
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparing software tools for optical chemical structure recognition
Aleksei
Krasnov
*a,
Shadrack J.
Barnabas
a,
Timo
Boehme
a,
Stephen K.
Boyer
b and
Lutz
Weber
 *ac
*ac
aOntoChem GmbH, Blücherstrasse 24, 06120, Halle (Saale), Germany. E-mail: aleksei.krasnov@ontochem.com; lutz.weber@molgenie.com
bCollabra Inc., San Jose, CA 95120, USA
cMolGenie GmbH, Felix-Dahn-Str. 4, 70597, Stuttgart, Germany
First published on 7th March 2024
Abstract
The extraction of chemical information from images, also known as Optical Chemical Structure Recognition (OCSR) has recently gained new attention. This new interest is ignited by various machine learning methods introduced over the last years and the new possibilities to train image models for specific tasks such as OCSR. In the present paper, we have compared 8 open access OCSR methods (DECIMER, ReactionDataExtractor, MolScribe, RxnScribe, SwinOCSR, OCMR, MolVec, and OSRA) using an independent test set of images from patents and patent applications as this is an application area of general interest – precision and recall are highly desired by those who are analysing the intellectual property of chemistry patents. As a result, the used methods have shown different strengths when predicting structures from different images containing different modalities and chemistry categories. These existing methodologies for image extraction overall remain unsatisfactory, indicating a need for further advancements in the field. Further, we have created a machine learning image classifier, classifying images into one out of four image categories and applying the best performing OCSR method for each category. This classifier, the image comparator tools, and datasets have been made available to the public as open access tools.
Introduction
Chemists very often communicate their scientific findings and knowledge by using images containing chemical structures instead of a pure textual description of their work. Whilst describing chemistry in text by using chemical nomenclature based names is a common standard in publishing, these names are often rather long for complex structures and therefore hard or slow to recognize even by trained chemists. As an alternative, readers will recognize depictions of those chemical structures or reactions much faster.Unfortunately, and to our best knowledge, only very few scientific journals or patent offices deliver the structures of published compounds in a computer readable chemical structure format. For example, the US Patent office is the only patent office that extracts and makes available chemical structures or chemical reactions as ChemDraw CDX as well as MDL Information Systems MOL files from respective patent images.
Nevertheless, chemical patents form the basis of the chemical and pharmaceutical industry – either claiming novel substances or their applications. It is therefore of the highest interest for such companies and national patent offices to collect all previously claimed or mentioned chemical structures in patents and publications to allow for novelty checks and freedom to operate opinions. The conversion of chemical names as found in text or chemical structures in images by specialised algorithms is used by several companies to create structure and substructure searchable content for the majority of scientific publications and patent documents. Whilst the conversion of text images using optical character recognition (OCR) into chemical rule based names, trivial names or hybrid names and finally into chemical structures from the resulting OCR text is an established procedure, but it is not the topic of this present work. In contrast, the conversion of images to chemical structures (optical chemical structure recognition, OCSR) still represents a significant challenge for established software tools like Kekulé, CLiDE, OSRA and others described by Rajan et al.1
In the recent past, the development of artificial intelligence (AI) such as transformer based machine learning tools has ignited a novel interest in image processing and the creation of generative models that lead to the rapid development of novel applications for predicting chemical structures from their images. Such AI based image-to-structure methods are MolScribe2 and RxnScribe,3 DECIMER,4 ReactionDataExtractor,5 Img2Mol,6 SwinOCSR7 and OCMR8 that have recently become available and that were shown to outperform previous rule based, analytical methods both in recall as well as in precision.
Nevertheless, older rule based, analytical algorithms like OSRA9 which was developed originally at the National Cancer Institute of the USA, followed later by a similar method called MolVec10 are constantly updated. OSRA is therefore still used by many groups and is also used by our company to extract chemical compounds and reactions from patent images of WO, EU, and US patents. These compounds are integrated into the search engine of the open access SciWalker application11 which implements a comprehensive chemical substructure search in preprints, publications, and patents.
It was therefore of great interest for us to evaluate which of those novel methods could replace our approach using the rule based OSRA. The mentioned recent publications on those AI methods provide performance data, comparing precision and recall. For example, in Qian et al.2 besides Img2Mol, SwinOCSR, OSRA, and MolVec also MSE-DUDL, ChemGrapher, Image2Graph were compared to MolScribe's structure prediction quality, all showing considerably lower performance. In the present work, we are comparing our current tool OSRA with MolScribe, RxnScribe, DECIMER, ReactionDataExtractor, SwinOCSR, and the most recently published OCMR8 which became available after our ChemRxiv preprint.12 MSE-DUDL, ChemGrapher, Image2Graph are not available publicly which was a requirement for our evaluation. Img2Mol was not evaluated as it does not predict stereochemistry and was extensively characterised in ref. 2 as being considerably inferior to MolScribe.
However, since the quality of any image to structure conversion is heavily dependent both on the image quality and its content modalities, we felt that we needed to develop and use our own task oriented dataset of images to perform an independent qualitative analysis on images from various patents. We also have created two new software tools to facilitate multi-curator quality analysis of OCSR predictions – a Java based tool “ImageComparator”13 to compare reactions and multistructure images as well as a Python based script “ExcelConstructor” that allows create Excel sheets for fast quality analysis of single molecules.14 Both ExcelConstructor and ImageComparator are described and available for download in the supplementary material. To convert predicted SMILES15–17 to images for manual inspection, the open access chemistry package CDK18,19 was used. Thus, the developed methods were created and designed to allow a faster manual inspection of the prediction results – they do not influence the respective quality criteria. Other interested users may take advantage of those when performing their own OCSR quality control efforts.
To this end, we manually selected 2702 images from patents and patent applications to contain chemical structures of different types, chemical reactions as well as images that do not contain interpretable chemical structures. This dataset is a new, independent test set for image-2-structure recognition methods that provides heterogeneous data including images with different resolutions and different chemistry content types. The dataset is not intended to represent all available images containing chemical structures but is rather inspired by our everyday task of identifying chemicals in patent images. Thus, each of the selected images was in the centre of interest for one of our chemistry clients – both from pharma as well as from chemistry – comprising a mixture of small to medium sized molecules, from inorganic complexes up to peptidic structures and typical heterocyclic structures. In most cases, these images were found to pose problems with a correct structure prediction using our OSRA tool. Thus, this image collection is rather an ad hoc collection of molecules with different modalities instead of a systematic collection following clearly defined principles.
Whilst the US patent office provides complex work units (CWU) with high, sufficient resolution images, especially older EP or WO patent images are often not of high quality. However, since much of the novel intellectual property often appears first as a WO application, those lower quality images are of specific interest to chemists in the industry.
An example from WO-2016199761-A1 represents a reaction as shown in Fig. 1. The reaction product is a plant disease control agent of interest for the agrochemical community and described in PubChem as CID 140317046, provided by the WIPO to PubChem but without reference to the respective patent. It is also part of SciWalker's chemistry compound database with its identifier OCID 190138015958 but not found as a compound in WO-2016199761-A1. Similarly, it was not found in Google Patents.
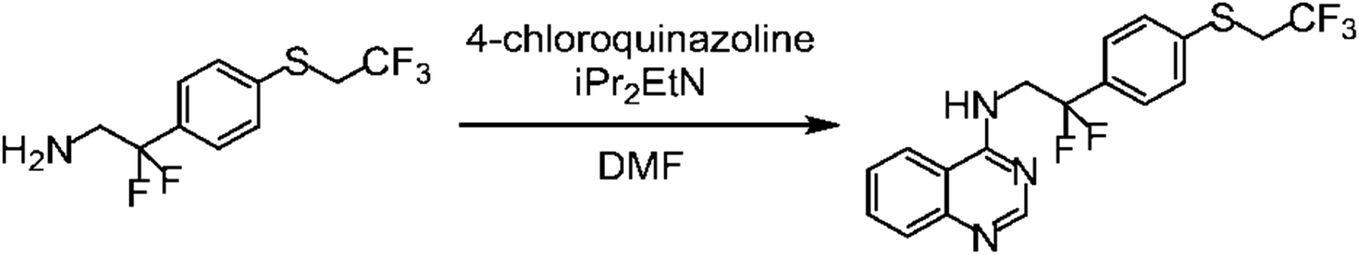 | ||
| Fig. 1 Image JPOXMLDOC01-appb-C000040.tif from WO-2016199761-A1. | ||
In another example from the United States Patent Office (USPTO), US-08680111-B2 is a patent of high interest for drug discovery – this Pfizer patent describes novel compounds that inhibit anaplastic lymphoma kinase (ALK) and was published in 2014. Its CWU files contain 1 sequence listing of a 13-mer peptide KKSRGDYNTMQIG in XML format, in total 238 TIF image files, as well as 234 Chemdraw CDX files and 234 MDL MOL files. In addition, 1 drawing with a crystal structure and 3 not interpretable image files are found. The chemical structure files were created by the USPTO from the images originally as Chemdraw files and then exported also as MDL MOL20 files.
Classifying those 238 images manually, 49 contain Markush like structures (for an example see Fig. 2) or collections of substituents or scaffolds as part of the claimed compounds.
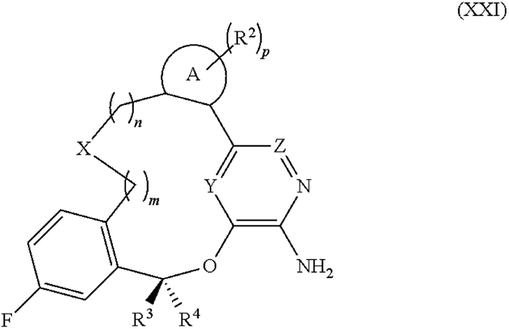 | ||
| Fig. 2 Image US08680111-20140325-C00029.TIF from US-08680111-B2. | ||
At the current development stage of image-2-structure conversion it is not yet possible to extract meaningful chemistry information from such images of Markush type structures.
Further, 187 images contain single or multistep reaction sequences that lead to the exemplified and claimed compounds of this patent. The claimed compounds are macrocyclic compounds with images that are most likely not very easy to translate into chemical structures. An example is shown in Image S1 of the supplementary material.14 The final reaction product of the synthesis sequence shown in S1 was published by PubChem as CID 89807863 and was found in a total of 8 patent documents. The underlying SureChEMBL (https://www.surechembl.org/search/) workflow uses, according to our information, the commercial program GLiDE21 for image-2-structure extraction. The same compound is also found by our SciWalker structure extraction, registered as OCID 190067469284 in 6 related patent documents, e.g. the related EP-2822953-B1 grant but not in application EP-2822953-A1 and grant EP-2822953-B9. Using OSRA version 2.1.3 to extract both reactions and compounds for SciWalker in production, we did extract 1539 unique chemical structures for EP-2822953-B9 (https://sciwalker.com) automatically. However, the OCID 190067469284 compound was missing among them.
We feel it is important to emphasise that this present work was not intended to investigate the underlying methodological reasons for specific strengths and weaknesses of each OCSR tool. In contrast, we were more interested in identifying an improved overall process for image-2-structure recognition to deliver improved compound and reaction information in SciWalker and databases such as Google Patents or PubChem.
Experimental
Image dataset
A dataset of hand selected 2702 images from patents and patent applications has been manually separated into 3 buckets (Table 1) and is available in the supplementary material.| Bucket | Image content | Number of images | Applied software |
|---|---|---|---|
| A | Single chemical structure | 1454 | Decimer, MolScribe, Molvec, OCMR, SwinOCSR and OSRA |
| B | Multiple chemical structures | 661 | Decimer, MolScribe, OCMR, SwinOCSR and OSRA |
| C | Single and multistep chemical reactions | 481 | ReactionDataExtractor, RxnScribe, and OSRA |
Image-to-structure software
Decimer v2.4.0, ReactionDataExtractor v2.0.0, MolScribe v1.1.1, RxnScribe v1.0, MolVec v0.9.8, OCMR, SwinOCSR, and OSRA version 2.1.5 have been downloaded and installed according to instructions as described in the original publications. These programs have been applied as meaningful to the images from the different buckets, as listed in Table 1.Quality control and scoring
The range of different chemistry modalities of interest as used in patent depictions is very broad – ranging from using text based descriptions such as peptide sequences or typical text abbreviations of solvents or reagents, up to partly hand drawn structures or combining structures together with depictions of polymer resins, biomolecules or other materials. We explicitly have to emphasise that the investigated OCSR methods were not able to extract correct chemical information from those modalities. If found in an image of our dataset they are typically rated as wrong prediction, resulting in a decreased precision and F score.Four chemists were involved in independent quality control procedures. We have used a simple scoring scheme: when the structures were correctly predicted, a score of 1 was given, otherwise, it was set to 0. Precision, recall, and F1-score were calculated as:
| Precision = TP/(TP + FP) × 100% |
| Recall = TP/(TP + FN) × 100% |
| F1 = 2 × (precision × recall)/(precision + recall) = 2 × TP/(2 × TP + FP + FN) |
Our evaluation method has used only an exact match of chemical structure connectivity tables as a scoring criterion – which is the same as used in the extensive evaluation provided by the MolScribe paper.2 Other publications have used the same exact prediction and the Tanimoto similarity as an additional metric.4,7 SwinOCSR also uses BLEU and ROUGE7 which are N-gram based precision methods developed for machine translation. OCMR has used the Levenshtein distance between the predicted SMILES string and the ground truth SMILES to quantify the dissimilarity of the predicted SMILES.8 The use of any such similarity metric like Tanimoto, Levenshtein, BLEU, or ROUGE is in our opinion not useful when one needs to perform a novelty check on a given molecule from any document, since any similar but not exactly the same molecular structure would not affect its novelty.
To compare results for bucket A we leveraged ExcelConstructor. In the created spreadsheet, the original image and the images generated from the predictions for each method are available in one row to facilitate quality control for the chemists (Fig. 3).
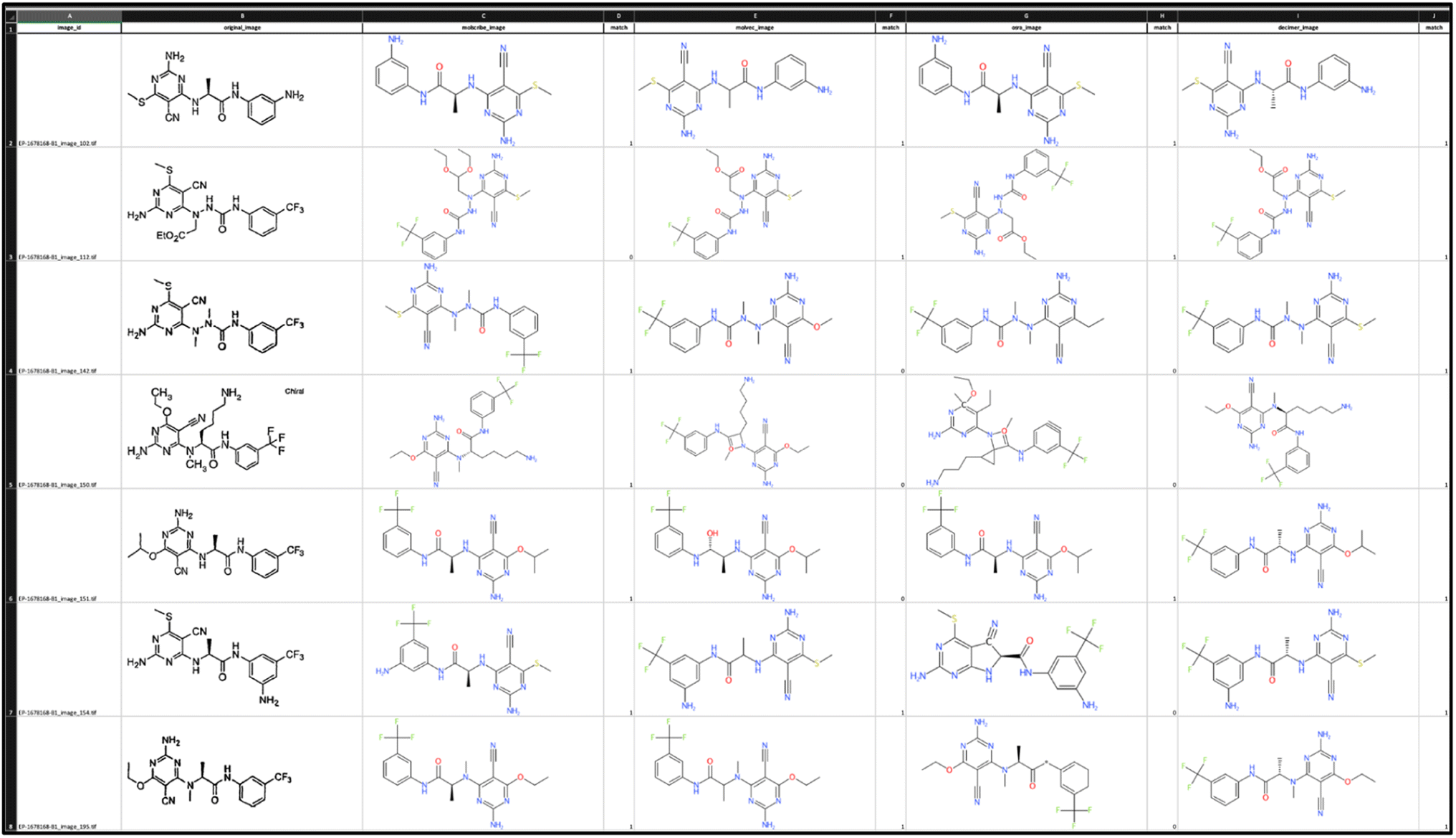 | ||
| Fig. 3 Excel file to compare and score the structure predictions of the various OCSR tools. | ||
To qualify single molecules, the Excel sheet is a good way but fails for multi structure (bucket B) and reaction images (bucket C) as they become very crowded. Therefore, a Java program ImageComparator was written.13,14 ImageComparator reads a CSV file with the image location in the first column and the respective SMILES15–17 output in the second column to generate a tabular view of input images and predicted structures. These table rows can be inspected and given a score that can be stored for later control, see Fig. 4.
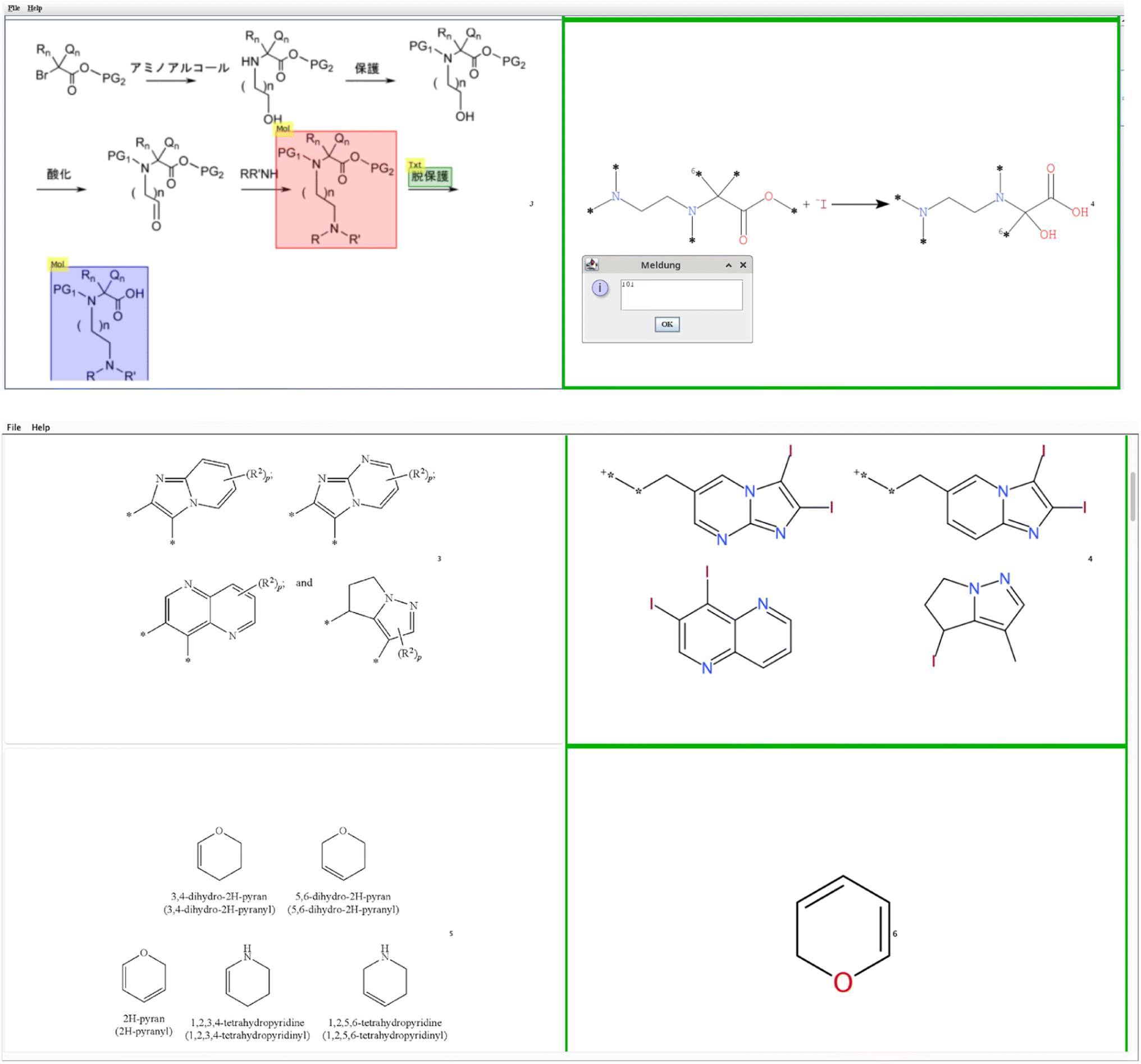 | ||
| Fig. 4 ImageComparator user interface to compare the original image with the predicted reactions (here a RxnScribe example is shown) or multiple chemical structures below (OSRA example). | ||
Results
Scoring results for the different predictions from the curators were checked for discrepancies and jointly corrected to achieve a commonly accepted result. Although it would be possible to introduce a graded scoring, for example by using a Tanimoto similarity score between the predicted and the correct chemical structure, we have preferred to use a simple metric which only accepts 100% correct predictions for the reasons outlined above.Thus, Table 2 contains the scoring results of 400 single structure images. These 400 images were randomly selected from bucket A.
| Method | TP | FP | FN | TP + FP + FN |
|---|---|---|---|---|
| SwinOCSR | 253 | 135 | 12 | 400 |
| OSRA | 256 | 144 | 0 | 400 |
| MolVec | 298 | 102 | 0 | 400 |
| OCMR | 308 | 92 | 0 | 400 |
| Decimer | 337 | 63 | 0 | 400 |
| MolScribe | 348 | 52 | 0 | 400 |
Whilst a true positive (TP) score was given for a correctly predicted structure, a false positive (FP) was a wrongly predicted structure. A false negative (FN) hit corresponds to an image where the software did not predict a chemical structure. For example, FN values are considered to be: missed predictions, none/NaN, ‘〈invalid〉’, ‘false’ values, and possible errors during the run of the OCSR program. A few false negative predictions were found in the complete set of predicted structures of bucket A (see supplementary material). However, in the evaluated sample set of 400 images, there were no FN, except for SwinOCSR, which had 12 FN values. From the numbers in Table 1, we have calculated the precision, recall, and F-score values as shown in Fig. 5.
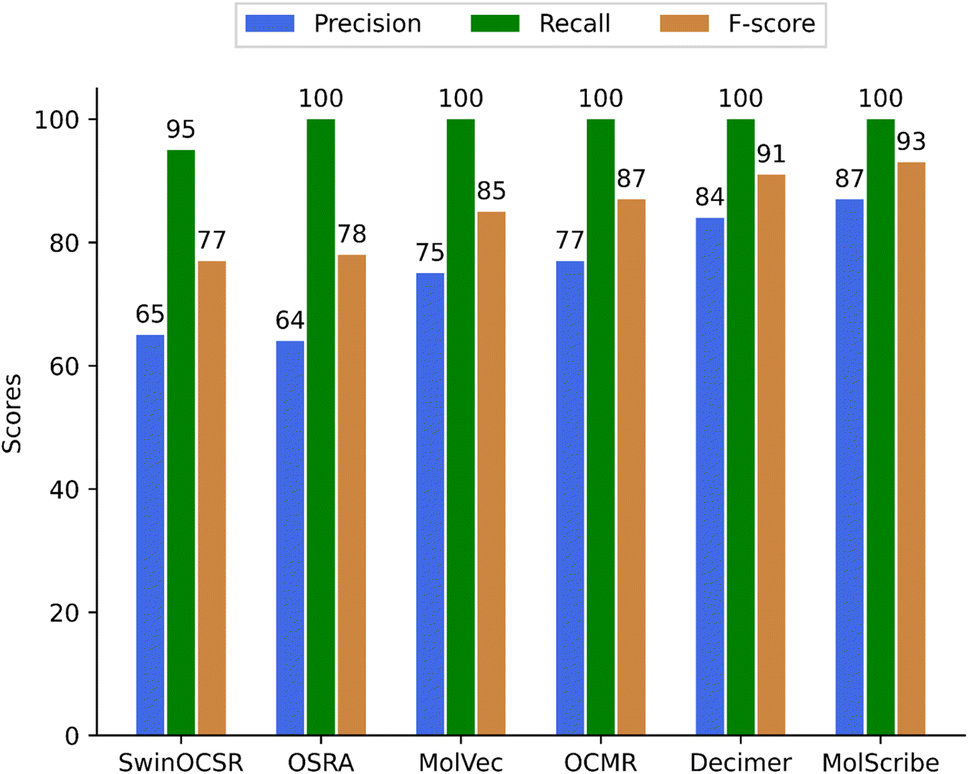 | ||
| Fig. 5 Precision, recall, and F-score of 6 different OCSR methods to predict single structures. | ||
For images with multiple structures, the situation is clearly less favourable. For this use case, OSRA outperformed all other AI based programs which is understandable since they were all trained on single structure images. For the multi structure prediction mode, OSRA has some in-built default limitations like the number of atoms in a single structure in such an image needs to be greater than five in order to be predicted, smaller compounds are neglected. For example in US-08680111-B2_image_702.TIF (Image S2 in the supplementary material) we see 20 five-membered rings and 4 six-membered heterocyclic rings systems – from those only the 4 six-membered compounds were predicted correctly.
For multi-structure images, we evaluated only a smaller fraction of structure predictions due to an overall poor prediction (Table 3). The precision was calculated as follows – for example, if three structures were correctly predicted from a total number of six predicted structures from such an image the precision is 50%. If only five out of 6 structures were predicted, the missing was counted as a false negative (Fig. 6). Among tested OCSR tools OCMR and SwinOCSR programs show the lowest metrics for multi-structure images and will not be discussed further (see the supplementary material). If better results will be obtained for such multi structure image modalities in the future, a more in depth quality assessment is indicated.
| Method | TP | FP | FN | TP + FP + FN |
|---|---|---|---|---|
| OSRA | 66 | 48 | 32 | 146 |
| Decimer | 59 | 204 | 23 | 286 |
| MolScribe | 38 | 92 | 26 | 156 |
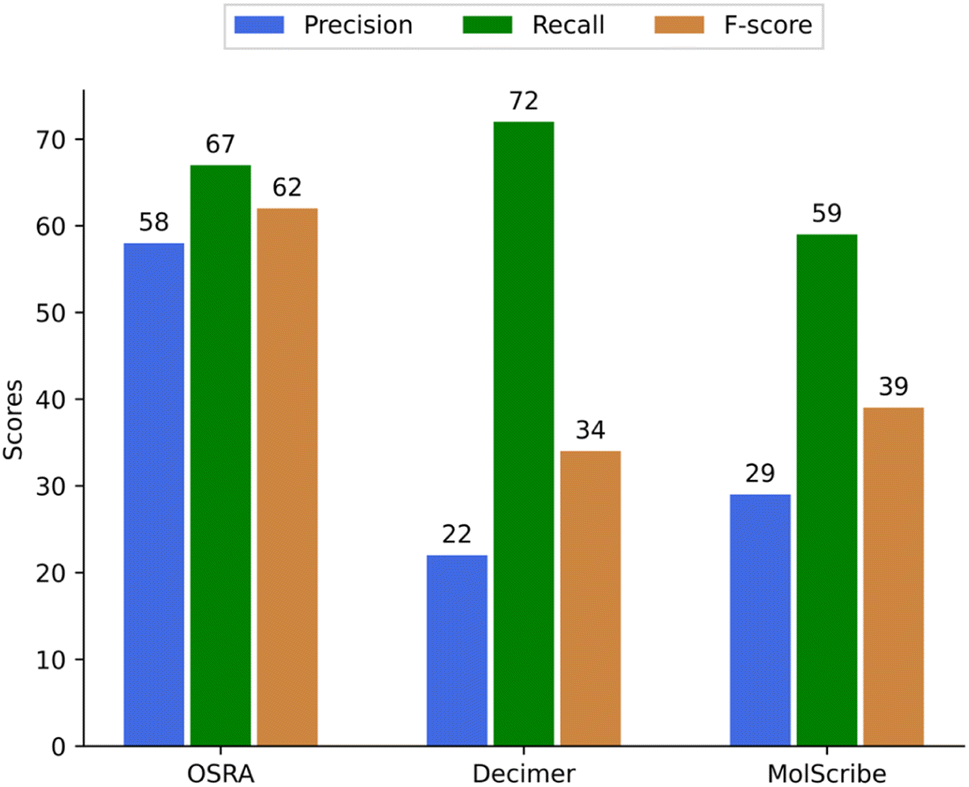 | ||
| Fig. 6 Precision, recall, and F-score of OSRA, Decimer, MolScribe for predicting multiple structures (for OCMR and SwinOCSR seesupplementary material). | ||
103 randomly selected reaction images were selected that contained 284 reactions or reaction steps in total for evaluating the quality of predicting reactions (Fig. 7). To compare predictions from OSRA, RxnScribe, and ReactionDataExtractor we have disregarded the output of all three programs for reaction reagents that are typically shown above or below the reaction arrow – these are often a mixture of text and chemical structure images. Thus, although not qualifying the correctness of such reagent extraction, both RxnScribe and ReactionDataExtractor were found to be able to extract more reagents than OSRA could (Table 4).
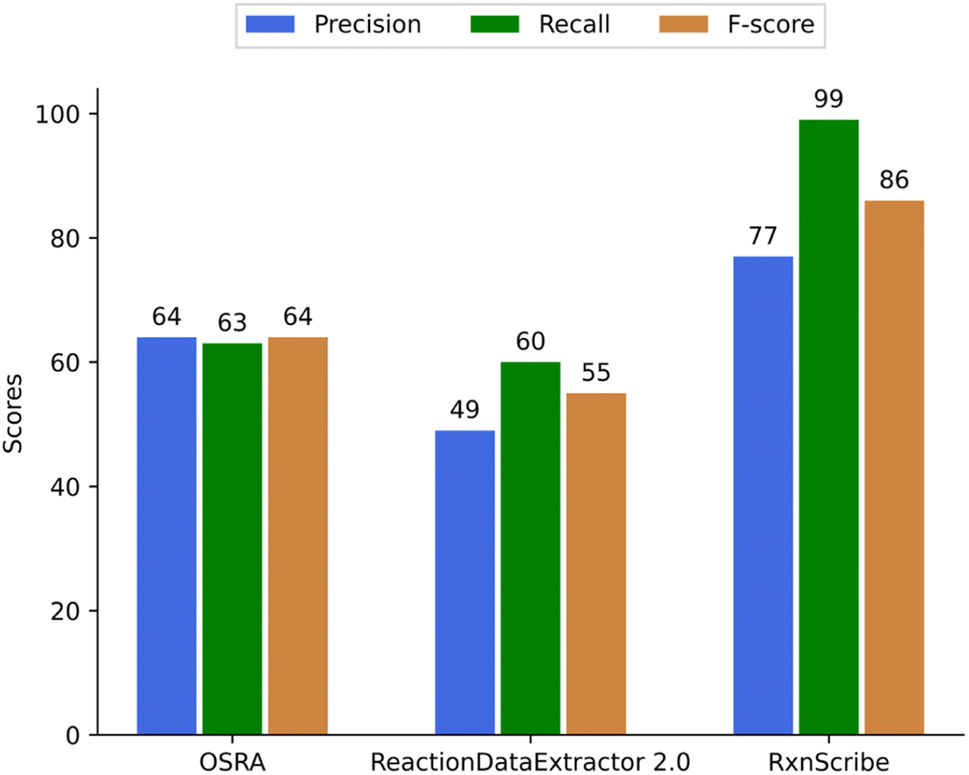 | ||
| Fig. 7 Precision, recall, and F-score of OSRA, RxnScribe, and ReactionDataExtractor to predict reactions. | ||
| Method | TP | FP | FN | TP + FP + FN |
|---|---|---|---|---|
| OSRA | 135 | 75 | 74 | 284 |
| ReactionDataExtractor 2.0 | 107 | 111 | 66 | 284 |
| RxnScribe | 219 | 67 | 6 | 288 |
The approach to obtain TP, FP, and FN values for reactions has been the same as for the single molecule images. It is interesting to note that RxnScribe has predicted more than 284 reactions; this seems to happen as any detected arrow is generally predicted as a chemical reaction.
The respective quality control files for Tables S1–S3 are found in the supplementary material.
Discussion
In general, the quality of structure predictions from images is highly dependent on the structure and image modalities of the selected image test set. There is no commonly accepted general or standard set of images for such quality measurements. Instead, we have selected a set of patent images that were of general interest to the chemical and pharmaceutical industry. Different test sets will produce different precision and recall results.Single molecules
In summary, our study showed that each of the methods has achieved a high recall rate of close to 100%, indicating that the methods were able to predict molecules for each or most of the chemistry containing images of small organic molecules. However, their precision rates varied considerably, with MolScribe achieving the highest precision at 87%, followed by DECIMER at 84%, MolVec at 74%, and OSRA at 64%. The F-scores also have reflected these trends, with MolScribe having the highest F-score at 93% and SwinOCSR having the lowest at 77% (see Fig. 5). These findings highlight the strengths and limitations of each method in accurately predicting molecules from images.Judging the quality of compound structure prediction is a complex task – we agreed on the following stringent scoring method using the following scoring principles:
• A compound structure was considered to be correct when all atoms, their valencies, and bonds were recognized correctly.
• Abbreviations of superatoms had to be recognized correctly.
• A variable group like R had to be translated into any heavy atom or group * for SMILES.
• Charge of the structure had to be recognized correctly.
• Correctness of stereochemistry prediction was not considered a scoring criterion.
Besides these overall results, we would like to stress certain other qualitative image modalities below that are relevant for a successful prediction and prediction quality.
All methods presented have been trained with small molecules that are organic small molecules. Therefore, in the following we would like to mention further properties of small molecules that have been captured correctly or not during a structure prediction.
The current data model of SMILES does not implement dative bonds – with the exception of the newest RDKit24 using a non-standard “→” arrow to designate dative bonds. Since MolScribe and OSRA are able to create MOL files as predictions, it would be possible to set a bond type to a dative bond in the output MOL files. Unfortunately, none of the evaluated OCSR tools were able to recognize those metal complexes and their coordinative bonds at a satisfactory level. To a certain degree, OSRA could extract dative bonds – OSRA version 2.1.5 has been developed aiming at generating MOL files that contain such bond types. When analysing such images of coordination complexes with MolScribe, conventional single bonds between the metal and the donor nitrogen atoms were predicted, increasing the charge for the nitrogen atom to +1 with its dative bond (see Fig. 8). In the SDF file prediction using OSRA four dative bonds were predicted, it remained unclear why nickel got a charge of +1. Also, with the same image a frequently occurring error of MolScribe was observed – converting nitriles to isonitriles which can be attributed to its novel logic of interpreting superatoms.
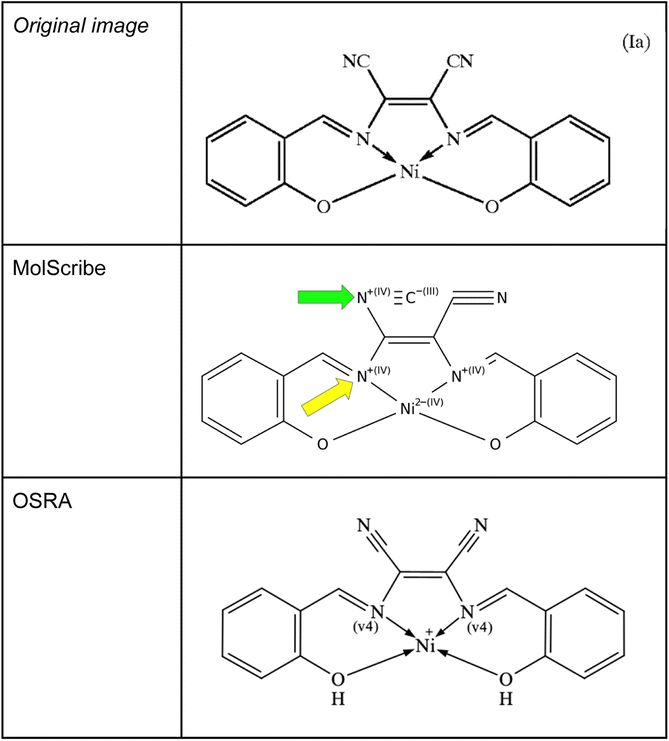 | ||
| Fig. 8 A common transition metal complex Ia from US-7001437-B2 above and MolScribe's prediction below. For MolScribe – green arrow: isonitrile group instead of nitrile group, yellow arrow: wrongly predicted valency of 4 at the nitrogen atoms participating in a dative bond. In contrast, OSRA is interpreting the dative bonds between the nitrogen atoms and nickel correctly but the ionic bonds between the oxygens and the metal are not understood. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CC(CC1[R3])[R1])[R2] whilst the right was incorrectly predicted as C1C(CC(CC1[Al])[R1])[Al]. MolScribe has recognized the left correctly as [1*]c1cc([2*])cc([3*])c1 but fails also with the right hand structure, predicting *c1cc(*)cc([1*])c. OSRA delivers *c1cc(cc(*)c1)* for the left and Nc1cc(*)cc(*)c1 for the right structure. These above labelled R-group Markush-type SMILES are not standard SMILES but dedicated extensions in CDK and RDKit. It is worthwhile to mention that MolScribe generates a standard MOL file which contains standard R1, R2, and R3 definitions. More should be possible in future methods when implementing version V3000 extended RGfiles (Rgroup files) formats. Thus, in order to correctly recognize simple Markush structures in the future it would certainly be needed to create a larger training set of such simple Markush structures. The development of dedicated models would open the avenue to also automatically enumerate simple Markush structures in the text and tables of patents.
C(CC(CC1[R3])[R1])[R2] whilst the right was incorrectly predicted as C1C(CC(CC1[Al])[R1])[Al]. MolScribe has recognized the left correctly as [1*]c1cc([2*])cc([3*])c1 but fails also with the right hand structure, predicting *c1cc(*)cc([1*])c. OSRA delivers *c1cc(cc(*)c1)* for the left and Nc1cc(*)cc(*)c1 for the right structure. These above labelled R-group Markush-type SMILES are not standard SMILES but dedicated extensions in CDK and RDKit. It is worthwhile to mention that MolScribe generates a standard MOL file which contains standard R1, R2, and R3 definitions. More should be possible in future methods when implementing version V3000 extended RGfiles (Rgroup files) formats. Thus, in order to correctly recognize simple Markush structures in the future it would certainly be needed to create a larger training set of such simple Markush structures. The development of dedicated models would open the avenue to also automatically enumerate simple Markush structures in the text and tables of patents.
 | ||
| Fig. 9 Two simple Markush structures. | ||
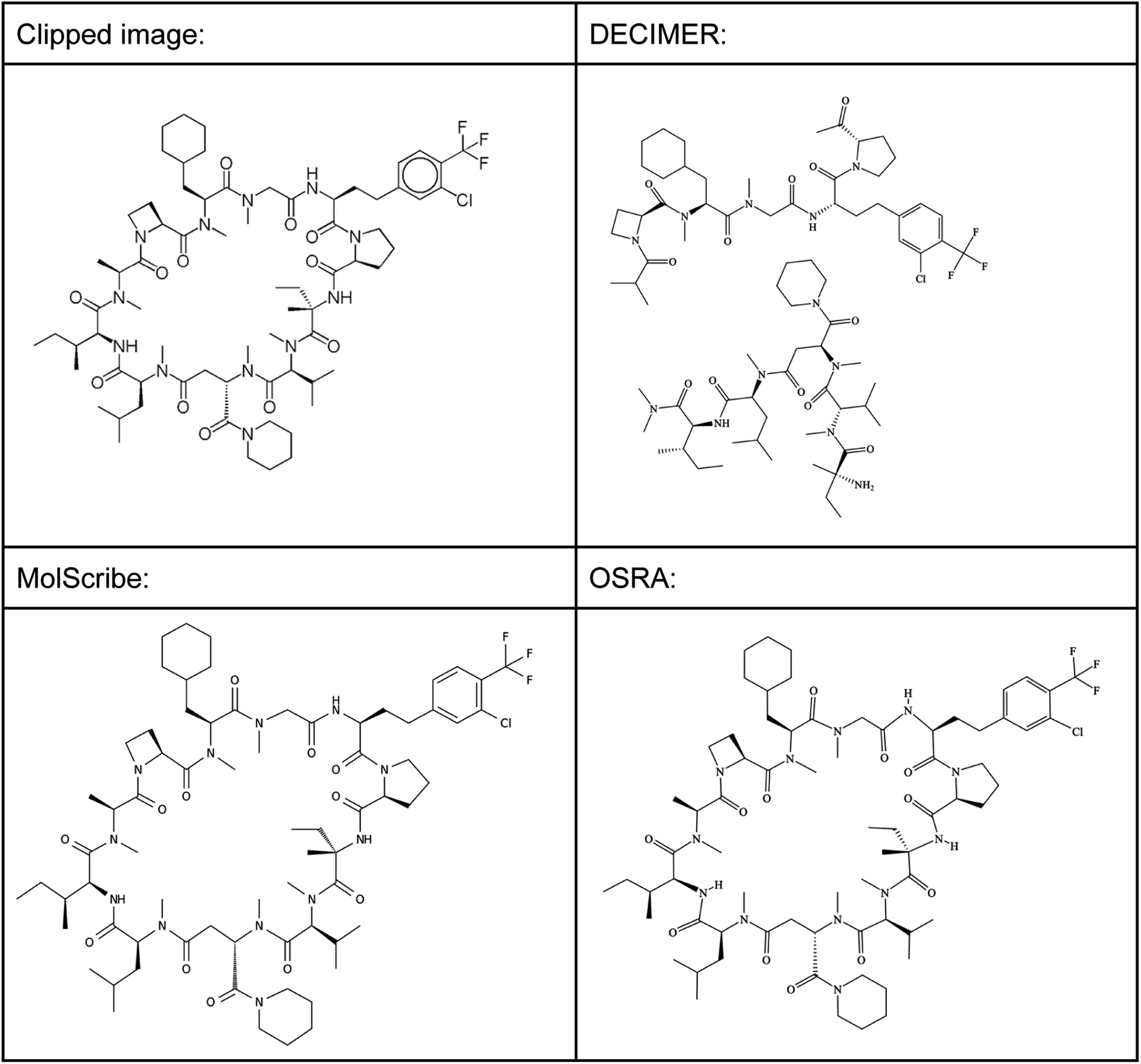 | ||
| Fig. 10 Prediction of large cyclic peptides. | ||
This structure has a molecular weight of 1416.18 Dalton, which means that also larger small molecules can be predicted well using MolScribe and OSRA. Both programs produce MOL file output formats with 3D coordinates that try to mimic the original image – a useful feature that allows a faster comparison of prediction results. However, when doubling the size of the molecule by creating a 2830.34 Dalton large dimer of the shown cyclic peptide (see supplementary material, Image S5), MolScribe throws an error while OSRA still predicts the respective structure well. MolScribe is resizing the image to 384 × 384 resolution for both training and inference, we suspect that this will reduce the needed information to a degree that large molecules will not be predicted correctly as a consequence.
In many cases however, the 2D structure of sugars, peptides, proteins, or oligonucleotides is described by letter codes for sugars, amino acids, or nucleotides and not with full structure image representations. Thus, in US patent documents we find special sequence files in complex work units (CWU) files. For example, RGD stands for the tripeptide H-Arg-Gly-Asp-OH or arginylglycylaspartic acid. However, none of the present OCSR tools were able to recognize such chemistry from images that contain such sequence codes. Instead, rule based systems together with optical character recognition are currently enabling the extraction of such polymeric structures from text but are outside the scope of this work.
Multiple molecules
All OCSR programs had difficulties achieving good results when predicting structures from multi structure containing images as evidenced by Table 2. This is not surprising as both DECIMER and MolScribe have been trained on images containing only single molecules.One question is how one should treat multiple molecular structures in one image – is it a substance where the image describes the composition of a substance or are the structures to be recognized as separate molecules? For example, MolScribe generates dot separated SMILES if it finds multiple compounds in an image. This is problematic as dot separation of chemical entities in SMILES typically represents different parts of a compound mixture if not connected via linking atom labels. Such mixtures are typical salt forms containing the cation and the anion separately.
Also, looking at the resulting SMILES from MolScribe, it had generated some obviously wrong or meaningless or hallucinated results like SMILES that consist of a series of asterisks, e.g. from EP-1678168-B1_212.tif *·*·*·*·*·* as a series of 6 asterisks. Cumulative asterisks were generated from EP-1678168-B1_423.tif with an output of:
| **·*O·[H]C(NO)C1C(SC)NC(N)NC1NCC(O)NC1CCCC(C(F)(F)F)C1 |
When trying to convert predicted SMILES into InChI or InChI keys,23 further problematic asterisk situations are discovered, for example atoms containing [*H], [Cn] or [*+]. Therefore, it seems advisable to include some sanity checks when using predicted chemical structures, filtering out invalid SMILES and chemical structures with hypervalent atoms or wrong isotopes. At the current stage of OCSR development, this task is left to the user of such tools.
Also, a simple conclusion is that retraining of structure predictions from multiple structures on an image is definitely needed. This is a rather unfortunate finding, as in patents multi structure images are found quite often. So far the best available method for this modality remains OSRA, provided we also have applied the error filters described above.
Reactions
Three methods were available to predict reactions from images: OSRA, RxnScribe, and ReactionDataExtractor 2.0. It is generally accepted that judging the quality of reaction prediction is a complex task – we have agreed and applied a simple, less stringent scoring method using the following principles:• A compound structure was considered to be correct when applying the rules for recognizing single structures as mentioned above.
• The reaction always needs to have at least one correct starting material and one correct product.
• The starting material(s) and the product(s) need to picture the main features/reacting atoms of the reaction.
• Reaction conditions are not required for correctness and were omitted in the evaluation.
• When small but wrong hallucinated single atoms were predicted as reactants or products, for example, C as a single carbon atom or Y like a yttrium atom, the reaction was still assumed to be correct provided larger reactants and products were present and the other criteria were met. Our reasoning for this rule was that one could potentially remove those single atoms in a post-prediction step with a rule based approach.
• If a reagent from reaction conditions (placed over the reaction arrow) was recognized as a reactant, the reaction was still considered as correct.
• Stoichiometry was not considered to be a required criterion.
• The recognition of complete multi step synthesis schemes is not required.
The following explicit exclusion criteria were applied and corresponding reaction predictions were assigned a score of 0:
• No reactants and/or products are recognized.
• A formally incorrect SMILES was created.
• Product is recognized as a reactant and vice versa.
• When in a multistep reaction a reactant or product from a different reaction step was used as a reactant or product in a given reaction step.
It was interesting to see that sometimes the depicted reactions input was wrong – as per an error of the chemist drawing the reaction arrow in the wrong direction – but the RSMI extraction of this incorrect content was still formally correct. For example, in image US-08680111-B2_765 (Fig. 11) – the direction of the reaction in the image is wrong. Also, the last reaction in this patent image is erroneous but the extracted reaction is correct and was therefore given a score of 1.
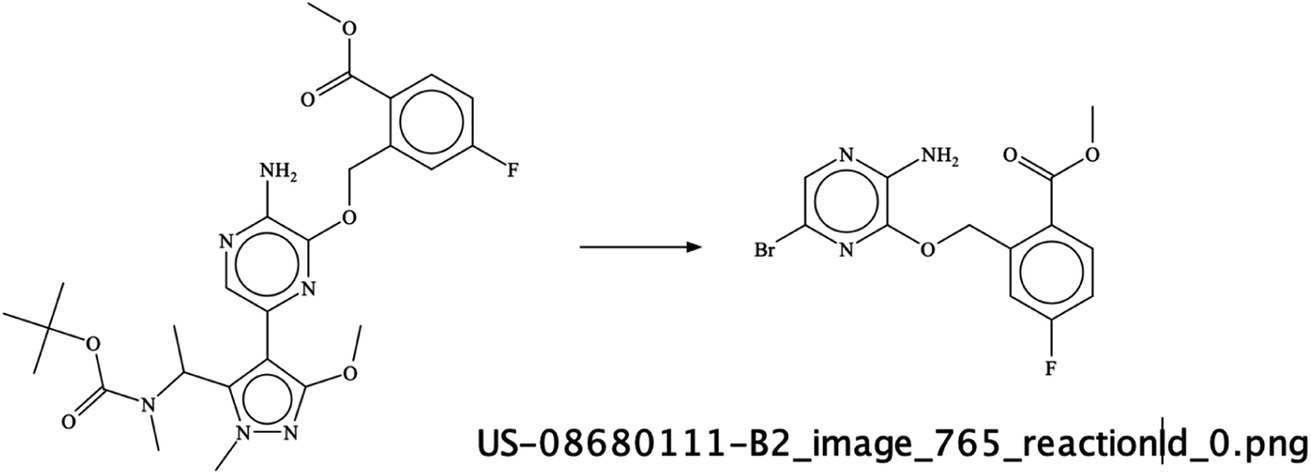 | ||
| Fig. 11 A wrongly drawn reaction direction in an example from US-8680111-B1 patent. | ||
Sometimes the reaction itself is wrong, Image S6 in the supplementary material gives an example where a Boc-protected amine was directly converted into a methyl-amino group using hydrochloric acid. However, as the formal reaction prediction was correct, the score was given as 1.
Compared to RxnScribe and OSRA, ReactionDataExtractor 2.0 had sometimes problems with correctly identifying the product and reactant role of compounds in a reaction – most likely since the arrow detection and image segmentation were not as good.
Similar to MolScribe, a frequently observed error in RxnScribe included a nitrile that was misrecognized as an isonitrile. In other cases, RxnScribe added some erroneous inert reactants, e.g. like ethane, whilst the main starting material and product were correctly recognized – in these cases we nevertheless considered this reaction as correct. The same forgiving procedure was used when in the image a polymeric resin holding a reagent/reactant was represented by a circle which was translated by RxnScibe as a methyl group.
A separate problem was the correct translation of superatoms to chemical structures. For example, “Tr” was not recognized as a trityl group, such as found in US-08680111-B2_894.TIF (Image S7). OSRA has the convenient option to define the structure of those superatoms in a separate file that is used during prediction whereas AI methods did not allow for such a feature.
Classifying images
Our current pipeline of predicting structures from images uses OSRA version 2.1.5. Assuming a hypothetical 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixture of single structure, multi structure and reaction images and with the found OSRA precision values for these three modalities we would end up with an average score of F1 = (78 + 62 + 64)/3 × 100% = 68% score. Using a hypothetical hybrid system with MolScribe for single images, OSRA for multi structure images and RxnScribe for reactions would yield a better overall F1 = (93 + 62 + 86)/3 × 100% = 80% score. To achieve this goal, we need to pre-process the images by a classifier that is able to distinguish between these 3 different image modalities. Also, classifying whether or not the image does or does not contain any chemistry can avoid processing time and costs. Therefore, we have developed a Chemical Image Classifier (ChemIC) that classifies any image into one of those 4 categories.
:1:1 mixture of single structure, multi structure and reaction images and with the found OSRA precision values for these three modalities we would end up with an average score of F1 = (78 + 62 + 64)/3 × 100% = 68% score. Using a hypothetical hybrid system with MolScribe for single images, OSRA for multi structure images and RxnScribe for reactions would yield a better overall F1 = (93 + 62 + 86)/3 × 100% = 80% score. To achieve this goal, we need to pre-process the images by a classifier that is able to distinguish between these 3 different image modalities. Also, classifying whether or not the image does or does not contain any chemistry can avoid processing time and costs. Therefore, we have developed a Chemical Image Classifier (ChemIC) that classifies any image into one of those 4 categories.
We used a pre-trained convolutional neural network (CNN) ResNet-50 model together with PyTorch.25 The dataset used for generating the image classifier consists of 16000 images that were collected from different sources:
(1) Chemical data images extracted from EP, US, and WO patents by OntoChem.
(2) Images from the MolScribe datasets2https://pubs.acs.org/doi/10.1021/acs.jcim.2c01480.
(3) DECIMER-hand-drawn molecule images dataset.22
(4) Images from the Rxnscribe training set.3
(5) Formulae images from the im2latex-100k dataset.26
The dataset for training the chemical image classifier consists of two directories. The “classified” directory contains manually labeled images. These images are divided into four distinct categories, with each category including 4000 images:
• One_molecule.
• Several_molecules.
• Reactions.
• Other.
In the “for_model” folder, we have collected the images for training, validation, and test steps by using train_test_split helper function from scikit-learn library:27
• Training_set: 12804 images.
• Test_set: 1604 images.
• Validation_set: 1604 images.
The model was trained on a high-performance machine with 40 Intel(R) Xeon(R) Gold 6226 CPUs, taking 6 hours to complete. Training was initially set to 100 epochs, each involving dataset processing and loss computation. As epochs progressed, loss decreased, indicating improved performance. Validation accuracy ranged from approximately 98.88% to 99.25%. Early stopping at epoch 26 prevented overfitting due to no improvement in validation accuracy. Upon completion of training, the model is evaluated on a separate test set, which the model has not seen. The metrics were calculated by averaging over the classes, weighted by the number of samples in each class. It achieves an impressive accuracy of 99.62% (see supplementary material for more information).
The expected overall performance improvement of 80% using a ChemIC classifier enabled hybrid system including OSRA, MolScribe and RxnScribe on a mixed dataset was not checked within the scope of this work but may be published later.
The performance of the image classifier might be further enhanced by expanding the dataset, and varying the architecture of the models and hyperparameters. Moreover, there are an extensive number of images where molecules are borderline cases – in most of these cases the model can classify the content on such images as one molecule/substance (Fig. 12). If these images are being sent to MolScribe or DECIMER, they both will recognize molecules and predict SMILES. Note, that in the example shown below DECIMER managed to predict correct SMILES, whilst MolScribe seems to be inaccurate due to the close location of the molecules on original images.
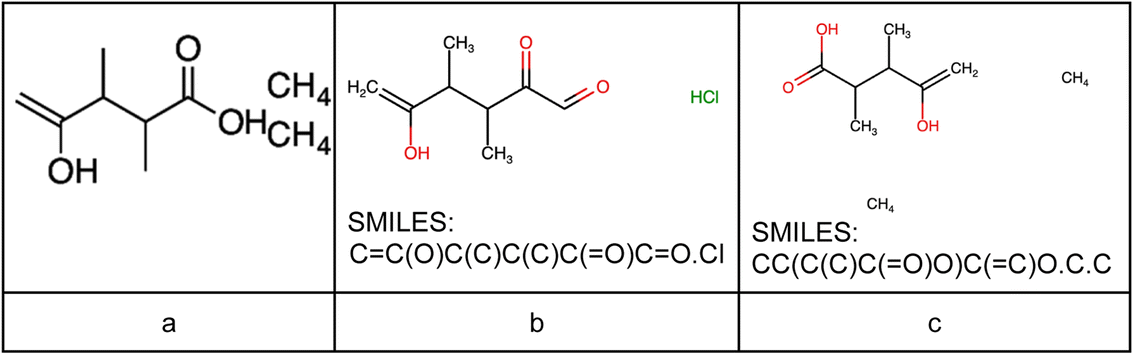 | ||
| Fig. 12 Borderline classification cases. (a) Original image US07314691-20080101-C00001.png. (b) MolScribe's prediction; (c) DECIMER's prediction. | ||
Files with the python code to perform model training and evaluation process as well as files to start using flask web service with trained model for classification images could be found in the folder ImageClassifier from the supplementary material (ref. 14) – detailed instructions are given in the included manual.
An alternative method to processing multi structure image files would be to segment those images into single structure images and subsequently apply OCSR on those segmented images. However, when processing large numbers of images from patent documents automatically one does not know a priori which image shall be regarded as multi structure image. In contrast, the US Patent office complex work (CWU) units contain manually created ChemDraw files that also contain multiple structures as provided by the patent applicant. Similarly, other patent offices clip chemical structures from the patent image files, but these clipped images also include multi structure images. Thus, it is not clear why these structures were not clipped or segmented – for example, segmenting compound images from mixture or complex salt forms would lead to a misinterpreted chemistry information.
Nevertheless, applying ChemIC on all images allows us to identify multi structure images that could be submitted to image segmentation and subsequent single image processing – with the caveat from above that we may generate more unwanted overtly granular information.
To demonstrate this, we have applied the DECIMER segmenter28 as an example segmentation method to the multi structure images used in Table 3 above. One problem of this segmentation method is shown in Fig. 13, where some of the atom labels at the border of the segmented images were lost during segmentation.
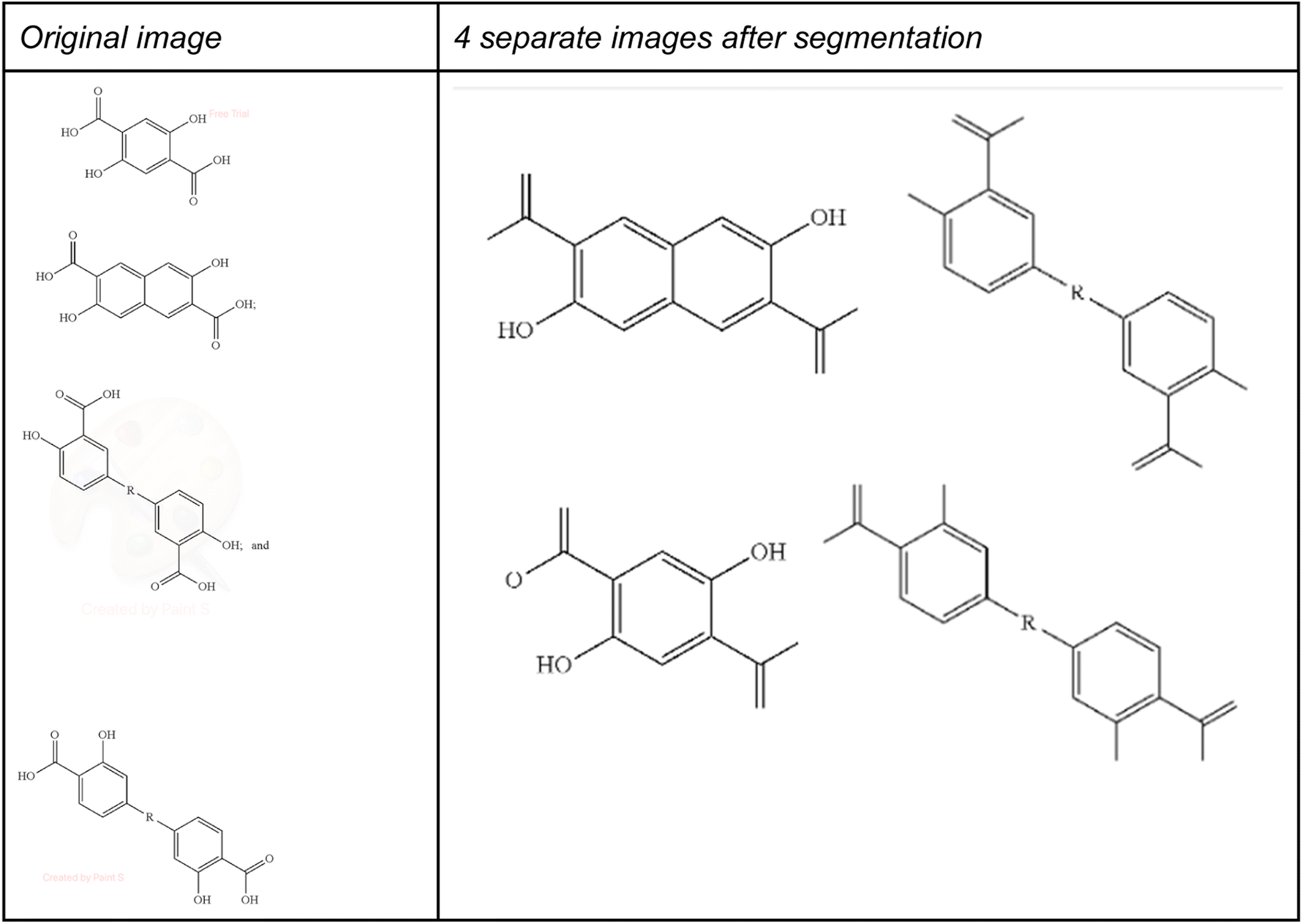 | ||
| Fig. 13 Segmentation of US-20220048929-A1_image_1674.TIF. | ||
Applying this segmentation method using the expand option set true to the 20 images from Table 3 together with the best performing MolScribe and DECIMER OCSR on the resulting 146 images lead to 120 correctly recognized structures for MolScribe and 101 for DECIMER, corresponding to precision of 82% (F1-score 90%) and 69% (F1-score 82%), respectively. Using the expand option set false we have got only 83 correctly recognized structures for MolScribe and 100 for DECIMER, corresponding to a precision of 57% (F1-score 72%) and 68% (F1-score 81%), respectively. Thus, the combination of expanded segmentation and MolScribe yielded better results than using OSRA on multi structure images, making it a good approach in a ChemIC driven modular OCSR pipeline.
Conclusions
This article compares image to structure prediction methods to aid decisions which algorithm is best to use for which image modality. MolScribe and RxnScribe were found to be superior to OSRA when extracting single structure and reaction information from images. For single, small organic molecule structures Decimer and MolScribe gave similar but better results than OSRA or MolVec. OSRA performed better on images of large molecules, transition metal complexes and images with multiple compound structures. As a result, we constructed a chemical image classifier (ChemIC) to classify images for different chemical modalities and funnel the image into the most appropriate image-2-structure method. Multi structure image segmentation followed by single structure OCSR turned out to be an interesting alternative to direct multi structure OCSR.However, some more immediate improvements of AI methods appear to be meaningful in the near future – for example improved resizing of images to allow for predictions of larger molecules as well as using training sets with multiple chemical structures. Although significant improvements could already be achieved with the novel AI based OCSR methods in a short time period, some serious problems are waiting to be solved by new approaches in the future. In most cases we are attributing those deficiencies to the limitations of the selected image learning sets that are missing for example label or caption resolution, images with multiple structure, more complex chemistries like oligomers, polymers and metal organic molecules. Thus, when separate OCSR models are trained for those modalities the chemical classifier idea as above could be implemented to integrate those different machine learning modules. Alternatively, a joint model with all these modalities could be considered.
In addition, we believe that any forthcoming new OCSR method should include a V2000 or V3000 RGfiles20 as an output – enabling the prediction of more complex Markush and polymeric structures using a commonly accepted standard chemistry structure format instead of creating non-standard smiles.
Data availability
Data and processing scripts for this paper, including image data sets, result sets, a supplementary material text in PDF format, python scripts and a java code mentioned in the publication are available at Zenodo at https://doi.org/10.5281/zenodo.10546827.Author contributions
SJB: investigation, software, data curation; AK: investigation, software, data curation, writing; SJB: investigation, software, data curation; TB: software, data curation; SKB: data curation; LW: conceptualization, data curation, writing.Conflicts of interest
AK, SJB, TB, SKB and LW declare that they have no conflicting interests.Acknowledgements
We would like to thank anonymous reviewers for their valuable comments and suggestions.References
- K. Rajan, A. Zielesny and C. Steinbeck, DECIMER: towards deep learning for chemical image recognition, J. Cheminf., 2020, 12, 65, DOI:10.1186/s13321-020-00469-w.
- Y. J. Guo, Z. Tu, Z. Li, C. W. Coley and R. Barzilay, MolScribe: Robust Molecular Structure Recognition with Image-to-Graph Generation, J. Chem. Inf. Model., 2023, 63(7), 1925–1934, DOI:10.1021/acs.jcim.2c01480.
- Y. Qian, J. Guo, Z. Tu, C. W. Coley and R. Barzilay, RxnScribe: A Sequence Generation Model for Reaction Diagram Parsing, 2023, arXiv:2305.11845v1, DOI:10.48550/arXiv.2305.11845.
- K. Rajan, H. O. Brinkhaus, M. I. Agea, A. Zielesny and C. Steinbeck, DECIMER.ai: an open platform for automated optical chemical structure identification, segmentation and recognition in scientific publications, Nat. Commun., 2023, 14, 5045, DOI:10.1038/s41467-023-40782-0.
- D. W. Wilary and J. M. Cole, ReactionDataExtractor 2.0: A Deep Learning Approach for Data Extraction from Chemical Reaction Schemes, J. Chem. Inf. Model., 2023, 63(19), 6053–6067, DOI:10.1021/acs.jcim.3c00422.
- D.-A. Clevert, T. Le, R. Winter and F. Montanari, Img2Mol – accurate SMILES recognition from molecular graphical depictions, Chem. Sci., 2021, 12, 14174–14181, 10.1039/D1SC01839F.
- Z. Xu, J. Li, Z. Yang, S. Li and H. Li, SwinOCSR: end-to-end optical chemical structure recognition using a Swin Transformer, J. Cheminf., 2022, 14, 41, DOI:10.1186/s13321-022-00624-5.
- Y. Wang, R. Zhang, S. Zhang, L. Guo, Q. Zhou, B. Zhao, X. Mo, Q. Yang, Y. Huang, K. Li, Y. Fan, L. Huang and F. Zhou, OCMR: A comprehensive framework for optical chemical molecular recognition, Comput. Biol. Med., 2023, 163, 107187, DOI:10.1016/j.compbiomed.2023.107187.
- I. V. Filippov and M. C. Nicklaus, Optical Structure Recognition Software To Recover Chemical Information: OSRA - an Open Source Solution, J. Chem. Inf. Model., 2009, 49(3), 740–743, DOI:10.1021/ci800067r , https://sourceforge.net/projects/OSRA/accessed 29 December 2022..
- T. Peryea, D. Katzel, T. Zhao, N. Southall and D.-T. Nguyen, MOLVEC: Open source library for chemical structure recognition, Abstracts of papers of the American Chemical Society, 2019, vol. 258, https://github.com/ncats/molvec/blob/master/README.md Search PubMed.
- SciWalker search application, please use and register yourself for free at https://sciwalker.com.
- A. Krasnov, S. J. Barnabas, T. Böhme, S. K. Boyer and L. Weber, Comparing Optical Chemical Structure Recognition Tools, ChemRxiv, 2023 DOI:10.26434/chemrxiv-2023-d6kmg.
- ImageComparator, see https://github.com/ontochem/ImageComparator.
- Supplementary materials: DOI: DOI:10.5281/zenodo.10546827.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Model., 1988, 28, 31–36 CrossRef CAS.
- Daylight Chemical Information Systems, Inc., SMILES - A Simplified Chemical Language, http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html, accessed 13 April 2019.
- Blue Obelisk, OpenSMILES Home Page, https://opensmiles.org/, accessed 14 November 2021.
- C. Steinbeck, Y. Han, S. Kuhn, O. Horlacher, E. Luttmann and E. L. Willighagen, The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics, J. Chem. Inf. Comput. Sci., 2003, 43, 493–500 CrossRef CAS PubMed.
- E. L. Willighagen, J. W. Mayfield, J. Alvarsson, A. Berg, L. Carlsson, N. S. Jeliazkova, S. Kuhn, T. Pluskal, M. Rojas-Chertó, O. Spjuth, G. Torrance, C. T. Evelo, R. Guha and C. Steinbeck, The Chemistry Development Kit (CDK) v2.0: atom typing, depiction, molecular formulas, and substructure searching, J. Cheminf., 2017, 9, 33 Search PubMed.
- Dassault Systèmes, BIOVIA CTfile formats, 2016, https://docplayer.net/145725575-Ctfile-formats-biovia-databases-2016.html Search PubMed.
- CLiDE, https://adventinformatics.com/portfolio/keymodule/, accessed 13 November 2023.
- H. O. Brinkhaus, A. Zielesny, C. Steinbeck and K. Rajan, DECIMER - hand-drawn molecule images dataset, J. Cheminf., 2022, 14, 36, DOI:10.1186/s13321-022-00620-9.
- S. Heller, A. McNaught, S. Stein, D. Tchekhovskoi and I. Pletnev, InChI - the worldwide chemical structure identifier standard, J. Cheminf., 2013, 5, 7 CAS.
- G. Landrum, RDKit, https://www.rdkit.org/, accessed 29 December 2022.
- Pytorch, https://pytorch.org/, accessed 16 Januar 2024.
- A prebuilt dataset for OpenAI's task for image-2-latex system, https://zenodo.org/record/56198#.YJjuCGZKgox, accessed 16 January 2024.
- Scikit-learn, https://scikit-learn.org/stable/index.html, accessed 12 February 2024.
- https://github.com/Kohulan/DECIMER-Image-Segmentation, accessed 5 February 2024.
| This journal is © The Royal Society of Chemistry 2024 |