
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective dehydrogenation of ammonia borane to borazine and derivatives by rhodium olefin complexes†
Pascal
Jurt
a,
Juan José
Gamboa-Carballo
 ab,
Clara
Schweinzer
a,
Daniel
Himmelbauer
c,
Debora
Thöny
a,
Thomas L.
Gianetti
*d,
Monica
Trincado
*ae and
Hansjörg
Grützmacher
*a
ab,
Clara
Schweinzer
a,
Daniel
Himmelbauer
c,
Debora
Thöny
a,
Thomas L.
Gianetti
*d,
Monica
Trincado
*ae and
Hansjörg
Grützmacher
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, CH-8049 Zurich, Switzerland. E-mail: hgruetzmacher@ethz.ch
bHigher Institute of Technologies and Applied Sciences (InSTEC), University of Havana, Ave. S. Allende 1110, 10600 Havana, Cuba
cInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163, A-1060 Vienna, Austria
dDepartment of Chemistry and Biochemistry, University of Arizona, University Blvd., Tucson, AZ 85719, USA. E-mail: tgianetti@email.arizona.edu
eDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: monica.trincado@chem.uzh.ch
First published on 13th August 2024
Abstract
This report presents a selective synthetic approach towards borazine from ammonia borane using a dinuclear rhodium olefin homogeneous catalyst. The synthesis and spectroscopic characterization of a dirhodium ammonia borane complex as an intermediate provides insight into a possible mode of activation.
Introduction
Borazine (B3N3H6), isoelectronic to benzene, is referred to as ‘inorganic benzene’ due to its structural resemblance and represents an important precursor to develop BN-ceramics.1,2 Both the electronic structure and reactivity patterns of B3N3H6 differ largely from those of benzene. Borazine undergoes addition reactions and is sensitive to ambient humidity, unstable over time and decomposes upon heating in the presence of oxygen, which complicates the development of efficient synthetic protocols.3 Recent research efforts have focused on materials derived from organoborazines, which in turn allow for the design of new gas storage and separation systems,4 materials with specific (opto)electronic properties (especially hybrid BCN materials) and new applications in supramolecular chemistry.5 However, practical applications of borazine and substituted borazines continue to be held back by the absence of convenient and selective synthetic routes. Principal constraints in current procedures include the utilization of harsh reaction conditions and excess organometallic hazardous reagents, boron halides or harmful solvents.6 Since the first synthesis of borazine by thermolysis of an ammonia diborane adduct, nearly one century ago by Stock and Pohland,7 several other procedures have been reported. Sneddon et al. reported a convenient synthesis of borazine by treating ammonia borane (AB) or a mixture of ammonium sulphate and sodium borohydride in tetraglyme at 120–140 °C.8 To avoid the rather harsh conditions, the reaction can be carried out at T < 50 °C in the presence of aluminium chloride as a catalyst, obtaining borazine in yields of about 67%, which to date represents the best protocol to obtain B3N3H6 on a gram scale.9 There are limited examples of transition metal catalysts that are able to convert ammonia borane AB selectively to borazine.10–13 The binuclear Ru complex known as Shvo's catalyst (1) and a nitrosyl Re(I) complex (2) (Fig. 1) allow the formation of borazine in good to excellent NMR yields (75%10 and 99%,11 respectively). However, relatively high catalyst loadings (5 mol%) were employed and the product was not isolated.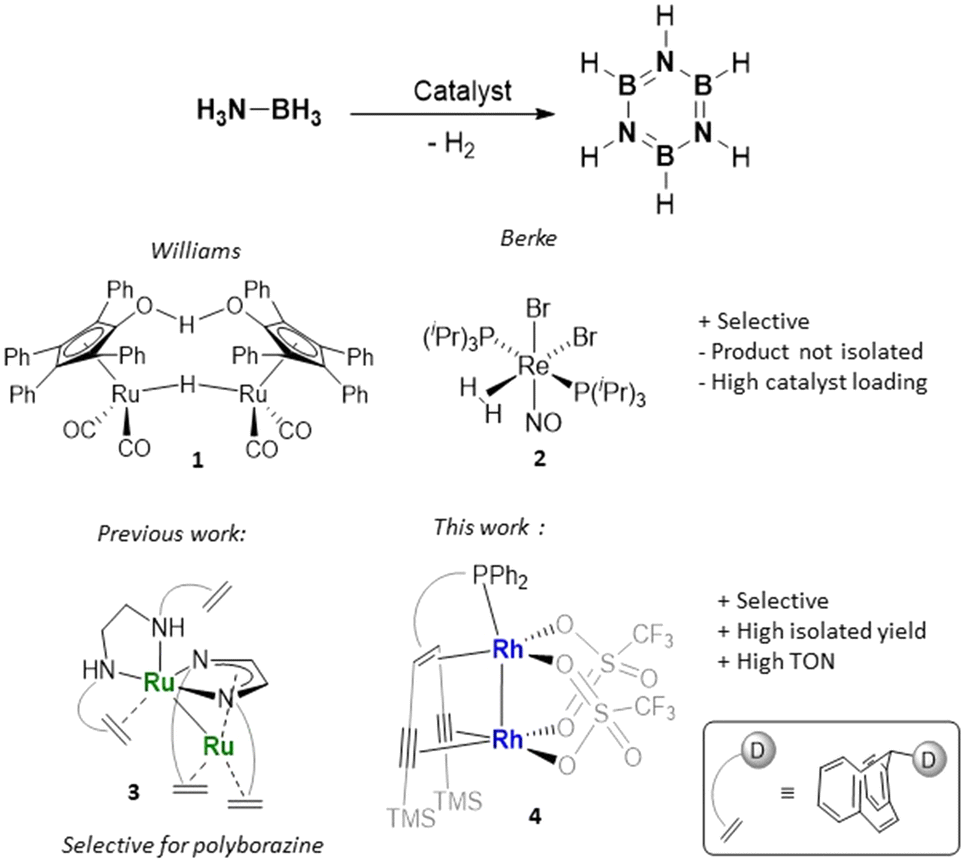 | ||
| Fig. 1 Previously reported catalysts for the dehydrogenation of H3N–BH3 to borazine or polyborazine and the new catalyst (D = amine or phosphine group). | ||
Manners et al. explored the photocatalytic dehydrogenation of AB with [CpFe(CO)2]2 as a catalyst (5 mol%), observing the formation of the oligomer B-(cyclodiborazanyl)amine-borane (BCDB) as an intermediate and its further dehydrogenation to borazine in 35% yield.12 Only once the isolation of borazine was reported from a transition metal-catalysed reaction, using [Rh2(μ-Cl)2(cod)2] (cod = 1,5-cyclooctadiene) (1 mol%) as a catalyst. The reaction proceeded with high selectivity, but the isolated yield was merely 10% and the reaction was found to be catalysed by rhodium nanoparticles or clusters.13 In our recent study, a ruthenium complex (3) with a Ru–Ru bond and a multidentate non-innocent ligand containing a diazadiene (dad) and olefins as bindings sites was able to catalyse the formation of soluble polycondensed borazine from ammonia borane.14 As a continuation of this investigation, we report here a dinuclear olefin Rh metal complex as an active and selective catalyst for the dehydrogenation of ammonia borane to borazine (Fig. 1). The catalyst was also employed in the dehydropolymerization of primary amine boranes to polymeric N-substituted borazines or BCN materials containing six-membered unsaturated B3N3 rings.
Results and discussion
Intrigued by the activity and selectivity of our previously reported catalyst (3) with two low-valent Ru centres, we investigated the reactivity of the dinuclear rhodium complexes 4 and 5 with two d8-Rh(I) centres (Scheme 1). In both complexes, the ligand {(TMS)C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C}2tropPPh2 containing a phosphane, an alkene, and two alkyne sites anchors two late transition metals in close proximity leading to complexes with dative intermetallic bonds.15 The resulting species are able to either undergo hydrogen transfer to the unsaturated ligand (4) or add hydrogen reversibly (5).15a The reaction of AB in 1,2-dimethoxyethane (DME) solution at 80 °C with either 4 or 5 as a catalyst did not lead to noteworthy formation of condensed borazine or polyaminoborane, but selectively produced borazine, B3N3H6. Catalyst 4 is superior to 5 concerning both activity and selectivity (entries 1 and 4, Table 1), affording almost exclusively borazine. A low catalyst loading of 4 (0.35 mol%) in a 1.1 M solution of ammonia borane in DME led to the formation of borazine in 95% yield (TOF = 18 h−1), with polyborazine as a side product. Although the isolated yield (72%) is somewhat lower than the NMR yield, this is to the best of our knowledge the highest reported for any borazine synthesis.9 By lowering the reaction temperature to 50 °C, dehydrogenation of ammonia borane with complex 4 formed borazine with low selectivity. Complete conversion of AB was achieved after 48 hours leading to a mixture of products. The related monometallic Rh(I) complex 6
C}2tropPPh2 containing a phosphane, an alkene, and two alkyne sites anchors two late transition metals in close proximity leading to complexes with dative intermetallic bonds.15 The resulting species are able to either undergo hydrogen transfer to the unsaturated ligand (4) or add hydrogen reversibly (5).15a The reaction of AB in 1,2-dimethoxyethane (DME) solution at 80 °C with either 4 or 5 as a catalyst did not lead to noteworthy formation of condensed borazine or polyaminoborane, but selectively produced borazine, B3N3H6. Catalyst 4 is superior to 5 concerning both activity and selectivity (entries 1 and 4, Table 1), affording almost exclusively borazine. A low catalyst loading of 4 (0.35 mol%) in a 1.1 M solution of ammonia borane in DME led to the formation of borazine in 95% yield (TOF = 18 h−1), with polyborazine as a side product. Although the isolated yield (72%) is somewhat lower than the NMR yield, this is to the best of our knowledge the highest reported for any borazine synthesis.9 By lowering the reaction temperature to 50 °C, dehydrogenation of ammonia borane with complex 4 formed borazine with low selectivity. Complete conversion of AB was achieved after 48 hours leading to a mixture of products. The related monometallic Rh(I) complex 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15a leads to a mixture of BN products, suggesting that the bimetallic Rh2 unit in 4 is essential for the activity and selectivity (entry 5, Table 1).
15a leads to a mixture of BN products, suggesting that the bimetallic Rh2 unit in 4 is essential for the activity and selectivity (entry 5, Table 1).
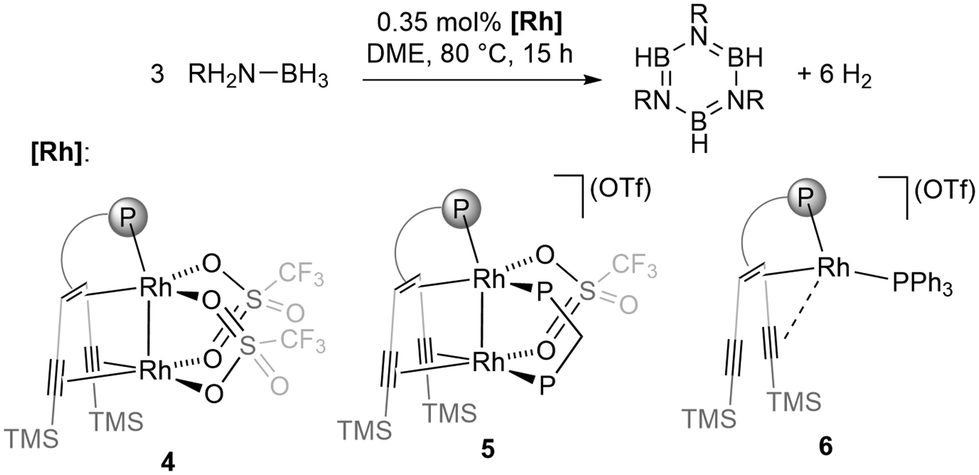 | ||
| Scheme 1 Dehydrogenation of ammonia borane with rhodium olefin complexes 4, 5 and 6 (P = PPh2). | ||
| Entry | Catalyst | R | Conv. [%] | Yielda [%] | TOF [h−1] | TON |
|---|---|---|---|---|---|---|
| a Yield of borazine determined by 11B NMR with NaBPh4 as an internal standard. b Isolated yield. c Catalyst loading 0.08 mol%. d Catalyst loading 0.8 mol%. e Catalyst loading 0.8 mol%, 35 h. | ||||||
| 1 | 4 | H | >99 | 95(72)b | 18 | 271 (938)c |
| 2 | 4 | Me | >99 | 65d | 6 | 81 |
| 3 | 4 | t Bu | >99 | 85e | 3 | 107 |
| 4 | 5 | H | >99 | 83 | 16 | 237 |
| 5 | 6 | H | >99 | 63 | 12 | 188 |
| 6 | — | H | >99 | 43 | — | — |
Thermal decomposition of AB in the absence of a catalyst leads to poor selectivity and low yield of borazine (entry 6, Table 1). The conversion rate of the reaction catalysed by 4 is not influenced by the addition of mercury while the addition of triphenylphosphine poisons the catalyst, indicating that the dehydrogenation is catalysed by a molecular species in homogeneous solution. This is in contrast to an earlier report in which the precatalyst [Rh2(μ-Cl2)(COD)2] was converted under the reaction conditions to colloidal Rh particles as catalytically active species.13b The solvent has a significant impact on the selectivity of the reaction. Changing from DME to diglyme lowers the yield from 95% to 75%, while more polyborazylene is observed (see Fig. S4†). In acetonitrile, only traces of borazine are observed and polyaminoborane is formed as the main product. N-Monosubstituted amine boranes can also be dehydrogenated to the corresponding N-substituted borazine analogues in high yields (entries 2 and 3, Table 1). N-Dimethylamine borane reacts rapidly and selectively forms the four-membered ring [Me2N–BH2]2 (quantitative yield after 4 h at 0.8% catalyst loading, TON = 125).
Cross-linked BCN materials are highly sought-after materials due to their remarkable properties such as wider HOMO–LUMO gaps than those of carbon analogues and the possibility of achieving coatings on metal surfaces.16 We investigated the dehydrogenation of N-propargylamine-borane 717 which contains a cross-linkable functional group using 4 as a catalyst (Fig. 2a). MAS NMR spectroscopic analysis of the resulting insoluble polymeric material formed after 15 h at 80 °C indicated that partial dehydrogenation occurred, and we denote the material as BCNHrcat (where Hr stands for residual hydrogen). 11B MAS NMR spectroscopy showed that the catalytic dehydrogenation led to the formation of B3N3 rings as cross-linking points, while also indicating the presence of residual BH3 (δ11B = −21 ppm) and BH2 groups (δ11B = 0 ppm) (Fig. S8†). When the parent N-propargylamine-borane is thermally dehydrogenated at 80 °C without a catalyst, significantly fewer B3N3 rings are formed. Further loss of hydrogen from BCNHrcat is achieved by heating at 200 °C affording a hydrogen-poor BCN material (BCNHpcat). This transformation to a hydrogen-poor BCN material at a rather low temperature only occurs from the previously partially dehydrogenated BCN polymer BCNHrcat, but not from heating N-propargylamine borane alone, as was shown by TGA (Fig. S10–S14†). It cannot be excluded that the transformation from BCNrcat to BCNpcat is catalyzed by residual Rh in the material (1.943 ± 0.023 mg Rh per g BCNrcat, determined by ICPMS). During the annealing process, all signals from νCC and νB–H vanished in the IR spectrum while two absorption peaks at 1256 and 846 cm−1 (associated with the B–N bonds) characteristic for BCN materials increased in intensity (Fig. S15†). Furthermore, the 11B-MAS-NMR spectrum showed a significant increase in the intensity of the signals corresponding to borazine units (Fig. S16†). Scanning electron microscopy (SEM) was used in order to investigate the change in morphology when the hydrogen-rich BCNHrcat was thermally dehydrogenated on a silicon support (Fig. 2b).
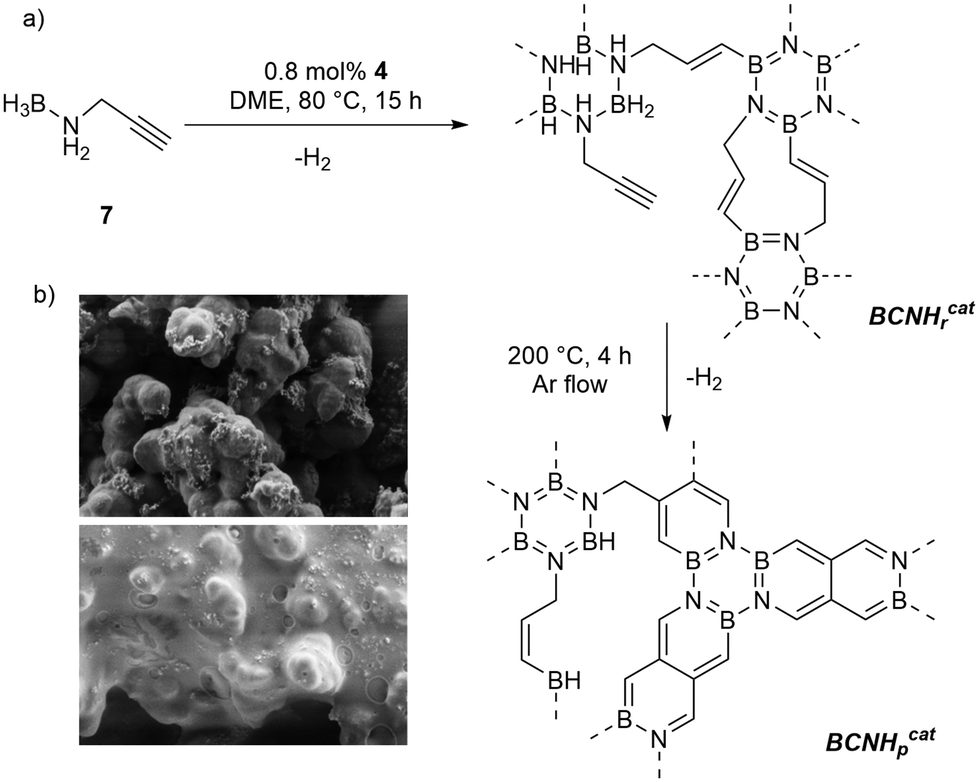 | ||
| Fig. 2 (a) Dehydrogenation of N-propargylamine-borane catalyzed by 4 forming a highly crosslinked polymeric material BCNHrcat and conversion to an amorphous BCN material by thermal treatment (BCNHpcat). (b) SEM image of BCNHrcat (top) and BCNHpcat (bottom). | ||
The crosslinked polymer, derived from BCNHrcat, is obtained as beige powder and forms small lumps on silicon as a support material. A much smoother and more extended layered structure of a BCN material is observed from the precursor material BCNHpcat (the material produced when BCNHrcat was further dehydrogenated at 200 °C for 12 h). The difference between both BCN@Si materials is shown by the SEM images in Fig. 2b.
To gain some insight into a possible reaction mechanism for the catalysed dehydrogenation of AB, the progress of the reaction was monitored by 11B NMR spectroscopy in the presence of complex 4. Previously reported compounds cyclotriborazane (CTB), B-(cyclodiborazanyl)amine-borane (BCDB), and the tetramer B-(cyclotriborazanyl)amine-borane (BCTB) were detected as intermediates (Fig. S21†).18 To demonstrate that these oligomers stem from the tri- or tetramerization of the highly reactive monomeric aminoborane H2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BH2, an excess of cyclohexene as a trapping reagent was added. Under these conditions, the disubstituted aminoborane H2NBCy2 was observed as the only boron-containing compound in quantitative yield. This result suggests indeed that H2NBH2 is formed and released from the catalyst in solution and serves as a source for the formation of CTB, BCDB, and BCTB. Likely, CTB with its saturated six-membered ring is simply dehydrogenated to give borazine and three equivalents of H2. Autrey et al. have shown that the constitutional isomer BCDB with a four-membered B2N2 ring can be converted to CTB at 80 °C. Consequently, we assume that BCDB is likewise converted to B3N3H6.19 The tetramer BCTB can thermally decompose to tetrahydroborazine, B3N3H10, and H3N–BH320 and this is likely the route by which this intermediate is converted to borazine. But it may also be responsible for the minor formation of polyborazines as side products. To probe this hypothesis, a catalytic reaction was carried out with BCTB as a starting material. In this reaction, the selectivity for borazine formation dropped to 63% yield (Fig. S22†). The salt [H2B(NH3)2]+[BH4]− (DADB) when used as a substrate led to even lower yields of B3N3H6 and considerable amounts of polyaminoborane are observed instead. Note that heating borazine for 15 h at 80 °C in the presence of 4 does not lead to any condensation reaction which explains the notable selectivity of this bimetallic complex in comparison with other catalysts.
BH2, an excess of cyclohexene as a trapping reagent was added. Under these conditions, the disubstituted aminoborane H2NBCy2 was observed as the only boron-containing compound in quantitative yield. This result suggests indeed that H2NBH2 is formed and released from the catalyst in solution and serves as a source for the formation of CTB, BCDB, and BCTB. Likely, CTB with its saturated six-membered ring is simply dehydrogenated to give borazine and three equivalents of H2. Autrey et al. have shown that the constitutional isomer BCDB with a four-membered B2N2 ring can be converted to CTB at 80 °C. Consequently, we assume that BCDB is likewise converted to B3N3H6.19 The tetramer BCTB can thermally decompose to tetrahydroborazine, B3N3H10, and H3N–BH320 and this is likely the route by which this intermediate is converted to borazine. But it may also be responsible for the minor formation of polyborazines as side products. To probe this hypothesis, a catalytic reaction was carried out with BCTB as a starting material. In this reaction, the selectivity for borazine formation dropped to 63% yield (Fig. S22†). The salt [H2B(NH3)2]+[BH4]− (DADB) when used as a substrate led to even lower yields of B3N3H6 and considerable amounts of polyaminoborane are observed instead. Note that heating borazine for 15 h at 80 °C in the presence of 4 does not lead to any condensation reaction which explains the notable selectivity of this bimetallic complex in comparison with other catalysts.
DFT calculations were performed on the reaction between 4 and AB (Scheme 2). In the first slightly exothermic step, a simple adduct 4(AB) is obtained in which a B–H binds terminally to Rh1 – the external rhodium centre of the Rh2 core – such that a Rh1–H–B bridge is formed in the trans-position to Rh2 – the internal rhodium centre. Adduct 4(AB) is almost iso-energetic with its isomer 4(AB)′ in which the Rh1–H–B bridge is in the cis-position to Rh2. Via the activated complex [TS1] one of the protons from the NH3 group is transferred to the α-carbon centre of the coordinated alkyne unit while one hydride is simultaneously transferred from the BH3 group to Rh1. This unusual intramolecular hydrogen transfer reaction gives complex 8 with an H2NBH2 molecule bound via a B–H–Rh bridge formed in the trans-position to Rh2 and is the rate-determining step. In a β-hydride insertion, 8 rearranges via a small barrier to 9, which contains a bridging carbene ligand between the rhodium centres Rh1 and Rh2 and a π-side-on coordinated H2NBH2 ligand bound to Rh1.
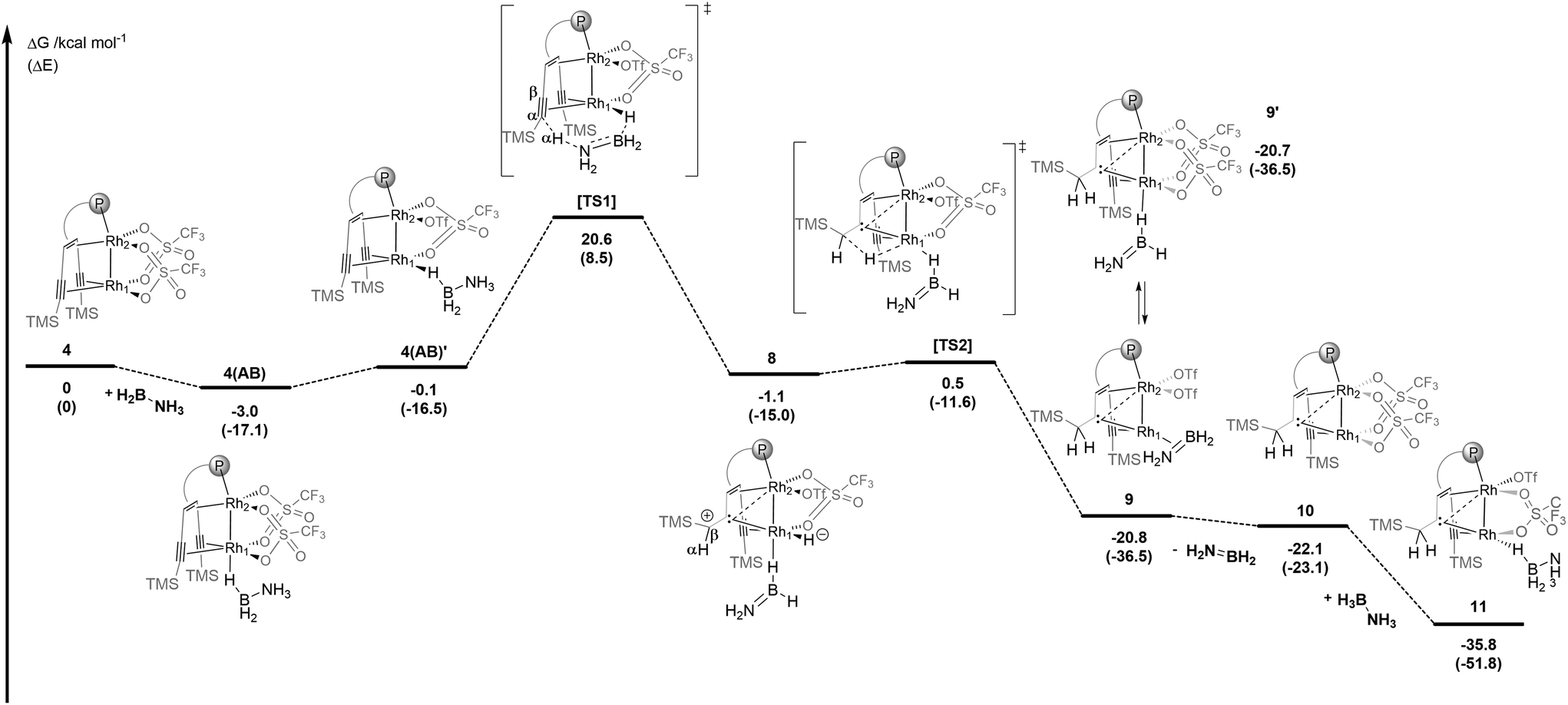 | ||
| Scheme 2 Proposed mechanism for the dehydrogenation of AB by complex 4 leading to 11 as the resting state calculated by DFT (Gaussian09, SMD/PBE0-D3/def2-SVP). Structures 4(AB)′ and 11 are stabilized by hydrogen bridges between the NH3 and the OTf− groups (not shown for clarity, see the optimized structures in the ESI†). | ||
Complex 9 is in equilibrium with isomer 9′, which contains a H2NBH2 moiety bound via a B–H–Rh unit to the external rhodium centre Rh1. The dissociation of H2NBH2 from this complex is a weakly exothermic reaction (ΔG = −1.2 kcal mol−1) to give the carbene bridged Rh2 complex 10 (vide infra). Finally, 10 reacts with AB in an exothermic reaction (ΔG = −11.6 kcal mol−1) to give complex 11 which is an adduct between 10 and an ammonia borane molecule that binds end-on via a B–H bond to Rh1 in the cis-position to Rh2 and is further stabilized by a hydrogen bond between the NH3 group and a coordinated triflate (OTf−). According to the DFT calculations, adduct 11 is the resting state of the catalytic system and might be an observable species. Therefore, 4 was reacted with 10 equivalents of ammonia borane in THF-d8 at room temperature (Fig. 3a). After a few minutes a new complex 11 is formed, and the proposed structure of which is based on multi-nuclear two-dimensional NMR spectroscopic analysis (Fig. 3b–d) combined with DFT calculations. The NMR resonance of the bridging carbene 13C nucleus is observed at δ13C = 184.0 ppm. The 13C chemical shifts of the coordinated alkyne unit are observed at δ13C = 80.7 and 107.6 ppm. A characteristic signal in the 1H NMR spectrum at δ1H = −11.37 ppm (Fig. 3b) with a large 1JHRh coupling constant of 25.3 Hz (determined by 1H–103Rh HMQC; Fig. 3c) indicates a direct Rh–H bond. A triplet at δ11B = −0.24 ppm with a 1JBH coupling constant of 1JBH = 118 Hz is observed in the 11B NMR spectrum (Fig. S29†). Furthermore, when 15N labeled ammonia borane, H3N15–BH3, is used as a substrate, a quartet at δ15N = 28.4 ppm (1JNH = 69.8 Hz) is observed, which proves the presence of an NH3 group in the molecule (Fig. 3d). In principle, these data would be consistent with a dinuclear rhodium complex in which the BH3 group has been oxidatively added to one Rh center. However, DFT calculations indicate that the formation of a boryl hydride Rh+3(H)(BH2–NH3) is rather endothermic (ΔE = 36.8 kcal mol−1) and therefore highly unlikely. Neither the formation of an anionic hydride complex with the formula [(THF)BH2(NH3)][Rh2(H)(OTf)2({(TMS)CC(TMS)CH2C}tropPPh2)] seems possible (20.0 kcal mol−1) (see the ESI† for details). Another possibility instead is that structure 11 (Fig. 3e) is a more plausible alternative, which contains a bridging Rh–H–B unit in the trans-position to the bridging carbene unit. Related coordination modes have been observed in aminoborane complexes with a dinuclear Rh2 core where either a BH2 group bridges both Rh centres to give a Rh–H–B–H–Rh unit21 or coordinates via both B–H bonds to one Rh centre.22 The coordination mode via only one B–H bond is rare and has so far only been observed for a Ru complex.23 Note that the 1JBH coupling in M–H–B bridges may be too small to observe.24,25
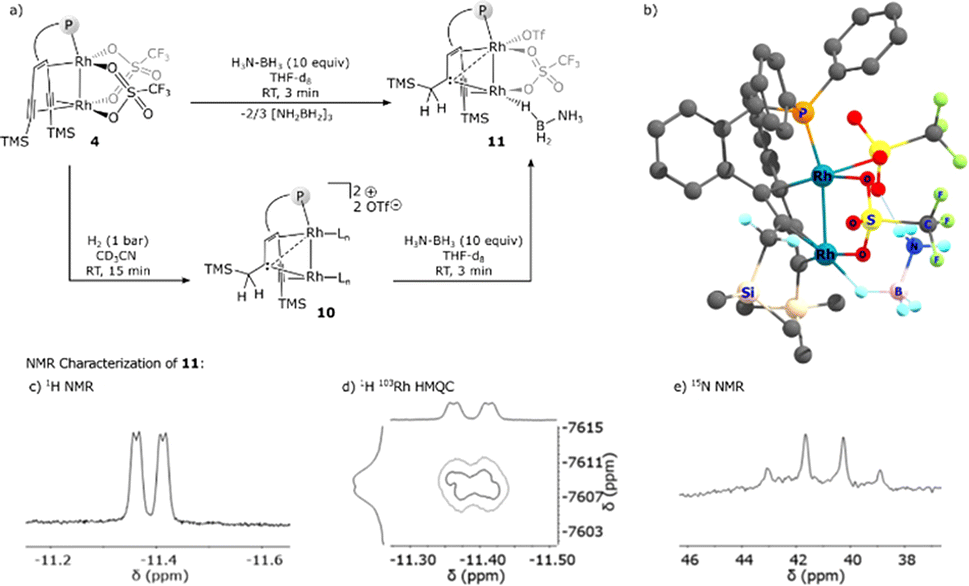 | ||
| Fig. 3 (a) Synthesis and proposed structure of 11 (Ln = CD3CN). (b) DFT calculated and geometry optimized structure of 11 (see the ESI†). (c) Hydride region of the 1H NMR spectrum of 11. (d) 1H 103Rh HMQC spectrum of 11; section showing the signal from the coupling of the hydridic proton with the rhodium center. (e) 15N NMR spectrum of 11 showing the presence of NH3. | ||
Complex 11 is closely related to compound 10 (δ13Ccarbene = 171.0 ppm; δ13Calkyne = 76.9, 101.7 ppm), which contains likewise a central Rh2 core bridged by a carbene. The alkyne group acts as a hydride reservoir. Similar hydrogen transfer to a single alkyne C atom and metal carbene formation was reported by Fürstner et al.26 Complex 10 was previously isolated from the reaction of complex 4 with hydrogen.15a In fact, complex 10 reacts cleanly with AB to give complex 11 as the sole product (Fig. 3a), which strongly supports our assignment of the structure of 11. In addition, complex 11 is likewise catalytically active (Fig. S35†) and was also detected in the reaction catalyzed by complex 4. Complex 11 is only observed in ethereal solvents, while deactivation occurs in acetonitrile or dichloromethane, in line with the observation that efficient catalysis fails in these solvents. Based on these experimental results, we propose a possible full catalytic cycle for the conversion of H3B–NH3 to H2BNH2 and H2, which is presented in Scheme 3. This is still a very simplified model and some ΔG0 values are too high, yet, to explain the comparatively high activity. It is likely that the reactions are even more complicated, and in some steps, more than one molecule of ammonia borane may be involved.
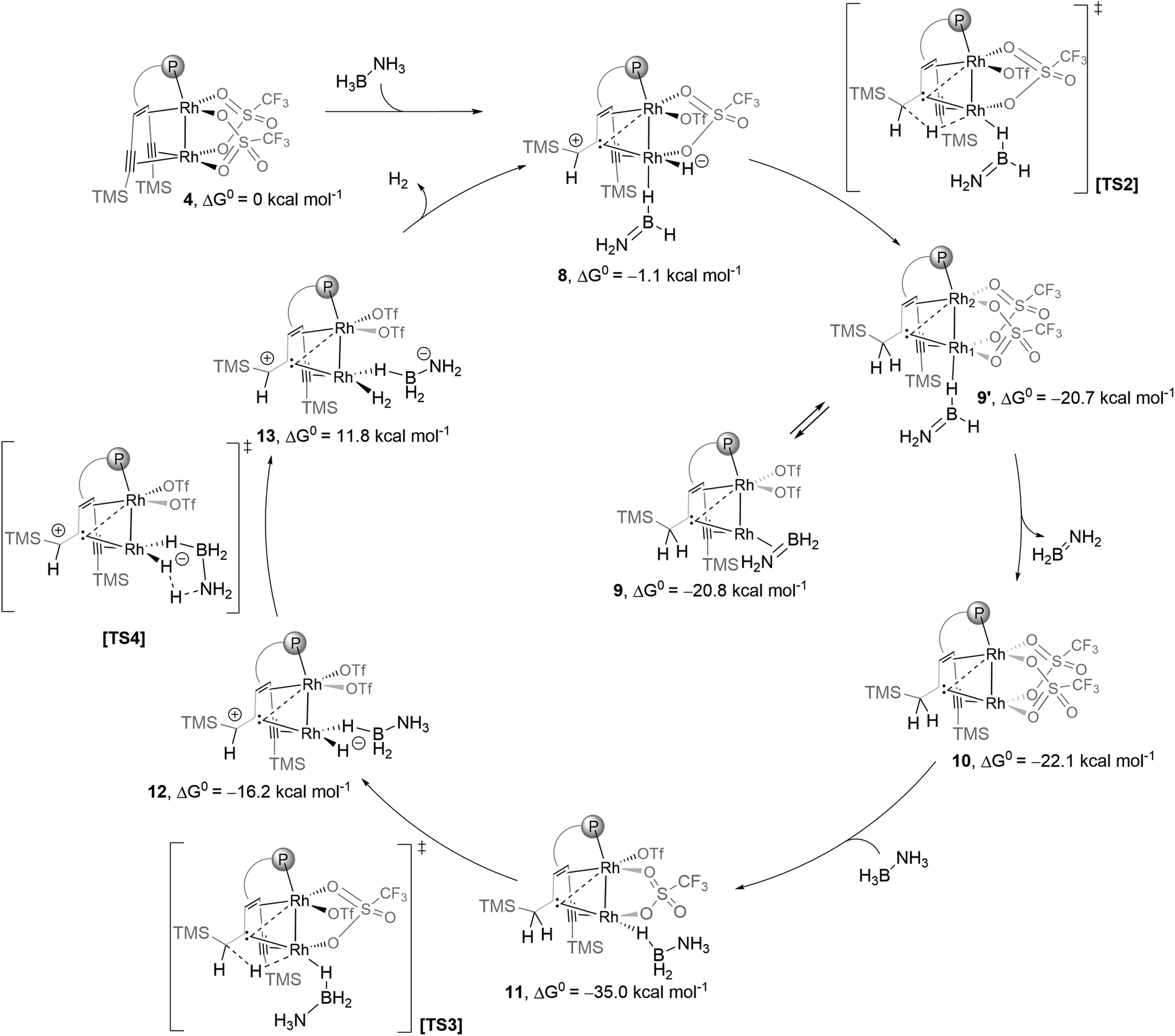 | ||
| Scheme 3 Proposed catalytic cycle according to DFT (Gaussian09, SMD/PBE0-D3/def2-SVP). Structures 11, 12, [TS3] and [TS4] are stabilized by hydrogen bonding between the NH3 and the OTf groups (not shown for clarity, see the optimized structures in the ESI†). | ||
Although the results of the DFT calculations can merely be regarded as preliminary in view of the complexity of the reaction, they indicate that the bis(alkynyl)trop platform – as an unsaturated molecular hydrocarbon support for the Rh2 unit – can act as an unusual cooperating ligand.15a,27 Specifically, protonation of one of the coordinated alkynyl units converts this into a bridging carbene ligand to which a carbenium unit is attached in the β-position to the metal centres. This electrophilic carbenium unit serves as an acceptor for a hydride ligand from a metal centre, such that in the simplified form the process M–M ← (RCCR′) + H2 ⇄ M–M(H−) ← :CR-C+HR′ ⇄ M ← :CR-CH2R′ is rather easily feasible (in the present example, the highest activation barrier is about 20 kcal mol−1; see the sequence 4(AB)′ ⇄ TS1 ⇄ 8 ⇄ TS2 ⇄ 9 in Scheme 3).
Conclusions
The dinuclear rhodium compound 4, which contains a direct metal–metal bond, is a highly selective catalyst for the dehydrogenation of ammonia borane, H3N–BH3, to borazine, B3N3H6. This leads to a synthetic protocol for the synthesis of borazine in high yield and purity from a homogeneously transition metal-catalysed reaction. The method could be extended to the preparation of organic borazines from primary amine boranes and highly cross-linked BNC amorphous polymers with a high content of six-membered B3N3 rings as cross-linkers. We assume that the dinuclear low-valent Rh(I) metal complexes investigated in this study lead to the formation of H2NBH2 as the first product of AB dehydrogenation. This assumption is bolstered by trapping experiments. Subsequently amino borane H2NBH2 as a highly reactive monomer forms cyclic oligomers, detected by NMR spectroscopy as intermediates, which finally converge selectively into borazine as a main product. Experiments combined with DFT calculations indicate that the bis(alkynyl)trop ligand in 4 and 5 is (i) converted into a bridging carbene ligand and (ii) the latter may play a cooperative role in the catalytic dehydrogenation of AB in the sense that the carbon centre adjacent to the μ2-carbene unit serves as a reservoir for a hydrogen centre. In combination with our previous observation that a low-valent dinuclear Ru complex allows for the selective production of polyborazine, the results reported in this paper show that dinuclear metal complexes may hold high potential for the syntheses of related BN and BNC materials and therefore should be further explored.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support by SNF (grants 192106, 181966) and ETH-Zürich is gratefully acknowledged. D. H. acknowledges the Austria Science Fund (FWF) (J-4571-N).References
- (a) E. Wiberg and A. Bolz, Ber. Dtsch. Chem. Ges. A/B, 1940, 73, 209 CrossRef; (b) R. T. Paine and C. K. Narula, Chem. Rev., 1990, 90, 73 CrossRef CAS.
- F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, Wiley-Interscience, New York, 5th edn, 1988, pp. 204 Search PubMed.
- (a) H. I. Schlesinger, L. Horvitz and A. B. Burg, J. Am. Chem. Soc., 1936, 58, 409 CrossRef CAS; (b) H. I. Schlesinger, D. M. Ritter and A. B. Burg, J. Am. Chem. Soc., 1938, 60, 1296 CrossRef CAS; (c) P. v. R. Schleyer, H. Jiao, N. J. R. van Eikema Hommes, V. G. Malkin and O. L. Malkina, J. Am. Chem. Soc., 1997, 119, 12669 CrossRef CAS.
- (a) T. Wilberforce, A. G. Olabi, E. T. Sayed, K. Elsaid and M. A. Abdelkareem, Sci. Total Environ., 2021, 761, 143203 CrossRef CAS; (b) J. K. Stolaroff, S. Bhattacharyya, C. A. Smith, W. L. Bourcier, P. J. Cameron-Smith and R. D. Aines, Environ. Sci. Technol., 2012, 46, 6455 CrossRef CAS PubMed; (c) J. O. Abe, A. P. I. Popoola, E. Ajenifuja and O. M. Popoola, Int. J. Hydrogen Energy, 2019, 44, 15072 CrossRef CAS.
- (a) A. Wakamiya, T. Ide and S. Yamaguchi, J. Am. Chem. Soc., 2005, 127, 14859 CrossRef CAS PubMed; (b) M. Lorenzo-García and D. Bonifazi, Chimia, 2017, 71, 550 CrossRef PubMed; (c) J. Dosso, T. Battisti, B. D. Ward, N. Demitri, C. E. Hughes, P. A. Williams, K. D. M. Harris and D. Bonifazi, Chemistry, 2020, 26, 6608 CrossRef CAS PubMed.
- For recent reviews of the synthesis of borazines, see: (a) I. Neogi and A. M. Szpilman, Synthesis, 2022, 54, 1877 CrossRef CAS; (b) D. Marchionni, S. Basak, A. N. Khodadadi, A. Marrocchi and L. Vaccaro, Adv. Funct. Mater., 2023, 33, 2303635 CrossRef CAS.
- A. Stock and E. Pohland, Chem. Ber., 1926, 59, 221 Search PubMed.
- T. Wideman and L. G. Sneddon, Inorg. Chem., 1995, 34, 1002 CrossRef CAS.
- J. S. Li, C. R. Zhang, B. Li, F. Cao and S. Q. Wang, Eur. J. Inorg. Chem., 2010, 1763 CrossRef CAS.
- Ru: (a) Z. Lu, B. L. Conley and T. J. Williams, Organometallics, 2012, 31, 6705–6714 CrossRef CAS PubMed; (b) X. Zhang, Z. Lu, L. K. Foellmer and T. J. Williams, Organometallics, 2015, 34, 3732 CrossRef CAS.
- Re: Y. Jiang and H. Berke, Chem. Commun., 2007, 3571 RSC.
- Fe: J. R. Vance, A. P. M. Robertson, K. Lee and I. Manners, Chem. – Eur. J., 2011, 17, 4099 CrossRef CAS PubMed.
- Rh: (a) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424 CrossRef CAS PubMed; (b) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, Chem. Commun., 2001, 962 RSC; (c) J. L. Fulton, J. C. Linehan, T. Autrey, M. Balasubramanian, Y. Chen and N. K. Szymczak, J. Am. Chem. Soc., 2007, 129, 11936 CrossRef CAS PubMed; (d) R. Rousseau, G. K. Schenter, J. L. Fulton, J. C. Linehan, M. H. Engelhard and T. Autrey, J. Am. Chem. Soc., 2009, 131, 10516 CrossRef CAS PubMed.
- D. Himmelbauer, F. Müller, C. Schweinzer, F. Casas, B. Pribanic, G. Le Corre, D. Thöny, M. Trincado and H. Grützmacher, Chem. Commun., 2024, 60, 885 RSC.
- (a) P. Jurt, O. G. Salnikov, T. L. Gianetti, N. V. Chukanov, M. G. Baker, G. Le Corre, J. E. Borger, R. Verel, S. Gauthier, O. Fuhr, K. V. Kovtunov, A. Fedorov, D. Fenske, I. V. Koptyug and H. Grützmacher, Chem. Sci., 2019, 10, 7937 RSC; (b) C. Schweinzer, P. Coburger and H. Grützmacher, Adv. Sci., 2024, 2400072 CrossRef CAS PubMed.
- (a) T. Lorenz, A. Lik, F. A. Plamper and H. Helten, Angew. Chem., Int. Ed., 2016, 55, 7236 CrossRef CAS PubMed; (b) A. W. Baggett, F. Guo, B. Li, S.-Y. Liu and F. Jäkle, Angew. Chem., Int. Ed., 2015, 54, 11191 CrossRef CAS PubMed; (c) L. Türker, Polycyclic Aromat. Compd., 2012, 32, 61 CrossRef; (d) N. Otero, K. E. El-kelany, C. Pouchan, M. Rérat and P. Karamanis, Phys. Chem. Chem. Phys., 2016, 18, 25315 RSC; (e) N. Otero, P. Karamanis, K. E. El-Kelany, M. Rérat, L. Maschio, B. Civalleri and B. Kirtman, J. Phys. Chem. C, 2017, 121, 709 CrossRef CAS; (f) T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, J. Am. Chem. Soc., 2011, 133, 18614 CrossRef CAS PubMed; (g) A. Wakamiya, T. Ide and S. Yamaguchi, J. Am. Chem. Soc., 2005, 127, 14859 CrossRef CAS PubMed; (h) N. Kalashnyk, P. Ganesh Nagaswaran, S. Kervyn, M. Riello, B. Moreton, T. S. Jones, A. De Vita, D. Bonifazi and G. Costantini, Chem. – Eur. J., 2014, 20, 11856 CrossRef CAS PubMed.
- For previous work on the synthesis of functionalized borazines as precursors of BCN polymers, see: (a) J. Haberecht, A. Krummland, F. Breher, B. Gebhardt, H. Rüegger, R. Nesper and H. Gruetzmacher, Dalton Trans., 2003, 2126 RSC; (b) J. Haberecht, F. Krumeich, R. Nesper and H. Gruetzmacher, Chem. Mater., 2004, 16, 418 CrossRef CAS; (c) J. Haberecht, R. Nesper and H. Gruetzmacher, Chem. Mater., 2005, 17, 2340 CrossRef CAS.
- (a) H. A. Kalviri, F. Gärtner, G. Ye, I. Korobkov and R. T. Baker, Chem. Sci., 2015, 6, 618 RSC; (b) S. Bhunya, P. M. Zimmerman and A. Paul, ACS Catal., 2015, 5, 3478 CrossRef CAS.
- W. J. Shaw, J. C. Linehan, N. K. Szymczak, D. J. Heldebrant, C. Yonker, D. M. Camaioni, R. T. Baker and T. Autrey, Angew. Chem., Int. Ed., 2008, 47, 7493 CrossRef CAS PubMed.
- V. Kumar, B. Roy and P. Sharma, Int. J. Hydrogen Energy, 2019, 44, 22022 CrossRef CAS.
- A. Kumar, N. A. Beattie, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2016, 55, 6651 CrossRef CAS PubMed.
- (a) D. Han, F. Anke, M. Trose and T. Beweries, Coord. Chem. Rev., 2019, 380, 260 CrossRef CAS; (b) A. Rossin and M. Peruzzini, Chem. Rev., 2016, 116, 8848 CrossRef CAS PubMed.
- A. E. W. Ledger, C. E. Ellul, M. F. Mahon, J. M. J. Williams and M. K. Whittlesey, Chem. – Eur. J., 2011, 17, 8704 CrossRef CAS PubMed.
- (a) T. M. Douglas, A. B. Chaplin and S. A. Weller, J. Am. Chem. Soc., 2008, 130, 14432 CrossRef CAS PubMed; (b) A. B. Chaplin and A. S. Weller, Angew. Chem., Int. Ed., 2010, 49, 581 CrossRef CAS PubMed.
- (a) R. D. Adams, Chem. Rev., 1989, 89, 1703 CrossRef CAS; (b) D. J. Cardin, B. Cetinkaya and M. F. Lappert, Chem. Rev., 1972, 72, 545 CrossRef CAS.
- M. Leutzsch, L. M. Wolf, P. Gupta, M. Fuchs, W. Thiel, C. Farès and A. Fürstner, Angew. Chem., 2015, 127, 12608 CrossRef PubMed.
- M. Trincado and H. Grützmacher, Cooperating Ligands in Catalysis, in Cooperative Catalysis, ed. R. Peters, Wiley-VCH, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01520g |
| This journal is © The Royal Society of Chemistry 2024 |