
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lithium chloride selective ion-pair recognition by heteroditopic [2]rotaxanes†
Vihanga K.
Munasinghe‡
a,
Hui Min
Tay‡
 a,
Dilhan
Manawadu
b,
Jessica
Pancholi
a,
Zongyao
Zhang
c and
Paul D.
Beer
*a
a,
Dilhan
Manawadu
b,
Jessica
Pancholi
a,
Zongyao
Zhang
c and
Paul D.
Beer
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory Mansfield Road, Oxford OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk
bDepartment of Chemistry, Physical and Theoretical Chemistry Laboratory, University of Oxford, Oxford, OX1 3QZ, UK
cChemistry Department, King's College London, Britannia House, London SE1 1DB, UK
First published on 2nd August 2024
Abstract
The first heteroditopic [2]rotaxane host systems capable of strong and selective binding of lithium chloride ion-pair species are described. Importantly, a cooperative ‘switch on’ mechanism was found to operate, in which complexation of a lithium metal cation enhances the halide anion affinity of the rotaxanes via a combination of favourable proximal electrostatic and preorganised allosteric effects. The mechanically bonded rotaxane host design features a macrocycle component possessing a 2,6-dialkoxy pyridyl cation binding motif and an isophthalamide anion binding group, as well as an axle component functionalised with either a halogen bonding (XB) iodotriazole or hydrogen bonding (HB) prototriazole moiety. Extensive quantitative 1H NMR titration studies in CD3CN/CDCl3 solvent mixtures determined enhanced ion-pair binding affinities for lithium halides over the corresponding sodium or potassium halide salts, with the axle prototriazole-containing HB rotaxane in particular demonstrating a marked selectivity for lithium chloride. Solid-state X-ray crystallographic studies and computational DFT investigations provide evidence for a [2]rotaxane host axle-separated ion-pair binding mode, in which complementary cation and anion binding motifs from both the macrocycle and axle components act convergently to recognise each of the charged guest species.
Introduction
The ubiquity and importance of charged species in a myriad of chemical, biological, industrial and environmental processes1–6 has stimulated ever increasing interest in the development of molecular receptors for their selective recognition. In particular, heteroditopic receptors capable of simultaneously binding cations and anions have demonstrated considerable advantages over their monotopic counterparts due to favourable allosteric and electrostatic effects associated with ion-pair binding.7–12 The use of mechanically interlocked molecules (MIMs) as molecular receptors has been effective in enhancing guest binding by exploiting the size and shape of their unique topological 3D binding cavities for complementary target guest recognition.13–17 Surprisingly, efforts to combine the advantages of MIMs and heteroditopicity are rare,18–22 with the majority of examples utilising hydrogen bonding (HB) donor motifs for anion binding, while the potential of halogen bonding (XB)-mediated anion recognition remains underexplored.23,24There is a growing need for receptors capable of recognising lithium salts due to the widespread use of lithium ion batteries25 and the potentially toxic effects of high concentrations of lithium in water sources.26,27 However, selective binding of lithium salts is challenging due to their high lattice enthalpies, with only a handful of acyclic and macrocyclic receptors capable of binding lithium chloride (LiCl) ion-pairs.28–32,41–43
We have recently reported a family of halogen bonding (XB) heteroditopic rotaxanes which selectively bind lithium bromide and iodide ion-pairs.33 However, the interlocked hosts were unable to recognise LiCl. Building on this work, herein, we present the synthesis and ion-pair binding properties of two new XB and HB heteroditopic rotaxanes (Fig. 1), wherein the integration of an isophthalamide anion binding motif into a dialkoxypyridyl containing macrocycle component and an XB iodotriazole or HB protic triazole axle facilitates the strong binding of lithium halide ion-pairs, notably including LiCl. The combination of extensive 1H NMR titration experiments, single-crystal X-ray structure analysis and computational DFT theoretical calculations provides substantial evidence for an axle-separated lithium halide ion-pair rotaxane binding mode (Fig. 1). Both rotaxanes display strong LiX (X = Cl, Br, I) binding, making them the first interlocked structures capable of selectively binding LiCl ion-pairs. Importantly, the HB axle rotaxane 9 showed notable selectivity for LiCl with respect to lithium halides and alkali metal chlorides, including NaCl and KCl.
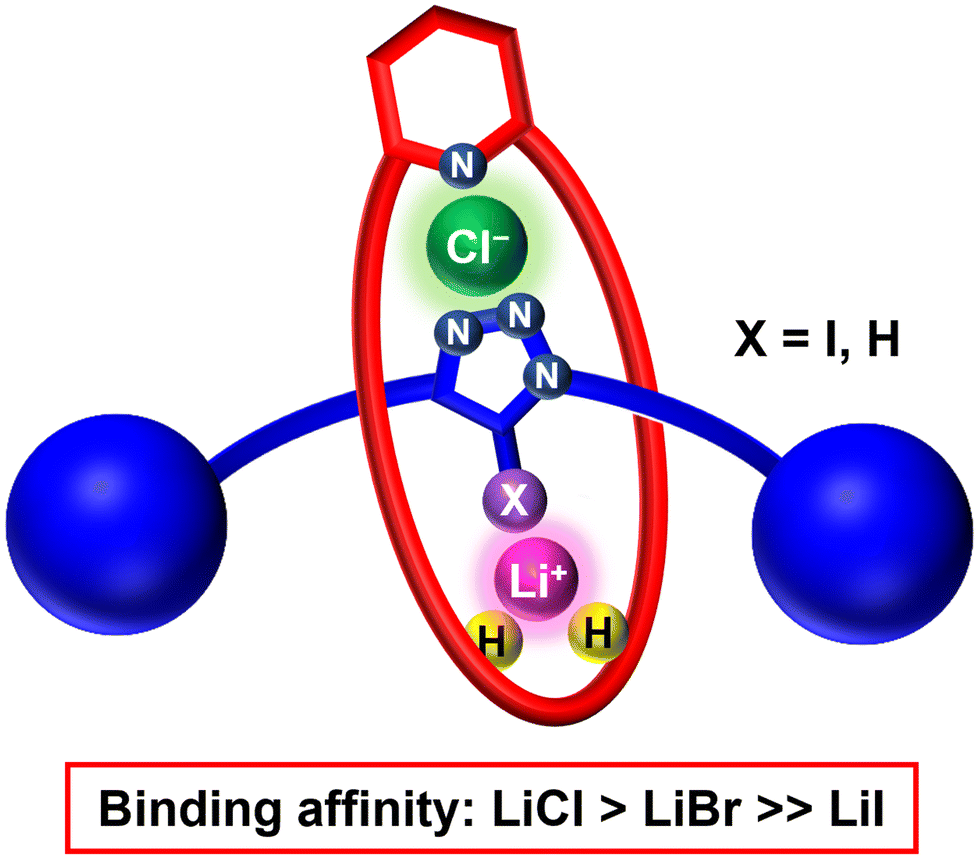 | ||
| Fig. 1 Cartoon representation of heteroditopic [2]rotaxanes for selective LiCl binding. | ||
Results and discussion
Synthesis and characterisation of [2]rotaxanes
The target isophthalamide-functionalised macrocycle 6 was prepared (Scheme 1a), and subsequently employed in a Cu(I)-catalysed azide–alkyne cycloaddition active metal template (CuAAC-AMT) [2]rotaxane synthetic reaction34,35 in conjunction with azide- and (iodo)alkyne-functionalised stopper precursors 7 and 8 to afford the heteroditopic [2]rotaxanes 9 and 10 in excellent yields of 77% and 74% respectively (Scheme 1b).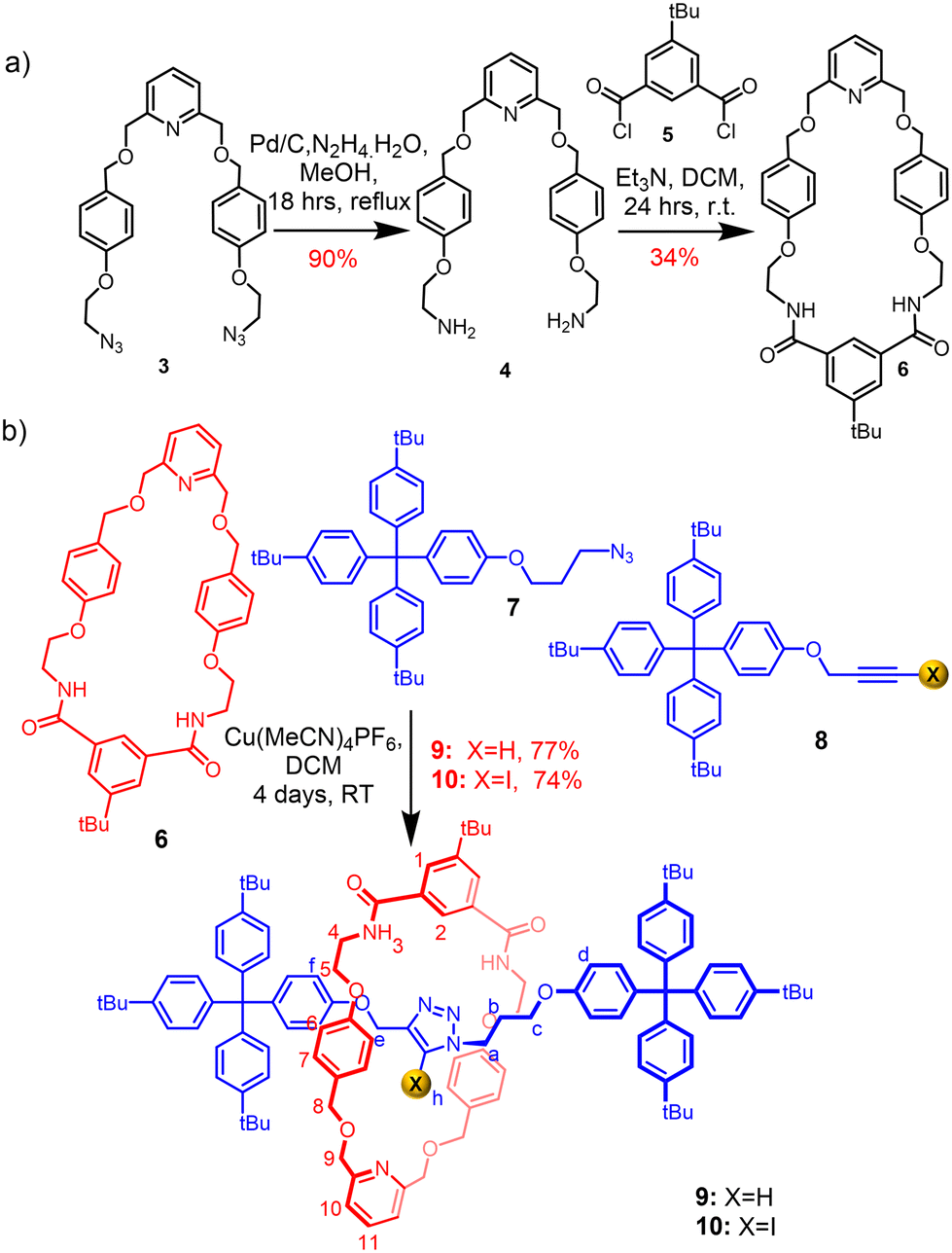 | ||
| Scheme 1 Synthesis of (a) macrocycle 6; (b) HB and XB [2]rotaxanes 9 and 10. | ||
Evidence for the interlocked nature of the [2]rotaxanes was obtained by comparing their 1H NMR spectra to those of the non-interlocked components (Fig. 2 and S1.10†), which revealed proton movements indicative of mechanical bond formation. The internal isophthalamide proton H2 and amide proton H3 of the macrocycle shifted downfield in the 1H NMR spectrum of the rotaxane relative to the free macrocycle, presumably due to HB interactions with the basic nitrogen atoms of the triazole axle. In addition, diagnostic upfield shifts were observed for the macrocycle aryl protons H6 and H7, indicative of aromatic donor–acceptor interactions between the electron-rich macrocycle aryl groups and the relatively electron-deficient axle triazole unit. Interestingly, the signal for methylene protons H8, which appeared as a singlet in the macrocycle, splits into a multiplet in the rotaxane spectrum, which was attributed to the loss of symmetry of the methylene protons upon formation of the interlocked structure. Further confirmation of the interlocked topology of the products was obtained by 1H–1H ROESY spectroscopy experiments, which showed cross-peaks arising from through-space interactions between the interlocked axle and the macrocycle components (Fig. S1.15†). Notably, weak cross-peaks were observed between triazole proton Hh of the axle and methylene proton signals H8 and H9 of the axle, which further supports the notion that the triazole moiety of the axle is oriented in a manner that facilitates intercomponent hydrogen bonding interactions between the triazole nitrogen atoms and isophthalamide NH donors of the macrocycle.
 | ||
| Fig. 2 Stacked 1H NMR spectra of prototriazole axle (top), rotaxane 9 (middle) and macrocycle 6 (bottom) (CDCl3, 500 MHz, 298 K). | ||
1H NMR binding studies
The alkali metal cation binding properties of rotaxane 9 were first studied by 1H NMR titration experiments in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 CD3CN/CDCl3 (section S3.2†). LiClO4 was used as the Li+ cation source while for Na+ and K+ tetrakis[3,5-bis(trifluoromethyl)phenyl] borate (BArF−) salts were employed due to the poor solubility of NaClO4 and KClO4. In a typical titration experiment, aliquots of the alkali metal salt were progressively added to a solution of the rotaxane. The addition of LiClO4 and NaBArF induced a notable downfield shift in macrocycle pyridyl proton H11, as well as pyridyl-proximal methylene protons H9 and H8, indicative of metal cation coordinating to the 2,6-dialkoxypyridyl motif. In addition, the axle methylene proton He undergoes an upfield shift, suggesting participation of the triazole nitrogen and adjacent oxygen atom in binding to the metal cation (Fig. S3.10†). In contrast, no significant changes were observed upon addition of KBArF, suggesting no binding of the larger potassium cation. Importantly, the protons near the anion binding site did not exhibit any significant perturbations, indicating that both BArF− and ClO4− are non-coordinating anions.
:7 CD3CN/CDCl3 (section S3.2†). LiClO4 was used as the Li+ cation source while for Na+ and K+ tetrakis[3,5-bis(trifluoromethyl)phenyl] borate (BArF−) salts were employed due to the poor solubility of NaClO4 and KClO4. In a typical titration experiment, aliquots of the alkali metal salt were progressively added to a solution of the rotaxane. The addition of LiClO4 and NaBArF induced a notable downfield shift in macrocycle pyridyl proton H11, as well as pyridyl-proximal methylene protons H9 and H8, indicative of metal cation coordinating to the 2,6-dialkoxypyridyl motif. In addition, the axle methylene proton He undergoes an upfield shift, suggesting participation of the triazole nitrogen and adjacent oxygen atom in binding to the metal cation (Fig. S3.10†). In contrast, no significant changes were observed upon addition of KBArF, suggesting no binding of the larger potassium cation. Importantly, the protons near the anion binding site did not exhibit any significant perturbations, indicating that both BArF− and ClO4− are non-coordinating anions.
The association constants for Li+ and Na+ were determined via a global Bindfit36 analysis of the binding isotherms generated by monitoring the movements of multiple proton signals proximal to the cation binding site, highlighting a notable preference for Li+ over weakly bound Na+ (K(Li) = 312 M−1, K(Na) = 69 M−1). This was attributed to the higher charge density and better size complementarity of the smaller lithium cation for the interlocked binding site.
The halide anion binding properties of rotaxanes 9 and 10 were subsequently investigated in the same solvent mixture 3:7 CD3CN/CDCl3. Addition of the halide anions as their tetrabutylammonium (TBA) salts did not induce any significant proton perturbations, suggesting that the interlocked receptors were incapable of binding halides in this solvent system. The weak halide binding of the neutral rotaxanes was attributed to the strong intercomponent HB interactions between the isophthalamide group and the triazole nitrogen atoms outcompeting intermolecular host–guest HB interactions.
Attention then turned to investigating the ion-pair binding properties of the heteroditopic rotaxanes. To this end, the rotaxanes were pre-complexed with 1 equivalent of LiClO4 in 3:7 CD3CN/CDCl3, to which increasing equivalents of TBAX salts (X = Cl, Br, I) were administered. The formation of a Li+-bound rotaxane complex (9/10·Li+) was evidenced by perturbations in the proton signals proximal to the cation binding site (H11, H9, He), which are consistent with the observed peak movements in the lithium cation titrations of the rotaxanes. In stark contrast to the anion binding studies conducted on the metal free neutral rotaxanes, addition of TBA halide salts to 9/10·Li+ caused protons in the vicinity of the respective rotaxane's macrocycle component isophthalamide anion binding site H2, H3, H4, H5 to undergo significant perturbations indicative of halide complexation (Fig. 3, S3.1 and S3.5†). Additionally, the axle triazole proton Hh in 9·Li+ broadens and shifts progressively downfield with increasing anion concentration, providing evidence for participation of the axle triazole HB donor group in anion binding.
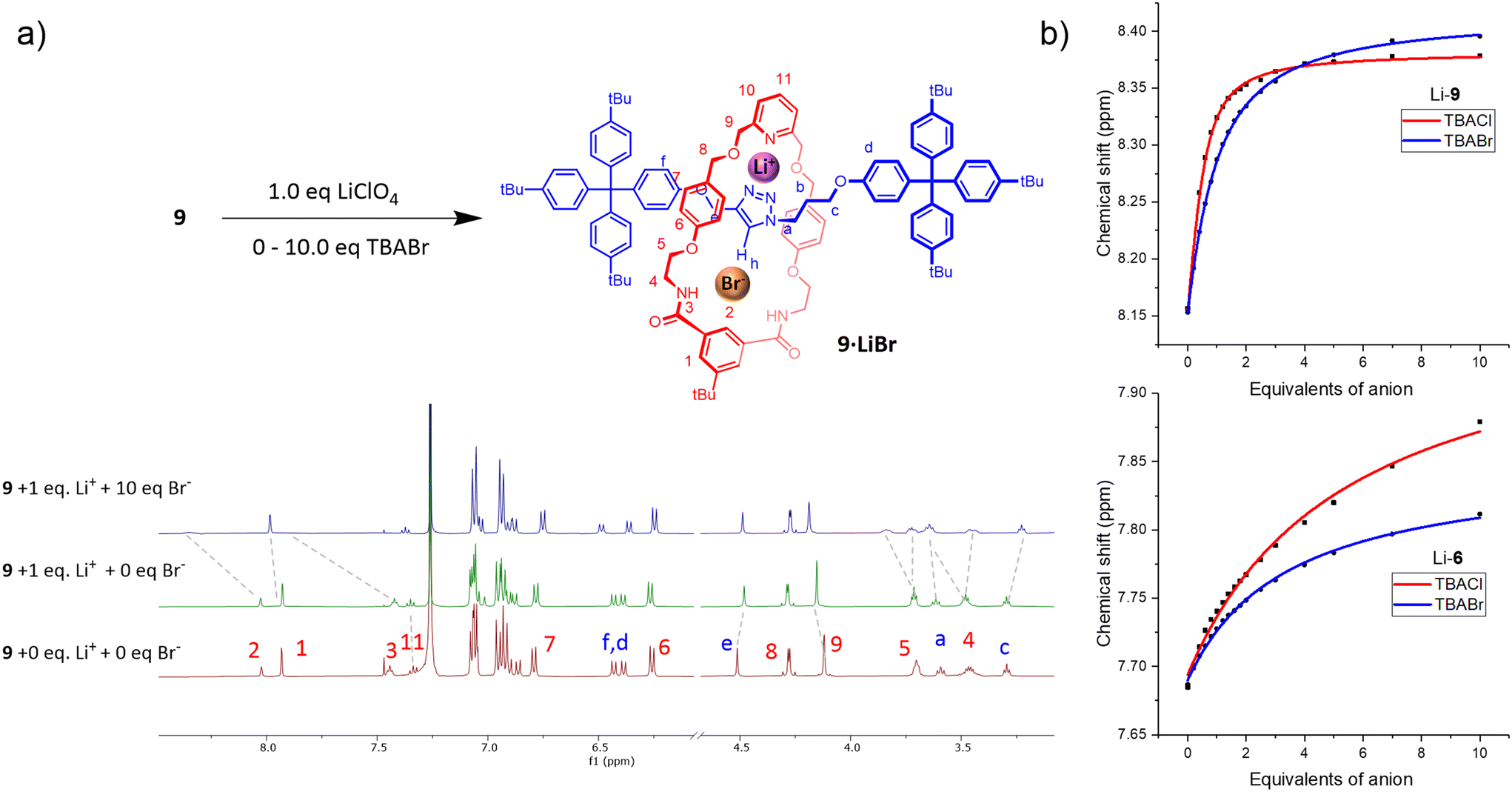 | ||
| Fig. 3 (a) Ion-pair binding studies of rotaxane 9, showing: (a) stacked 1H NMR spectra of rotaxane 9 in the absence of guest ions (bottom), after addition of 1 eq. Li+ (middle) and 10 eq. Br− (3:7 CD3CN/CDCl3, 500 MHz, 298 K); (b) binding isotherms of 9·Li+ (top) and macrocycle 6 (bottom), upon progressive addition of halide anions, constructed by monitoring chemical shift perturbations of isophthalamide proton H2. | ||
The notable 1H NMR perturbations observed in the ion-pair titrations of 9/10·Li+ indicate that the halide binding affinities of both rotaxanes are ‘switched on’ by pre-complexation to Li+, likely due to favourable proximal electrostatic interactions between the co-bound ions as well as a co-conformational change in the mechanically interlocked host upon Li+ complexation, as cation coordination by the nitrogen atoms of the triazole conceivably disrupts their intramolecular HB interactions with the macrocycle isophthalamide NH groups, allowing the latter to participate in halide anion binding. Importantly, the proton signals perturbed by Li+ binding did not return to their original peak positions upon addition of the halide anions, confirming that LiX ion-pair binding to the receptor outcompetes salt recombination.
The apparent halide anion association constants of 9 and 10 in the presence of 1 eq. LiClO4 were determined by a global Bindfit36 analysis of monitoring the 1H NMR signals (H1, H3, Hf, He) near the anion binding site with increasing halide concentration (Table 1). It is noteworthy that due to the modest Li+ binding affinity (Ka = 312 M−1) of the rotaxanes, only a fraction of the rotaxanes (approx. 20%, see ESI section 3.2† for details) exists as the Li+-bound complexes 9·Li+ and 10·Li+ at the beginning of the titration, therefore the determined Kapp values necessarily underestimate the ‘true’ anion association constant of the rotaxane-Li+ complexes. Nonetheless, the determined Kapp values of both rotaxanes indicated high affinities for lithium halide ion-pair binding in 3:7 CD3CN/CDCl3. The apparent association constants for LiCl and LiBr in 3:7 CD3CN/CDCl3 are >6-fold higher than that of LiI, attributed to the lower basicity of iodide as well as size complementarity between the rotaxanes’ anion binding cavity and the smaller halides. As the LiCl binding constants were too high to be reliably determined by 1H NMR (>104 M−1) in 3:7 CD3CN/CDCl3, the binding studies were repeated in a more competitive 4:6 CD3CN/CDCl3 solvent system (Fig. S3.2 and S3.6†). In this solvent system, HB rotaxane 9·Li+ displayed >4-fold selectivity for LiCl over LiBr, and even greater selectivity over LiI. The marked preference for LiCl ion-pairs is particularly impressive given its high lattice energy, which typically favours salt recombination over ion-pair binding. Analogous 1H NMR ion-pair binding studies conducted on 9·Na+ in 4:6 CD3CN/CDCl3 showed no binding of NaCl (Fig. S3.3†), crucially demonstrating a remarkable selectivity of the rotaxane for binding LiCl over other alkali metal halide ion-pairs.
| Cation | Anion | R·9 | R·10 | R·9 | R·10 | M·6 |
|---|---|---|---|---|---|---|
| 3:7 CD3CN/CDCl3 |
4:6 CD3CN/CDCl3 |
|||||
|
a
K
a values were calculated using Bindfit software using 1:1 binding model. Errors (%) are in parenthesis. All anions were added as their TBA salts. [Receptor] = 1 mM, T = 298 K.
b
1H NMR perturbations too small to reliably determine Ka values (<0.05 ppm).
c Binding studies not conducted.
|
||||||
| Li+ | Cl− | >104 | >104 | >104 | 5081(8) | 289(7) |
| Li+ | Br− | 9291(10) | 7496(10) | 2144(3) | 2480(4) | 439(5) |
| Li+ | I− | 1408(3) | 1117(3) | |||
| H+ | Cl− | 8011(7) | >104 | |||
| H+ | Br− | 3291(2) | >104 | |||
| H+ | I− | 725(2) | 3712(7) | |||
Macrocycle 6 displayed significantly diminished lithium halide association constants relative to rotaxanes 9 and 10, alongside a slight preference for LiBr over LiCl binding (Table 1 and Fig. S3.9†), highlighting the salient role of the interlocked rotaxane topology in enhancing the ion-pair binding affinities and dictating the LiCl selectivity of the receptors.
The lithium precomplexed XB rotaxane 10·Li+ displayed lower anion association constant values relative to the HB rotaxane 9·Li+ in 3:7 CD3CN/CDCl3 solvent mixtures, and also with chloride in 4:6 CD3CN/CDCl3 (Table 1). This contrasts with previous comparative anion binding studies conducted on monotopic XB iodotriazole donor versus HB prototriazole-containing acyclic, macrocyclic and MIM receptor systems, which commonly display stronger XB-mediated halide anion recognition.37 To investigate whether MIM lithium cation complexation involving the nitrogen donor atom of the iodotriazole axle component of rotaxane 10·Li+ may be responsible, studies were conducted on protonated rotaxanes 9·H+/10·H+ using HBF4 as the proton source, in which the small H+ cation is expected to interact primarily with the pyridyl nitrogen donor over the less basic triazole nitrogen donor.
Addition of 1 eq. of HBF4 to the rotaxanes in 4:6 CD3CN/CDCl3 caused significant downfield shifts in the macrocycle protons (H11, H10, H8, and H9) indicative of protonation occurring at the pyridyl motif as anticipated. The subsequent addition of TBA halide salts led to shifts in macrocycle aryl and amide protons H2 and H3, as well as axle triazole proton Hh in rotaxane 9, indicating the involvement of both the axle and the macrocycle in halide binding (Fig. S3.4 and S3.7†).
Halide anion binding constants determined by fitting the chemical shifts of protons H2 and H3 to a global 1:1 stoichiometric host–guest binding model in Bindfit (Table 1) revealed protonated XB rotaxane 10·H+ displayed significantly larger halide binding constant magnitudes than HB rotaxane 9·H+. This observation, which displays the opposite trend to that observed in the lithium ion-precomplexed rotaxanes, indicates the lithium cation coordination binding mode of 10·Li+ indeed affects XB donor capability. The ion-pair binding modes of the protonated and lithium-complexed rotaxanes were therefore investigated in greater detail via solid state crystallographic analysis and computational study (vide infra).
Solid-state X-ray crystallographic structural analysis§
Single crystal X-ray diffraction analysis of 10·H+Cl− obtained from the chloride anion 1H NMR titration investigation provided solid state evidence for an axle-separated ion-pair binding mode. Analysis of the structure (Fig. 4a) shows a 1:1:1 stoichiometric host–cation–anion complex. The Cl− guest is located in the predicted rotaxane's anion binding site, where it forms HB interactions with amide groups of the macrocycle component. Importantly, the axle iodotriazole XB donor is oriented in a suitable conformation to form a strong XB interaction with the chloride guest, as demonstrated by a C–I⋯Cl− bond angle of 173° (consistent with the stringent linear directionality of XB interactions)38 and a short I⋯Cl distance (79% of the sum of the van der Waals radii of chloride and iodine).
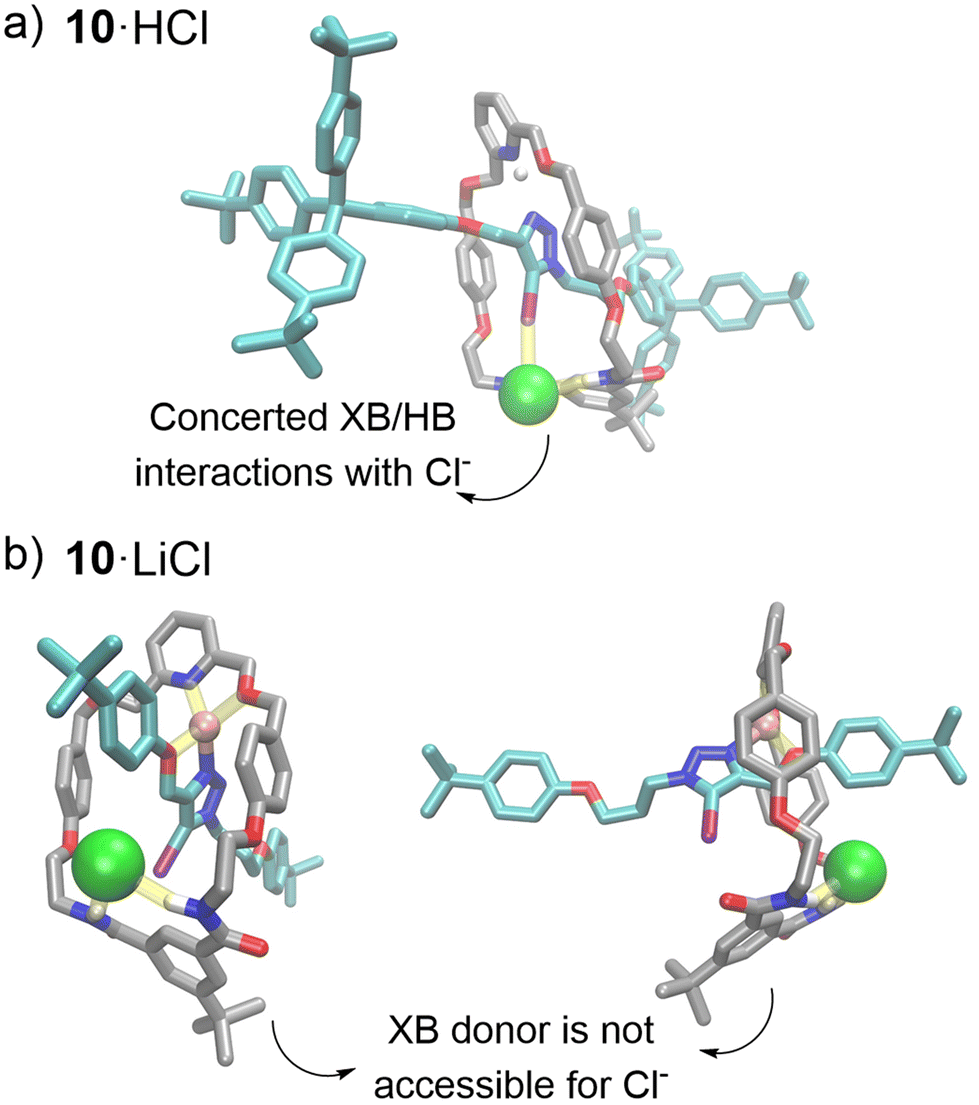 | ||
| Fig. 4 Structures of ion-pair bound rotaxane 10; (a) crystal structures of axle-separated HCl-bound 10 (b) front view and side view of DFT optimised LiCl-bound rotaxane 10, colour code of atoms: H (white), O (red), N (blue), I (purple), Cl (green), Li (pink), C(axle) (teal), C(MC) (grey). Non-covalent host–guest interactions are shown in yellow. | ||
Computational studies
Density Functional Theory (DFT) studies of the lithium complexed rotaxanes reveal that in addition to macrocycle pyridyl coordination, Li+ interacts with both the triazole nitrogen and oxygen in the respective rotaxane's axle component, restricting the rotation of the axle triazole moiety (Fig. 4b, section S4†). Therefore, in the geometry-optimised structure of 10·Li+, the XB iodine donor atom of the axle is unable to adopt the optimal linear geometry required to form XB interactions with the halide guest. In contrast, in the optimised structure of LiCl-bound HB rotaxane 9, an overlap of the ionic and van der Waals radii of the anion and the triazole HB donor was observed (Fig. S4.2†). This is due in part to the reduced steric bulk of the HB donor atom, as well as the less stringent requirement for a linear donor–acceptor binding geometry in HB interactions relative to XB. This enables the HB donor in the axle of rotaxane 9 to participate in Cl− binding despite the sub-optimal non-linear binding geometry. However, no overlap of van der Waals and ionic radii of the triazole HB donor and Br−/I− was observed, which may be attributed to the steric inaccessibility of the larger halide anions to the interlocked binding cavity (Fig. S4.1/S4.2 and Table S4.1†). These findings are consistent with the experimental 1H NMR binding studies, where LiCl binding to the HB rotaxane 9 was significantly stronger than the XB rotaxane 10, compared to LiBr and LiI binding, making rotaxane 9 highly selective for LiCl.In the case of the protonated rotaxanes, DFT studies predicted the H+ cation to interact primarily with the basic pyridyl nitrogen of the macrocycle. Hence, unlike 10·Li+, the triazole group of the axle in 10·H+ is able to adopt the geometry required to form linear XB interactions with anionic guests, as shown in the crystal structure 10·HCl (Fig. 3a, S4.3 and S4.4†). This is concordant with the solution-state 1H NMR titrations, which indicate stronger halide anion binding to protonated XB rotaxane 10·H+ compared to HB rotaxane 9·H+.
Electrostatic potential maps were used to investigate the role of electrostatic interactions in the ‘switching on’ of the anion binding affinities of the rotaxanes by pre-complexation of a Li+ and H+ cation (Fig. S4.6†). Binding of Li+ and H+ was found to increase the electrostatic potential of axle HB/XB donors, demonstrating the potential of cation complexation to augment the anion binding potency of the HB/XB motifs via electrostatic effects.
Conclusions
In conclusion, XB and HB heteroditopic rotaxanes 9 and 10 were synthesised and their cation, anion and ion-pair binding properties studied via1H NMR titrations, X-ray crystallography and computational studies. Binding of Li+ was found to ‘switch on’ the halide anion binding affinities of both rotaxanes, making rotaxanes 9 and 10, to the best of our knowledge, the first reported heteroditopic MIMs capable of overcoming the high lattice enthalpy of LiCl. In particular, rotaxane 9 demonstrated remarkable selectivity for LiCl ion-pairs with respect to other lithium halides and group I metal chlorides, highlighting the potential efficacy of exploiting the confined environments of interlocked binding cavities to engineer selectivity for smaller cationic and anionic guests. The differences in the ion-pair binding affinities of rotaxanes 9 and 10 were rationalised by computational and crystallographic studies, which revealed that Li+ complexation to 10 rendered the axle iodotriazole XB donor motif unable to optimally participate in a linear XB⋯X− anion binding fashion, whereas protonation of the pyridyl group in 10 facilitated the formation of concerted XB/HB–anion interactions involving both the macrocycle and axle anion binding motifs. This was consistent with the significantly enhanced halide anion binding affinities of the protonated XB rotaxane 10·H+ relative to both 9·H+ and 10·Li+, which may serve to inform the design of future interlocked heteroditopic receptors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. K. M thanks the E. P. A Cephalosporin Scholarship fund, Linacre College for financial support. H. M. T acknowledges the Clarendon Fund and the Oxford Australia Scholarships Fund for a postgraduate research scholarship. D. M thanks the EPSRC Centre for Doctoral Training, Theory and Modelling in Chemical Sciences, under Grant No. EP/L015722/1 and Linacre College for a Carolyn and Franco Giantruco Scholarship and the Department of Chemistry, University of Oxford for financial support. J. P thanks the EPSRC for postdoctoral funding (EPSRC Grant No. EP/P033490/1). Z. Z. thanks the University of Oxford and China Scholarship Council for postgraduate studentship funding. Authors would like to acknowledge the use of the University of Oxford Advanced Research Computing (ARC) facility in carrying out this work.References
- M. P. Anderson, R. J. Gregory, S. Thompson, D. W. Souza, S. Paul, R. C. Mulligan, A. E. Smith and M. J. Welsh, Science, 1991, 253, 202 CrossRef CAS PubMed.
- V. H. Smith and D. W. Schindler, Trends Ecol. Evol., 2009, 24, 201–207 CrossRef PubMed.
- J. H. P. Watson and D. C. Ellwood, Nucl. Eng. Des., 2003, 226, 375–385 CrossRef CAS.
- F. Delange, Thyroid, 1994, 4, 107–128 CrossRef CAS PubMed.
- J. L. Way, Annu. Rev. Pharmacol. Toxicol., 1984, 24, 451–481 CrossRef CAS PubMed.
- P. K. Dasgupta, J. V. Dyke, A. B. Kirk and W. A. Jackson, Environ. Sci. Technol., 2006, 40, 6608–6614 CrossRef CAS PubMed.
- A. J. Taylor, A. Docker and P. D. Beer, Chem. – Asian J., 2023, 18, e202201170 CrossRef CAS PubMed.
- J. L. Sessler, S. K. Kim, D. E. Gross, C.-H. Lee, J. S. Kim and V. M. Lynch, J. Am. Chem. Soc., 2008, 130, 13162–13166 CrossRef CAS PubMed.
- A. J. McConnell and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 5052–5061 CrossRef CAS PubMed.
- B. Qiao, A. Sengupta, Y. Liu, K. P. McDonald, M. Pink, J. R. Anderson, K. Raghavachari and A. H. Flood, J. Am. Chem. Soc., 2015, 137, 9746–9757 CrossRef CAS PubMed.
- Q. He, G. I. Vargas-Zúñiga, S. H. Kim, S. K. Kim and J. L. Sessler, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS PubMed.
- A. J. McConnell, A. Docker and P. D. Beer, ChemPlusChem, 2020, 85, 1824–1841 CrossRef CAS PubMed.
- K. M. Bąk, K. Porfyrakis, J. J. Davis and P. D. Beer, Mater. Chem. Front., 2020, 4, 1052–1073 RSC.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC.
- E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128–5142 RSC.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- J. T. Wilmore and P. D. Beer, Adv. Mater., 2024, 36, 2309098 CrossRef CAS PubMed.
- R. C. Knighton and P. D. Beer, Chem. Commun., 2014, 50, 1540–1542 RSC.
- M. Denis, L. Qin, P. Turner, K. A. Jolliffe and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 5315–5319 CrossRef CAS PubMed.
- D.-H. Li and B. D. Smith, J. Org. Chem., 2019, 84, 2808–2816 CrossRef CAS PubMed.
- J. R. Romero, G. Aragay and P. Ballester, Chem. Sci., 2017, 8, 491–498 RSC.
- A. Arun, A. Docker, H. M. Tay and P. D. Beer, Chem. – Eur. J., 2023, 29, e202301446 CrossRef CAS PubMed.
- H. M. Tay, Y. C. Tse, A. Docker, C. Gateley, A. L. Thompson, H. Kuhn, Z. Zhang and P. D. Beer, Angew. Chem., Int. Ed., 2023, 62, e202214785 CrossRef CAS PubMed.
- H. M. Tay, A. Docker, Y. C. Tse and P. D. Beer, Chem. – Eur. J., 2023, 29, e202301316 CrossRef PubMed.
- G. Martin, L. Rentsch, M. Höck and M. Bertau, Energy Storage Mater., 2017, 6, 171–179 CrossRef.
- L. A. Kszos and A. J. Stewart, Ecotoxicology, 2003, 12, 439–447 CrossRef CAS PubMed.
- F. Harari, A. Åkesson, E. Casimiro, Y. Lu and M. Vahter, Environ. Res., 2016, 147, 1–7 CrossRef CAS PubMed.
- J. M. Mahoney, A. M. Beatty and B. D. Smith, Inorg. Chem., 2004, 43, 7617–7621 CrossRef CAS PubMed.
- Q. He, Z. Zhang, J. T. Brewster, V. M. Lynch, S. K. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 9779–9782 CrossRef CAS PubMed.
- Q. He, N. J. Williams, J. H. Oh, V. M. Lynch, S. K. Kim, B. A. Moyer and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 11924–11928 CrossRef CAS PubMed.
- Z. Kokan and M. J. Chmielewski, J. Am. Chem. Soc., 2018, 140, 16010–16014 CrossRef CAS PubMed.
- Y. C. Tse, A. Docker, Z. Zhang and P. D. Beer, Chem. Commun., 2021, 57, 4950–4953 RSC.
- V. K. Munasinghe, J. Pancholi, D. Manawadu, Z. Zhang and P. D. Beer, Chem. – Eur. J., 2022, 28, e202201209 CrossRef CAS PubMed.
- V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed.
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS.
- J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- H. Piotrowski, K. Polborn, G. Hilt and K. Severin, J. Am. Chem. Soc., 2001, 123(11), 2699–2700 CrossRef CAS PubMed.
- H. Piotrowski and K. Severin, Proc. Natl. Acad. Sci., 2002, 99(8), 4997–5000, DOI:10.1073/pnas.062663899.
- H. Piotrowski, G. Hilt, A. Schulz, P. Mayer, K. Polborn and K. Severin, Chem. – Eur. J., 2001, 7(15), 3196–3208 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2164638. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01807a |
| ‡ These authors contributed equally to the work. |
| § Low temperature single crystals X-ray diffraction data were collected using an Oxford Diffraction Supernova X-ray diffractometer and reduced using CrysAlisPro. The structures were solved using SHELXS39 and refined using SHELXL.40. Full details are included in the accompanying ESI (CIF). The data can be obtained from the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service https://www.ccdc.cam.ac.uk/structures with deposition number 2164638.† |
| This journal is © The Royal Society of Chemistry 2024 |