
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated quantum chemistry for estimating nucleophilicity and electrophilicity with applications to retrosynthesis and covalent inhibitors†
Nicolai
Ree
 a,
Andreas H.
Göller
*b and
Jan H.
Jensen
*a
a,
Andreas H.
Göller
*b and
Jan H.
Jensen
*a
aDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, 2100, Copenhagen Ø, Denmark. E-mail: jhjensen@chem.ku.dk
bBayer AG, Pharmaceuticals, R&D, Computational Molecular Design, 42096, Wuppertal, Germany. E-mail: andreas.goeller@bayer.com
First published on 8th January 2024
Abstract
Reactivity scales such as nucleophilicity and electrophilicity are valuable tools for determining chemical reactivity and selectivity. However, prior attempts to predict or calculate nucleophilicity and electrophilicity are either not capable of generalizing well to unseen molecular structures or require substantial computing resources. We present a fully automated quantum chemistry (QM)-based workflow that automatically identifies nucleophilic and electrophilic sites and computes methyl cation affinities and methyl anion affinities to quantify nucleophilicity and electrophilicity, respectively. The calculations are based on r2SCAN-3c SMD(DMSO) single-point calculations on GFN1-xTB ALPB(DMSO) geometries that, in turn, derive from a GFNFF-xTB ALPB(DMSO) conformational search. The workflow is validated against both experimental and higher-level QM-derived data resulting in very strong correlations while having a median wall time of less than two minutes per molecule. Additionally, we demonstrate the workflow on two different applications: first, as a general tool for filtering retrosynthetic routes based on chemical selectivity predictions, and second, as a tool for determining the relative reactivity of covalent inhibitors. The code is freely available on GitHub under the MIT open source license and as a web application at https://www.esnuel.org.
Introduction
A fundamental principle covered in most chemistry textbooks is that electrophiles (electron-seeking) react with nucleophiles (nucleus-seeking), and one textbook even states that:In essence, organic chemistry is all about the interaction between electron-rich atoms or molecules and electron-deficient atoms or molecules.1
The concept of nucleophiles and electrophiles dates back to 1933,2 when it was introduced by Ingold based on Lewis's electronic theory of valency and the acid–base theory of Brøndsted and Lowry.3–5 Over the years, nucleophilicity and electrophilicity have been quantified to create reactivity scales that explain why some atoms or molecules are more reactive than others. Most noteworthy is the work by Mayr and co-workers, who experimentally studied the reactivity of organic molecules that led to the Mayr–Patz equation;6
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k20°C = sN(N + E) k20°C = sN(N + E) | (1) |
Essential to eqn (1) is that k20°C can span from non-observable reactions (k20°C < 10−5 M−1 s−1) to diffusion-controlled reactions (k20°C > 109 M−1 s−1),7,8 and its applicability has been confirmed for various molecules resulting in Mayr's database that currently holds experimental reactivity parameters for 352 electrophiles and 1261 nucleophiles.7 However, measuring reaction rates to extract reactivity parameters is generally time-consuming and often difficult at the extremes of the reactivity scales. Thus, several in silico methods have been proposed to circumvent this problem.8,9 This includes estimating the rate constant using the Eyring equation10,11 and computing the reactivity parameters from frontier molecular orbital (FMO) energies12,13 or chemical affinities.14–16 Furthermore, a lot of new ML approaches based on Mayr's database have recently emerged.17–23
In this work, we will focus on the studies by Van Vranken and Baldi showing that calculated methyl cation affinities (MCAs) and methyl anion affinities (MAAs) of structurally different molecules correlate with Mayr's N × sN and E, respectively, when considering solvent effects.15,16 Based on these findings, they created two QM-derived datasets with reactivity parameters for 1232 nucleophiles and 1113 electrophiles (we have excluded 76 duplicates) covering ∼50 orders of magnitude in each case.24 The datasets were used to train different ML models by treating the affinities as either atomic, functional group, or molecular properties. The best-performing architecture was an atom-based graph attention network (GAT) achieving 10-fold cross-validation R2 coefficients of 0.92 ± 0.02 and 0.94 ± 0.02 for MCAs and MAAs, respectively.
However, it is worth noting that the two datasets contain molecules that are not in Mayr's database, and the ML models have not been validated against experimental reactivity parameters. Furthermore, the atom sites are identified by hand, which makes it difficult to apply their method to arbitrary molecules in an automated fashion. This work will address these limitations by introducing a fast and fully automated quantum chemistry-based workflow that detects relevant sites and calculates associated MCAs and MAAs. In addition, we will apply the workflow to different tasks to highlight the applicability of MCAs and MAAs as quantitative measures of chemical reactivity.
Methods
Automated quantum chemistry-based workflow
We have previously presented fully automated quantum chemistry-based workflows for predicting the regioselectivity of EAS reactions25 and palladium-catalyzed Heck reactions.26 Following this work, we introduce a workflow that, given a SMILES string as input, identifies possible nucleophilic and electrophilic sites and individually attaches methyl cations or anions to calculate associated MCA or MAA. The input SMILES string is modified using a set of SMIRKS and RDKit,27 which can easily be adjusted to include/exclude certain functional groups. The nucleophilic sites include double/triple-bonded atoms, singly charged anions, atoms with lone pairs, and specific functional groups such as aldehydes, amides, amines, carbanions, carboxylic acids, cyanoalkyl/nitrile anions, enolates, esters, ethers, imines, isonitriles, ketones, nitranions, nitriles, and nitronates. The electrophilic sites include double/triple-bonded atoms, singly charged cations, and specific functional groups such as acyl halides, aldehydes, amides, anhydrides, boranes, carbocations, esters, imines, iminium ions, ketones, Michael acceptors, and oxonium ions.The calculated MCAs and MAAs are defined in eqn (2) and (3) as the negative energy difference (−ΔE) of a nucleophile (Nuc) reacting with a methyl cation (CH3+) and an electrophile (Elec) reacting with a methyl anion (CH3−), respectively.
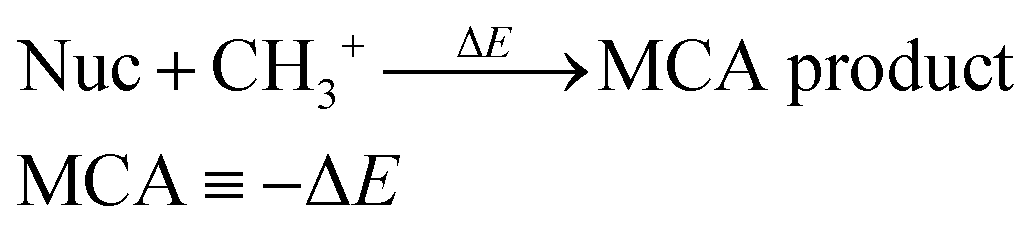 | (2) |
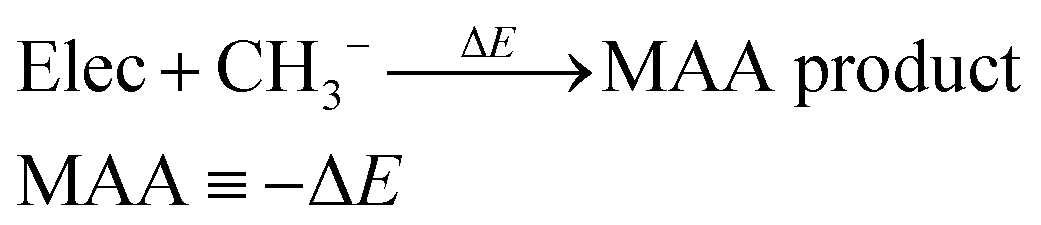 | (3) |
Datasets
The automated quantum chemistry-based workflow is tested against both experimental and higher-level QM-derived datasets. The former refers to subsets of Mayr's database,7 which were used in the initial work of Van Vranken and Baldi,15,16 and contain 90 nucleophiles and 74 electrophiles. The latter involves the two QM-derived datasets by Tavakoli et al.24 containing 1232 nucleophiles and 1113 electrophiles with calculated MCAs and MAAs at the PBE0-D3(BJ)/DEF2-TZVP COSMO(∞) level of theory. The workflow failed for two nucleophiles and one electrophile due to atomic connectivity changes during the geometry optimizations, and these are therefore excluded in the following data analysis.The workflow is also applied to two different tasks. The first task employs experimental data from the work of Caputo et al.34 and a retrosynthetic route by Manifold from PostEra35 for the synthesis of Raltegravir. The data are used to highlight the applicability of the workflow within computer-aided synthesis planning (CASP) by predicting the selectivity of chemical reactions. The second task involves reactivity data for different covalent inhibitors including 249 acrylamides, 9 propargylamides, and 238 2-chloroacetamides.13 The dataset contains calculated activation energies for the reaction of the warheads with methanethiolate (CH3S−) and FMO-derived electrophilicities at the ωB97XD/cc-pVDZ CPCM(H2O) level of theory.
Results and discussion
Comparison to experimental and higher-level QM-derived data
In Fig. 1, we present a comparison of the automated quantum chemistry-based workflow against the experimental and higher-level QM-derived data. The reference data are provided for a single atom in each molecule, so the calculated MCAs and MAAs are only evaluated for these labeled atom sites. However, such labels are not provided for the data in Fig. 1b. In this case, we use the highest calculated MAAs, although some associated sites have been compared to the molecular coordinates provided by Mood et al.15 and confirmed as the actual reaction centers.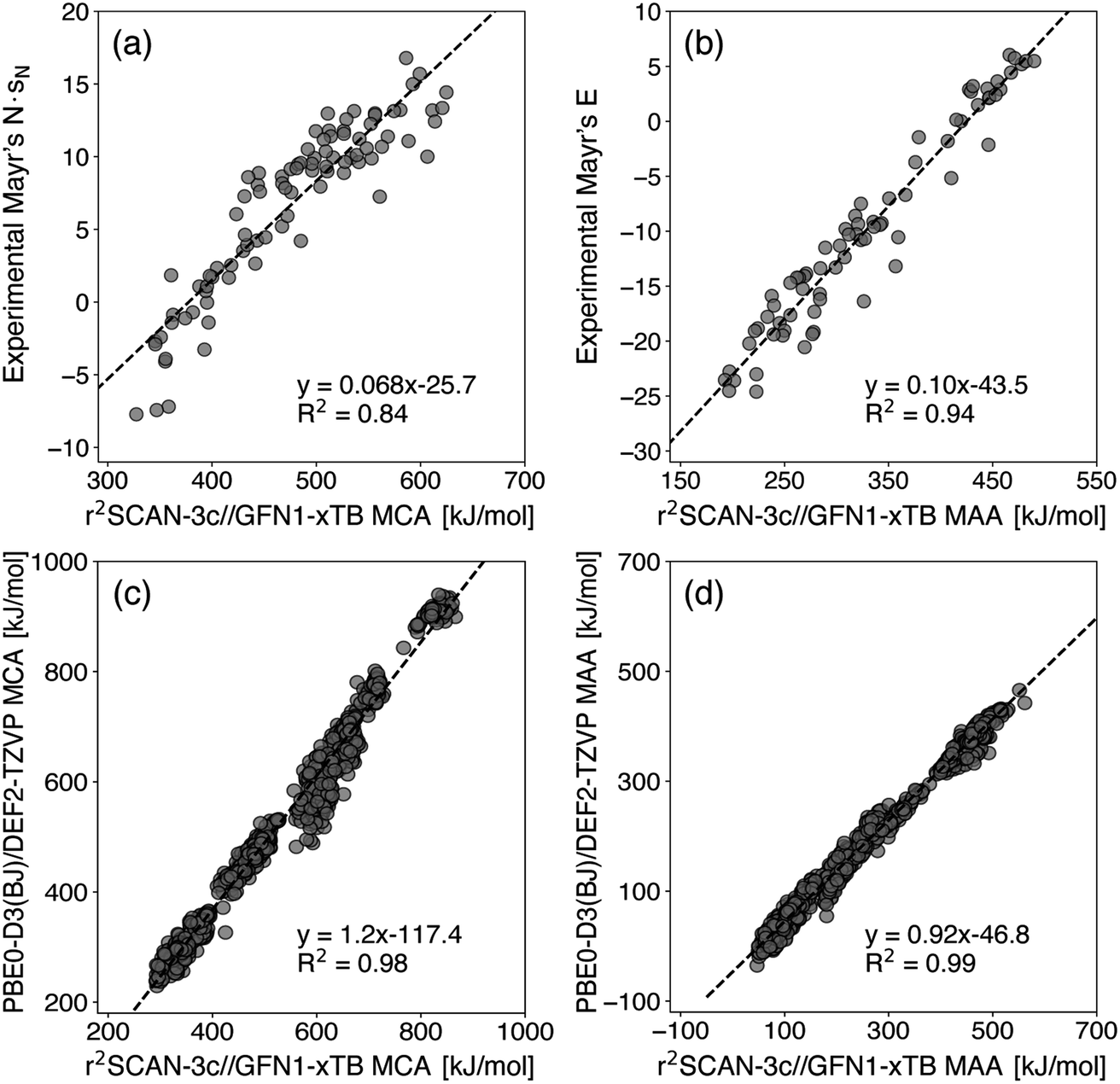 | ||
| Fig. 1 Correlation plots between (a) experimental Mayr's N × sN and calculated MCAs, (b) experimental Mayr's E and calculated MAAs, (c) calculated MCAs at different levels of theory, and (d) calculated MAAs at different levels of theory. Calculations on the x- and y-axes are performed at the r2SCAN-3c SMD(DMSO)//GFN1-xTB ALPB(DMSO) and PBE0-D3(BJ)/DEF2-TZVP COSMO(∞) level of theory, respectively. | ||
The top panels in Fig. 1 show that the workflow can reproduce a strong correlation between Mayr's experimental reactivity parameters and QM-calculated MCAs and MAAs as previously observed by Van Vranken and Baldi.15,16 Specifically, the R2 coefficients of 0.84 and 0.94 in Fig. 1a and b are somewhat similar to 0.89 and 0.96 for MCAs and MAAs, respectively, with the latter based on Gibbs free energies at the PBE0-D3(BJ)/DEF2-TZVP COSMO(∞) level of theory as reported by Van Vranken and Baldi.15,16
The ability to replicate higher-level results is further supported by the very strong correlation in the bottom panels of Fig. 1 with R2 coefficients of 0.98 and 0.99 for MCAs and MAAs, respectively. These results actually outperform the ML models by Tavakoli et al.,24 which achieved 10-fold cross-validation R2 coefficients of 0.92 ± 0.02 and 0.94 ± 0.02 for MCAs and MAAs, respectively. Of course, the better performance comes with a higher computational cost, although the median wall time for this data is less than two minutes using eight CPU cores (Intel(R) Xeon(R) CPU X5550@2.67 GHz). In fact, the timings can be further improved due to the handling of structures and conformers being embarrassingly parallel. Alternatively, omitting the single-point r2SCAN-3c SMD(DMSO) calculations will greatly reduce the computational cost without impacting the above results significantly as seen in Fig. S2 in the ESI.† It should be noted that the molecules in the bottom panels of Fig. 1 have on average ∼10 heavy atoms and ∼6 identified electrophilic and nucleophilic sites.
Post-filtering of retrosynthetic routes
To highlight the applicability of the workflow, we will first demonstrate how MCAs and MAAs can provide insights into the selectivity of chemical reactions. In Fig. 2, we show two examples of synthesizing the antiretroviral drug Raltegravir that is used to treat HIV infections. The first example is an experimentally reported procedure from the work of Caputo et al.34 and the second example is a predicted retrosynthetic route by Manifold, which is a CASP tool from PostEra.35 The most nucleophilic and electrophilic sites for each structure are highlighted in green and blue, respectively, and additional values can be found in the ESI.†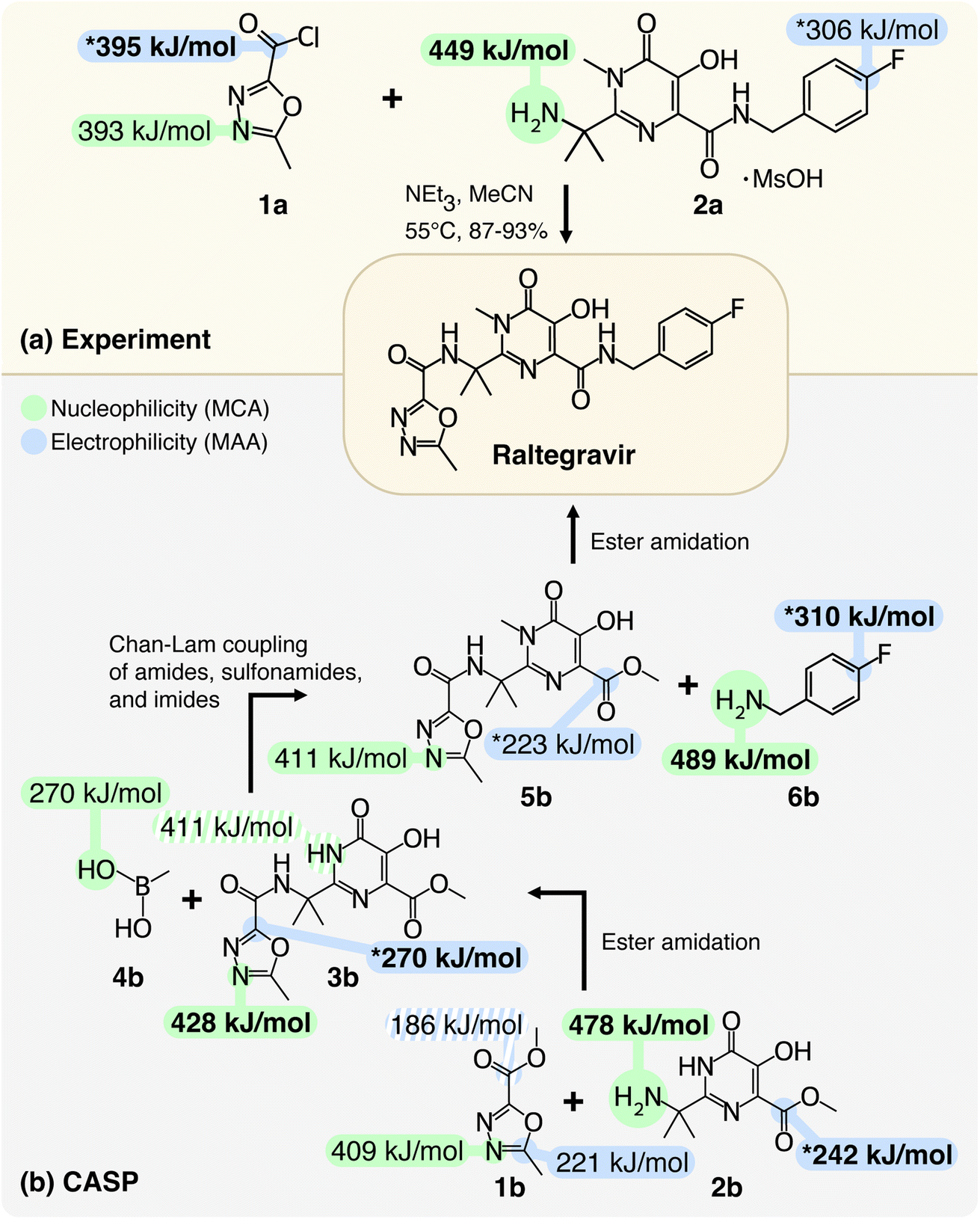 | ||
| Fig. 2 Chemical selectivity predictions for the synthesis of Raltegravir with respect to (a) experimental work by Caputo et al.34 and (b) a computer-aided synthesis planning (CASP) route by Manifold from PostEra.35 The most nucleophilic and electrophilic sites for each structure are highlighted in green and blue, respectively. The shaded highlights are predicted reaction centers with lower MCAs and MAAs, and the starred highlights are discussed in the text. The MCAs and MAAs are obtained at the r2SCAN-3c SMD(DMSO)//GFN1-xTB ALPB(DMSO) level of theory and additional values can be found in the ESI.† | ||
The results in Fig. 2a show that by locating the highest MCA and MAA among the two reactants (the values marked in bold), it is possible to predict the selectivity of the reaction. The most electrophilic site is the carbonyl carbon of 1a with a MAA of 395 kJ mol−1, which is 89 kJ mol−1 higher than the second-highest MAA. The MAA of the carbonyl carbon is marked with a star (“*”) as the chlorine atom acts as a leaving group during the geometry optimization when attaching the methyl anion to the carbonyl carbon. The most nucleophilic site in Fig. 2a is the primary amine of 2a with a MCA of 449 kJ mol−1, which is 56 kJ mol−1 higher than the second-highest MCA. Hence, the experimentally observed nucleophilic acyl substitution reaction can be correctly predicted by locating the highest MCA and MAA despite the two reactants 1a and 2a having a total of 27 nucleophilic and 20 electrophilic sites identified by the workflow.
Now, we will turn to Fig. 2b to analyze and validate the reaction steps proposed by Manifold and demonstrate how the workflow can be applied as a post-filtering tool. A CASP tool like Manifold usually outputs many different retrosynthetic routes, but some of the steps in the retrosynthetic routes may not be feasible due to selectivity issues. We propose using MCAs and MAAs as a tool to predict chemical selectivity and flag steps that are potentially incorrect. The retrosynthetic routes can then be ranked based on the number of warning flags similar to how retrosynthetic routes are commonly ranked based on the number of synthetic steps and the total price of building blocks.
The first reaction step in Fig. 2b involves an ester amidation between 1b and 2b. However, based on the highest MCA and MAA, a more favorable reaction would be an ester amidation between two 2b compounds with MCA and MAA of 478 and 242 kJ mol−1 for the primary amine and the carbonyl carbon, respectively. The latter is marked with a star (“*”) as the proton from the phenol group moves to the carbonyl oxygen during the geometry optimization when attaching the methyl anion to the carbonyl carbon. The MAA of the carbonyl carbon in 1b is 186 kJ mol−1, which is 56 kJ mol−1 lower than the highest MAA. In fact, another site in 1b has a MAA that is 35 kJ mol−1 higher than the MAA of the carbonyl carbon. This reaction step should therefore be flagged as the chance of this reaction step being a success is low. Instead, one could imagine a similar retrosynthetic route starting with a nucleophilic acyl substitution similar to the one shown in Fig. 2a by replacing 1b with 1a.
The second reaction step involves a Chan–Lam coupling reaction between the product of the first reaction step (3b) and methylboronic acid (4b). This reaction employs a copper catalyst, which makes the reaction mechanism more complex, and validating the reaction solely based on the highest MCA and MAA is probably not sufficient. However, looking at the MCAs and MAAs we see that none of the proposed reaction centers have the highest MCA and MAA. In terms of the MAAs, the workflow did not identify any electrophilic sites in 4b, and the highest MAA is therefore found in 3b. This MAA is marked with a star (“*”) as the attached proton moves to the neighboring nitrogen atom during the geometry optimization. The highest MCA is 428 kJ mol−1, which is only 17 kJ mol−1 higher than the MCA of the proposed reaction center. Removing the proton from the proposed reaction center would make it the most nucleophilic site. However, based on predicted pKa values by MarvinSketch from ChemAxon the phenol group is more acidic, and a deprotonation of this phenol group would lower the MCA of the proposed reaction center as seen in the ESI.†
The last reaction step involves an ester amidation reaction between the product of the second reaction step (5b) and 4-fluorobenzylamine (6b). The most nucleophilic site is the primary amine of 6b with a MCA of 489 kJ mol−1, and this site is also the proposed reaction center of 6b. The most electrophilic site is the highlighted carbon atom next to the fluorine atom of 6b with a MAA of 310 kJ mol−1. The MAA is marked with a star (“*”) as the fluorine atom acts as a leaving group during the geometry optimization when attaching the methyl anion to the highlighted carbon atom. The second most electrophilic site is the carbonyl carbon in 5b, which is the other proposed reaction center. This site is marked with a star (“*”) as the proton from the phenol group moves to the carbonyl oxygen during the geometry optimization when attaching the methyl anion to the carbonyl carbon.
In summary, the use of MCAs and MAAs to flag retrosynthetic steps for further inspection seems promising but will require further work. For example, the effect of using other reactivity probes than methyl anion and cation.
Covalent inhibitor reactivity predictions
A second application of the automated quantum chemistry-based workflow is the ability to predict the reactivity of covalent inhibitors. In the work of Hermann et al.,13 calculated activation energies of different warheads reacting with CH3S− were used to estimate the reactivity towards cysteine. As a faster alternative, they also showed that a warhead-associated electrophilicity index could be used to predict the reactivity of some warhead classes. The electrophilicity index (ω) is defined as36 | (4) |
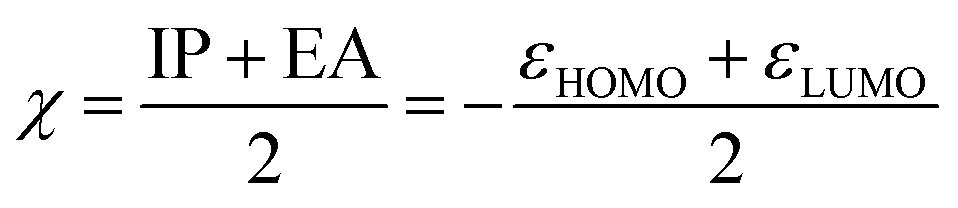 | (5) |
| η = IP − EA = εLUMO − εHOMO | (6) |
 | ||
| Fig. 3 Covalent inhibitor reactivity prediction for various acrylamides (top), propargylamides (middle), and 2-chloroacetamides (bottom). The calculated MAAs are obtained at the r2SCAN-3c SMD(DMSO)//GFN1-xTB ALPB(DMSO) level of theory and otherwise ωB97XD/cc-pVDZ CPCM(H2O). The black regression lines consider all points, whereas the blue regression lines are defined in the text. The red dots are considered outliers in the work of Hermann et al.13 | ||
In Fig. 3, we compare the calculated activation energies to the warhead-associated electrophilicity index and calculated MAAs for various acrylamides (top), propargylamides (middle), and 2-chloroacetamides (bottom). The calculated MAAs are obtained for the leftmost carbon atom in each of the depicted structures with the chlorides (Cl−) being removed for the 2-chloroacetamides.
The results of the acrylamides (Fig. 3a and b) show strong correlations for both the warhead-associated electrophilicity index and MAAs with R2 coefficients of 0.87 and 0.80, respectively. However, computing the warhead-associated electrophilicity index requires an analysis of the FMOs to select suitable orbitals, whereas the MAAs are straightforward to calculate. In fact, the R2 coefficient for the acrylamides without adjusting the electrophilicity index to the warhead-associated HOMO and LUMO energies (i.e., simply relying on the canonical MO-based HOMO and LUMO energies) is only 0.60,13 which is significantly worse than calculating MAAs.
The propargylamides (Fig. 3c and d) only include nine different structures shown in Fig. S11.† When considering all of them, the R2 coefficients are 0.64 and 0.67 for the warhead-associated electrophilicity index and calculated MAAs, respectively. Excluding the red entries in Fig. 3c, viewed as outliers in the work of Hermann et al.,13 results in a significantly better R2 coefficient of 0.89 for the warhead-associated electrophilicity index. Yet, the MAAs for these structures align well with the other entries. On the other hand, the blue entry in Fig. 3d is originally relatively far from the black regression line. This entry corresponds to a structure with bulky groups on both sides of the triple bond, and the transition vector could be pointing toward the neighboring SP-hybridized carbon atom. Unfortunately, the transition state structures are not available. Instead, we can calculate the MAA for this neighboring carbon atom resulting in a strong correlation with an R2 coefficient of 0.85. This approach is only possible for the atom-specific MAAs as the warhead-associated electrophilicity index uses FMOs primarily localized on both SP-hybridized carbon atoms.
The results of the 2-chloroacetamides (Fig. 3e and f) show no correlation for both the warhead-associated electrophilicity index and MAAs. This behavior is extensively studied in the work of Hermann et al.,13 and their arguments reflect the change from the Michael-type nucleophilic additions to an SN2 reaction. Specifically, they show that the LUMO energy correlates with the bond strength of both the C–Cl and C–SMe bonds. Thus, the electrophilicity index fails to capture the energetics of the reaction due to the simultaneous bond formation and rupture. However, the calculated MAAs behave similarly despite not depending on FMO energies, which raises questions about their reasonings. Furthermore, when using CH3S− instead of CH3−, we again find no correlation with an R2 coefficient of 0.01 as seen in the ESI.† This result is surprising as it contradicts the Bell–Evans–Polanyi principle (i.e., the change in activation energy for similar reactions being proportional to the change in reaction enthalpy), indicating that further analysis of the transition state structures should be carried out.
Conclusions and outlook
We present a fully automated quantum chemistry (QM)-based workflow that automatically identifies nucleophilic and electrophilic sites and computes methyl cation affinities (MCAs) and methyl anion affinities (MAAs) to quantify nucleophilicity and electrophilicity, respectively. The workflow shows strong correlations against experimental data from Mayr's database with R2 coefficients of 0.84 and 0.94 for the comparison of MCAs and MAAs to experimental N × sN and E values, respectively. Furthermore, the workflow achieves similar performance as higher-level PBE0-D3(BJ)/DEF2-TZVP COSMO(∞) calculations with R2 coefficients of 0.98 and 0.99 for MCAs and MAAs, respectively, while having a median wall time of less than two minutes per molecule.Additionally, we highlight two different applications of the workflow. The first application is within computer-aided synthesis planning (CASP), where the workflow can be used to predict chemical selectivity and detect potential problems in a retrosynthetic route. This is demonstrated using experimental data from the work of Caputo et al.34 and a retrosynthetic route by Manifold from PostEra35 for the synthesis of Raltegravir. The workflow correctly predicts the reported reaction from the work of Caputo et al.34 by locating the highest MCA and MAA despite the two reactants having a total of 27 nucleophilic and 20 electrophilic sites. However, some of the steps in the retrosynthetic route by Manifold are found problematic suggesting that another route should be preferred.
In the second application, we show that the workflow can be used to predict the reactivity of covalent inhibitors. We report a strong correlation between MAAs and calculated activation energies for various acrylamides and propargylamides similar to the work by Hermann et al.13 using a warhead-associated electrophilicity index. The results of the 2-chloroacetamides showed no correlation for both the warhead-associated electrophilicity index and MAAs, which could be due to errors in the calculated activation energies. The advantage of the MAAs over the warhead-associated electrophilicity index is that the MAAs are atom-specific and completely straightforward to calculate. Whereas, the warhead-associated electrophilicity index requires a selection of the highest occupied and lowest unoccupied orbitals that are associated with the warhead to match the performance of the MAAs.
Future work will use the QM-based workflow to calculate MCAs and MAAs for a large set of diverse molecules and train an atom-based ML model for each property similar to the one presented in Ree et al.37 The ML models will then be used to predict the chemical selectivity for a large set of experimentally reported reactions to provide a statical basis for using the MCAs and MAAs to predict chemical selectivity.
Data availability
The code for the automated workflow and results of the analyzed data are available at https://github.com/jensengroup/ESNUEL.Author contributions
All authors contributed to the conceptualization and method development. NR wrote all the code and performed all the calculations. All authors contributed to the data analysis. All authors read and approved the final manuscript.Conflicts of interest
The authors declare that there are no competing interests.Acknowledgements
This work was supported by Bayer AG.References
- P. Y. Bruice, Essential Organic Chemistry, Pearson, Pearson New International Edition, 2nd edn, 2013 Search PubMed.
- C. K. Ingold, Significance of tautomerism and of the reactions of aromatic compounds in the electronic theory of organic reactions, J. Chem. Soc., 1933, 1120–1127 RSC.
- G. N. Lewis, Valence and the structure of atoms and molecules, ACS Monogr.; Chemical Catalog Company, Incorporated, 1923, p. 142 Search PubMed.
- J. N. Brønsted, Einige bemerkungen über den begriff der säuren und basen, Recl. Trav. Chim. Pays-Bas, 1923, 42, 718–728 CrossRef.
- T. M. Lowry, The uniqueness of hydrogen, J. Soc. Chem. Ind., 1923, 42, 43–47 CrossRef.
- H. Mayr and M. Patz, Scales of nucleophilicity and electrophilicity: A system for ordering polar organic and organometallic reactions, Angew. Chem., Int. Ed., 1994, 33, 938–957 CrossRef.
- Mayr, H. and Ofial, A. R., Mayr's database of reactivity parameters, accessed: Mar. 08, 2023.
- M. Vahl and J. Proppe, The computational road to reactivity scales, Phys. Chem. Chem. Phys., 2023, 25, 2717–2728 RSC.
- K. Jorner, A. Tomberg, C. Bauer, C. Sköld and P.-O. Norrby, Organic reactivity from mechanism to machine learning, Nat. Rev. Chem., 2021, 5, 240–255 CrossRef PubMed.
- C. Wang, Y. Fu, Q.-X. Guo and L. Liu, First-principles prediction of nucleophilicity parameters for π nucleophiles: Implications for mechanistic origin of Mayr's equation, Eurasian J. Chem., 2010, 16, 2586–2598 CrossRef PubMed.
- K. Jorner, T. Brinck, P.-O. Norrby and D. Buttar, Machine learning meets mechanistic modelling for accurate prediction of experimental activation energies, Chem. Sci., 2021, 12, 1163–1175 RSC.
- L.-G. Zhuo, W. Liao and Z.-X. Yu, A frontier molecular orbital theory approach to understanding the Mayr equation and to quantifying nucleophilicity and electrophilicity by using HOMO and LUMO energies, Asian J. Org. Chem., 2012, 1, 336–345 CrossRef.
- M. R. Hermann, A. Pautsch, M. A. Grundl, A. Weber and C. S. Tautermann, Covalent inhibitor reactivity prediction by the electrophilicity index—in and out of scope, J. Comput.-Aided Mol. Des., 2020, 35, 531–539 CrossRef PubMed.
- C. Schindele, K. N. Houk and H. Mayr, Relationships between carbocation stabilities and electrophilic reactivity parameters, E: Quantum mechanical studies of benzhydryl cation structures and stabilities, J. Am. Chem. Soc., 2002, 124, 11208–11214 CrossRef.
- A. Mood, M. Tavakoli, E. Gutman, D. Kadish, P. Baldi and D. L. V. Vranken, Methyl anion affinities of the canonical organic functional groups, J. Org. Chem., 2020, 85, 4096–4102 CrossRef PubMed.
- D. Kadish, A. D. Mood, M. Tavakoli, E. S. Gutman, P. Baldi and D. L. V. Vranken, Methyl cation affinities of canonical organic functional groups, J. Org. Chem., 2021, 86, 3721–3729 CrossRef PubMed.
- G. Hoffmann, M. Balcilar, V. Tognetti, P. Héroux, B. Gaüzère, S. Adam and L. Joubert, Predicting experimental electrophilicities from quantum and topological descriptors: A machine learning approach, J. Comput. Chem., 2020, 41, 2124–2136 CrossRef.
- B. Lee, J. Yoo and K. Kang, Predicting the chemical reactivity of organic materials using a machine-learning approach, Chem. Sci., 2020, 11, 7813–7822 RSC.
- S. Boobier, Y. Liu, K. Sharma, D. R. J. Hose, A. J. Blacker, N. Kapur and B. N. Nguyen, Predicting solvent-dependent nucleophilicity parameter with a causal structure property relationship, J. Chem. Inf. Model., 2021, 61, 4890–4899 CrossRef.
- V. Saini, A. Sharma and D. Nivatia, A machine learning approach for predicting the nucleophilicity of organic molecules, Phys. Chem. Chem. Phys., 2022, 24, 1821–1829 RSC.
- W. Nie, D. Liu, S. Li, H. Yu and Y. Fu, Nucleophilicity prediction using graph neural networks, J. Chem. Inf. Model., 2022, 62, 4319–4328 CrossRef PubMed.
- S. A. Cuesta, M. Moreno, R. A. López, J. R. Mora, J. L. Paz and E. A. Márquez, ElectroPredictor: An application to predict Mayr's electrophilicity E through implementation of an ensemble model based on machine learning algorithms, J. Chem. Inf. Model., 2023, 63, 507–521 CrossRef PubMed.
- Y. Liu, Q. Yang, J. Cheng, L. Zhang, S. Luo and J.-P. Cheng, Prediction of nucleophilicity and electrophilicity based on a machine-learning approach, ChemPhysChem, 2023, 24, e20230016 Search PubMed.
- M. Tavakoli, A. Mood, D. V. Vranken and P. Baldi, Quantum mechanics and machine learning synergies: Graph attention neural networks to predict chemical reactivity, J. Chem. Inf. Model., 2022, 62, 2121–2132 CrossRef PubMed.
- N. Ree, A. H. Göller and J. H. Jensen, RegioSQM20: improved prediction of the regioselectivity of electrophilic aromatic substitutions, J. Cheminf., 2021, 13, 10 Search PubMed.
- N. Ree, A. H. Göller and J. H. Jensen, What the Heck? – Automated regioselectivity calculations of palladium-catalyzed Heck reactions using quantum chemistry, ACS Omega, 2022, 7, 45617–45623 CrossRef PubMed.
- RDKit, Open-source cheminformatics, version 2021.09.2, http://www.rdkit.org.
- S. Spicher and S. Grimme, Robust atomistic modeling of materials, organometallic, and biochemical systems, Angew. Chem., Int. Ed., 2020, 59, 15665–15673 CrossRef PubMed.
- S. Ehlert, M. Stahn, S. Spicher and S. Grimme, Robust and efficient implicit solvation model for fast semiempirical methods, J. Chem. Theory Comput., 2021, 17, 4250–4261 CrossRef PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Extended tight-binding quantum chemistry methods, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 11, e1493 Search PubMed.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, r2SCAN-3c: A “Swiss army knife” composite electronic-structure method, J. Chem. Phys., 2021, 154, 064103 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef.
- F. Neese, Software update: The ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- F. Caputo, S. Corbetta, O. Piccolo and D. Vigo, Seeking for selectivity and efficiency: New approaches in the synthesis of raltegravir, Org. Process Res. Dev., 2020, 24, 1149–1156 CrossRef.
- Manifold by PostEra, https://app.postera.ai, accessed: Jun. 30, 2023.
- R. G. Parr, L. V. Szentpály and S. Liu, Electrophilicity index, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef.
- N. Ree, A. H. Göller and J. H. Jensen, RegioML: Predicting the regioselectivity of electrophilic aromatic substitution reactions using machine learning, Digital Discovery, 2022, 1, 108–114 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00224a |
| This journal is © The Royal Society of Chemistry 2024 |