsorption energy of HCOOH on CuCoCo-LDH (−0.023 eV) was lower than that of CoCo-LDH (−1.03 eV) (Fig. 9f), which accelerated the desorption of HCOOH, avoiding its over-oxidation and increasing the selectivity (Fig. 9g).
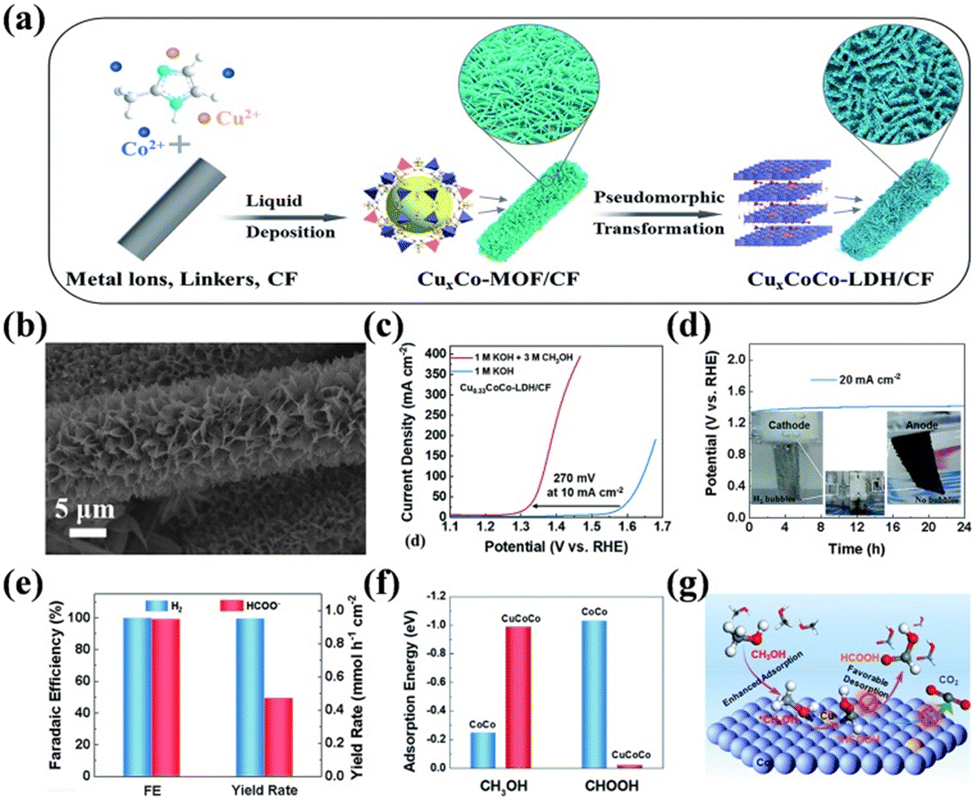 | ||
| Fig. 9 (a) Schematic illustration of the fabrication of CuxCoCo-LDHs/CF. (b) SEM image of Cu0.33CoCo-LDHs/CF. (c) Polarization curves of Cu0.33CoCo-LDH/CF in 1 M KOH or 1 M KOH + 3 M CH3OH solution. (d) Chronopotentiometry curve of Cu0.33CoCo-LDH/CF in 1 M KOH + 3 M CH3OH solution at 20 mA cm−2. (e) Comparison of the FE and yield rate for formate and H2 on Cu0.33CoCo-LDH/CF. (f) Calculated adsorption energy of methanol and formate molecules on the surfaces of CoCo-LDH and CuCoCo-LDH. (g) Schematic process for the transformation of CH3OH molecules into HCOOH instead of CO2 on the surface of CuCoCo-LDH.154 Reproduced from ref. 154 with permission from the Royal Society of Chemistry, Copyright 2022. | ||
Metal carbides have been reported to have the superiority of flexibility, anisotropy, excellent conductivity, and chemical stability.156 In 2020, Cui's group first proved the feasibility of using the non-precious metal catalyst Ni3C for SMOR to realize near 100% FEformate.157 They utilized in situ infrared spectroscopy and NMR to identify the adsorbed and dissolved intermediates/products, and no CO adsorption and carbonate production were found, which confirmed the extremely high selectivity. Metal nitrides, acting as interstitial compounds, exhibit the characteristics of covalent compounds, ionic crystals, and transition metals because of the integration of N atoms into the parent metal atoms.158 Meanwhile, the lattice parameter and d-band of the metal element can be modulated to generate good electrocatalytic performances.159,160 For instance, Cu3N nanosheets were fabricated via the nitridation of a Cu-containing precursor (Fig. 10a).161 Benefiting from the surface reconstruction of Cu3N, an FEformate of over 90% was obtained in the wide potential window of 1.4 to 1.8 V vs. RHE (Fig. 10b). In the X-ray absorption near-edge structure (XANES) profiles, the edge position of the used Cu3N was located at a higher energy value than that of the fresh Cu3N (Fig. 10c), indicating the formation of high-valent Cu species. In the meantime, the coordination number of Cu–O/N was revealed to increase, whereas the Cu–Cu coordination number decreased, representing the partial oxidation of the Cu3N surface (Fig. 10d). HRTEM demonstrated the existence of an interface between the CuO layer and Cu3N (Fig. 10e). In situ Raman detected the generation of a vibrational mode belonging to CuO with an increase in the potential during the SMOR (Fig. 10f). Moreover, in situ infrared spectroscopy (IR) confirmed the symmetric and antisymmetric stretching vibration of OCO in formate (Fig. 10g). All the aforementioned results evidenced that Cu3N underwent surface reconstruction with the formation of a Cu(II) shell as the active face and the Cu3N core ensuring electron transport. Metal phosphides have been extensively used as catalysts in various electrocatalytic reactions owing to their semiconducting property, diverse phase structure, and high activity.162–164 In the case of SMOR, an Os-doped NixP/N-doped carbon composite on NF (Os–NixP/N–C/NF) was assembled to simultaneously harvest value-added formate and H2.165 The FEformate and FEhydrogen reached 92% and 100%, respectively. A cell voltage of 1.43 V was required to produce 10 mA cm−2 in alkaline methanol electrolysis. Amorphous phosphide was also investigated for SMOR. An amorphous of Ni–Co–Fe ternary phosphide (NixCoyFez–Ps) was obtained via the electrodeposition method.166 Ni2Co2Fe1–P was screened as the most suitable catalyst for SMOR. Under the catalysis of Ni2Co2Fe1–P, the cell voltage was reduced by 110 mV to deliver 20 mA cm−2 by combining SMOR-HER compared to conventional water splitting.
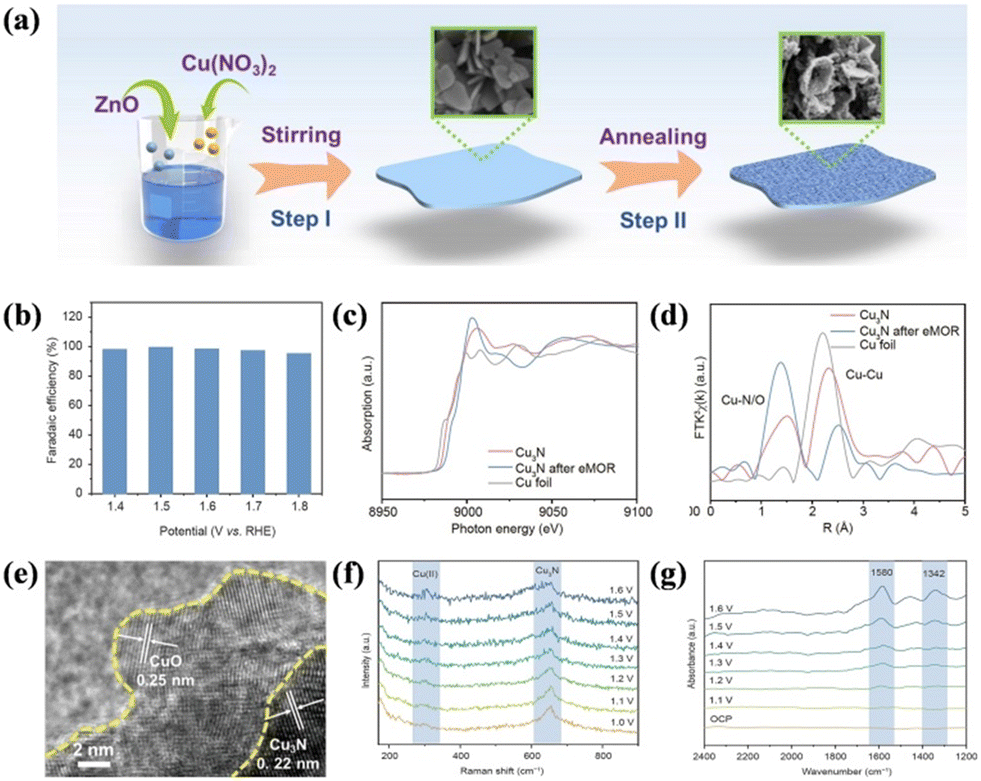 | ||
| Fig. 10 (a) Schematic of the synthesis of Cu3N nanosheets. (b) FE of Cu3N for selective methanol conversion reactions. (c) XANES profiles and (d) FT-EXAFS spectra of Cu foil, Cu3N, and Cu3N after eMOR. (e) HRTEM image of Cu3N after the reaction. (f) In situ Raman spectroscopy and (g) in situ IRAS of Cu3N electrode.161 Reproduced from ref. 161 with permission from Springer Nature, Copyright 2023. | ||
Metal chalcogenides are a significant category of catalysts that have been widely studied. Among them, metal sulfides have attracted significant attention on account of its suitable band gap and adjustable electronic structures.167 Taking NiS as an example, the presence of S was found to greatly influence the electronic properties of the in situ-formed NiOOH species, which improved the electroconductivity and accelerated the electron transfer, thereby boosting the SMOR performance.168 The valence electron states and spin of the d electrons of metal sulfides were also proven to be significant. Fu's group electrodeposited NiCo2S4 on carbon cloth (CC@NiCo2S4) as a bifunctional catalyst for energy-methanol hybrid electrolysis.169 A high current density and long-term stability were achieved by modulating the magnetism on the surface of CC@NiCo2S4. XANES revealed a positive shift in the absorption edge of the used CC@NiCo2S4, indicating a reduction in the electron density of the Ni species after the reaction (Fig. 11a). Moreover, the local atomic structures involving bond length and type of the catalyst were probed through extended X-ray absorption fine structure (EXAFS) (Fig. 11b) and wavelet transform EXAFS (WT-EXAFS) spectra (Fig. 11c–f). The results proved that partial oxidation occurred on the catalyst surface. DFT calculations confirmed that the d electrons in Ni(II) and Co(III) of the fresh catalyst were all in the state of low spin (Fig. 11g). However, when adsorbed with OH, the surface Ni and Co turned into high spins with the bulk Ni and Co maintaining low spins (Fig. 11h and i). This change in spin state contributed to the enhancement in SMOR activity. Metal selenides possess similar characteristics to the corresponding metal sulfides, but display higher electron transfer ability and a lower band gap.170,171 Se atoms are inclined to form strong covalent bonds with metal atoms,172 thus enhancing the metallic property and resulting in a unique band structure, which favors the SMOR catalytic activity.173,174 For example, Ni0.75Fe0.25Se2 could generate 0.47 mmol cm−2 h−1 of formate with an FEformate of 99% due to its large ECSA, fast electron transfer kinetics, and high proton diffusivity.175
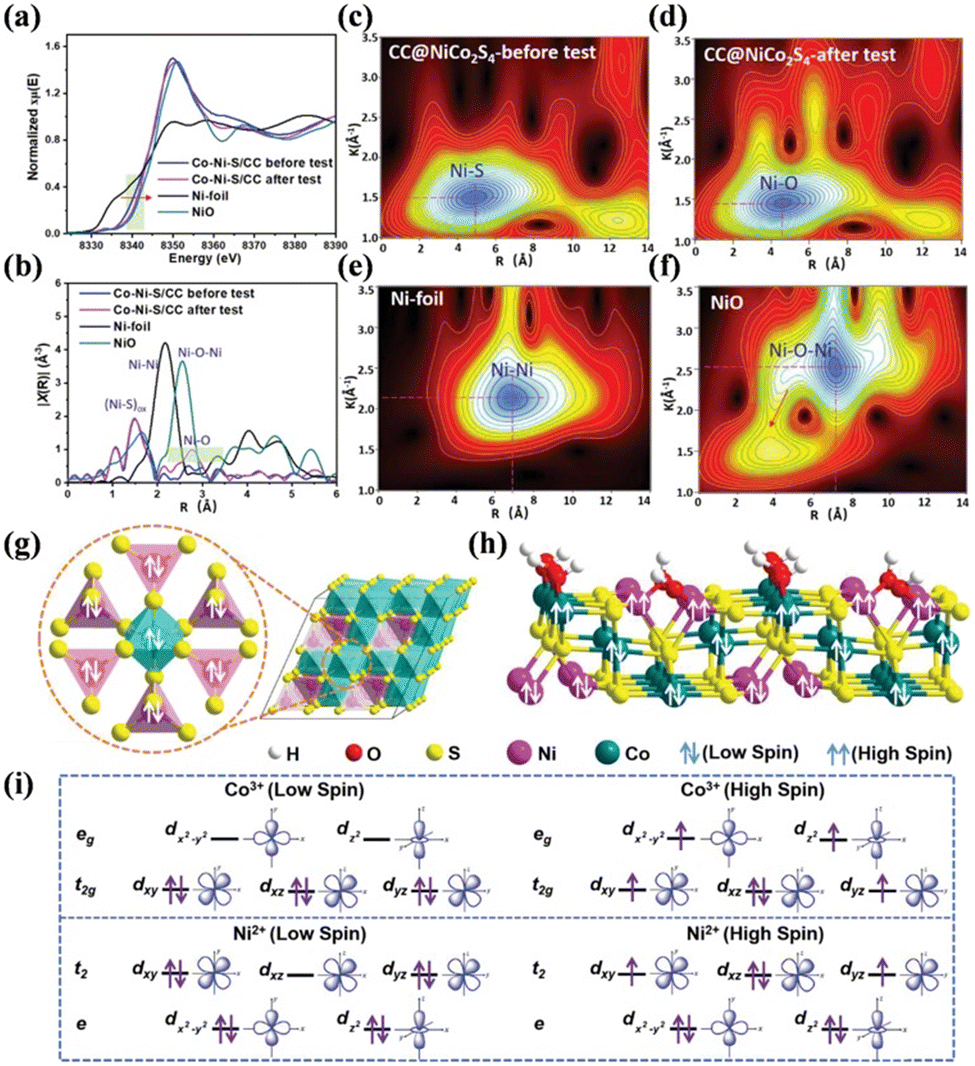 | ||
| Fig. 11 (a) Ni K-edge XANES spectra of fresh CC@NiCo2S4, used CC@NiCo2S4, pristine Ni foil, and NiO. (b) Fourier transformation of the EXAFS spectra in R space. WT-EXAFS for (c) CC@NiCo2S4-before the test, (d) CC@NiCo2S4 after the test, (e) pristine Ni foil, and (f) NiO. (g) Illustration of spin for the crystalline structure of NiCo2S4. (h) Illustration of the spin of NiCo2S4. (i) Occupation of d electrons for the high-spin and low-spin Ni2+ and Co3+, respectively.169 Reproduced from ref. 169 with permission from John Wiley and Sons, Copyright 2022. | ||
In SMOR, phosphide- and hydroxide-based heterostructure catalysts are the most studied. For instance, a heterostructure Ni2P-CoP/NF catalyst was prepared via a successive electrodeposition and phosphidation method.127 An obvious phase boundary was observed between Ni2P and CoP, which induced an interfacial bonding effect and exposed more active area (Fig. 12a). When used as a bifunctional catalyst in the two-electrode system, Ni2P-CoP/NF delivered 10 mA cm−2 at a cell voltage of 1.35 V (Fig. 12b). Meanwhile, an FEformate of 100% was achieved at the given current densities (Fig. 12c). In the case of hydroxide-based heterostructure catalysts, a core–shell structured Cu(OH)2@NiFe(OH)x catalyst was built via a facile ion exchange method.128 The heterointerface induced a strong synergistic effect due to the hydroxide and optimized the adsorption energy for the reactants and intermediates, thereby improving the SMOR performance. In another study, NiFe-LDH was assembled on an SnO2 supported NF layer by layer (NiFe LDH@SnO2/NF).184 The semiconductor SnO2 layer not only regulated the growth of NiFe-LDH but also altered the electron distribution around the interface, leading to an increase in the valence state of Fe and Ni. The SnO2 layer also acted as an electron transport layer, which accelerated the electron transfer from NiFe-LDH to the NF electrode. Due to these properties, enhanced SMOR activity was obtained. It is known that there exists a competition zone of MOR and OER in the conventional OER potential window, thus suppressing OER is beneficial to maintain efficient MOR. To achieve this goal, the strategy of constructing heterostructure catalysts has been employed. As reported, heterostructure NiFe-LDH and NiFe-hexylaminobenzene loaded on carbon fibers (NiFe-LDH/NiFe-HAB/CF) exhibited near 100% of FEformate with the suppression of OER activity (Fig. 12d–f).129 DFT calculations revealed that 0.43 e− transferred from NiFe-HAB to NiFe-LDH across the interface (Fig. 12g), leading to the NiFe-HAB being a passivation layer for OER. Moreover, the adsorption energy value of methanol molecules on NiFe-LDH/NiFe-HAB/CF was found to be more negative than that on NiFe-LDH (Fig. 12h), indicating the favorable methanol adsorption and activation, which significantly improved the activity and selectivity of SMOR. The heterostructure composed of phosphide and hydroxide was also proven to be efficient for selective methanol oxidation. Fu's group coated NiCo-LDH on CoxP nanosheet arrays to build a heterostructure catalyst (CoxP/NiCo-LDH).185 The ultrathin NiCo-LDH provided a large electrochemical active area and CoxP endowed the catalyst with fast charge transfer at the interface. Therefore, enhanced catalytic activity, kinetics, and stability were achieved. Similarly, a heterostructure catalyst assembled by binary metal phosphides NiCoxP and NiCo-LDH also showed an improved SMOR performance due to the synergism of the two hetero-components.186
 | ||
| Fig. 12 (a) HRTEM image of Ni2P–CoP/NF. (b) Cell LSV curve comparison of MOR‖HER for the phosphide samples. (c) Corresponding FE of formate at current densities of 10, 20, 50, and 100 mA cm−2.127 Reproduced from ref. 127 with permission from John Wiley and Sons, Copyright 2021. (d) HRTEM images of NiFe-LDH/NiFe-HAB/CF. (e) Comparison of FE and yield rate for formate on NiFe-LDH/CF and NiFe-LDH/NiFe-HAB/CF. (f) Chronopotentiometry curve at 20 mA cm−2, FE, yield, and yield rate for H2 and HCOO− on NiFe-LDH/NiFe-HAB/CF. (g) Differential charge density image of NiFe-LDH/NiFe-HAB/CF. (h) Adsorption energy of methanol on the surfaces of NiFe-LDH and NiFe-LDH/NiFe-HAB.129 Reproduced from ref. 129 with permission from John Wiley and Sons, Copyright 2023. | ||
5. Conclusions and perspective
Coupling water electrolysis with selective methanol oxidation is an efficient energy conversion strategy to simultaneously obtain value-added formate and green hydrogen. It not only lowers the overpotential but also harvests valuable chemicals at both the anode and cathode, which is superior to conventional water splitting. Here, we reviewed the recent advances in the electrocatalysts for selective methanol oxidation for the co-production of value-added formate and green hydrogen, aiming to inspire more advanced research. In this work, the mechanism of SMOR was introduced and the catalyst design strategy was discussed in terms of chemical bond adjustment, electronic structure tuning, construction of dual active sites, and exposure of active sites. Then, the catalytic performance descriptors and catalyst development were summarized. It was demonstrated that noble metals tend to completely oxidize methanol to CO2, while non-noble metals are more favorable to SMOR due to their moderate activity. The active sites of non-noble metals are widely recognized as the in situ-formed high-valent species originating from surface reconstruction. Hydroxide-based catalysts are the most extensively studied due to their easy fabrication, facile active phase formation, and high feasibility of being modified. The reaction routine mainly follows the surface adsorption mechanism (SAM) or lattice oxygen participation mechanism (LOM). By activating or inhibiting the chemical bond of methanol molecules, the process of methanol oxidation can be well controlled at the stage of formate. The strategies of foreign atom doping, defect creation, and heterostructure construction can all tune the electronic distribution of catalysts, thereby optimizing the adsorption/desorption energy of the reactants/intermediates and reducing the energy barrier. Constructing dual active sites can greatly improve the bifunctional ability of methanol electrolysis, especially in acid media. Increasing the available active sites may enlarge the electrochemical surface area and expose more active sites, hence improving the activity.However, although many momentous discoveries and remarkable progress have been achieved in improving SMOR, there are still some ambiguities, restrictions, and challenges that need to be addressed.
(1) Mechanism study: (i) it is known that dehydrogenation can occur on two sites of the methanol molecule (C–H bond or O–H bond) at the initial stage. Thus, it is necessary to identify which path SMOR follows based on more direct and convincing evidence. (ii) The prevailing mechanisms used to analyze SMOR nowadays are the adsorption mechanism and lattice oxygen mechanism, and the in situ-formed NiOOH is commonly regarded as the real active phase. Actually, the conclusion of the active sites being Ni3+ species derived from surface reconstruction in many electrocatalytic reactions was raised many years ago and has been adopted to date. Attractively, in recent years, some new mechanisms have been proposed for OER and UOR, which share the same high-valent active Ni species with methanol oxidation. In the case of OER, the new mechanism of light-triggered coupled oxygen evolution mechanism (COM) and oxide path mechanism (OPM) was proposed.194,195 Regarding UOR, the active site of Ni4+ species and a new two-stage reaction pathway involving ammonia formation and its subsequent oxidation to N2 was revealed, which is different from most other Ni-based catalysts with NiOOH as the active phase.196,197 Therefore, more in-depth study is needed to determine whether there are other mechanisms involved in SMOR to make a breakthrough. (iii) In addition to pure Ni-based catalysts, other catalysts with multi-component metallic compounds including Ni element have also been reported to possess high-valent Ni species as the active sites. The valence state and role of other metals should also be explored. (iv) An increasing number of reports on heterostructure catalysts attributed the enhanced SMOR performance to the in situ-formed hetero-interface. However, the investigation of hetero-interfaces is mostly a qualitative description, lacking quantitative insight. Thus, more quantitative properties of hetero-interfaces should be identified to better understand the catalytic mechanism, such as the crystal phase composition, lattice distortion, interface length, spacing, and area, the origin of heterostructure catalyst activity, and surface structure evolution. (v) Although the overpotential of SMOR is lower than that of OER, there still exists a certain competitive relationship between them. Specifically, two types of oxidation currents coexist within a certain potential window. Therefore, identifying the cross-competition region and determining the origin of the current are significant to optimize the potential window and promote the understanding of the anode coupling process in hybrid water electrolysis. (vi) DFT calculations and in situ characterizations have been widely used to probe the catalytic mechanism. However, it is difficult to simulate the changes in the external conditions in catalytic reactions, such as the electrolyte concentration and mass transfer, via DFT. In situ characterizations have their own limitation and application range. Thus, machine learning can be a good supplement by building models from data to better understand the structure–activity relationship.
(2) Performance evaluation: (i) selectivity is the most significant descriptor to evaluate the SMOR activity. Generally, the selectivity of methanol to formate is described by FE. A few studies calculated the production rate of formate. Nevertheless, the conversion rate of formate is rarely mentioned in the literature. Thus, it is suggested to detect the consumption of methanol to examine the conversion rate of formate. (ii) It is known that 100% of FE means all the input electrons are devoted to generating the corresponding product. Thus, an FE below 100% indicates there are byproducts generated. However, lots of reports on FE below 100% did not explain whether any other byproducts exists. Thus, this issue should be further addressed. (iii) Stability is an indispensable concern in practical use. Thus far, the main durability evaluation method used is the steady-state stability test by conducting CA and CP measurements for tens of hours. The dynamic stability evaluation by comparing the CV or LSV curves before and after cycling tests will be an important supplement to catalytic stability. Moreover, the time for the stability test should be further extended to estimate the potential of practical use. (iv) Moreover, the change in the morphology, structure, and element composition of the catalyst during electrocatalysis should also be given attention. They are significant in the study of surface reconstruction, catalyst degradation, and inactivation mechanism.
(3) Large-scale production and practical application. (i) Restricted by the experimental instrument size and precise operation, the catalysts prepared in the laboratory have a small scale with several mg or g. Alternatively, their large-scale synthesis may lead to the problem of unevenness and instability. Thus, techniques for the scaled-up fabrication of catalysts that balance activity and stability should be developed for practical application. (ii) It can be seen that the electrochemical tests of SMOR and SMOR&HER were nearly all conducted in three-electrode or two-electrode systems throughout the literature. Few reports implemented SMOR in MEA, which is more closely related to the actual working conditions. Loading catalysts and evaluating their performance in MEA can explore the feasibility of practical applications and provide a scientific basis for engineering amplification. (iii) A critical issue that must be considered is how to collect the produced formate. Formate and methanol are both liquids, and thus certain separation techniques must be used to extract formic acid. Also, the difficulty and cost of the post-treatment should be appraised.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (22090030).References
- H. Zhang, Y. Fu, H. T. Nguyen, B. Fox, J. H. Lee, A. K. Lau, H. Zheng, H. Lin, T. Ma and B. Jia, Coord. Chem. Rev., 2023, 494, 215272 CrossRef CAS.
- J. Na, H. Yu, S. Jia, J. Chi, K. Lv, T. Li, Y. Zhao, Y. Zhao, H. Zhang and Z. Shao, J. Energy Chem., 2024, 91, 370–382 CrossRef CAS.
- L. Ding, Y. Zhang, T. Wang, P. Li and K. Chang, Catal. Sci. Technol., 2024, 14, 1730–1755 RSC.
- Y. Kuang, R. He, X. Gu, F. Yang, X. Tian and L. Feng, Chem. Eng. J., 2023, 456, 141055 CrossRef CAS.
- Y. Tsubonouchi, Z. N. Zahran, D. Chandra, N. Hoshino and M. Yagi, Catal. Sci. Technol., 2024, 14, 3287–3319 RSC.
- W. Zhu, Z. Yue, W. Zhang, N. Hu, Z. Luo, M. Ren, Z. Xu, Z. Wei, Y. Suo and J. Wang, J. Mater. Chem. A, 2018, 6, 4346–4353 RSC.
- A. Shankar, S. Marimuthu and G. Maduraiveeran, J. Mater. Chem. A, 2024, 12, 121–127 RSC.
- X. Wei, Y. Li, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 3148–3155 CrossRef CAS.
- Y. Aykut and A. Bayrakçeken Yurtcan, Electrochim. Acta, 2023, 471, 143335 CrossRef CAS.
- H. Wang, Q. Hu, J. Qiu, R. Guo and X. Liu, Catal. Sci. Technol., 2023, 13, 6102–6125 RSC.
- Z. Li, L. Sun, Y. Zhang, Y. Han, W. Zhuang, L. Tian and W. Tan, Coord. Chem. Rev., 2024, 510, 215837 CrossRef CAS.
- E. Lee, J. H. Kim, J. Choi, Y. Hong, D. Shin, H. Yun, J. Kim, G. Bak, S. Hong and Y. J. Hwang, J. Mater. Chem. A, 2023, 11, 16559–16569 RSC.
- J. Li, S. Sun, Y. Yang, Y. Dai, B. Zhang and L. Feng, Chem. Commun., 2022, 58, 9552–9555 RSC.
- F. Arshad, T. u. Haq, I. Hussain and F. Sher, ACS Appl. Energy Mater., 2021, 4, 8685–8701 CrossRef CAS.
- L. Yu, X. Pang, Z. Tian, S. Wang and L. Feng, Electrochim. Acta, 2023, 440, 141724 CrossRef CAS.
- T. V. M. Sreekanth, K. Prasad, J. Yoo, J. Kim and K. Yoo, Catal. Sci. Technol., 2023, 13, 3445–3455 RSC.
- T. Wang, X. Cao and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202213328 CrossRef CAS.
- Q. Qian, Y. Zhu, N. Ahmad, Y. Feng, H. Zhang, M. Cheng, H. Liu, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2024, 36, 2306108 CrossRef CAS.
- K. Lu, Y. Zhang, Y. Shen and H. Li, Catal. Sci. Technol., 2024, 14, 2973–2990 RSC.
- K. Yan, Y. Liu, Y. Lu, J. Chai and L. Sun, Catal. Sci. Technol., 2017, 7, 1622–1645 RSC.
- W. Zhang, Y. Chao and S. Guo, Energy Lab, 2023, 1, 220004 Search PubMed.
- D. Bellotti, L. Cassettari, M. Mosca and L. Magistri, J. Cleaner Prod., 2019, 240, 117947 CrossRef CAS.
- B. C. Yang, S. E. Jo, T. Kim, G. Park, D. Go, T. M. Gür and J. An, J. Alloys Compd., 2022, 921, 166078 CrossRef CAS.
- X. Du, M. Tan, T. Wei, H. Kobayashi, J. Song, Z. Peng, H. Zhu, Z. Jin, R. Li and W. Liu, Chem. Eng. J., 2023, 452, 139404 CrossRef CAS.
- J. Chang, L. Feng, C. Liu, W. Xing and X. Hu, Energy Environ. Sci., 2014, 7, 1628–1632 RSC.
- J. Li, S. Wang, B. Zhang, W. Wang and L. Feng, Int. J. Hydrogen Energy, 2017, 42, 12236–12245 CrossRef CAS.
- T. Xia, K. Zhao, Y. Zhu, X. Bai, H. Gao, Z. Wang, Y. Gong, M. Feng, S. Li, Q. Zheng, S. Wang, R. Wang and H. Guo, Adv. Mater., 2023, 35, 2206508 CrossRef CAS.
- S. Yu, S. Xu, R. Khan, H. Zhao and C. Li, Catal. Sci. Technol., 2024, 14, 820–834 RSC.
- Y. Zhou, Q. Wang, X. Tian, J. Chang and L. Feng, J. Energy Chem., 2022, 75, 46–54 CrossRef CAS.
- W. Qiao, L. Yu, J. Chang, F. Yang and L. Feng, Chin. J. Catal., 2023, 51, 113–123 CrossRef CAS.
- Y. Kuang, W. Qiao, S. Wang, F. Yang and L. Feng, ACS Mater. Lett., 2024, 6, 1722–1731 CrossRef CAS.
- B. Zhu, B. Dong, F. Wang, Q. Yang, Y. He, C. Zhang, P. Jin and L. Feng, Nat. Commun., 2023, 14, 1686 CrossRef CAS PubMed.
- S. Wu, N. Salmon, M. M. Li, R. Bañares Alcántara and S. C. E. Tsang, ACS Energy Lett., 2022, 7, 1021–1033 CrossRef CAS.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536–1551 RSC.
- D. A. Bulushev and J. R. H. Ross, ChemSusChem, 2018, 11, 821–836 CrossRef CAS PubMed.
- W. Wei, Z. Wei, R. Li, H. Yuan, J. Chen, J. Lv and S. Ouyang, Green Carbon, 2024 DOI:10.1016/j.greenca.2024.05.003.
- R. Li, Y. Li, Z. Li, W. Wei, Q. Hao, Y. Shi, S. Ouyang, H. Yuan and T. Zhang, ACS Catal., 2022, 12, 5316–5326 CrossRef CAS.
- R. Li, Y. Li, Z. Li, W. Wei, S. Ouyang, H. Yuan and T. Zhang, Sol. RRL, 2021, 5, 2000488 CrossRef CAS.
- Y. Li, R. Li, Z. Li, W. Wei, S. Ouyang, H. Yuan and T. Zhang, Chem. Res. Chin. Univ., 2020, 36, 1006–1012 CrossRef CAS.
- Y. X. Chen, A. Miki, S. Ye, H. Sakai and M. Osawa, J. Am. Chem. Soc., 2003, 125, 3680–3681 CrossRef CAS.
- W. Qiao, X. Huang and L. Feng, Chin. J. Struct. Chem., 2022, 41, 2207016–2207034 CAS.
- X. Zhang, L. Hui, D. Yan, J. Li, X. Chen, H. Wu and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202308968 CrossRef CAS.
- X. Yang, J. Xue and L. Feng, Chem. Commun., 2019, 55, 11247–11250 RSC.
- H. Chi, J. Lin, S. Kuang, M. Li, H. Liu, Q. Fan, T. Yan, S. Zhang and X. Ma, J. Energy Chem., 2023, 85, 267–275 CrossRef CAS.
- J. Shi, H. He, S. Zhou, J. Li and W. Cai, Green Chem., 2024, 26, 2638–2644 RSC.
- J. Li, L. Li, J. Wang, A. Cabot and Y. Zhu, ACS Energy Lett., 2024, 9, 853–879 CrossRef CAS.
- T. Iwasita, Electrochim. Acta, 2002, 47, 3663–3674 CrossRef CAS.
- D. Cao, G. Q. Lu, A. Wieckowski, S. A. Wasileski and M. Neurock, J. Phys. Chem. B, 2005, 109, 11622–11633 CrossRef CAS PubMed.
- P. Ferrin and M. Mavrikakis, J. Am. Chem. Soc., 2009, 131, 14381–14389 CrossRef CAS.
- J. Wang, B. Zhang, W. Guo, L. Wang, J. Chen, H. Pan and W. Sun, Adv. Mater., 2023, 35, 2211099 CrossRef CAS PubMed.
- N. Ullah, S. Ullah, S. Khan, D. Guziejewski and V. Mirceski, Int. J. Hydrogen Energy, 2023, 48, 3340–3354 CrossRef CAS.
- J. Wu, J. Wen and Y. X. Yu, Appl. Surf. Sci., 2021, 565, 150567 CrossRef CAS.
- J. M. Huo, Y. Wang, J. N. Xue, W. Y. Yuan, Q. G. Zhai, M. C. Hu, S. N. Li and Y. Chen, Small, 2024, 20, 2305877 CrossRef CAS.
- X. Pang, H. Zhao, Y. Huang, B. Luo, H. Bai and W. Fan, Appl. Surf. Sci., 2023, 608, 155152 CrossRef CAS.
- H. Zhang, H. Wang, X. Tong, L. Zhou, X. Yang, Y. Wang, M. Zhang and Z. Wu, Chem. Eng. J., 2023, 452, 139582 CrossRef CAS.
- K. Deng, P. Liu, X. Liu, H. Li, W. Tian and J. Ji, Green Chem., 2023, 25, 9837–9846 RSC.
- Q. Qian, X. He, Z. Li, Y. Chen, Y. Feng, M. Cheng, H. Zhang, W. Wang, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2023, 35, 2300935 CrossRef CAS.
- W. Chen, Y. Wang, B. Wu, J. Shi, Y. Li, L. Xu, C. Xie, W. Zhou, Y.-C. Huang, T. Wang, S. Du, M. Song, D. Wang, C. Chen, J. Zheng, J. Liu, C.-L. Dong, Y. Zou, J. Chen and S. Wang, Adv. Mater., 2022, 34, 2105320 CrossRef CAS.
- B. Zhao, J. Liu, C. Xu, R. Feng, P. Sui, L. Wang, J. Zhang, J. Luo and X. Fu, Adv. Funct. Mater., 2021, 31, 2008812 CrossRef CAS.
- S. Ganguly, S. Paul, D. Khurana, T. S. Khan, P. K. Giri, C. Loha and S. Ghosh, ACS Appl. Energy Mater., 2023, 6, 5331–5341 CrossRef CAS.
- Y. Yi, J. Li and C. Cui, Chin. Chem. Lett., 2022, 33, 1006–1010 CrossRef CAS.
- G. Solomon, A. Landström, R. Mazzaro, M. Jugovac, P. Moras, E. Cattaruzza, V. Morandi, I. Concina and A. Vomiero, Adv. Energy Mater., 2021, 11, 2101324 CrossRef CAS.
- F. Meng, Q. Wu, K. Elouarzaki, S. Luo, Y. Sun, C. Dai, S. Xi, Y. Chen, X. Lin, M. Fang, X. Wang, D. Mandler and Z. J. Xu, Sci. Adv., 2023, 9, eadh9487 CrossRef CAS PubMed.
- Y. Zhou, Z. Wang, W. Fang, R. Qi, Z. Wang, C. Xia, K. Lei, B. You, X. Yang, Y. Liu, W. Guo, Y. Su, S. Ding and B. Y. Xia, ACS Catal., 2023, 13, 2039–2046 CrossRef CAS.
- B. Zhao, J. Liu, X. Wang, C. Xu, P. Sui, R. Feng, L. Wang, J. Zhang, J. Luo and X. Fu, Nano Energy, 2021, 80, 105530 CrossRef CAS.
- L. Yang, J. Fan, B. Xiao and W. Zhu, Chem. Eng. J., 2023, 468, 143823 CrossRef CAS.
- H. Chen, Q. Wu, Y. Wang, Q. Zhao, X. Ai, Y. Shen and X. Zou, Chem. Commun., 2022, 58, 7730–7740 RSC.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- B. Zhao, J. W. Liu, Y. R. Yin, D. Wu, J. L. Luo and X. Z. Fu, J. Mater. Chem. A, 2019, 7, 25878–25886 RSC.
- Y. Li, Z. Yin, M. Cui, X. Liu, J. Xiong, S. Chen and T. Ma, J. Mater. Chem. A, 2021, 9, 2070–2092 RSC.
- Y. Zhu, X. Wang, X. Zhu, Z. Wu, D. Zhao, F. Wang, D. Sun, Y. Tang, H. Li and G. Fu, Small, 2023, 19, 2206531 CrossRef CAS.
- Y. Kuang, W. Qiao, F. Yang and L. Feng, J. Energy Chem., 2023, 85, 447–454 CrossRef CAS.
- H. Adamu, Z. H. Yamani and M. Qamar, Mater. Renew. Sustain. Energy, 2022, 11, 169–213 CrossRef.
- R. Yu, R. Shao, F. Ning, Y. Yu, J. Zhang, X.-Y. Ma, R. Zhu, M. Li, J. Lai, Y. Zhao, L. Zeng, J. Zhang and Z. Xia, Small, 2024, 20, 2305817 CrossRef CAS.
- J. M. Martínez de la Hoz and P. B. Balbuena, J. Phys. Chem. C, 2011, 115, 21324–21333 CrossRef.
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- B. Zheng, J. Fan, B. Chen, X. Qin, J. Wang, F. Wang, R. Deng and X. Liu, Chem. Rev., 2022, 122, 5519–5603 CrossRef CAS.
- A. A. Tedstone, D. J. Lewis and P. O'Brien, Chem. Mater., 2016, 28, 1965–1974 CrossRef CAS.
- N. B. Saleh, D. J. Milliron, N. Aich, L. E. Katz, H. M. Liljestrand and M. J. Kirisits, Sci. Total Environ., 2016, 568, 926–932 CrossRef CAS.
- C. Rao, H. Wang, K. Chen, H. Chen, S. Ci, Q. Xu and Z. Wen, Small, 2024, 20, 2303300 CrossRef CAS.
- C. Xiao, L. Cheng, Y. Wang, J. Liu, R. Chen, H. Jiang, Y. Li and C. Li, J. Mater. Chem. A, 2022, 10, 1329–1335 RSC.
- C. Xie, D. Yan, H. Li, S. Du, W. Chen, Y. Wang, Y. Zou, R. Chen and S. Wang, ACS Catal., 2020, 10, 11082–11098 CrossRef CAS.
- H. Yun, M. Topsakal, A. Prakash, B. Jalan, J. S. Jeong, T. Birol and K. A. Mkhoyan, Sci. Adv., 2021, 7, eabd4449 CrossRef CAS PubMed.
- W. Li, D. Wang, Y. Zhang, L. Tao, T. Wang, Y. Zou, Y. Wang, R. Chen and S. Wang, Adv. Mater., 2020, 32, 1907879 CrossRef CAS.
- Y. Jia, K. Jiang, H. Wang and X. Yao, Chem, 2019, 5, 1371–1397 CAS.
- M. Khan, A. Hameed, A. Samad, T. Mushiana, M. I. Abdullah, A. Akhtar, R. S. Ashraf, N. Zhang, B. G. Pollet, U. Schwingenschlögl and M. Ma, Commun. Chem., 2022, 5, 109 CrossRef CAS PubMed.
- B. Liu, X. Wang, S. Wang, H. Peng, T. Xiao, G. Liu, S. Bai, Y. Zhao, W. Zhang and Y. Song, Mater. Today Energy, 2022, 28, 101082 CrossRef CAS.
- T. R. Naveen Kumar, S. Kamalakannan, M. Prakash, B. Viswanathan and B. Neppolian, ACS Appl. Energy Mater., 2022, 5, 2104–2111 CrossRef.
- J. Chen, M. Wang, B. Liu, Z. Fan, K. Cui and Y. Kuang, J. Phys. Chem. B, 2006, 110, 11775–11779 CrossRef CAS.
- F. Liu and Z. Fan, Chem. Soc. Rev., 2023, 52, 1723–1772 RSC.
- H. Qin, W. Tong, Q.-X. Pei, Z. Wang, G. Zhang, Y. Chen, P. Li, J. Liu, Z. Xu and Y. Liu, Adv. Funct. Mater., 2023, 33, 2300210 CrossRef CAS.
- W. Wei, Z. Wei, R. Li, Z. Li, R. Shi, S. Ouyang, Y. Qi, D. L. Philips and H. Yuan, Nat. Commun., 2022, 13, 3199 CrossRef PubMed.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C.-L. Dong, Y.-C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 CAS.
- Y. Qi, Y. Zhang, L. Yang, Y. Zhao, Y. Zhu, H. Jiang and C. Li, Nat. Commun., 2022, 13, 4602 CrossRef CAS.
- S. Ni, H. Qu, Z. Xu, X. Zhu, H. Xing, L. Wang, J. Yu, H. Liu, C. Chen and L. Yang, Appl. Catal., B, 2021, 299, 120638 CrossRef CAS.
- C. Li, Y. Liu, Z. Zhuo, H. Ju, D. Li, Y. Guo, X. Wu, H. Li and T. Zhai, Adv. Energy Mater., 2018, 8, 1801775 CrossRef.
- X. Yue, S. Liping, H. Lihua and Z. Hui, ACS Appl. Nano Mater., 2023, 6, 10312–10321 CrossRef CAS.
- Y. Hao, D. Yu, S. Zhu, C. Kuo, Y. Chang, L. Wang, H. Chen, M. Shao and S. Peng, Energy Environ. Sci., 2023, 16, 1100–1110 RSC.
- B. Zhao, J. Liu, C. Xu, R. Feng, P. Sui, J. Luo, L. Wang, J. Zhang, J. Luo and X. Fu, Appl. Catal., B, 2021, 285, 119800 CrossRef CAS.
- S. Li, R. Ma, J. Hu, Z. Li, L. Liu, X. Wang, Y. Lu, G. E. Sterbinsky, S. Liu, L. Zheng, J. Liu, D. Liu and J. Wang, Nat. Commun., 2022, 13, 2916 CrossRef CAS PubMed.
- J. Xue, X. Wu and L. Feng, Chem. Commun., 2022, 58, 2371–2374 RSC.
- Y. A. Awoke, M. C. Tsai, D. B. Adam, A. A. Ayele, S. C. Yang, W. H. Huang, J. L. Chen, C. W. Pao, C. Y. Mou, W. N. Su and B. J. Hwang, Electrochim. Acta, 2022, 432, 141161 CrossRef CAS.
- M. Li, D. Zhang, Y. Yi, B. Xue and B. Liu, Electrochim. Acta, 2022, 423, 140566 CrossRef CAS.
- N. Kakati, J. Maiti, S. H. Lee, S. H. Jee, B. Viswanathan and Y. S. Yoon, Chem. Rev., 2014, 114, 12397–12429 CrossRef CAS PubMed.
- Y. Wang, X. Yang, K. Wang, Z. Liu, X. Sun, J. Chen, S. Liu, X. Sun, J. Xie and B. Tang, Green Chem., 2023, 25, 8216–8225 RSC.
- Y. Zhou, K. Zhao, C. Shi, H. Ma, D. Yuan and Z. Yi, J. Electrochem. Soc., 2024, 171, 056506 CrossRef.
- Y. Zhang, X. Wu, G. Fu, F. Si, X. Fu and J. Luo, Int. J. Hydrogen Energy, 2022, 47, 17150–17160 CrossRef CAS.
- N. Cai, L. Fu, Z. Yang and D. Huang, Curr. Opin. Electrochem., 2022, 34, 100994 CrossRef CAS.
- Y. Zhang, J. Yang, R. Ge, J. Zhang, J. M. Cairney, Y. Li, M. Zhu, S. Li and W. Li, Coord. Chem. Rev., 2022, 461, 214493 CrossRef CAS.
- G. Fu, X. Kang, Y. Zhang, X. Yang, L. Wang, X. Fu, J. Zhang, J. Luo and J. Liu, Nano-Micro Lett., 2022, 14, 200 CrossRef CAS PubMed.
- D. Li, H. Liu and L. Feng, Energy Fuels, 2020, 34, 13491–13522 CrossRef CAS.
- J. Li, S. Wang, J. Chang and L. Feng, Adv. Powder Mater., 2022, 1, 100030 CrossRef.
- M. I. Abdullah, A. Hameed, N. Zhang, M. H. Islam, M. Ma and B. G. Pollet, ACS Appl. Mater. Interfaces, 2021, 13, 30603–30613 CrossRef CAS PubMed.
- J. Luo, D. A. Vermaas, D. Bi, A. Hagfeldt, W. A. Smith and M. Grätzel, Adv. Energy Mater., 2016, 6, 1600100 CrossRef.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS.
- Y. Luo, Z. Zhang, M. Chhowalla and B. Liu, Adv. Mater., 2022, 34, 2108133 CrossRef CAS PubMed.
- Y. Wang, G. Qian, Q. Xu, H. Zhang, F. Shen, L. Luo and S. Yin, Appl. Catal., B, 2021, 286, 119881 CrossRef CAS.
- J. Hao, J. Liu, D. Wu, M. Chen, Y. Liang, Q. Wang, L. Wang, X. Fu and J. Luo, Appl. Catal., B, 2021, 281, 119510 CrossRef CAS.
- F. Meng, C. Dai, Z. Liu, S. Luo, J. Ge, Y. Duan, G. Chen, C. Wei, R. R. Chen, J. Wang, D. Mandler and Z. J. Xu, eScience, 2022, 2, 87–94 CrossRef.
- K. Xiang, Z. Song, D. Wu, X. Deng, X. Wang, W. You, Z. Peng, L. Wang, J. Luo and X. Fu, J. Mater. Chem. A, 2021, 9, 6316–6324 RSC.
- Y. H. Fang and Z. P. Liu, ACS Catal., 2014, 4, 4364–4376 CrossRef CAS.
- G. T. Burstein, Corros. Sci., 2005, 47, 2858–2870 CrossRef CAS.
- F. Guo, K. Ye, M. Du, X. Huang, K. Cheng, G. Wang and D. Cao, Electrochim. Acta, 2016, 210, 474–482 CrossRef CAS.
- N. Meddings, M. Heinrich, F. Overney, J. Lee, V. Ruiz, E. Napolitano, S. Seitz, G. Hinds, R. Raccichini, M. Gaberšček and J. Park, J. Power Sources, 2020, 480, 228742 CrossRef CAS.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- M. Li, X. Deng, K. Xiang, Y. Liang, B. Zhao, J. Hao, J. Luo and X. Fu, ChemSusChem, 2020, 13, 914–921 CrossRef CAS PubMed.
- D. Wu, J. Hao, W. Wang, Y. Yu, X. Fu and J. Luo, ChemSusChem, 2021, 14, 5450–5459 CrossRef CAS PubMed.
- Y. Liang, Z. Song, Y. Zhang, B. Zhao, X. Wang, K. Xiang, Z. Ge, X. Fu and J. Luo, ACS Appl. Nano Mater., 2021, 4, 8723–8732 CrossRef CAS.
- S. Jiang, T. Xiao, C. Xu, S. Wang, H. Peng, W. Zhang, B. Liu and Y. Song, Small, 2023, 19, 2208027 CrossRef CAS PubMed.
- R. Yuan, J. Wang, X. Luo, G. Wang, D. Wang, S. Liu and Z. Zhao, Energy Fuels, 2024, 38, 1496–1507 CrossRef CAS.
- J. Li, Electrochem. Commun., 2023, 146, 107416 CrossRef CAS.
- C. Cao, D. Ma, J. Jia, Q. Xu, X. Wu and Q. Zhu, Adv. Mater., 2021, 33, 2008631 CrossRef CAS.
- C. Zhou, H. Yuan, P. Hu and H. Wang, Chem. Commun., 2020, 56, 15201–15204 RSC.
- Y. Xu, M. Liu, M. Wang, T. Ren, K. Ren, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2022, 300, 120753 CrossRef CAS.
- M. Zhou, C. Li and J. Fang, Chem. Rev., 2021, 121, 736–795 CrossRef CAS.
- L.-W. Chen and L. Wang, Catal. Sci. Technol., 2023, 13, 4590–4614 RSC.
- F. Arshad, A. Tahir, T. u. Haq, H. Duran, I. Hussain and F. Sher, Energy Fuels, 2023, 37, 14161–14170 CrossRef CAS.
- F. Arshad, A. Tahir, T. u. Haq, H. Duran, I. Hussain and F. Sher, Int. J. Hydrogen Energy, 2022, 47, 36118–36128 CrossRef CAS.
- J. Chen, M. Ahmad, Y. Zhang, H. Ye, L. Wang, J. Zhang, X. Fu and J. Luo, Chem. Eng. J., 2023, 454, 140056 CrossRef CAS.
- S. A. Krishna, N. Noble, N. Radhika and B. Saleh, J. Manuf. Process., 2024, 109, 583–606 CrossRef.
- S. Wang, L. Zhao, J. Li, X. Tian, X. Wu and L. Feng, J. Energy Chem., 2022, 66, 483–492 CrossRef CAS.
- J. Li, Y. Chang, D. Li, L. Feng and B. Zhang, Chem. Commun., 2021, 57, 7035–7038 RSC.
- J. Li, S. Wang, S. Sun, X. Wu, B. Zhang and L. Feng, J. Mater. Chem. A, 2022, 10, 9308–9326 RSC.
- J. Zhang, Y. Hua, H. Li, X. Zhang, C. Shi, Y. Li, L. Di and Z. Wang, Chem. Eng. J., 2023, 478, 147288 CrossRef CAS.
- X. Wang, C. Xiao, Y. Li, T. Murayama, T. Ishida, M. Lin and G. Xiu, Appl. Catal., A, 2023, 664, 119341 CrossRef CAS.
- S. Royer, D. Duprez, F. Can, X. Courtois, C. Batiot-Dupeyrat, S. Laassiri and H. Alamdari, Chem. Rev., 2014, 114, 10292–10368 CrossRef CAS.
- T. Q. N. Tran, S. W. Yoon, B. J. Park and H. H. Yoon, J. Electroanal. Chem., 2018, 818, 76–83 CrossRef CAS.
- L. Gong, N. Xuan, G. Gu, P. Lv, N. Huang, C. Song, M. Zheng, J. Wang, P. Cui, G. Gu, Y. Jia, G. Cheng and Z. Du, Nano Energy, 2023, 107, 108124 CrossRef CAS.
- L. Ming, X. Wu, S. Wang, W. Wu and C. Lu, ChemElectroChem, 2022, 9, e202200522 CrossRef CAS.
- Y. Fan, X. Yang, E. Wei, Y. Dong, H. Gao, X. Luo and W. Yang, Appl. Catal., B, 2024, 345, 123716 CrossRef CAS.
- L. Yang, Z. Liu, S. Zhu, L. Feng and W. Xing, Mater. Today Phys., 2021, 16, 100292 CrossRef CAS.
- Y. Wang, M. Zhang, Y. Liu, Z. Zheng, B. Liu, M. Chen, G. Guan and K. Yan, Adv. Sci., 2023, 10, 2207519 CrossRef CAS.
- D. P. Sahoo, K. K. Das, S. Mansingh, S. Sultana and K. Parida, Coord. Chem. Rev., 2022, 469, 214666 CrossRef CAS.
- B. Liu, T. Xiao, X. Sun, H. Peng, X. Wang, Y. Zhao, W. Zhang and Y. Song, J. Mater. Chem. A, 2022, 10, 19649–19661 RSC.
- J. Masa and W. Schuhmann, ChemCatChem, 2019, 11, 5842–5854 CrossRef CAS.
- T. Qin, Z. Wang, Y. Wang, F. Besenbacher, M. Otyepka and M. Dong, Nano-Micro Lett., 2021, 13, 183 CrossRef CAS.
- J. Li, R. Wei, X. Wang, Y. Zuo, X. Han, J. Arbiol, J. Llorca, Y. Yang, A. Cabot and C. Cui, Angew. Chem., 2020, 59, 20826–20830 CrossRef CAS.
- H. Wang, J. Li, K. Li, Y. Lin, J. Chen, L. Gao, V. Nicolosi, X. Xiao and J. Lee, Chem. Soc. Rev., 2021, 50, 1354–1390 RSC.
- S. Dong, X. Chen, X. Zhang and G. Cui, Coord. Chem. Rev., 2013, 257, 1946–1956 CrossRef CAS.
- S. V. Didziulis, K. D. Butcher and S. S. Perry, Inorg. Chem., 2003, 42, 7766–7781 CrossRef CAS.
- L. Zhao, Q. Sun, M. Li, Y. Zhong, P. Shen, Y. Lin and K. Xu, Sci. China Mater., 2023, 66, 1820–1828 CrossRef CAS.
- J. Chang, L. Feng, C. Liu, W. Xing and X. Hu, Angew. Chem., Int. Ed., 2014, 53, 122–126 CrossRef CAS PubMed.
- J. Luo, K. Liang, Q. Zeng, L. Tang, Y. Yang, J. Song, S. Liu, S. Li, L. Hu and Y. Fang, Appl. Surf. Sci., 2024, 654, 159433 CrossRef CAS.
- X. Li, Z. Niu, M. Niu, J. Wang, D. Cao and X. Zeng, Small, 2024, 20, 2311335 CrossRef CAS PubMed.
- Z. Duan, T. Ren, Q. Mao, H. Yu, K. Deng, Y. Xu, Z. Wang, L. Wang and H. Wang, J. Mater. Chem. A, 2022, 10, 18126–18131 RSC.
- J. Chang, W. Wang, D. Wu, F. Xu, K. Jiang, Y. Guo and Z. Gao, J. Colloid Interface Sci., 2023, 648, 259–269 CrossRef CAS PubMed.
- B. Zhao, C. Xu, M. Shakouri, R. Feng, Y. Zhang, J. Liu, L. Wang, J. Zhang, J. Luo and X. Fu, Appl. Catal., B, 2022, 305, 121082 CrossRef CAS.
- J. Li, X. Tian, X. Wang, T. Zhang, M. C. Spadaro, J. Arbiol, L. Li, Y. Zuo and A. Cabot, Inorg. Chem., 2022, 61, 13433–13441 CrossRef CAS.
- F. Si, J. Liu, Y. Zhang, B. Zhao, Y. Liang, X. Wu, X. Kang, X. Yang, J. Zhang, X. Fu and J. Luo, Small, 2023, 19, 2205257 CrossRef CAS PubMed.
- X. Peng, Y. Yan, X. Jin, C. Huang, W. Jin, B. Gao and P. K. Chu, Nano Energy, 2020, 78, 105234 CrossRef CAS.
- C. Yang, Y. Lu, W. Duan, Z. Kong, Z. Huang, T. Yang, Y. Zou, R. Chen and S. Wang, Energy Fuels, 2021, 35, 14283–14303 CrossRef CAS.
- X. Xia, L. Wang, N. Sui, V. L. Colvin and W. W. Yu, Nanoscale, 2020, 12, 12249–12262 RSC.
- R. A. Hussain and I. Hussain, J. Solid State Chem., 2019, 277, 316–328 CrossRef CAS.
- T. Lu, S. Dong, C. Zhang, L. Zhang and G. Cui, Coord. Chem. Rev., 2017, 332, 75–99 CrossRef CAS.
- J. Li, C. Xing, Y. Zhang, T. Zhang, M. C. Spadaro, Q. Wu, Y. Yi, S. He, J. Llorca, J. Arbiol, A. Cabot and C. Cui, Small, 2021, 17, 2006623 CrossRef CAS PubMed.
- W. Liao, S. Wang, H. Su and Y. Zhang, Nano Res., 2023, 16, 1984–1991 CrossRef CAS.
- Y. Zhang, F. Gao, D. Wang, Z. Li, X. Wang, C. Wang, K. Zhang and Y. Du, Coord. Chem. Rev., 2023, 475, 214916 CrossRef CAS.
- A. Song, S. Song, M. Duanmu, H. Tian, H. Liu, X. Qin, G. Shao and G. Wang, Small Sci., 2023, 3, 2300036 CrossRef CAS.
- G. Zhang, B. Wang, L. Li, S. Yang, J. Liu and S. Yang, J. Mater. Chem. A, 2020, 8, 18945–18954 RSC.
- Y. Duan, X. L. Zhang, F. Y. Gao, Y. Kong, Y. Duan, X. T. Yang, X. X. Yu, Y. R. Wang, S. Qin, Z. Chen, R. Wu, P. P. Yang, X. S. Zheng, J. F. Zhu, M. R. Gao, T. B. Lu, Z. Y. Yu and S. H. Yu, Angew. Chem., 2023, 62, e202217275 CrossRef CAS PubMed.
- X. Ding, R. Jiang, J. Wu, M. Xing, Z. Qiao, X. Zeng, S. Wang and D. Cao, Adv. Funct. Mater., 2023, 33, 2306786 CrossRef CAS.
- Y. Qi, Y. Zhu, H. Jiang and C. Li, Chin. J. Catal., 2024, 56, 139–149 CrossRef CAS.
- S. Liu, S. Yin, L. Cui, H. Yu, K. Deng, Z. Wang, Y. Xu, L. Wang and H. Wang, Energy Lab, 2023, 1, 220005 CrossRef.
- C. Wan, J. Jin, X. Wei, S. Chen, Y. Zhang, T. Zhu and H. Qu, J. Mater. Sci. Technol., 2022, 124, 102–108 CrossRef CAS.
- M. Li, X. Deng, Y. Liang, K. Xiang, D. Wu, B. Zhao, H. Yang, J. Luo and X. Fu, J. Energy Chem., 2020, 50, 314–323 CrossRef.
- Y. Zhang, X. Wu, G. Fu, X. Fu and J. Luo, J. Alloys Compd., 2022, 906, 164305 CrossRef CAS.
- M. Jiang, H. Zhai, L. Chen, L. Mei, P. Tan, K. Yang and J. Pan, Adv. Funct. Mater., 2023, 33, 2302621 CrossRef CAS.
- A. H. Al Naggar, N. M. Shinde, J. S. Kim and R. S. Mane, Coord. Chem. Rev., 2023, 474, 214864 CrossRef CAS.
- M. Ahmad, M. B. Hussain, J. Chen, Y. Yang, X. Wu, H. Chen, S. Afzal, W. Raza, Z. Zeng, F. Ye, X. Zhao, J. Zhang, R. Feng, X. Fu and J. Luo, ACS Catal., 2024, 14, 9134–9143 CrossRef CAS.
- Q. Yang, C. Zhang, B. Dong, Y. Cui, F. Wang, J. Cai, P. Jin and L. Feng, Appl. Catal., B, 2021, 296, 120359 CrossRef CAS.
- M. Khan, M. I. Abdullah, A. Samad, Z. Shao, T. Mushiana, A. Akhtar, A. Hameed, N. Zhang, U. Schwingenschlögl and M. Ma, Small, 2023, 19, 2205499 CrossRef CAS PubMed.
- J. Tian, C. Cao, D. Ma, S. Han, Y. He, X. Wu and Q. Zhu, Small Struct., 2022, 3, 2100134 CrossRef CAS.
- M. Zhang, J. Zhu, R. Wan, B. Liu, D. Zhang, C. Zhang, J. Wang and J. Niu, Chem. Mater., 2022, 34, 959–969 CrossRef CAS.
- X. Wang, S. Xi, P. Huang, Y. Du, H. Zhong, Q. Wang, A. Borgna, Y. W. Zhang, Z. Wang, H. Wang, Z. G. Yu, W. S. V. Lee and J. Xue, Nature, 2022, 611, 702–708 CrossRef CAS PubMed.
- C. Lin, J. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S. H. Kim, D. H. Kim, S. S. Shinde, Y. F. Li, Z. P. Liu, Z. Jiang and J. H. Lee, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS.
- L. Zhang, L. Wang, H. Lin, Y. Liu, J. Ye, Y. Wen, A. Chen, L. Wang, F. Ni, Z. Zhou, S. Sun, Y. Li, B. Zhang and H. Peng, Angew. Chem., Int. Ed., 2019, 58, 16820–16825 CrossRef CAS PubMed.
- S. K. Geng, Y. Zheng, S. Q. Li, H. Su, X. Zhao, J. Hu, H. B. Shu, M. Jaroniec, P. Chen, Q. H. Liu and S. Z. Qiao, Nat. Energy, 2021, 6, 904–912 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00727a |
| This journal is © The Royal Society of Chemistry 2024 |