
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraph neural networks for identifying protein-reactive compounds†
Victor Hugo
Cano Gil
 and
Christopher N.
Rowley
*
and
Christopher N.
Rowley
*
Department of Chemistry, Carleton University, 1125 Colonel By Dr, Ottawa, ON K1S 5B6, Canada. E-mail: christopherrowley@cunet.carleton.ca; Tel: +1(613) 520-2600 x 1647
First published on 25th July 2024
Abstract
The identification of protein-reactive electrophilic compounds is critical to the design of new covalent modifier drugs, screening for toxic compounds, and the exclusion of reactive compounds from high throughput screening. In this work, we employ traditional and graph machine learning (ML) algorithms to classify molecules being reactive towards proteins or nonreactive. For training data, we built a new dataset, ProteinReactiveDB, composed primarily of covalent and noncovalent inhibitors from the DrugBank, BindingDB, and CovalentInDB databases. To assess the transferability of the trained models, we created a custom set of covalent and noncovalent inhibitors, which was constructed from the recent literature. Baseline models were developed using Morgan fingerprints as training inputs, but they performed poorly when applied to compounds outside the training set. We then trained various Graph Neural Networks (GNNs), with the best GNN model achieving an Area Under the Receiver Operator Characteristic (AUROC) curve of 0.80, precision of 0.89, and recall of 0.72. We also explore the interpretability of these GNNs using Gradient Activation Mapping (GradCAM), which shows regions of the molecules GNNs deem most relevant when making a prediction. These maps indicated that our trained models can identify electrophilic functional groups in a molecule and classify molecules as protein-reactive based on their presence. We demonstrate the use of these models by comparing their performance against common chemical filters, identifying covalent modifiers in the ChEMBL database and generating a putative covalent inhibitor based on an established noncovalent inhibitor.
1 Introduction
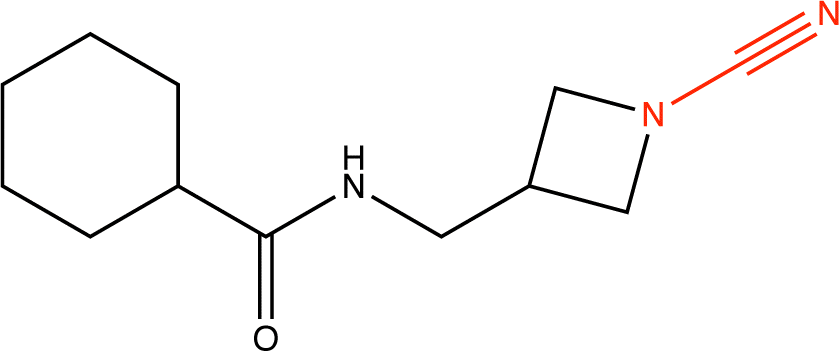
Proteins can undergo a range of chemical reactions with endogenous and exogenous molecules.1–3 The amino acids cysteine, serine, lysine, threonine, and tyrosine can act as nucleophiles in reactions with electrophilic compounds. The covalent linkage formed through these reactions provides a more durable connection to the ligand than intermolecular interactions alone, so these reactions are often used to inhibit or label proteins.4,5 These reactions typically occur between the amino acid side chain and a reactive moiety of the molecule, referred to as the covalent “warhead.” Michael acceptors like acrylamides and α-haloacetamides commonly modify cysteine residues, while epoxides and lactones often target serine residues. In recent years, many warheads have been identified, including alkynes, cyclopropanes, chloropyridines, and benzaldehydes. The reactive warhead of a variety of covalent inhibitors is highlighted in Fig. 1. The number of types of covalent warheads is large and growing; a database of covalent inhibitors (CovalentInDB) is organized into 63 different warhead categories.6 Additional protein-reactive electrophilic functional groups are still being identified.7 | ||
| Fig. 1 Examples of protein-reactive inhibitors. The substructure that reacts with the protein side chain (a.k.a., the warhead) is indicated in red. | ||
Some covalent inhibitors are highly promiscuous and will inactivate a broad number of enzymes; however, Backus et al. showed that there was a surprising degree of specificity for a covalent inhibitor to specific proteins in whole cells and lysates.8 This is consistent with the theory that the covalent modification of a protein often requires that the inhibitor has favorable non-covalent interactions with its target but can also form a covalent linkage with a complementary reactive amino acid in the target protein. This dual covalent-noncovalent binding is the basis for the development of Targeted Covalent Inhibitors (TCI).9
While covalent inhibitors have significant therapeutic uses, there are other instances where it is important to detect protein reactivity because it is a liability in a specific application. Protein-reactive compounds can have off-target activity due to promiscuous reactions with other cellular components10 and can be metabolized at faster rates due to higher electrophilicity.11 Likewise, the development of noncovalent inhibitors now routinely uses high-throughput screening of the compounds in large chemical datasets to a protein target.12,13 Alternatively, generative AI methods are now being used to design new compounds optimized to bind to a target.14,15 In both of these cases, protein-reactive compounds should generally be excluded from the searches for non-covalent inhibitors. Conversely, AI development of covalent inhibitors will require models trained to select for protein reactivity. These applications would benefit from an efficient, automatic approach for identifying protein-reactive compounds.
There have been several efforts to predict the reactivity of compounds towards proteins using quantum chemistry.16,17 Some model the reaction of a specific covalent inhibitor with its target,18,19 while others attempt to predict the intrinsic reactivity of a warhead to model thiols.20–23 These limitations require 3D structures of the warheads to be constructed and for quantum chemical calculations to be performed. Thio-Michael additions are a uniquely challenging chemical reaction for conventional DFT models,24,25 and these methods have been limited to narrow classes of warheads and are not amenable to automated high-throughput screening. For example, the BIreactive method can predict the DFT activation energy for the reaction of glutathione with a warhead using DFT-calculated descriptors, which were correlated to the halflives of glutathione addition to an electrophile using multiple linear regression.26 The reaction halflife between a test set of acrylamides was predicted with an R2 value of 0.69, although this declined an R2 for the 2-chloroacetamide test set.
One approach to identifying protein-reactive compounds would be to search for warhead substructures in a molecule. The Pan-Assay INterference compoundS (PAINS) criteria include some electrophilic motifs because compounds that promiscuously modify proteins can be false positives in high-throughput screening campaigns. Methods have been developed to automatically check if a compound matches the criteria set for PAINS compounds, such as the PAINSfilter27 set of SMARTS search strings. Pearce et al. have also published a deck of substructure filters to identify promiscuous inhibitors, including those that act through covalent modification of a protein.28 Lastly, the Eli Lilly medicinal chemistry29 rules are another automated method for screening viable drug candidates and include query patterns for many protein-reactive substructures.
Although approaches that search for substructures using defined patterns are efficient, these filters are not entirely effective for detecting protein reactive compounds; only 7% of the CovalentInDB are identified as PAINS compounds and 41% are rejected by the Eli Lilly rules, so many modes of protein reactivity are missed by these searches. The diversity of warheads means it is less practical to define search patterns for all variations individually. Further, the neighboring atoms in a molecule can amplify or attenuate the reactivity of an electrophile group; for example, certain acrylamides that are normally non-reactive become potent covalent inhibitors of S6 kinase RSK2 if they are β-substituted with cyano groups.30 This effect is difficult to capture through pattern-based substructure searches.
A machine learning classifier could provide a more general approach for predicting protein reactivity without requiring a researcher to define specific warhead substructures individually. These methods can leverage large quantities of data to define algorithms to classify molecules or predict their properties. This type of “data-driven” approach would allow protein-reactive structures to be identified using only structures of inhibitors that are known to be reactive or non-reactive.
These methods require a method to encode the molecular structure into a representation that is amenable to machine learning methods. Chemical fingerprints are a popular input to ML algorithms.31–33 These fingerprints are vectors that contain ordered elements encoding for physical, chemical, and structural properties. A widely used class of chemical fingerprints is Extended Connectivity Fingerprints (ECFP),34 which is based on the Morgan algorithm.35 This produces binary sequences of fixed length, where a positive bit at a given position indicates the presence of a chemical substructure inside the molecule. Morgan fingerprints have been successfully used as a molecular representation in numerous chemical machine learning applications.32,36–38
Graph representations of molecules are an alternative to these fingerprint methods.39 In these models, atoms are represented as nodes of a graph and the bonds between them are represented as edges. Atomic and bond properties can be added as features to the graph nodes and edges, respectively. This allows extensive chemical data to be encoded in the graph. These graphs can be used as the inputs to Graph Neural Networks,40 which can be trained for both classification and regression tasks.39–41
GNNs have been used to predict some modes of protein–molecule reactions. For example, Xenosite is a machine learning method that can predict if a compound can undergo a bioorganic transformation like epoxidation, glutathione conjugation, or alkylation.42 A drawback of this model is that it was trained using data from the Accelrys Metabolite Database, which cannot be distributed openly, so neither the model nor the training set are widely available. Generally, an open and extensible model for protein reactivity will require the use of publicly available datasets that can be extended as new compounds are synthesized and their modes of inhibition are reported. In this paper, we use machine learning techniques to develop a classifier to designate a molecule as being reactive towards proteins or non-reactive. We impose three criteria in our development so that the method is general and can be used without restrictions:
(1) The method should use only existing, open software without modification.
(2) The training set should be sourced from public molecular data sets.
(3) The method will not require any user-defined criteria or substructure patterns for protein-reactivity.
As noted, covalent-modifier inhibitors have both covalent and non-covalent interactions with their targets. As a consequence, searchers for reactive substructures in the inhibitor can only provide limited information as to its ability to react with a specific protein target. Instead, these models predict if a molecule has the potential to react with a protein.
2 Methods
Fig. 2 illustrates the overall project workflow. The methods developed in this paper take the molecular structure of a molecule as its input and output a classification of the molecule as being protein-reactive (positive class) or non-protein-reactive (negative class). These methods are trained using machine learning methods from datasets of molecules that are labeled as protein-reactive or non-reactive. A separate test set was curated to assess the transferability of these models. The construction of the training and test sets are described in the following sections. The training data and source code for all our models are deposited on GitHub.43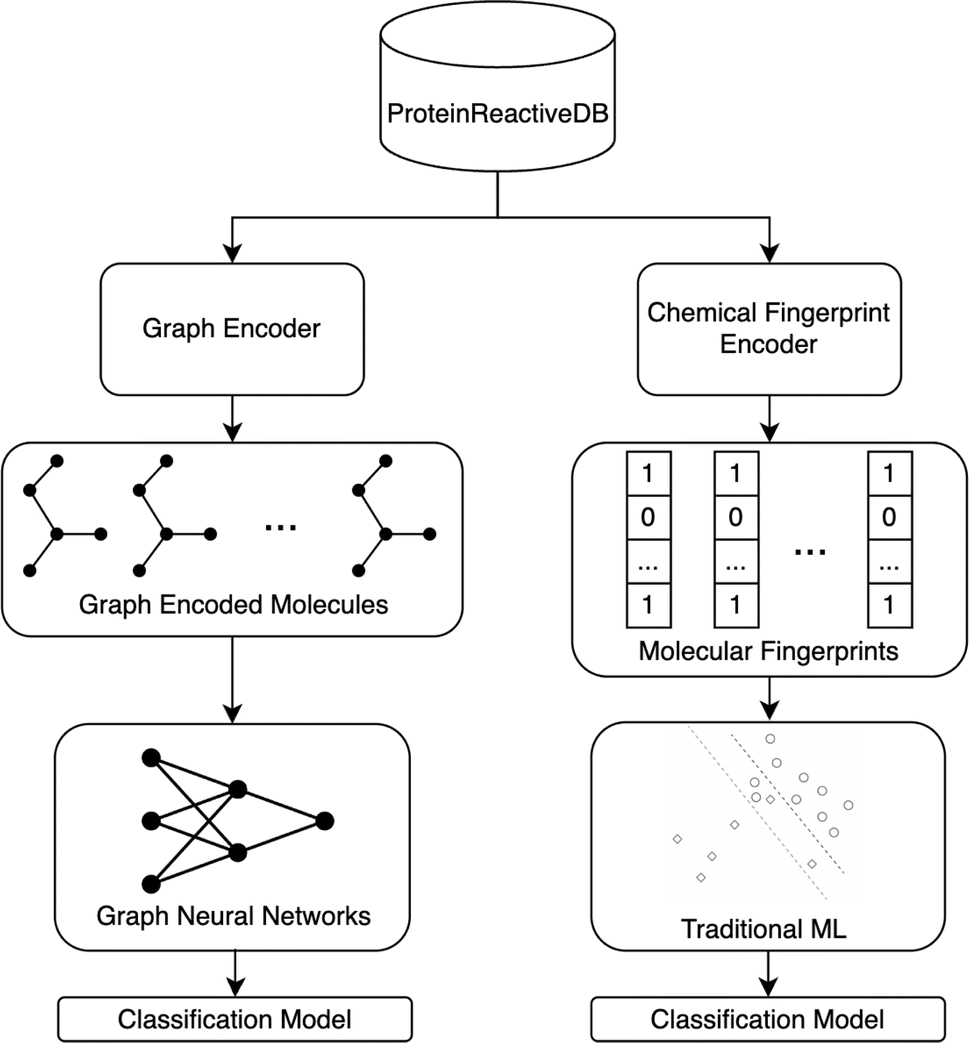 | ||
| Fig. 2 Schematics for the workflow of the ML protein reactivity classifiers. The models are trained from ProteinReactiveDB, which includes sets of inhibitors in public databases of covalent and noncovalent inhibitors. This labeled data is used to train models to classify molecules as being covalent or non-covalent using GNN and fingerprint-based ML models. | ||
2.1 Data – training
For training, we have built a new dataset, ProteinReactiveDB. This dataset was constructed from the data in three publicly available datasets: DrugBank,44 BindingDB,45 and CovalentInDB.6 The DrugBank is predominantly composed of drug molecules. The BindingDB contains a broader set of molecules reported in the medicinal chemistry literature. These two datasets served as the bulk of the negative (non-protein-reactive) set of molecules in the training set. The CovalentInDB is a database of inhibitors that have been determined to inhibit their targets by covalently modifying them. This dataset includes 4511 covalent inhibitors with 280 different protein targets, although in this approach compounds were only classed as being protein-reactive or non-reactive, irrespective of their target or rate of inactivation. This dataset was collected from the PubChem,46 ChEMBL,47 DrugBank,44 PDB,48 and UniProt49 database. The compounds were manually verified to act through a covalent mechanism based on published reports. This dataset served as the bulk of the positive (protein-reactive) set of molecules in the training set.The molecules from the datasets above were curated and combined into a dataset appropriate for the available representations. If this library failed to generate a structure for a compound, it was not included in the data set. All compounds containing inorganic components were excluded (i.e., containing only the elements H, B, C, N, S, O, F, Cl, Br). Phosphorous-containing compounds are not currently supported because the positive component of the training set had a small number P-containing compounds and many of the phosphorylated compounds in the dataset are prodrugs. The RDKit (version 2023.03.2) toolkit50 was used to convert the database entry into a molecular representation.
An immediate challenge was that both the DrugBank and BindingDB contain some compounds that are covalent modifiers. Any compound that was present in CovalentInDB was removed from the negative class so that it only appeared in the positive class. Further, an extensive manual effort was made to identify these compounds and move them from the non-protein-reactive class training set to the protein-reactive class. This included 88 compounds in the DrugBank database that were annotated as DNA alkylating agents, insecticides, or broad-spectrum antibacterial compounds. Additional compounds that were believed to be misannotated or were not suitable for the representations used in these models (e.g., metal-containing compounds, antibodies, medical adhesives, etc.) were removed from the training set entirely (n = 291). Compounds annotated as prodrugs were also excluded (n = 64).
Compounds were moved to the protein-reactive set if there was experimental evidence in the literature that they act through a covalent mechanism. All compounds that were present in both the DrugBank and the CovalentInDB were categorized as protein-reactive. We performed an additional search of the compounds in our non-protein-reactive set that our first models classified as positive to determine if they act through a covalent mechanism. For these compounds, we searched the Protein-Databank for crystallographic structures of protein–ligand complexes and searched the macromolecular Crystallographic Information File (mmCIF) file for a covalent linkage between the compounds and the proteins. Lastly, a literature search was performed to identify any published studies where the enzyme kinetics were analyzed to determine if the mode of inhibition was reversible or irreversible. 162 compounds in the DrugBank and 285 compounds from the BindingDB dataset were added to the covalent set through this process.
In total, the training set used in this study was composed of 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 740 noncovalent inhibitors and 6487 covalent inhibitors. The dataset and lists of compounds included from the source databases are included in our GitHub repository.43
740 noncovalent inhibitors and 6487 covalent inhibitors. The dataset and lists of compounds included from the source databases are included in our GitHub repository.43
2.2 Data – testing
The models presented in this paper are evaluated using two test sets. The first test set is generated by extracting 5% of the compounds in ProteinReactiveDB using stratified sampling. We will refer to this set as the Internal Test Set. A second test set was constructed to test the transferability of these models to the types of compounds that might be evaluated in a modern medicinal chemistry campaign. This set will be referred to as the External Test Set, which is composed of covalent and non-covalent inhibitors that were not present in the training set (Table 1). These compounds were manually curated from the recent chemical literature, and are split into three groups: covalent inhibitors, first disclosures, and nonreactive decoys.| Class | Type | Count |
|---|---|---|
| Noncovalent | First disclosures | 139 |
| Nonreactive decoys | 47 | |
| Covalent | Aldehyde | 10 |
| Alkenes | 217 | |
| Alkyne | 13 | |
| Aziridine | 6 | |
| Atypical | 27 | |
| Boronic | 7 | |
| Epoxides | 21 | |
| Furan | 4 | |
| Haloacetamides | 14 | |
| Lactam | 11 | |
| Lactone | 18 | |
| Nitrile | 9 | |
| Quinone | 3 | |
| Sulfonyl | 49 | |
| Thiocyanate | 2 | |
| Thioketone | 7 |
 | ||
| Fig. 3 Examples of compounds from the non-reactive “decoy” set that contain a deactivated warhead (red). These compounds have been determined experimentally to react with nucleophiles at a slow rate. | ||
2.3 Metrics
To measure the performance of our models, we employ common classification metrics such as precision, recall, and area under receiver operating characteristic (AUROC). For the external test set, we also review the accuracy of each prediction for each group. The classification metrics are defined as: | (1) |
 | (2) |
 | (3) |
The ROC curve is the plot of recall against false positive rate (FPR), defined as:
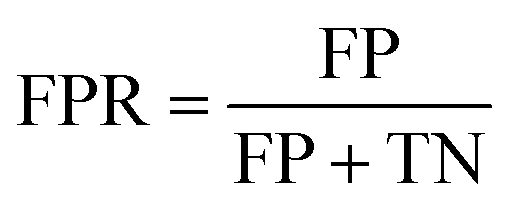 | (4) |
The integral of this curve provides the AUROC.59 This metric indicates how confident a given model is when making a classification. An AUROC of 1 indicates that the model is capable of perfectly distinguishing between different classes. A binary classifier with an AUROC of 0.5 indicates that the model is no better than a random chance, i.e. is very uncertain when assigning a predicted class.
2.4 Models and features
 | (5) |
 , Fl is the convolutional activations at the layer l, F0 = X,Ã = A + IN is the adjacency matrix with added self-connections where IN is the identity matrix, Wl are the trainable convolutional weights,
, Fl is the convolutional activations at the layer l, F0 = X,Ã = A + IN is the adjacency matrix with added self-connections where IN is the identity matrix, Wl are the trainable convolutional weights, 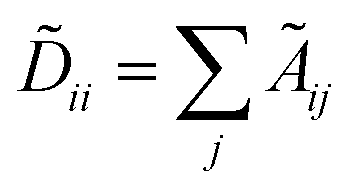 , and σ is the nonlinear activation function.
, and σ is the nonlinear activation function.
In this work, the GNNs were implemented using the Molgraph library67 (version 0.5.8), which also provides a wrapper to RDKit descriptors that were used to generate atomic and bond features. The atomic and bond features include common chemical descriptors such as chemical symbol, total number of hydrogens, being a part of aromatic system, etc. A full list of the atomic and bond features is presented in the ESI.† Additionally, Conceptual Density Functional Theory (CDFT) derived Fukui functions and electrophilicity indices were calculated and used as part of the atomic features in some models. Molecules were converted into 3D structures using RDKit and then the Fukui functions were calculated using AIMNET.68
 | (6) |
Then, using eqn (5), we can define LGradCAMc as the heatmap from layer l:
 | (7) |
These values can be presented visually as a heatmap where the nodes are colored according to the magnitude of L for a node. In this work, the heatmaps were produced using LGradCAMc[l,n] Avg, defined by
 | (8) |
3 Results and discussion
3.1 Morgan fingerprint models
The performance of the models trained using the Morgan fingerprints of the inhibitors as features is summarized in Table 2. For each model, optimal hyperparameters were determined using a random search with 10-fold cross validation. The optimal models for all five classifiers performed reasonably well on the internal test set, with AUCROCs ranging from 0.74 to 0.95; however, the transferability of these models to the external test set was modest, with the AUCROCs between 0.58 and 0.74. These models have very high precisions on the external test set, ranging from 0.89 to 1, but have recalls that range from 0.16 to 0.54. This indicates that these models are skewed such that they are prone to classifying a candidate molecule as negative. We also explored models using MACCS-augmented fingerprints. The performance of the model on the internal test set is better, but the performance on the external test set was only incrementally improved and the best-performing model (HGB) was not improved (Table 3).| Model | Internal test AUROC | External test AUROC | External test precision | External test recall | Nonreactive decoy FPR |
|---|---|---|---|---|---|
| SVC | 0.94 | 0.64 | 0.89 | 0.42 | 0.20 |
| HGB | 0.95 | 0.73 | 0.95 | 0.53 | 0.17 |
| LR | 0.97 | 0.74 | 0.95 | 0.54 | 0.11 |
| RF | 0.74 | 0.58 | 1.0 | 0.16 | 0.00 |
| MLP | 0.95 | 0.64 | 0.94 | 0.33 | 0.10 |
| Model | Internal test AUROC | External test AUROC | External test precision | External test recall | Nonreactive decoy FPR |
|---|---|---|---|---|---|
| SVC | 0.96 | 0.74 | 0.94 | 0.56 | 0.28 |
| HGB | 0.97 | 0.73 | 0.91 | 0.59 | 0.46 |
| LR | 0.97 | 0.74 | 0.93 | 0.58 | 0.23 |
| RF | 0.77 | 0.58 | 1.0 | 0.15 | 0.00 |
| MLP | 0.97 | 0.71 | 0.93 | 0.50 | 0.23 |
All these models showed significantly poorer performance on the external test set than on the internal test set. In general, these models show that the approach of using Morgan fingerprints has limited transferability to compounds outside the training set. A chemical substructure indicated by a specific Morgan fingerprint bit can be connected to protein reactivity, but these models fail to generalize in cases where the specific substructure is lost but protein-reactive activity is still present. The limited performance of these fingerprint based models led us to explore more advanced methods using molecular graphs.
3.2 Graph neural network models
Several variants of GNN were evaluated as classifiers. For each model, optimal hyperparameters were found using a random search with 10-fold cross-validation. The full details of the hyperparameter search, best hyperparameters for each type of model, and feature impact analysis are described in the ESI.† The performance of GNN models is summarized in Table 4. All these GNNs performed better than the fingerprint models on classifications of compounds in the external test set. Most significantly, these methods consistently had much higher recall rates, which ranged from 0.70 to 0.76. These results suggest that GNNs are significantly better at classifying protein-reactive compounds distinct from those in the training set. These models all had significant false positive rates on the decoy set (false positive rates that ranged from 0.35 to 0.57).| Graph architecture | Internal test AUROC | External test AUROC | External test precision | External test recall | Nonreactive decoy FPR | Ref. |
|---|---|---|---|---|---|---|
| GCN | 0.98 | 0.84 | 0.90 | 0.70 | 0.51 | 66 |
| GCNII | 0.95 | 0.80 | 0.89 | 0.72 | 0.35 | 72 |
| GraphSage | 0.98 | 0.84 | 0.89 | 0.73 | 0.57 | 73 |
| GAT | 0.97 | 0.83 | 0.89 | 0.76 | 0.49 | 74 |
| GatedGCN | 0.96 | 0.82 | 0.90 | 0.71 | 0.53 | 75 |
| GIN | 0.97 | 0.84 | 0.90 | 0.70 | 0.51 | 76 |
| GT | 0.98 | 0.84 | 0.90 | 0.74 | 0.49 | 77 |
| GMM | 0.97 | 0.83 | 0.90 | 0.72 | 0.55 | 78 |
| GATv2 | 0.96 | 0.84 | 0.89 | 0.74 | 0.49 | 79 |
All graph models have similar AUCROCs for the external test set, although there were some differences in the external test set recall rates, with values ranging from 0.7 to 0.76. Models with higher recalls also had higher nonreactive decoy FPRs, indicating these models are biased towards positive classification. For our immediate use, false positives are a greater concern than false negatives, so we have chosen the Graph Convolutional via Initial residue and Identity mapping72 (GCNII) model based on its lower FPR. The GCNII model was developed to address issues with oversmoothing,72 which is an advantage in these systems where a covalent substructure can span 3–4 bonds (edges) and deactivation of these substructures involves even more distant atoms.
3.3 Comparison to existing filters
One application of this classifier is to identify compounds that may react promiscuously with proteins. Currently, the Eli Lilly medicinal chemistry rules and PAINSfilter criteria are commonly used to identify protein-reactive compounds. These filters include additional criteria to screen for non-drug-like and assay-interference properties other than covalent reactivity, such as solubility, metabolism, permeability, dyes, etc. Where possible, these queries were removed from the filter so that it would only return positives based on protein reactivity. The queries removed from each filter are listed in ESI† and the modified implementation of Eli Lilly rules is available in the GitHub repository.43 We note that some molecules of the test set could not be processed by Eli Lilly filters, reducing the test size from 610 to 596 samples.The performance of GCNII models and the filters mentioned above are shown in Table 5. The GCNII models with default cutoff performs worse than Eli Lilly filters (F1 scores of 0.78 versus 0.84 respectively), but significantly better than PAINSFilter (F1 score of 0.07), which suffer from very low recall (0.02). A significant advantage of these ML classifier methods over conventional filters is that they allow the researcher to choose the decision threshold at which a candidate molecule is classified as reactive or nonreactive. The original model used the default threshold of 0.5, but adjusting the decision cutoff of the GCNII to 0.17 increases the recall of the model to 0.84 with only a small decrease in precision (0.89 to 0.87). This adjusted model now outperforms the Eli Lilly filter (F1 scores of 0.85 versus 0.84) while maintaining a better nonreactive decoy FPR (0.62 versus 0.74). It should also be noted that the Eli Lilly rules were developed over an 18 years period within Lilly Research Laboratories and patterns for any new warheads must be defined “by hand” by researchers, while the GNN model was trained automatically using only datasets of the molecular structures of covalent and non-covalent inhibitors. We have also performed GradCAM analysis of the external test set to determine whether the heatmaps generated consistently indicated the atoms in a molecule that were part of the warhead. To that end, we have compared the atomic selections calculated using the set of SMARTS patterns to that of atoms in positively-classified molecules where the normalized GradCAM value was greater than 0.3, which corresponds to strong importance attribution by the model. Examples are presented in the ESI.† We found that 179 of 217 compounds in the positive component of the test set had the atoms of this warhead that were selected by one of the filter SMARTS strings also had high values in the GradCAM map (threshold > 0.3). This indicates that the positive GradCAM map generally indicates the atoms that are part of the protein-reactive region, although there is not a strict correspondence.
| Precision | Recall | Nonreactive decoy FPR | F 1 score | |
|---|---|---|---|---|
| GCNII (DT = 0.5) | 0.89 | 0.70 | 0.42 | 0.78 |
| GCNII (DT = 0.17) | 0.87 | 0.84 | 0.62 | 0.85 |
| Eli Lilly | 0.81 | 0.87 | 0.74 | 0.84 |
| Pearce | 0.99 | 0.34 | 0.37 | 0.51 |
| PAINSfilter | 0.83 | 0.04 | 0.02 | 0.07 |
3.4 Conceptual density functional theory features
Conceptual Density Functional Theory (CDFT) is often used to rationalize chemical reactivity.80–82 The Fukui function is one of the most significant CDFT concepts. The electrophilic Fukui function (f+) describes the rate at which the electron density at a point in space will change when an electron is added to the molecule. Electrophiles transfer electron density to the molecule, so the points where this function has a high magnitude have a high propensity for an electrophilic attack. The nucleophilic Fukui function (f−) is defined as the rate that electron density at a point in space changes as an electron is removed from the molecule. Nucleophiles transfer electron density from the molecule, so the points where this function has a high magnitude have a high propensity for nucleophilic attack. These functions can be condensed onto individual atoms to define atomic Fukui functions by calculating the partial atomic charges of the neutral, anionic, and cation states of a molecule and estimating the Fukui functions by finite difference. These condensed Fukui functions can be multiplied by the CDFT molecular electrophilicity to provide the positive (ω+) and negative (ω−) electrophilicity indices, which have been noted as useful descriptors for the prediction of warhead reactivity.83,84CDFT features like the Fukui functions could be useful as atomic node features in GNNs for predicting chemical reactivity, but traditionally, calculating these terms would require a quantum chemical calculation. Isayev and coworkers have implemented CDFT predictions into AIMNET, a message-passing neural network approach that approximates ωB97x/def2-TZVPP minimal basis iterative stockholder charge analysis, without a quantum chemical calculation.68 Using AIMNET, the nucleophilic and electrophilic Fukui functions can be calculated from these data with a very small computational cost, making it practical to include these charges as features in high-throughput GNN models.
To test whether GNNs with CDFT features perform better for predicting protein-reactivity, we trained a second GNN classifier with AIMNET-calculated atomic charges, positive Fukui function and negative condensed Fukui functions, and positive and negative condensed electrophilicity indices functions included as atomic features. Calculation of the AIMNET CDFT features requires the generation of a 3D structure, search for an optimal conformation, optimization of the structure, and calculation of CDFT properties using a message passing NN. The AIMNET NN currently only allows CDFT properties to be calculated for neutral molecules and other failures in this workflow reduced the training set to 5875 covalent inhibitors and 43373 non-covalent inhibitors. For comparison, a second GNN classifier was trained using this dataset but without the CDFT features. The metrics for both models are presented in Table 6. Both GNNs used the same architecture as the GCNII from Table 4.
| Architecture | Internal test AUROC | External test AUROC | External test F1 score | Nonreactive decoy FPR |
|---|---|---|---|---|
| Without CDFT | 0.95 | 0.85 | 0.80 | 0.49 |
| With CDFT | 0.96 | 0.84 | 0.81 | 0.78 |
The CDFT model performed similarly to the non-CDFT model across classification metrics; however, it performed significantly worse on the decoy set (false positive rate of 0.78 compared to 0.49). This is surprising because CDFT properties like the Fukui function are standard quantum chemical methods for quantifying the electrophilicity of an atom in a molecule. Hughes et al. also investigated the utility of CDFT features in their Xenosite GNN classifier for mechanisms of biomolecular reaction and metabolism and found that they did not result in a large improvement.85 We suspect that the existing atomic features defined based on the bonding connectivity are sufficient for the GNN to make predictions of protein reactivity that are already near the limit of these graph architectures given the limited training data, so CDFT features do not provide data that can improve upon this.
There are several drawbacks associated with including CDFT features vs. our main GNN classifier. Calculation of the AIMNET CDFT features requires the generation of a 3D structure, generation of an optimal conformation, optimization of the structure, and calculation of CDFT properties. In contrast, all the features in our previous model can be calculated from the 2D structure alone. Calculating a 3D structure is computationally intensive and occasionally fails, so adding these features significantly complicates an automated workflow.
3.5 Gradient activation maps
Like any neural network architecture, GNNs are not directly interpretable. As we have constructed the positive and negative classes of our datasets from different sources, there is some risk that the trained network would make classifications based on characteristics that are not generalizable. When adapted to graph inputs, GradCAM is capable of producing graph heatmaps (see eqn (7)). To assess whether the models developed here classify based on generalizable criteria, we calculated the GradCAM heatmaps for a variety of molecules (Table 7), allowing us to visualize which atoms in a molecule are contributing most to its classification as a protein-reactive or non-protein-reactive molecule.





G12Si-5 features a lactone warhead, which forms a covalent bond with the mutant Serine-12 residue in KRAS G12S.92 The GNN correctly classifies it as being a covalent inhibitor with a classifier confidence score of 99.9%. The heatmap highlights the lactone warhead, indicating that classification is correctly based on the presence of this electrophile in the molecule. Likewise, the covalent inhibitors NVP-DPP-728, dimethyl fumarate, futibatinib, and ganfeborole are all correctly classified as being protein-reactive. The heatmaps highlight their nitrile, acrylate, acrylamide, and cyclic borate warheads, respectively, indicating that their positive classification was correctly based on the presence of these motifs.
Futibatinib is a notable example because it contains two nominally electrophilic groups: an acrylamide and an alkyne. The heatmap indicates that the acrylamide group was the most significant class for the positive classification. This is in keeping with the mode of action of this inhibitor, which inhibits FGFR1–4 through the chemical modification of a P-loop cysteine and the acrylamide, while the alkyne is unmodified.93 This demonstrates that the GNN model can recognize that the acrylamide is activated while the reactivity of the alkyne is muted by conjugation with two aromatic rings.
The trans-stilbene oxide is an instance of false positive classification where the classifier categorizes the compound as protein reactive with high confidence (85.2%). Although the compound contains an epoxide, the mechanism of action is believed to be through induction of metabolic enzyme Cyp2B94 and estrogenic activity of its hydroxylated metabolite rather than the covalent modification of a protein.95 The failure of the classifier to classify this compound as non-reactive likely reflects the larger number of covalent modifiers with epoxide warheads and an insufficient number of inert epoxides in the training set. Additional training data or atomic features may address this issue.
3.6 Limitations
Although the metrics of the GNN classifier are good for a chemical application of this type, there are some areas where the performance is weaker. This is evident when the true and false classifications of the external test set are grouped by type for the GCNII model (Table 8). The compounds in the first disclosure set are non-covalent inhibitors that have been reported in the literature recently, so they are not present in the training sets. The GCNII classifier is generally effective in classifying them as non-covalent inhibitors, with 90% of structures being classified correctly. The decoy compounds are a set of molecules that contain electrophilic functional groups, but their reactivity has been determined to be very slow or insignificant by experimental measurements. The classifier predicts correctly 57% of structures, indicating that it has marginal ability to exclude non-reactive compounds and frequently misclassifies them.| Class | Type | Total samples | Predicted correctly (%) |
|---|---|---|---|
| Noncovalent | First disclosures | 139 | 90 |
| Nonreactive decoys | 47 | 57 | |
| Covalent | Aldehyde | 10 | 80 |
| Alkenes | 217 | 76 | |
| Alkyne | 13 | 69 | |
| Aziridine | 6 | 33 | |
| Atypical | 27 | 33 | |
| Boronic | 7 | 100 | |
| Epoxides | 21 | 67 | |
| Furan | 4 | 50 | |
| Haloacetamides | 14 | 93 | |
| Lactam | 11 | 82 | |
| Lactone | 18 | 56 | |
| Nitrile | 9 | 11 | |
| Quinone | 3 | 100 | |
| Sulfonyl | 49 | 84 | |
| Thiocyanate | 2 | 100 | |
| Thioketone | 7 | 57 |
The model shows good performance on compounds with a warhead featuring an unsaturated bond, which can be explained by them being well represented in training data – CovalentInDB contains a large number of this type of Michael acceptor warheads. It performed poorer on “atypical” warhead portion of the test set, which includes novel functional groups that have only recently been identified as protein reactive (e.g., isoxazoline-based electrophiles96); less than half were classified correctly. This is likely due to very limited training data and a lack of transferability. For several other groups, the model underperforms either because those groups are not well represented in the training data. There are only 10 aziridines in the positive training set, so the network is likely undertrained in recognizing when these structures will be protein-reactive. Further, both epoxides and aziridines have triangular elements, which are not amenable to graph convolutional methods.97 Hughes et al. introduced a special epoxide atomic feature and additional training data to train their GCN Xenosite model to predict the reactivity of epoxides correctly, which may also be needed to improve the performance of this model on epoxides.85
To investigate this further, we performed GradCAM analysis on the nonreactive decoy and atypical sets (see ESI†). In some cases, an atypical Michael acceptor warhead is successfully identified by the GNN, such as the tyrosine-conjugating cyclic imine Mannich electrophiles reported by Krusemark and coworkers.98 In contrast, the novel ring-strain bicyclobutane carboxylic amides warheads were not indicated by the GradCAM map, so many of these compounds were incorrectly classified as non-reactive. In the cases where these compounds were positively classified, it appears to be a case of being “right for the wrong” reason because the GradCAM heatmap highlighted marginally electrophilic groups such as alkynes and alkenes rather than the warhead substructure. Based on this, it is unlikely that the current GNN models can reliably discover novel warheads, although there are some suggestions that these GNNs have some capability out of their domain and further elaboration of these methods might be able to generate truly original warheads.
The reaction between a protein side chain and a molecule is often mediated by the environment inside the protein, such as neighboring charged residues and hydrogen bonding networks inside the active site of an enzyme.99 While some covalent inhibitors are promiscuous,100,101 Kuljanin et al., found that there was a high level of selectivity for a particular covalent inhibitor in whole-cell assays.102 Our categorization of inhibitors as either covalent or noncovalent ignores these distinctions, so this classifier cannot predict if a compound will covalently modify a specific protein, but rather it predicts whether the molecule could covalently modify a protein provided there is a protein with a binding site that can accommodate the ligand in an orientation that will put its warhead in contact with a reactive side chain.
The false positive rate of the GCN models is one of the more significant limitations. This is apparent in both the modest recall rate on the external test set and false positive rate on the decoy set (0.72 and 0.35, respectively for the GCNII model). This indicates that the GCN models as implemented here struggle to distinguish between molecules that possess various modestly reactive groups but are not sufficiently reactive to covalently-modify a protein. Simple approaches, such as adjusting the network architecture and adding QM features had limited success in improving these metrics. It is evident that these models perform better on regions of the chemical space where there is extensive training data (e.g., acrylamides) but have limited transferability to more exotic areas of chemical space (e.g., compounds in the atypical set). The inherently small size of the training data limits the performance of direct, data-based approaches like those used here, so more advanced chemically-aware AI methods may be needed to improve these models further.
Generally, the composition of our training set imposes significant limitations on our methods. All three source datasets are based on compounds where inhibition experiments have been performed. Many highly electrophilic compounds would not be present in the training set because they are too unstable to perform inhibition studies of. Further, novel covalent warheads that employ unprecedented chemical motifs are still being identified. These structures are inherently absent from the training sets and these models have only limited abilities to predict reactivity in compounds dissimilar to those in their training set. More generally, the labeling of the data is “noisy” because currently covalent inhibitors must be manually separated from the non-covalent training sets and is not always apparent when an inhibitor acts through a covalent mechanism103–105 The expansion of the training set and developments to make these models more transferable to new chemical substrates may help address this issue. As part of an effort to help the end user assess whether our models can be applied to a particular structure and/or dataset, we have included a script that calculates the average Tanimoto similarity and average pairwise distance for a given structure or set of structures.
3.7 Protein-reactive molecules in the ChEMBL database
Libraries of chemical structures are often used in high-throughput screening campaigns. Generally, these screenings are intended to identify non-covalent inhibitors, so molecules likely to react with proteins would create risks of off-target inhibition or toxicity. Generally, protein-reactive molecules should be excluded from these searches. The ChEMBL database is a widely used library of drug-like compounds collected from medicinal chemistry journals and patents,47,106,107 but it is not currently separated into covalent and non-covalent inhibitors.This led us to apply the GNN classifier developed in this work to the ChEMBL database to identify how many potentially protein-reactive molecules are in this set. The GNN classifier developed in the previous section was used with a threshold of 0.9 to minimize false positives. 5.1% of the ChEMBL database was flagged as potentially being protein-reactive by these criteria. Eight examples are presented in Table 9. These compounds were confirmed to be covalent-modifier inhibitors through a literature search. Researchers using the ChEMBL database to search for non-covalent inhibitors may consider testing if the compounds are protein-reactive using this classifier to exclude these molecules from the search because there is a risk that they will react with a protein other than the target. The full distribution of prediction confidences of the GCNII can be found in the ESI.† The classification of each molecule in the CheEMBL dataset using the GCNII classifier is available for download from ref. 115.





The Eli Lilly filters and the GNN classifier scores do not consistently identify the same compounds as being reactive or non-reactive when applied to the CheEMBL dataset (see Fig. S3 in ESI†). This indicates the GNN classifier developed here makes distinct predictions to the Eli Lilly filters when applied to diverse datasets. The GNN classifier is specifically trained to identify features molecules in datasets of inhibitors that do or do not impart protein reactivity, while the filters are searching for substructures that are undesirable within medicinal chemistry campaigns for a broader set of criteria. As such, if the models developed here are used to screen databases, they should be used in conjunction with existing filter-based methods rather than in place of them.
3.8 Generative models
Another potential application of these methods is in the AI generation of covalent inhibitors. As a proof of concept, we have explored using the GNN classifier developed here in conjunction with generative models. Commonly, a non-covalent inhibitor of a target is known and the molecular scaffold of this molecule is modified to introduce a covalent warhead. For example, gefitinib is a non-covalent inhibitor of the Epidermal Growth Factor Receptor (EGFR), which contains a phenyl(amino)quinazoline group that binds to the ATP-binding pocket of the kinase domain.116 The covalent inhibitor afatinib was developed by introducing an acrylamide group in one of the pendant substituents, while preserving the phenyl(amino)quinazoline core.117 Prospectively, this process could be automated by generative AI models, where covalent variants are generated from a non-covalent scaffold.To explore whether the GNN classifier developed here could be used to automatically generate covalent inhibitors, we used the STONED algorithm118 to generate 200000 variations of gefitinib that preserve the phenyl(amino)quinazoline core. These compounds were filtered to select neutral, organic inhibitors and ranked according to their difficulty-of-synthesis using the SYBA classifier.119 The GNN covalent classifier was used to identify the top ranked inhibitors with a classifier score of >0.99. The 20 top-ranked compounds were docked to the crystallographic structure of the EGFR kinase domain using MOE.120 One of these compounds was predicted to bind in a mode where its terminal acrolein group formed a close non-bonded contact with Cys797 (r(Cβ–S) = 3.8 Å). This non-covalently-bound structure was modified to form a covalent adduct, where the cysteine has undergone a 1,2-conjugate addition to the warhead. After minimization, the structure of the covalent adduct holds a similar pose to the experimental X-ray crystallographic structure of covalent-bound afatinib (Fig. 4). A general implementation of this workflow could generate candidates for covalent variants of non-covalent inhibitors in a fully automatic process.
 | ||
| Fig. 4 Top: Crystallographic structures of gefitinib (green, PDB ID: 4WKQ) and afatinib (blue, PDB ID: 4G5J) bound to EGFR along with the modelled covalent-adduct pose of the generated compound (red). The generated compound and afatinib form a covalent adduct with Cys797 (yellow). Bottom: The chemical structures of the three compounds. The conserved (phenylamino)quinazoline core is shown in grey. The β-carbon site of the covalent warheads of the generated compound and afatinib is indicated with an asterisk. | ||
4 Conclusions
Machine learning methods for predicting if a molecule is protein-reactive were developed. A new dataset, ProteinReactiveDB was constructed from public datasets of molecular inhibitors. These data were used to train classifiers to designate a molecule as being protein-reactive or not protein reactive. To test the transferability of these models, an external test set was constructed from compounds that are not present in these sets, as well as a non-reactive decoy test set of compounds that contain functional groups that can be protein-reactive but are not reactive in the chemical context of that molecule.Conventional ML methods using Morgan fingerprints as features had limited transferability and performed poorly in identifying protein-reactive molecules in the external test set. The HGB and LR classifier was the best-performing models of this type, both with an AUCROC of 0.95 on the internal test set. This performance on the external test set degraded to 0.73 and 0.74. The primary limitation of these models is a high false negative rate; both HGB and LR have poor recalls of 0.53 and 0.54, respectively.
The GNNs showed improved performance over the models based on Morgan fingerprints, with GCNII model performing the best across most metrics. This model had an AUCROC of 0.95 for the internal test set and 0.80 for the external test set. Notably, the recall of these models was much improved. Analysis of the GNN using the gradient activation map indicates that these models successfully identify the relevant reactive regions of these inhibitors and can distinguish electrophilic groups that are made less electrophilic by their environment.
The GNN can also be compared to other pattern-based filters that have been developed to screen for protein-reactive inhibitors. The calibrated GCNII classifier outperformed PAINsfilter and Eli Lilly Medchem rules on the external test set. These pattern-based filters were developed by cheminformaticians over many years by defining specific substructure patterns for each electrophilic group, while the GNN classifier was built over a much shorter time period using only public databases of covalent and non-covalent inhibitors. The GNN classifier can be updated to recognize new warheads simply by adding new compounds into the training set. The GNN is also better able to discern if a potentially protein-reactive group is deactivated by the molecule, while pattern-based filters would flag the presence of these substructures indiscriminately. The GNN architecture can also be used for transfer learning to other ML problems and generative AI methods in a way that pattern-based filters cannot be.
These models were effective using only basic atomic and bond properties as features and adding more sophisticated CDFT properties did not provide a model that was systematically improved. Analysis of the GradCAM heatmaps showed that these models can successfully identify the electrophilic warhead of the compound, indicating the classification being made based on chemically sensible criteria.
The GNN models may have a small but significant false-positive rate, so when these are applied to large databases, there will be a significant number of compounds incorrectly classified as protein-reactive. This can be partially rectified by calibrating the decision threshold cutoff.
These models show the ability to recognize “decoy” compounds that contain similar functional groups as covalent inhibitors but are not sufficiently reactive to be considered a practical covalent inhibitor. This is generally more challenging because it requires the degree of electrophilicity to be estimated rather than just the presence or absence of a reactive motif. However, these models had limited success in identifying protein-reactive compounds with newly developed warheads that are not well-represented in the training set.
Despite these limitations, this study demonstrates the remarkable ability of GNNs to learn to recognize reactive chemical substructures based exclusively on the classification of compounds as covalent and noncovalent inhibitors. This suggests that the substantial libraries of covalent and non-covalent inhibitors are an effective training set for machine perception of electrophilicity. Currently, there are only a modest number of experimental chemical datasets that have the quality and extent that is suitable for machine learning, so the success of these models using these data opens new possibilities in chemical reaction prediction. There are also possibilities to improve these classifiers by adding new features and more advanced ML techniques.
Data availability
The ProteinReactiveDB, the external test set, and our complete code for both the fingerprint and graph models are distributed on our GitHub repository: https://github.com/RowleyGroup/covalent-classifier. The version of the code employed for this study is version july2024. The classifier scores of the GNN model and the Eli Lilly model for the compounds in the CheMBL dataset are distributed on FigShare: https://figshare.com/articles/dataset/ChEMBL_with_GNN_preds_and_Eli_Lilly/25853467.Author contributions
Conceptualization: V. H. C. and C. N. R.; data curation: V. H. C. and C. N. R.; formal analysis: V. H. C. and C. N. R.; funding acquisition: C. N. R.; investigation: V. H. C. and C. N. R.; methodology: V. H. C. and C. N. R.; project administration: C. N. R.; resources: C. N. R.; software: V. H. C. and C. N. R.; supervision: C. N. R.; validation: V. H. C. and C. N. R.; visualization: V. H. C. and C. N. R.; writing – original draft: V. H. C. and C. N. R.; writing – review & editing: V. H. C. and C. N. R.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank NSERC of Canada for funding through the Discovery Grants program (RGPIN-05795-2016). VHCG thanks Dr Liqin Chen for a scholarship. Computational resources were provided by Compute Canada (RAPI: djk-615-ab). We gratefully acknowledge the support of NVIDIA Corporation with the donation of the Titan Xp GPU used for this research.References
- B. Coles, Drug Metab. Rev., 1984, 15, 1307–1334 CrossRef CAS PubMed.
- T. K. Rudolph and B. A. Freeman, Sci. Signaling, 2009, 2, re7 CrossRef PubMed.
- S. J. Enoch, C. M. Ellison, T. W. Schultz and M. T. D. Cronin, Crit. Rev. Toxicol., 2011, 41, 783–802 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- T. A. Baillie, Angew. Chem., Int. Ed., 2016, 55, 13408–13421 CrossRef CAS PubMed.
- H. Du, J. Gao, G. Weng, J. Ding, X. Chai, J. Pang, Y. Kang, D. Li, D. Cao and T. Hou, Nucleic Acids Res., 2021, 49, D1122–D1129 CrossRef CAS PubMed.
- N. Péczka, Z. Orgován, P. Ábrányi Balogh and G. M. Keserű, Expert Opin. Drug Discovery, 2022, 17, 413–422 CrossRef PubMed.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Nature, 2016, 534, 570–574 CrossRef CAS PubMed.
- R. Lonsdale and R. A. Ward, Chem. Soc. Rev., 2018, 47, 3816–3830 RSC.
- U. P. Dahal, R. S. Obach and A. M. Gilbert, Chem. Res. Toxicol., 2013, 26, 1739–1745 Search PubMed.
- Y. Shibata and M. Chiba, Drug Metab. Dispos., 2015, 43, 375–384 CrossRef PubMed.
- A. Clyde, S. Galanie, D. W. Kneller, H. Ma, Y. Babuji, B. Blaiszik, A. Brace, T. Brettin, K. Chard, R. Chard, L. Coates, I. Foster, D. Hauner, V. Kertesz, N. Kumar, H. Lee, Z. Li, A. Merzky, J. G. Schmidt, L. Tan, M. Titov, A. Trifan, M. Turilli, H. Van Dam, S. C. Chennubhotla, S. Jha, A. Kovalevsky, A. Ramanathan, M. S. Head and R. Stevens, J. Chem. Inf. Model., 2022, 62, 116–128 CrossRef CAS PubMed.
- O. Garland, A.-T. Ton, S. Moradi, J. R. Smith, S. Kovacic, K. Ng, M. Pandey, F. Ban, J. Lee, M. Vuckovic, L. J. Worrall, R. N. Young, R. Pantophlet, N. C. J. Strynadka and A. Cherkasov, J. Chem. Inf. Model., 2023, 63, 2158–2169 CrossRef CAS PubMed.
- C. Arnold, Nat. Med., 2023, 29, 1292–1295 CrossRef CAS PubMed.
- D. D. Martinelli, Comput. Biol. Med., 2022, 145, 105403 CrossRef PubMed.
- E. Awoonor-Williams, A. G. Walsh and C. N. Rowley, Biochim. Biophys. Acta, Proteins Proteomics, 2017, 1865, 1664–1675 CrossRef CAS PubMed.
- A. T. Voice, G. Tresadern, R. M. Twidale, H. van Vlijmen and A. J. Mulholland, Chem. Sci., 2021, 12, 5511–5516 RSC.
- E. Awoonor-Williams and C. N. Rowley, J. Chem. Inf. Model., 2021, 61, 5234–5242 CrossRef CAS PubMed.
- S. Martí, K. Arafet, A. Lodola, A. J. Mulholland, K. Świderek and V. Moliner, ACS Catal., 2022, 12, 698–708 CrossRef PubMed.
- J. A. H. Schwöbel, D. Wondrousch, Y. K. Koleva, J. C. Madden, M. T. D. Cronin and G. Schüürmann, Chem. Res. Toxicol., 2010, 23, 1576–1585 Search PubMed.
- E. Awoonor-Williams, J. Kennedy and C. N. Rowley, The Design of Covalent-Based Inhibitors, Academic Press, 2021, vol. 56, pp. 203–227 Search PubMed.
- R. Lonsdale, J. Burgess, N. Colclough, N. L. Davies, E. M. Lenz, A. L. Orton and R. A. Ward, J. Chem. Inf. Model., 2017, 57, 3124–3137 CrossRef CAS PubMed.
- J. M. Smith and C. N. Rowley, J. Comput.-Aided Mol. Des., 2015, 29, 725–735 CrossRef CAS PubMed.
- J. M. Smith, Y. Jami Alahmadi and C. N. Rowley, J. Chem. Theory Comput., 2013, 9, 4860–4865 CrossRef CAS PubMed.
- E. Awoonor-Williams, W. C. Isley III, S. G. Dale, E. R. Johnson, H. Yu, A. D. Becke, B. Roux and C. N. Rowley, J. Comput. Chem., 2020, 41, 427–438 CrossRef CAS PubMed.
- F. Palazzesi, M. R. Hermann, M. A. Grundl, A. Pautsch, D. Seeliger, C. S. Tautermann and A. Weber, J. Chem. Inf. Model., 2020, 60, 2915–2923 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- B. C. Pearce, M. J. Sofia, A. C. Good, D. M. Drexler and D. A. Stock, J. Chem. Inf. Model., 2006, 46, 1060–1068 CrossRef CAS PubMed.
- R. F. Bruns and I. A. Watson, J. Med. Chem., 2012, 55, 9763–9772 CrossRef CAS PubMed.
- I. M. Serafimova, M. A. Pufall, S. Krishnan, K. Duda, M. S. Cohen, R. L. Maglathlin, J. M. McFarland, R. M. Miller, M. Frödin and J. Taunton, Nat. Chem. Biol., 2012, 8, 471–476 CrossRef CAS PubMed.
- D. Baptista, J. Correia, B. Pereira and M. Rocha, J. Integr. Bioinform., 2022, 19, 20220006 CrossRef PubMed.
- Z. Qiao, L. Li, S. Li, H. Liang, J. Zhou and R. Q. Snurr, AIChE J., 2021, 67, e17352 CrossRef CAS.
- M. Yang, B. Tao, C. Chen, W. Jia, S. Sun, T. Zhang and X. Wang, J. Chem. Inf. Model., 2019, 59, 5002–5012 CrossRef CAS PubMed.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- H. L. Morgan, J. Chem. Doc., 1965, 5, 107–113 CrossRef CAS.
- S. Riniker, N. Fechner and G. A. Landrum, J. Chem. Inf. Model., 2013, 53, 2829–2836 CrossRef CAS PubMed.
- P. Banerjee and R. Preissner, Front. Chem., 2018, 6, 93 CrossRef PubMed.
- X. Zhu, V. R. Polyakov, K. Bajjuri, H. Hu, A. Maderna, C. A. Tovee and S. C. Ward, J. Chem. Inf. Model., 2023, 63, 2948–2959 CrossRef CAS PubMed.
- O. Wieder, S. Kohlbacher, M. Kuenemann, A. Garon, P. Ducrot, T. Seidel and T. Langer, Drug Discovery Today: Technol., 2020, 37, 1–12 CrossRef PubMed.
- Z. Wu, S. Pan, F. Chen, G. Long, C. Zhang and P. S. Yu, IEEE Transact. Neural Networks Learn. Syst., 2021, 32, 4–24 Search PubMed.
- V. P. Dwivedi, C. K. Joshi, A. T. Luu, T. Laurent, Y. Bengio and X. Bresson, Benchmarking Graph Neural Networks, arXiv, 2022, preprint, arXiv:2003.00982, DOI:10.48550/arXiv.2003.00982.
- T. B. Hughes, G. P. Miller and S. J. Swamidass, ACS Cent. Sci., 2015, 1, 168–180 CrossRef CAS PubMed.
- GitHub, 2023, https://github.com/RowleyGroup/covalent-classifier Search PubMed.
- D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li, Z. Sayeeda, N. Assempour, I. Iynkkaran, Y. Liu, A. Maciejewski, N. Gale, A. Wilson, L. Chin, R. Cummings, D. Le, A. Pon, C. Knox and M. Wilson, Nucleic Acids Res., 2018, 46, D1074–D1082 CrossRef CAS PubMed.
- T. Liu, Y. Lin, X. Wen, R. N. Jorissen and M. K. Gilson, Nucleic Acids Res., 2007, 35, D198–D201 CrossRef CAS PubMed.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He, B. A. Shoemaker, J. Wang, B. Yu, J. Zhang and S. H. Bryant, Nucleic Acids Res., 2015, 44, D1202–D1213 CrossRef PubMed.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, Nucleic Acids Res., 2011, 40, D1100–D1107 CrossRef PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235 CrossRef CAS PubMed.
- The Uniprot Consortium, Nucleic Acids Res., 2007, 36, D190–D195 CrossRef PubMed.
- G. Landrum, RDKit: Open-Source Chemoinformatics, 2023, https://www.rdkit.org Search PubMed.
- Covalent Modifiers, ed. C. N. Rowley, 2023, https://covalentmodifiers.blogspot.com/ Search PubMed.
- Drug Hunter, ed. R. McAtee, 2023, https://drughunter.com/ Search PubMed.
- C. Avonto, O. Taglialatela-Scafati, F. Pollastro, A. Minassi, V. Di Marzo, L. De Petrocellis and G. Appendino, Angew. Chem., Int. Ed., 2011, 50, 467–471 CrossRef CAS PubMed.
- A. Böhme, D. Thaens, A. Paschke and G. Schüürmann, Chem. Res. Toxicol., 2009, 22, 742–750 Search PubMed.
- A. Birkholz, D. J. Kopecky, L. P. Volak, M. D. Bartberger, Y. Chen, C. M. Tegley, T. Arvedson, J. D. McCarter, C. Fotsch and V. J. Cee, J. Med. Chem., 2020, 63, 11602–11614 CrossRef CAS PubMed.
- K. E. Gilbert, A. Vuorinen, A. Aatkar, P. Pogány, J. Pettinger, E. K. Grant, J. M. Kirkpatrick, K. Rittinger, D. House, G. A. Burley and J. T. Bush, ACS Chem. Biol., 2023, 18, 285–295 CrossRef CAS PubMed.
- D. Wade, S. Airy and J. Sinsheimer, Mutat. Res., Genet. Toxicol., 1978, 58, 217–223 CrossRef CAS PubMed.
- U. Blaschke, A. Paschke, I. Rensch and G. Schüümann, Chem. Res. Toxicol., 2010, 23, 1936–1946 Search PubMed.
- J. A. Hanley and B. J. McNeil, Radiology, 1982, 143, 29–36 CrossRef CAS PubMed.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 CrossRef CAS PubMed.
- L. Xie, L. Xu, R. Kong, S. Chang and X. Xu, Front. Pharmacol, 2020, 11, 606668 CrossRef PubMed.
- C. Cortes and V. Vapnik, Mach. Learn., 1995, 20, 273–297 Search PubMed.
- J. H. Friedman, Ann. Stat., 2001, 29, 1189–1232 Search PubMed.
- S. Pal and S. Mitra, IEEE Trans. Neural Netw. Learn. Syst., 1992, 3, 683–697 CrossRef CAS PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- T. N. Kipf and M. Welling, International Conference on Learning Representations, 2017 Search PubMed.
- A. Kensert, G. Desmet and D. Cabooter, MolGraph: a Python package for the implementation of molecular graphs and graph neural networks with TensorFlow and Keras, arXiv, 2022, preprint, arXiv:2208.0994, DOI:10.48550/arXiv.2208.0994.
- R. Zubatyuk, J. S. Smith, J. Leszczynski and O. Isayev, Sci. Adv., 2019, 5, eaav6490 CrossRef CAS PubMed.
- Y. Zhang, P. Tiño, A. Leonardis and K. Tang, CoRR, 2020, abs/2012.14261 Search PubMed.
- Z. Liu and F. Xu, Front. Artif. Intell., 2023, 6, 974295 CrossRef PubMed.
- P. E. Pope, S. Kolouri, M. Rostami, C. E. Martin and H. Hoffmann, 2019 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), 2019, pp. 10764–10773 Search PubMed.
- M. Chen, Z. Wei, Z. Huang, B. Ding and Y. Li, Simple and Deep Graph Convolutional Networks, arXiv, 2020, preprint, arXiv:2007.02133, DOI:10.48550/arXiv.2007.02133.
- W. L. Hamilton, R. Ying and J. Leskovec, Inductive Representation Learning on Large Graphs, arXiv, 2018, preprint, arXiv:1706.02216, DOI:10.48550/arXiv.1706.02216.
- P. Veličković, G. Cucurull, A. Casanova, A. Romero, P. Liò and Y. Bengio, Graph Attention Networks, arXiv, 2018, preprint, arXiv:1710.10903, DOI:10.48550/arXiv.1710.10903.
- X. Bresson and T. Laurent, Residual Gated Graph ConvNets, arXiv, 2018, preprint, arXiv:1711.07553, DOI:10.48550/arXiv.1711.07553.
- K. Xu, W. Hu, J. Leskovec and S. Jegelka, How Powerful are Graph Neural Networks?, arXiv, 2019, preprint, arXiv:1810.00826, DOI:10.48550/arXiv.1810.00826.
- L. Müller, M. Galkin, C. Morris and L. Rampášek, Attending to Graph Transformers, arXiv, 2023, preprint, arXiv:2302.04181, DOI:10.48550/arXiv.2302.04181.
- F. Monti, D. Boscaini, J. Masci, E. Rodolà, J. Svoboda and M. M. Bronstein, Geometric deep learning on graphs and manifolds using mixture model CNNs, arXiv, 2016, preprint, arXiv:1611.08402, DOI:10.48550/arXiv.1611.08402.
- S. Brody, U. Alon and E. Yahav, How Attentive are Graph Attention Networks?, arXiv, 2022, preprint, arXiv:2105.14491, DOI:10.48550/arXiv.2105.14491.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1874 CrossRef CAS PubMed.
- P. Geerlings, E. Chamorro, P. K. Chattaraj, F. De Proft, J. L. Gázquez, S. Liu, C. Morell, A. Toro-Labbé, A. Vela and P. Ayers, Theor. Chem. Acc., 2020, 139, 36 Search PubMed.
- N. Flores-Holguín, J. Frau and D. Glossman-Mitnik, BMC Res. Notes, 2019, 12, 442 Search PubMed.
- F. Palazzesi, M. A. Grundl, A. Pautsch, A. Weber and C. S. Tautermann, J. Chem. Inf. Model., 2019, 59, 3565–3571 CrossRef CAS PubMed.
- M. R. Hermann, A. Pautsch, M. A. Grundl, A. Weber and C. S. Tautermann, J. Comput.-Aided Mol. Des., 2021, 35, 531–539 CrossRef CAS PubMed.
- T. B. Hughes, N. L. Dang, G. P. Miller and S. J. Swamidass, ACS Cent. Sci., 2016, 2, 529–537 CrossRef CAS PubMed.
- Z. Zhang, K. Z. Guiley and K. M. Shokat, Nat. Chem. Biol., 2022, 18, 1177–1183 CrossRef CAS PubMed.
- C. Rummey and G. Metz, Proteins: Struct., Funct., Bioinf., 2007, 66, 160–171 CrossRef CAS PubMed.
- H. Sootome, H. Fujita, K. Ito, H. Ochiiwa, Y. Fujioka, K. Ito, A. Miura, T. Sagara, S. Ito, H. Ohsawa, S. Otsuki, K. Funabashi, M. Yashiro, K. Matsuo, K. Yonekura and H. Hirai, Cancer Res., 2020, 80, 4986–4997 CrossRef CAS PubMed.
- J. L. Andersen, B. Gesser, E. D. Funder, C. J. F. Nielsen, H. Gotfred-Rasmussen, M. K. Rasmussen, R. Toth, K. V. Gothelf, J. S. C. Arthur, L. Iversen and P. Nissen, Nat. Commun., 2018, 9, 4344 CrossRef PubMed.
- X. Li, V. Hernandez, F. L. Rock, W. Choi, Y. S. L. Mak, M. Mohan, W. Mao, Y. Zhou, E. E. Easom, J. J. Plattner, W. Zou, E. Pérez-Herrán, I. Giordano, A. Mendoza-Losana, C. Alemparte, J. Rullas, I. Angulo-Barturen, S. Crouch, F. Ortega, D. Barros and M. R. K. Alley, J. Med. Chem., 2017, 60, 8011–8026 CrossRef CAS PubMed.
- J. Seidegård and J. DePierre, Chem.-Biol. Interact., 1982, 40, 15–25 CrossRef PubMed.
- Z. Zhang, K. Z. Guiley and K. M. Shokat, Nat. Chem. Biol., 2022, 18, 1177–1183 CrossRef CAS PubMed.
- F. Meric-Bernstam, R. Bahleda, C. Hierro, M. Sanson, J. Bridgewater, H.-T. Arkenau, B. Tran, R. K. Kelley, J. O. Park, M. Javle, Y. He, K. A. Benhadji and L. Goyal, Cancer Discovery, 2022, 12, 402–415 CrossRef CAS PubMed.
- A. L. Slitt, N. J. Cherrington, M. Z. Dieter, L. M. Aleksunes, G. L. Scheffer, W. Huang, D. D. Moore and C. D. Klaassen, Mol. Pharmacol., 2006, 69, 1554–1563 CrossRef CAS PubMed.
- K. Sugihara, S. Kitamura, S. Sanoh, S. Ohta, N. Fujimoto, S. Maruyama and A. Ito, Toxicol. Appl. Pharmacol., 2000, 167, 46–54 CrossRef CAS PubMed.
- S. Bruno, A. Pinto, G. Paredi, L. Tamborini, C. De Micheli, V. La Pietra, L. Marinelli, E. Novellino, P. Conti and A. Mozzarelli, J. Med. Chem., 2014, 57, 7465–7471 CrossRef CAS PubMed.
- A. Tolmachev, A. Sakai, M. Todoriki and K. Maruhashi, Bermuda Triangles: GNNs Fail to Detect Simple Topological Structures, arXiv, 2021, preprint, arXiv:2105.00134, DOI:10.48550/arXiv.2105.00134.
- S. Wang, M. Hadisurya, W. A. Tao, E. Dykhuizen and C. Krusemark, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-tvgn-1.
- E. Awoonor-Williams and C. N. Rowley, J. Chem. Theory Comput., 2016, 12, 4662–4673 CrossRef CAS PubMed.
- C. Jöst, C. Nitsche, T. Scholz, L. Roux and C. D. Klein, J. Med. Chem., 2014, 57, 7590–7599 CrossRef PubMed.
- S. Rao, D. Gurbani, G. Du, R. A. Everley, C. M. Browne, A. Chaikuad, L. Tan, M. Schröder, S. Gondi, S. B. Ficarro, T. Sim, N. D. Kim, M. J. Berberich, S. Knapp, J. A. Marto, K. D. Westover, P. K. Sorger and N. S. Gray, Cell Chem. Biol., 2019, 26, 818–829 CrossRef CAS PubMed.
- M. Kuljanin, D. C. Mitchell, D. K. Schweppe, A. S. Gikandi, D. P. Nusinow, N. J. Bulloch, E. V. Vinogradova, D. L. Wilson, E. T. Kool, J. D. Mancias, B. F. Cravatt and S. P. Gygi, Nat. Biotechnol., 2021, 39, 630–641 CrossRef CAS PubMed.
- G. J. Roth, N. Stanford and P. W. Majerus, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 3073–3076 CrossRef CAS PubMed.
- E. Ortlund, M. W. Lacount, K. Lewinski and L. Lebioda, Biochemistry, 2000, 39, 1199–1204 CrossRef CAS PubMed.
- H. Su, S. Yao, W. Zhao, Y. Zhang, J. Liu, Q. Shao, Q. Wang, M. Li, H. Xie, W. Shang, C. Ke, L. Feng, X. Jiang, J. Shen, G. Xiao, H. Jiang, L. Zhang, Y. Ye and Y. Xu, Nat. Commun., 2021, 12, 3623 CrossRef CAS PubMed.
- A. P. Bento, A. Gaulton, A. Hersey, L. J. Bellis, J. Chambers, M. Davies, F. A. Krüger, Y. Light, L. Mak, S. McGlinchey, M. Nowotka, G. Papadatos, R. Santos and J. P. Overington, Nucleic Acids Res., 2013, 42, D1083–D1090 CrossRef PubMed.
- M. Davies, M. Nowotka, G. Papadatos, N. Dedman, A. Gaulton, F. Atkinson, L. Bellis and J. P. Overington, Nucleic Acids Res., 2015, 43, W612–W620 CrossRef CAS PubMed.
- J.-P. Falgueyret, R. M. Oballa, O. Okamoto, G. Wesolowski, Y. Aubin, R. M. Rydzewski, P. Prasit, D. Riendeau, S. B. Rodan and M. D. Percival, J. Med. Chem., 2001, 44, 94–104 CrossRef CAS PubMed.
- Y. F. Shealy, C. A. Krauth, R. F. Struck and J. A. Montgomery, J. Med. Chem., 1983, 26, 1168–1173 CrossRef CAS PubMed.
- Z. Xiao, Z. Zhou, C. Chu, Q. Zhang, L. Zhou, Z. Yang, X. Li, L. Yu, P. Zheng, S. Xu and W. Zhu, Eur. J. Med. Chem., 2020, 203, 112511 CrossRef CAS PubMed.
- M. E. Prime, O. A. Andersen, J. J. Barker, M. A. Brooks, R. K. Y. Cheng, I. Toogood-Johnson, S. M. Courtney, F. A. Brookfield, C. J. Yarnold, R. W. Marston, P. D. Johnson, S. F. Johnsen, J. J. Palfrey, D. Vaidya, S. Erfan, O. Ichihara, B. Felicetti, S. Palan, A. Pedret-Dunn, S. Schaertl, I. Sternberger, A. Ebneth, A. Scheel, D. Winkler, L. Toledo-Sherman, M. Beconi, D. Macdonald, I. Muñoz-Sanjuan, C. Dominguez and J. Wityak, J. Med. Chem., 2012, 55, 1021–1046 CrossRef CAS PubMed.
- X. Li, V. Hernandez, F. L. Rock, W. Choi, Y. S. L. Mak, M. Mohan, W. Mao, Y. Zhou, E. E. Easom, J. J. Plattner, W. Zou, E. Pérez-Herrán, I. Giordano, A. Mendoza-Losana, C. Alemparte, J. Rullas, I. Angulo-Barturen, S. Crouch, F. Ortega, D. Barros and M. R. K. Alley, J. Med. Chem., 2017, 60, 8011–8026 CrossRef CAS PubMed.
- G.-F. Zha, S.-M. Wang, K. Rakesh, S. Bukhari, H. Manukumar, H. Vivek, N. Mallesha and H.-L. Qin, Eur. J. Med. Chem., 2019, 162, 364–377 CrossRef CAS PubMed.
- R. Kozaki, T. Yoshizawa, S. Tohda, T. Yasuhiro, S. Hotta, Y. Ariza, Y. Ueda, M. Narita and K. Kawabata, Blood, 2011, 118, 3731 CrossRef.
- V. Cano and C. Rowley, ChEMBL with GNN preds and Eli Lilly, 2024, https://figshare.com/articles/dataset/ChEMBL_with_GNN_preds_and_Eli_Lil_ly/25853467 Search PubMed.
- R. S. Herbst, M. Fukuoka and J. Baselga, Nat. Rev. Cancer, 2004, 4, 956–965 CrossRef CAS PubMed.
- D. Li, L. Ambrogio, T. Shimamura, S. Kubo, M. Takahashi, L. R. Chirieac, R. F. Padera, G. I. Shapiro, A. Baum, F. Himmelsbach, W. J. Rettig, M. Meyerson, F. Solca, H. Greulich and K.-K. Wong, Oncogene, 2008, 27, 4702–4711 CrossRef CAS PubMed.
- A. Nigam, R. Pollice, M. Krenn, G. d. P. Gomes and A. Aspuru-Guzik, Chem. Sci., 2021, 12, 7079–7090 RSC.
- M. Voršilák, M. Kolář, I. Čmelo and D. Svozil, J. Cheminf., 2020, 12, 35 Search PubMed.
- Molecular Operating Environment (MOE), Chemical Computing Group Inc., 2013.08, 1010 Sherbooke St. West Suite #910 Montreal Q, Canada H3A 2R7, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the hyperparameter optimization, list of GNN atomic and bond features, filters removed from pattern-based tools, additional figures of data set similarity and comparision of filters to GNN scores. See DOI: https://doi.org/10.1039/d4dd00038b |
| This journal is © The Royal Society of Chemistry 2024 |